
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effect of aluminum diethylphosphinate/sodium stearate modified vermiculite on flame retardant and smoke suppression properties of amino coatings†
Siwei Li‡
 ,
Jihu Wang‡,
Shaoguo Wen*,
Yabo Chen,
Jijia Zhang and
Changrui Wang
,
Jihu Wang‡,
Shaoguo Wen*,
Yabo Chen,
Jijia Zhang and
Changrui Wang
College of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, Shanghai, 201620, China. E-mail: sgwen1@sues.edu.cn
First published on 20th October 2021
Abstract
Various inorganic fillers are proved to be desirable synergists to improve the fire resistance of fire-retardant coatings. Herein, a functional filler (ANE) with flame retardant property was prepared by intercalating aluminum diethylphosphinate into microwave expanded vermiculite and grafting sodium stearate on its surface. The structure of ANE was fully characterized by FTIR, XRD, XPS and SEM analyses. Then ANE was applied to melamine modified urea-formaldehyde resin to produce fire-retardant coatings. The fire resistance test, TGA and cone calorimeter test demonstrate that ANE imparts great heat insulation, thermal stability, and flame retardancy to the coatings. Moreover, the introduction of ANE exhibits an excellent synergistic effect on reducing the heat release and smoke emission of the coatings. Specifically, with the addition of 3 wt% ANE, the heat release rate and smoke density grade of the coatings are decreased by 25.24% and 60.32%, respectively, compared to that without ANE. The excellent flame retardancy and smoke suppression performances of the coatings are mainly attributed to the formation of more cross-linking structures in the carbon layers, resulting in a more stable and compact char structure. In addition, the good hydrophobicity of ANE coatings can ensure the durability of flame retardancy.
1. Introduction
Damage and injuries from fire lead to enormous loss of life and property each year.1 The low strength of steel structures at high temperature and the inherent flammability of wood, fabric and polymers make it necessary to study fire retardants to improve safety and preserve lives. As pyrolysis is inevitable in extreme heat situations, it is not always realistic for flame retardants to completely negate ignition. The primary objectives of flame retardants are to impede pyrolysis and increase time to ignition, prevent flame spread and suppress the production of toxic smoke, which provides time for people to safely evacuate the premises.2The most typical intumescent flame retardant (IFR) used in coatings mainly consists of ammonium polyphosphate, pentaerythritol, and melamine.3–7 Meanwhile, various inorganic fillers such as montmorillonite, layered double hydroxides are exploited as synergistic flame retardants for IFR.8,9 Among them, the layered silicas show desirable synergistic effect in improving the thermal property and fire resistance of intumescent systems.4,10 Yan11 and Xu12 et al. found that the layered silicates including organically modified montmorillonite and layered double hydroxides were used as a highly efficient synergist for amino fire-retardant coatings. Vermiculite (EV) is a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 layered silicate with high layer charge density. The thermal stability and interlayer ion capacity of vermiculite is much higher than that of montmorillonite.13,14 Moreover, it can be expanded to 8–12 times of its original volume after calcination at high temperature. After expansion, vermiculite has good heat insulation with its tiny air interlayer.15 Natural vermiculite can be used as a fire-retardant synergist in some matrix. Ren16 et al. found that the formulation with 3 wt% exfoliated vermiculate had the best performance for decreasing heat release and smoke production parameters of flame-retardant polypropylene. Since intumescent fire-retardant coatings are normally used as decorative coatings and endure for a long time before a fire actually breaks out, the durability of the coatings is quite important.17 However, due to large surface area and abundant capillaries, vermiculite has strong water absorption.18 Based on the water sensibility of IFR, low compatibility and dispersibility of vermiculite within resin, the addition of origin vermiculite will reduce the durability and expected fire protection property of the coatings.19,20
:1 layered silicate with high layer charge density. The thermal stability and interlayer ion capacity of vermiculite is much higher than that of montmorillonite.13,14 Moreover, it can be expanded to 8–12 times of its original volume after calcination at high temperature. After expansion, vermiculite has good heat insulation with its tiny air interlayer.15 Natural vermiculite can be used as a fire-retardant synergist in some matrix. Ren16 et al. found that the formulation with 3 wt% exfoliated vermiculate had the best performance for decreasing heat release and smoke production parameters of flame-retardant polypropylene. Since intumescent fire-retardant coatings are normally used as decorative coatings and endure for a long time before a fire actually breaks out, the durability of the coatings is quite important.17 However, due to large surface area and abundant capillaries, vermiculite has strong water absorption.18 Based on the water sensibility of IFR, low compatibility and dispersibility of vermiculite within resin, the addition of origin vermiculite will reduce the durability and expected fire protection property of the coatings.19,20
Most researches have been done using organic cation like amine salt intercalation to enhance the hydrophobicity of vermiculite.21 Aluminum diethylphosphinate (ADP) is a kind of alkyl-substituted phosphinate. Due to the P–C bonds in its molecule, it has good hydrophobicity. Therefore, the intercalation of ADP may reduce the water absorption of vermiculite. Moreover, it is reported that the flame retardancy outcome of combining ADP with montmorillonite results in an enhanced effect when compared with their application on a separate basis.22 With the cooperation of ADP, the composite has a combined condensed-phase and gaseous-phase activity.23 Up to now, there is less report concerning modified vermiculite and its application as a synergist into IFR coatings. Therefore, it may be a novel and advisory research direction to modify vermiculite and study its synergistic effect on flame retardants.
In this work, ADP was intercalated into vermiculite interlayer to improve the flame retardancy of composite, then the surfactants sodium stearate (NaSt) was grafted on the surface of vermiculite to reduce water absorption and enhance its compatibility with resin. Afterwards, the aluminum diethylphosphinate/sodium stearate modified vermiculite (ANE) was applied to amino resin to produce fire-retardant coatings. The synergistic effect of thermal behavior and smoke suppression performance of flame-retardant coatings filled with ANE was evaluated, and the flame retardant and smoke suppression mechanism of ANE coatings were proposed.
2. Experimental
2.1 Materials
Vermiculite (EV) with an average size of 30 μm was purchased from Hebei Jingpeng Material Co., Ltd. Aluminum diethyl-phosphinate (ADP, purity: 99.5%) was supplied by Jiangsu Liside New Material Co., Ltd. Sodium stearate (NaSt, purity: 98%) and ethanol (purity: 99.7%) were purchased from Shanghai Titanchem Co., Ltd. Pentaerythritol (PER, purity: 99.5%) were purchased from Shanghai Lingfeng Chemical Reagent Co., Ltd. Glacial acetic acid was purchased from Shanghai Kuling Fine Chemical Co., Ltd. Polydimethylsiloxane (PDMS, Mn = 3000) was supplied by Guangzhou Silok Polymer Co., Ltd. Melamine modified urea-formaldehyde resin (MUF, 37% in water) was purchased from Shandong Dengnuo New Material Co., Ltd. Zinc borate (6–10 μm) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Phosphorylated polyvinyl alcohol (PPVA, esterification degree is 85.6%) was homemade by using polyvinyl alcohol 1788 (PVA, degree of polymerization: 1700) and phytic acid (PA, 70% in water) in laboratory.2.2 Preparation of AEV
Aluminum diethylphosphinate (ADP) was intercalated into the interlayer of microwave expanded vermiculite to produce ADP modified vermiculite named AEV. Firstly, vermiculite (EV, 10 g) was weighed and heated in a microwave oven at 800 W for 2 min to obtain microwave expanded vermiculite named MEV. Then ADP (0.2 g) and ethanol (10 wt%) were fed into a 500 ml three-necked flask with stirring at 90 °C for 30 min. Afterwards, MEV was added into the flask after ADP was completely dissolved. The pH of the mixed solution was adjusted to 4–5 with glacial acetic acid and a stirring at 100 °C for 2 h. Finally, the mixture was filtrated, rinsed with deionized water and ethanol for three times and dried in an oven at 100 °C for 24 h to obtain AEV.2.3 Preparation of ANE
Sodium stearate (NaSt) was grafted on the surface of the AEV to produce organic modified vermiculite (ANE) via the reaction between –COOH in NaSt and Si–OH in AEV. Firstly, NaSt (0.2 g) and ethanol (5 wt%) were fed into a 500 ml three-necked flask with stirring at 60 °C to dissolve. Then a certain amount of AEV was added into the flask and stirring at 100 °C for 1 h. Finally, the mixture was filtrated, rinsed with deionized water and ethanol for three times, and dried in an oven at 100 °C for 24 h to obtain ANE. The synthesis process is shown in Scheme 1.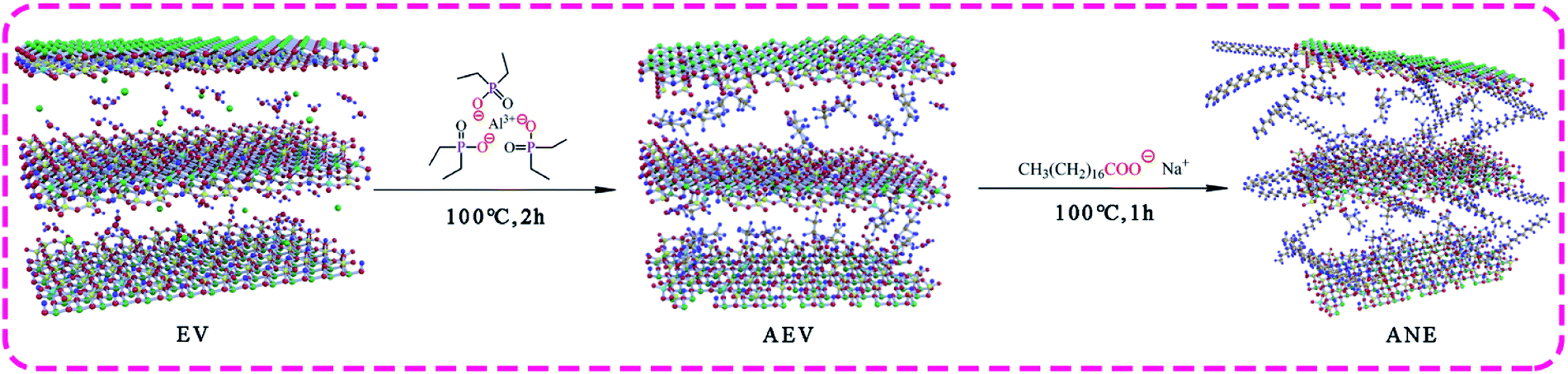 | ||
| Scheme 1 The preparation of organic modified vermiculite. | ||
2.4 Preparation of intumescent fire-retardant coatings
Phosphorylated polyvinyl alcohol (PPVA, 18 wt%) and pentaerythritol (PER, 12 wt%) were ground by sand mill in advance. Then the mixture was fully compounded with a certain amount of ANE, polydimethylsiloxane (PDMS, 6 wt%), zinc borate (1 wt%), deionized water and amino resin to produce fire-retardant coatings. Finally, the coatings were painted with a brush on the polished steel and solid wood boards (100 mm × 100 mm × 3 mm). The dry film thickness was controlled at 2 ± 0.2 mm. According to the different content of ANE, the coatings are named as 1 wt% ANE, 3 wt% ANE and 5 wt% ANE, respectively. In the same way, the coatings named as 1 wt% EV, 3 wt% EV and 5 wt% EV are made from origin vermiculite as a comparison. The coating without vermiculite is named as unadded.2.5 Characterizations
Fourier Transform Infrared (FTIR) spectra of the samples were recorded by an iS 10 spectrometer (Thermo Fisher, America) using the KBr disk.The crystal structure of vermiculite was performed on X-ray diffractometer D8 Advance (Brook, Germany) with Cu Kα radiation source (λ = 0.15406 nm).
X-ray Photoelectron Spectroscopy (XPS) spectra of modified vermiculite were recorded by an ESCALAB 250 XI (Thermo Scientific, America) using Al Kα excitation radiation (hν = 1486.6 eV), and calibrated by assuming the electron binding energy of carbonaceous carbon to be 284.8 eV.
The morphologies of vermiculite and chars were observed by a SU8010 scanning electron microscopy (SEM) (Hitachi, Japan).
The water contact angles of vermiculite and coatings were measured with a drop-shape analysis system (JC2000DF, China). Vermiculite powders were pressed into tablets by tableting machine (BP-1, Dandong, China) under 20 MPa.
The water absorption of vermiculite was tested as follows. The vermiculite was dried in an oven at 105 °C with the recorded mass of M1, and then placed to a constant weight at room temperature (25 °C) and humidity (60%), with the recorded mass of M2. The moisture absorption rate (R) of the sample was calculated by the following equation:
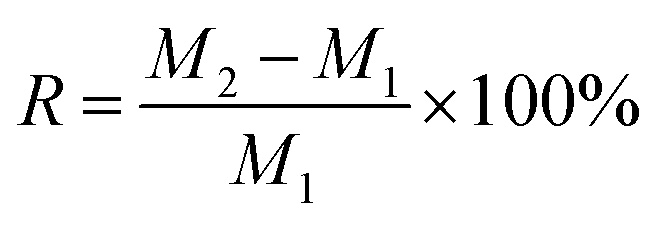 | (1) |
The sedimentation test was used to evaluate the dispersion stability of vermiculite. The vermiculite particles were dispersed in water for 30 min by ultrasonic treatment to make them fully dispersed. Then the sedimentation observation was performed at different time under room temperature. The solute/solution mass percentage is 10%.
The fire resistance test was used to evaluate the heat shielding performance of different intumescent formulations. The coating on the steel substrate was exposed to a flame from the alcohol blowlamp. The back temperature of steel panel was measured by a k-type thermocouple and recorded by a data acquisition device.
Thermo gravimetric analysis (TGA) was carried out on a TGA55 thermal analyzer (TA, America), at a heating rate of 20 °C min−1 under the nitrogen atmosphere in the temperature range from 25 to 800 °C.
Cone calorimeter test (CCT) was applied to assess the fire protection and smoke suppression properties of the coatings according to ISO5660-1. It was performed on a Fire Testing Technology apparatus (VOUCH-6810, Jiangsu) under a heat flux of 35 kW m−2 according to EN45545. The specimen (100 mm × 100 mm × 2 mm) was measured horizontally without any grids. The test was performed for three times.
3. Results and discussion
3.1 Characterization of modified vermiculite
FTIR spectra of ADP, NaSt, EV, AEV and ANE are presented in Fig. 1. In the FTIR spectra of AEV and ANE, the absorption peaks at 3600 cm−1 to 3700 cm−1 are attributed to the stretching vibrations of –OH groups attached to the octahedral Mg2+ ions and coordinated water molecules of vermiculite.24 The strong and sharp bands at 2939 cm−1, 2900 cm−1 and 2833 cm−1 are the stretching vibrations of CH3(CH2)– groups of NaSt and ADP.25 In the spectra of AEV, the peaks at 1258 cm−1, 1135 cm−1 and 763 cm−1 are attributed to the bending vibrations of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, infrared absorption of PO2− anion and P–O groups of ADP, respectively.26,27 This indicates that ADP was successfully introduced into vermiculite. After modification with NaSt, the stretching vibrations of CO groups at 1569 cm−1 and C–O groups at 1135 cm−1 reveal that the carboxyl groups of NaSt are bonded with –OH groups on the surface of vermiculite.28 All the analyses above verify the successful introduction of ADP and NaSt.
O groups, infrared absorption of PO2− anion and P–O groups of ADP, respectively.26,27 This indicates that ADP was successfully introduced into vermiculite. After modification with NaSt, the stretching vibrations of CO groups at 1569 cm−1 and C–O groups at 1135 cm−1 reveal that the carboxyl groups of NaSt are bonded with –OH groups on the surface of vermiculite.28 All the analyses above verify the successful introduction of ADP and NaSt.
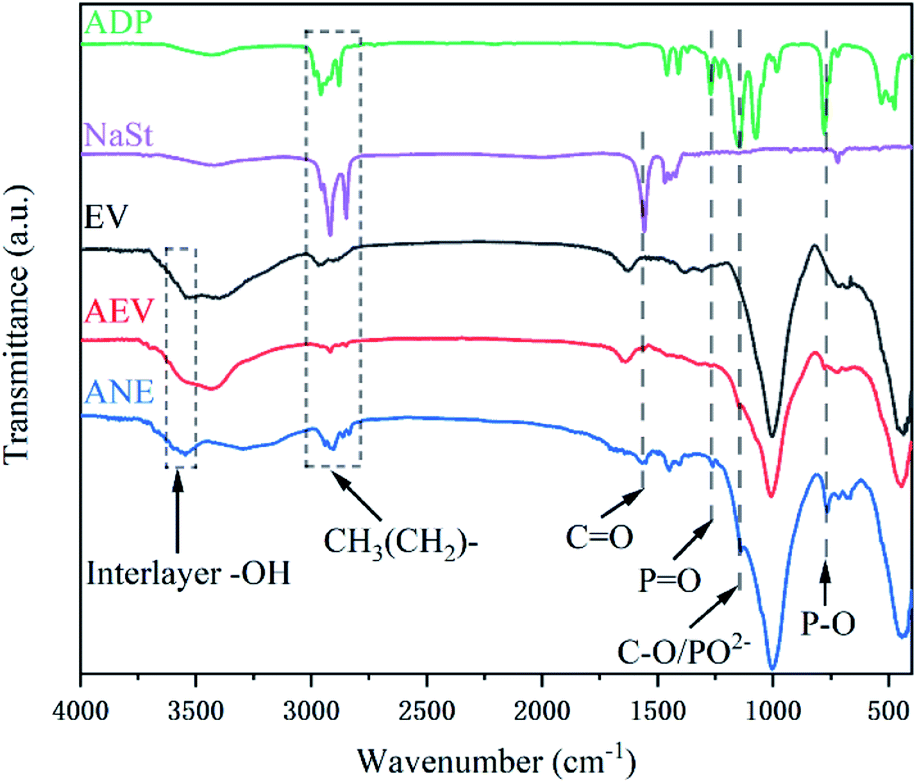 | ||
| Fig. 1 FTIR spectra of ADP, NaSt, EV, AEV and ANE. | ||
In order to analyze the crystal structure of vermiculite, XRD patterns of EV, AEV, ANE and related modified agents were presented in Fig. 2. Through retrieval, it is found that the main diffraction lines of origin vermiculite are consistent with those of phlogopite (Phlogopite-1M PDF #10-0495), which are 8.889° (001), 26.603° (003), 34.276° (−131), 45.043° (005), 54.828° (−135) and 60.283° (060).29 This is because vermiculite is an interlayer mineral formed by phlogopite, biotite and vermiculite crystal layers. The new characteristic diffraction peaks of ANE at 23.974°, 31.681° and 47.277° indicate that the crystal structure of AEV was changed due to the grafting of NaSt.
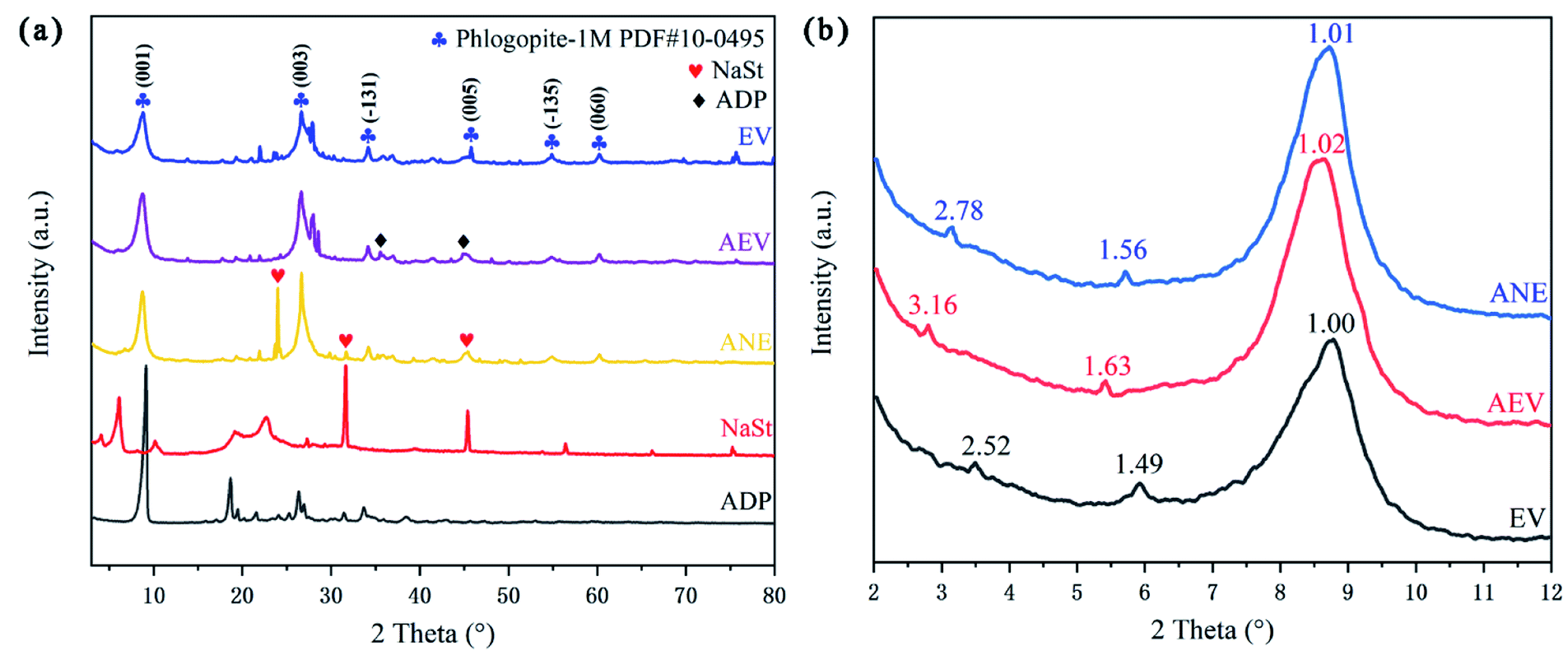 | ||
| Fig. 2 XRD patterns (a) and small-angle XRD patterns (b) of EV, AEV, ANE and related modified agents. | ||
From Fig. 2b, the diffraction peak at 1.49 nm is the (001) characteristic peak of vermiculite, and the peak near 3.53° (2.52 nm) is the (001) characteristic peak of vermiculite-mica mixed-layer minerals. Reflections at 1.49 and 2.52 nm for EV were shift to 1.63 and 3.16 nm, which indicates that ADP is successfully intercalated into vermiculite. After further modification, the shifting of reflections at 1.56 and 2.78 nm may be due to the bonding between the –COOH groups formed by NaSt hydrolysis and reactive –OH groups of AEV, which makes the introduction of long chain alkyl.18 Compared to EV, the larger interlayer spacing is helpful to delay the diffusion of small molecules and exert a better barrier effect.
XPS analysis could further certificate the chemical structure of ANE. The wide scan and high-solution XPS spectra of ANE are shown in Fig. 3. It can be found that C, O, Si, Al, P and Mg elements are detected in ANE. In the C1s XPS spectra, the peak at 284.8 eV is assigned to the C–C bonds in aliphatic groups, and the peaks at 286.1 eV and 288.5 eV are assigned to C–O/C–O–Si and CO groups of NaSt, respectively.5 In the O1s XPS spectra of Fig. 3c, the peak at 530.7 eV is assigned to hydroxide, Mg–O and Al–O groups of vermiculite,30 the peak at 531.5 eV is assigned to O in phosphate and carbonyl groups (PO and CO), and the peak at 532.3 eV is corresponding to OC–O groups of NaSt, P–O groups of ADP and Si–O groups of vermiculite, respectively.31 The Si2p peaks centered at 101.5 eV belong to Si–O, and the peak at 102.1 eV is corresponding to Si–O–C, indicating the interaction between NaSt and vermiculite.32 As shown in Fig. 3e, the P2p XPS spectra shows two peaks at 131.7 eV and 132.6 eV are assigned to P–C and P–O groups in hypophosphite, respectively.33 In the Al2p XPS spectrum of Fig. 3f, the two peaks at 73.4 eV and 73.8 eV are assigned to Al–O and hypophosphite groups, indicating ADP was successfully introduced into vermiculite.34 Combined with FTIR and XRD analyses, ANE was successfully prepared as shown in Scheme 1.
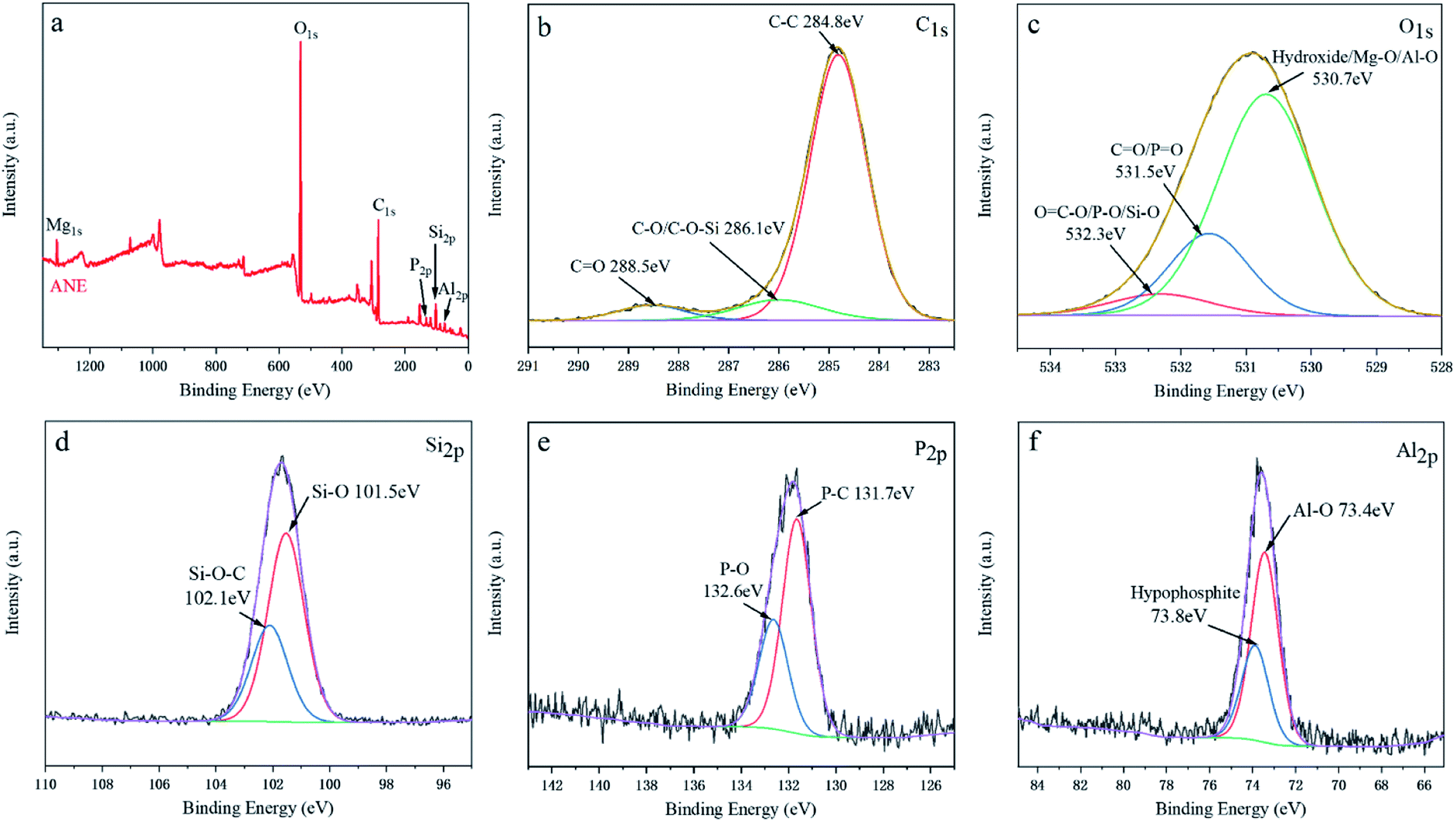 | ||
| Fig. 3 XPS spectra of ANE: (a) a full scan, (b) C1s, (c) O1s, (d) Si2p, (e) P2p, (f) Al2p. | ||
The microstructures of EV, AEV and ANE were observed by SEM. An even surface of EV can be observed in Fig. 4a. Through microwave expansion, MEV exhibits more obvious separation layers (Fig. S1†). After acidification and intercalation of ADP, the dissolution of Si atoms and cations destroyed the lamellar structure of EV and formed a scale-like structure, resulting in an increased porosity and high specific surface area (Fig. 4b).35 This can improve the compatibility between the mineral surface and organic species.36 After modification with NaSt, the scale-like structure of AEV was transformed into a porous structure, with an average pore diameter of 0.2 μm–0.5 μm. Furthermore, a rough surface morphology can be observed in Fig. 4c. This indicates that the long alkyl chains of NaSt were attached to the surface of AEV and entangled with each other to form tiny pores. The organic molecule layer can form tiny hydrophobic regions to improve the hydrophobicity and compatibility of vermiculite.18
 | ||
| Fig. 4 SEM images of EV (a), AEV (b) and ANE (c). | ||
3.2 Wettability
Fig. 5 shows the results of water contact angle and moisture absorption rate of EV, AEV and ANE. Compared to EV, it can be noticed that the water contact angle of AEV is increased to 54.6°. However, the periphery of the water droplet is still wetted. The moisture absorption of AEV only exhibits a slight decrease when it reaches to the equilibrium state. This indicates that the modification with ADP only delays the penetration of water, but could not significantly enhance the hydrophobicity of vermiculite. For ANE, it exhibits the largest water contact angle of 86.7° and the lowest moisture absorption rate of 0.524%. This verifies that the combination of ADP and NaSt results in an enhanced effect on improving the hydrophobicity of vermiculite. Combined with SEM analysis, the high hydrophobicity of ANE may be mainly attributed to its microstructure which imparts a high interfacial resistance to prevent it from wetting by water.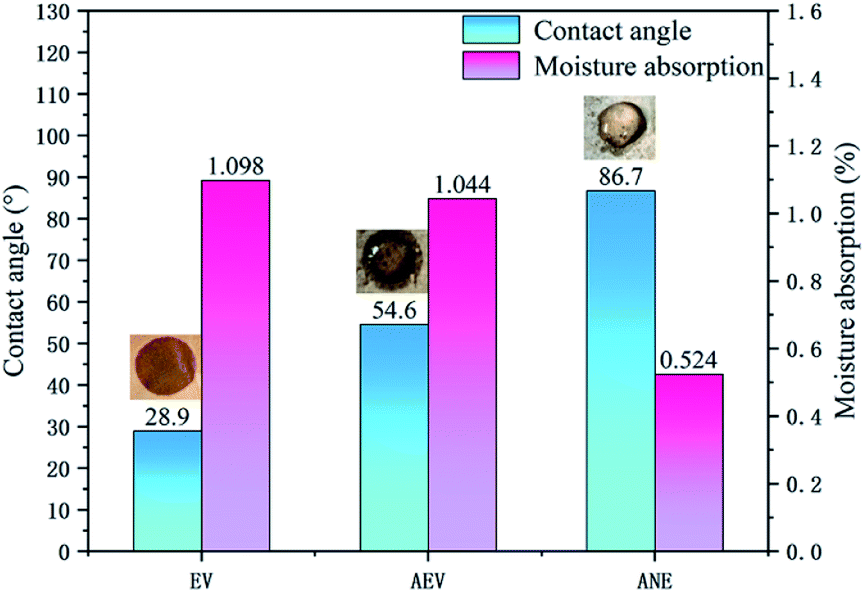 | ||
| Fig. 5 Contact angles and moisture absorption of EV, AEV and ANE. | ||
3.3 Dispersion stability
Fig. 6 shows the dispersion capability of EV, AEV and ANE in water after various storage time. It can be observed that most of the EV particles were deposited on the bottom of the bottle immediately. However, the ANE solution has no obvious stratification after storage for 3 h. After storage for 72 h, a uniform ANE solution can still be observed in Fig. 6c. Compared to other particles, the dispersion stability of ANE is significantly improved due to AEV wrapped by NaSt, thereby improving its protection performance. | ||
| Fig. 6 Water dispersion stability of EV, AEV and ANE at different time: (a) 0 h, (b) 3 h, (c) 72 h. | ||
3.4 Fire retardancy property
The fire resistance results of coatings during combustion are presented in Fig. 7 and Table 1. It can be found that the backside temperature of bare steel increased sharply to 570.5 °C within 300 s. After being coated with IFR coatings, the substrate can maintain a low temperature for a long period. For EV, the steel coated with 5 wt% EV coating exhibits the best fire resistance performance with the lowest final backside temperature (Tmax) of 338.3 °C.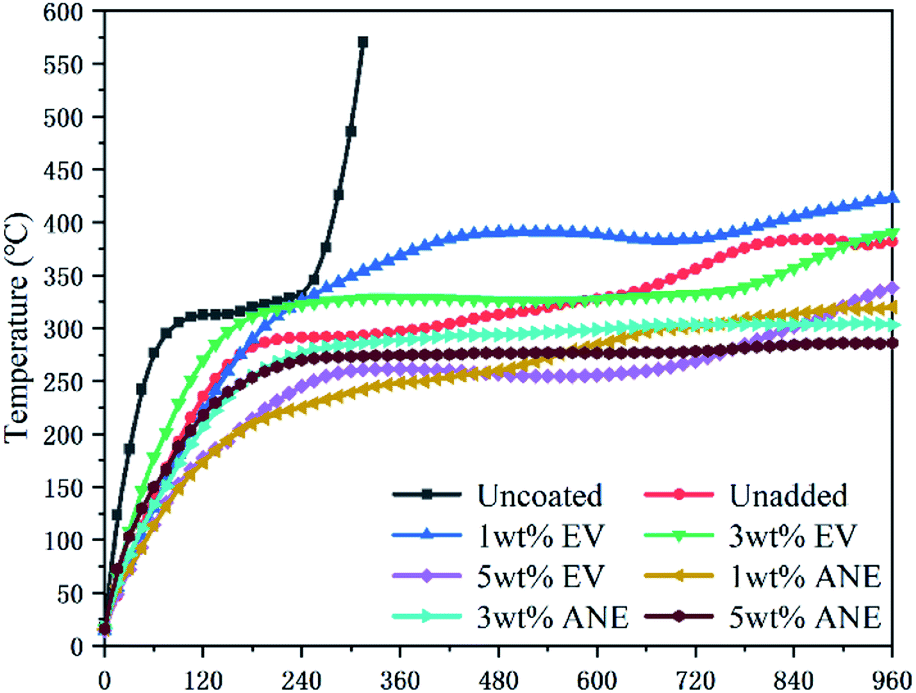 | ||
| Fig. 7 Backside time–temperature curves of different coatings. | ||
| Samples | Uncoated | Unadded | 1 wt% EV | 3 wt% EV | 5 wt% EV | 1 wt% ANE | 3 wt% ANE | 5 wt% ANE |
|---|---|---|---|---|---|---|---|---|
| a Tmax: final backside temperature at 960 s. | ||||||||
| Tmax (°C) | 570.5 ± 4.8 | 382.0 ± 3.3 | 423.6 ± 2.5 | 390.1 ± 3.2 | 338.3 ± 3.7 | 320.1 ± 3.6 | 303.5 ± 2.5 | 286.2 ± 2.5 |
By contrast, coatings containing ANE exhibit a much lower Tmax than others. With the increasing content of ANE, the Tmax of coatings are decreased to 320.1, 303.5 and 286.2 °C, respectively. This indicates that the good dispersibility of ANE can better improve the thermal insulation performance of the coatings. It is noteworthy that only 1 wt% ANE can exhibit superior synergistic effect with flame retardants on improving the fire resistance property of the coating.
The carbon residue on the surface of each sample after the fire resistance test is shown in Fig. 8. It can be observed that both 1 wt% and 3 wt% EV samples exhibit large pores and cracks on the char residue. This allows the flame to act directly on the substrate, resulting in higher Tmax than other samples. But for ANE samples, the char with fewer pores and cracks can still maintain a dense structure after combustion. Since vermiculite has high thermal stability and insulation property, when ANE was uniformly dispersed in the coatings, it can effectively enhance the compactness and strength of the carbon layer to delay its thermal oxidative decomposition, thus stabilizing the carbon skeleton and providing longer protection for the substrate.37
 | ||
| Fig. 8 Char residue of different coatings obtained from fire resistance test. | ||
Cone calorimeter test (CCT) was employed to evaluate the synergistic effect of ANE on fire protection and smoke suppression properties. The corresponding data are illustrated in Fig. 9 and Table 2.
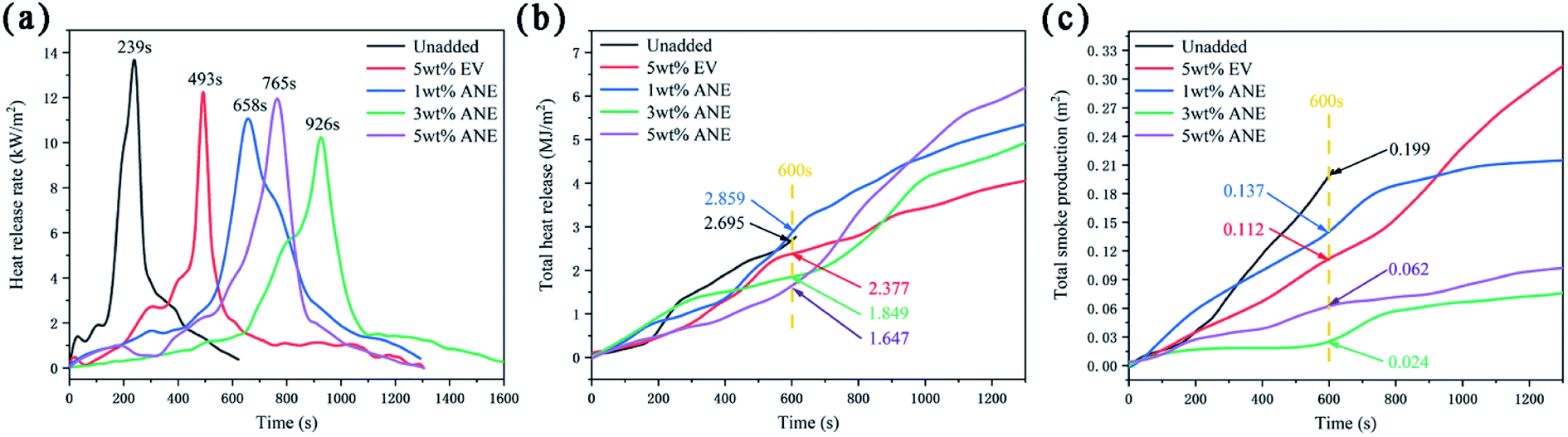 | ||
| Fig. 9 HRR (a) THR (b) and TSP (c) curves of different coatings. | ||
| Samples | TTI (s) | PHRR (kW m−2) | THR (MJ m−2) | PSPR (m2 s−1) | TSP (m2) | PMLR (g s−1) | Residue mass (%) |
|---|---|---|---|---|---|---|---|
| a TTI: time to ignition; PHRR: peak of heat release rate; THR: total heat release at 600 s; PSPR: peak of smoke produce rate; TSP: total smoke production; PMLR: peak of mass loss rate. | |||||||
| Unadded | 179 | 13.675 | 2.695 | 1.643 × 10−3 | 0.199 | 0.129 | 35.45 |
| 5 wt% EV | — | 12.238 | 2.377 | 0.877 × 10−3 | 0.112 | 0.065 | 63.18 |
| 1 wt% ANE | — | 11.061 | 2.859 | 1.386 × 10−3 | 0.137 | 0.097 | 59.66 |
| 3 wt% ANE | — | 10.223 | 1.849 | 0.652 × 10−3 | 0.042 | 0.078 | 65.03 |
| 5 wt% ANE | — | 11.951 | 1.647 | 1.074 × 10−3 | 0.062 | 0.082 | 63.06 |
Heat release rate (HRR) and total heat release (THR) are applied to reflect the heat feedback from the combustion surface of material, which accelerates the pyrolysis of the material and produces more volatile combustibles, resulting in faster flame spread and higher fire risk.38 According to Fig. 9a and Table 2, 3 wt% ANE sample exhibits the lowest peak of HRR (PHRR) with 10.223 kW m−2, and its PHRR appeared in the latest at 926 s. This peak is mainly due to the rupture of the carbon layer under continuous thermal radiation, thus releasing gases and a large amount of heat.39 In addition, the THR of 3 wt% ANE and 5 wt% ANE samples at 600 s are much lower than other samples, indicating that ANE protects the substrate from fast degradation, which can play a key role in isolating oxygen and blocking heat transfer in the subsequent process of continuous heat radiation.40
Although HRR and THR can assess the latent fire hazard for the materials, materials must be ignited by external heat sources before burning. Therefore, the time to ignition (TTI) is also an important index to evaluate the fire hazard of materials.2 It can be seen from Table 2 that the sample without vermiculite was ignited at 179 s, simultaneously, HRR increased sharply at this stage. In contrast, neither EV nor ANE samples were ignited during the whole process, which fully verifies that the addition of vermiculite can effectively reduce the overflowing of combustible gases against ignition.
Besides heat release, smoke production is another dominant hazard parameter in a fire.40 Combined with Fig. 9c and Table 2, it can be found that 3 wt% ANE sample exhibits the lowest smoke produce rate (PSPR) and total smoke production (TSP) of 0.652 × 10−3 m2 s−1 and 0.042 m2, which is 78.89% and 62.50% lower than that of unadded and EV samples. The results demonstrate that an appropriate amount of ANE can better inhibit smoke release and reduce smoke hazards.
During the thermal expansion of the coatings, flue gases can pass through the larger space between the polymer molecular chains and escape into the air to form smoke. When ANE is uniformly dispersed in the coatings, due to the microstructure of ANE (Fig. 4c) as a barrier in the matrix, these small molecules can only diffuse along the gap and surface between the ANE sheets. Owing to the increase of interlayer spacing and capillary effect of ANE, small molecules have to diffuse through a longer and more tortuous path.38 During this process, most flue gases and solid particles are deposited and adsorbed in the pore walls and gaps of ANE. Meanwhile, the main decomposition products of ADP can act as the gas phase flame retardant mechanism to help reduce the smoke generation ability of coatings.34
The peak of mass loss rate (PMLR) and residue mass reflect the pyrolysis rate and behavior of materials under certain radiation intensity.41 Combined with the appearance of char residue in Fig. 10, large cracks can be observed on the char surface of EV, while ANE retains compact char residue, thus exhibiting better char stability and lower HRR and TSP during combustion. Because of the decomposition of organic layer on the surface of ANE, the final THR and PMLR of ANEs are higher than that of 5 wt% EV. However, it can be seen from Fig. S4† that the coatings containing ANE tend to form a more compact char with smaller pores, implying the synergistic effect of ANE in the formation of high quality char residue during combustion process.42
 | ||
| Fig. 10 The side and top view of the char residues after cone calorimeter test: (a) unadded, (b) 5 wt% EV, (c) 1 wt% ANE, (d) 3 wt% ANE and (e) 5 wt% ANE. | ||
Due to barrier effect, EV exhibits a remarkable performance of PMLR and char residue.16 However, the uneven distribution and lamellar structure of origin vermiculite are not conducive to the expansion of carbon layer, corresponding to the final expansion height of 18.0 mm. The low height of intumescent carbon layer does not have a good thermal insulation performance and is more likely to cause the material to lose strength under high temperature.43 This is also the reason that EV coatings exhibit higher Tmax (Table 1) than ANE coatings in the fire resistance test. In contrast, the expansion height of 3 wt% ANE reaches to 29.0 mm, and the expansion ratio is 14.5 times. As ANE is uniformly distributed in the coatings, it can effectively improve the compactness and strength of carbon layer. But excessive content of 5 wt% ANE may solidify the molten char layer then inhibit the intumescence of the coatings as shown in Fig. 10, thus resulting in the decrease of synergistic effect on fire performance.44
Based on the above analyses, it can be verified that the addition of an appropriate amount of ANE into the coatings contributes to generating more crosslinking structures in the condensed phase that produces a more compact and thermally stable char against the release of heat and smoke, thus exhibiting excellent flame retardancy and smoke suppression properties. However, an excess of 5 wt% ANE will decrease this synergistic effect.
To analyze the thermal decomposition of ANE coatings during combustion, the TG and DTG curves of the coatings under nitrogen atmosphere are presented in Fig. 11. The unadded and 5 wt% EV samples were also tested as comparison. According to the initial decomposition and final decomposition temperature of DTG peaks, all the coatings exhibit four similar decomposition stages in the temperature ranges of 25–120 °C, 120–180 °C, 180–550 °C and 550–800 °C. The first stage at 25–120 °C corresponding to a strong DTG peak is ascribed to the removal of water and other small molecules in the coatings with a mass loss of 9.68–12.36 wt%.45 The second stage at 120–180 °C is mainly ascribed to the evaporation of residual solvents and volatile organic compounds.46,47 For coatings filled with vermiculite, the exclusion of interlayer water from vermiculite are also contributed to the total mass loss of 5.39–8.09 wt% at this stage.48 The third stage at 180–550 °C is ascribed to the thermal pyrolysis of fire retardants, amino resin and organic hydrophobic layer on the surface of ANE. The samples exhibit three decomposition regions, accompanied by a great mass loss about 16.68–20.04 wt%. The first region from 180 °C to 380 °C is attributed to the crystal water loss of zinc borate and pyrophosphates liberated from PPVA.49 During this process, the phosphoric acid derivatives were released from pyrophosphates, which esterify and dehydrate PER to form a molten char layer. Meanwhile, the incombustible gases released from amino resin promote the expansion of molten char.50 In the second region from 380 °C to 430 °C, PDMS was decomposed into cyclo-siloxane, and its side groups were oxidized to release water vapor and carbon dioxide to form crosslinking structure.51 At the same time, zinc borate was further decomposed into boron oxide. ADP was also decomposed to produce polyphosphate and small molecular gaseous substances containing phosphorus free radicals, which continued promoting the growth of carbon layer.34 The third decomposition region from 430 °C to 550 °C is attributed to the further degradation or aromatization of unstable double bonds and heterocyclic rings.52 In the meantime, the stearic acid ester on the surface of vermiculite was completely decomposed into carbon dioxide, thus the active Si–O bonds were exposed to form a dense protective layer via cyclization and crosslinking. The fourth stage at 550–800 °C is attributed to the further decomposition of unstable carbonized backbones in the char. The weight loss of samples showed nearly no change in this stage.
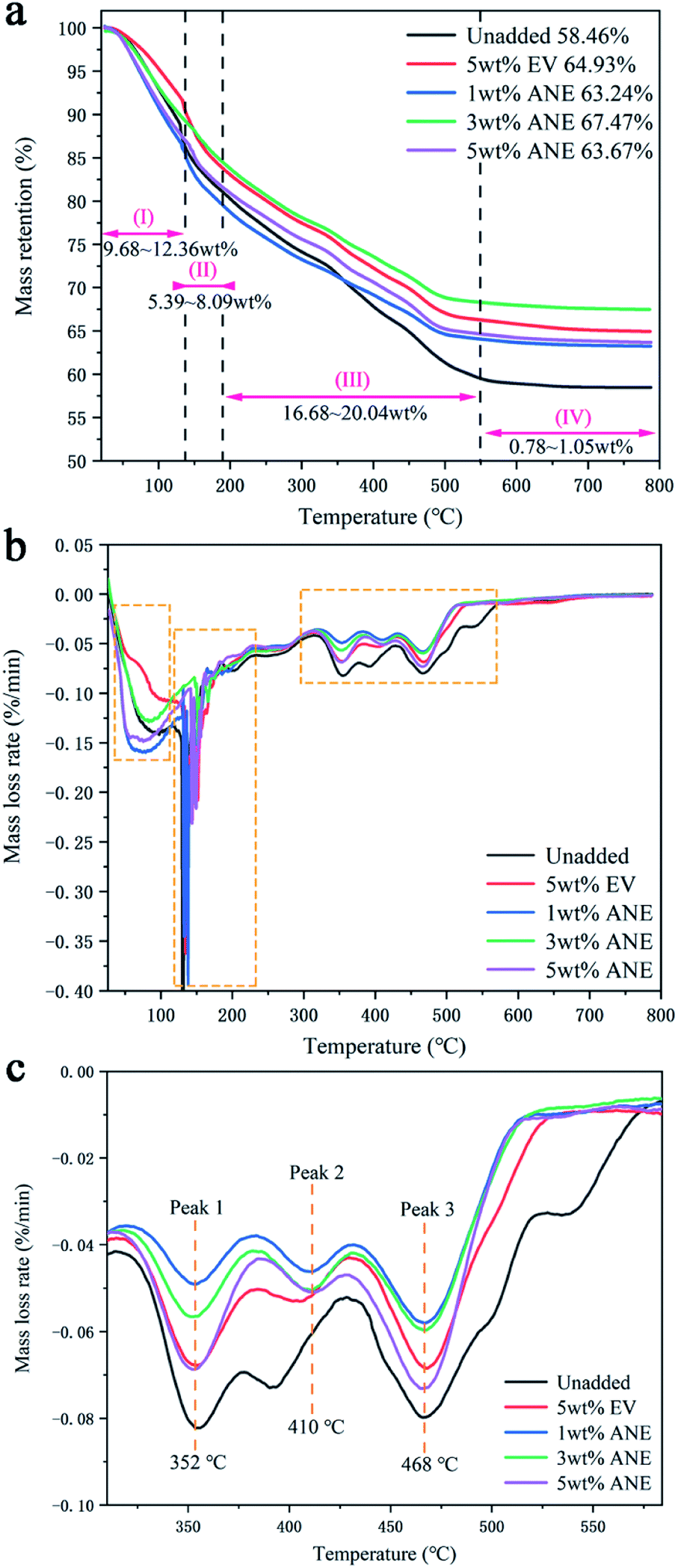 | ||
| Fig. 11 TG (a) and DTG (b) and (c) curves of different coatings. | ||
By further comparison, it can be found that the residue weight of ANE coatings is higher than others. Especially, 3 wt% ANE exhibits the highest residual weight of 67.47 wt% among all coatings. Moreover, the temperature of maximum thermal degradation rate of ANE coatings are delayed to 410 °C, indicative of the enhancement of thermal stability by incorporation of ANE.44
The mechanism for the enhancement of thermal stability of coatings is the integration of several positive factors of ANE. The intercalated ADP and grafted NaSt improve the compatibility between ANE and matrix. The strong interfacial interactions, such as entanglement between ANE and polymer chains, can increase the thermal degradation activation energy of coatings by restricting the thermal motion of polymer chains. Besides, the low thermal conductivity of ANE can insulate the external heat effectively and uniformly, thereby delays the thermal degradation of carbon layer and improves the thermal stability of IFR coatings.
3.5 Fire retardant mechanism
According to the previous results, 3 wt% ANE coating exhibits the best fire retardant and smoke suppression properties. As the coating morphology and distribution of ANE are very important when explaining mechanism. Therefore, the SEM cross-section image of 3 wt% ANE char residue coupled with elemental mapping analysis is exhibited in Fig. 12. Fig. 12b and c show the elemental mapping of Si and Mg, respectively. The mapping images confirm the presence and dispersion of Si and Mg elements, which further indicates that ANE is uniformly distributed in the carbon layer to effectively exert its synergistic effect (Fig. 13). | ||
| Fig. 12 (a) SEM image of 3 wt% ANE char residues and element mapping of (b) Si and (c) Mg. | ||
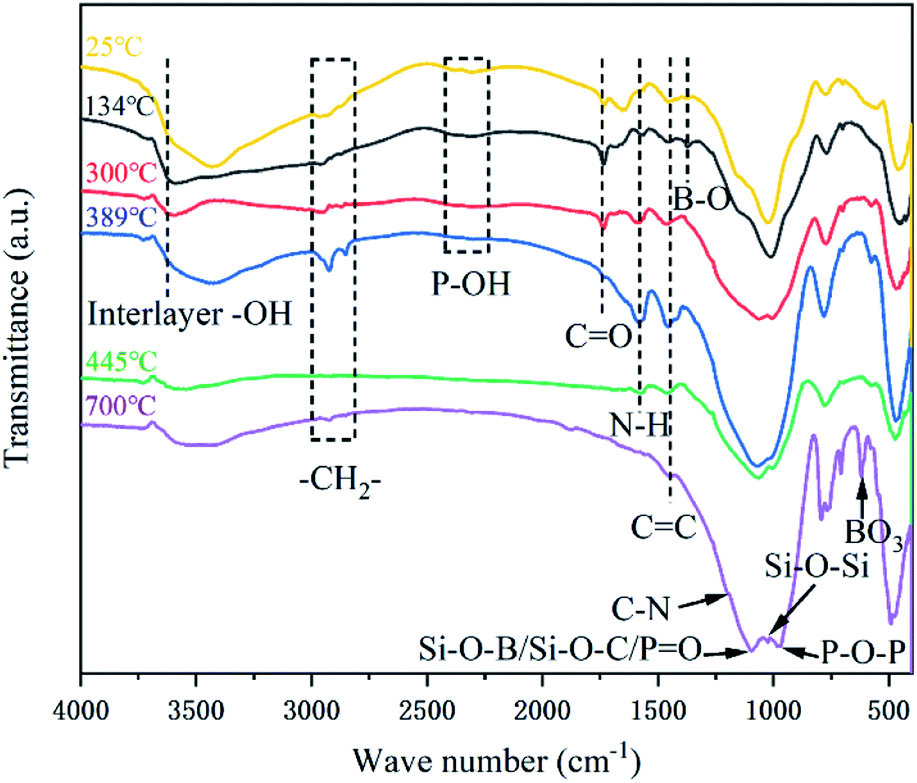 | ||
| Fig. 13 FTIR spectra of char residues from 3 wt% ANE coating at different temperatures. | ||
In order to illustrate the flame retardant mechanism of ANE fire-retardant coatings, according to the temperature at the end of the corresponding peaks of DTG curves in Fig. 11, the 3 wt% ANE coating was calcined in a muffle furnace at 134 °C, 300 °C, 389 °C, 445 °C and 700 °C for 30 min, respectively. Then FTIR and SEM of the residuals were tested to analyse the changes of chemical composition and microstructure of the coatings during combustion.
At 134 °C, the peaks at 3600 cm−1, 3100–3400 cm−1 and 2300 cm−1 are attributed to the stretching vibrations of interlayer water (–OH) of vermiculite, C–OH groups of PER and phosphate groups (P–OH) of PPVA, respectively.11,12 From Fig. 14b, a compact structure can still be observed at this time. When the temperature increased to 300 °C, the (C–OH) peak at 3100 cm−1 to 3400 cm−1 disappeared, and the intensity of (P–OH) peak at 2350 cm−1 was weakened. On the contrary, the intensity of peaks at 990 cm−1 (P–O–P/P–O–C) and 490 cm−1 (OP–OH) kept increasing, indicating that PPVA was decomposed into pyrophosphoric acid, polymetaphosphoric acid and its derivatives during the pyrolysis.40,53 Meanwhile, PER was esterified under the acid catalysis and interacted with the incombustible gases released from amino resin to form honeycomb-like voids in the coating (Fig. 14c). At 389 °C, the peak at 3400 cm−1 is attributed to the stretching vibration of –OH groups of PDMS and ANE, indicating that the Si–C bonds of PDMS were broken to form Si–OH groups after oxidation.54 More importantly, the increased intensity of characteristic peaks at 2930 cm−1, 2850 cm−1 and 1574 cm−1 which were relevant to –CH2– and N–H groups demonstrates that the amino resin was further decomposed to generate triazine compounds and release organic small molecules containing –CH3 and –CH2 groups.23,41 At 445 °C, the disappearance of CO peak at 1736 cm−1 indicates that the stearic acid of ANE was completely decomposed into CO2. The peaks at 1574 cm−1 and 1449 cm−1 are ascribed to the bending vibration of N–H and CC respectively.22 Moreover, the characteristic peak around 1000 cm−1 is split into two peaks at 1069 cm−1 and 1002 cm−1 (Si–O and P–O structures), revealing that the above-mentioned compounds were interacted to convert into a crosslinking structure.12 Combined with Fig. 14e, it can be observed that the carbon layer continuously expanded and became thicker during this process. Over 700 °C, the remaining characteristic peaks are mainly attributed to the Si–O–B, Si–O–C, PO, Si–O–Si, Si–O–P and P–O–C structures, indicating the composition of stable intumescent char is consist of C, O, N, Si, P and B elements.11,15 Besides, the peaks at 1637 cm−1 and 707 cm−1 verifies the existence of anthracene and triazine structures. A compact char structure can be found in the condensed phase (Fig. 14f) that enhances the barrier effect and effectively reduces the release of heat and smoke.
 | ||
| Fig. 14 SEM images of char residues from 3 wt% ANE coating at different temperatures (a) 25 °C, (b) 134 °C, (c) 300 °C, (d) 389 °C, (e) 445 °C, (f) 700 °C. | ||
Combined with TGA and the above results, the flame retardant mechanism of modified vermiculite fire-retardant coatings is illustrated in Fig. 15. During combustion, organic macro-molecules such as amino resin, PPVA, PDMS are gradually degraded to release incombustible gases and small molecules like NH3 and H2O. Then, PER is esterified and dehydrated under the catalysis of phosphoric acid-like compounds to generate spirophosphate. The main decomposition products are triazine compounds, cyclosiloxane, polyphosphate and its derivatives.41,46 With the temperature rising, these decomposition products, boron trioxide and ANE form the precursor of intumescent char via aromatization, cyclization and crosslinking reactions. After that, the release of incombustible gases results in the swelling of char layer. Meanwhile, the concentration of oxygen and combustible gases on the coating surface is also diluted. The ultimate stable carbon layer is the combination of phosphorus, silicon and boron-containing crosslinking structure and aromatic structure. Since the porosity and specific surface area of vermiculite were enhanced, the improved adsorption capacity and bond strength of ANE can not only adsorb more flue gases, but also promote the formation of denser char layer.55 Thereby exhibiting an effective performance on segregating the transmission of heat, oxygen and combustible gases.
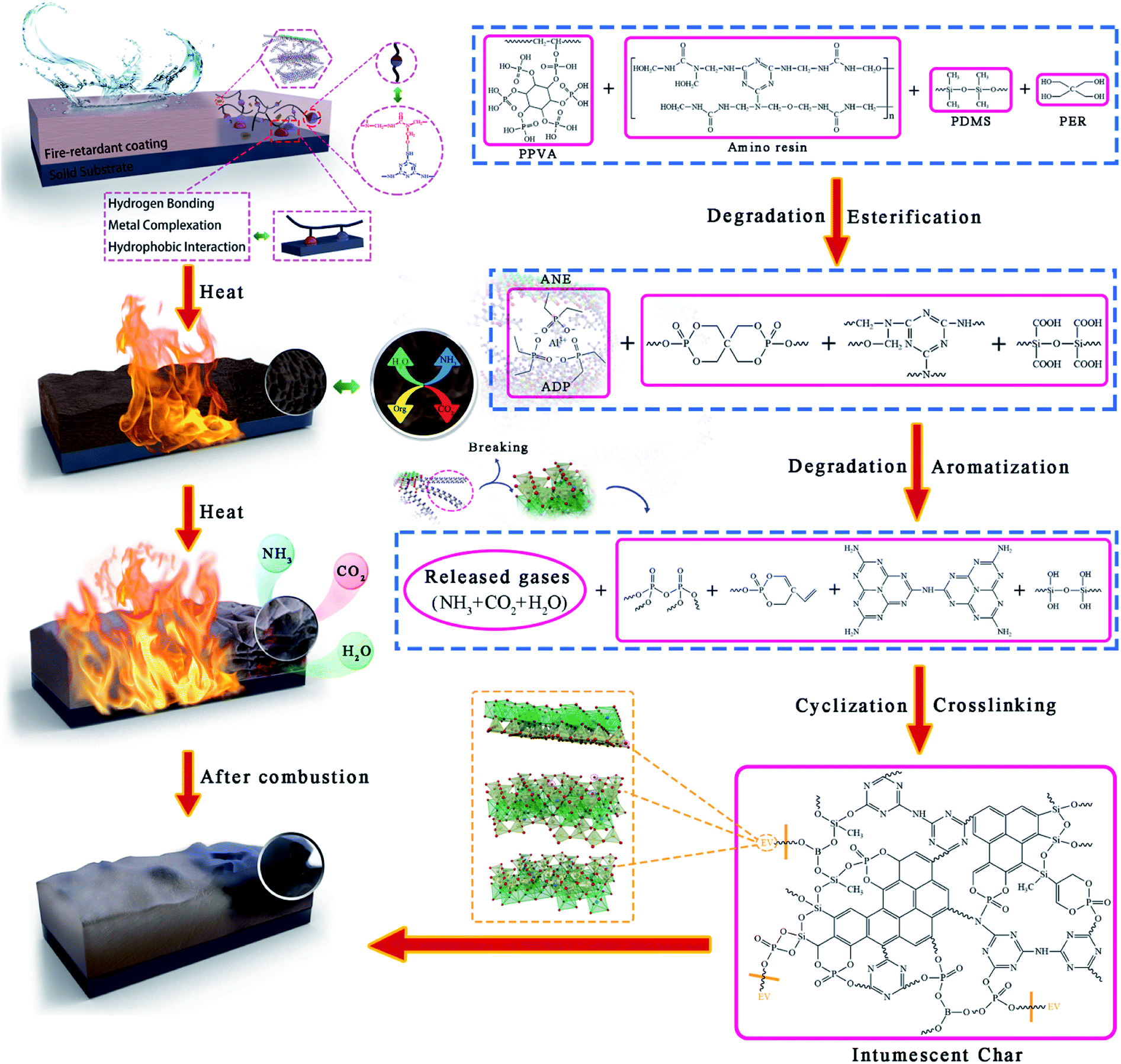 | ||
| Fig. 15 Flame-retardant and smoke suppression mechanism of ANE fire-retardant coatings. | ||
4. Conclusions
In summary, using vermiculite as a matrix, a functional filler ANE with both flame retardant and hydrophobic property is successfully prepared via the intercalation of ADP and grafting of NaSt to improve the flame retardant and smoke suppression properties of amino coatings. After modification, ANE exhibits good dispersion stability and exerts a remarkable synergistic effect on flame retardant and smoke suppression. Especially, 3 wt% ANE coating exhibits the lowest heat release rate of 10.223 kW m−2 and smoke produce rate of 0.652 × 10−3 m2 s−1, which is 25.24% and 60.32% lower than the unadded vermiculite coating, respectively. Besides, ANE improves the high-temperature stability of coatings with the final residual weight of 67.47 wt% at 800 °C. The excellent synergistic effect of ANE on the coatings is mainly ascribed to the uniform distribution and formation of more crosslinking structures to increase the density of char and inhibit the release of heat and combustible gases. More importantly, ANE exhibits a high hydrophobicity to improve the water resistance of the films, thus ensuring the durability and expected fire protection property of the fire-retardant coatings.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Science and Technology Ministry of China (2018YFC1801503).References
- S. T. Lazar, T. J. Kolibaba and J. C. Grunlan, Nat. Rev. Mater., 2020, 5, 259–275 CrossRef CAS.
- J. Green, J. Fire Sci., 1996, 14, 426–442 CrossRef CAS.
- S. C. de Sá, M. M. de Souza, R. S. Peres, A. V. Zmozinski, R. M. Braga, D. M. de Araújo Melo and C. A. Ferreira, Prog. Org. Coat., 2017, 113, 47–59 CrossRef.
- H. Hazwani Dzulkafli, F. Ahmad, S. Ullah, P. Hussain, O. Mamat and P. S. M. Megat-Yusoff, Appl. Clay Sci., 2017, 146, 350–361 CrossRef CAS.
- M. Zia-ul-Mustafa, F. Ahmad, S. Ullah, N. Amir and Q. F. Gillani, Prog. Org. Coat., 2017, 102, 201–216 CrossRef CAS.
- S. Yang, J. Wang, S. Huo, M. Wang, J. Wang and B. Zhang, Polym. Degrad. Stab., 2016, 128, 89–98 CrossRef CAS.
- L. Li, Y. Qian and C. M. Jiao, Iran. Polym. J., 2012, 21, 557–568 CrossRef CAS.
- A. Gupta, C. S. Manohar and B. S. Kumar, Water Sci. Technol.: Water Supply, 2019, 19, 1686–1694 CAS.
- M. C. Yew, N. H. Ramli Sulong, M. K. Yew, M. A. Amalina and M. R. Johan, Prog. Org. Coat., 2015, 81, 116–124 CrossRef CAS.
- P. Kiliaris and C. D. Papaspyrides, Prog. Polym. Sci., 2010, 35, 902–958 CrossRef CAS.
- L. Yan, Z. Xu and X. Wang, Prog. Org. Coat., 2018, 122, 107–118 CrossRef CAS.
- Z. Xu, N. Deng and L. Yan, J. Coat. Technol. Res., 2020, 17, 157–169 CrossRef CAS.
- Z. Miao, T. Peng, Y. Xi and W. Yang, Adv. Mater. Res., 2010, 96, 155–160 CAS.
- S. Williams-Daryn and R. K. Thomas, J. Colloid Interface Sci., 2002, 255, 303–311 CrossRef CAS PubMed.
- S. Letaïef, M. A. Martín-Luengo, P. Aranda and E. Ruiz-Hitzky, Adv. Funct. Mater., 2006, 16, 401–409 CrossRef.
- Q. Ren, Y. Zhang, J. Li and J. C. Li, J. Appl. Polym. Sci., 2010, 120, 1225–1233 CrossRef.
- Z. Liu, M. Dai, Y. Zhang, X. Gao and Q. Zhang, Prog. Org. Coat., 2016, 95, 100–106 CrossRef CAS.
- Y. Xie and A. Wang, J. Compos. Mater., 2009, 43, 2401–2417 CrossRef CAS.
- G. Wang and J. Yang, Prog. Org. Coat., 2011, 70, 150–156 CrossRef CAS.
- Z. Liu, M. Dai, Q. Hu, S. Liu, X. Gao, F. Ren and Q. Zhang, J. Coat. Technol. Res., 2019, 16, 135–145 CrossRef CAS.
- S. Williams-Daryn, R. K. Thomas, M. A. Castro and A. Becerro, J. Colloid Interface Sci., 2002, 256, 314–324 CrossRef CAS.
- A. Ramani and A. E. Dahoe, Polym. Degrad. Stab., 2014, 105, 1–11 CrossRef CAS.
- Z. M. Zhu, W. H. Rao, A. H. Kang, W. Liao and Y. Z. Wang, Polym. Degrad. Stab., 2018, 154, 1–9 CrossRef CAS.
- F. J. Del Rey-Perez-Caballero and G. Poncelet, Microporous Mesoporous Mater., 2000, 37, 313–327 CrossRef CAS.
- Y. Li, S. Wu, Y. Dai, L. Pang, Q. Liu, J. Xie and D. Kong, Constr. Build. Mater., 2018, 172, 509–518 CrossRef CAS.
- X. Han, L. Yang, C. Han, L. Li and B. Zhang, Adv. Mater. Res., 2011, 183–185, 35–39 CAS.
- L. Yang, X. Y. Han, X. J. Tang, C. X. Han, Y. X. Zhou and B. G. Zhang, Chin. Chem. Lett., 2011, 22, 385–388 CrossRef CAS.
- L. Yu, S. Sun, D. Hao and X. Hou, Surf. Rev. Lett., 2020, 27, 1950224 CrossRef.
- V. Hojatollah, Clays Clay Miner., 1992, 40, 240–245 CrossRef.
- S. S. Li, M. Jiang, T. J. Jiang, J. H. Liu, Z. Guo and X. J. Huang, J. Hazard. Mater., 2017, 338, 1–10 CrossRef CAS PubMed.
- Z. Xu, Z. Chu, L. Yan, H. Chen, H. Jia and W. Tang, Polym. Compos., 2019, 40, 2712–2723 CrossRef CAS.
- T. P. Nguyen and S. Lefrant, J. Phys.: Condens. Matter, 1989, 1, 5197–5204 CrossRef CAS.
- T. Zheng, L. Yang, P. Dai and C. Qin, Mater. Sci. Eng., 2019, 544, 012003 CAS.
- M. Xu, J. Wang, Y. Ding and B. Li, Chin. J. Polym. Sci., 2015, 33, 318–328 CrossRef CAS.
- N. Wu, L. Wu, L. Liao and G. Lv, J. Colloid Interface Sci., 2015, 457, 264–271 CrossRef CAS.
- R. Zhu, Q. Zhou, J. Zhu, Y. Xi and H. He, Clays Clay Miner., 2015, 63, 199–221 CrossRef CAS.
- B. Tang, W. Feng, J. Guo, J. Sun, S. Zhang, X. Gu, H. Li and W. Yang, Prog. Org. Coat., 2020, 138, 105391 CrossRef CAS.
- H. Pan, W. Wang, Y. Pan, L. Song, Y. Hu and K. M. Liew, ACS Appl. Mater. Interfaces, 2014, 7, 101–111 CrossRef.
- E. Akdogan, M. Erdem, M. E. Ureyen and M. Kaya, Polym. Compos., 2020, 41, 1749–1762 CrossRef CAS.
- N. Zhang, M. Zhang, J. Zhang, X. Guo, H. Wang, B. Niu and H. Yan, Prog. Org. Coat., 2019, 137, 105322 CrossRef CAS.
- L. Yan, Z. Xu and N. Deng, Prog. Org. Coat., 2019, 135, 123–134 CrossRef CAS.
- Z. Xu, Z. Chu and L. Yan, J. Therm. Anal. Calorim., 2018, 133, 1241–1252 CrossRef CAS.
- Z. Yan, X. Liao, G. He, S. Li, F. Guo, F. Zou and G. Li, Ind. Eng. Chem. Res., 2020, 59, 19244–19251 CrossRef CAS.
- L. Yan, Z. Xu and X. Wang, Prog. Org. Coat., 2017, 112, 319–329 CrossRef CAS.
- W. Zhan, L. Chen, F. Cui, Z. Gu and J. Jiang, Prog. Org. Coat., 2020, 140, 105491 CrossRef CAS.
- S. Liu, C. Wang, Q. Hu, S. Huo, Q. Zhang and Z. Liu, Prog. Org. Coat., 2020, 140, 105494 CrossRef CAS.
- A. Beyler-Çiğil, Prog. Org. Coat., 2020, 148, 105850 CrossRef.
- J. Feng, M. Liu, L. Fu, K. Zhang, Z. Xie, D. Shi and X. Ma, RSC Adv., 2020, 10, 7635–7642 RSC.
- J. Shen, P. Zhang, L. Song, J. Li, B. Ji, J. Li and L. Chen, Composites, Part B, 2019, 179, 107545 CrossRef CAS.
- M. Jimenez, S. Duquesne and S. Bourbigot, Surf. Coat. Technol., 2006, 201, 979–987 CrossRef CAS.
- G. Fei, L. Sun, H. Wang, F. Gohar, Y. Ma and Y. Kang, Prog. Org. Coat., 2020, 140, 105484 CrossRef CAS.
- Z. Xu, D. Liu, L. Yan and X. Xie, Prog. Org. Coat., 2020, 141, 105572 CrossRef CAS.
- A. I. Balabanovich, Thermochim. Acta, 2005, 435, 188–196 CrossRef CAS.
- G. Camino, S. M. Lomakin and M. Lazzari, Polymer, 2001, 42, 2395–2402 CrossRef CAS.
- G. Abate, L. B. O. dos Santos, S. M. Colombo and J. C. Masini, Appl. Clay Sci., 2006, 32, 261–270 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05731f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |