
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of dithienofurans via cascade copper catalysed dual C–S coupling and ring closure reactions under mild conditions†
Lu Zhou,
Zhaopeng Chen ,
Jiahui Li and
Baolin Li*
,
Jiahui Li and
Baolin Li*
School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, P. R. China. E-mail: libl@ucas.ac.cn
First published on 21st October 2021
Abstract
We have developed a mild catalytic approach for the synthesis of new dithienofuran derivatives via cascade copper catalysed dual C–S coupling and subsequent ring closure reactions. Sonogashira coupling between perbromofuran and terminal alkynes produced 3,4-dibromo-2,5-dialkynylfuran (1) in good yields. Next, copper catalysed C–S coupling between 1 and Na2S·9H2O and a subsequent ring-closure reaction afforded dithienofuran compounds (2) under mild conditions. We found that this strategy shows broad substrate scope and can be used to prepare not only aryl and heteroaryl but also alkyl substituted dithienofuran derivatives in up to 70% yields. Furthermore, we proposed a mechanism including two catalytic cycles: a typical Cu(I)/Cu(III) catalytic cycle and a subsequent Cu(II) induced cyclization mechanism.
Fused-ring conjugated organic compounds have been widely researched and applied in organic optoelectronic devices in the past decades.1 In particular, thienoacenes have attracted wide interest due to their good stability, high planarity, strong intermolecular π–π interaction and excellent carrier transport properties.2 Prominent examples are benzo[1,2-b:5,4-b′]dithiophene (BDT), and benzothieno[3,2-b]benzothiophene (BTBT) derivatives.3
Furan is the simplest oxygen-containing five-membered heterocyclic compound. It is different from other types of conjugated materials and can be obtained from renewable resources.4 Furan has a similar chemical structure and exhibits similar electronic properties to thiophene. In general, fused-ring furan compounds exhibit less aromaticity, higher solubility and higher fluorescence quantum efficiency compared with their thiophene analogues.5 Thienofuran compounds may inherit some advantages from both thiophene and furan, thus becoming potential organic semiconductor materials applicable in optoelectronic devices. Therefore, they have attracted great interest from researchers, and their design and synthesis as well as physical properties have gradually been reported.6 Typical examples of thienofuran compounds and their applications are shown in Scheme 1. Bisbenzo[d,d′]furo[2,3-d;2′,3′-d′]benzo[1,2-b;4,5-b′]dithiophene (BFBDT) was used as a new p-type organic semiconductors with a hole mobility of 0.04 cm2 V−1 s−1 in organic-field-effect transistors (OFET).7 PO2T can be used as a potential high-efficiency sensitizer for dye-sensitized solar cells (DSSC).8 TBF derivatives usually exhibit ferroelectric liquid crystal (FLP) properties.9 5-Methyl-5′-(thieno[3,2-b]furan-2-yl)-2,2′-bipyridine (TFBP) was used as a fluorescent indicator for zinc(II) ions in laser confocal microscopic imaging experiments.10 Recently, our group have synthesized benzo[4,5]thieno[3,2-b]benzofurans (BTBFs) and benzo[4,5]thieno[3,2-b]naphtho[2,3-d]furan (BTNF) by intramolecular C–O coupling method, and investigated their hole transporting and fluorescent properties.11 We found that BTNF emits violet fluorescence with the highest quantum yield of 72% in solution.
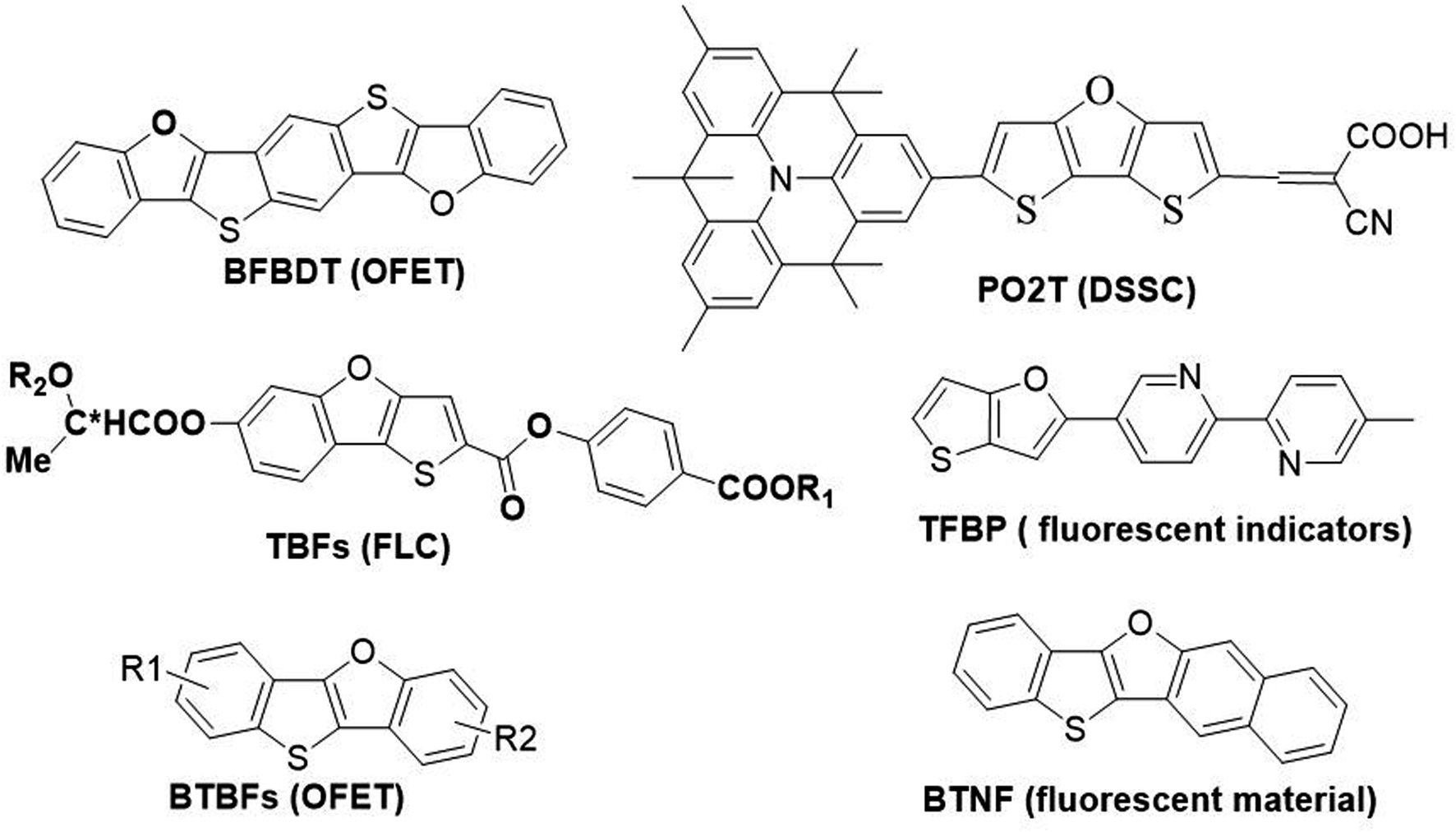 | ||
| Scheme 1 Chemical structures of thienofuran compounds and their applications. | ||
Although a variety of synthetic approaches to dithieno[3,2-b:2′,3′-d]thiophene (DTT) compounds have been reported, to date only three reports on synthesis of dithienofuran (DTF) compounds are found despite their similar central skeletons.12 Karminski-Zamola group reported the first synthesis of DTF derivatives (DTFDC) (Scheme 2).13 Through multistep reactions, i.e., esterification, Vilsmeier–Haack formylation, Knoevenagel aldol condensation and subsequent chlorination, 3,5-dichlorodithieno[3,2-b:2′,3′-d]furan-2,6-dicarbonyl dichloride (DTFDC) was obtained in a low total yield of 6.5%. In 2015, Svoboda group reported synthesis of DTF by lithiation, formylation, cyclization and decarboxylation reactions using 3,4-dibromofuran as starting substrate.14 In 2017, Suga group reported three-step synthesis of benzodithienofuran ([1]benzothieno[3,2-b]thieno[2,3-d]furan; BDTF) using 3-bromobenzo[b]thiophene 1,1-dioxide and thiophen-3-ol as starting materials.15 However, all these methods require expensive metal catalysts or harsh conditions, multistep transformations for the synthesis of specific DTFs, and the synthesis of their derivatives needs further functionalization.14 Therefore, it is necessary to develop a mild and more efficient method to synthesize DTFs from simple and easily available raw materials.
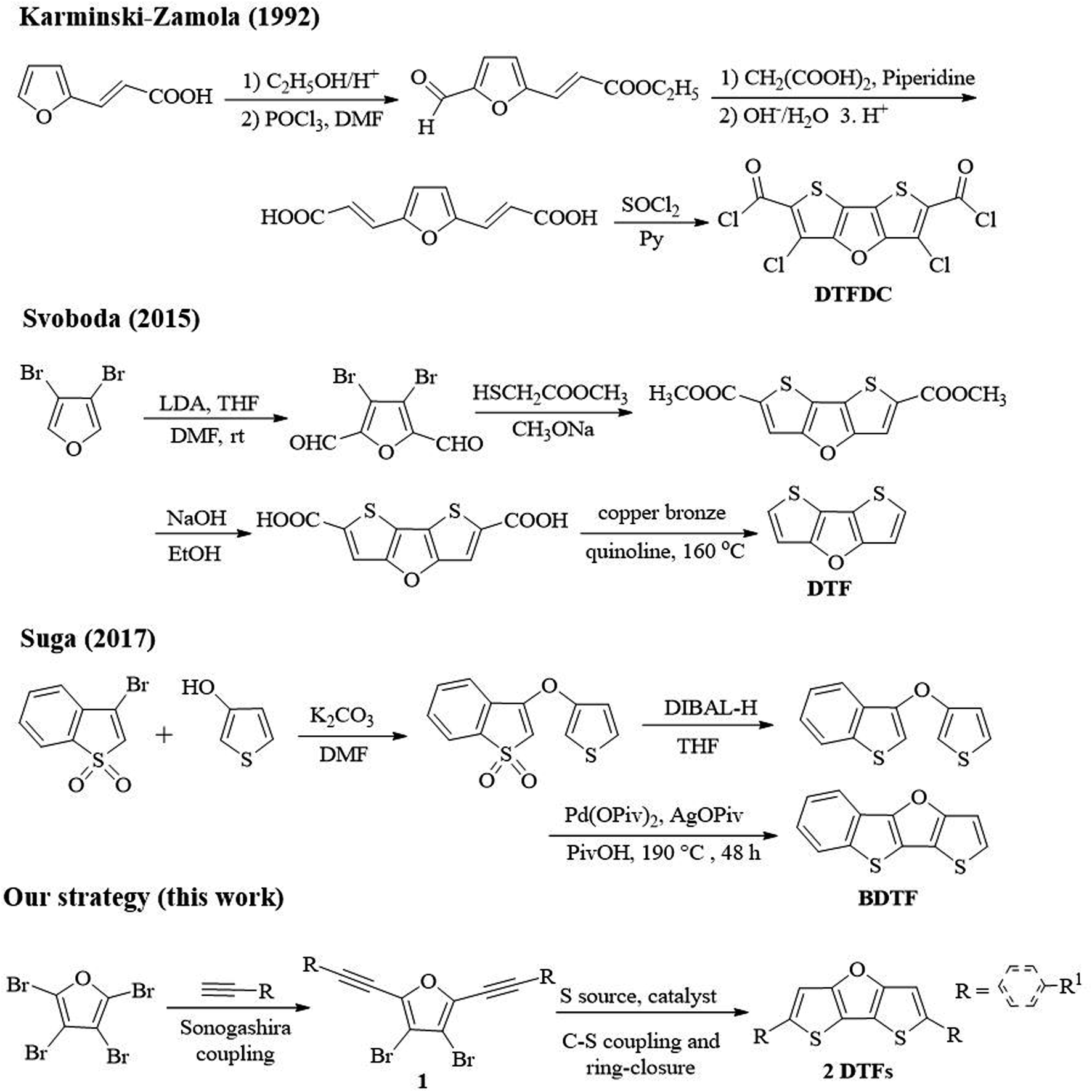 | ||
| Scheme 2 Previously reported synthetic strategies toward DFT derivatives and our strategy. | ||
Our recent interest in the synthesis of novel fused-ring furan compounds prompted us to develop a novel strategy for the synthesis of DTFs, which is depicted in Scheme 2. Herein, we report a novel copper catalysed synthesis of DTFs under mild conditions, and a variety of new DTFs can be constructed using our strategy. Perbromofuran was used as the starting substrate,16 and Sonogashira coupling between perbromofuran and terminal alkynes produced 3,4-dibromo-2,5-dialkynylfuran (1) as key intermediate in good yields. Next, copper catalysed C–S coupling between 1 and Na2S·9H2O and subsequent ring-closure reaction afforded DTFs (2). Notably, we found that this strategy can be applied to prepare not only aryl, heteroaryl but also alkyl substituted DTFs under mild reaction conditions.
Our investigation commenced with the synthesis of 3,4-dibromo-2,5-dialkynylfuran (1) via Sonogashira coupling between perbromofuran and terminal alkynes (Scheme 3). As expected, Sonogashira reaction majorly occurs at α-position of perbromofuran and minorly at β-position.17 Thus the reaction mixture contains α-dialkynylated furan (1) as the major product, and α-monoalkynylated and trialkynylated furans as byproducts, and the target compound 1 could be isolated by silica gel column chromatography. It was found that a moderate to high yield (1a–1f, 40–78%) can be obtained whether introduction of electron-withdrawing group or electron-donating group on the phenyl group. Generally speaking, the bigger substituent on the phenyl group, the lower yield. Heteroaryl alkyne such as 3-ethynylthiophene and 2-ethynylpyridine could be also used in this reaction: electron-rich alkynes afforded much higher yield than electron-deficient alkynes (1g 76% vs. 1h 32%). The similar electron effect in Sonogashira coupling had been previously reported.18 Not unexpectedly, alkyl alkynes afford moderate yields (1i–1k, 41–63%).
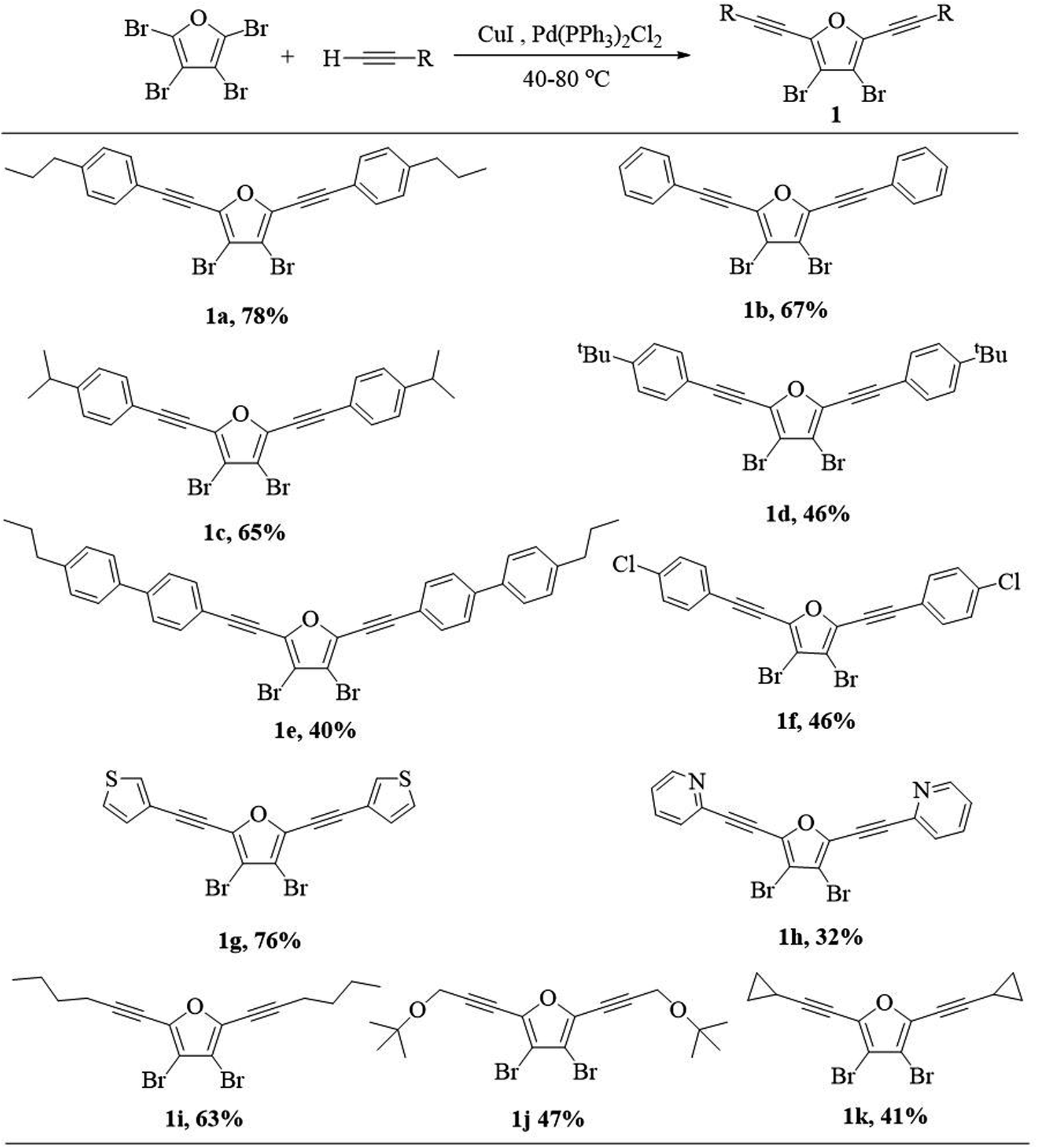 | ||
| Scheme 3 Reaction scope for 3,4-dibromo-2,5-dialkynylfuran 1 via Sonogashira coupling. | ||
For the second step ring-closure reaction, we selected synthesis of 2,6-bis(4-propylphenyl)dithieno[3,2-b:2′,3′-d]furan (2a) as model reaction to optimize the reaction conditions. The reaction yields were determined by nuclear magnetic resonance (NMR) method using 1,3,5-trimethoxybenzene as an internal reference, as summarized in Table 1. Considering the relatively low price of iron salts, firstly we tested iron salts as catalyst together with 1,10-phenanthroline (Phen) as ligand, K3PO4·H2O as the base, N-methylpyrrolidone (NMP) as solvent, and Na2S·9H2O as sulfur source and the reaction mixture was stirred at 80 °C for 12 hours under nitrogen. Unfortunately, it was found that both iron reagents tested can catalyse the ring-closure reaction, but the reaction yields were very low (trace, Table 1, entries 1 and 2). Next we tested nickel and palladium catalysts, the yields were also low (trace to 8%, entries 3–5). Without metal catalyst, the yield was only 3% (entry 6). After many efforts to screen reaction conditions, we finally found that the copper reagents tested can catalyse the ring-closure reaction, affording 2a in moderate yield (38–51%, entries 7–17). Because Cu(OAc)2·H2O as catalyst afforded the highest yield (51%, entry 17), we used it as the catalyst for further screening. Then we studied the effect of ligand on reaction yields (Table 1, entries 18–20). The experimental results showed that the NMR yield dropped to 28% when there was no ligand involved in the reaction (entry 18), which was nearly half of the yield when the ligand was involved in the reaction. When Phen ligand was replaced by 5-nitro-1,10-phenanthroline (NPhen), the yield dropped to 34% (entry 19). To our delight, when the ligand was replaced by 4,7-diphenyl-1,10-phenanthroline (DPPhen), the yield increased to 70% (entry 20). Therefore, DPPhen was the best ligand in this method, and was used for further screening.
|
||||
|---|---|---|---|---|
| Entry | Catalyst/ligand | Base | Solvent | Yieldb (%) |
| a Reaction conditions: in a 25 mL Schlenk tube, compound 1a (0.2 mmol), base (0.4 mmol), Na2S·9H2O (0.6 mmol), catalyst (5 mol%) and ligand (10 mol%) were mixed in 5 mL of corresponding solvent and stirred for 12 h at 80 °C under nitrogen atmosphere.b NMR yield: using 1,3,5-trimethoxybenzene as an internal standard.c CuTc: copper(I) thiophene-2-carboxylate. | ||||
| 1 | FeSO4·7H2O/Phen | K3PO4·H2O | NMP | Trace |
| 2 | FeCl3/Phen | K3PO4·H2O | NMP | Trace |
| 3 | Ni(acac)2/Phen | K3PO4·H2O | NMP | Trace |
| 4 | Pd(OOCCF3)2/Phen | K3PO4·H2O | NMP | Trace |
| 5 | Pd(OAc)2/Phen | K3PO4·H2O | NMP | 8 |
| 6 | —/Phen | K3PO4·H2O | NMP | 3 |
| 7 | Cu/Phen | K3PO4·H2O | NMP | 43 |
| 8 | CuI/Phen | K3PO4·H2O | NMP | 49 |
| 9 | CuCl/Phen | K3PO4·H2O | NMP | 46 |
| 10 | Cu2S/Phen | K3PO4·H2O | NMP | 45 |
| 11 | CuTc/Phen | K3PO4·H2O | NMP | 48c |
| 12 | CuOAc/Phen | K3PO4·H2O | NMP | 38 |
| 13 | CuBr2/Phen | K3PO4·H2O | NMP | 40 |
| 14 | CuCl2/Phen | K3PO4·H2O | NMP | 42 |
| 15 | CuSO4/Phen | K3PO4·H2O | NMP | 47 |
| 16 | Cu(OAc)2/Phen | K3PO4·H2O | NMP | 50 |
| 17 | Cu(OAc)2·H2O/Phen | K3PO4·H2O | NMP | 51 |
| 18 | Cu(OAc)2·H2O | K3PO4·H2O | NMP | 28 |
| 19 | Cu(OAc)2·H2O/NPhen | K3PO4·H2O | NMP | 34 |
| 20 | Cu(OAc)2·H2O/DPPhen | K3PO4·H2O | NMP | 70 |
| 21 | Cu(OAc)2·H2O/DPPhen | K3PO4 | NMP | 57 |
| 22 | Cu(OAc)2·H2O/DPPhen | CS2CO3 | NMP | 61 |
| 23 | Cu(OAc)2·H2O/DPPhen | K2CO3 | NMP | 50 |
| 24 | Cu(OAc)2·H2O/DPPhen | KOAc | NMP | 43 |
| 25 | Cu(OAc)2·H2O/DPPhen | tBuCOONa | NMP | 63 |
| 26 | Cu(OAc)2·H2O/DPPhen | — | NMP | 16 |
| 27 | Cu(OAc)2·H2O/DPPhen | K3PO4·H2O | DMF | 53 |
| 28 | Cu(OAc)2·H2O/DPPhen | K3PO4·H2O | Toluene | 0 |
| 29 | Cu(OAc)2·H2O/DPPhen | K3PO4·H2O | 1,4-Dioxane | 0 |
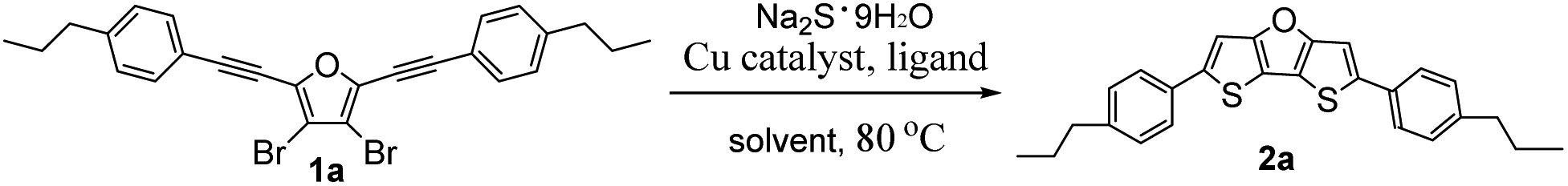
Using Cu(OAc)2·H2O as the catalyst and DPPhen as the ligand, we continued to screen different bases (Table 1, entries 21–25): potassium phosphate, cesium carbonate, potassium carbonate, sodium carbonate, potassium acetate and sodium pivalate. The yields were 57%, 61%, 50%, 43% and 63%, respectively, which were all lower than the yield using K3PO4·H2O as the base. However, all the yields are much higher than the yield without using base (entry 26, 16%). We suppose that the base should increase the polarity of NMP solution, that is, dielectric constants, which favours C–S coupling and thus improve the reaction yields. Finally, we screened different solvents, including N,N-dimethylformamide (DMF), toluene and 1,4-dioxane (entries 27–29). It was found that when the solvent was replaced by DMF, the yield dropped to 53%. When the solvent was toluene or 1,4-dioxane, no target product was detected. These results suggest that the suitable solvent for this reaction should be polar amide type solvent.
Through a series of screening conditions, we finally got the optimal reaction conditions (as shown in entry 20): under nitrogen, 5 mol% of Cu(OAc)2·H2O as catalyst, 10 mol% of DPPhen as ligand, 3 equivalents of Na2S·9H2O as sulfur source, 2 equivalents of K3PO4·H2O as base, NMP as solvent, and the reaction mixture was stirred at 80 °C for 12 hours.
After obtaining the optimal reaction conditions, we explored the generalizability of the synthetic method (Scheme 4). We investigated the effect of substituents on the reactivity, and found that no matter electron-donating groups or electron-withdrawing groups, target products can be obtained. In general, compounds with electron-rich substituents afford higher yield than those compounds with electron-deficient substituents. Compound 2b without alkyl group on the phenyl group was afforded in low isolation yield (26%) partly due to its poor solubility resulting in loss of part of compound on silica gel column chramotography. When the alkyl group was introduced on the phenyl group, the isolation yields of compound 2a (70%), 2c (42%) and 2d (58%) were improved partly due to improved solubility. However, the separation yield of 2e with p-propylbiphenyl substituents decreased to 35%. It might be because the p-propylbiphenyl substituents slow down the molecular motion and increase the difficulty for ring closure reaction, thus leading to lower yield. Compound 2f with p-chlorophenyl substituents was afforded in slightly lower yield (42%) when compared with those compounds containing p-alkylphenyl substituents, probably due to electron-withdrawing nature of p-chloro substituents. The compounds containing heteroaryl substituents such as 3-thiophenyl or 2-pyridinyl group could also be isolated in reasonable yields (2g, 49%; 2h, 30%). The comparable low yield of 2h was due to the electron-withdrawing nature of pyridinyl group.
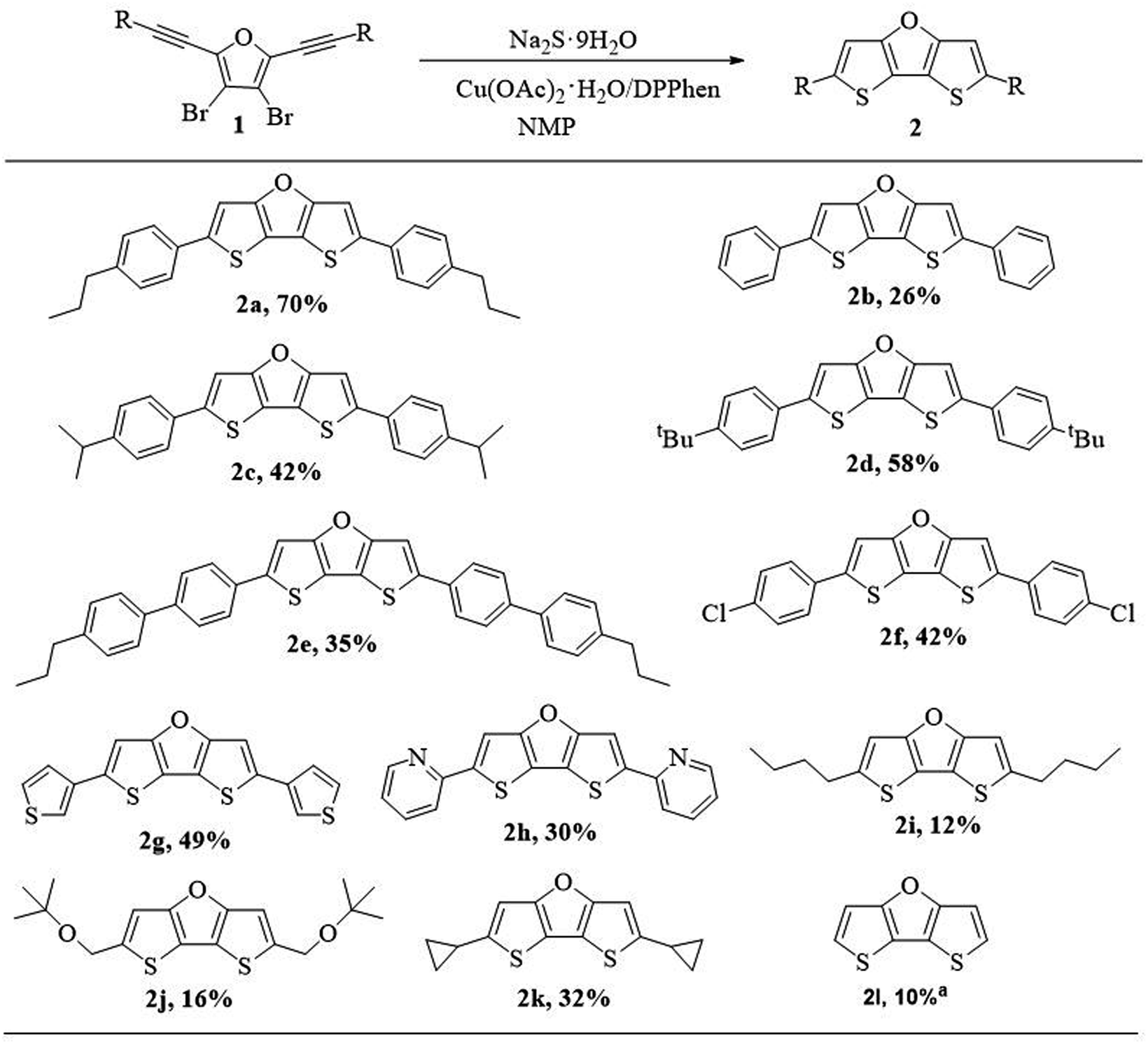 | ||
| Scheme 4 Substrate scope for the ring closure reaction for DTFs (2). aCompound 2l was prepared in a one-pot method using perbromofuran and trimethylsilylacetylene as starting materials. | ||
However, compounds with alkyl substituents or without substituent (2i–2l, 10–32%) were obtained in much lower yield, compared with the yields of compounds with phenyl or other aryl substituents (2a–2h, 26–70%). This is probably on account of the fact that phenyl and other aryl substituents are more electron-rich than alkyl groups, and electron-rich group could better stabilize the reaction intermediate for the ring closure reaction (see Scheme 5, proposed mechanism). To be noted, the relatively higher yield of 2k (32%) is attributed to the cyclopropyl group which is partly like an alkenyl group and thus could also contribute to stabilizing the reaction intermediate through hyperconjugation.
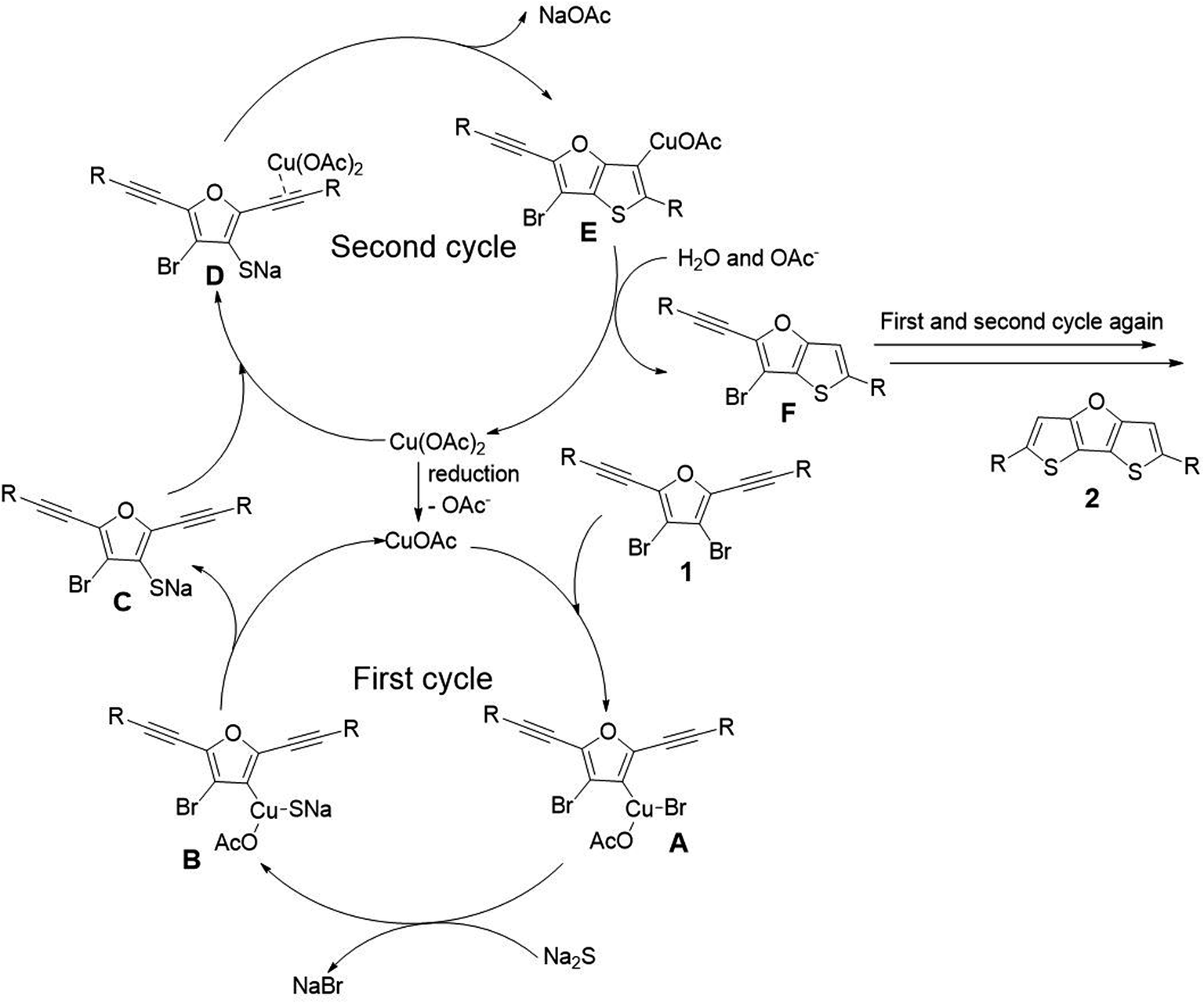 | ||
| Scheme 5 Proposed mechanism. | ||
Compounds 2a–2h with aryl substituents are stable toward air under ambient conditions and thermally stable under nitrogen atmosphere (see Fig. S1† for TGA traces). However, compounds 2i–2l with alkyl substituents or without substituent are not so stable under ambient conditions. They are partly decomposed after storage for several days, possibly because the C–H bond on the DTF backbone is slowly oxidized by air. The stability of 2k is relatively better than that of 2i, 2j and 2l. We suppose the different stability toward air is originated from different steric hindrance of different substituents: phenyl > cyclopropyl > alkyl > H.
The molecular structures of 2b (CCDC 2094066) and 2c (CCDC 2094067) were clearly revealed by single crystal X-ray diffraction analysis. Both compounds show good planarity, and are stacked in typical herringbone patterns with C–H⋯π interaction (Fig. 1 and 2), which are favourable for charge carrier transport in optoelectronic devices. The single crystal structure of the 2b molecule is disordered: the whole molecule has two possible positions with the same probability (50%–50%) (Fig. 1a). Two kinds of intermolecular interactions are observed: the interaction distance of C–H⋯π is 2.738–2.866 Å, and the interaction distance of C⋯S is 3.463 Å (Fig. 1b). The distance between two parallel molecules is relatively long, indicating no π⋯π interaction. The difference between compounds 2c and 2b is that 2c has an isopropyl chain attached to the benzene ring on both sides, which results in different packing behavior. For 2c, three kinds of intermolecular interactions were observed: the C–H⋯π interaction distance is 2.780–2.858 Å, the C⋯O interaction distance is 3.151 Å, and the C–H⋯S interaction distance is 2.852 Å (Fig. 2a).
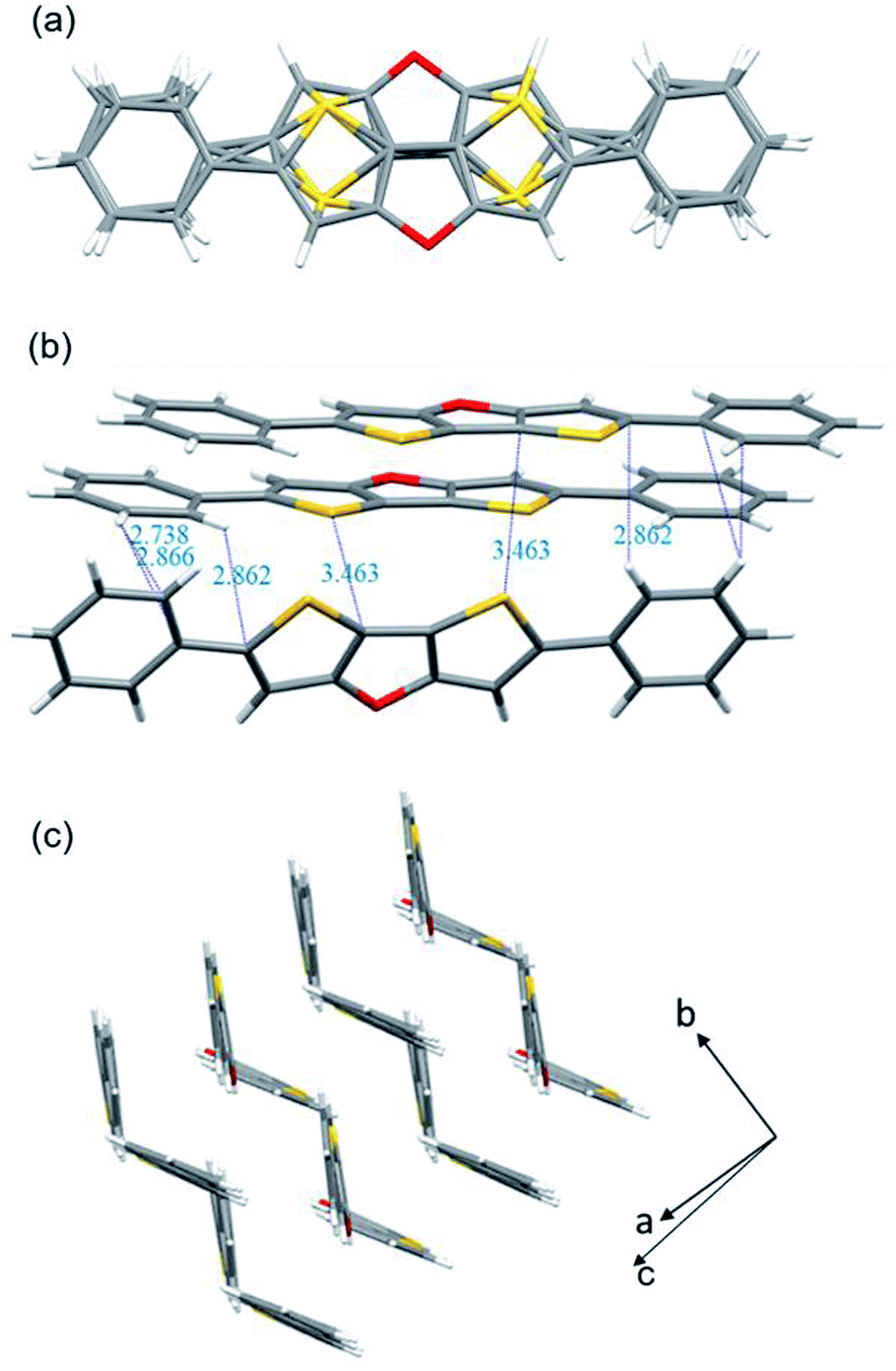 | ||
| Fig. 1 X-ray crystallographic analysis of 2b: (a) molecular structure with disorder (50%–50% probability); (b) intermolecular interactions; (c) stacking motif. For clarity, only one molecular structure is retained in (b) and (c). | ||
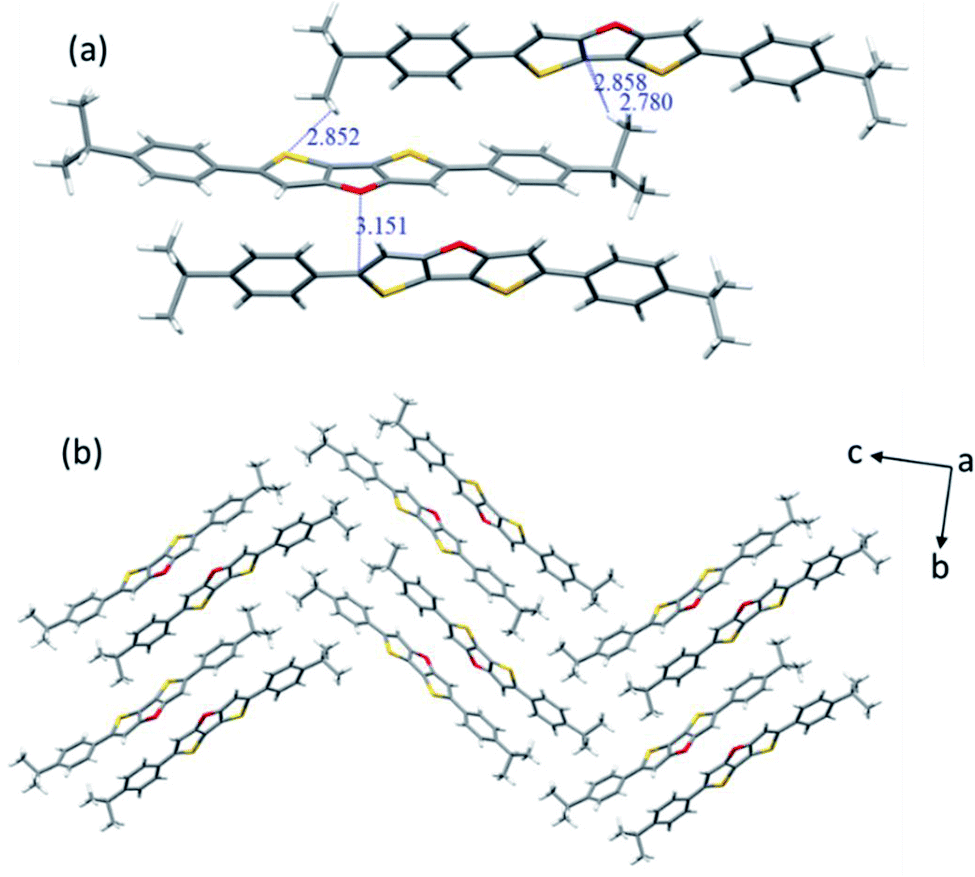 | ||
| Fig. 2 X-ray crystallographic analysis of 2c: (a) intermolecular interaction; (b) stacking motif. | ||
To gain mechanistic insights, 2 equivalent of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) and butylated hydroxytoluene (BHT) were added as radical scavenger under the optimized condition. The formation of desired product 2a was not inhibited, suggesting that there is no radical pathway involved in the reaction. Therefore, we propose a mechanism including two catalytic cycles as shown in Scheme 5.19 The first catalytic cycle is a typical Cu(I)/Cu(III) catalytic cycle. A small amount of Cu(II) is reduced to Cu(I) at first, and then Cu(I) is subjected to oxidative addition with substrate 1 to afford intermediate A. Then transmetallization with sodium sulfide affords intermediate B. Finally, intermediate C is obtained through reductive elimination. The second catalytic cycle is Cu(II) induced cyclization mechanism. The triple bond of intermediate C is activated by coordination with Cu(OAc)2, and the subsequent attack by sulfur anion affords the ring closure intermediate E. Finally, intermediate E abstracts proton from the coordination water of sodium sulfide to obtain intermediate F. Thus one thiophene ring has been constructed. The second thiophene ring of target product is formed via the similar mechanism as described above, and thus the target product 2 is obtained.
In summary, we have developed a novel copper-catalysed synthesis of new DTFs under mild reaction conditions. Copper catalysed C–S coupling between 3,4-dibromo-2,5-dialkynylfuran (1) and Na2S·9H2O and subsequent ring-closure reaction afforded DTFs (2) in reasonable yields. This strategy shows broad substrate scope and can be used to prepare not only aryl, heteroaryl but also alkyl substituted DTF derivatives with up to 70% yield. The yields are not high, but it is understandable due to dual C–S coupling and dual ring-closure reaction involved in the synthesis. Furthermore, we proposed a mechanism including two catalytic cycles: a typical Cu(I)/Cu(III) catalytic cycle and subsequent Cu(II) induced cyclization mechanism. By the way, we have also tried to synthesize bisthieno[3,2-b:2′,3′-d]thiophene compounds under the optimal reaction conditions, e.g., 2,6-bis(4-propylphenyl)dithieno[3,2-b:2′,3′-d]thiophene, however, we failed to isolate the target compound due to very low yield.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the financial support from the National Natural Science Foundation of China (21402194), Fundamental Research Funds for the Central Universities, and University of Chinese Academy of Sciences.Notes and references
- (a) U. Scherf, J. Mater. Chem., 1999, 9, 1853–1864 RSC; (b) D. J. Gundlach, J. A. Nichols, L. Zhou and T. N. Jackson, Appl. Phys. Lett., 2002, 80, 2925–2927 CrossRef CAS; (c) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed; (d) M.-C. Chen, C. Kim, S.-Y. Chen, Y.-J. Chiang, M.-C. Chung, A. Facchetti and T. J. Marks, J. Mater. Chem., 2008, 18, 1029–1036 RSC; (e) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed; (f) H. Usta, A. Facchettti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501–510 CrossRef CAS PubMed; (g) J. S. Wu, S. W. Cheng, Y. J. Cheng and C. S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC; (h) M. Stepien, E. Gonka, M. Zyla and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef CAS PubMed; (i) G.-J. Deng, Org. Lett., 2019, 21, 8630–8634 CrossRef PubMed.
- (a) K. Xiao, Y. Liu, T. Qi, W. Zhang, F. Wang, J. Gao, W. Qiu, Y. Ma, G. Cui, S. Chen, X. Zhan, G. Yu, J. Qin, W. Hu and D. Zhu, J. Am. Chem. Soc., 2005, 127, 13281–13286 CrossRef CAS PubMed; (b) Y. M. Sun, Y. Q. Ma, Y. Q. Liu, Y. Y. Lin, Z. Y. Wang, Y. Wang, C. A. Di, K. Xiao, X. M. Chen, W. F. Qiu, B. Zhang, G. Yu, W. P. Hu and D. B. Zhu, Adv. Funct. Mater., 2006, 16, 426–432 CrossRef CAS; (c) T. Osaka, T. Abe, S. Shinamura, E. Miyazaki and K. Takimiya, J. Am. Chem. Soc., 2010, 132, 5000–5001 CrossRef PubMed; (d) T. Li, S. Dai, Z. Ke, L. Yang, J. Wang, C. Yan, W. Ma and X. Zhan, Adv. Mater., 2018, 30, 1705969 CrossRef PubMed; (e) X. Shi, J. Chen, K. Gao, L. Zuo, Z. Yao, F. Liu, J. Tang and A. K. Y. Jen, Adv. Energy Mater., 2018, 8, 1702831 CrossRef.
- (a) K. Takimiya, H. Ebata, K. Sakamoto, T. Izawa, T. Otsubo and Y. Kunugi, J. Am. Chem. Soc., 2006, 128, 12604–12605 CrossRef CAS PubMed; (b) T. Mori, T. Nishimura, T. Yamamoto, I. Doi, E. Miyazaki, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 13900–13913 CrossRef CAS; (c) H. Usta, D. Kim, R. Ozdemir, Y. Zorlu, S. Kim, M. C. Ruiz Delgado, A. Harbuzaru, S. Kim, G. Demirel, J. Hong, Y.-G. Ha, K. Cho, A. Facchetti and M.-G. Kim, Chem. Mater., 2019, 31, 5254–5263 CrossRef CAS.
- (a) J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed; (b) O. Gidron, A. Dadvand, Y. Sheynin, M. Bendikov and D. F. Perepichka, Chem. Commun., 2011, 47, 1976–1978 RSC.
- (a) O. Gidron, Y. D. Posner and M. Bendikov, J. Am. Chem. Soc., 2010, 132, 2148–2150 CrossRef CAS PubMed; (b) B. Li, Chin. J. Org. Chem., 2015, 35, 2487–2506 CrossRef CAS; (c) Y. Liu, J. Yuan, Y. Zou and Y. Li, Acta Chim. Sin., 2017, 75, 257–270 CrossRef CAS.
- (a) J. T. Henssler and A. J. Matzger, J. Org. Chem., 2012, 77, 9298–9303 CrossRef CAS PubMed; (b) J. S. d. Melo, F. Elisei and R. S. Becker, J. Chem. Phys., 2002, 117, 4428–4435 CrossRef; (c) V. Shirinian, A. Shimkin, S. Tipikin and M. Krayushkin, Synthesis, 2009, 22, 3803–3806 CrossRef.
- Y. S. Yang, T. Yasuda and C. Adachi, Bull. Chem. Soc. Jpn., 2012, 85, 1186–1191 CrossRef CAS.
- W. Li, F.-Q. Bai, J. Chen, J. Wang and H.-X. Zhang, J. Power Sources, 2015, 275, 207–216 CrossRef CAS.
- K. Černovská, J. Svoboda, I. Stibor, M. Glogarová, P. Vaněk and V. Novotná, Ferroelectrics, 2000, 241, 231–238 CrossRef.
- Z. Yuan, A. H. Younes, J. R. Allen, M. W. Davidson and L. Zhu, J. Org. Chem., 2015, 80, 5600–5610 CrossRef CAS PubMed.
- (a) D. Chen, D. Yuan, C. Zhang, H. Wu, J. Zhang, B. Li and X. Zhu, J. Org. Chem., 2017, 82, 10920–10927 CrossRef CAS PubMed; (b) W. Ma, J. Huang, C. Li, Y. Jiang, B. Li, T. Qi and X. Zhu, RSC Adv., 2019, 9, 7123–7127 RSC.
- (a) F. D. Jong and M. J. Janss, J. Org. Chem., 1971, 36, 1645–1648 CrossRef; (b) F. Allared, J. Hellberg and T. Remonen, Tetrahedron Lett., 2002, 43, 1553–1554 CrossRef CAS; (c) J. Frey, A. D. Bond and A. B. Holmes, Chem. Commun., 2002, 20, 2424–2425 RSC; (d) M.-C. Chen, Y.-J. Chiang, C. Kim, Y.-J. Guo, S.-Y. Chen, Y.-J. Liang, Y.-W. Huang, T.-S. Hu, G.-H. Lee, A. Facchetti and T. J. Marks, Chem. Commun., 2009, 14, 1846–1849 RSC; (e) P. Oechsle, P. Hou, U. Flçrke and J. Paradies, Adv. Synth. Catal., 2016, 358, 3770–3776 CrossRef CAS.
- G. M. Karminski-Zamola, M. Malešević, M. Bajić and G. Golja, Croat. Chem. Acta, 1992, 65, 847–849 CAS.
- V. Kozmík, M. Pozník, J. Svoboda and P. Frére, Tetrahedron Lett., 2015, 56, 6251–6253 CrossRef.
- K. Mitsudo, Y. Kurimoto, H. Mandai and S. Suga, Org. Lett., 2017, 19, 2821–2824 CrossRef CAS PubMed.
- C. W. Shoppee, J. Chem. Soc., Perkin Trans. 1, 1985, 1, 45–52 RSC.
- I. Malik, Z. Ahmad, S. Reimann, M. Nawaz, T. Patonay and P. Langer, Synlett, 2012, 23, 1463–1466 CrossRef CAS.
- (a) K. Sonogashira, Handbook of Organopalladium Chemistry for Organic Synthesis, John Wiley & Sons, Inc, 2002, pp. 493–529 Search PubMed; (b) J. A. Marsden and M. M. Haley, Metal-Catalyzed Cross-Coupling Reactions, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2nd edn, 2004, pp. 317–394 Search PubMed.
- (a) D. Ma, S. Xie, P. Xue, X. Zhang, J. Dong and Y. Jiang, Angew. Chem., Int. Ed. Engl., 2009, 48, 4222–4225 CrossRef CAS PubMed; (b) L. L. Sun, C. L. Deng, R. Y. Tang and X. G. Zhang, J. Org. Chem., 2011, 76, 7546–7550 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, thermogravimetric data, crystal structure data, and other additional information. CCDC 2094066 and 2094067. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06881d |
| This journal is © The Royal Society of Chemistry 2021 |