DOI:
10.1039/D1RA05525A
(Paper)
RSC Adv., 2021,
11, 34086-34094
Regioselective formylation of rhenium-oxo and gold corroles: substituent effects on optical spectra and redox potentials †
Received
19th July 2021
, Accepted 29th September 2021
First published on 28th October 2021
Abstract
Vilsmeier–Haack formylation of ReO and Au meso-triarylcorroles over 16–18 hours affords moderate to good yields (47–65%) of the ReO-3-formyl and Au-3,17-diformyl derivatives in a highly regioselective manner. Formylation was found to effect substantial upshifts for redox potentials (especially the reduction potentials) as well as significant to dramatic redshifts for both the Soret and Q bands.
Introduction
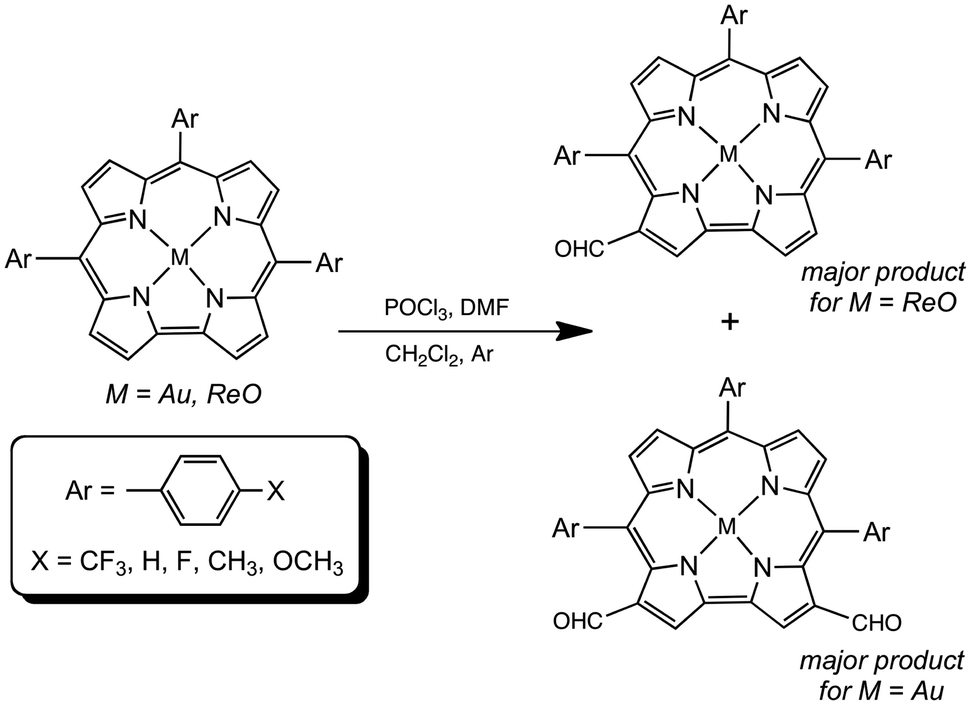
Our research on 5d metallocorroles1–4 began a decade ago, with two publications documenting the first examples of gold corroles. The first of these reported gold insertion into β-octabromo-meso-triarylcorroles,5 which was also accomplished at about the same time by Gross and coworkers.6 The second paper described Au insertion into simple meso-triarylcorroles with Au(III) acetate7 – a significant breakthrough, considering the steric mismatch between the large ionic radii of 5d transition metals and the relatively constricted central cavity of corroles. The ‘acetate method’ has since become the method of choice for the synthesis of Au corroles. Understandably, we were intrigued by the possibility of inserting other heavier transition metals into corroles and, to our satisfaction, the first 99TcO,8 ReO,9 ReNAr,10 RuN,11 OsN,12 and Pt13,14 triarylcorroles (as well as Mo and W biscorroles15–17 and quadruple-bonded Re corrole dimers18) were synthesized in our Tromsø laboratory. Several of the 5d metallocorroles19–24 were found to exhibit room-temperature NIR phosphorescence, leading to applications as oxygen sensors and as photosensitizers in photodynamic therapy and dye-sensitized solar cells.25–27 In contrast (compared to 3d metallocorroles,28,29 especially Cu corroles30–34), the chemical reactivity of the 5d complexes remains poorly explored. Peripheral functionalizations accomplished so far include β-perchlorination35 and perbromination,36,37 as well as polyiodinations.38,39 Metal-centered transformations include the generation of Mo and Re Viking-helmet dichlorido derivatives from the corresponding oxido precursors.40,41 In an alternative approach, the use of water-soluble, axial phosphine ligands has led to water-soluble iridium corroles.42 A final, more esoteric example consists of the use of an OsN corrole as a π-acceptor ligand in formation of an OsVIN–PtII heterobimetallic complex.35 Here we document our efforts to formylate ReO and Au corroles via the Vilsmeier–Haack method,43 which has previously been used for free-base, aluminum and gallium corroles.44–47 As before, the reaction was found to proceed in a highly regioselective manner, with ReO and Au corroles affording mono- and diformyl derivatives, respectively (Fig. 1). Optical and electrochemical characterization of the products permitted a moderately detailed, experimental picture of the substantial electronic effects of β-formyl groups, as recounted below.
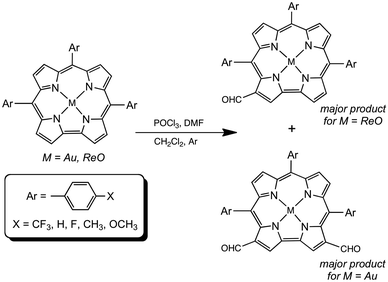 |
| | Fig. 1 Formylation of Au and ReO corroles. | |
Results and discussion
(a) Synthesis and proof of composition
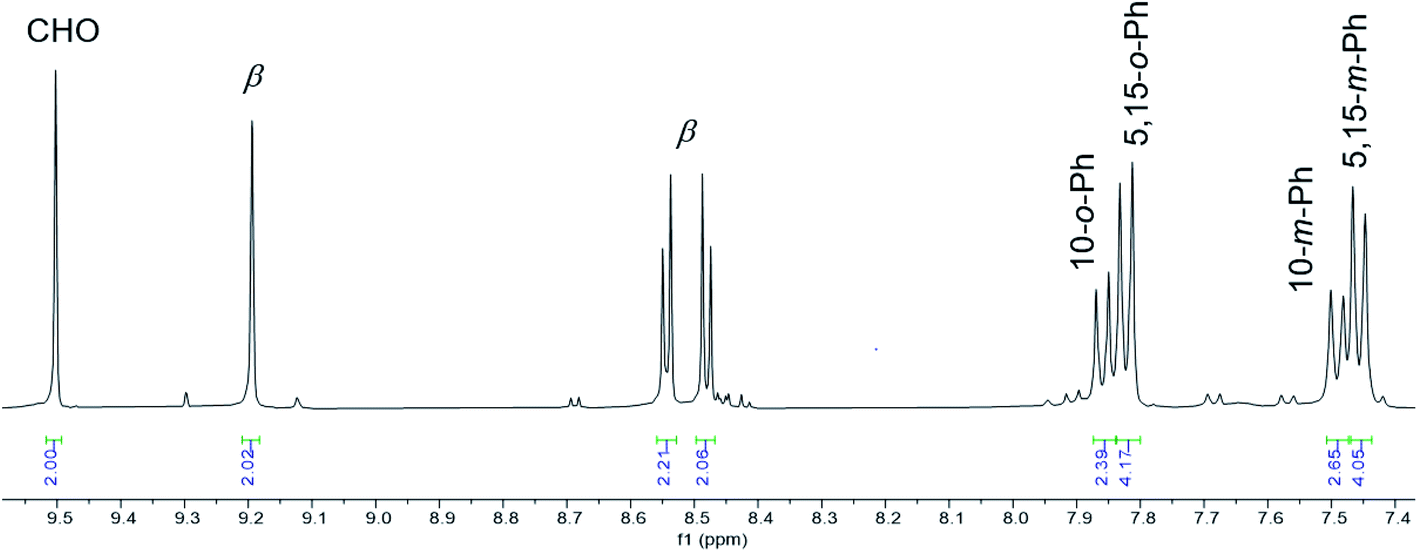
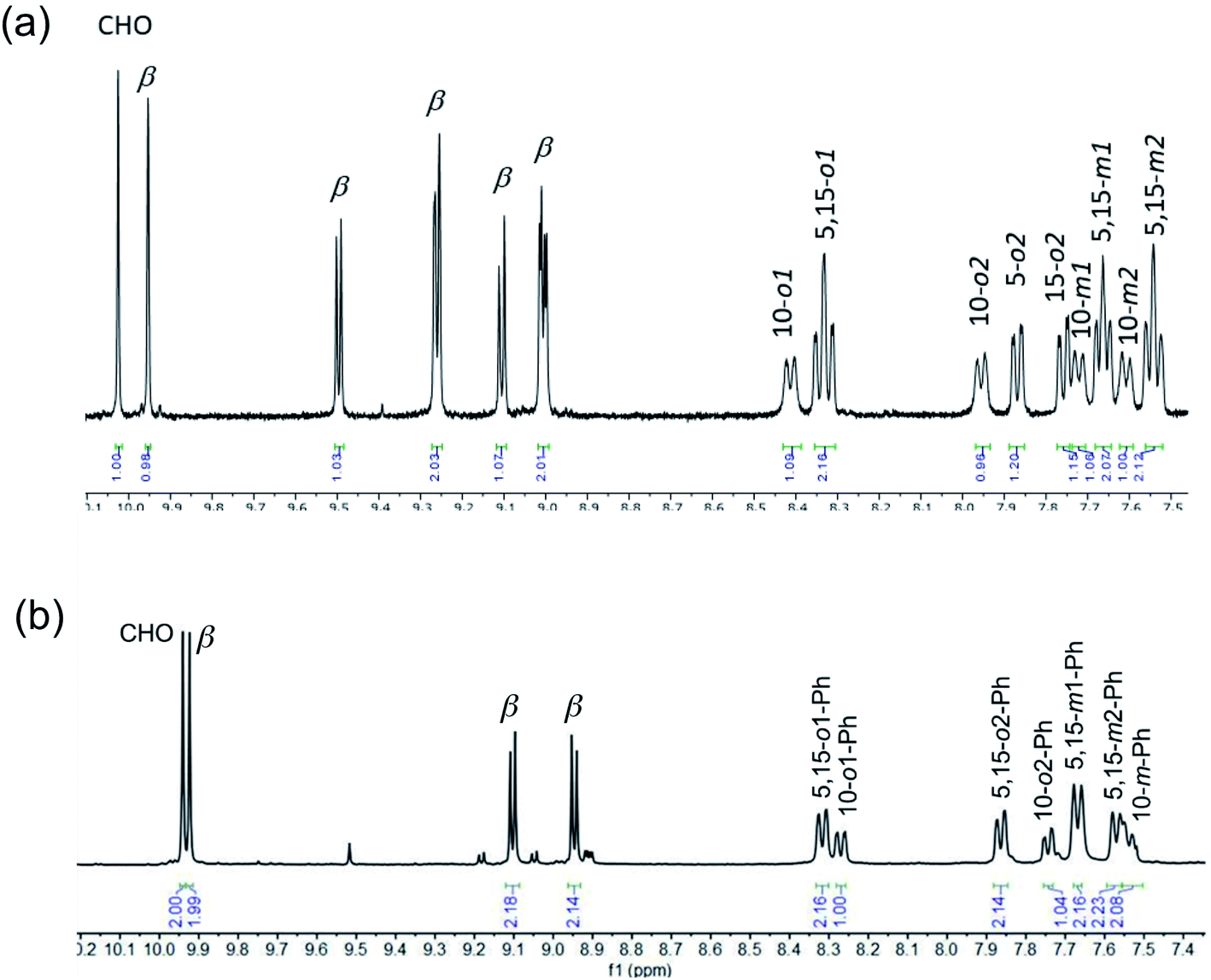
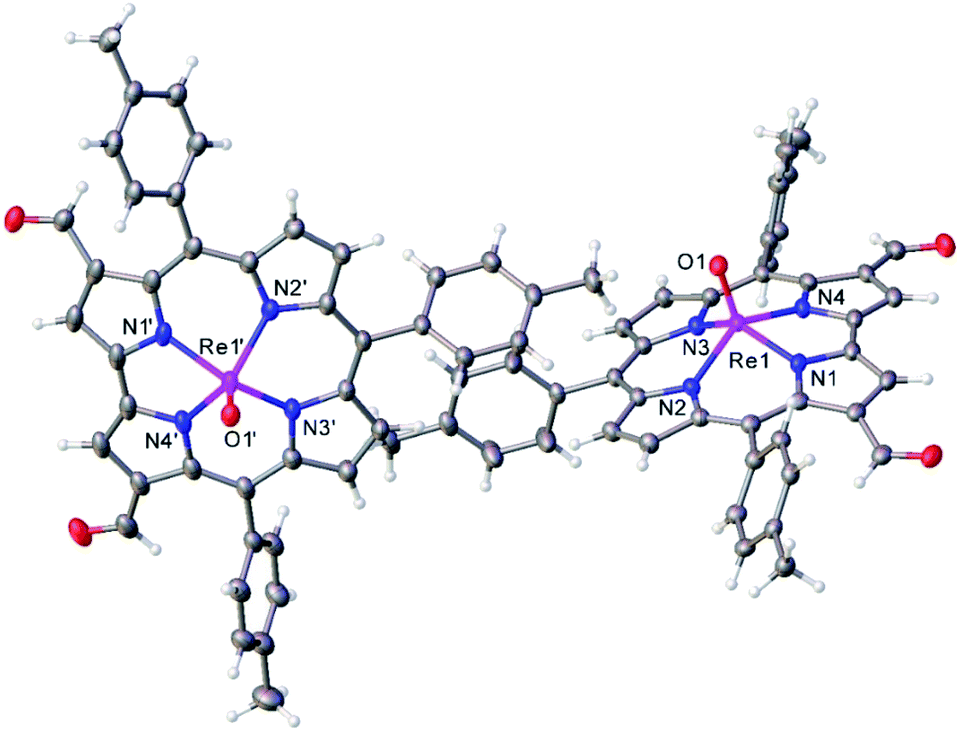
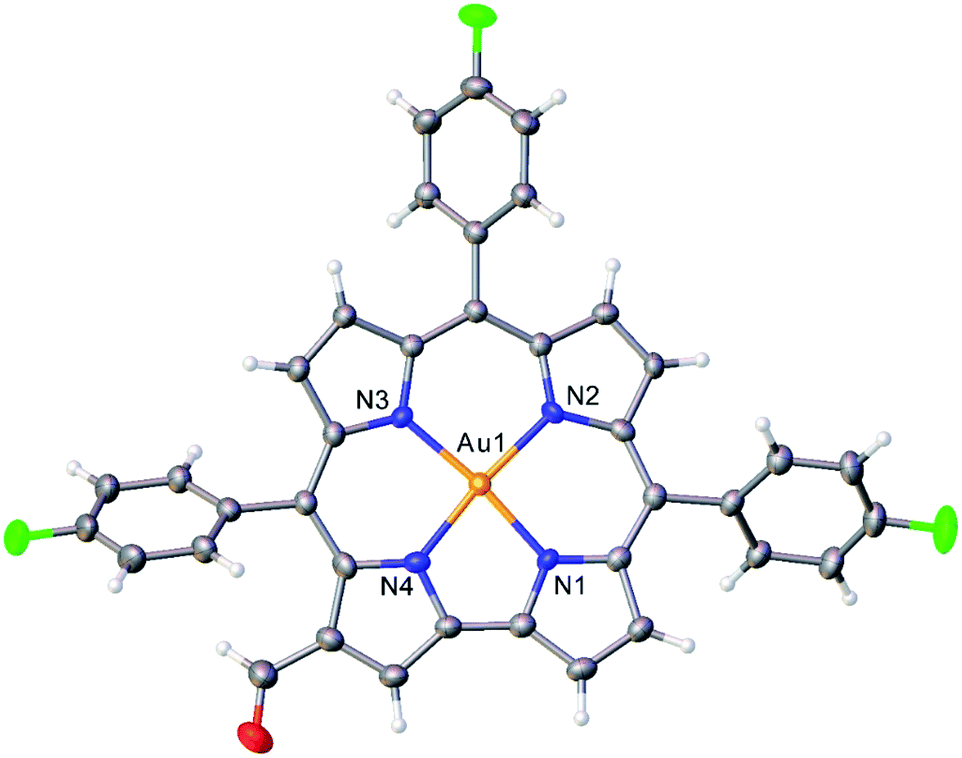
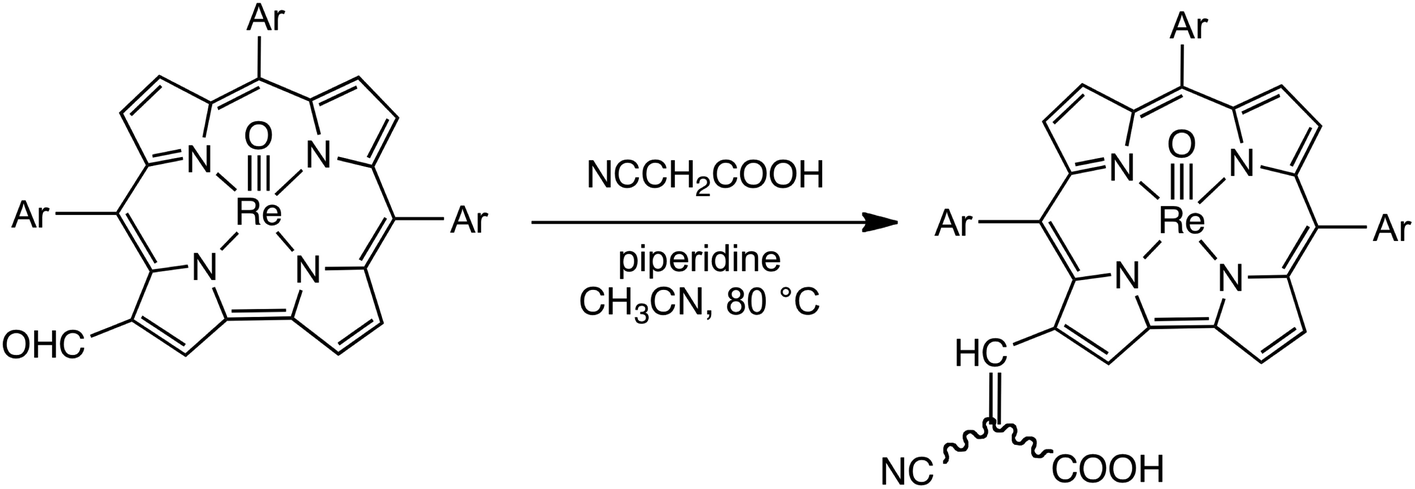
Vilsmeier–Haack formylation, carried out over 16–18 h at 0 °C to room temperature, proceeded smoothly for both ReO and Au meso-tris(para-X-phenyl)corroles, M[TpXPC] (M = ReO, Au), regioselectively affording good yields of the 3-monoformyl (∼50%) and 3,17-diformyl (∼60%) derivatives for the two metals, respectively. The major products, hereafter denoted as Re[TpXPC-3-CHO](O) and Au[TpXPC-3,17-(CHO)2], were fully characterized and the regiochemistry of formylation was established largely via 1H NMR spectroscopy. In particular, the simplicity of the 1H NMR of the Au-diformyl derivatives (relative to the ReO-monoformyl derivatives) immediately suggested a regiochemistry consistent with time-averaged C2v symmetry (Fig. 2 and 3). The minor products – the ReO-diformyl and Au-monoformyl derivatives – generally could not be fully characterized. Surprisingly, two minor products – Re[TpCH3PC-3,17-(CHO)2](O) and Au[TpCH3PC-3-CHO] – proved amenable to single-crystal X-ray structure determination (Table 1 and Fig. 4 and 5). The structures established that the formyl groups are essentially coplanar with the mean plane of the pyrrole rings they are attached to, but aside from that, they proved largely unremarkable. Finally, in a brief examination of their reactivity, ReO 3-formylcorroles were found to undergo facile Knoevenagel condensation with active methylene compounds (Fig. 6). While a full exposition of reactivity studies is outside the scope of the present study, a few examples of protocols involving cyanoacetic acid are given in the experimental section.
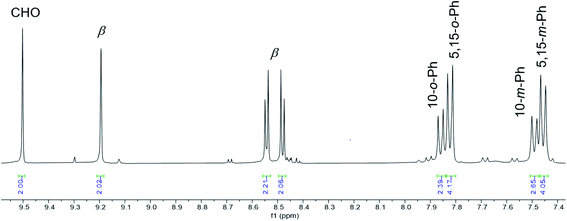 |
| | Fig. 2 Aromatic region of the 1H NMR spectrum (400 MHz, CDCl3, 293 K) of Au[TpFPC-3,17-(CHO)2]. | |
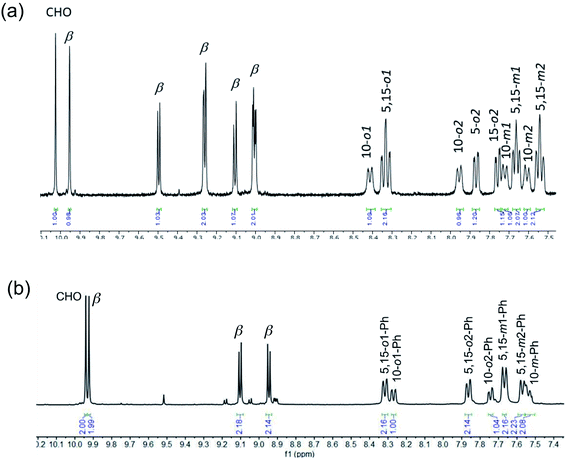 |
| | Fig. 3 Aromatic region of the 1H NMR spectra (400 MHz, CDCl3, 273 K) of (a) Re[TpCH3PC-3-CHO](O) and (b) Re[TpCH3PC-3-17-(CHO)2](O). Note the different numbers of β protons in the two complexes. Note also that the most downfield singlet has been tacitly assigned as the formyl proton. | |
Table 1 Selected crystallographic data for the complexes analyzed
| |
Re[TpCH3PC-3,17-(CHO)2](O) |
Au[TpFPC-3-CHO] |
| Chemical formula |
C45H36N4O3Re |
C38H20F3N4OAu |
| Formula mass |
866.98 |
802.55 |
| Crystal system |
Triclinic |
Monoclinic |
| Crystal size (mm3) |
0.230 × 0.080 × 0.020 |
0.100 × 0.020 × 0.010 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c |
| λ (Å) |
0.7288 |
0.7288 |
| a (Å) |
9.5807(4) |
20.9037(12) |
| b (Å) |
12.6649(6) |
19.7169(12) |
| c (Å) |
29.1467(13) |
15.8376(10) |
| α (deg) |
95.279(2) |
90 |
| β (deg) |
91.627(2) |
91.467(3) |
| γ (deg) |
90.341(2) |
90 |
| Z |
4 |
8 |
| V (Å3) |
3520.1(3) |
6525.4(7) |
| Temperature (K) |
100(2) |
100(2) |
| Density (g cm−3) |
1.636 |
1.634 |
| Measured reflections |
131![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 064 064 |
84580 |
| Unique reflections |
21566 |
10078 |
| Parameters |
975 |
424 |
| Restraints |
40 |
0 |
| Rint |
0.0354 |
0.0697 |
| θ Range (deg.) |
1.744–31.439 |
2.913–31.600 |
| R1, wR2 all data |
0.0361, 0.0667 |
0.0453, 0.1001 |
| S (GooF) all data |
1.092 |
1.070 |
| Max/min res. dens. (e Å−3) |
2.379/−2.218 |
1.925/−1.512 |
 |
| | Fig. 4 Thermal ellipsoid plot for Re[TpCH3PC-3,17-(CHO)2](O). Selected distances (Å): Re1–N1 2.007(2), Re1–N2 2.012(2), Re1–N3 2.013(2), Re1–N4 1.999(2), and Re1–O1 1.681(2), Re1′–N1′ 1.999(2), Re1′–N2′ 2.016(2), Re1′–N3′ 2.013(2), Re1′–N4′ 2.000(2), Re1′–O1′ 1.685. | |
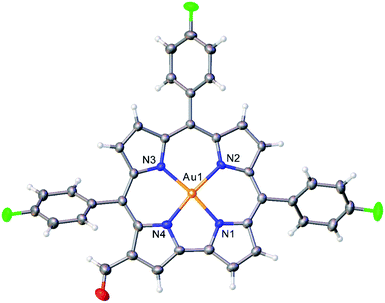 |
| | Fig. 5 Thermal ellipsoid plot for Au[TpFPC-3-CHO]. Selected distances (Å): Au1–N1 1.939, Au1–N2 1.973, Au1–N3 1.958, Au1–N4 1.950. | |
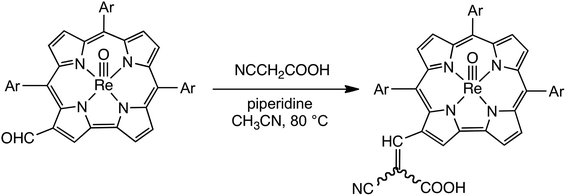 |
| | Fig. 6 Knoevenagel condensation of ReO 3-formylcorroles with cyanoacetic acid. | |
(b) Electronic absorption spectra and redox potentials
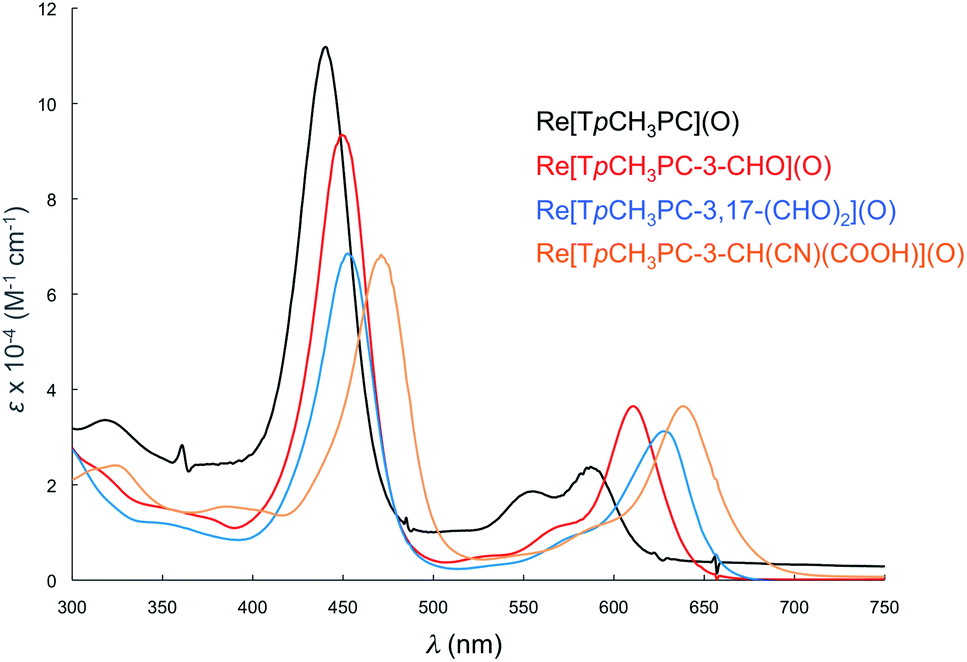
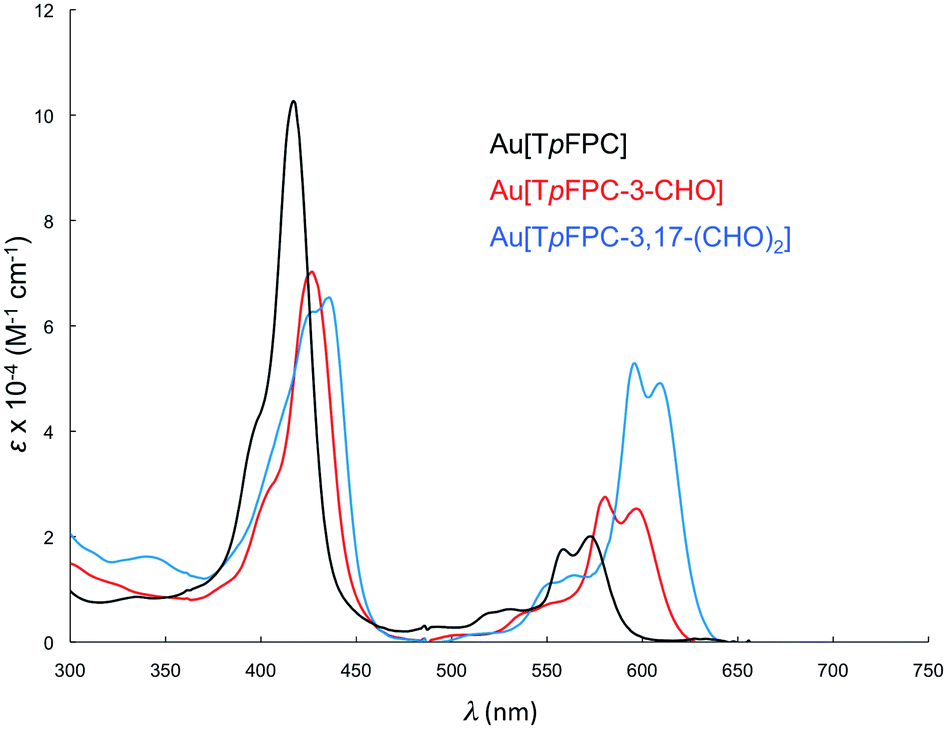
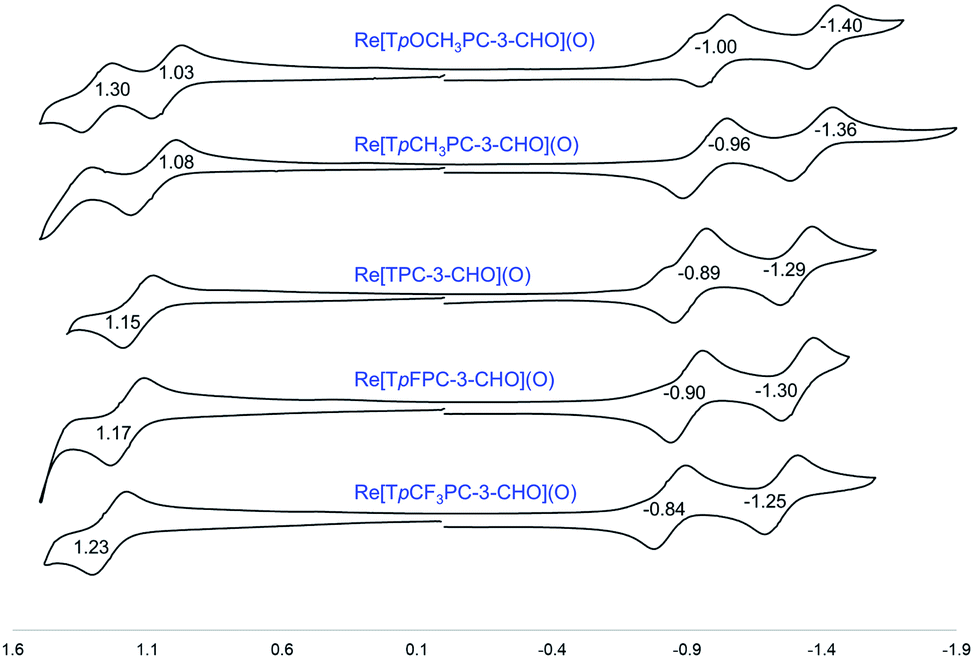
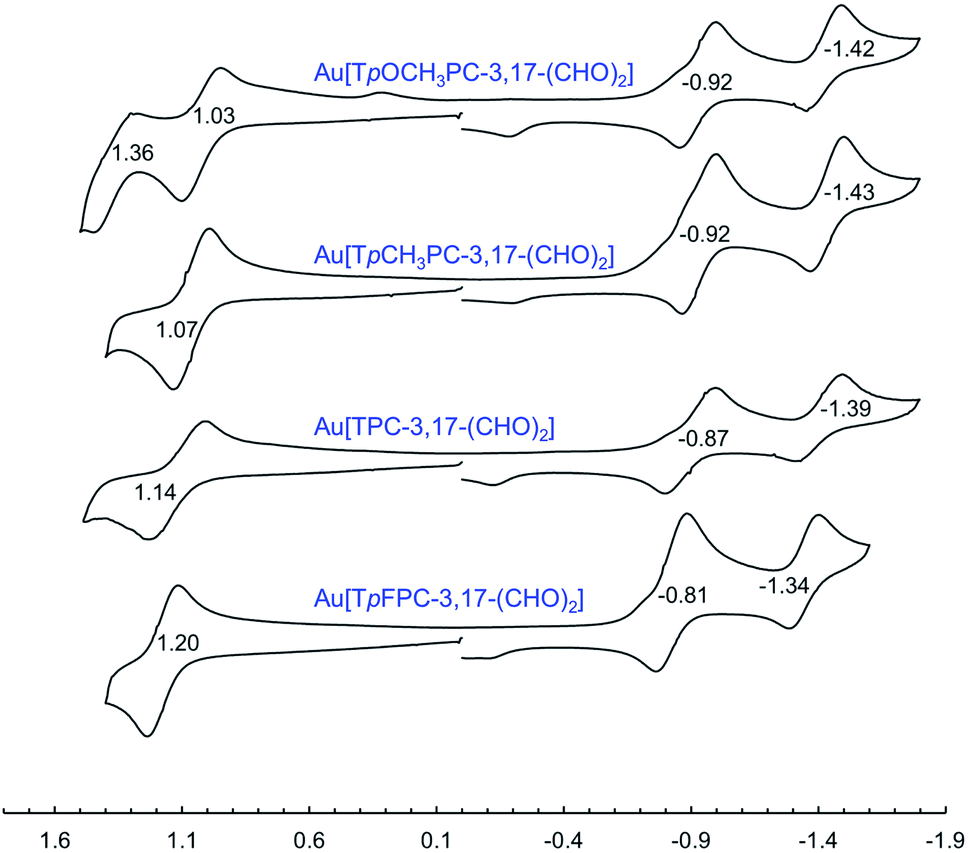
Formylation engenders significant redshifts for both the Soret bands and double-humped Q bands of the ReO and Au corroles (Table 2, Fig. 7 and 8). Thus, 3-monoformylation redshifts the Soret maxima by about 8 nm (for either metal), while diformylation brings about redshifts about one-and-a-half (for ReO) to twice (for Au) that magnitude. For the lowest-energy Q bands (for either metal), mono- and diformylation result in redshifts of 20–25 and 37–41 nm, respectively. Interestingly, formylation appears to weaken the intensity of the Soret band, suggesting a certain weakening of macrocyclic aromaticity, while strengthening the Q band, potentially indicating corrole to formyl charge transfer character for the latter feature.
Table 2 UV-vis absorption maxima (nm), redox potentials (V) and electrochemical HOMO–LUMO gaps (ΔE, V) for selected compounds
| Compound |
Soret |
Q |
E1/2−ox2 |
E1/2−ox1 |
E1/2−red1 |
E1/2−red2 |
ΔE |
Ref. |
| Au[TpOCH3PC] |
420 |
560, 580 |
1.32 |
0.76 |
−1.57 |
— |
2.33 |
6 |
| Au[TpCH3PC] |
420 |
560, 576 |
1.35 |
0.78 |
−1.42 |
— |
2.20 |
6 |
| Au[TPC] |
418 |
560, 575 |
1.35 |
0.80 |
−1.38 |
— |
2.18 |
6 |
| Au[TpFPC] |
419 |
559, 573 |
1.38 |
0.85 |
−1.37 |
— |
2.22 |
6 |
| Au[TpCH3PC-3-CHO] |
428 |
581, 602 |
— |
— |
— |
— |
— |
tw |
| Au[TpFPC-3-CHO] |
427 |
580, 597 |
— |
1.03 |
−1.02 |
−1.46 |
2.05 |
tw |
| Au[TpOCH3PC-3,17-(CHO)2] |
439 |
597, 618 |
1.36 |
1.03 |
−0.92 |
−1.42 |
1.95 |
tw |
| Au[TpCH3PC-3,17-(CHO)2] |
439 |
596, 615 |
— |
1.07 |
−0.92 |
−1.43 |
1.99 |
tw |
| Au[TPC-3,17-(CHO)2] |
437 |
596, 611 |
— |
1.14 |
−0.87 |
−1.39 |
2.01 |
tw |
| Au[TpFPC-3,17-(CHO)2] |
436 |
597, 609 |
— |
1.20 |
−0.81 |
−1.34 |
2.01 |
tw |
|
| Re[TpOCH3PC](O) |
441 |
556, 592 |
— |
0.93 |
−1.29 |
— |
2.22 |
8 |
| Re[TpCH3PC](O) |
440 |
555, 587 |
— |
0.94 |
−1.29 |
— |
2.23 |
8 |
| Re[TPC](O) |
439 |
552, 585 |
— |
0.98 |
−1.26 |
— |
2.24 |
8 |
| Re[TpFPC](O) |
438 |
553, 585 |
— |
1.01 |
−1.23 |
— |
2.24 |
8 |
| Re[TpCF3PC](O) |
438 |
552, 585 |
— |
1.10 |
−1.16 |
— |
2.26 |
8 |
| Re[TpOCH3PC-3-CHO](O) |
449 |
609 |
1.30 |
1.03 |
−1.00 |
−1.40 |
2.03 |
tw |
| Re[TpCH3PC-3-CHO](O) |
449 |
611 |
1.41 |
1.08 |
−0.96 |
−1.36 |
2.04 |
tw |
| Re[TPC-3-CHO](O) |
452 |
613 |
— |
1.15 |
−0.89 |
−1.29 |
2.04 |
tw |
| Re[TpFPC-3-CHO](O) |
448 |
608 |
— |
1.17 |
−0.90 |
−1,30 |
2.07 |
tw |
| Re[TpCF3PC-3-CHO](O) |
448 |
607 |
— |
1.23 |
−0.84 |
−1,25 |
2.07 |
tw |
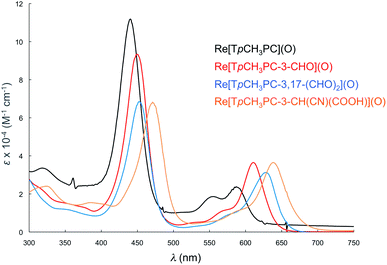 |
| | Fig. 7 Comparative UV-vis spectra in dichloromethane for Re[TpCH3PC](O) (black), Re[TpCH3PC-3-CHO](O) (red), (c) Re[TpCH3PC-3,17-(CHO)2](O) (blue), (d) Re[TpCH3PC-3-CH(CN)(COOH)](O) (saffron). | |
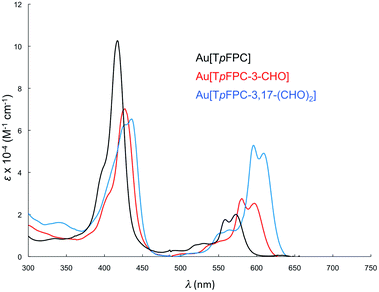 |
| | Fig. 8 Comparative UV-vis spectra in dichloromethane for Au[TpFPC] (black), Au[TpFPC-3-CHO] (red), (c) Au[TpFPC-3,17-(CHO)2] (blue). | |
Formylation has a substantial impact on the redox potentials of both ReO and Au corroles (which all correspond to ligand-centered processes1,3,7,9,48), the impact on reduction potentials being particularly dramatic (Table 2). Thus, 3-formylation upshifts oxidation potentials of ReO triarylcorroles by 100–170 mV and the reduction potentials by ∼300 mV (Fig. 9). Likewise, 3,17-diformylation upshifts oxidation potentials of Au triarylcorroles by ∼260 mV and the reduction potentials by 500–650 mV (Fig. 10). The larger shifts for the reduction potentials may be qualitatively understood in terms of the ability of the strongly electron-withdrawing formyl substituents to stabilize a negative charge. The effect also translates to smaller electrochemical HOMO–LUMO gaps (i.e., the algebraic difference between the first oxidation and reduction potentials) for the formylated metallocorroles relative to their β-unsubstituted counterparts. For one of the Au corroles, Au[TpFPC], we were able to determine the effect of both mono- and diformylation. For both oxidation and reduction potentials, the second formyl group was found to have a distinctly smaller effect relative to the first one – i.e., a nonadditive effect (Fig. 11).
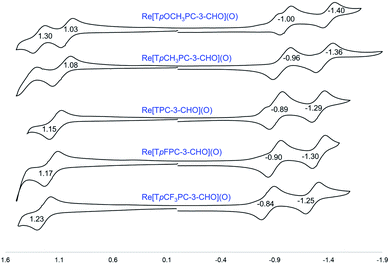 |
| | Fig. 9 Cyclic voltammograms (V vs. SCE) for Re[TpXPC-3-CHO](O) derivatives in dichloromethane containing 0.1 M TBAP. | |
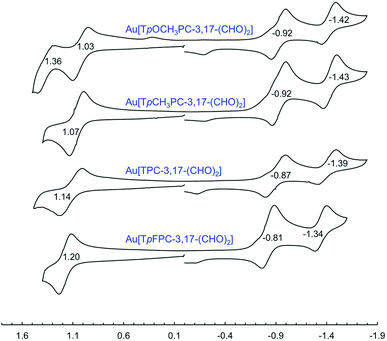 |
| | Fig. 10 Cyclic voltammograms (V vs. SCE) for Au[TpXPC-3,17-(CHO)2] derivatives in dichloromethane containing 0.1 M TBAP. | |
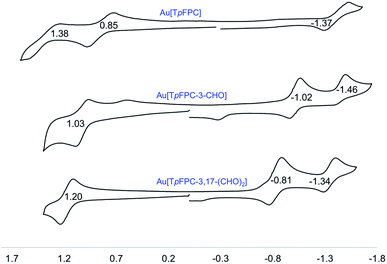 |
| | Fig. 11 Cyclic voltammograms (V vs. SCE) for Au[TpXPC-(CHO)n] (n = 0, 1, 2) derivatives in dichloromethane containing 0.1 M TBAP. | |
The redox potentials of the compounds studied also provide a potential explanation for the observed tendency toward monoformylation for ReO corroles and toward diformylation for Au corroles. The oxidation potentials of β-unsubstituted Au triarylcorroles are about 200 mV lower than those of analogous ReO complexes, suggesting that the former are more nucleophilic. (It may be noted that the higher oxidation potentials in the Re case correlates with the higher oxidation state of the Re center, +V, compared to +III for Au. A similar metal oxidation state dependence has long been known for metalloporphyrins.49) Moreover, the oxidation potentials of the ReO–monoformyl complexes are about the same as those of analogous Au-diformyl complexes, potentially explaining why formylation stops at the monoformyl stage for ReO triarylcorroles.
Conclusion
Vilsmeier–Haack formylation has been found to proceed in a highly regioselective manner for ReO and Au triarylcorroles, affording 3-formyl and 3,17-diformyl derivatives, respectively, as the major products. The difference in reactivity between the two metals appears at least partially attributable to a difference in oxidation potential, and hence nucleophilicity, between the two families of 5d metallocorroles. The formyl groups engender significant to dramatic redshifts of the Soret and Q bands and large upshifts for the redox potentials for both series of metallocorroles. A preliminary study demonstrating successful Knoevenagel condensations suggests that the formylated products should serve as versatile starting materials for a wide range of functionalized, potentially water-soluble 5d metallocorroles.
Experimental section
Materials and instruments
Except for solvents, all chemicals were obtained from Sigma-Aldrich and used as such. Dichloromethane used in syntheses was dried over 3 Å molecular sieves. CHROMASOLV HPLC-grade n-hexane and dichloromethane were used for column chromatography. Silica gel 60 (0.04–0.063 mm particle size; 230–400 mesh, Sigma) was used for column chromatography, with columns about 8 cm in height and 3 cm in diameter. UV-vis spectra were recorded on an HP 8454 spectrophotometer at room temperature. High-resolution electrospray ionization (ESI) mass spectra were recorded on an LTQ Orbitrap XL spectrometer. 1H NMR spectra were recorded on a Bruker Avance III HD 400 MHz spectrometer. Cyclic voltammetry analyses were carried out in dry dichloromethane containing 0.1 M TBAP as a supporting electrolyte. An EG&G 263A potentiostat with a standard three-electrode set-up, consisting of a glassy carbon working electrode (3 mm i.d.), a platinum wire counter electrode, and saturated calomel reference electrode (SCE), was used for cyclic voltammetry.
General procedure for formylation of ReVO triarylcorroles
To a 50 mL round-bottom flask equipped with a magnetic stir-bar, a rubber septum, and an argon in/outlet, and placed in an ice/water bath, were added dichloromethane (5.0 mL), POCl3 (20 mmol, 1.8 mL), and DMF (23 mmol, 1.8 mL) were added. The metallocorrole7,9 starting material (0.075 mmol, ∼55 mg) was dissolved in dichloromethane (10 mL) and added via syringe to the chilled solution. The mixture was left to stir under a constant flow of argon at 0 °C for 15 minutes, whereupon it was allowed to warm to room temperature and left to stir overnight. The following morning, the reaction was quenched with water (3 × 100 mL). The organic phase was dried and subjected to column chromatography on silica gel with 3:1 n-hexane/dichloromethane as eluent. Further elution with dichloromethane with up to 0–5% methanol led to product isolation.
Re[TPC-3-CHO](O). Yield 34.5 mg (62.8%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 449 (8.47), 609 (3.19). 1H NMR (400 MHz, 20 °C) δ: 10.06 (s, 1H, CHO); 9.96 (s, 1H, β-H); 9.52 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.25 (d overlapping, 2H, 3JHH = 4.3 Hz, β-H); 9.09 (d, 1H, 3JHH = 5.0 Hz, β-H); 9.00 (d overlapping, 2H, 3JHH = 4.9 Hz, β-H); 8.55 (d, 1H, 3JHH = 7.6 Hz, 10-o1-Ph); 8.45 (d overlapping, 2H, 3JHH = 8.2 Hz, 5,15-o1-Ph); 8.07 (d, 1H, 3JHH = 6.6 Hz, 10-o2-Ph); 7.99 (d, 2H, 3JHH = 7.6 Hz, 5/15-o2-Ph); 7.82 (m, 10H, 5/15-o2-Ph + 5,10,15-m-Ph + 5,10,15-p-Ph). MS (ESI): M+ = 754.1383 (expt), 754.1375 (calcd for C38H23N4O2Re). Elemental analysis (%) calcd for C38H23N4O2Re: C 60.55, H 3.08, N 7.43, found C 60.93, H 3.36, N 7.59. IR νReO: 992 cm−1, νCHO: 1659 cm−1
Re[TpCH3PC-3-CHO](O). Yield 31.1 mg (56.6%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 449 (9.34), 611 (3.65). 1H NMR (400 MHz, 0 °C) δ: 10.03 (s, 1H, CHO); 9.95 (s, 1H, β-H); 9.50 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.26 (d, 2H, 3JHH = 4.5 Hz, β-H); 9.11 (d, 1H, 3JHH = 5.1 Hz, β-H); 9.01 (dd, 2H, 3JHH = 4.9, 1.9 Hz, β-H); 8.41 (d, 1H, 3JHH = 7.5 Hz, 10-o1-Ph); 8.33 (t, 2H, 3JHH = 9.3 Hz, 5,15-o1-Ph); 7.96 (d, 1H, 3JHH = 7.5 Hz, 10-o2-Ph); 7.87 (d, 1H, 3JHH = 7.6 Hz, 5/15-o2-Ph); 7.76 (d, 1H, 3JHH = 6.7 Hz, 5/15-o1-Ph); 7.72 (d, 1H, 3JHH = 7.7 Hz, 10-m1-Ph); 7.66 (t, 2H, 3JHH = 6.7 Hz, 5,15-m1-Ph); 7.61 (d, 1H, 3JHH = 7.7 Hz, 10-m2-Ph); 7.54 (t, 2H, 3JHH = 7.5 Hz, 5,15-m2-Ph); 2.72 (s, 3H, 10-p-CH3); 2.70 (s, 6H, 5,15-p-CH3). MS (ESI): M+ = 796.1853 (expt), 796.1845 (calcd for C41H29N4O2Re). Elemental analysis (%) calcd for C41H29N4O2Re: C 61.87, H 3.67, N 7.04; found C 61.63, H 3.62, N 7.34. IR (cm−1): νReO 992, νCHO 1659.
Re[TpOCH3PC-3-CHO](O). Yield 36 mg (65.4%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 452 (11.06), 613 (4.14). 1H NMR (400 MHz, −20 °C, dichloromethane-d2) δ: 9.97 (s, 1H, CHO); 9.93 (s, 1H, β-H); 9.50 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.32 (d, 1H, 3JHH = 4.9 Hz, β-H); 9.25 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.15 (d, 1H, 3JHH = 5.0 Hz, β-H); 9.06 (t, 2H, 3JHH = 4.3 Hz, β-H); 8.44 (d, 1H, 3JHH = 6.5 Hz, 10-o1-Ph); 8.34 (d, 2H, 3JHH = 8.3 Hz, 5,15-o1-Ph); 8.02 (d, 1H, 3JHH = 6.6 Hz, 10-o2-Ph); 7.89 (d, 1H, 3JHH = 6.2 Hz, 5/15-o2-Ph); 7.82 (d, 1H, 3JHH = 6.2 Hz, 5/15-o2-Ph); 7.43 (d, 1H, 3JHH = 5.9 Hz, 10-m1-Ph); 7.39–7.31 (m, 3H, 10-m2-Ph & 5,15-m1-Ph); 7.27–7.23 (m, 2H, 5,15-m2-Ph); 4.06 (s, 6H, 5,15-p-OCH3); 4.04 (s, 3H, 10-p-OCH3). MS (ESI): M+ = 844.1695 (expt), 844.1692 (calcd for C41H29N4O5Re). Elemental analysis (%) calcd for C41H29N4O5Re: C 58.35, H 3.46, N 6.64; found C 58.13, H 3.62, N 6.54. IR (cm−1): νReO 992, νCHO 1655.
Re[TpFPC-3-CHO](O). Yield 35 mg (63.8%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 448 (8.87), 608 (2.93). 1H NMR (400 MHz, CDCl3, 20 °C) δ: 10.07 (s, 1H, CHO); 10.05 (s, 1H, β-H); 9.52 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.22 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.21 (d, 1H, 3JHH = 4.9 Hz, β-H); 9.06 (d, 1H, 3JHH = 5.0 Hz, β-H); 8.975 (d, 1H, 3JHH = 4.9 Hz, β-H); 8.972 (d, 1H, 3JHH = 5.1 Hz, β-H); 8.46–8.38 (m, 3H, Ph-H); 7.93 (m, 2H, Ph-H); 7.81 (m, 2H, Ph-H); 7.54–7.59 (m, 3H, Ph-H); 7.42–7.59 (m, 2H, Ph-H). MS (ESI): M+ = 808.1107 (expt), 808.1103 (calcd for C38H20F3N4O2Re). Elemental analysis (%) calcd for C38H20F3N4O2Re: C 56.50, H 2.50, N 6.94; found C 56.23, H 2.62, N 6.63 IR νReO: 1007 cm−1, νCHO: 1730, 1666 cm−1.
Re[TpCF3PC-3-CHO](O). Yield 25 mg (47.6%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 448 (9.21), 607 (3.17). 1H NMR (400 MHz, −20 °C, CDCl3) δ: 10.14 (s, 1H, CHO); 9.99 (s, 1H, β-H); 9.61 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.27 (dd, 2H, 3JHH = 9.4, 4.7 Hz, β-H); 9.06 (d, 1H, 3JHH = 5.1 Hz, β-H); 9.01 (dd overlapping, 2H, 3JHH = 4.9, 3.4 Hz, β-H); 8.68 (d, 1H, 3JHH = 8.0 Hz, 10-m1-Ph); 8.63 (dd overlapping, 2H, 3JHH = 12.0, 8.1 Hz, 5,15-m1-Ph); 8.22–8.14 (m, 5H, 10-m2-Ph & 5,15-m2-Ph & 10-o-Ph); 8.10–8.00 (m, 4H, 5,15-o-Ph). MS (ESI): M− = 958.1015 (expt), 958.1008 (calcd for C41H20F9N4O2Re). Elemental analysis (%) calcd for C41H20F9N4O2Re: C 51.41, H 2.10, N 5.85; found C 51.13, H 2.32, N 5.54. IR νReO: 992 cm−1, νCHO: 1663 cm−1.
Re[TpCH3PC-3,17-(CHO)2](O). Small quantities of this minor product were combined from multiple syntheses to derive the following spectroscopic data. UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 452 (6.84), 628 (3.12). 1H NMR (400 MHz, −20 °C, CDCl3) δ: 9.94 (s, 2H, CHO); 9.92 (s, 2H, β-H); 9.10 (d, 2H, 3JHH = 5.0 Hz, β-H); 8.95 (d, 2H, 3JHH = 5.0 Hz, β-H); 8.32 (d, 2H, 3JHH = 8.6 Hz, 5,15-o1-Ph); 8.27 (d, 1H, 3JHH = 7.7 Hz, 10-o1-Ph); 7.86 (d, 2H, 3JHH = 8.8 Hz, 5,15-o2-Ph); 7.74 (d, 1H, 3JHH = 7.6 Hz, 10-o2-Ph); 7.67 (d, 2H, 3JHH = 7.7 Hz, 5,15-m1-Ph); 7.57 (d, 2H, 3JHH = 7.6 Hz, 5,15-m2-Ph); 7.99 (d, 2H, 3JHH = 8.0 Hz, 10-m-Ph); 2.71 (s, 6H, 5,15-p-CH3); 2.70 (s, 3H, 10-p-CH3). MS (ESI): M+ = 824.1807 (expt), 824.1792 (calcd for C42H29N4O3Re).
General synthesis of cyanoacetic-acid (Knoevenagel) adducts
To a two-necked 50 mL round-bottom flask equipped with a rubber septum, a reflux condenser, a magnetic stir-bar, and an argon inlet/outlet, was added the desired ReVO-corrole-3-CHO derivative (0.04 mmol, ∼30 mg), dissolved in acetonitrile (10 mL). To this solution, cyanoacetic acid (30 mg, 0.04 mmol) and piperidine (two drops) were added. The reaction mixture was heated to reflux (∼80 °C) and left to stir for ∼1 hour under argon. After cooling, the reaction was quenched by washing with water (3 × 100 mL), prior to purification via column chromatography on silica gel using dichloromethane with 5% methanol as mobile phase. Unreacted monoformyl starting material eluted first, followed by the desired product.
Re[TPC-3-CH(CN)(CO2H)](O). Yield 21 mg (64.3%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 470 (7.32), 637 (3.98). 1H NMR (400 MHz, −20 °C) δ: 10.62 (s, 1H, β-H); 9.54 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.25 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.22 (d, 1H, 3JHH = 4.9 Hz, β-H); 9.00 (d, 1H, 3JHH = 5.0 Hz, β-H); 8.96 (d, 2H, 3JHH = 4.5 Hz, β-H); 8.75 (s, 1H, C–H); 8.53 (d, 1H, 3JHH = 7.4 Hz, 10-o1-Ph); 8.42 (t, 2H, 3JHH = 8.6 Hz, 5,15-o1-Ph); 8.06 (d, 1H, 3JHH = 6.7 Hz, 10-m1-Ph); 7.86 (m, 10H, 10-o2/m2/p-Ph, 5,15-o2/m/p-Ph). MS (ESI): M+ = 821.1360 (expt), 821.1366 (calcd for C41H24N5O3Re). Elemental analysis (%) calcd for C41H24N5O3Re: C 59.99, H 2.95, N 8.53; found C 59.73, H 2.62, N 8.59. IR (cm−1): νReO 996, νCHO 1570 cm−1, νCN: 1704 cm−1.
Re[TpCH3PC-3-CH(CN)(CO2H)](O). Yield 23.9 mg (79.74%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 471 (6.82), 638 (3.65). 1H NMR (400 MHz, 0 °C) δ: 10.58 (s, 1H, β-H); 9.52 (d, 1H, 3JHH = 4.6 Hz, β-H); 9.23 (dd, 2H, 3JHH = 6.8, 4.6 Hz, β-H); 9.03 (d, 1H, 3JHH = 4.8 Hz, β-H); 8.96 (d, 2H, 3JHH = 5.1 Hz, β-H); 8.73 (s, 1H, CH); 8.40 (d, 1H, 3JHH = 7.6 Hz, 10-o1-Ph); 8.30 (d, 1H, 3JHH = 7.8 Hz, 5/15-o1-Ph); 8.26 (d, 1H, 3JHH = 7.8 Hz, 5/15-o1-Ph); 7.96 (d, 1H, 3JHH = 7.4 Hz, 10-o2-Ph); 7.83 (d, 1H, 3JHH = 8.1 Hz, 10-m1-Ph); 7.74 (t, 3H, 3JHH = 8.7 Hz, 10-m2-Ph & 5,15-o2-Ph); 7.69–7.59 (m, 3H, 5,15-m1-Ph & 5/15-m2-Ph); 7.55 (d, 1H, 3JHH = 7.7 Hz, 5/15-m2-Ph); 2.80 (s, 3H, 10-p-CH3); 2.71 (s, 6H, 5,15-p-CH3). MS (ESI): M+ = 863.1831 (expt), 863.1836 (calcd for C44H30N5O3Re). Elemental analysis (%) calcd for C44H30N5O3Re: C 61.24, H 3.50, N 8.12; found C 61.13, H 3.62, N 8.54.
Re[TpFPC-CH(CN)(CO2H)](O). Yield 19.7 mg (66.2%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 470 (4.31), 637 (2.19). 1H NMR (400 MHz, THF, 20 °C) δ: 10.63 (s, 1H, β-H); 9.79 (d, 1H, 3JHH = 4.4 Hz, β-H); 9.29 (dd, 2H, 3JHH = 4.68, 2.4 Hz, β-H); 9.09 (d, 1H, 3JHH = 5.0 Hz, β-H); 9.03 (dd, 2H, 3JHH = 7.0, 5.0 Hz, β-H); 8.66 (s, 1H, CH); 8.50 (ddd, 1H, 3JHH = 8.1, 5.4, 2.3 Hz, 10-m1-Ph); 8.43 (ddd, 1H, 3JHH = 8.1, 5.4, 2.3 Hz, 5/15-m1-Ph); 7.97–7.86 (m, 3H, 5/15-m1-Ph, 10-m2, o1-Ph); 7.74–7.48 (m, 7H, Ph-H). MS (ESI): M+ 875.1163 (expt), 875.1163 (calcd for C41H21F3N5O3Re). Elemental analysis (%) calcd for C41H21F3N5O3Re: C 56.29, H 2.42, N 8.01; found C 56.13, H 2.62, N 8.34. IR νReO: 996 cm−1, νCHO: 1328 cm−1.
Re[TpCF3PC-CH(CN)(CO2H)](O). Yield 11 mg (46.7%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 466 (8.45), 631 (4.36). 1H NMR (400 MHz, CDCl3, 20 °C) δ: 10.58 (s, 1H, β-H); 9.55 (d, 1H, 3JHH = 4.6 Hz, β-H); 9.18 (d, 1H, 3JHH = 4.7 Hz, β-H); 9.13 (d, 1H, 3JHH = 4.7 Hz, β-H); 8.93 (d, 1H, 3JHH = 5.0, β-H); 8.90–8.86 (m, 2H, β-H); 8.52–8.46 (m, 4H, m1-Ph + CH); 8.11 (dd, 4H, 3JHH = 12.4, 8.8 Hz, 5,10,15-m2-Ph/10-o1-Ph); 8.02 (s broad, 3H, 10-o2-Ph/5,15-o1-Ph); 7.95 (d, 2H, 3JHH = 3.8 Hz, 5,15-o2-Ph). MS (ESI): M− 1025.1069 (expt), 1025.1064 (calcd for C44H21F9N5O3Re). Elemental analysis (%) calcd for C44H21F9N5O3Re: C 51.57, H 2.07, N 6.83; found C 51.33, H 2.32, N 6.54. IR νReO: 996 cm−1, νCHO: 1328 cm−1.
General procedure for formylation of Au triarylcorroles
To a 50 mL round-bottom flask equipped with a magnetic stir-bar, a rubber septum, and an argon inlet/outlet and placed in an ice/water bath, were added dichloromethane (5 mL), POCl3 (20 mmol, 1.8 mL), and DMF (23 mmol, 1.8 mL). Metallocorrole (0.025 mmol, ∼20 mg) was dissolved in DCM (10 mL) and added via syringe to the cold solution. The mixture was left to stir under a constant flow of argon at 0 °C for 15 minutes upon heating to room temperature. The reaction was left to stir overnight. The following morning the reaction was quenched via washing with water (3 × 100 mL) before purification through silica gel column chromatography using a mixture of n-hexane/dichloromethane (3:1) as eluent to isolate any unreacted starting material. The desired product was isolated by further elution with dichloromethane or a mixture with dichloromethane and methanol (5%).
Au[TPC-3,17-(CHO)2]. Yield 20 mg (61.17%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 426 (7.53), 437 (8.33), 596 (6.78), 611 (6.66). 1H NMR (400 MHz, 20 °C) δ: 9.65 (s, 2H, CHO); 9.53 (s, 2H, β-H); 8.66 (d, 2H, 3JHH = 5.1 Hz, β-H); 8.58 (d, 2H, 3JHH = 5.1 Hz, β-H); 8.10 (m, 5H, 5,15-o-Ph + 10-p-Ph); 7.77 (m, 10H, 5,10,15-m-Ph + 10-o-Ph + 5,15-p-Ph). MS (ESI): M+ = 776.1583 (expt), 776.1579 (calcd for C39H23N4O2Au). Elemental analysis (%) calcd for C39H23N4O2Au: C 60.32, H 2.99, N 7.21; found C 60.13, H 3.12, N 7.54.
Au[TpCH3PC-3-CHO]. Yield 2.3 mg (11.3%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 428 (9.84), 581 (4.02), 602 (4.21). 1H NMR (400 MHz, 20 °C) δ: 9.76 (s, 1H, CHO); 9.54 (s, 1H, β-H); 8.97 (d, 1H, 3JHH = 4.2 Hz, β-H); 8.90 (d, 1H, 3JHH = 4.9 Hz, β-H); 8.70 (d overlapping, 2H, 3JHH = 8.0 Hz, β-H); 8.64 (d overlapping, 2H, 3JHH = 4.8 Hz, β-H); 8.10 (d, 2H, 3JHH = 7.8 Hz, 10-o-Ph); 7.99 (d, 4H, 3JHH = 7.7 Hz, 5,15-o-Ph); 7.62 (d, 2H, 3JHH = 7.8 Hz, 10-m-Ph); 7.54 (dd, 4H, 3JHH = 12.6, 7.7 Hz, 5,15-m-Ph); 2.70 (s, 3H, p-CH3), 2.68 (s, 6H, p-CH3). MS (ESI): M+ = 791.2080 (expt), 791.2079 (calcd for C41H29N4OAu). Elemental analysis (%) calcd for C41H29N4OAu: C 62.28, H 3.70, N 7.09; found C 62.53, H 3.62, N 7.43. IR νCHO: 1655 cm−1.
Au[TpCH3PC-3,17-(CHO)2]. Yield 10.7 mg (53.5%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 425 (4.35), 439 (4.77), 596 (3.53), 615 (3.73). 1H NMR (400 MHz, 20 °C) δ: 9.58 (s, 2H, CHO); 9.27 (s, 2H, β-H); 8.62 (d, 2H, 3JHH = 5.0 Hz, β-H); 8.56 (d, 2H, 3JHH = 5.1 Hz, β-H); 7.94 (d overlapping, 2H, 3JHH = 8.0 Hz, 10-o-Ph); 7.91 (d overlapping, 4H, 3JHH = 7.8 Hz, 5,15-o-Ph); 7.57 (d, 2H, 3JHH = 7.8 Hz, 10-m-Ph); 7.53 (d, 4H, 3JHH = 7.7 Hz, 5,15-m-Ph); 2.68 (s, 9H, p-CH3). MS (ESI): M+ = 819.2029 (expt), 818.2029 (calcd for C42H29N4O2Au). Elemental analysis (%) calcd for C42H29N4O2Au: C 61.62, H 3.57, N 6.84; found C 61.23, H 3.62, N 6.56. IR νCHO: 1663 cm−1.
Au[TpOCH3PC-3,17-(CHO)2]. Yield 10.2 mg (51.1%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 426 (5.63), 439 (5.70), 597 (4.29), 618 (4.44). 1H NMR (400 MHz, 20 °C, CD2Cl2) δ: 9.70 (s, 2H, CHO); 9.35 (s, 2H, β-H); 8.67 (d, 2H, 3JHH = 5.0 Hz, β-H); 8.60 (d, 2H, 3JHH = 5.0 Hz, β-H); 7.99 (d broad, 6H, 3JHH = 8.0 Hz, m-Ph); 7.32–7.27 (m, 6H, o-Ph); 4.08 (s, 6H, 5,15-p-OCH3); 4.06 (s, 3H, 10-p-OCH3). MS (ESI): M+ = 866.1806 (expt), 866.1807 (calcd for C42H29N4O5Au). Elemental analysis (%) calcd for C42H29N4O5Au: C 58.21, H 3.37, N 6.46; found C 58.43, H 3.82, N 6.01.
Au[TpFPC-3-CHO]. Yield 4.1 mg (20.5%). UV-vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 427 (7.02), 580 (2.75), 597 (2.53). 1H NMR (20 °C, 400 MHz) δ: 9.68 (s, 1H, CHO); δ 9.42 (S, 1H, β-H); 8.86 (d, 1H, 3JHH = 4.5 Hz, β-H); 8.80 (d, 1H, 3JHH = 4.9 Hz, β-H); 8.59–8.55 (m, 4H, β-H); 8.11–8.07 (m, 2H, Ph-H); 8.00–7.94 (m, 4H, Ph-H); 7.47–7.34 (m, 6H, Ph-H). MS (ESI): M+ = 802.1257 (expt), 802.1260 (calcd for C38H20F3N4OAu). Elemental analysis was not performed.
Au[TpFPC-3,17-(CHO)2]. Yield 10 mg (52.8%). UV-vis (CH2Cl2): λmax (nm), [ε × 10−4 (M−1 cm−1)]: 436 (6.53), 597 (4.29), 609 (4.91). 1H NMR (400 MHz, CDCl3, 20 °C) δ: 9.52 (s, 2H, CHO); 9.16 (s, 2H, β-H); 8-53-8.54 (d, 2H, 3JHH = 5.0 Hz, β-H); 8.49 (d, 2H, 3JHH = 5.1 Hz, β-H); 7.97–7.94 (m, 2H, Ph-H); 7.92–7.88 (m, 4H, Ph-H); 7.35–7.45 (m, 6H, Ph-H). MS (ESI): M+ = 830.1207 (expt), 830.1209 (calcd for C39H20F3N4O2Au). Elemental analysis (%) calcd for C39H29F3N4O2Au: C 56.40, H 2.43, N 6.75; found: C 56.76, H 2.62, N 7.11.
X-ray crystallography
X-ray quality crystals were obtained via slow diffusion of methanol vapor into concentrated dichloromethane solutions of the compounds in question. X-ray data were collected (as previously described37) on beamline 12.2.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory. Samples were mounted on MiTeGen Kapton loops and placed in a 100(2)-K nitrogen cold stream provided by an Oxford Cryostream 800 Plus low-temperature apparatus on the goniometer head of a Bruker D8 diffractometer equipped with a PHOTON II CPAD detector operating in shutterless mode. Diffraction data were collected using synchrotron radiation monochromated using silicon(111) to a wavelength of 0.7288(1)Å. An approximate full-sphere of data was collected using a combination of φ and ω scans with scan speeds of one second per degree for the φ scans and one and three seconds per degree for the ω scans at 2θ = 0 and −20°, respectively. The structures were solved by intrinsic phasing (SHELXT50) and refined by full-matrix least-squares on F2 (SHELXL-2014 (ref. 51)). All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were geometrically calculated and refined as riding atoms. Additional crystallographic information has been summarized in Table 1.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Research Council of Norway (grant no. 262229 and 324139 to AG) and the Arctic Center for Sustainable Energy at UiT – The Arctic University of Norway. The work also used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.
References
- A. Ghosh, Chem. Rev., 2017, 117, 3798–3881 CrossRef CAS PubMed.
- S. Nardis, F. Mandoj, M. Stefanelli and R. Paolesse, Coord. Chem. Rev., 2019, 388, 360–405 CrossRef CAS.
- Y. Fang, Z. Ou and K. M. Kadish, Chem. Rev., 2017, 117, 3377–3419 CrossRef CAS PubMed.
- A. B. Alemayehu, K. E. Thomas and R. F. Einrem, Acc. Chem. Res., 2021, 54, 3095–3107 CrossRef CAS PubMed.
- A. B. Alemayehu and A. Ghosh, J. Porphyrins Phthalocyanines, 2011, 15, 106–110 CrossRef CAS.
- E. Rabinovitch, I. Goldberg and Z. Gross, Chem.–Eur. J, 2011, 17, 12294–12301 CrossRef PubMed.
- K. E. Thomas, A. B. Alemayehu, J. Conradie, C. Beavers and A. Ghosh, Inorg. Chem., 2011, 50, 12844–12851 CrossRef CAS PubMed.
- R. F. Einrem, H. Braband, T. Fox, H. Vazquez-Lima, R. Alberto and A. Ghosh, Chem.–Eur. J, 2016, 22, 18747–18751 CrossRef CAS PubMed.
- R. F. Einrem, K. J. Gagnon, A. B. Alemayehu and A. Ghosh, Chem.–Eur. J, 2016, 22, 517–520 CrossRef CAS PubMed.
- A. B. Alemayehu, S. J. Teat, S. M. Borisov and A. Ghosh, Inorg. Chem., 2020, 59, 6382–6389 CrossRef CAS PubMed.
- A. B. Alemayehu, H. Vazquez-Lima, K. J. Gagnon and A. Ghosh, Inorg. Chem., 2017, 56, 5285–5294 CrossRef CAS PubMed.
- A. B. Alemayehu, K. J. Gagnon, J. Terner and A. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 14411–14414 CrossRef CAS PubMed.
- A. B. Alemayehu, H. Vazquez-Lima, C. M. Beavers, K. J. Gagnon, J. Bendix and A. Ghosh, Chem. Commun., 2014, 50, 11093–11096 RSC.
- A. B. Alemayehu, L. J. McCormick, K. J. Gagnon, S. M. Borisov and A. Ghosh, ACS Omega, 2018, 3, 9360–9368 CrossRef CAS PubMed.
- A. B. Alemayehu, H. Vazquez-Lima, K. J. Gagnon and A. Ghosh, Chem.–Eur. J, 2016, 22, 6914–6920 CrossRef CAS PubMed.
- A. B. Alemayehu, H. Vazquez-Lima, L. J. McCormick and A. Ghosh, Chem. Commun., 2017, 53, 5830–5833 RSC.
- C. Schies, A. B. Alemayehu, H. Vazquez-Lima, K. E. Thomas, T. Bruhn, G. Bringmann and A. Ghosh, Chem. Commun., 2017, 53, 6121–6124 RSC.
- A. B. Alemayehu, L. J. McCormick-McPherson, J. Conradie and A. Ghosh, Inorg. Chem., 2021, 60, 8315–8321 CrossRef CAS PubMed.
- J. H. Palmer, A. C. Durrell, Z. Gross, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 9230–9231 CrossRef CAS PubMed.
- W. Sinha, L. Ravotto, P. Ceroni and S. Kar, Dalton Trans., 2015, 44, 17767–17773 RSC.
- S. M. Borisov, A. B. Alemayehu and A. Ghosh, J. Mater. Chem. C, 2016, 4, 5822–5828 RSC.
- C. M. Lemon, D. C. Powers, P. J. Brothers and D. G. Nocera, Inorg. Chem., 2017, 56, 10991–10997 CrossRef CAS PubMed.
- A. B. Alemayehu, N. U. Jae Day, T. Mani, A. B. Rudine, K. E. Thomas, O. A. Gederaas, S. A. Vinogradov, C. C. Wamser and A. Ghosh, ACS Appl. Mater. Interfaces, 2016, 8, 18935–18942 CrossRef CAS PubMed.
- S. M. Borisov, R. F. Einrem, A. B. Alemayehu and A. Ghosh, Photochem. Photobiol. Sci., 2019, 18, 1166–1170 CrossRef CAS PubMed.
- R. D. Teo, J. Y. Hwang, J. Termini, Z. Gross and H. B. Gray, Chem. Rev., 2017, 117, 2711–2729 CrossRef CAS PubMed.
- A. Mahammed and Z. Gross, Coord. Chem. Rev., 2019, 379, 121–132 CrossRef CAS.
- C. M. Lemon, Pure Appl. Chem., 2020, 92, 1901–1919 CrossRef CAS.
- G. Golubkov, et al., Angew. Chem., Int. Ed., 2001, 40, 2132–2134 CrossRef CAS PubMed.
- J. F. B. Barata, M. G. P. M. S. Neves, M. A. F. Fautino, A. C. Tomé and J. A. Cavaleiro, Chem. Rev., 2017, 117, 3192–3253 CrossRef CAS PubMed.
- J. H. Palmer, A. C. Durrell, Z. Gross, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2008, 130, 7786–7787 CrossRef CAS PubMed.
- K. E. Thomas, J. Conradie, L. K. Hansen and A. Ghosh, Eur. J. Inorg. Chem., 2011, 1865–1870 CrossRef CAS.
- S. Berg, K. E. Thomas, C. M. Beavers and A. Ghosh, Inorg. Chem., 2012, 51, 9911–9916 CrossRef CAS PubMed.
- K. E. Thomas, L. J. McCormick, D. Carrié, H. Vazquez-Lima, G. Simmoneaux and A. Ghosh, Inorg. Chem., 2018, 57, 4270–4276 CrossRef CAS PubMed.
- I. K. Thomassen, L. J. McCormick and A. Ghosh, ACS Omega, 2018, 3, 5106–5110 CrossRef CAS PubMed.
- A. Reinholdt, A. B. Alemayehu, K. J. Gagnon, J. Bendix and A. Ghosh, Inorg. Chem., 2020, 59, 5276–5280 CrossRef CAS PubMed.
- J. H. Palmer, A. C. Durrell, Z. Gross, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2008, 130, 7786–7787 CrossRef CAS PubMed.
- A. B. Alemayehu, R. F. Einrem, L. J. McCormick-McPherson, N. S. Settineri and A. Ghosh, Sci. Rep., 2020, 10, 19727 CrossRef CAS PubMed.
- M. Soll, et al., Org. Lett., 2016, 18, 5840–5843 CrossRef CAS PubMed.
- K. Sudhakar, et al., Angew. Chem., Int. Ed., 2017, 56, 9837–9841 CrossRef CAS PubMed.
- P. Schweyen, K. Brandhorst, M. Hoffmann, B. Wolfram, M.-K. Zaretzke and M. Bröring, Chem.–Eur. J, 2017, 23, 13897–13900 CrossRef CAS PubMed.
- H. Vazquez-Lima, J. Conradie, M. A. L. Johansen, S. R. Martinsen, A. B. Alemayehu and A. Ghosh, Dalton Trans., 2021, 50, 12843–12849 RSC.
- I. K. Thomassen, D. Rasmussen, R. F. Einrem and A. Ghosh, ACS Omega, 2021, 6, 16683–16687 CrossRef CAS PubMed.
- A. Vilsmeier and A. Haack, Ber. Dtsch. Chem. Ges., 1927, 27, 119–122, DOI:10.1002/cber.19270600118.
- R. Paolesse, L. Jaquinod, M. O. Senge and K. M. Smith, J. Org. Chem., 1997, 62, 6193–6198 CrossRef CAS.
- I. Saltsman, A. Mahammed, I. Goldberg, E. Tkachenko, M. Botoshansky and Z. Gross, J. Am. Chem. Soc., 2002, 124, 7411–7420 CrossRef CAS PubMed.
- R. Paolesse, S. Nardis, M. Venanzi, M. Mastroianni, M. Russo, F. Fronczek and M. G. H. Vicente, Chem.–Eur. J, 2003, 9, 1192–1197 CrossRef CAS PubMed.
- K. Sorasaenee, P. Taqavi, L. M. Henling, H. B. Gray, E. Tkachenko, A. Mahammed and Z. Gross, J. Porphyrins Phthalocyanines, 2007, 11, 189–197 CrossRef CAS.
- S. Ganguly and A. Ghosh, Acc. Chem. Res., 2019, 52, 2003–2014 CrossRef CAS PubMed.
- J.-H. Fuhrhop, K. M. Kadish and D. G. Davis, J. Am. Chem. Soc., 1973, 95, 5140–5147 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2091847 and 2091848. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05525a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Nicholas S. Settineri
b,
Nicholas S. Settineri