
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of 1T@2H MoS2 heterostructures in situ from natural molybdenite with enhanced electrochemical performance for lithium-ion batteries†
ChengLong Peng ,
Mingming Shi,
Fei Li,
Yang Wang,
Xueqin Liu,
HuaSheng Liu and
Zhen Li*
,
Mingming Shi,
Fei Li,
Yang Wang,
Xueqin Liu,
HuaSheng Liu and
Zhen Li*
Engineering Research Center of Nano-Geomaterials of Ministry of Education, Faculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, China. E-mail: zhenli@cug.edu.cn
First published on 13th October 2021
Abstract
Natural molybdenite, an inexpensive and naturally abundant material, can be directly used as an anode material for lithium-ion batteries. However, how to release the intrinsic capacity of natural molybdenite to achieve high rate performance and high capacity is still a challenge. Herein, we introduce an innovative, effective, and one-step approach to preparing a type of heterostructure material containing 1T@2H MoS2 crafted from insertion and expansion of natural molybdenite. The metallic 1T phase formed in situ can significantly improve the electronic conductivity of MoS2. At the same time, 1T@2H MoS2 heterostructures can provide an internal electric field (E-field) to accelerate the migration rate of electrons and ions, promote the charge transfer behaviour, and ensure the reaction reversibility and lithium storage kinetics. Such worm-like 1T@2H MoS2 heterostructures also have a large specific surface area and a large number of defects, which will help shorten the lithium-ion transmission distance and provide more ion transmission channels. As a result, it exhibits a discharge capacity of 788 mA h g−1 remarkably at 100 mA g−1 after 485 cycles and stable cycling performance. It also shows excellent magnification performance of 727 mA h g−1 at 1 A g−1, compared to molybdenite concentrate. Briefly, this work's heterostructure architectures open up a new avenue for applying natural molybdenite in lithium-ion batteries, which has the potential to achieve large-scale commercial applications.
Introduction
As one of the most critical energy storage systems, lithium-ion batteries (LIBs) have advanced energy storage and conversion systems. Exploring electrode materials with functional electrochemical performance and large-scale application is the current research hotspot.1,2 Transition metal sulfides have been considered as promising anode materials for LIBs because of their graphite-like layered structure, adjustable interlayer distance and bandgap, diverse composition, and good electrochemical reversibility in their high specific capacity. Among them, MoS2, as the most representative, has a higher theoretical capacity (669 mA h g−1) and a larger interlayer spacing (0.615 nm), which can achieve rapid transmission of alkali metal ions. Meanwhile, the capacity of MoS2 in actual research can even be as high as 1000 mA h g−1. Mo atoms can accommodate a large number of Li ions in the cycle process, which can provide additional capacity.3 The high lithium storage mechanism facilitates the development of energy storage equipment with high capacity and power density.So far, MoS2 are synthesized by chemical methods in most studies, such as hydrothermal,4 chemical vapor deposition,5 solvothermal,6,7 other way,8–10 etc. Although the above chemical synthesis methods can prepare MoS2 on a large scale, their industrial application is still limited by strict reaction conditions and environmental pollution. In nature, MoS2 exists in the form of natural molybdenite and is generally obtained by physical purification. Therefore, preparing MoS2 material directly from natural molybdenite concentrate can eliminate many intermediate complex steps and reduce synthetic pollutants. Compared with synthetic MoS2, natural molybdenite also has the advantages of abundant resources and low cost, which makes it become a potential candidate of commercial energy storage material for sustainable development. Unfortunately, during the charging and discharging process of natural molybdenite and synthetic MoS2, the active material will be crushed and loosely contacted due to their low internal electronic conductivity and significant volume changes, and eventually leads to rapid capacity degradation.11,12 Improving the electronic conductivity of MoS2 to achieve its high capacity and cycle stability is still a challenge. There are roughly two general strategies to overcome the issues: (1) composite with carbon-based materials to improve the material's electrical conductivity, such as MoS2/graphite heterostructures, or with graphene to form MoS2@C@RGO composites.12,13 However, this strategy also has drawbacks such as complex processes, high cost, and reduced specific capacity; (2) accomplish few layers through the insertion/exfoliation method, which will effectively shorten the transmission distance of lithium ions and ensure transmission efficiency.11,14 Simultaneously, a part of the triangular 2H phase usually transforms into the octahedral 1T phase during the insertion/exfoliation process to make Mo and S coordinate differently, giving it a different electronic structure. Noted, because of its metallicity (107 times more conductive than 2H), hydrophilicity, and larger layer spacing, 1T MoS2 can be used in lithium-ion batteries to effectively reduce charge transfer resistance and accelerate the ion transfer rate.15–18 In recent research, the chemical intercalation exfoliation19 and electrochemical intercalation exfoliation20 would have realized the few layers and crystal phase transformation of bulky MoS2. Still, it has drawbacks, including complex processes, low yield, low tap density, and easy aggregation of the nanostructures. Therefore, we need a simple design that can guarantee the bulky natural molybdenite's advantages of high tap density and non-agglomeration, improve its electronic conductivity, release its inherent capacity, and achieve high rate performance.
To better resolve the issues, we designed a MoS2 heterostructures anode material composed of abundant 1T MoS2 and 2H MoS2 (denoted as 1T@2H MoS2). The 1T@2H MoS2 heterostructures were fabricated by a novel and simple expansion method. Due to its metallic properties, 1T MoS2 synthesized in situ from natural molybdenite could enhance its electrical conductivity and decrease the kinetic barrier.17 The 1T@2H MoS2 heterostructures generated an internal E-field, dramatically accelerating the migration rate of ions and electrons and improved the reaction kinetics. Simultaneously, the natural molybdenite was worm-like, which retained the high tap density of the bulk material, which significantly increased the specific surface areas and pore volumes, and provided more ion channels and exposed active sites. Since there is enough void space between the adjacent layers of MoS2, a worm-like structure is beneficial to accommodate the volume change during the intercalation of lithium ions. Furthermore, 1T MoS2 and the designed E-field can initially solve carriers' transport, electrons transfer, and ions diffusion in the dense MoS2 structure. As a result, the worm-like 1T@2H MoS2 heterostructures had higher electronic conductivity and Li+ diffusion rate than natural molybdenite, indicating a higher specific capacity, good cycle stability, and excellent magnification performance. 1T@2H MoS2 heterostructures will improve the lithium storage performance. All these features make it a promising material for practical application.
Experimental section
Preparation of 1T@2H MoS2
All the reagents were purchased from Sinopharm and used without other purification. Molybdenite concentrate (MoS2 content: ∼78%, Luanchuan City, Henan Province, China) was obtained from raw ore through flotation and grinding. In the experiment, 1T@2H MoS2 heterostructures were received by the expansion method. Molybdenite concentrate (1.5 g), sodium hydroxide (20 g), hydrazine hydrate (20 mL, wt% = 85%), and magnetons were sealed in a PTFE autoclave, heated, and stirred in an oil bath at 150 °C for 24 h. The bulky molybdenite concentrate was put into the autoclave at 150 °C and stirred by magnetic force. The worm-like 1T@2H MoS2 was washed with deionized water and ethanol until it was neutral. Finally, the samples were placed in an oven at 60 °C overnight to obtain 1T@2H MoS2. The reaction temperature, reaction time, and the concentration of sodium hydroxide all have essential effects on the worm-like 1T@2H MoS2 heterostructures. Table S1† listed the different reaction conditions such as reaction temperature, reaction time, and sodium hydroxide concentration to prepare 1T@2H MoS2 heterostructures.Characterization
Crystal structures were measured using X-ray diffraction (XRD, Bruker AXS D8 Advance) with Cu K radiation (=1.5406 Å) in a scan range of 5–70°. The element contents of the sample were analyzed by X-ray fluorescence (XRF, XRF-AXIOSmax). Raman (ThermoFisher Scientific) was a non-destructive and effective method for collecting sample structure and molecular rotation information with a test range of 400–4000 cm−1. The morphology and microstructure of the samples were characterized by HITACHI SU8010 scanning field-emission electron microscope (FESEM). A transmission electron microscope (TEM, JEOL 2100), high-resolution (HRTEM), and scanning transmission electron microscopy (STEM) was used to observe the fine structure of the samples. X-ray photoelectron spectroscopy (XPS, Thermo Scientific, Al Kα X-ray) equipped with Al Kα X-ray source was used to study the sample's chemical state and valence state. The diffuse reflection spectra of ultraviolet-visible (UV-Vis) were investigated by Shimadzu UV-2600 spectrophotometer. N2 adsorption and desorption isotherm (BET, ASAP2460) was used to measure the sample's specific surface area and pore size distribution characteristics. The BT-9300ST laser particle size analyzer analyzed the particle size of the samples. A four-probe tester (RTS-9, 10−5 ∼ 105 S cm−1) measured the electronic conductivity of the model.Electrochemical measurement
During the battery test, the active substance, conductive agent acetylene black, and binder sodium alginate were mixed in an aqueous solution at a mass ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 to prepare the working electrode. Then the slurry mixture was uniformly coated on the copper foil. Finally, the powder-coated copper foil was placed in a vacuum oven at 80 °C for 12 hours and used as the final working electrode. The mass density of the active material of each electrode sheet is 1.5–2.0 mg cm−2. LiR2032 half cells were assembled in a glove box filled with argon (Mikrouna, O2, and H2O ≤ 0.01 ppm). The prepared electrode was used as anode material, and the lithium metal sheet was used as counter-electrode. 1 M LIPF6 in a 50:50 (w/w) mixture of dimethyl carbonate (DMC) and ethylene carbonate (EC) was used as the electrolytes. A CT2001A cell tester (Land, China) was used to perform Galvanostatic cycle stability within the potential range of 0.01–3.0 V (vs. Li+/Li) and test rate performance at different current densities. Electrochemical impedance spectroscopy (EIS) tests ranged from 0.01 Hz to 100 kHz, and cyclic voltammetry (CV) studies were conducted at the CHI660E Electrochemical Workstation (CHI-Instrument).
:2:1 to prepare the working electrode. Then the slurry mixture was uniformly coated on the copper foil. Finally, the powder-coated copper foil was placed in a vacuum oven at 80 °C for 12 hours and used as the final working electrode. The mass density of the active material of each electrode sheet is 1.5–2.0 mg cm−2. LiR2032 half cells were assembled in a glove box filled with argon (Mikrouna, O2, and H2O ≤ 0.01 ppm). The prepared electrode was used as anode material, and the lithium metal sheet was used as counter-electrode. 1 M LIPF6 in a 50:50 (w/w) mixture of dimethyl carbonate (DMC) and ethylene carbonate (EC) was used as the electrolytes. A CT2001A cell tester (Land, China) was used to perform Galvanostatic cycle stability within the potential range of 0.01–3.0 V (vs. Li+/Li) and test rate performance at different current densities. Electrochemical impedance spectroscopy (EIS) tests ranged from 0.01 Hz to 100 kHz, and cyclic voltammetry (CV) studies were conducted at the CHI660E Electrochemical Workstation (CHI-Instrument).
Results and discussion
Due to the low electronic conductivity of natural molybdenite, it hinders the transport of ions and electrons. This enables natural molybdenite to adopt a reasonable and universal strategy to accelerate the transmission of charge carriers. Heterostructures were constructed to allow the formation of an internal E-field in the bulk MoS2 to accelerate reaction kinetics and transport charge carriers. In theory, the strength of the electric field is crucial to the carrier transmission rate, and the bandgap difference of the components shows a positive linear relationship.21–23 The natural molybdenite belongs to 2H MoS2 with an indirect bandgap of about 1.17 eV and a low inherent electronic conductivity.11 However, 1T MoS2 has a metallic property with a bandgap difference of about 0 eV.24 When the 1T phase was in contact with the 2H phase, a relatively strong electric field could be formed, providing rapid charge transfer dynamics (Fig. 1). Electrons move from the 2H MoS2 at the high Fermi level to the 1T MoS2 at the low Fermi level, and an internal electric field is generated to balance this transfer. The key is to transform part of the 2H phase in situ into 1T phase in natural molybdenite and construct 1T@2H MoS2 heterostructures. To verify the heterostructure properties can enhance the electron migration rate, the powder electrical conductivity of 1T@2H MoS2 was tested to be 10 S cm−1. It showed more than 106 of the molybdenite concentrate and ∼4.5 × 104 times of the commercial MoS2 (Table S2†).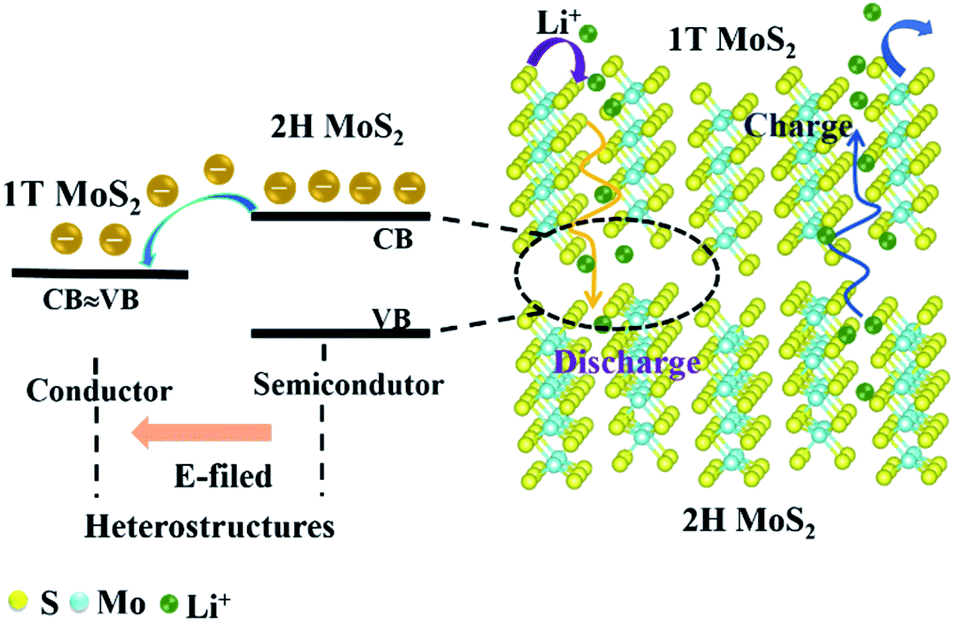 | ||
| Fig. 1 Schematic of the internal E-field of the 1T@2H MoS2 heterostructures. | ||
Fig. 2 illustrates the new synthesis method of 1T@2H MoS2 heterostructures with worm-like prepared from natural molybdenite via the expansion. The 1T@2H MoS2 heterostructures were caused by the synergistic effect of hydrazine hydrate and sodium hydroxide through insertion and expansion. Hydrazine and sodium hydroxide overcome the force between the MoS2 layers to expand in a high-pressure and high-temperature environment. The redox rearrangement model can explain the formation of 1T@2H MoS2 heterostructures. N2H4 was oxidized to N2H5+ upon intercalation. Under heating conditions, N2H5+ could be decomposed and gasified into N2, NH3, and H2 because it was not heat-resistant.25,26 These gases overcame the van der Waals force between MoS2 layers to expand, significantly boosting the volume of MoS2 and forming many pores. Simultaneously, MoS2 was reduced and negatively charged, and Na+ was first adsorbed on the crystal surface. The van Waals forces of the first two layers continued to weaken and penetrated to the next layer to form NaxMoS2+, increasing the electronic energy in the MoS2 layer. This structural change from the 2H to 1T phase is crucial. Note that the transformation of the trigonal prismatic coordinated 2H MoS2 into the octahedral coordinated 1T MoS2 could effectively improve the electronic conductivity of the MoS2. In addition, sodium hydroxide has another vital role in removing other impurities in molybdenite concentrate components to obtain high purity MoS2. The expansion method has a low cost and simple process for the scalable production of 1T@2H MoS2 heterostructures.
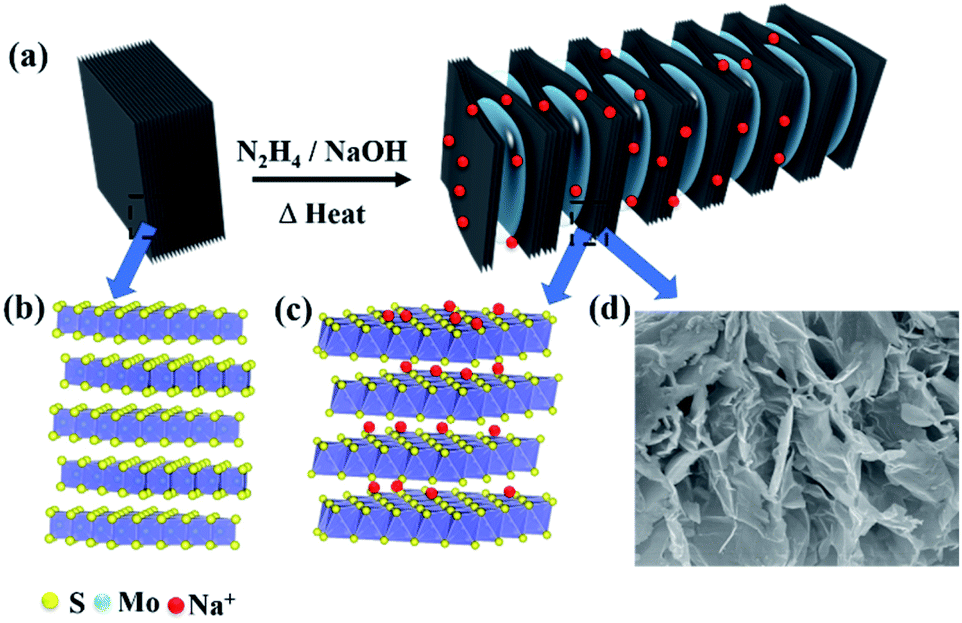 | ||
| Fig. 2 (a) Schematic diagram of the preparation process of 1T@2H MoS2 heterostructures with worm-like. (b) 2H MoS2 crystal structure model. (c) 1T MoS2 crystal structure model of Na+ embedded 2H MoS2. (d) Local FESEM image of 1T@2H MoS2. | ||
The crystal phase and structure of molybdenite concentrate and 1T@2H MoS2 samples were detected by XRD and Raman (Fig. 3). As shown in Fig. 3a, the prominent diffraction peaks of molybdenite concentrate are at 2θ = 14.47°, 32.71° and 60.18° corresponding to the (002), (100) and (110) crystal planes of 2H MoS2 (PDF: 65-0160), respectively. Talc (PDF: 83-1768) and quartz (PDF: 75-0443) are also exist with molybdenite concentrate. Through XRD semi-quantitative analysis, the purity of molybdenite concentrate is about 78.51%. After expansion, the impurity phase disappeared, and the characteristic peaks (2θ = 14.50°) of MoS2 shifted to a high angle, which was attributed to the production of 1T MoS2.27,28 A new peak appeared at a low angle of 2θ = 7.26°, corresponding to the (001) crystal plane, with Na+ insertion to form NaxMoS2.29,30 The characteristic peak intensity of 1T@2H MoS2 is lower than that of molybdenite concentrate, indicating the synergistic effect of hydrazine and sodium hydroxide expanded successfully natural molybdenite.20,31 The XRF test (Table S3†) further verified that after the expansion of molybdenite concentrate, the content of Mo and S elements increased significantly, while other impurities decreased greatly. Meanwhile, the content of 2.52% Na2O appeared, proving that Na+ was embedded in MoS2 layers, causing the transition from the 2H to the 1T phase. According to different process conditions such as reaction temperature, time, and sodium hydroxide concentration, 1T@2H MoS2 was prepared and subjected to an XRD test (Fig. S1†). The structure shows that the best expansion process is a reaction at 150 °C for 24 h with 25 mol L−1 sodium hydroxide.
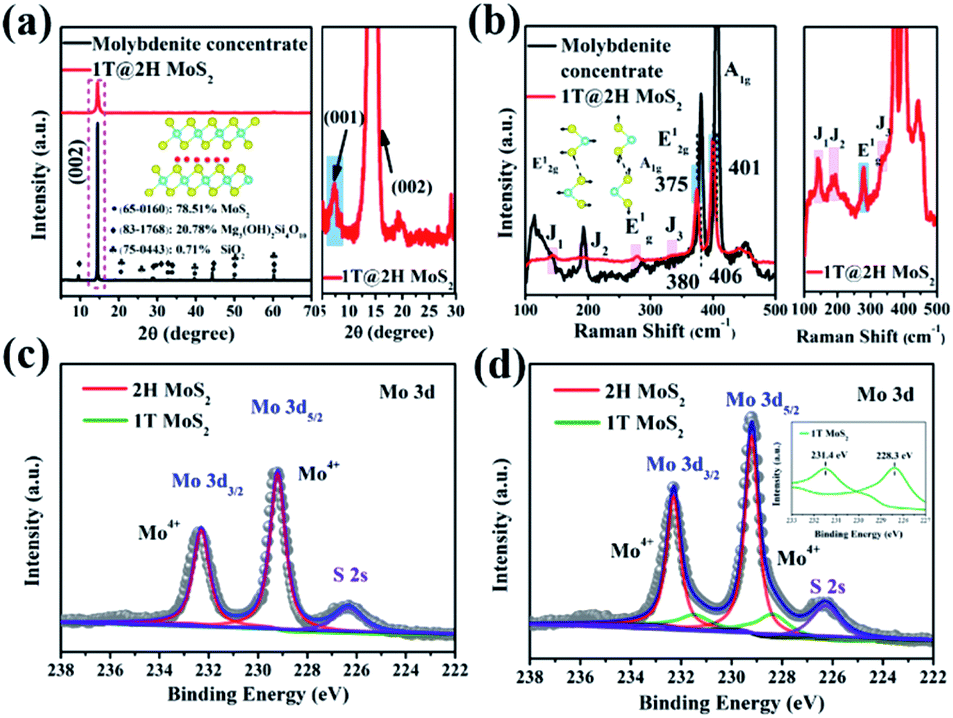 | ||
| Fig. 3 (a) XRD patterns of molybdenite concentrate and 1T@2H MoS2. (b) Raman patterns of molybdenite concentrate and 1T@2H MoS2. (c) Mo 3d spectra of molybdenite concentrate. (d) Mo 3d spectra of 1T@2H MoS2. | ||
Raman spectroscopy is a sensitive method to identify the generation of 1T MoS2.28 Fig. 3b shows that molybdenite concentrate has two significant characteristic peaks at 380 cm−1 (E12g, Mo–S in-plane vibration mode) and 406 cm−1 (A1g, Mo–S out-of-plane vibration mode).32 After expansion, in addition to E12g (375 cm−1) and A1g (401 cm−1), a series of additional Raman signals appeared at 144 cm−1 (J1), 197 cm−1 (J2), 279 cm−1 (E1g), and 332 cm−1 (J3).33–35 The E1g band is associated with the octahedral coordination of Mo in 1T MoS2.33 These new characteristic peaks prove the formation of 1T MoS2. Remarkably, compared with molybdenite concentrate, the characteristic fingerprinting peaks of MoS2 after expansion slightly shifted to lower wavenumbers, indicating the formation amount 1T@2H MoS2 heterostructures. The red-shift of the Raman spectrum provides adequate evidence for the heterostructures interaction between 1T and 2H MoS2.11,36,37 UV-Vis absorption spectra tested the molybdenite concentrate and 1T@2H MoS2 prepared under different conditions, and the results are shown in Fig. S2†. In contrast, after the expansion of molybdenite concentrate, the dispersion was gray, and the exciton peaks at A and B were not apparent, which also proved the existence of 1T MoS2.38,39 Moreover, XPS semi-quantitative method was used to verify that the 1T phase was produced in 1T@2H MoS2, and the relative content ratio of the 2H and 1T phase was determined by analysis.34 AS shown in Fig. 3c and d, the Mo 3d5/2 and Mo 3d3/2 peaks for 1T@2H MoS2 exhibit a shift of 0.1 eV to lower binding energy, attributed to the production of the 1T phase.15,34 In the Mo 3d region, Mo4+ 3d5/2 and Mo4+ 3d3/2 are at 229.2 eV and 232.3 eV, respectively (calibrated with C 1s peak at 284.5 eV), which belong to 2H MoS2.15,40 The corresponding peak of Mo in 1T MoS2 is located at a lower position near 228.3 and 231.4 eV.29,40 The same trend exists in the S 2p XPS area (Fig. S3†). By calculating the peak area of Mo 3d, the 1T phase composed about 20% in the 1T@2H MoS2.
According to XPS analysis, it can be concluded that the change of molybdenite concentrate and 1T@2H MoS2 are originated from the electronic migration synergistic effect between 1T and 2H phase heterostructures.
FESEM and TEM were used to examine the morphology and structure of the samples. In particular, HRTEM can effectively prove the existence of 1T MoS2. As a comparison, natural molybdenite is a compact lamellar accumulation structure with a thickness of fewer than 5 μm in most cases (Fig. S4(a–c)†), accompanied by its average volume diameter of 42 μm (Fig. S4d†). The 1T@2H MoS2 appears worm-like with the interlayer spacing opening and the formation of multilevel pore structures on the lamellar surface (Fig. 3a–c). This increases the contact area between the electrode materials and the electrolyte. Worm-like structures are beneficial for accommodating the enlargement of volume during the Li+ intercalation owing to enough void space between neighboring sheets. The nitrogen adsorption–desorption isotherm results (Fig. S5†) validated that 1T@2H MoS2 has a high specific surface area and pore volume, respectively 46.91 m2 g−1 and 0.145 cm3 g−1, 38 and 29 times that of molybdenite concentrate. Moreover, the pore size distribution results showed that abundant micropores of 1T@2H MoS2 mainly concentrated at 2 nm by Barrett–Joyner–Halenda (BJH) analysis (inset of Fig. S5b†). 1T@2H MoS2 has a high specific surface area and rich micropores, which is beneficial to accelerate electrolyte penetration and provide more Li-ion channels. The FSSEM images of 1T@2H MoS2 prepared under different process conditions are shown in Fig. S6–S8.† When the oil bath was at 150 °C for 24 h with 25 mol L−1 sodium hydroxide, the expansion was the most sufficient. This is consistent with the XRD conclusions. The TEM image (Fig. 4d) reveals that several micro-sized few layers of MoS2 constructed the bulky worm-like 1T@2H MoS2 with many holes on the surface. HRTEM image in Fig. 4e, the interlayer spacing of MoS2 at the defect sites increased in varying degrees. The interlayer spacing is 0.55–0.78 nm, suggesting Li+ is easier to diffuse into the expanded interlayer with vacancies.41 The layer spacing is consistent with XRD results. HRTEM images (Fig. 4f) showed that intermittent lattice fringes appeared on the MoS2 sheets, indicating the existence of S defects. The abundant defects could also realize the 3D diffusion of lithium ions.42,43 Fig. 4g and h displayed two amplified HRTEM images intercepted from Fig. 4f, showing some hexagonal lattice regions with 2H MoS2 and triangular lattice regions 1T MoS2, which directly proved the existence of 1T MoS2.44 The illustrations in the upper right corner of Fig. 4g and h were obtained by Fourier transformation of the D and H regions, corresponding to 2H and 1T phases, respectively. Fig. 4i, STEM-EDX confirmed that Mo, S, and Na were uniformly distributed on the 1T@2H MoS2 sheets, which further ensured Na+ insert into the MoS2 interlayer.
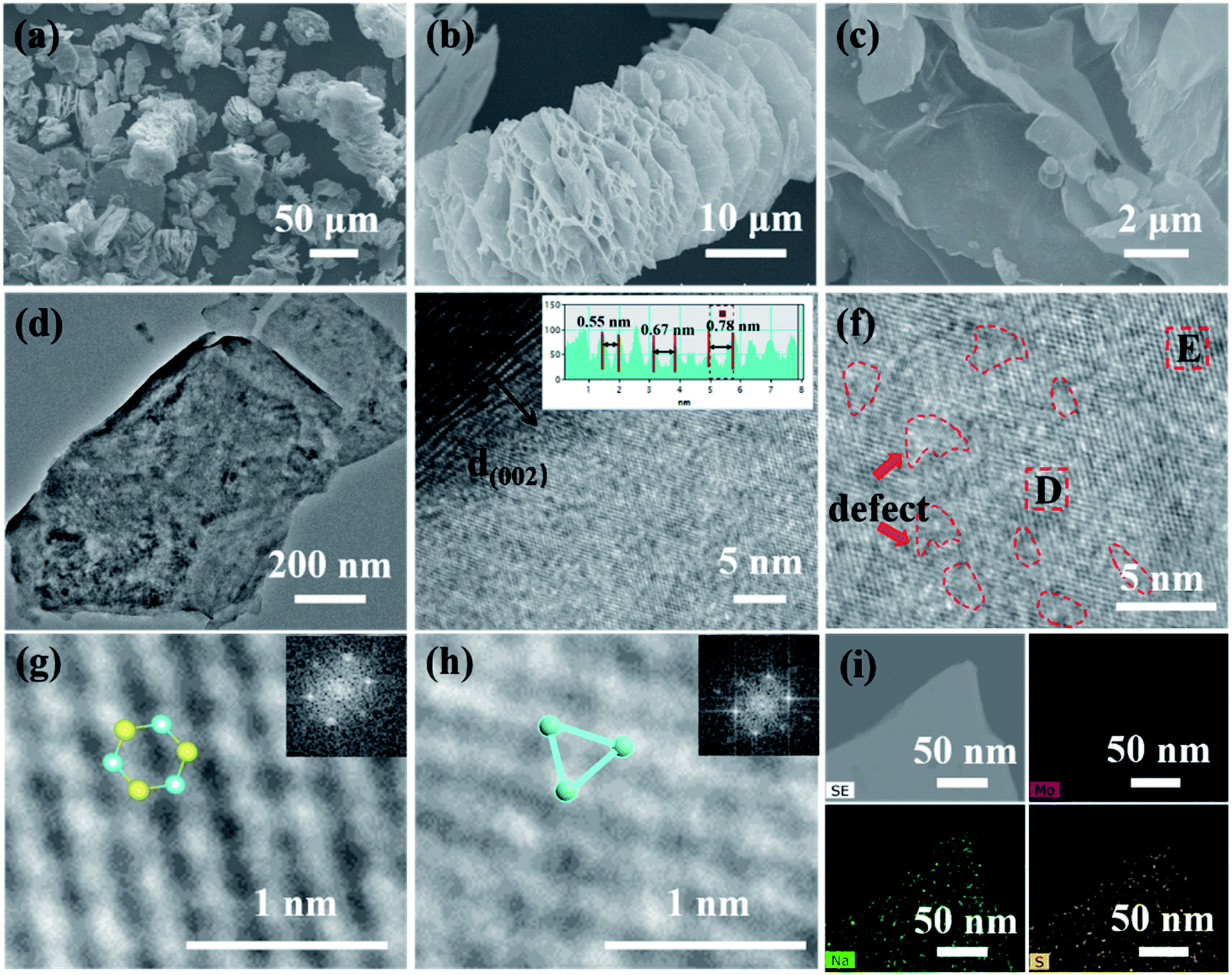 | ||
| Fig. 4 (a–c) SEM images of 1T@2H MoS2. (d) TEM, and (e–h) high-resolution TEM images of 1T@2H MoS2. The insets in (g) and (h) are atomic models and fast-Fourier-transform patterns for 2H MoS2 and 1T MoS2. The illustrations in (g) and (h) are the atomic models of 2H MoS2 and 1T MoS2. (i) STEM-EDX element mapping, Mo (red), Na (yellow), S (blue), respectively. | ||
The metallic 1T MoS2 with high electron conductivity formed in situ could include an internal E-field with 2H MoS2 to accelerate the transport of lithium ions and electrons. Fig. 5a shows the initial three cyclic voltammograms (CV) curves of the 1T@2H MoS2 heterostructures anode at a scan rate of 0.2 mV s−1 to explore the occurred reaction's details. Molybdenite concentrate has a similar changing trend as reference material (Fig. S9a†). Typical redox reaction characteristics were observed in the CV curve of MoS2, which exhibited two reduction peaks at ∼0.8 V and ∼0.4 V in the first cathodic scan. The former represents Li+ embedded in MoS2 to form LixMoS2 (i.e., MoS2 + xLi+ → LixMoS2), while the latter is attributed to Li+ embedded in LixMoS2 transform into metallic Mo particles and Li2S (i.e., LixMoS2 + (4 − x)Li+ + (4 − x)e− → Li2S + Mo).45,46 In addition, Li2S and S from a reversible redox couple at around 1.9/2.3 V.47,48 It is worth noting that the coincidence degree of the first three cycles of 1T@2H MoS2 is better than that of natural molybdenite, suggesting good cycling reversibility.49
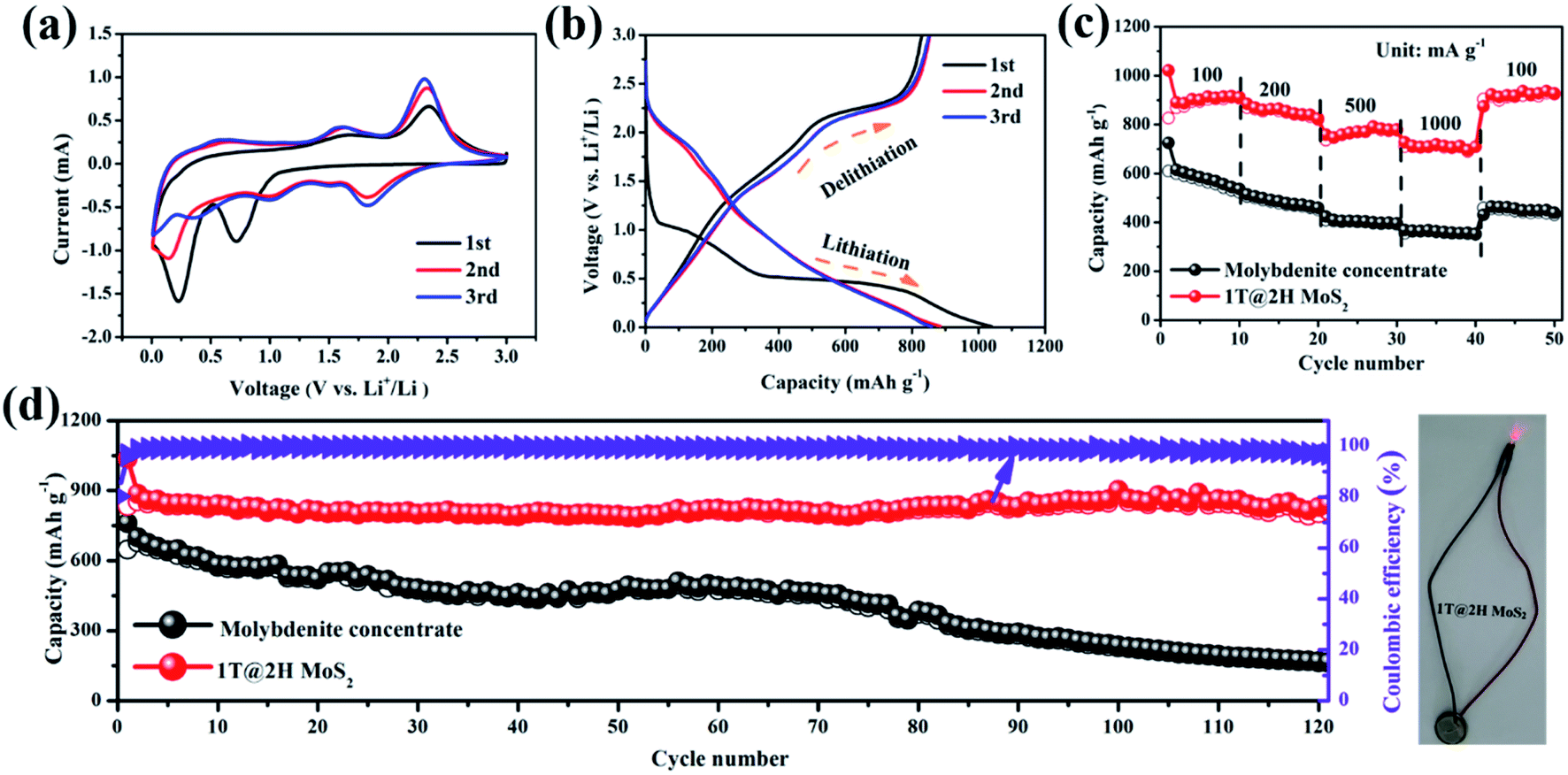 | ||
| Fig. 5 Electrochemical performance: (a) CV curve of 1T@2H MoS2; (b) charge and discharge curve of 1T@2H MoS2; (c) rate capabilities of 1T@2H MoS2; (d) cyclic performance of molybdenite concentrate and 1T@2H MoS2. | ||
As a comparison, the 1T@2H MoS2 delivers a high discharging capacity of 1036 mA h g−1, which is much higher than that of molybdenite concentrate (762 mA h g−1), at a current density of 100 mA g−1 (Fig. 5b and S9b†). As shown in Fig. S9c†, 1T@2H MoS2 is significantly shortened on the ∼1.1 V platform, indicating that part of the triangular prism 2H phase is transformed into the octahedral 1T phase because this platform corresponds to Li+ embedded MoS2 to form LixMoS2.50,51 Compared with the discharge platform of MoS2 prepared under different conditions, sample C20 showed a relatively higher platform, indicating that it has better lithium-ion diffusion kinetics (Fig. S9(d–f)†).41 Fig. 5c, 1T@2H MoS2 has excellent rate performance (727 mA h g−1) and is superior over molybdenite concentrate, especially at 1 A g−1 high current density. When the current density is back to 100 mA g−1, the capacity can be recovered to 927 mA h g−1 again, indicating that the electrode materials can achieve rapid charge–discharge performance. The 1T@2H MoS2 has a worm-like structure and expanded layer spacing, which increases the specific capacity and effectively alleviates the capacity attenuation caused by the volume expansion effect in the cycle process. As shown in Fig. 5d, within 120 cycles, the capacity of 1T@2H MoS2 decays by only 0.04% per cycle after the second cycle compared to the capacity of molybdenite concentrate of 169 mA h g−1. Hence, 1T@2H MoS2 has excellent cycling stability performance. Similarly, after 120 cycles, the molybdenite concentrate electrode suffered damage severely, while the 1T@2H MoS2 electrode maintained a fine integrity structure, which further proved that the heterostructures material has better structural stability (Fig. S10(a and b)†). In addition, after 120 cycles, the separator color of the 1T@2H MoS2 electrode is relatively lighter, which indicates that the electrode can inhibit the shuttle of polysulfide and more effectively maintain electrode stability (Fig. S10c†). After 482 cycles, 1T@2H MoS2 still has a 788 mA h g−1, even at the lowest specific capacity of 311 mA h g−1, which is close to the theoretical capacity of commercial graphite (Fig. S11†). Based on the comprehensive comparison in Table S4†, the 1T@2H MoS2 heterostructures deliver high specific capacity and long cycle life among the most reported works for LIBs. The rate performance and cycling stability of 1T@2H MoS2 prepared under other conditions are shown in Fig. S12 and S13†.
To better understand excellent electrochemical performance, a series of CV at scan rates 0.1–0.8 mV s−1 (Fig. 6a and d) and EIS measurements (Fig. 6e and f) were used to study the reaction kinetics of the 1T@2H MoS2 electrode. In general, the battery capacity can be divided into two parts: the diffusion-controlled battery capacity and the surface pseudocapacitance.49 Compared with the capacitance contribution, the battery contribution behaviour (i.e., insertion mechanism) is a process that takes place in the bulk of the electrode and is not conducive to long cycle life. The charge storage mechanism is evaluated according to the formula: i = avb, and the parameter b values are determined by log(v) ∼ log(i).52 When the value of b is close to 1 or more than 1, indicating capacitive-controlled plays a leading role, whereas the b-value approaches 0.5, suggesting that diffusion-controlled are dominant in the electrochemical reactions.53 Fig. 6b shows the b value are 0.89 and 1.16 for anodic and cathodic peaks, respectively, suggesting a significant capacitive contribution. The b values of 1T@2H MoS2 are larger than that of molybdenite concentrate (Fig. S15b†), which means faster reaction kinetics. According to eqn (1),44,54 the capacitance contribution ratio can be quantified at a given scanning rate.
| i = k1ν + k2ν1/2 | (1) |
| Z′ = Re + Rct + σω−1/2 | (2) |
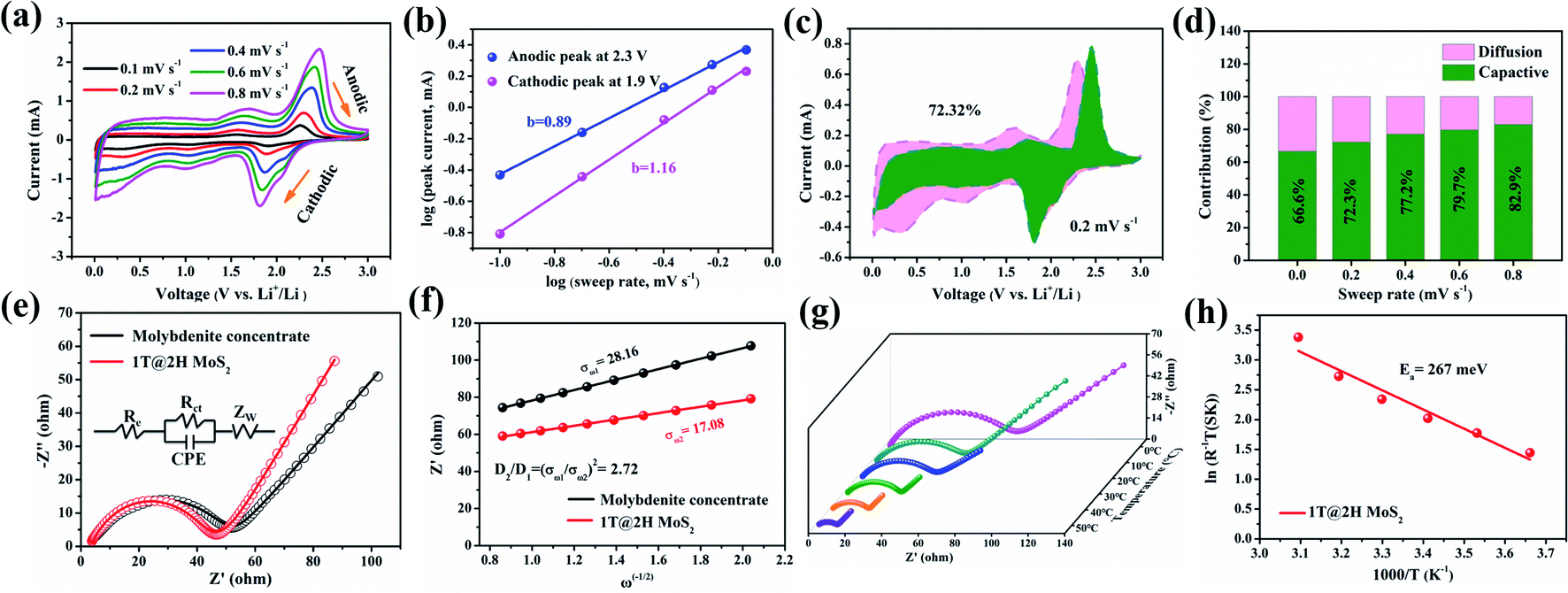 | ||
| Fig. 6 Electrochemical kinetic analysis diagram: (a) CV curves of scanning rate from 0.1 to 0.2 mV s−1; (b) log(i) vs. log(v) plots at oxidation and reduction state; (c) capacitance and diffusion control contribute to the CV curve at 0.2 mV s−1; (d) the contribution ratio of pseudocapacitive and diffusion-controlled contribution of the1T@2H MoS2 electrode at different scan rates; (e) EIS Nyquist plots of molybdenite concentrate and 1T@2H MoS2 (the inset of equivalent circuit); (f) the relationship between Z′ and ω−1/2 for molybdenite concentrate and 1T@2H MoS2 electrodes. (g) Temperature-dependent Nyquist plots of 1T@2H MoS2; (h) reciprocal bulk and interface impedance as a function of the reciprocal temperature of 1T@2H MoS2. | ||
Then, the Warburg factor σ obtained by fitting is substituted into eqn (3)50 to calculate the D value.
| D = R2T2/2n4F4C2σ2A2 | (3) |
| σT = A exp(−Ea/kBT) | (4) |
Conclusions
In conclusion, we used an innovative expansion method to construct worm-like 1T@2H MoS2 heterostructures in situ on natural molybdenite, which could effectively improve lithium-ion storage and reaction performance kinetics. With the advantage of the internal E-field, the 1T@2H MoS2 heterostructures electrode displays a high reversible capacity of 788 mA h g−1 at a current density of 0.1 A g−1 over 485 cycles (corresponding to 92% capacity retention of the second cycle). More importantly, the 1T@2H MoS2 heterostructures electrode exhibits high rate capability performance (727 mA h g−1 at 1 A g−1) for lithium-ion batteries. Electrochemical kinetic analysis shows that 1T@2H MoS2 has lower charge transfer resistance and activation energy, and pseudocapacitance dominates the storage mechanism of lithium-ions. These fast dynamics aid high magnification and high capacity capabilities. This work not only solves the significant challenges of the low inherent electrical conductivity of MoS2 but also provides a feasible way to use low-cost molybdenite concentrate as raw materials for high-performance next-generation commercial energy storage devices.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Cooperation Foundation of Key Laboratory of Mineralogy and Metallogeny, Chinese Academy of Sciences, Guangzhou Institute of Geochemistry (2020B1212060055), the National Natural Science Foundation of China (52002361).References
- W. T. Li, X. W. Guo, Y. Lu, L. Wang, A. L. Fan, M. L. Sui and H. J. Yu, Energy Storage Materials, 2017, 7, 203–208 CrossRef.
- S. J. Li, H. H. Tang, P. Ge, F. Jiang, J. H. Zhou, C. Y. Zhang, H. S. Hou, W. Sun and X. B. Ji, ACS Appl. Mater. Interfaces, 2018, 10, 6378–6389 CrossRef CAS.
- L. L. Wang, Q. F. Zhang, J. Y. Zhu, X. D. Duan, Z. Xu, Y. T. Liu, H. G. Yang and B. G. Lu, Energy Storage Materials, 2018, 16, 37–45 CrossRef.
- J. Shao, Q. T. Qu, Z. M. Wan, T. Gao, Z. C. Zuo and H. H. Zhe, ACS Appl. Mater. Interfaces, 2015, 7, 22927–22934 CrossRef CAS PubMed.
- K. K. Liu, W. J. Zhang, Y. H. Lee, Y. C. Lin, M. T. Chang, C. Y. Su, C. S. Chang, H. Li, Y. M. Shi, H. Zhang, C. S. Lai and L. J. Li, Nano Lett., 2012, 12, 1538–1544 CrossRef CAS PubMed.
- J. X. Guo, H. F. Zhu, Y. F. Sun, L. Tang and X. Zhang, J. Mater. Chem. A, 2016, 4, 4783–4789 RSC.
- H. Y. Wang, B. Y. Wang, D. Wang, L. Lu, J. G. Wang and Q. C. Jiang, RSC Adv., 2015, 5, 58084–58090 RSC.
- Y. C. Lin, D. O. Dumcenco, Y. S. Huang and K. Suenga, Nat. Nanotechnol., 2014, 9, 391–396 CrossRef CAS PubMed.
- Y. Katagiri, T. Nakamura, A. Ishii, C. Ohata, M. Hasegawa, S. Katsumoto, T. Cusati, A. Fortunelli, G. Iannaccone, G. Fiori, S. Roche and J. Haruyama, Nano Lett., 2016, 16, 3788–3794 CrossRef CAS PubMed.
- Y. Nagamine, J. Sato, Y. Qian, T. Inoue, T. Nakamura, S. Maruyama, S. Katsumoto and J. Haruyama, Appl. Phys. Lett., 2020, 117, 043101 CrossRef CAS.
- C. Z. Zhang, F. Han, F. Wang, Q. D. Liu, D. W. Zhou, F. Q. Zhang, S. H. Xu, C. L. Fan, X. K. Li and J. S. Liu, Energy Storage Materials, 2020, 24, 208–219 CrossRef.
- D. Sun, D. L. Ye, P. Liu, Y. G. Tang, J. Guo, L. Z. Wang and H. Y. Wang, Adv. Energy Mater., 2018, 8, 1702383 CrossRef.
- S. C. Li, P. Liu, X. B. Huang, Y. G. Tang and H. Y. Wang, J. Mater. Chem. A, 2019, 7, 10988–10997 RSC.
- J. Bai, B. C. Zhao, J. F. Zhou, Z. T. Fang, K. Z. Li, H. Y. Ma, J. M. Dai, X. B. Zhu and Y. P. Sun, ChemElectroChem, 2019, 6, 1930–1938 CrossRef CAS.
- D. Sun, D. Huang, H. Y. Wang, G. L. Xu, X. Y. Zhang, R. Zhang, Y. G. Tang, D. Abd EI-Hady, W. Alshitari, A. S. AL-Bogami, K. Amine and M. H. Shao, Nano Energy, 2019, 61, 361–369 CrossRef CAS.
- Y. Zhou, Y. Liu, W. X. Zhao, R. M. Xu, D. H. Wang, B. J. Li, X. Zhou and H. Shen, Electrochim. Acta, 2016, 211, 1048–1055 CrossRef CAS.
- Z. D. Lei, L. Q. Xu, Y. L. Jiao, A. J. Du, Y. Zhang and H. J. Zhang, Small, 2018, 14, 1704410 CrossRef.
- S. Das, G. Swain and K. Parida, Mater. Chem. Front., 2021, 5, 2143–2172 RSC.
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. W. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS PubMed.
- A. Ejigu, L. A. Kinloch, E. Prestat and R. A. W. Dryfe, J. Mater. Chem. A, 2017, 5, 11316–11330 RSC.
- Y. Zheng, T. F. Zhou, C. F. Zhang, J. F. Mao, H. K. Liu and Z. P. Guo, Angew. Chem., Int. Ed., 2016, 55, 3408–3413 CrossRef CAS PubMed.
- J. J. Deng, X. L. Yu, X. Y. Qin, D. Zhou, L. H. Zhang, H. Duan, F. Y. Kang, B. H. Li and G. X. Wang, Adv. Energy Mater., 2019, 9, 1803612 CrossRef.
- C. Z. Zhang, F. Han, J. M. Ma, Z. Li, Z. Li, F. Q. Zhang, S. H. Xu, H. B. Liu, X. K. Li, J. S. Liu and A. H. Lu, J. Mater. Chem. A, 2019, 7, 11771–11781 RSC.
- X. F. Qian, J. W. Liu, L. Fu and J. Li, Science, 2014, 346, 1344–1347 CrossRef CAS PubMed.
- N. D. Scott, J. F. Walker and V. L. Hansley, J. Am. Chem. Soc., 1936, 58, 2442 CrossRef CAS.
- J. Zheng, H. Zhang, S. H. Dong, Y. P. Liu, C. T. Nai, H. S. Shin, H. Y. Jeong, B. Liu and K. P. Loh, Nat. Commun., 2014, 5, 2995 CrossRef PubMed.
- Y. F. Yu, G. H. Nam, Q. Y. He, X. J. Wu, K. Zhang, Z. Z. Yang, J. Z. Wang, H. Li, X. Huang, B. Li, Q. H. Xiong, Q. Zhang, Z. Liu, L. Gu, Y. H. Du, W. Huang and H. Zhang, Nat. Chem., 2018, 10, 638–643 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. S. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- N. Joseph, P. M. Shafi and A. C. Bose, New J. Chem., 2018, 42, 12082–12090 RSC.
- K. Chang, X. Hai, H. Pang, H. B. Zhang, L. Shi, G. G. Liu, H. M. Liu, G. X. Zhao, M. Li and J. H. Ye, Adv. Mater., 2016, 28, 10033–10041 CrossRef CAS PubMed.
- G. S. Bang, K. W. Nam, J. Y. Kim, J. Y. Kim, J. Shin, J. W. Choi and S. Y. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 7084–7089 CrossRef CAS PubMed.
- D. S. Kong, H. T. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS PubMed.
- H. M. Fan, R. Wu, H. Y. Liu, X. Yang, Y. F. Sun and C. Chen, J. Mater. Sci., 2018, 53, 10302–10312 CrossRef CAS.
- X. Y. Chen, Z. M. Wang, Y. Z. Wei, X. Zhang, Q. H. Zhang, L. Gu, L. J. Zhang, N. L. Yang and R. B. Yu, Angew. Chem., Int. Ed., 2019, 58, 17621–17624 CrossRef CAS PubMed.
- S. J. R. Tan, I. Abdelwahab, Z. J. Ding, X. X. Zhao, T. S. Yang, G. Z. J. Loke, H. Lin, I. Verzhbitskiy, S. M. Poh, H. Xu, C. T. Nai, W. Zhou, G. Eda, B. H. Jia and K. P. Loh, J. Am. Chem. Soc., 2017, 139, 2504–2511 CrossRef CAS PubMed.
- C. T. Zhao, C. Yu, B. Qiu, S. Zhou, M. D. Zhang, H. W. Huang, B. Q. Wang, J. J. Zhao, X. L. Sun and J. S. Qiu, Adv. Mater., 2018, 30, 1702486 CrossRef PubMed.
- Y. Y. Wang, W. P. Kang, D. W. Cao, M. H. Zhang, Z. X. Kang, Z. Y. Xiao, R. M. Wang and D. F. Sun, J. Mater. Chem. A, 2018, 6, 4776–4782 RSC.
- S. Reshmi, M. V. Akshaya, B. Satpati, P. K. Basu and K. Bhattacharjee, Nanotechnology, 2018, 29, 205604 CrossRef CAS PubMed.
- X. M. Geng, W. W. Sun, W. Wu, B. Chen, A. Al-Hilo, M. Benamara, H. L. Zhu, F. Watanabe, J. B. Cui and T. P. Chen, Nat. Commun., 2016, 7, 10672 CrossRef CAS PubMed.
- T. Xiang, Q. Fang, H. Xie, C. Q. Wu, C. D. Wang, Y. Zhou, D. B. Liu, S. M. Chen, A. Khalil, S. Tao, Q. Liu and L. Song, Nanoscale, 2017, 9, 6975–6983 RSC.
- K. Yao, Z. W. Xu, J. F. Huang, M. Ma, L. C. Fu, X. T. Shen, J. Li and M. S. Fu, Small, 2019, 15, 1805405 CrossRef.
- Y. Yin, J. C. Han, Y. M. Zhang, X. H. Zhang, P. Xu, Q. Yuan, L. Samad, X. J. Wang, Y. Wang, Z. H. Zhang, P. Zhang, X. Z. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- D. Su, S. X. Dou and G. X. Wang, Adv. Energy Mater., 2014, 5, 1401205 CrossRef.
- W. Ye, F. F. Wu, N. X. Shi, H. Zhou, Q. Q. Chi, W. H. Chen, S. Y. Du, P. Gao, H. B. Li and S. L. Xiong, Small, 2019, 16, 1906607 CrossRef PubMed.
- Z. Y. Li, A. Ottmann, T. Zhang, Q. Sun, H. P. Meyer, Y. Vaynzof, J. H. Xiang and R. Klingeler, J. Mater. Chem. A, 2017, 5, 3987–3994 RSC.
- F. Y. Xiong, Z. Y. Cai, L. B. Qu, P. F. Zhang, Z. F. Yuan, O. K. Asare, W. W. Xu, C. Lin and L. Q. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 12625–12630 CrossRef CAS.
- J. W. Zhou, J. Qin, X. Zhang, C. S. Shi, E. Z. Liu, J. J. Li, N. Q. Zhao and C. N. He, ACS Nano, 2015, 9, 3837–3848 CrossRef CAS.
- X. X. Zuo, K. Chang, J. Zhao, Z. Z. Xie, H. W. Tang, B. Li and Z. R. Chang, J. Mater. Chem. A, 2016, 4, 51–58 RSC.
- X. Y. Li, K. K. Li, S. C. Zhu, K. Fan, L. L. Lyu, H. M. Yao, Y. Y. Li, J. L. Hu, H. T. Huang, Y. W. Mai and J. B. Goodenough, Angew. Chem., Int. Ed., 2019, 58, 6239–6243 CrossRef CAS PubMed.
- J. Bai, B. C. Zhao, J. F. Zhou, J. G. Si, Z. T. Fang, K. Z. Li, H. Y. Ma, J. M. Dai, X. B. Zhu and Y. P. Sun, Small, 2019, 15, 1805420 CrossRef PubMed.
- Y. C. Jiao, A. Mukhopadhyay, Y. Ma, L. Yang, A. M. Hafez and H. L. Zhu, Adv. Energy Mater., 2018, 8, 1702779 CrossRef.
- J. Bai, B. C. Zhao, S. Lin, K. Z. Li, J. F. Zhou, J. M. Dai, X. B. Zhu and Y. P. Sun, Nanoscale, 2020, 12, 1144–1154 RSC.
- M. M. Yin, X. T. Feng, D. Zhao, Y. Zhao, H. S. Li, W. Zhou, H. B. Liu, X. P. Bai, H. X. Wang, C. H. Feng and Q. Z. Jiao, ACS Sustainable Chem. Eng., 2019, 7, 6122–6130 CrossRef CAS.
- Y. J. Fang, D. Y. Luan, Y. Chen, S. Y. Gao and X. W. Lou, Angew. Chem., Int. Ed., 2020, 59, 7178–7183 CrossRef CAS PubMed.
- G. Barik and S. Pal, Adv. Theory Simul., 2020, 3, 2000157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05565h |
| This journal is © The Royal Society of Chemistry 2021 |