
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceViologen–cucurbituril host/guest chemistry – redox control of dimerization versus inclusion†
Parastoo Dalvanda,
Katia Nchimi Nonob,
Dinesh Shetty c,
Farah Benyettoud,
Zouhair Asfarie,
Carlos Platas-Iglesiasf,
Mark A. Olsong,
Ali Trabolsi*d and
Mourad Elhabiri*a
c,
Farah Benyettoud,
Zouhair Asfarie,
Carlos Platas-Iglesiasf,
Mark A. Olsong,
Ali Trabolsi*d and
Mourad Elhabiri*a
aUniversité de Strasbourg, Université de Haute-Alsace, CNRS, LIMA, UMR 7042, Equipe Chimie Bioorganique et Médicinale, ECPM, 25 Rue Becquerel, 67000 Strasbourg, France. E-mail: elhabiri@unistra.fr
bDepartment of Inorganic Chemistry, Faculty of Science, University of Yaoundé 1, Yaoundé, Cameroon
cDepartment of Chemistry & Center for Catalysis and Separation (CeCaS), Khalifa University, Abu Dhabi, United Arab Emirates
dNew York University Abu Dhabi (NYUAD), Experimental Research Building, Building C1, Saadiyat Island, Abu Dhabi, United Arab Emirates. E-mail: ali.trabolsi@nyu.edu
eEquipe de Synthèse Pour l'Analyse (SynPA), Institut Pluridisciplinaire Hubert Curien (IPHC), UMR 7178 CNRS/Université de Strasbourg, ECPM, Bâtiment R1N0, 25-rue Becquerel, 67087 Strasbourg Cedex 2, France
fUniversidade da Coruña, Centro de Investigacións Científicas Avanzadas (CICA), Departamento de Química, Facultade de Ciencias, 15071 A Coruña, Galicia, Spain
gNorthwestern University, Department of Chemistry, 2145 Sheridan Rd, Evanston, Illinois, USA
First published on 2nd September 2021
Abstract
Two calix[4]arene systems, C234+ and C244+ – where 2 corresponds to the number of viologen units and 3–4 corresponds to the number of carbon atoms connecting the viologen units to the macrocyclic core – have been synthesized and led to the formation of [3]pseudorotaxanes when combined with either CB[7] or CB[8]. The [3]pseudorotaxanes spontaneously dissociate upon reduction of the bipyridinium units as the result of intramolecular dimerization of the two face-to-face viologen radical cations. CB[7] and CB[8]-based [2]pseudorotaxanes containing monomeric viologen guest model compounds, MC32+ and MC4+, do not undergo decomplexation and dimerization following electrochemical reduction of their bipyridinium units.
Introduction
The stimuli–responsive properties of a molecule with redox-active units can be advantageously used in a broad range of applications including drug delivery and development, electronic displays, electronic memory, batteries, and multi-state switchable materials.1–11 Among these redox-active molecules, for example metallocenes, tetrathiafulvalene derivatives, naphthalene diimides, and benzoquinones, bipyridinium derivatives12 (also known as viologens) are of particular interest because of their vibrant electrochromic properties. Bipyridinium dications (BIPY2+), for example, are typically colorless whereas BIPY˙+ radical monocations13 are deep blue in color.14 Furthermore, these radical cations spontaneously undergo π-dimerization, a process known as pimerization15 that is driven by radical–radical interactions that favor the formation of the singlet state (i.e., panchromatic absorption)14–16 to afford the (BIPY˙+)2 dimer in aqueous solution. Pimerization of BIPY˙+ radical cations has been used in supramolecular and mechanostereochemical systems to generate translational and rotational molecular movement in many recently reported mechanically interlocked molecules.17–19 Furthermore, the rich redox chemistry of viologens also makes them attractive for use in electrochromic, catalytic, and bioanalytical applications.20–22However, one major complication that limits the use of viologen-based pimerization to develop stimuli–responsive materials is that the system suffers from low dimerization constants (e.g., log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KDim = 2.70).23 This results in loosely associated π-dimers in solution, and thus requires the use of high concentrations of the constituent molecular components to drive the binding. The use of low temperatures, micelle environments,24 or macrocyclic hosts with large cavity sizes such as cucurbiturils (e.g., CB[8]), cyclodextrins (e.g., β-CD), or Stoddart's Blue Box17,18,25 can help favor the formation of intermolecular π-dimer species. A. E. Kaifer, K. Kim and their coworkers described for the first time the interaction of MV2+ with cucurbit[n]uril hosts such as CB[7] and CB[8].26–28 With CB[8], it was found that MV2+ strongly binds in a 1:1 stoichiometry inside the macrocycle cavity, the major driving force being ion–dipole interactions between the positively charged viologen and the oxygen rich host portals atoms. One electron reduction of methyl viologen MV2+ allowed the formation of a bisradical species ((MV˙+)2⊂CB[8]). The dimerization constant of the MV˙+ in the presence of the CB[8] was estimated to be ∼2 × 107 M−1 which is about 105 times larger than MV˙+ alone.28,29 Supramolecular assemblies based on CB[8] have been reviewed recently by A. E. Kaifer, M. D. Garcia et al.30
KDim = 2.70).23 This results in loosely associated π-dimers in solution, and thus requires the use of high concentrations of the constituent molecular components to drive the binding. The use of low temperatures, micelle environments,24 or macrocyclic hosts with large cavity sizes such as cucurbiturils (e.g., CB[8]), cyclodextrins (e.g., β-CD), or Stoddart's Blue Box17,18,25 can help favor the formation of intermolecular π-dimer species. A. E. Kaifer, K. Kim and their coworkers described for the first time the interaction of MV2+ with cucurbit[n]uril hosts such as CB[7] and CB[8].26–28 With CB[8], it was found that MV2+ strongly binds in a 1:1 stoichiometry inside the macrocycle cavity, the major driving force being ion–dipole interactions between the positively charged viologen and the oxygen rich host portals atoms. One electron reduction of methyl viologen MV2+ allowed the formation of a bisradical species ((MV˙+)2⊂CB[8]). The dimerization constant of the MV˙+ in the presence of the CB[8] was estimated to be ∼2 × 107 M−1 which is about 105 times larger than MV˙+ alone.28,29 Supramolecular assemblies based on CB[8] have been reviewed recently by A. E. Kaifer, M. D. Garcia et al.30
Alternatively, it has been shown that pre-organizing two or more organic radicals with well-tailored anchoring platforms and appropriate chain lengths can strengthen the intramolecular binding of the π-dimers in solution.31 We have previously demonstrated that a hexavalent phosphazene30 fulfilled most of the prerequisites including ease of functionalization, and exhibited efficient and fast pimerization. This compound was applicable towards electrochromic material research and possessed specific recognition properties as a function of the viologen redox state. In addition, alkyl,32–37 porphyrinic,38,39 ferrocenyl,40 aryl,32–44 C60,45 and calixarene46,47 linkers, as well as dendritic48,49 and covalent organic polymeric50,51 viologen-based materials (Scheme S1 in the ESI†) have also been demonstrated to promote intramolecular dimerization of their corresponding bipyridinium radical cations.
In line with our ongoing research on viologen-based systems for generating redox-active devices, we turned our attention to calix[4]arene as an inert anchoring platform. Calix[4]arenes can be easily chemically modified and their structural and conformational properties make for an excellent foundation upon which to build increasingly complex architectures. Calix[4]arene can exist in four conformations in solution, the cone, the partial cone, the 1,2-alternate, and the 1,3-alternate. Furthermore, the alkylation selectivity, in regard to the number of substituted phenols, can be controlled by the reaction conditions, thereby giving rise to a broad range of conformers and cavity sizes.52
Herein, we report the synthesis and physicochemical characterization of a series of host–guest monomeric and calix[4]arene-functionalized dimeric viologen⊂CB-based pseudorotaxane complexes and the structure–property relationships that enable their electrochemical-triggered decomplexation. Calix[4]arene was functionalized with two terminal viologens using spacers of varying length (Fig. 1).45 The use of synthetic receptors that have both a high affinity and a high selectivity for the binding of guests in water is indeed a very interesting prospect.53–56 The recognition properties of the viologen-based systems (oxidized and reduced) with CB[7] and CB[8] were analyzed using a set of complementary analytical methods which include ESI-MS, square wave and cyclic voltammetry, UV-visible-near infrared (NIR) spectrophotometry, 1H-NMR, and electron paramagnetic resonance (EPR).
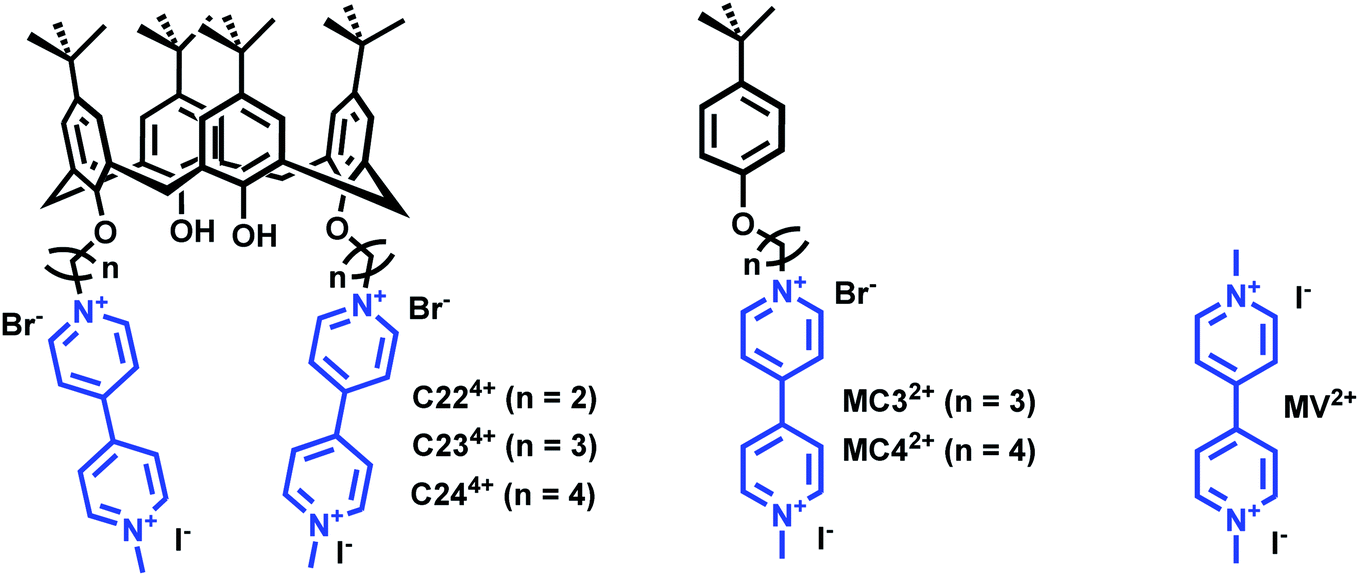 | ||
| Fig. 1 Chemical structures of the calix[4]arene viologen-based systems C234+ and C244+ and the reference compounds MC32+ and MC44+. The number 2 stands for the number of viologens while 3–4 correspond to the number of carbons of the chain connecting the terminal viologens to the calix[4]arene backbone. Methyl-viologen (MV2+) has been used as a model compound in this work. C224+ (PF6− salt) was not synthesized in this work (see ref. 45) and only considered for DFT calculations. | ||
Experimental section
Synthesis
Results and discussion
Synthesis
Two calix[4]arene systems, namely C234+ and C244+, whereby 2 stands for the number of viologens and 3–4 correspond to the number of carbons of the chain connecting the viologens to the calix[4]arene core (Scheme 1), were synthesized. For a deeper understanding of the host–guest complexation properties of the viologen derivatives with either CB[7] or CB[8], two model compounds, namely MC32+ and MC42+, were also prepared. 4,4′-bipyridine was first monomethylated using 1 equivalent of methyl iodide in acetone leading to I1 with 63% yield. Tert-butyl-phenol was reacted either with 1,3-dibromo-propane or 1,4-dibromo-butane in acetone in the presence of 1 equivalent of potassium carbonate to provide intermediates I2 or I3, respectively. The synthesis of MC32+ and MC42+ was then achieved following the reaction of I2 or I3 with 1-methyl-4,4′-bipyridinium (I1) in CH3CN. The synthesis of the tetrakis-p-tert-butyl-calix[4]arene I4 (63% yield) was achieved by reaction of p-tert-butyl-phenol with excess of formaldehyde in the presence of NaOH. As described above, I4 was then reacted with 1,3-dibromo-propane and 1,4-dibromo-butane to lead to intermediates I5 (92% yield) or I6 (65% yield), respectively. Reacting these two calix[4]arene intermediates with I1 then leads to the targeted bis-viologen systems C234+ (85% yield) and C244+ (67% yield). All these viologen derivatives were isolated in the form of mixed iodide/bromide salts.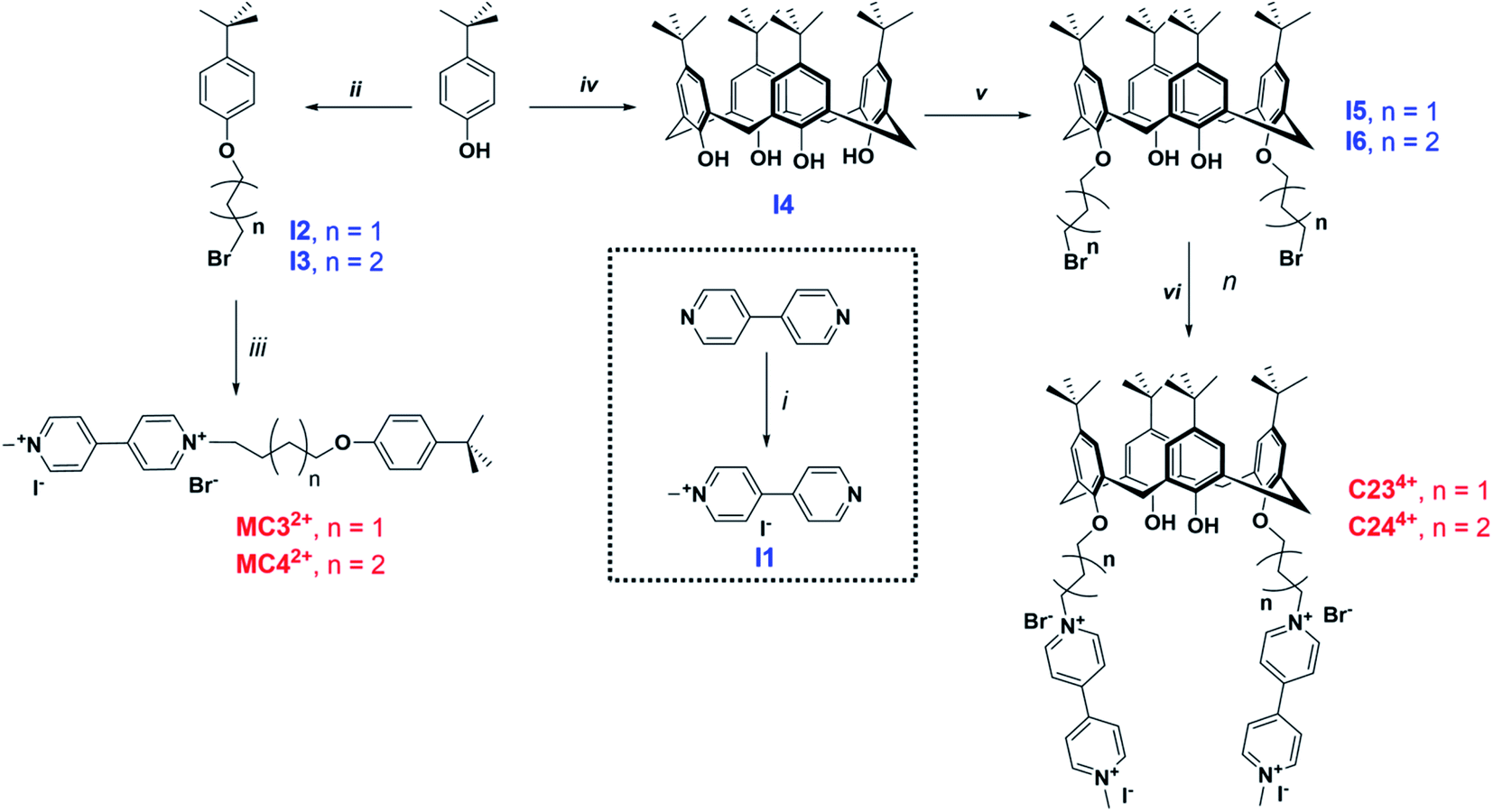 | ||
| Scheme 1 Synthetic route to the MC32+ and MC42+ monomers as well as the calix[4]arene-bis-viologen systems C234+ and C244+. Conditions: (i) CH3I, acetone, reflux, 24 h, 63%; (ii) I2: 1,3-dibromopropane, K2CO3, acetone, reflux, 48 h, yield 70%; I3: 1,4-dibromobutane, K2CO3, acetone, reflux, 72 h, yield 27%; (iii) MC32+: I1, CH3CN, reflux, 72 h, 31%; MC42+: I1, CH3CN, reflux, 72 h, 71%; (iv) NaOH, HCHO, 100 °C, then heating at 195–250 °C in diphenyl ether, toluene, xylene, yield 63%; (v) I5: 1,3-dibromopropane, K2CO3, acetone, reflux, 96 h, yield 92%; I6: 1,4-dibromobutane, K2CO3, acetone, reflux, 96 h, yield 65%; (vi) (iii) C234+: I1, CH3CN, reflux, 72 h, 85%; C244+: I1, CH3CN, reflux, 72 h, 67%. | ||
Characterization of the thread/CB[7] [n]pseudorotaxanes
:1 stoichiometric host–guest complexes. Both MC32+ and MC42+ are characterized by two main absorptions at ∼225 nm and 260 nm (Table 1) that are attributed to the π–π* transitions of the tert-butyl-phenyl and BIPY2+ chromophoric units, respectively. It was observed that complexation with CB[7] induces a hypochromic shift of the absorption band attributed to the BIPY2+ transitions, while those of the tert-butyl-phenyl units remain almost unaltered. This suggested that CB[7] is bound to the BIPY2+ subunit rather than the apparently more hydrophobic alkoxy-tert-butyl-phenyl group. This binding behavior is most likely explained by the weaker affinity67 of the alkoxy-tert-butyl-phenyl moiety for CB[7] and steric hindrance of its bulky tert-butyl group. The calculated binding constants (logKMC32+⊂CB[7] = 4.51(6) and logKMC42+⊂CB[7] = 4.68(5) determined in 0.1 M phosphate buffer, pH 7.0) were found to be lower than that determined for MV2+, most likely as a consequence of statistical effects. In this case, the two pyridinium aromatic faces of MV2+ are not sterically blocked to CB[7], while one side of the bipyridinium units of MC3+ and MC4+ are capped with a tert-butyl-phenyl bulky stopper. Consequently, the KMC32+/KMV2+ (0.16) and KMC42+/KMV2+ (0.24) ratios are close to the statistical value of 0.25.68 At the higher concentrations that were used for 1H NMR (Fig. S12 and S13 in the ESI†) and the higher CB[7]/MC32+ or CB[7]/MC42+ host–guest ratios employed for ESI-MS measurements (Fig. S8 and S9 in the ESI†), other host–guest complexes were observed, namely the [3]pseudorotaxanes, MC32+⊂(CB[7])2 and MC42+⊂(CB[7])2. For these complexes, the 1H NMR titrations suggested that the second CB[7] macrocycle is sitting in close proximity to the tert-butyl-phenyl unit (Fig. 2a). Binding of the first CB[7] indeed mainly affects the β and β′ protons of the BIPY2+ unit, while recognition of the second CB[7] macrocycle influences the protons of the tert-butyl-phenyl residue (Fig. S12 and S13 in the ESI†). The inability to quantify the second recognition event leading to the formation of MC32+⊂(CB[7])2 or MC42+⊂(CB[7])2 by absorption spectrophotometry likely resulted from weak spectral variations and their much lower stability constants.
| Viologen V | logKV⊂CB[7] |
V | V⊂CB[7] |
|---|---|---|---|
| λ (ε) nm (104 M−1 cm−1) | λ (ε) nm (104 M−1 cm−1) | ||
| a Solvent: water buffered at pH 7.0 with 0.1 M Na2H2PO4/NaH2PO4); l = 1 cm; T = 25.0(1) °C. The error (indicated in brackets) on the stability constants correspond to 3σ with σ = standard deviation. sh = shoulder. | |||
| MV2+ | 5.30 (2) | 227 (2.96)/257 (2.06) | 226 (3.13)/281 (sh) |
| MC32+ | 4.51 (6) | 224 (2.29)/259 (2.17) | 223 (2.49)/255 (1.51) |
| MC42+ | 4.68 (5) | 224 (2.17)/260 (2.29) | 224 (2.48)/252 (1.40) |
| C234+ | 4.5 (1) | 224 (7.42)/262 (3.20) | 225 (7.50)/275 (2.10) |
| C244+ | 4.44 (8) | 220 (7.16)/263 (2.99) | 220 (7.29)/280 (1.83) |
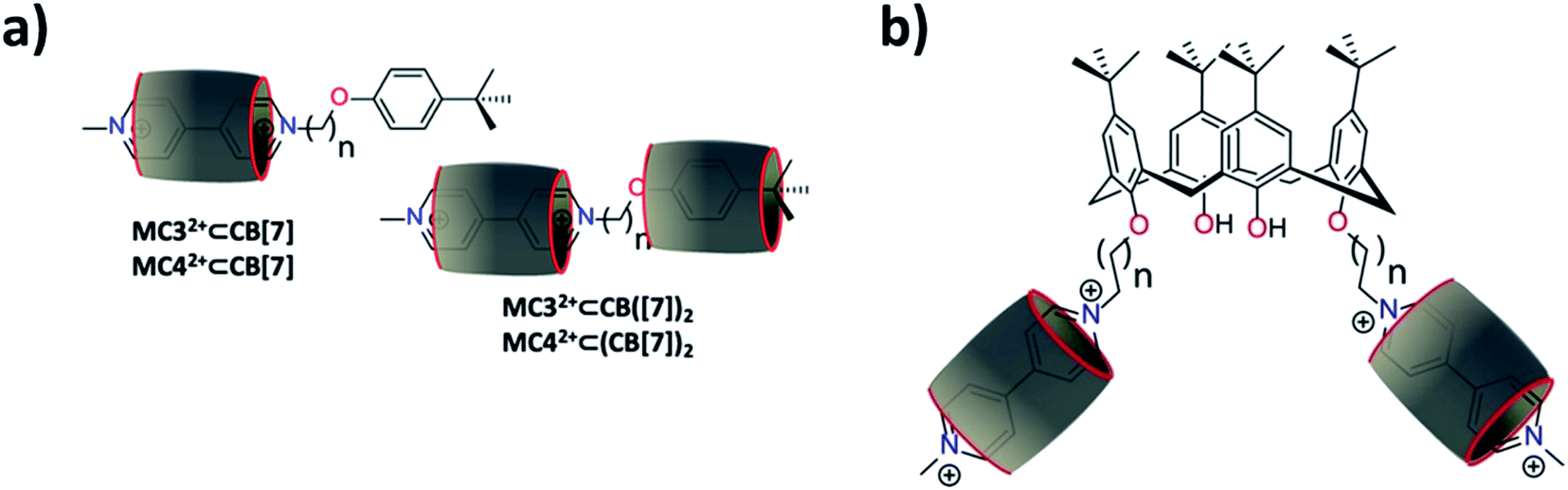 | ||
| Fig. 2 Schematic representation of (a) the [2]pseudorotaxanes MC32+⊂CB[7] and MC42+⊂CB[7] (CB[7] mainly resides on the bipyridinium group) and of MC32+⊂(CB[7])2 and MC42+⊂(CB[7])2 (i.e., the hydrophobic cavity of the second CB[7] is sitting close to the terminal phenyl group); and (b) the [3]pseudorotaxanes C234+⊂(CB[7])2 and C244+⊂(CB[7])2 (i.e., the CB[7] mainly resides on the bipyridinium groups). n = 2, or 3. CB[7] is represented with the grey cylinder. | ||
To gain further insight into the molecular recognition properties, diffusion coefficients were determined by 1H NMR-DOSY for MC32+ and MC42+ both in the absence and in the presence of increasing amounts of CB[7] (Table 2). Firstly, a comparison of the diffusion coefficients of the free MC32+ and MC42+ revealed similar values, which indicated that both model systems share common structural properties (i.e., comparable volume of 303 cm3 mol−1 for MC32+ and 318 cm3 mol−1 for MC42+ estimated with DFT) notwithstanding the slightly elongated carbon spacer for MC42+. Secondly, the values of the diffusion coefficients decreased as a function of the number of equivalents of CB[7], demonstrating the successive formation of host–guest species of increasing volume and weight (e.g., volume of 928 cm3 mol−1 obtained with DFT for MC32+⊂CB[7]). Lastly, the diffusion coefficients measured for MC32+ and MC42+ using the same amount of CB[7] are comparable to one another, which suggested that the [2]pseudorotaxanes, MC32+⊂CB[7] and MC42+⊂CB[7], and the [3]pseudorotaxanes, MC32+⊂(CB[7])2 and MC42+⊂(CB[7])2, are roughly structurally equivalent. The inability to determine the stability constant of [3]pseudorotaxane prevents the evaluation of the diffusion coefficients specific to each inclusion complex (i.e., [2]- and [3]pseudorotaxanes).
| D (×10−6 cm2 s−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| No CB[7] | +1 eq. CB[7] | +2 eq. CB[7] | No CB[7] | +2 eq. CB[7] | |||||
| MC32+ | MC42+ | MC32+ | MC42+ | MC32+ | MC42+ | C234+ | C244+ | C234+ | C244+ |
| a Measured by DOSY experiments on a 600 MHz spectrometer, D2O, 298 K. The errors on these values are estimated to less than 5%. | |||||||||
| 6.51 | 6.57 | 4.89 | 4.17 | 3.56 | 3.29 | 2.58 | 4.30 | 3.85 | 3.11 |
K*CB234+⊂(CB[7])2 = 4.5(1) and logK*CB244+⊂(CB[7])2 = 4.44(8)) were found to be close to those evaluated for the corresponding models, MC32+ and MC42+ (Table 1). Again, these stability constant values were found to be lower than those measured for MV2+ (logKMV2+⊂CB[7] = 5.30(2)) as a consequence of the presence of the bulky calix[4]arene core (Fig. 2b).Diffusion coefficients were also determined for C234+ and C244+ both in the absence and the presence of CB[7]. In contrast to the values measured for the model ligands, the D value measured for C234+ was found to be lower than that of C244+. One would expect that the extended spacer of C244+ should increase the global volume of the molecule (i.e., DFT calculated volumes of 824 cm3 mol−1 for C234+ and 873 cm3 mol−1 for C244+), thereby causing it to diffuse slower in solution. This unexpected result thus might be explained by the slightly more flexible arms of C244+ which should minimize the steric interactions of the viologen units with the bulky calix[4]arene core, thereby resulting in markedly different solvation properties with respect to C234+. As a consequence, the diffusion coefficient decreases in the presence of CB[7] for C244+, while it increases for C234+, so that both converge to a comparable value, the volumes of host–guest species with C234+ and C244+ being then globally equivalent in the presence of CB[7].
Characterization of the radical cations
KDim value of 3.4. This value is very similar to the value previously determined for the benzyl methyl viologen radical cation BMV˙+ (logKDim = 3.46(5)),30 but much higher than that reported for the methyl viologen radical cation MV˙+ (logKDim ∼2.5–2.9).69,70 The values of KDim are also similar to those measured for viologens decorated with hydrophobic alkyl substituents.15 This suggested that increasing the hydrophobicity around the BIPY˙+ unit with aryl/alkyl substituents favors the intermolecular pimerization in aqueous solution. Moreover, we could again demonstrate that the extended spacer for MC4˙+ does not drastically alter its pimerization since similar KDim values were calculated for MC3˙+ and MC4˙+. These two radical cations are characterized by intense and structured absorption bands in the visible region (MC3˙+: λmax ∼602 nm, ε602 = 1.01 × 104 M−1 cm−1; MC4˙+: λmax ∼602 nm, ε602 = 9.56 × 103 M−1 cm−1, Fig. S21 and S22 in the ESI†), in agreement with the spectroscopic parameters determined for MV˙+ (600–606 nm)15,23,67,71 and BMV˙+ (600 nm).30 Formation of the radical cation dimers induced a significant hypsochromic shift of the absorption band at ∼600 nm (Δλ ∼50 nm) and gave rise to intense absorption bands corresponding to radical–radical transitions in the NIR region (λmax > 850–900 nm).15 These UV-vis absorption studies also demonstrated that no significant pimerization occurs unless the concentration of the radical cations is high enough (>10−3 M, Fig. S21 and S22 in the ESI†). As previously reported,31–45 preorganization of the BIPY2+ electrophores to within close proximity of one another around a robust and inert molecular platform can enhance their pimerization, leading to very stable π-dimers in aqueous solution.Upon chemical generation of the radical cations, C232(˙+) and C242(˙+) (Fig. S27 and S28 in the ESI†), intense absorption bands appeared at ∼530 nm (Fig. 3). An additional intense absorption band centered at 1072 nm for C232(˙+) and at 925 nm for C242(˙+) were observed and are unambiguously assigned to the intramolecular charge resonance that occurs in the dimerized viologen radical cation species. This feature indicated that a two-electron reduction of C234+ and C244+ leads to two radical divalent cations, C232(˙+) and C242(˙+), that spontaneously pimerize intramolecularly, leading to a stable radical dimeric species.
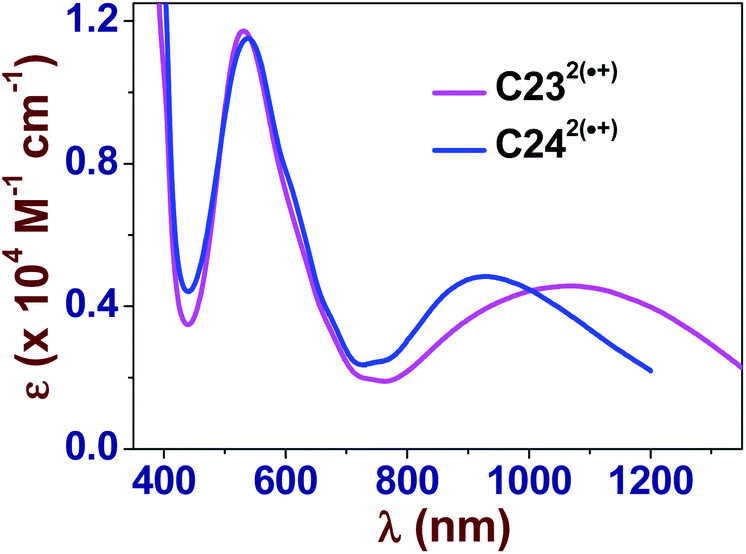 | ||
| Fig. 3 Electronic UV-vis-NIR absorption spectra of C232(˙+) and C242(˙+) in water. Solvent: water buffered at pH 7.0 with 0.1 M Na2HPO4/NaH2PO4. T = 25.0(1) °C. | ||
The NIR absorption spectroscopic signatures of the π-dimerized complexes were demonstrated to be closely related to the extent of overlapping between both π-orbitals of the BIPY˙+ radicals.45 When a propyloxy chain links the viologens to the calix[4]arene moiety (C232(˙+): λmax ∼960 nm in CH3CN45 and λmax ∼1070 nm in water), DFT calculations at the BLYP-D3/DZVP level suggested that only one pyridinium ring per viologen radical is involved in the intramolecular π-dimerization process. This result, which is imposed by geometric constraints that is inherent to the length and structure of the propyl chain, is not observed with the ethyloxy-derived calix[4]arene-bis-viologen (C222(˙+): λmax ∼860 nm in CH3CN45), which affords a fully face-to-face stacked arrangement. DFT calculations performed at the M06-2X/6-311G(d,p) level (Fig. 4) provided a minimum energy conformation for C222(˙+) in which the two BIPY˙+ units display an almost perfect face-to-face interaction. Similarly, the BIPY˙+ units in C232(˙+) are twisted with respect to each other by an angle of ∼78°, reducing the overlap between the corresponding π-orbitals. Increasing the length from 3 to 4 carbons (butyloxy derivative, C242(˙+): λmax ∼925 nm in water) does not completely reinstate the face-to-face dimerized π-stacking orientation, but it does increase the overlap of the π-orbitals with respect to C232(˙+) with a twist angle of ∼60°. Thus, the nature and size of the linker between the calix[4]arene platform and the terminal BIPY˙+ radical cations, as well as the nature of the platform, with the calix[4]arene displaying a cone conformation, appear to be crucial factors that govern the arrangement of the π-dimers. It is noteworthy that preorganization of the designed systems apparently allows for efficient intramolecular dimerization of the two viologens upon reduction in a range of solvents (e.g., water and CH3CN45).
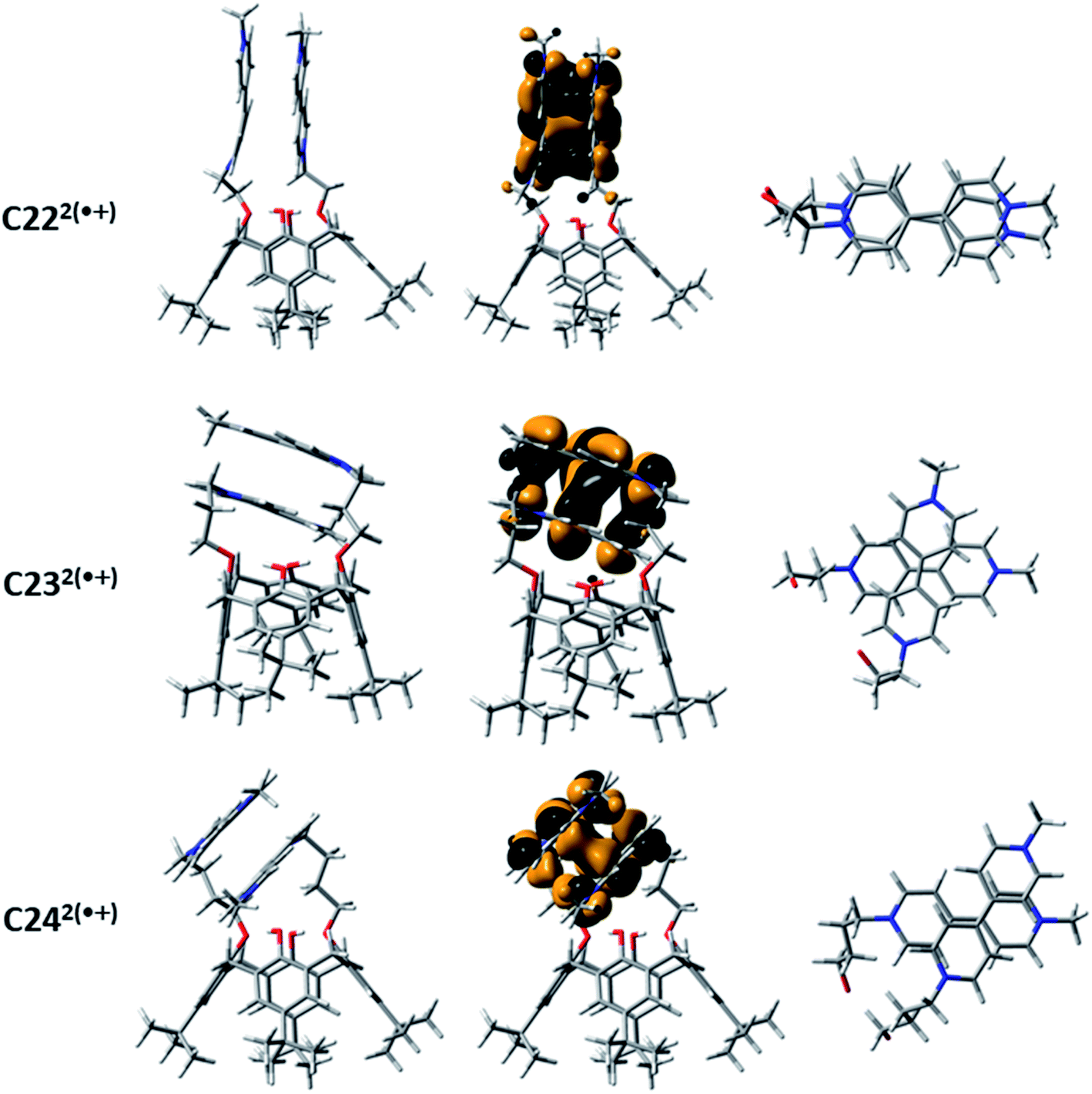 | ||
| Fig. 4 Geometries obtained with DFT (M062X/6-311G(d,p)) (left panel), the corresponding HOMOs (central panel) and detail of the arrangements of the redox-active BIPY˙+ units within the two electrons reduced calix[4]arene-bis-viologens C222(˙+), C232(˙+) and C242(˙+) that undergo intramolecular pimerization. | ||
Reduction of the [n]pseudorotaxanes with CB[7]
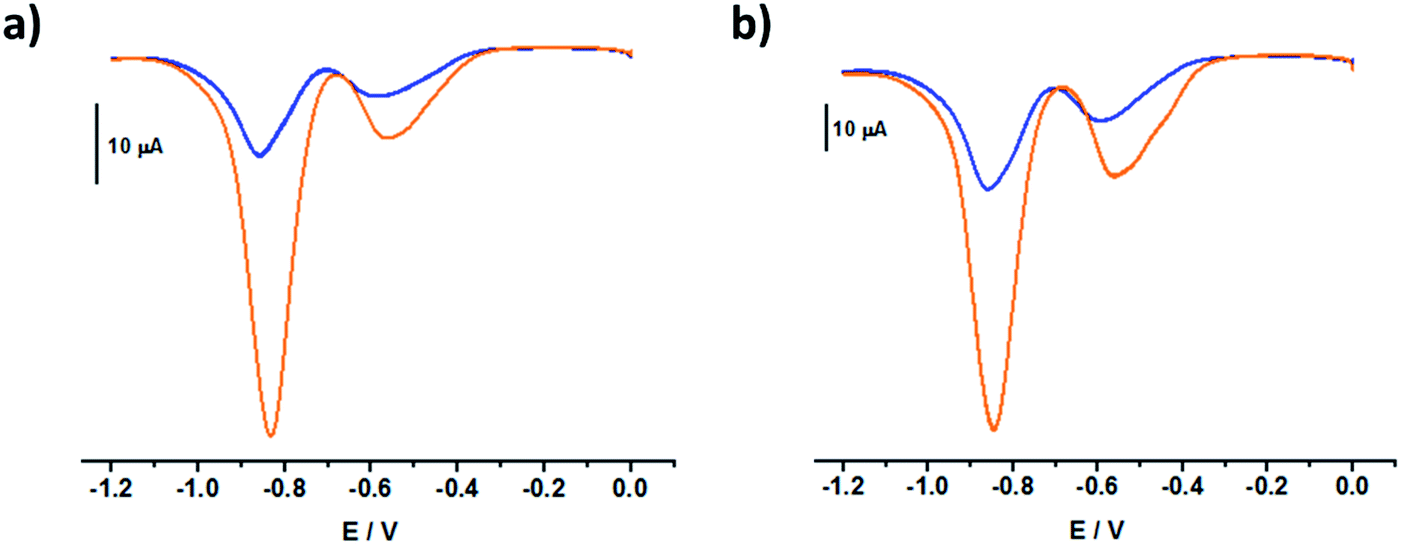 | ||
| Fig. 5 Square wave voltammograms of (a) MC32+, [MC32+] = 0.05 mM and b) MC42+, [MC42+] = 0.06 mM in the absence (orange) and the presence of CB[7] (blue, 3 equivalents of CB[7]). All voltammograms were recorded in argon-purged phosphate buffer solutions (pH 7) at 298 K (E versus Ag/AgCl). | ||
| Species | E1/21 (CV) | E1/21 (SW) | E1/22 (CV) | E1/22 (SW) |
|---|---|---|---|---|
| MC32+ | −0.56 V | −0.56 V | −0.83 V | −0.83 V |
| MC32+ + 3 eq. CB[7] | −0.60 V | −0.59 V | −0.85 V | −0.86 V |
| MC42+ | −0.56 V | −0.56 V | −0.83 V | −0.84 V |
| MC42+ + 3 eq. CB[7] | −0.58 V | −0.58 V | −0.83 V | −0.86 V |
| C234+ | −0.42 V | −0.43 V | −0.84 V | −0.85 V |
| C234+ + 3 eq. CB[7] | −0.44 V | −0.46 V | −0.84 V | −0.85 V |
| C244+ | −0.45 V | −0.47 V | −0.85 | −0.85 |
| C244+ + 4 eq. CB[7] | −0.50 | −0.51 | −0.85 | −0.85 |
These shifts corresponded to the signatures of the relative affinities of CB[7] for the different redox states of the model systems. Assuming a logKMC32+⊂CB[7] value of 4.51(6) for MC32+⊂CB[7] (Fig. S1 in the ESI†), a logKMC3˙+⊂CB[7] of ∼4–4.2 and a logKMC3(0)⊂CB[7] of ∼3.5–3.9 values were accordingly calculated.26,29 Similarly, using a logKMC42+⊂CB[7] value of 4.68(5) (Fig. S3 in the ESI†), a logKMC4˙+⊂CB[7] of ∼4.2 and a logKMC40⊂CB[7] of ∼3.8 were calculated. Using an electrochemical approach, these calculated values were in reasonably good agreement, within error, with those derived from direct absorption binding titrations (logKMC3˙+⊂CB[7] = 3.8(4) (Fig. S24 in the ESI†) and logKMC4˙+⊂CB[7] = 4.54(5) (Fig. S26 in the ESI†). In contrast to BMV2+, where translational motion of the CB[7] macrocycle occurred upon electrochemical reduction of the bipyridinium unit,30 the CB[7] macrocycle remained firmly bound to the BIPY2+ electroactive units of the MC4+ derivatives regardless of their oxidation state.
Assuming that only the first one-electron reduction process dominates from 0 V to −0.7 V (Fig. S16 in the ESI† and Fig. 5), the diffusion coefficients of MC32+ and MC42+ in the absence and in the presence of CB[7] (Table 4) can be evaluated by chronocoulometric experiments. The differences observed between the values of the diffusion coefficients of MC3˙+/MC32+ and MC4˙+/MC42+ could be an indication of the dimerization of the model systems upon the one -electron reduction reactions. Addition of CB[7] suppresses the pimerization process in favor of the inclusion complexes. As a consequence, the inclusion complexes with CB[7] have lower diffusion coefficients than the corresponding free viologens.
| No CB[7]/D | +CB[7]/D |
|---|---|
| MC3˙+/0.7 × 10−5 | MC3˙+ + 2 eq. CB[7]/1.9 × 10−5 |
| MC32+/5.4 × 10−5 | MC32 + 2 eq. CB[7]/14.2 × 10−5 |
| MC4˙+/3.1 × 10−5 | MC4˙+ + 2 eq. CB[7]/6.2 × 10−5 |
| MC42+/81.0 × 10−5 | MC42+ + 2 eq. CB[7]/22 × 10−5 |
| C23˙+/1.41 × 10−5 | C23˙+ + 3 eq. CB[7]/2.9 × 10−5 |
| C232+/7.58 × 10−5 | C232 + 3 eq. CB[7]/3.91 × 10−5 |
| C24˙+/0.81 × 10−5 | C24˙+ + 4 eq. CB[7]/0.16 × 10−5 |
| C242+/20.1 × 10−5 | C242+ + 4 eq.CB[7]/42.1 × 10−5 |
| C23˙+/1.41 × 10−5 | C23˙+ + 3 eq. CB[7]/2.9 × 10−5 |
| C232+/7.58 × 10−5 | C232 + 3 eq. CB[7]/3.91 × 10−5 |
| C24˙+/0.81 × 10−5 | C24˙+ + 4 eq. CB[7]/0.16 × 10−5 |
| C242+/20.1 × 10−5 | C242+ + 4 eq. CB[7]/42.1 × 10−5 |
Investigations of the UV-vis-NIR absorption spectra of MC32+ and MC42+ in water at pH 7.0 in the absence or in the presence of a reducing agent were carried out to evaluate the effect of CB[7] addition (Fig. S23 and S25 in the ESI†). MC3˙+⊂CB[7] and MC4˙+⊂CB[7] both clearly exhibited a significant hypochromic shift of the visible absorption band associated with the radical cation with increasing concentration of CB[7], strong evidence for the inclusion of the BIPY˙+ radical cation within the hydrophobic cavity of CB[7]. This binding event effectively hampers radical cation pimerization.
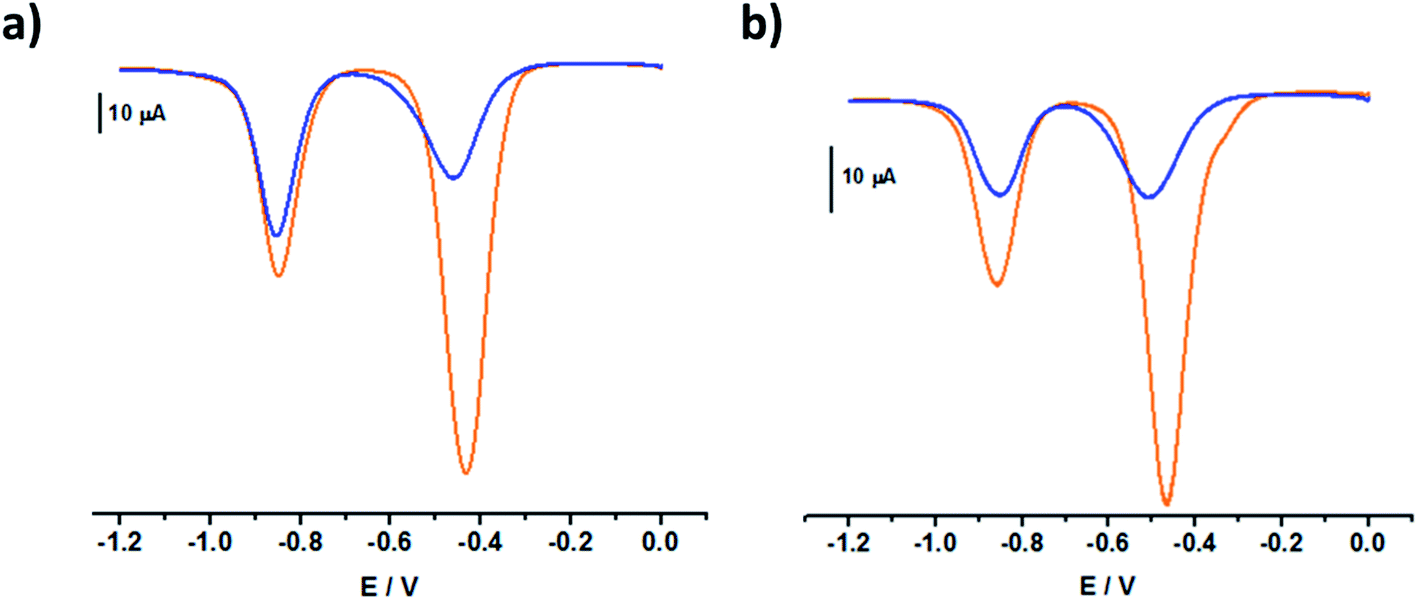 | ||
| Fig. 6 Square wave voltammograms of (a) C234+, [C234+] = 0.05 mM and (b) C244+, [C244+] = 0.05 mM in the absence (orange line) and the presence of CB[7] (blue line, respectively 3 and 4 equivalents of CB[7]). All voltammograms were recorded in argon-purged phosphate buffer solutions (pH 7) at 298 K (E versus Ag/AgCl). | ||
The square wave voltammograms of C234+ and C244+ (Fig. 6) displayed two reversible two-electron reduction processes: E11/2 (C234+ → C232(˙+)) = −0.43 V and E1/22 (C232(˙+) → C230) = −0.85 V; E1/21 (C244+ → C242(˙+)) = −0.47 V and E1/22 (C242(˙+) → C240) = −0.85 V. Interestingly, for the [3]pseudorotaxanes, C234+⊂(CB[7])2 and C244+⊂(CB[7])2, no shifts were observed for the second reduction wave (Table 3), which would have corresponded to the following electrochemical processes: C232(˙+)⊂(CB[7])2 → C230⊂(CB[7])2 and C242(˙+)⊂(CB[7])2 → C240⊂(CB[7])2. Rather, our observations indicated that after the first two-electron reduction process, the threads behave as if they are unbound C232(˙+) and C242(˙+). In other words, the first reduction of C234+⊂(CB[7])2 and C244+⊂(CB[7])2 induced dethreading of the CB[7] macrocycles, which is spontaneously followed by intramolecular dimerization of the two terminal BIPY˙+ groups, leading to the formation of intramolecular dimeric species, C232(˙+) and C242(˙+). These results were consistent with the absorption spectrophotometric analyses (Fig. S27 and S28 in the ESI†) and EPR behavior (Fig. S19 and S20 in the ESI†) of the C234+/CB[7] and C244+/CB[7] [3]pseudorotaxanes.
Chronocoulometry was also used to evaluate the diffusion coefficients of the fully oxidized and radical cationic states of the different species in aqueous solution. During the two electrons reduction process (0 V → −0.7 V) the fully oxidized species predominate and diffuse to the electrode surface where they are reduced. By measuring the reduction rates, the diffusion constants for C234+ and C244+ species can be calculated in the absence and in the presence of CB[7] (Table 4). Alternatively, by setting the voltage to −0.7 V and performing the oxidation process, the rates of oxidation and the diffusion coefficients of the corresponding radical cationic species can be measured as well. In the absence of CB[7], larger diffusion coefficients were observed for C234+ and C244+ than for their corresponding fully reduced forms. This could be explained by the ability of higher charged species to diffuse faster towards or away from an electrode. In the presence of CB[7], larger diffusion coefficients were measured for the [3]pseudorotaxanes C234+⊂(CB[7])2 and C234+⊂(CB[7])2 than the corresponding reduced species (i.e., upon reduction, a dethreading of the CB[7] occurs as a consequence of favored intramolecular pimerization with respect to the recognition process). Here also, one would expect that the [3]pseudorotaxanes C234+⊂(CB[7])2 and C234+⊂(CB[7])2 display larger hydrodynamic size by comparison with C232(˙+) or C242(˙+). Markedly different solvation of the latter species is suggested to explain these peculiar properties. The close diffusion coefficients of the radical cation species in the absence and the presence of CB[7] indicate that the dimerization of the viologen radical cations induce a dethreading of the CB[7] macrocycles.
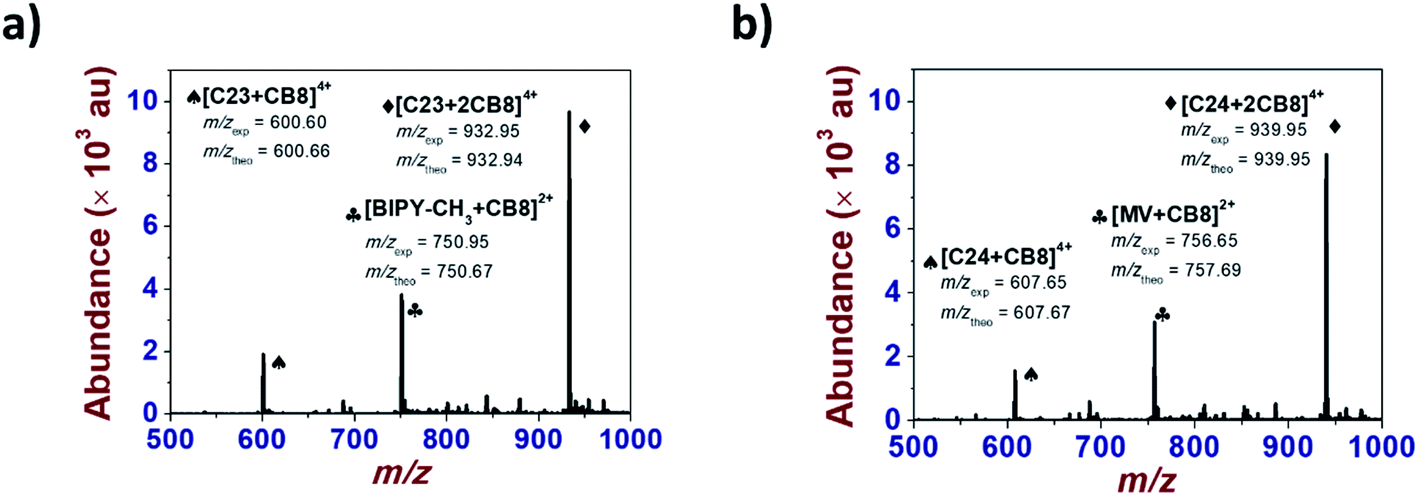 | ||
| Fig. 7 ESI mass spectra of inclusion complexes of a) C234+ and b) C244+ (bottom) with CB[8]. (a) [C234+]0 = 5 × 10−5 M; [CB[7]]0 = 10−4 M. Solvent: H2O; positive mode. Vc = 200 V. (b) [C244+]0 = 5 × 10−5 M; [CB[7]]0 = 10−4 M. Solvent: H2O; positive mode. Vc = 100 V. | ||
For the latter [3]pseudorotaxanes, chemical reduction of the BIPY2+ electroactive units led to dethreading of the CB[8] macrocyclic host despite its larger cavity size (Fig. S31 and S32 in the ESI†). This suggests significant steric constraints with the calix[4]arene platform and/or poor flexibility of the designed systems even though longer and apparently more flexible spacers were introduced. However, the MC32+ and MC42+ models clearly led (Fig. S29 and S30 in the ESI†) to the expected [3]pseudorotaxanes (MC3˙+)2⊂CB[8] and (MC4˙+)2⊂CB[8] thus emphasizing the detrimental role of the anchoring platform rather than the length of the spacers. The occurrence of [3]pseudorotaxanes (MC3˙+)2⊂CB[8] and (MC4˙+)2⊂CB[8] upon reduction is in agreement with the published reports26–28 that showed that the stability of methyl viologen dimer (MV˙+)2⊂CB[8]) was significantly increased in the presence of CB[8] following the formation of a 2:1 (MV˙+)2⊂CB[8]) supramolecular complex in water.
Conclusions
Two calix[4]arene-bis-viologen systems, namely C234+ and C244+ were synthesized. The host–guest properties were extensively studied using a large set of analytical methods and led to the characterization of [3]pseudorotaxanes in combination with either CB[7] or CB[8]. For each of these host–guest species, CB[7] or CB[8] was demonstrated to reside in the middle of the BIPY2+ cations as a result of steric interactions with the anchoring platform. Upon reduction of the terminal BIPY2+ cations, these [3]pseudorotaxanes spontaneously dissociate as the result of a strong intramolecular dimerization of the two face-to-face viologen radical cations. The arrangement of the BIPY˙+ radical cations within the dimeric species seemingly relies on the length of the spacer that links the electroactive units to the anchoring moiety. Dethreading and dimerization do not occur in experiments involving CB[7] (or CB[8]) and either of the two monomeric viologen guests MC32+ and MC42+, which were used a models. On the other hand, the model compounds provided unexpected and valuable properties. Thanks to the presence of a alkoxy-phenyl substitution, [3]pseudorotaxanes such as MC32+⊂(CB[7])2 and MC42+⊂(CB[7])2 were characterized. For the MC32+⊂CB[7] and MC42+⊂CB[7] [2]pseudorotaxanes, no redox-triggered translocation of CB[7] between the two binding stations were observed indicating that the BIPY2+ unit remained the favored recognition site whatever its redox state. In the presence of CB[8], similar properties were emphasized with the oxidized form of MC32+ and MC42+ threads. However, [3]pseudorotaxanes (MC3(˙+))2⊂CB[8] and (MC4(˙+))2⊂CB[8]) were predominantly formed as a consequence of the larger cavity size of the CB[8] host. This preliminary study provided interesting information for the further development of functional electroactive systems and will be extended to tetrakis analogues displaying either a cone or 1,3-alternate arrangement of the anchoring calix[4]arene unit.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. E. thank the CNRS (LIMA, UMR 7042) and the University of Strasbourg for funding this research work. C. P.-I. acknowledges Centro de Supercomputación de Galicia (CESGA) for providing access to supercomputing facilities. A. T. thank NYUAD for its generous support of the research program at NYUAD.Notes and references
- (a) T. Fukino, H. Yamagishi and T. Aida, Adv. Mater., 2016, 29, 1603888 CrossRef PubMed; (b) K. Madasamy, D. Velayutham, V. Suryanarayanan, M. Kathiresan and K. C. Ho, J. Mater. Chem. C, 2019, 7, 4622–4637 RSC; (c) A. F. Greene, M. K. Danielson, A. O. Delawder, K. P. Liles, X. Li, A. Natraj, A. Wellen and J. C. Barnes, Chem. Mater., 2017, 29, 9498–9508 CrossRef CAS; (d) K. C. Ho, H. C. Lu and H. F. Yu, RSC Smart Mater., 2019, 33, 372–405 CAS; (e) J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- K. Wadhwa, S. Nuryyeva, A. C. Fahrenbach, M. Elhabiri, C. Platas-Iglesias and A. Trabolsi, J. Mater. Chem. C, 2013, 1, 2302–2307 RSC.
- (a) R. J. Mortimer, Electrochim. Acta, 1999, 44, 2971–2981 CrossRef CAS; (b) L. C. Cao, M. Mou and Y. Wang, J. Mater. Chem., 2009, 19, 3412–3418 RSC; (c) S. Asaftei, M. Ciobanu, A. M. Lepadatu, E. Song and U. Beginn, J. Mater. Chem., 2012, 22, 14426–14437 RSC.
- M. Kuroboshi, T. Shiba and H. Tanaka, Tetrahedron Lett., 2013, 54, 3666–3668 CrossRef CAS.
- K. Ciepluch, N. Katir, A. El Kadib, A. Felczak, K. Zawadzka, M. Weber, B. Klajnert, K. Lisowska, A. M. Caminade, M. Bousmina, M. Bryszewska and J. P. Majoral, Mol. Pharmaceutics, 2012, 9, 448–457 CrossRef CAS PubMed.
- S. Berger, A. Ghicov, Y. C. Nah and P. Schmuki, Langmuir, 2009, 25, 4841–4844 CrossRef CAS PubMed.
- D. Cummins, G. Boschloo, M. Ryan, D. Corr, S. N. Rao and D. Fitzmaurice, J. Phys. Chem. B, 2000, 104, 11449–11459 CrossRef CAS.
- G. De Filpo, F. P. Nicoletta and G. Chidichimo, Chem. Mater., 2006, 18, 4662–4666 CrossRef CAS.
- R. Cinnsealach, G. Boschloo, S. Nagaraja Rao and D. Fitzmaurice, Sol. Energy Mater. Sol. Cells, 1998, 55, 215–223 CrossRef CAS.
- P. Bonhôte, E. Gogniat, M. Grätzel and P. V. Ashrit, Thin Solid Films, 1999, 350, 269–275 CrossRef.
- P. Sidorov, I. Desta, M. Chessé, D. Horvath, G. Marcou, A. Varnek, E. Davioud-Charvet and M. Elhabiri, ChemMedChem, 2016, 11, 1339–1351 CrossRef CAS PubMed.
- P. M. S. Monk, The Viologens: Physicochemical Properties, Synthesis and Applications of the Salts of 4, 4’-Bipyridine, Chichester, 1998 Search PubMed.
- (a) J. Bruinink, C. G. A. Kregting and J. J. Ponjeé, J. Electrochem. Soc., 1977, 124, 1854–1858 CrossRef CAS; (b) A. G. Evans, J. C. Evans and M. W. Baker, J. Am. Chem. Soc., 1977, 99, 5882–5884 CrossRef CAS; (c) D. Meisel, W. A. Mulac and M. S. Matheson, J. Phys. Chem., 1981, 85, 179–187 CrossRef CAS; (d) E. Adar, Y. Degani, Z. Goren and I. Willner, J. Am. Chem. Soc., 1986, 108, 4696–4700 CrossRef CAS; (e) A. Yasuda, H. Mori and J. Seto, J. Appl. Electrochem., 1987, 17, 567–573 CrossRef CAS.
- (a) A. Trabolsi, M. Hmadeh, N. M. Khashab, D. C. Friedman, N. Humbert, M. Elhabiri, H. A. Khatib, M. E. Belowich, A. Coskun, A. M. Albrecht-Gary and J. F. Stoddart, New J. Chem., 2009, 33, 254–263 RSC; (b) W. Geuder, S. Hünig and A. Suchy, Tetrahedron, 1986, 42, 1665–1677 CrossRef CAS.
- E. M. Kosower and J. L. Cotter, J. Am. Chem. Soc., 1964, 86, 5524–5527 CrossRef CAS.
- E. M. Kosower and J. Hajdu, J. Am. Chem. Soc., 1971, 93, 2534–2535 CrossRef CAS.
- A. C. Fahrenbach, J. C. Barnes, D. A. Lanfranchi, H. Li, A. Coskun, J. J. Gassensmith, Z. Liu, D. Benítez, A. Trabolsi, W. A. Goddard, M. Elhabiri and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 3061–3072 CrossRef CAS PubMed.
- Y. Wang, J. Sun, Z. Liu, M. S. Nassar, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2017, 8, 2562–2568 RSC.
- (a) A. F. Greene, M. K. Danielson, A. O. Delawder, K. P. Liles, X. Li, A. Natraj, A. Wellen and J. C. Barnes, Chem. Mater., 2017, 29, 9498–9508 CrossRef CAS; (b) Z. Qian, X. Huang and Q. Wang, Dyes Pigm., 2017, 145, 365–370 CrossRef CAS.
- G. C. Granqvist, Handbook of Inorganic Electrochromic Materials, Elsevier, Amsterdam, 1995 Search PubMed.
- P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge University Press, Cambridge, UK, 2007 Search PubMed.
- J. R. J. J. Platt, J. Chem. Phys., 1961, 34, 862–863 CrossRef CAS.
- J. W. Park, N. H. Choi and J. H. Kim, J. Phys. Chem., 1996, 100, 769–774 CrossRef CAS.
- P. A. Quintela and A. E. Kaifer, Langmuir, 1987, 3, 769–773 CrossRef CAS.
- A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Cotí, D. Benítez, E. Tkatchouk, J. C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, Nat. Chem., 2010, 2, 42–49 CrossRef CAS PubMed.
- H. J. Kim, W. S. Jeon, Y. H. Ko and K. Kim, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5007–5011 CrossRef CAS PubMed.
- W. Ong, M. Gomez-Kaifer and A. E. Kaifer, Org. Lett., 2002, 4, 1791–1794 CrossRef CAS PubMed.
- W. S. Jeon, H. J. Kim, C. Lee and K. Kim, Chem. Commun., 2002, 1828–1829 RSC.
- J. W. Lee, S. Samal, N. Selvapalam, H. J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- E. Pazos, P. Novo, C. Peinador, A. E. Kaifer and M. D. García, Angew. Chem., Int. Ed., 2019, 58, 403–416 CrossRef CAS PubMed.
- (a) K. Nchimi Nono, P. Dalvand, K. Wadhwa, S. Nuryyeva, S. Alneyadi, A. Fahrenbach, J. C. Olsen, Z. Asfari, C. Platas-Iglesias, M. Elhabiri and A. Trabolsi, Chem.–Eur. J., 2014, 20, 7334–7344 CrossRef CAS PubMed; (b) K. Wadhwa, S. Nuryyeva, A. C. Fahrenbach, M. Elhabiri, C. Platas-Iglesias and A. Trabolsi, J. Mater. Chem. C, 2013, 1, 2302–2307 RSC.
- R. Kannappan, C. Bucher, E. Saint-Aman, J. C. Moutet, A. Milet, M. Oltean, E. Métay, S. Pellet-Rostaing, M. Lemaire and C. Chaix, New J. Chem., 2010, 34, 1373–1386 RSC.
- W. Geuder, S. Huenig and A. Suchy, Tetrahedron, 1986, 42, 1665–1667 CrossRef CAS.
- S. J. Atherton, K. Tsukahara and R. G. Wilkins, J. Am. Chem. Soc., 1986, 108, 3380–3385 CrossRef CAS.
- W. S. Abdul-Hassan, D. Roux, C. Bucher, S. Cobo, F. Molton, E. Saint-Aman and G. Royal, Chem.–Eur. J., 2018, 24, 12961–12969 CrossRef CAS PubMed.
- M. Berville, S. Choua, C. Gourlaouen, C. Boudon, L. Ruhlmann, C. Bailly, S. Cobo, E. Saint-Aman, J. A. Wytko and J. Weiss, ChemPhysChem, 2017, 18, 796–803 CrossRef CAS PubMed.
- M. Berville, L. Karmazin, J. A. Wytko and J. Weiss, Chem. Commun., 2015, 51, 15772–15775 RSC.
- A. Iordache, M. Retegan, F. Thomas, G. Royal, E. Saint-Aman and C. Bucher, Chem.–Eur. J., 2012, 18, 7648–7653 CrossRef CAS PubMed.
- S. Chowdhury, Y. Nassar, L. Guy, D. Frath, F. Chevallier, E. Dumont, A. P. Ramos, G. J. F. Demets and C. Bucher, Electrochim. Acta, 2019, 316, 79–92 CrossRef CAS.
- A. Iordache, M. Oltean, A. Milet, F. Thomas, E. Saint-Aman and C. Bucher, J. Am. Chem. Soc., 2012, 134, 2653–2671 CrossRef CAS PubMed.
- M. Mohammad, Electrochim. Acta, 1988, 33, 417–419 CrossRef CAS.
- T. G. Zhan, T. Y. Zhou, F. Lin, L. Zhang, C. Zhou, Q. Y. Qi, Z. T. Li and X. Zhao, Org. Chem. Front., 2016, 3, 1635–1645 RSC.
- K. Madasamy and M. Kathiresan, ChemistrySelect, 2016, 1, 354–359 CrossRef CAS.
- Y. Wang, M. Frasconi, W. G. Liu, Z. Liu, A. A. Sarjeant, M. S. Nassar, Y. Y. Botros, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 876–885 CrossRef CAS PubMed.
- J. Iehl, M. Frasconi, H. P. Jacquot de Rouville, N. Renaud, S. M. Dyar, N. L. Strutt, R. Carmieli, M. R. Wasielewski, M. A. Ratner, J. F. Nierengarten and J. F. Stoddart, Chem. Sci., 2013, 4, 1462–1469 RSC.
- (a) A. Iordache, R. Kanappan, E. Métay, M. C. Duclos, S. Pellet-Rostaing, M. Lemaire, A. Milet, E. Saint-Aman and C. Bucher, Org. Biomol. Chem., 2013, 11, 4383–4389 RSC; (b) C. Kahlfuss, E. Métay, M. C. Duclos, M. Lemaire, M. Oltean, A. Milet, E. Saint-Aman and C. Bucher, C. R. Chim., 2014, 17, 505–511 CrossRef CAS.
- C. Kahlfuss, E. Metay, M. C. Duclos, M. Lemaire, A. Milet, E. Saint-Aman and C. Bucher, Chem.–Eur. J., 2015, 21, 2090–2106 CrossRef CAS PubMed.
- M. Elancheziyan, K. Theyagarajan, D. Saravanakumar, K. Thenmozhi and S. Senthilkumar, Mater. Today Chem., 2020, 16, 100274 CrossRef CAS.
- C. M. Ronconi, J. F. Stoddart, V. Balzani, M. Baroncini, P. Ceroni, C. Giansante and M. Venturi, Chem.–Eur. J., 2008, 14, 8365–8373 CrossRef CAS PubMed.
- G. Das, T. Prakasam, S. Nuryyeva, D. S. Han, A. Abdel-Wahab, J. C. Olsen, K. Polychronopoulou, C. Platas-Iglesias, F. Ravaux, M. Jouiad and A. Trabolsi, J. Mater. Chem. A, 2016, 4, 15361–15369 RSC.
- M. Marchini, M. Baroncini, G. Bergamini, P. Ceroni, M. D'Angelantonio, P. Franchi, M. Lucarini, F. Negri, T. Szreder and M. Venturi, Chem.–Eur. J., 2017, 23, 6380–6390 CrossRef CAS PubMed.
- I. Thondorf, A. Shivanyuk and V. Bohmer, in Calixarene, 2001, ed. Z. Asfari, V. Bohmer, J. Harrowfield and J. Vicens, Kluwer Academic publisher, Dordrecht, 2001, vol. 2 Search PubMed.
- Functional Synthetic Receptors, ed. T. Schrader and A. D. Hamilton, Wiley-VCH, Weinheim, 2005 Search PubMed.
- K. Ariga and T. Kunitake Supramolecular Chemistry-Fundamentals and Applications, Springer, Berlin, 2005 Search PubMed.
- Highlights in Bioorganic Chemistry: Methods and Applications, ed. C. Schmuck and H. Wennemers, Wiley-VCH, Weinheim, 2004 Search PubMed.
- S. Kubik, C. Reyheller and S. Stuwe, J. Inclusion Phenom. Macrocyclic Chem., 2005, 52, 137–187 CrossRef CAS.
- H. C. Ko, S. A. Park, W. K. Paik and H. Lee, Synth. Met., 2002, 132, 15–20 CrossRef CAS.
- (a) S. Luan, Q. Ge, Y. Chen, M. Dai, J. Yang, K. Li, D. Liu and L. Zhao, Bioorg. Med. Chem. Lett., 2017, 27, 1943–1948 CrossRef CAS PubMed; (b) W. Wang, C. Sheng, X. Che, H. Ji, Y. Cao, Z. Miao, J. Yao and W. Zhang, Bioorg. Med. Chem. Lett., 2009, 19, 5965–5969 CrossRef CAS PubMed.
- W.-W. Gu, W.-J. Chen and C.-G. Yan, Supramol. Chem., 2015, 27, 407–413 CrossRef CAS.
- R. Pomecko, PhD thesis University of Strasbourg, 2007.
- Z. T. Li, G. Z. Ji, C. X. Zhao, S. D. Yuan, H. Ding, C. Huang, A. L. Du and M. Wei, J. Org. Chem., 1999, 64, 3572–3584 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed; (b) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- A. S. Jalilov, S. Patwardhan, A. Singh, T. Simeon, A. A. Sarjeant, G. C. Schatz and F. D. Lewis, J. Phys. Chem. B, 2014, 118, 125–133 CrossRef CAS PubMed.
- A. Alberti, in Organic Photochromic and Thermochromic Compounds, ed. J. C. Crano and R. J. Guglielmetti, Kluwer Academic publisher, 2002, 4, pp. 211–240 Search PubMed.
- W. L. Mock and N.-Y. Shih, J. Org. Chem., 1986, 51, 4440–4446 CrossRef CAS.
- B. Perlmutter-Hayman, Acc. Chem. Res., 1986, 19, 90–96 CrossRef CAS.
- C. Lee, M. S. Moon and J. W. Park, J. Electroanal. Chem., 1996, 407, 161–167 CrossRef.
- J. F. Stargardt and F. M. Hawkridge, Anal. Chim. Acta, 1983, 146, 1–8 CrossRef CAS.
- P. M. S. Monk, N. M. Hodgkinson and S. A. Ramzan, Dyes Pigm., 1999, 43, 207–217 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Physicochemical characterisation, spectrophotometric titrations, Job's plots, NMR, MS and EPR spectra, and DFT structures. See DOI: 10.1039/d1ra05488k |
| This journal is © The Royal Society of Chemistry 2021 |