
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, cytotoxicity evaluation and molecular docking studies on 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone derivatives†
Nopawit Khamto ab,
Lada Chaichuangab,
Puracheth Rithchumponab,
Worrapong Phupongc,
Phuangthip Bhoopongd,
Suriya Tateinge,
Wilart Pompimonf,
Natthawat Semakulah,
Ni-orn Chomsrig and
Puttinan Meepowpan*ah
ab,
Lada Chaichuangab,
Puracheth Rithchumponab,
Worrapong Phupongc,
Phuangthip Bhoopongd,
Suriya Tateinge,
Wilart Pompimonf,
Natthawat Semakulah,
Ni-orn Chomsrig and
Puttinan Meepowpan*ah
aDepartment of Chemistry, Faculty of Science, Chiang Mai University, 239 Huay Kaew Road, Chiang Mai 50200, Thailand. E-mail: pmeepowpan@gmail.com
bGraduate School, Chiang Mai University, 239 Huay Kaew Road, Chiang Mai 50200, Thailand
cSchool of Science, Walailak University, 222 Thaiburi, Nakhon Si Thammarat 80161, Thailand
dSchool of Allied Health Science, Walailak University, 222 Thaiburi, Nakhon Si Thammarat 80161, Thailand
eDepartment of Plant and Soil Sciences, Faculty of Agriculture, Chiang Mai University, 239 Huay Kaew Road, Chiang Mai 50200, Thailand
fLaboratory of Natural Products, Centre for Innovation in Chemistry, Faculty of Science, Lampang Rajabhat University, Lampang 52100, Thailand
gAgricultural Technology Research Institute (ATRI), Rajamangala University of Technology Lanna, 202 Pichai District, Lampang 52100, Thailand
hCenter of Excellence in Materials Science and Technology, Chiang Mai University, 239 Huay Kaew Road, Chiang Mai 50200, Thailand
First published on 22nd September 2021
Abstract
2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone (DMC, 1) was isolated from seeds of Syzygium nervosum A.Cunn. ex DC. exhibiting intriguing biological activities. Herein, thirty three DMC derivatives including 4′-O-monosubstituted-DMC (2), 7-O-acylated-4-hydroxycoumarin derivatives (3), stilbene–coumarin derivatives (4), 2′,4′-disubstituted-DMC (5), and flavanone derivatives (6), were synthesised through acylation, alkylations, and sulfonylation. These semi-synthetic DMC derivatives were evaluated for in vitro cytotoxicity against six carcinoma cell lines. It was found that most derivatives exhibited higher cytotoxicity than DMC. In particular, 4′-O-caproylated-DMC (2b) and 4′-O-methylated-DMC (2g) displayed the strongest cytotoxicity against SH-SY5Y with IC50 values of 5.20 and 7.52 μM, respectively. Additionally, 4′-O-benzylated-DMC (2h) demonstrated the strongest cytotoxicity against A-549 and FaDu with IC50 values of 9.99 and 13.98 μM, respectively. Our structure–activity relationship (SAR) highlights the importance of 2′-OH and the derivatisation pattern of 4′-OH. Furthermore, molecular docking simulation studies shed further light on how these bioactive compounds interact with cyclin-dependent kinase 2 (CDK2).
Introduction
Natural products are known nowadays as important sources for the discovery and development of new drugs.1 Over 33 years from 1981 to 2014, more than 26% of FDA-approved drugs (320 of 1211) were natural products (NPs) or NP-derived pharmacophores.2 However, there has been quite a challenge to apply NPs to medication due to undesirable properties such as low oral bioavailability,3 metabolic instability,4 and less solubility in aqueous media5 that meant a low permeation rate from gut to blood system.6Cancer is an uncontrolled proliferation of cells and a widespread cause of death among the worldwide population.7 According to reports from the World Health Organization, it is expected that the number of cancer patients will have dramatically increased by more than 50% in 2020. Numerous methods, such as surgery, chemotherapy, and radiation therapy, targeted therapy, and checkpoint inhibitors,8 are potentially used for the treatment of cancer. Nevertheless, a large number of undesirable side effects due to the low selectivity of a chemotherapeutic agent for tumour cells is a major problem, which may be the cause of death in patients.9 Currently, an abundance of natural substances and their derivatives have been used as anticancer agents, such as vincristine, vinblastine, docetaxel, etoposide, and seliciclib.10
Syzygium nervosum A.Cunn. ex DC. or Cleistocalyx operculatus (Roxb.) Merr & L. M. Perry, aka Ma-kiang in Thai, is distributed over tropical countries11 such as Thailand, Vietnam, China, Myanmar, and Malaysia. Its fresh fruits are valuable for the Thai food and drink manufacturing industry for jam, juice, tea, yogurt, and wine. Its leaves and buds were traditionally used to treat colds, fever, inflammation,12 gastrointestinal disorders, and for antisepsis.11 Phytochemical investigations of S. nervosum disclosed a number of natural products, including terpenes,13 flavanone,14 flavone,15 flavonol,16 chalcone,17 anthocyanin,18 and phloroglucinol derivatives.19 These derivatives have been reported to possess a wide range of anticancer and cytotoxic activities.19–21
More specifically, chalcone (1,3-diphenyl-2-propen-1-ones) is an open-chain flavonoid containing an α,β-unsaturated carbonyl moiety. This class of compound is a secondary metabolite typically found in natural products and serves as a common precursor for flavone, flavanone, flavonol, and aurone syntheses.22 Recently, the chalcone scaffold has attracted considerable interest from researchers in the field of drug discovery and development.23 Chalcone derivatives exhibited a variety of pharmaceutical activities such as anti-inflammatory,24 antiviral, antimalarial, antibacterial, anticancer, antifungal, antioxidant, anti-inflammation, anti-HIV-1 reverse transcriptase, tyrosinase inhibitor, anti-plasmodial, α-glucosidase inhibitory, and protein kinase C inhibitor effects.25 In particular, metochalcone and sofalcone are clinically approved drugs for the treatment of choleretic and gastrointestinal diseases, respectively.23 The actions of chalcone derivatives against malignant cells are exerted through multiple mechanisms such as cell cycle arrest induction,26 tubulin polymerisation inhibition,27 epidermal growth factor receptor tyrosine kinase (EGFR-TK) inhibition,28 DNA and mitochondrial damage, cell apoptosis induction,29 blockade of the nuclear factor-kappa B (NF-κb) signalling pathway,30 and angiogenesis inhibition.31 These pathways are associated with the cell proliferation process, which may contribute to the further development of new anticancer drugs.32
2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone (DMC, 1) is a major bioactive compound isolated from S. nervosum seeds, which exhibits various bioactivities, such as anticancer, antimicrobial,33 AMP-dependent protein kinase (AMPK) activator,34 anti-influenza,35,36 and antiviral effects.37 More specifically, DMC shows moderate to excellent anticancer activities in various cancer cell lines, such as human leukaemia (K562),38 pancreatic cancer (PANC-1),39 cervical cancer (HeLa), hepatocellular carcinoma (SMMC-7721),40 oral nasopharyngeal carcinoma (KB), breast cancer (MCF-7), and lung cancer (A-549).37 Furthermore, drug combination studies involving DMC and the 5-fluorouracil (5-FU) anticancer drug revealed a dramatic increase in the anticancer activity of 5-FU against 5-FU-selected drug-resistant human hepatocellular carcinoma (BEL-7402/5-FU) by inducing apoptosis and cell cycle arrest through the PI3K/AKT pathway.41 Moreover, DMC increased the sensitivity of the tumour cells and showed reversal effects on drug-resistant KB-A1 cells both in vitro and in vivo.42
Herein, the structure of DMC was modified through methylation, allylation, benzoylation, benzylation, acylation, and sulfonylation under basic conditions yielding several semi-synthetic DMC derivatives, including 4′-O-monosubstituted-DMC (2a–2n), 7-O-acylated-4-hydroxycoumarin (3a–3e), stilbene–coumarin derivatives (4a–4e), 2′,4′-O-disubstituted-DMC (5a–5n), and flavanone derivatives (6g and 6m). These semi-synthetic DMC derivatives were evaluated for cytotoxicity against six cancer cell lines, KKU-M213, FaDu, HT-29, MDA-MB-231, A-549, and SH-SY5Y, as well as CL as a normal cell line. The anticancer activity of bioactive DMC derivatives was further studied by in silico molecular docking with cyclin-dependent kinase 2 (CDK2) to understand the plausible binding modes and the corresponding key interactions with amino acid residues in protein pockets. Furthermore, the antimicrobial activities of DMC derivatives were also evaluated against three bacterial strains: Escherichia coli, Staphylococcus aureus and Pseudomonas aeruginosa.
Results and discussion
Isolation and characterisation of DMC
Air-dried and ground seeds of S. nervosum were extracted with EtOAc. The extract was subjected to flash column chromatography (FCC) on silica gel, which was eluted by gradient eluents of n-hexane/EtOAc. The DMC-containing fractions were further chromatographed on silica gel and eluted by isocratic elution of n-hexane/acetone. The DMC-containing fractions were additionally crystallised in CH2Cl2/n-hexane to obtain orange needle-shaped crystals. DMC (1), orange needle crystals, had a melting point value of 124.5–126.4 °C. The HRMS-ESI experiment indicated that the (M + Na)+ ion at m/z 321.1100, belonged to a molecular formula of C18H18O4Na (calcd: 321.1103). The FTIR spectrum indicated characteristic absorption bands of the hydroxy group and conjugated carbonyl ketone at 3545 and 1632 cm−1, respectively. The 1H-NMR spectrum exhibited proton signals of a monosubstituted aromatic ring resonating at δ 7.36–7.45 and 7.60–7.68 ppm, trans-double bond protons at δ 7.84 and 7.99 ppm with a coupling constant value (J) of 15.7 Hz, a methoxy group at δ 3.67 ppm, and benzylic methyl groups at δ 2.13 and 2.15 ppm. Additionally, the spectrum indicated the presence of two hydroxy groups resonating at δ 5.39 and 13.61 ppm which were assigned to free hydroxy and chelated hydroxy groups, respectively. These spectroscopic data were similar to the published values.37Structural modifications of DMC
The DMC (1) has two hydroxy groups at C-2′ and C-4′ on aromatic ring A. Typically, the 2′-OH is less reactive than the 4′-OH due to intramolecular hydrogen bonding with an adjacent carbonyl group. The modification was conducted under weak basic conditions through a nucleophilic substitution reaction with various acylating, alkylating, and sulfonylating agents to facilely provide several semi-synthetic DMC derivatives (Scheme 1).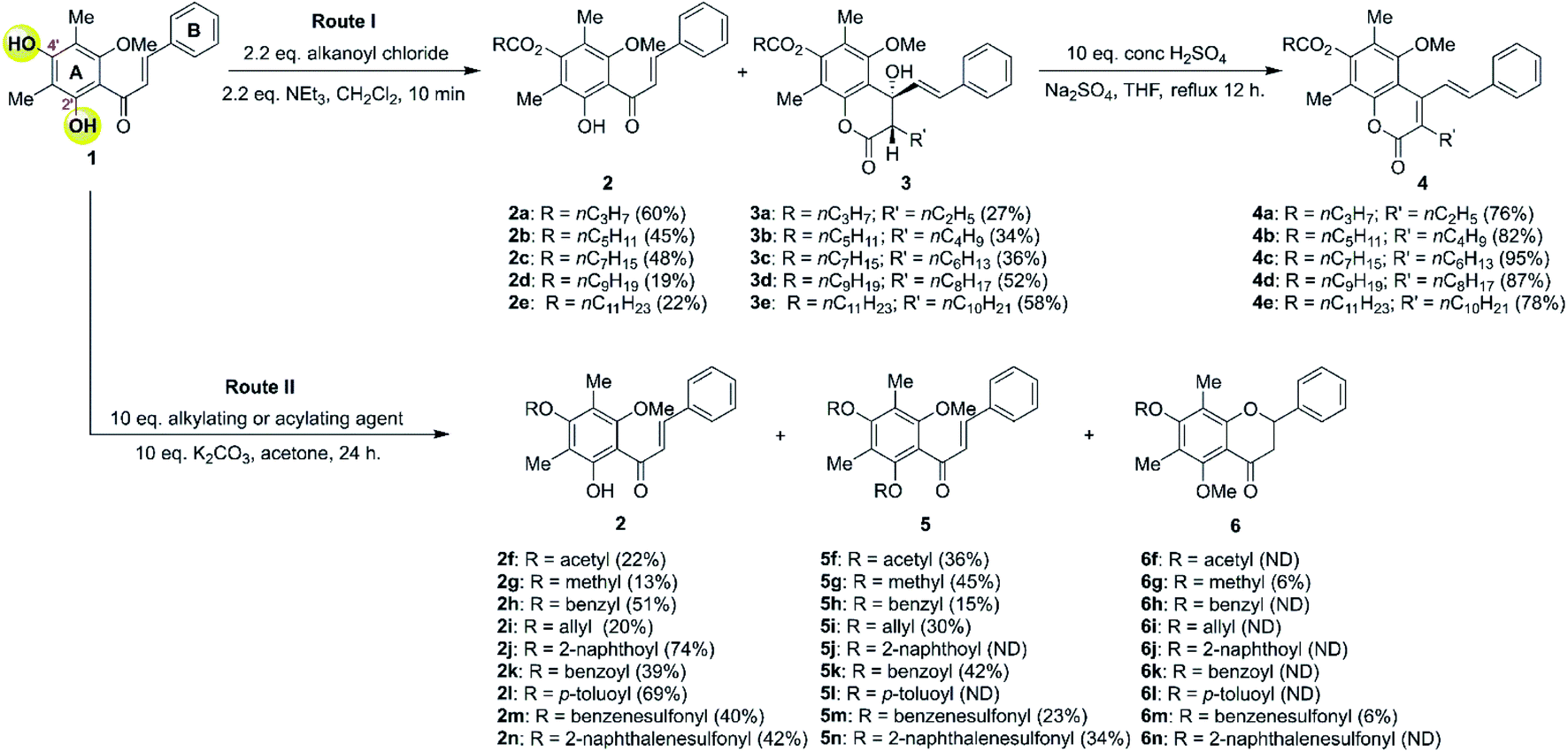 | ||
| Scheme 1 Derivatisations of DMC. ND = not detected. | ||
We began the investigation by performing the O-acylation of DMC with various alkanoyl chlorides, as shown in Scheme 1, route I. The DMC readily reacts with alkanoyl chlorides under basic conditions which provide a mixture of 4′-O-acylated-DMC derivatives (2a–2e) and 7-O-acylated-4-hydroxycoumarins (3a–3e). The monoacylation product 2 was thought to proceed via the acylation at more reactive 4′-OH thus providing 4′-O-acylated-DMC derivatives (2a–2e) in moderate yields. However, compound 2 can also undergo acylation at the less reactive 2′-OH and subsequent intramolecular Claisen-cyclisation to afford 7-O-acylated-4-hydroxycoumarins (3a–3e) in moderate yields. It should be noted that the product derived from monoacylation at 2′-OH was not observed in all cases.
The example FTIR spectrum of 4′-O-caprinoylated-DMC (2d) evidently indicated a new strong absorption band at 1767 cm−1 which was assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carbonyl ester. The 1H-NMR spectrum showed an absence of a 4′-OH signal of DMC and it became apparent that the α-methylene proton signal of the attached alkanoyl moiety resonating at δ 2.63 ppm. Furthermore, the 13C-NMR spectrum displayed the signal of a CO ester carbon resonating at δ 171.0 ppm, which confirmed the completed acylation reaction.
O stretching of carbonyl ester. The 1H-NMR spectrum showed an absence of a 4′-OH signal of DMC and it became apparent that the α-methylene proton signal of the attached alkanoyl moiety resonating at δ 2.63 ppm. Furthermore, the 13C-NMR spectrum displayed the signal of a CO ester carbon resonating at δ 171.0 ppm, which confirmed the completed acylation reaction.
In contrast, 7-O-caprinoylated-4-hydroxycoumarin (3d) showed the characteristic peaks of hydroxy and carbonyl ester groups at 3470 and 1779 cm−1, respectively. The 1H-NMR spectrum showed the signals of a hydroxy group at δ 5.68 ppm and a methine proton at δ 2.84 ppm. The 13C-NMR spectrum showed the peaks at δ 168.7 and 171.1 ppm which indicated the presence of lactone and carbonyl ester groups, respectively. To understand the relative configuration of the Claisen-cyclisation product, a NOESY experiment (see Fig. 24S†) was performed which suggests key correlations between 4-OH and 5-OCH3, 6-CH3, and 2′-CH. Furthermore, correlations between 3-CH and 1′-CH and 2′-CH were also observed as evidence of the anti-configuration between 3-CH and 4-OH, which are in agreement with the simulated structure performed by DFT calculation at B3LYP/6-311++G(d,p) level of theory in the gas phase, as depicted in Fig. 1.
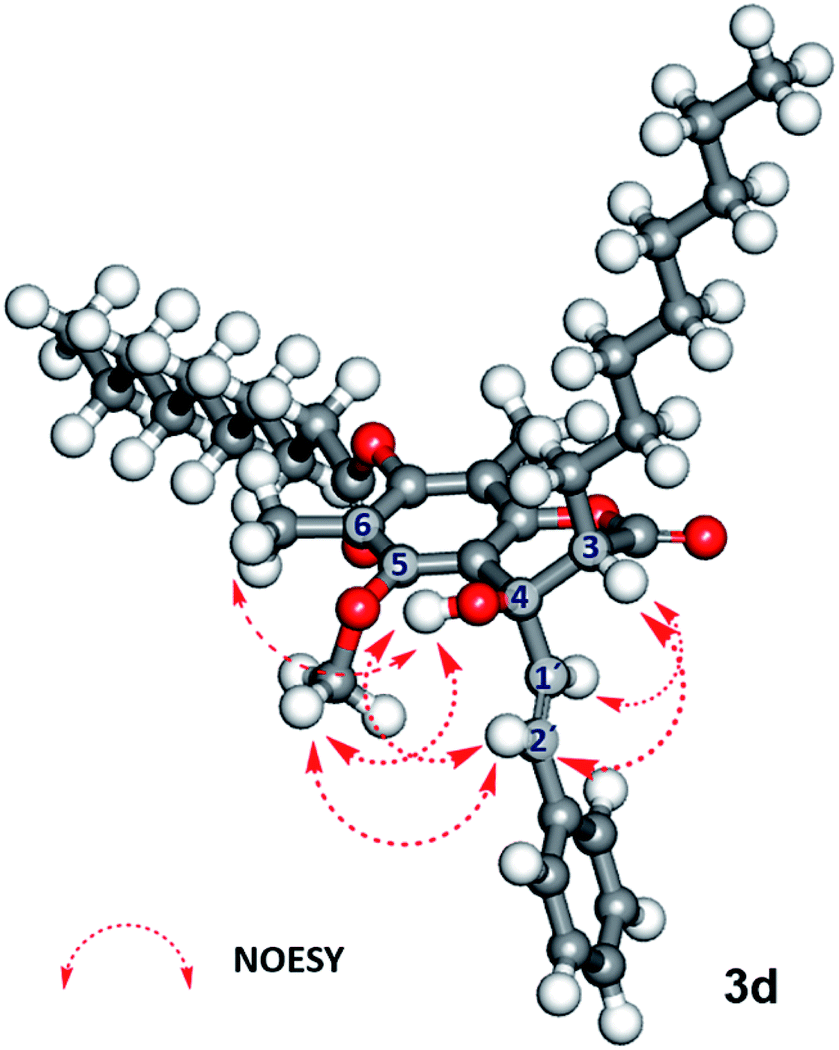 | ||
| Fig. 1 Geometrically optimised structure of 7-O-caprinoylated-4-hydroxycoumarin (3d) performed by B3LYP/6-311++G (d,p) level of theory and its key NOESY experiment. | ||
Controlled experiments (as shown in Table 1) were performed to further understand the plausible mechanisms for product formation. The use of caprinoyl chloride (1.2 equiv.) and NEt3 (1.2–2.2 equiv.) only provided 4′-O-caprinoylated-DMC (2d) as the sole product (entries 1 and 2). However, increasing the equivalents of caprinoyl chloride (2.2 equiv.) and NEt3 (2.2 equiv.) yielded a mixture of 4′-O-caprinoylated-DMC (2d) and 7-O-capryloylated-4-hydroxycoumarin (3d) products in 19 and 52%, respectively. The use of excess caprinoyl chloride (5 equiv.) and NEt3 (5 equiv.) exclusively provided Claisen-cyclisation product 3d in 91% yield.
| Entry | Caprinoyl chloride (equiv.) | NEt3 (equiv.) | Yield (%) | |
|---|---|---|---|---|
| 2d | 3d | |||
| a ND = not detected. | ||||
| 1 | 1.2 | 1.2 | 75 | ND |
| 2 | 2.2 | 1.2 | 74 | ND |
| 3 | 2.2 | 2.2 | 19 | 52 |
| 4 | 5 | 5 | ND | 91 |
The plausible mechanism for the formation of Claisen-cyclised product is shown in Scheme 2. Our results suggested that 2′,4′-O-diacylated DMC (INT) was presumably the key intermediate for the Claisen-cyclised product 3. However, attempts to spectroscopically detect and isolate a diacylated intermediate were unsuccessful. Once 2′,4′-O-diacylated DMC (INT) was formed, the α-proton of INT was rapidly deprotonated by an NEt3 base to generate an enolate anion (A and B). The anti-alignment (B) between the ketone carbonyl group and the enolate anion was favoured to minimise the steric repulsion between the ketone and methoxy group. The enolate anion (in structure B) then undergoes an intramolecular cyclisation with α,β-unsaturated ketone via more favourable 6-(enolendo)-exo-trig cyclisation39 thus providing a stereospecific 7-O-acylated-4-hydroxycoumarin (3).
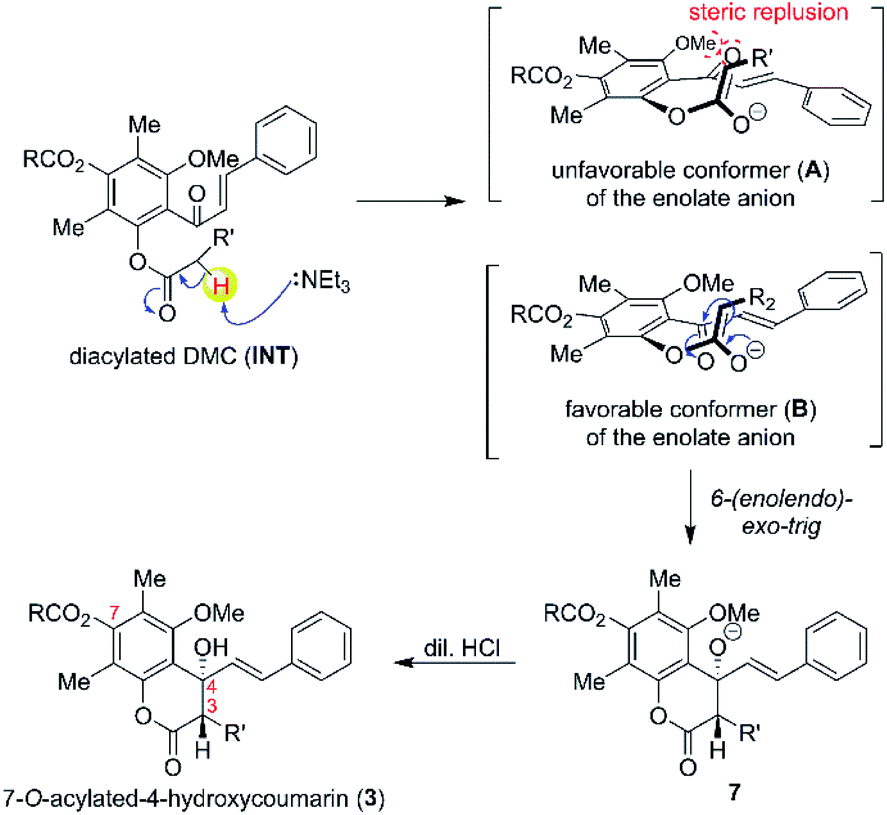 | ||
| Scheme 2 Plausible mechanism for the synthesis of 7-O-acylated-4-hydroxycoumarin derivatives (3). | ||
According to previous literature, the stilbene–coumarin hybrid scaffold exhibited strong cytotoxicity toward several cancer cell lines.43 In our studies, 7-O-acylated-4-hydroxycoumarin derivatives (3) can serve as valuable precursors to access stilbene–coumarin derivatives. To our delight, 7-O-acylated-4-hydroxycoumarin derivatives (3) underwent dehydration by refluxing in THF with H2SO4 as the catalyst that delivered stilbene–coumarin derivatives 4 in excellent yields.
Spectroscopically, the FTIR spectrum of stilbene–coumarin 4d indicated strong absorption bands at 1755 and 1712 cm−1 corresponding to CO stretching of the ester and conjugated CO of lactone, respectively. The 1H-NMR spectrum exhibited the disappearance of both hydroxy and methine proton signals. In addition, the 13C-NMR spectrum showed the signals of the quaternary carbons of the double bond of the lactone ring as evidence of a completed dehydration reaction.
Next, we turn our attention to modifying the DMC core with the alkylation, acylation or sulfonylation, as shown in Scheme 1, route II. The DMC was dissolved in anhydrous acetone in the presence of anhydrous K2CO3 and then subjected to several alkylating, acylating, and sulfonylating agents. A mixture of products, including 4′-O-monosubstituted-DMC (2f–2n), 2′,4′-O-disubstituted-DMC (5f–5n), and flavanone derivatives 6g and 6m, was obtained. The 4′-O-monosubstituted-DMC derivatives (2f–2n) were obtained in moderate yield, while derivatives 5f–5n were obtained in low yield, presumably due to intramolecular hydrogen bonding with 2′-OH.
To rationalise the product formation, the DMC was first reacted at more reactive 4′-OH with alkylating, acylating or sulfonylating agents to yield 4′-O-monosubstituted-DMC (2). Next, the substitution at the less reactive 2′-OH further provided 2′,4′-O-disubstituted-DMC (5). To our surprise, only flavanones 6g and 6m can be isolated in 6% yield. This flavanone product was derived from the intramolecular Michael addition of 2′-OH to α,β-unsaturated ketone via 6-exo-trig cyclisation. Moreover, the observed low yield of flavanone formation may result from the intramolecular hydrogen bonding between 2′-OH and the carbonyl group that decreases the acidity of the hydroxy proton. As a result, the 2′-OH was difficult to deprotonate by the weak base K2CO3.41 In addition, optical rotation experiments suggested that racemic flavanones 6 were formed.
In vitro cytotoxicity
Our semi-synthetic DMC derivatives, including 4′-O-monosubstituted-DMC derivatives (2a–2n), 7-O-acylated-4-hydroxycoumarins (3a–3e), stilbene–coumarins (4a–4e), 2′,4′-O-disubstituted-DMC derivatives (5f–5n), and flavanone derivatives (6g and 6m), were further evaluated for cytotoxicity against six cancer cell lines, KKU-M213, FaDu, HT-29, MDA-MB-231, A-549, and SH-SY5Y with CL as a normal cell line, using sulforhodamine B (SRB) assay. DMC and anticancer drugs used in general medicine such as ellipticine were taken as the reference and positive control, respectively. Cytotoxicity was expressed as IC50 (the concentration which indicated 50% cell-growth inhibition) in μM units of concentration as listed in Table 2, while the residues are given in Table 1S.†| Compound | Cytotoxicity (IC50, μM) | ||||||
|---|---|---|---|---|---|---|---|
| KKU-M213 | FaDu | HT-29 | MDA-MB-231 | A-549 | SH-SY5Y | CL | |
| a Ellipticine was used as a positive control for the cytotoxicity assay. | |||||||
| 2a | 30.05 | 31.16 | 35.77 | 38.24 | >50 | 27.55 | 32.82 |
| 2b | 21.26 | 18.92 | 26.63 | 18.13 | 24.84 | 5.20 | 23.53 |
| 2c | 24.43 | 20.63 | 26.62 | 25.86 | >50 | 18.04 | 25.16 |
| 2d | >50 | 34.71 | >50 | 36.70 | >50 | 24.79 | 41.87 |
| 2f | 42.98 | 36.49 | 38.63 | 40.28 | >50 | 28.56 | 31.02 |
| 2g | 23.88 | 24.84 | 19.69 | 26.22 | 25.35 | 7.52 | 25.42 |
| 2h | 23.19 | 13.98 | 27.47 | 26.85 | 9.99 | 16.45 | 22.58 |
| 2i | 31.26 | 28.96 | 34.63 | 39.10 | 36.67 | 28.25 | 31.68 |
| 2k | >50 | 37.89 | 32.53 | >50 | 41.30 | 20.25 | >50 |
| 2l | 28.86 | 22.09 | 26.75 | 25.09 | 22.43 | 15.56 | 25.36 |
| 2m | 23.76 | 27.00 | 26.66 | 26.11 | 26.29 | 20.43 | 29.01 |
| 2n | 27.84 | 21.10 | 24.73 | 39.75 | >50 | 16.37 | 30.38 |
| 3c | >50 | >50 | >50 | >50 | >50 | 20.86 | 35.90 |
| 4a | 29.58 | >50 | 30.87 | >50 | >50 | >50 | >50 |
| 5f | 28.01 | 33.65 | 36.51 | 29.78 | 15.40 | 18.75 | 27.35 |
| 5g | 23.47 | 19.30 | 21.94 | 29.78 | 22.67 | 12.56 | 18.69 |
| 5k | 31.55 | 24.54 | 33.76 | >50 | 30.93 | 25.31 | 25.31 |
| 6m | >50 | >50 | 45.49 | >50 | 37.86 | 44.69 | >50 |
| DMC (1) | 29.4 | 37.57 | 38.58 | 37.64 | 19.9 | 13.1 | 15.6 |
| Ellipticine (+)a | 2.40 | 2.03 | 2.15 | 2.80 | 1.14 | 2.64 | 2.07 |
4′-O-Monosubstituted-DMC (2) exhibited the strongest cytotoxicity among the DMC derivatives. 4′-O-Caproylated-DMC (2b) and 4′-O-methylated-DMC (2g) showed the strongest cytotoxicity toward the SH-SY5Y cell line with IC50 values of 5.20 μM and 7.52 μM, respectively. 4′-O-Benzylated-DMC (2h) showed the strongest cytotoxicity toward A-549 and FaDu cell lines with IC50 values of 9.99 and 13.98 μM, respectively. Furthermore, the cytotoxicity of the other DMC derivatives toward HT-29 and MDA-MB-231 cell lines displayed moderate activity with IC50 values higher than 19.69 and 18.13 μM, respectively. The cytotoxicity on other cell lines was quite similar to that of these cell lines, which were classified as having moderate cytotoxicity. Moreover, most DMC derivatives displayed low cytotoxicity toward the normal cell line (CL) with the selectivity index (SI) to the cancer cell lines being higher than that of DMC with SI values up to 4.53 (see Table 1S†) for SH-SY5Y in compound 2b. Overall, our structural modifications enhanced the cytotoxicity and selectivity of DMC toward cancer cell lines.
Generally, 2′,4′-O-disubstituted-DMC (5) displayed lower cytotoxicity than that of 4′-O-monosubstituted-DMC (2). In particular, 2′,4′-O-dimethylated-DMC (5g) showed moderate cytotoxicity on all tested cancer cell lines with IC50 value ranging from 12.56 to 29.78 μM, while other derivatives such as 2′,4′-diacetylated-DMC (5f) and 2′,4′-O-dibenzoylated-DMC (5k) displayed poor cytotoxicity. It should be noted that there was no cytotoxicity when derivative 5 was derivatised with allyl, benzenesulfonyl, 2-naphthalenesulfonyl, and benzyl moieties. Most 7-O-acylated-4-hydroxycoumarin (3), stilbene–coumarin (4), and flavanone (6) derivatives lacked cytotoxicity for all cell lines under tested concentrations.
Because of their intriguing cytotoxicity, it is required to build a complete structure–activity relationship (SAR) for these molecules. The summarisation of SAR studies is shown in Fig. 2. Our findings showed that functionalising DMC on 4′-OH significantly increased cytotoxicity while decreasing cytotoxicity when both 2′- and 4′-OH were derivatised. Importantly, our results suggest 2′-OH is required for anticancer activity. Moreover, among 4′-O-acylated-DMC derivatives (2a–2e), the cytotoxicity increased with the increment of the number of carbon atoms and reached a maximum at six carbons, whereas a shorter or longer chain resulted in a decrease in activity. Additionally, methylation, benzylation, benzenesulfonylation, naphthalenesulfonylation, and naphthoylation on 4′-OH showed remarkable cytotoxicity.
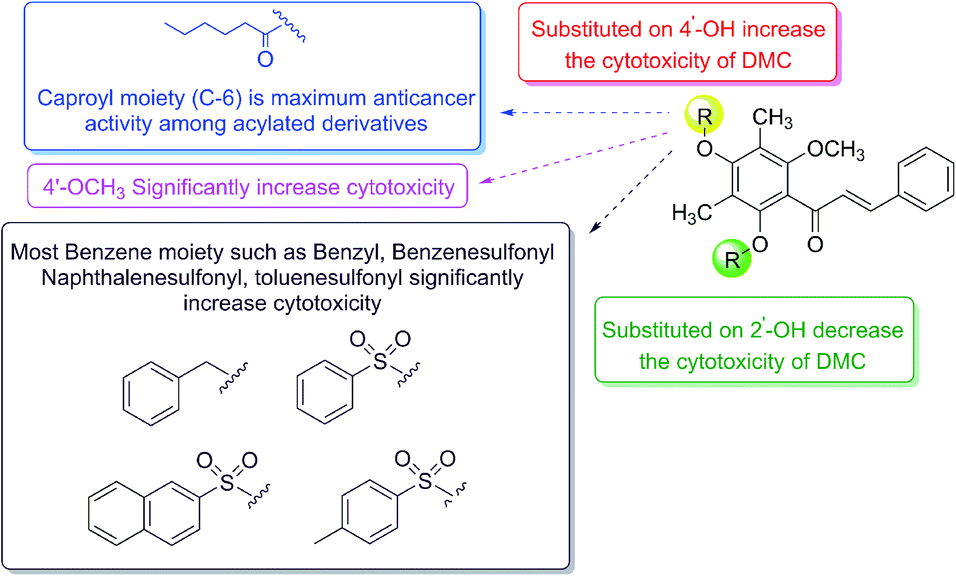 | ||
| Fig. 2 The structure–activity relationships (SAR) study of 2′,4′-disubstituted-6′-methoxy-3′,5′-dimethylchalcone derivatives on the anticancer activity. | ||
In silico molecular docking study
The molecular docking study was performed on AutoDock Vina to understand the binding modes of DMC derivatives 2b, 2g, 2h, and 5g on the cyclin-dependent kinase 2 (CDK2) which is related to cell cycle regulation to rationalise their promising cytotoxicity. According to previous reports, key amino acid residues of CDK2 are located at Thr14, Lys33, Glu81, Leu83, Asp86, Asp127, Asn132, and Asp145 of the ATP binding pocket.44,45CDK2 in complex with inhibitor RC-3-89 (PDB ID 4GCJ) was employed first. The molecular docking protocol was validated by redocking of a co-crystallised ligand to the binding pocket, as indicated by an RMSD value of 0.976 Å between the docking pose and the co-crystallised ligand with a binding energy value of −9.7 kcal mol−1 as well as all key interactions being identical (see Fig. 1S†).
The docking score and the protein–ligand interactions are shown in Table 3. The molecular docking study revealed that our compounds can accommodate the binding pocket of CDK2 with binding energies ranging from −7.1 to −9.6 kcal mol−1, which included H-bonding interactions with key amino acid residues of the receptor. Moreover, the predicted binding interactions are shown in Fig. 3.
| Compound | Binding energy (kcal mol−1) | Protein–ligand interactions | ||
|---|---|---|---|---|
| Hydrogen bonding | Electrostatic | Hydrophobic | ||
| 2b | −8.2 | Leu83, Asp86, Gln131 | — | Ile10, Val18, Ala31, Lys33, Phe80, Leu134, Ala144 |
| 2g | −9.4 | Leu83, Gln131, Asp145 | — | Ile10, Val18, Ala31, Phe80, Leu134, Ala144 |
| 2h | −9.6 | Asp86, Gln131 | Glu12 | Val18, Ala31, Val64, Phe80, Leu134, Ala144 |
| 5g | −7.1 | Glu12, Thr14, Gly16, Lys33, Asp127, Gln131 | Asp127, Asp145 | Val18, Ala144 |
| DMC (1) | −8.4 | Asp86, Gln131, Asn132, Asp145 | — | Ile10, Ala31, Leu134 |
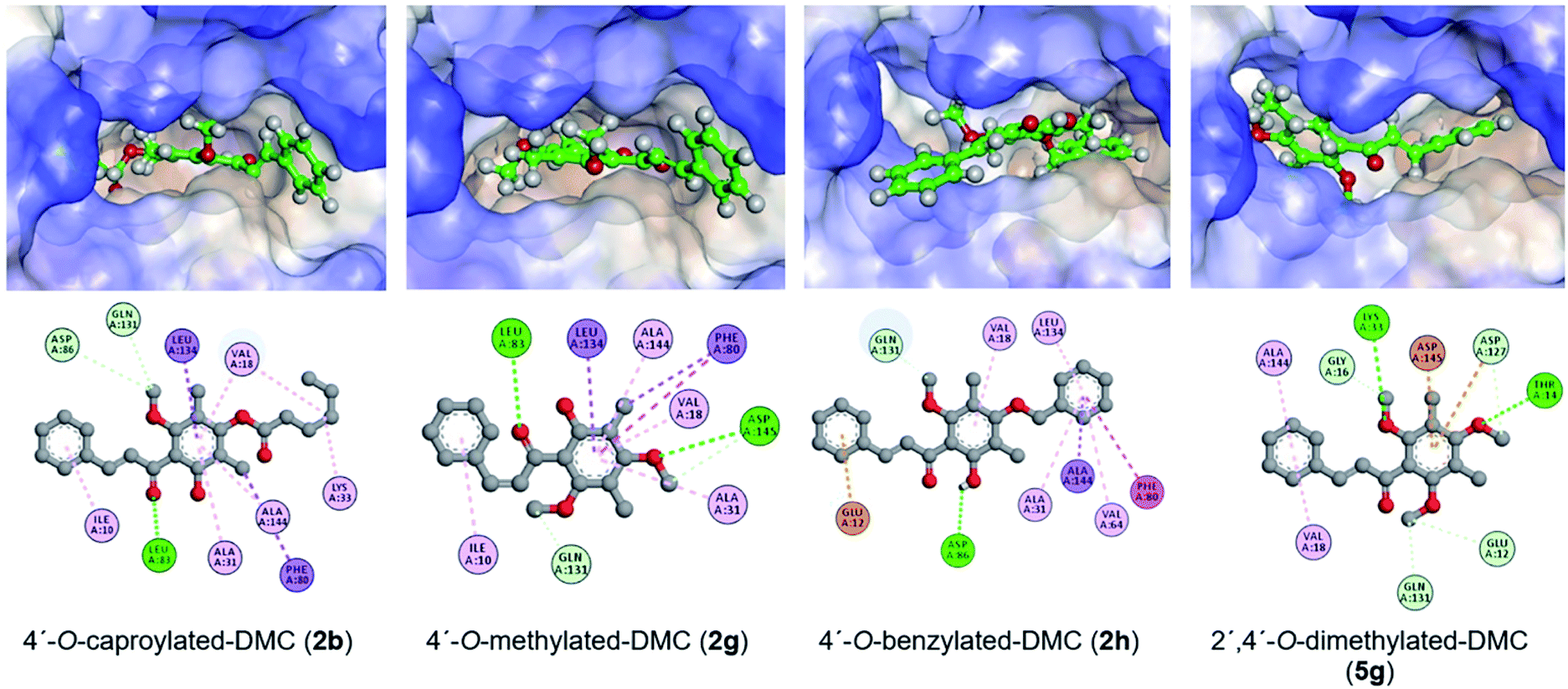 | ||
| Fig. 3 Docking poses of compounds 2b, 2g, 2h and 5g at the ATP binding site of CDK2 (PDB ID 4GCJ) and ligand binding interactions. | ||
As a control, DMC can accommodate the same ATP binding site as the co-crystallised inhibitor RC-3-89 with a binding energy value of −8.4 kcal mol−1. Hydrogen bonding interactions of 2′-OH with the key amino acid Asn132 (OH⋯OC–Asn132, 2.62 Å) and Asp145 (OH⋯OC–Asp145, 2.57 Å) backbones are crucial for the interactions (see Fig. 2S†).
The binding energy of 4′-O-caproylated-DMC (2b) with CDK2 was −8.2 kcal mol−1. Several hydrogen bonding interactions can be deduced from the docking pose, including the interaction between α,β-unsaturated ketone and Leu83 residue (CO⋯HN–Leu83, 4.61 Å), and carbon–hydrogen interactions with Asp86 (C⋯OC–Asp86, 3.63 Å) and Gln131 (C⋯OC–Gln131, 3.69 Å) residues. Furthermore, the docking studies also suggest hydrophobic interactions between the alkanoyl moiety and various residues such as Val18 and Lys33 which may play an important role. The binding energy of 4′-O-methylated-DMC (2g) with CDK2 of −9.4 kcal mol−1 could be calculated. Hydrogen bonding interactions between α,β-unsaturated CO ketone and the key amino acid Leu83 residue (CO⋯HN–Leu83, 2.33 Å), and the methoxy group with Asp145 residue (CO⋯HN–Asp145, 3.03 Å) were observed. Additionally, the interactions with Ile10, Val18, Ala31, Phe80, Leu134, and Ala144 residues through hydrophobic interaction were also noted. 4′-O-Benzylated-DMC (2h) showed the lowest binding energy among DMC derivatives with a value of −9.6 kcal mol−1. The hydrogen bonding interaction between 2′-OH and key amino acid Asp86 residue (OH⋯OC–Asp86, 2.58 Å) was noticed. Besides, it was found that the phenyl ring B interacted with Glu12 through π–anion interaction. The attached benzyl group also interacted with Phe80, Ala31, Val64, Ala144, and Leu134 residues. In 2′,4′-O-dimethylated-DMC (5g), the oxygen atom of the methoxy groups interacted with key amino acids Thr14 (O⋯HN–Thr14, 4.14 Å) and Lys33 (O⋯HN–Lys33, 4.52 Å) through hydrogen bonding interactions. Furthermore, multiple interactions were observed including carbon–hydrogen interactions between the incorporated methoxy carbon and Asp127 residue (C⋯OC–Asp127, 3.26 Å) and electrostatic interactions between the phenyl ring and the key amino acids i.e. Asp 127 and Asp145 through π–anion interactions with bond distances of 4.66 and 3.17 Å, respectively.
We also found that 7-O-acylated-4-hydroxycoumarin derivatives (3) cannot accommodate the binding pocket of CDK2. The two new chiral positions resulted in a loss of flat geometry and sterically hindered access to the binding site, which is consistent with the lack of cytotoxicity (see Fig. 3S†). Most stilbene–coumarin derivatives showed excellent binding energy values to the CDK2 binding site. However, their diminished anticancer activities may be due to their low solubility (see Fig. 4S†).
In vitro antimicrobial activity
The DMC derivatives were further evaluated for antimicrobial activities against S. aureus, E. coli, and P. aeruginosa. In the broth microdilution method, DMC demonstrated moderate antimicrobial activity against all bacterial strains, with MIC values of 2.50, 1.25, and 1.25 mg mL−1 for S. aureus, E. coli, and P. aeruginosa, respectively, while the MBC values were higher than 5.00 mg mL−1. In addition, the DMC displayed a zone of inhibition (ZOI) with diameter values of 9, 8, and 8 mm for S. aureus, E. coli, and P. aeruginosa, respectively. However, most DMC derivatives may decrease in antimicrobial activity in comparison to unmodified DMC and they were lacking in activity in some derivatives. For more details see Table 2S.†Experimental section
General experimental methods
All melting points were determined on a Gallenkamp Electrothermal apparatus. Optical rotations were measured at room temperature in a 1 cm cell on a Rudolph research analytical automatic polarimeter at the sodium D-line (589 nm) reported as [α]T589 (c in g per 100 mL, solvent). FTIR spectra were recorded on a Bruker TENSER 27 spectrometer and the wavenumbers of the maximum absorption peaks are reported in cm−1. The high-resolution mass spectra (HRMS, m/z values) were obtained from an ESI-QTOF-MS/MS Agilent model QTOF 6540 UHD mass spectrometer. 1H-NMR and 13C-NMR were recorded on a Bruker DRX-400 or NEO™ 500 spectrometer. Where necessary, 2D NMR experiments (HMBC and HBQC) were carried out to assist the structural elucidation and signal assignments. Chemical shifts (δ) were reported in ppm down-field from TMS as an internal reference or with the solvent resonance as the internal standard (CHCl3 impurity in CDCl3, δ 7.26 and 77.16 ppm for 1H-NMR and 13C-NMR, respectively); data are reported in the following order: chemical shift, multiplicity and coupling constants (J) are given in hertz. Flash column chromatography was performed employing Merck silica gel 60H. The progress of chemical reactions was monitored by a thin layer chromatography (TLC), using Merck silica gel PF254 aluminum plates. All the reactions were conducted in oven-dried glassware, under N2, with anhydrous solvents, which were dried and distilled before use according to standard procedures, and acid chlorides were freshly distilled under reduced pressure prior to use.Extraction and isolation of DMC (1)
A previous study on the seeds of S. nervosum illustrated isolation and structural elucidation of 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone (DMC).37 A similar process was conducted to isolate DMC for the starting material in chemical modification. Briefly, 13.38 kg of air-dried ground seeds of S. nervosum were macerated with 40 L of EtOAc at ambient temperature (performed three times, each time for three days). The seed residues were filtered out and the filtrate was collected. Then it was concentrated under reduced pressure to afford 266.49 g of crude EtOAc extract as a dark-green viscous liquid. The extract was subjected to flash column chromatography on silica gel eluted with gradient eluents of n-hexane/EtOAc = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 80:20. DMC-containing fractions were collected and further purified by flash column chromatography on silica gel with n-hexane/acetone = 90:10 as an eluent to give the orange precipitates. The DMC was crystallised with CH2Cl2/n-hexane to obtain 4.3615 g of orange needle-shaped crystals. The chemical structure was confirmed by comparison of its spectral data with the published values.37
O), 1610 (CC), 1423 (–CH3), 1232 (C–O–C), 1170 (C–O–C) cm−1; δH (400 MHz, CDCl3) 2.13 (s, 3H, Me), 2.15 (s, 3H, Me), 3.67 (s, 3H, OMe), 5.39 (s, 1H, OH), 7.36–7.45 (m, 3H, ArCH), 7.60–7.68 (m, 2H, ArCH), 7.84 (AB system, d, J = 15.7 Hz, 1H, CH), 7.99 (AB system, d, J = 15.7 Hz, 1H, CH), 13.61 (s, 1H, OH); δc (100 MHz, CDCl3) 7.6, 8.2, 62.4, 106.6, 108.9, 109.0, 126.7, 128.4, 128.9, 130.2, 135.3, 142.9, 158.8, 159.2, 162.0, 193.4; HRMS (ESI) calcd for C18H18O4Na (M + Na)+: m/z 321.1103, found 321.1100.
:0 to 80:20. DMC-containing fractions were collected and further purified by flash column chromatography on silica gel with n-hexane/acetone = 90:10 as an eluent to give the orange precipitates. The DMC was crystallised with CH2Cl2/n-hexane to obtain 4.3615 g of orange needle-shaped crystals. The chemical structure was confirmed by comparison of its spectral data with the published values.37
O), 1610 (CC), 1423 (–CH3), 1232 (C–O–C), 1170 (C–O–C) cm−1; δH (400 MHz, CDCl3) 2.13 (s, 3H, Me), 2.15 (s, 3H, Me), 3.67 (s, 3H, OMe), 5.39 (s, 1H, OH), 7.36–7.45 (m, 3H, ArCH), 7.60–7.68 (m, 2H, ArCH), 7.84 (AB system, d, J = 15.7 Hz, 1H, CH), 7.99 (AB system, d, J = 15.7 Hz, 1H, CH), 13.61 (s, 1H, OH); δc (100 MHz, CDCl3) 7.6, 8.2, 62.4, 106.6, 108.9, 109.0, 126.7, 128.4, 128.9, 130.2, 135.3, 142.9, 158.8, 159.2, 162.0, 193.4; HRMS (ESI) calcd for C18H18O4Na (M + Na)+: m/z 321.1103, found 321.1100.General procedure for the synthesis of DMC derivatives
:20:76 as eluent to obtain compounds 2a and 3a in 42% (0.0548 g) and 45% (0.0698 g) yields, respectively.O), 1636 (CO), 1605 (CC), 1191 (C–O–C), 1143 (C–O–C) cm−1; δH (500 MHz, CDCl3) 1.08 (t, J = 7.4 Hz, 3H, Me), 1.79–1.88 (m, 2H, CH2), 2.04 (s, 3H, Me), 2.05 (s, 3H, Me), 2.62 (t, J = 7.4 Hz, 2H, CH2), 3.67 (s, 3H, OMe), 7.45–7.37 (m, 3H, ArCH), 7.62–7.68 (m, 2H, ArCH), 7.87 (AB system, d, J = 15.7 Hz, 1H, CH), 7.95 (AB system, d, J = 15.7 Hz, 1H, CH), 12.96 (s, 1H, OH); δC (125 MHz, CDCl3) 9.1, 9.3, 13.9, 18.6, 36.0, 62.7, 113.3, 115.3, 115.8, 126.5, 128.7, 129.1, 130.6, 135.2, 143.9, 154.3, 158.3, 160.9, 170.8, 194.5; HRMS (ESI) calcd for C22H24O5 (M)+: m/z 368.1624, found 368.1616.O), 1603 (CC), 1202 (C–O–C), 1149 (C–O–C), 1102 (C–O–C) cm−1; δH (500 MHz, CDCl3) 1.00 (t, J = 7.5 Hz, 3H, CH3), 1.09 (t, J = 7.5 Hz, 3H, CH3), 1.28–1.38 (m, 1H, CH), 1.80–1.89 (m, 2H, CH2) 2.06 (s, 3H, Me), 2.10 (s, 3H, Me), 2.12–2.24 (m, 1H, CH), 2.63 (t, J = 7.4 Hz, 2H, CH2), 2.78 (dd, J = 11.1, 4.0 Hz, 1H, CH), 3.73 (s, 3H, OMe), 5.66 (br s, 1H, OH), 6.36 (d, J = 15.8 Hz, 1H, CH), 6.46 (br d, J = 15.8 Hz, 1H, CH), 7.20–7.36 (m, 5H, ArCH); δC (125 MHz, CDCl3) 9.7, 9.9, 13.9, 14.0, 20.4, 24.8, 34.2, 52.0, 62.1, 75.3, 115.8, 116.5, 120.7, 127.1, 128.1, 128.7, 129.5, 133.4, 135.8, 146.3, 149.4, 154.7, 168.8, 171.3; HRMS (ESI) calcd for C26H30O6 (M)+: m/z 438.2042, found 438.2039.O), 1637 (CO), 1607 (CC), 1570 (CC), 1284 (C–O–C), 1135 (C–O–C), 1115 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.94 (t, J = 6.8 Hz, 3H, Me), 1.34–1.49 (m, 4H, CH2), 1.75–1.86 (m, 2H, CH2), 2.04 (s, 3H, Me), 2.05 (s, 3H, Me), 2.63 (t, J = 7.3 Hz, 2H, CH2), 3.67 (s, 3H, OMe), 7.39–7.47 (m, 3H, ArCH), 7.62–7.69 (m, 2H, ArCH), 7.87 (AB system, d, J = 15.7 Hz, 1H, CH), 7.95 (AB system, d, J = 15.7 Hz, 1H, CH), 12.97 (s, 1H, OH); δC (125 MHz, CDCl3) 9.1, 9.3, 14.0, 22.4, 24.8, 31.5, 34.1, 62.7, 113.4, 115.3, 115.8, 126.5, 128.7, 129.1, 130.7, 135.2, 144.0, 154.3, 158.3, 160.9, 171.0, 194.5.; HRMS (ESI) calcd for C24H28O5 (M)+: m/z 396.1937, found 396.1935.O), 1602 (CC), 1219 (C–O–C), 1145 (C–O–C), 1102 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.87 (t, J = 7.0 Hz, 3H, Me), 0.95 (t, J = 6.9 Hz, 3H, Me), 1.18–1.51 (m, 9H, CH2), 1.76–1.86 (m, 2H, CH2), 2.07 (s, 3H, Me), 2.10 (s, 3H, Me), 2.10–2.16 (m, 1H, CH), 2.64 (t, J = 7.5 Hz, 2H, CH2), 2.84 (dd, J = 10.7, 3.8 Hz, 1H, CH), 3.73 (s, 3H, OMe), 5.68 (br s, 1H, OH), 6.36 (AB system, d, J = 15.8 Hz, 1H, CH), 6.46 (AB system, br d, J = 15.8 Hz, 1H, CH), 7.19–7.40 (m, 5H, ArCH); δC (125 MHz, CDCl3) 9.7, 9.9, 13.9, 14.0, 22.4, 22.5, 24.8, 25.8, 29.6, 31.5, 34.0, 52.0, 62.1, 75.3, 115.8, 116.4, 120.7, 127.0, 128.2, 128.7, 129.5, 133.4, 135.9, 146.3, 149.3, 154.7, 168.8, 171.2; HRMS (ESI) calcd for C30H38O6 (M)+: m/z 494.2668, found 494.2666.O), 1637 (CO), 1607 (CC), 1570 (CC), 1284 (C–O–C), 1136 (C–O–C), 1115 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.90 (t, J = 6.7 Hz, 3H, Me), 1.28–1.51 (m, 8H, CH2), 1.76–1.86 (m, 2H, CH2), 2.04 (s, 3H, Me), 2.05 (s, 3H, Me), 2.63 (t, J = 7.5 Hz, 2H, CH2), 3.67 (s, 3H, OMe), 7.38–7.50 (m, 3H, ArCH), 7.62–7.69 (m, 2H, ArCH), 7.88 (AB system, d, J = 15.7 Hz, 1H, CH), 7.95 (AB system, d, J = 15.7 Hz, 1H, CH), 12.97 (s, 1H, OH); δC (125 MHz, CDCl3) 9.1, 9.3, 14.2, 22.7, 25.2, 29.0, 29.3, 29.8, 31.8, 34.1, 62.8, 113.4, 115.3, 115.8, 126.5, 128.7, 129.1, 130.7, 135.2, 144.0, 154.3, 158.3, 160.9, 171.0, 194.5; HRMS (ESI) calcd for C26H32O5 (M)+: m/z 424.2250, found 424.5546.O), 1603 (CC), 1206 (C–O–C), 1144 (C–O–C), 1103 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.80–0.95 (m, 6H, Me), 1.17–1.50 (m, 17H, CH2), 1.75–1.85 (m, 2H, CH2), 2.07 (s, 3H, Me), 2.08–2.15 (m, 1H, CH), 2.10 (s, 3H, Me), 2.64 (t, J = 7.5 Hz, 2H, CH2), 2.85 (dd, J = 10.7, 3.7 Hz, 1H, CH), 3.72 (s, 3H, OMe), 5.67 (br s, 1H, OH), 6.36 (AB system, d, J = 15.8 Hz, 1H, CH), 6.46 (AB system, br d, J = 15.8 Hz, 1H, CH), 7.18–7.36 (m, 5H, ArCH); δC (125 MHz, CDCl3) 9.7, 9.8, 14.1, 14.2, 22.6, 22.7, 25.1, 26.1, 27.4, 29.0, 29.1, 29.3, 31.6, 31.7, 34.0, 52.0, 62.0, 75.3, 115.7, 116.4, 120.7, 127.0, 128.2, 128.6, 129.5, 133.4, 135.8, 146.3, 149.3, 154.7, 168.8, 171.2; HRMS (ESI) calcd for C34H46O6 (M)+: m/z 550.3294, found 550.3295.O), 1637 (CO), 1607 (CC), 1570 (CC), 1135 (C–O–C), 1114 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.89 (t, J = 6.7 Hz, 3H, Me), 1.21–1.49 (m, 12H, CH2), 1.75–1.86 (m, 2H, CH2), 2.03 (s, 3H, Me), 2.05 (s, 3H, Me), 2.63 (t, J = 7.6 Hz, 2H, CH2), 3.67 (s, 3H, OMe), 7.38–7.46 (m, 3H, ArCH), 7.61–7.69 (m, 2H, ArCH), 7.88 (AB system, d, J = 15.7 Hz, 1H, CH), 7.94 (AB system, d, J = 15.7 Hz, 1H, CH), 12.97 (s, 1H, OH); δC (125 MHz, CDCl3) 9.1, 9.3, 14.2, 22.8, 25.2, 29.3, 29.4, 29.4, 29.6, 32.0, 34.1, 62.7, 113.4, 115.3, 115.8, 126.5, 128.7, 129.1, 130.7, 135.2, 144.0, 154.3, 158.3, 160.9, 171.0, 194.5; HRMS (ESI) calcd for C28H36O5 (M)+: m/z 452.2563, found 452.2558.O), 1603 (CC), 1198 (C–O–C), 1143 (C–O–C), 1101 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.81–0.92 (m, 6H, Me), 1.15–1.50 (m, 25H, CH2), 1.75–1.85 (m, 2H, CH2), 2.06 (s, 3H, Me), 2.07–2.14 (m, 1H, CH) 2.10 (s, 3H, Me), 2.64 (t, J = 7.5 Hz, 2H, CH2), 2.84 (dd, J = 10.7, 3.8 Hz, 1H, CH), 3.73 (s, 3H, OMe), 5.68 (br s, 1H, OH), 6.36 (AB system, d, J = 15.7 Hz, 1H, CH), 6.46 (AB system, br d, J = 15.7 Hz, 1H, CH), 7.18–7.38 (m, 5H, ArCH); δC (125 MHz, CDCl3) 9.8, 9.9, 14.2, 22.7, 22.8, 25.1, 26.1, 27.4, 29.2, 29.3, 29.4, 29.5, 29.6, 31.9, 32.0, 34.0, 52.0, 62.1, 75.2, 115.6, 116.3, 120.6, 126.8, 128.1, 128.6, 129.4, 133.3, 135.7, 146.1, 149.2, 154.6, 168.7, 171.1; HRMS (ESI) calcd for C38H54O6 (M)+: m/z 606.3920, found 606.3923.O), 1637 (CO), 1607 (CC), 1570 (CC), 1136 (C–O–C), 1113 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.88 (t, J = 6.7 Hz, 3H, Me), 1.18–1.50 (m, 16H, CH2), 1.75–1.86 (m, 2H, CH2), 2.04 (s, 3H, Me), 2.05 (s, 3H, Me), 2.63 (t, J = 7.6 Hz, 2H, CH2), 3.67 (s, 3H, OMe), 7.38–7.46 (m, 3H, ArCH), 7.61–7.69 (m, 2H, ArCH), 7.88 (AB system, d, J = 15.7 Hz, 1H, CH), 7.94 (AB system, d, J = 15.7 Hz, 1H, CH), 12.97 (s, 1H, OH); δC (125 MHz, CDCl3) 9.1, 9.3, 14.3, 22.8, 25.2, 29.4, 29.5, 29.6, 29.7, 32.0, 34.1, 62.7, 113.3, 115.3, 115.8, 126.5, 128.7, 129.1, 130.6, 135.2, 144.0, 154.3, 158.3, 160.9, 171.0, 194.5; HRMS (ESI) calcd for C30H40O5 (M)+: m/z 480.2876, found 480.2873.O), 1604 (CC), 1204 (C–O–C), 1143 (C–O–C), 1107 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.83–0.93 (m, 6H, Me), 1.15–1.50 (m, 33H, CH2), 1.75–1.85 (m, 2H, CH2), 2.06 (s, 3H, Me), 2.07–2.14 (m, 1H, CH), 2.10 (s, 3H, Me), 2.64 (t, J = 7.5 Hz, 2H, CH2), 2.84 (dd, J = 10.7, 3.8 Hz, 1H, CH), 3.72 (s, 3H, OMe), 5.67 (br s, 1H, OH), 6.36 (AB system, d, J = 16.1 Hz, 1H, CH), 6.46 (AB system, br d, J = 16.1 Hz, 1H, CH), 7.17–7.37 (m, 5H, ArCH); δC (125 MHz, CDCl3) 9.7, 9.8, 14.2, 22.7, 22.8, 25.1, 26.1, 27.4, 29.3, 29.3, 29.4, 29.4, 29.5, 29.6, 29.6, 29.7, 29.8, 32.0, 32.1 34.0, 52.0, 62.1, 75.3, 115.7, 116.4, 120.7, 126.9, 128.2, 128.6, 129.4, 133.4, 135.8, 146.2, 149.2, 154.7, 168.8, 171.1; HRMS (ESI) calcd for C42H62O6 (M)+: m/z 662.4546, found 662.4542.:10 as eluent to obtain compounds 2f and 5f in 22% yield (0.0276 g) and 36% (0.0507 g) yields, respectively.O), 1635 (CO), 1607 (CC), 1562 (CC), 1211 (C–O–C), 1112 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.05 (s, 3H, Me), 2.06 (s, 3H, Me), 2.38 (s, 3H, Me), 3.67 (s, 3H, OMe), 7.37–7.46 (m, 3H, ArCH), 7.62–7.69 (m, 2H, ArCH), 7.88 (AB system, d, J = 15.7 Hz, 1H, CH), 7.94 (AB system, d, J = 15.7 Hz, 1H, CH), 12.95 (s, 1H, OH); δC (125 MHz, CDCl3) 9.1, 9.3, 20.6, 62.8, 113.5, 115.3, 115.8, 126.5, 128.8, 129.2, 130.7, 135.2, 144.0, 154.2, 158.3, 160.9, 168.2, 194.5; HRMS (ESI) calcd for C20H20O5 (M)+: m/z 340.1311, found 340.1308.O), 1653 (CO), 1607 (CC), 1190 (C–O–C), 1180 (C–O–C), 1105 (C–O–C) cm−1; δH (500 MHz, CDCl3) 1.94 (s, 3H, Me), 2.10 (s, 3H, Me), 2.17 (s, 3H, Me) 2.38 (s, 3H, Me), 3.70 (s, 3H, OMe), 7.02 (d, J = 16.1 Hz, 1H, CH), 7.36–7.42 (m, 3H, ArCH), 7.46 (d, J = 16.1 Hz, 1H, CH), 7.53–7.58 (m, 2H, ArCH); δC (125 MHz, CDCl3) 9.9, 10.3, 20.5, 20.6, 62.7, 120.8, 123.0, 125.8, 127.4, 128.8, 129.1, 130.9, 134.5, 144.9, 146.3, 150.2, 154.8, 168.3, 168.7, 192.7; HRMS (ESI) calcd for C22H22O6 (M)+: m/z 382.1416, found 382.1414.O), 1567 (CC), 1453 (–CH3), 1145 (C–O–C), 1114 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.17 (s, 3H, Me), 2.18 (s, 3H, Me), 3.66 (s, 3H, OMe), 3.76 (s, 3H, OMe), 7.39–7.46 (m, 3H, ArCH), 7.62–7.68 (m, 2H, ArCH), 7.87 (AB system, d, J = 15.6 Hz, 1H, CH), 7.97 (AB system, d, J = 15.6 Hz, 1H, CH), 13.09 (s, 1H, OH); δC (125 MHz, CDCl3) 8.7, 8.8, 60.1, 62.3, 111.9, 115.7, 115.8, 126.6, 128.4, 129.0, 130.4, 135.2, 143.4, 158.7, 161.7, 163.6, 194.1; HRMS (ESI) calcd for C19H20O4 (M)+: m/z 312.1362, found 312.1359.O), 1584 (CC), 1456 (–CH3), 1197 (C–O–C), 1151 (C–O–C), 1110 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.21 (s, 6H, Me), 3.69 (s, 6H, OMe), 3.76 (s, 3H, OMe), 7.04 (d, J = 16.1 Hz, 1H, CH), 7.35–7.38 (m, 4H, ArCH and CH), 7.52–7.55 (m, 2H, ArCH); δC (125 MHz, CDCl3) 9.4, 60.2, 62.2, 120.9, 125.0, 128.6, 128.7, 129.0, 130.7, 134.7, 145.7, 154.6, 159.4, 195.1; HRMS (ESI) calcd for C20H22O4 (M)+: m/z 326.1518, found 326.1517.O), 1591 (CC), 1203 (C–O–C), 1157 (C–O–C), 1116 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.17 (s, 6H, Me), 2.85 (ABX system, dd, J = 16.6, 3.1 Hz, 1H, CH), 2.99 (ABX system, dd, J = 16.6, 13.1 Hz, 1H, CH), 3.75 (s, 3H, OMe), 3.82 (s, 3H, OMe), 5.42 (ABX system, dd, J = 13.1, 3.1 Hz, 1H, CH), 7.35–7.51 (m, 5H, ArCH); δC (125 MHz, CDCl3) 8.7, 9.2, 45.9, 60.2, 61.2, 78.7, 111.9, 115.8, 118.9, 125.9, 128.6, 128.9, 139.3, 157.8, 159.8, 163.6, 190.4; HRMS (ESI) calcd for C19H20O4 (M)+: m/z 312.1362, found 312.1360.O), 1603 (CC), 1567 (CC), 1152 (C–O–C), 1114 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.17 (s, 3H, Me), 2.19 (s, 3H, Me), 3.67 (s, 3H, OMe), 4.85 (s, 2H, CH2), 7.34–7.51 (m, 8H, ArCH), 7.63–7.69 (m, 2H, ArCH), 7.87 (AB system, d, J = 15.7 Hz, 1H, CH), 7.98 (AB system, d, J = 15.7 Hz, 1H, CH), 13.11 (s, 1H, OH); δC (125 MHz, CDCl3) 9.2, 9.4, 62.5, 74.8, 112.1, 116.1, 116.2, 126.7, 128.1, 128.4, 128.7, 128.8, 129.1, 130.5, 135.4, 137.0, 143.6, 158.9, 161.8, 162.5, 194.2; HRMS (ESI) calcd for C25H24O4 (M)+: m/z 388.1675, found 388.1672.O), 1564 (CC), 1170 (C–O–C), 1114 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.21 (s, 3H, Me), 2.24 (s, 3H, Me), 3.72 (s, 3H, OMe), 4.84 (s, 2H, CH2), 4.85 (s, 2H, CH2), 7.06 (d, J = 16.0 Hz, CH), 7.22–7.56 (m, 16H, ArCH and CH); δC (125 MHz, CDCl3) 9.8, 10.1, 62.3, 74.5, 76.8, 121.5, 121.7, 125.6, 128.0, 128.2, 128.2, 128.3, 128.5, 128.6, 128.7, 128.9, 130.6, 134.7, 137.0, 137.2, 145.7, 153.3, 154.8, 158.1, 195.1; HRMS (ESI) calcd for C32H30O4 (M)+: m/z 478.2144, found 478.2140.O), 1606 (CC), 1568 (CC), 1412 (–CH3), 1153 (C–O–C), 1113 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.16 (s, 3H, Me), 2.18 (s, 3H, Me), 3.66 (s, 3H, OMe), 4.34 (d, J = 5.6 Hz, 2H, CH2), 5.30 (d, J = 10.4 Hz, 1H, CH2), 5.45 (m, 1H, CH2), 6.10 (m, 1H, CH), 7.38–7.45 (m, 3H, ArCH), 7.61–7.70 (m, 2H, ArCH), 7.87 (AB system, d, J = 15.8 Hz, 1H, CH), 7.97 (AB system, d, J = 15.8 Hz, 1H, CH), 13.09 (s, 1H, OH); δC (125 MHz, CDCl3) 9.2, 9.3, 62.5, 73.8, 112.0, 116.0, 116.1, 118.0, 126.7, 128.7, 129.1, 130.5, 133.5, 135.4, 143.6, 158.8, 161.8, 162.7, 194.2; HRMS (ESI) calcd for C21H22O4 (M)+: m/z 338.1518, found 338.1520.O), 1631 (CC), 1605 (CC), 1454 (–CH3), 1189 (C–O–C), 1136 (C–O–C), 1112 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.20 (s, 6H, Me), 3.69 (s, 3H, OMe), 4.31 (d, J = 5.6 Hz, 2H, CH2), 4.34 (d, J = 5.4 Hz, 2H, CH2), 5.15 (d, J = 10.6 Hz, 1H, CH2), 5.24–5.35 (m, 2H, CH2), 5.46 (d, J = 17.1 Hz, 1H, CH2), 5.89–6.01 (m, 1H, CH), 6.05–6.17 (m, 1H, CH), 7.03 (d, J = 16.1 Hz, 1H, CH), 7.33–7.40 (m, 4H, ArCH and CH), 7.50–7.56 (m, 2H, ArCH); δC (125 MHz, CDCl3) 9.7, 10.0, 62.3, 73.6, 75.7, 117.7, 121.2, 121.4, 125.2, 128.6, 128.7, 129.0, 130.6, 133.7, 133.8, 134.8, 145.6, 153.5, 154.6, 158.2, 195.1; HRMS (ESI) calcd for C24H26O4 (M)+: m/z 378.1831, found 378.1835.O), 1638 (CO), 1606 (CC), 1564 (CC), 1206 (C–O–C), 1136 (C–O–C), 1114 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.13 (s, 3H, Me), 2.14 (s, 3H, Me) 3.72 (s, 3H, OMe), 7.40–7.49 (m, 3H, ArCH), 7.59–7.63 (m, 1H, ArCH), 7.64–7.70 (m, 3H, ArCH), 7.88–8.06 (m, 5H, ArCH and CH), 8.19–8.26 (m, 1H, ArCH), 8.85 (br s, 1H, ArCH), 13.01 (s, 1H, OH); δC (125 MHz, CDCl3) 9.3, 9.4, 62.8, 113.6, 115.6, 116.2, 125.6, 126.0, 126.6, 127.2, 128.1, 128.7, 128.8, 129.0, 129.2, 129.7, 130.7, 132.3, 132.7, 135.3, 136.2, 144.1, 154.5, 158.4, 161.1, 164.1, 194.6; HRMS (ESI) calcd for C29H24O5 (M)+: m/z 452.1624, found 452.1622.O), 1637 (CO), 1606 (CC), 1568 (CC), 1137 (C–O–C), 1115 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.10 (s, 3H, Me), 2.11 (s, 3H, Me), 3.71 (s, 3H, OMe), 7.40–7.47 (m, 3H, ArCH), 7.56 (m, 2H, ArCH), 7.64–7.72 (m, 3H, ArCH), 7.91 (AB system, d, J = 15.7 Hz, 1H, CH), 7.98 (AB system, d, J = 15.7 Hz, 1H, H), 8.25 (d, J = 7.7 Hz, 2H, ArCH), 12.99 (s, 1H, OH); δC (125 MHz, CDCl3) 9.2, 9.4, 62.8, 113.5, 115.6, 116.1, 126.5, 128.7, 128.8, 128.9, 129.1, 130.4, 130.7, 134.1, 135.2, 144.0, 154.4, 158.4, 161.0, 163.9, 194.5; HRMS (ESI) calcd for C25H22O5 (M)+: m/z 402.1467, found 402.1465.O), 1655 (CO), 1606 (CC), 1250 (C–O–C), 1182 (C–O–C), 1103 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.03 (s, 3H, Me), 2.19 (s, 3H, Me), 3.76 (s, 3H, OMe), 6.98 (d, J = 16.2 Hz, 1H, CH), 7.32–7.72 (m, 12H, ArCH and CH), 8.05 (d, J = 7.6 Hz, 2H, ArCH), 8.27 (d, J = 7.6 Hz, 2H, ArCH); δC (125 MHz, CDCl3) 10.1, 10.5, 62.7, 121.3, 123.4, 126.1, 127.6, 128.6, 128.7, 128.8, 128.8, 128.9, 130.4, 130.5, 130.8, 133.8, 134.1, 134.6, 145.1, 146.8, 150.3, 154.8, 164.0, 164.4, 192.9; HRMS (ESI) calcd for C32H26O6 (M)+: m/z 506.1729, found 506.1727.O), 1654 (CO), 1604 (CC), 1507 (CC), 1185 (C–O–C), 1137 (C–O–C) cm−1; δH (500 MHz, CDCl3) 2.08 (s, 3H, Me), 2.09 (s, 3H, Me), 2.47 (s, 3H, Me), 3.70 (s, 3H, OMe), 7.35 (d, J = 7.6 Hz, 2H, ArCH), 7.40–7.45 (m, 3H, ArCH), 7.63–7.70 (m, 2H, ArCH), 7.90 (AB system, d, J = 15.6 Hz, 1H, CH), 7.98 (AB system, d, J = 15.6 Hz, 1H, CH), 8.13 (d, J = 7.6 Hz, 2H, ArCH), 13.00 (s, 1H, OH); δC (125 MHz, CDCl3) 9.2, 9.4, 21.9, 62.8, 113.4, 115.6, 116.1, 126.0, 126.6, 128.7, 129.1, 129.6, 130.5, 130.7, 135.2, 144.0, 145.0, 154.5, 158.3, 161.0, 163.9, 194.5; HRMS (ESI) calcd for C26H24O5 (M)+: m/z 416.1624, found 416.1619.O), 1602 (CC),1566 (CC), 1347 (SO), 1195 (C–O–C), 1114 (C–O–C) cm−1; δH (500 MHz, CDCl3) 1.89 (s, 3H, Me), 2.08 (s, 3H, Me), 3.66 (s, 3H, OMe), 7.39–7.46 (m, 3H, ArCH), 7.57–7.73 (m, 5H, ArCH), 7.86–7.95 (m, 2H, CH), 7.99 (d, J = 7.5 Hz, 2H, ArCH), 12.84 (s, 1H, OH); δC (125 MHz, CDCl3) 10.2, 10.5, 62.7, 114.0, 117.3, 117.8, 126.2, 128.3, 128.8, 129.2, 129.5, 130.9, 134.5, 135.1, 137.1, 144.5, 152.5, 158.5, 160.9, 194.5; HRMS (ESI) calcd for C24H22O6S (M)+: m/z 438.1137, found 438.1135.O), 1566 (CC), 1453 (CH3), 1348 (SO), 1134 (C–O) cm−1; δH (500 MHz, CDCl3) 1.91 (s, 3H, Me), 2.17 (s, 3H, Me), 3.66 (s, 3H, OMe), 6.72 (d, J = 16.0 Hz, 1H, CH), 7.35–7.50 (m, 7H, ArCH and CH), 7.55–7.66 (m, 5H, ArCH), 7.79–7.84 (m, 2H, ArCH) 7.97–8.01 (m, 2H, ArCH); δC (125 MHz, CDCl3) 11.3, 12.1, 62.5, 124.9, 126.9, 127.3, 127.7, 128.3, 128.4, 128.5, 128.6, 128.9, 129.1, 129.6, 130.8, 133.6, 134.3, 134.6, 143.1, 143.2, 145.0, 149.0, 155.0, 190.6; HRMS (ESI) calcd for C30H26O8S2 (M)+: m/z 578.1069, found 578.1065.O), 1587 (CC), 1302 (SO), 1197 (C–O–C), 1146 (C–O–C), 1120 (C–O–C) cm−1; δH (400 MHz, CDCl3) 1.94 (s, 3H, Me), 2.02 (s, 3H, Me), 2.88 (ABX system, dd, J = 16.6, 3.0 Hz, 1H, CH), 3.03 (ABX system, dd, J = 16.6, 13.2 Hz, 1H, CH), 3.80 (s, 3H, OMe), 5.42 (ABX system, dd, J = 13.2, 3.0 Hz, 1H, CH), 7.35–7.51 (m, 5H, ArCH), 7.57–7.64 (m, 2H, ArCH), 7.69–7.76 (m, 1H, ArCH), 7.96–8.02 (m, 2H, ArCH); δC (125 MHz, CDCl3) 10.1, 10.9, 45.8, 61.5, 79.0, 113.9, 118.0, 120.3, 126.0, 128.3, 128.8, 128.9, 129.5, 134.6, 137.0, 138.8, 152.5, 157.6, 159.4, 190.4; HRMS (ESI) calcd for C24H22O6S (M)+: m/z 438.1137, found 438.1135.O), 1602 (CC), 1567 (CC), 1349 (SO), 1186 (C–O–C), 1113 (C–O–C) cm−1; δH (500 MHz, CDCl3) 1.89 (s, 3H, Me), 2.08 (s, 3H, Me) 3.65 (s, 3H, OMe), 7.40–7.46 (m, 3H, ArCH), 7.62–7.69 (m, 3H, ArCH), 7.72 (m, 1H, ArCH), 7.86–8.08 (m, 6H, ArCH, and CH), 8.53 (s, 1H, ArCH), 12.84 (s, 1H, OH); δC (125 MHz, CDCl3) 10.4, 10.6, 62.7, 114.0, 117.4, 117.9, 122.9, 126.2, 128.1, 128.2, 128.8, 129.2, 129.7, 129.9, 130.0, 130.8, 132.1, 134.0, 135.1, 135.6, 144.5, 152.6, 158.5, 160.9, 194.5; HRMS (ESI) calcd for C28H24O6S (M)+: m/z 488.1294, found 488.1291.O), 1604 (CC), 1382 (SO), 1185 (C–O–C), 1114 (C–O–C) cm−1; δH (500 MHz, CDCl3) 1.98 (s, 3H, Me), 2.18 (s, 3H, Me) 3.60 (s, 3H, OMe), 6.57 (d, J = 16.0 Hz, 1H, CH), 7.08 (d, J = 16.0 Hz, 1H, CH), 7.16–7.41 (m, 5H, ArCH), 7.46–7.63 (m, 2H, ArCH), 7.63–7.92 (m, 6H, ArCH), 7.93–8.10 (m, 4H, ArCH), 8.32 (s, 1H, ArCH), 8.55 (s, 1H, ArCH); δC (125 MHz, CDCl3) 11.5, 12.5, 62.5, 122.8, 122.9, 125.2, 126.5, 127.3, 127.8, 128.0, 128.1, 128.2, 128.5, 128.6, 128.9, 129.5, 129.6, 129.7, 129.8, 129.9, 130.0, 130.2, 130.4, 130.8, 131.8, 132.1, 133.2, 133.7, 134.2, 135.5, 135.7, 143.6, 144.8, 149.3, 155.1, 190.5; HRMS (ESI) calcd for C38H30O8S2 (M)+: m/z 678.7700, found 678.7701.:85 as the eluent to obtain compound 4a in 92% yield (0.0239 g).O), 1719 (CO), 1597 (CC), 1118 (C–O–C) cm−1; δH (500 MHz, CDCl3) 1.08 (t, J = 7.4 Hz, 3H, Me), 1.22 (t, J = 7.4 Hz, 3H, Me), 1.80–1.89 (m, 2H, CH2), 2.10 (s, 3H, Me), 2.22 (s, 3H, Me), 2.63 (t, J = 7.4 Hz, 2H, CH2), 2.76 (q, J = 7.4 Hz, 2H, CH2), 3.47 (s, 3H, OMe), 6.64 (d, J = 16.5 Hz, 1H, CH), 7.30–7.44 (m, 4H, ArCH and CH), 7.50–7.56 (m, 2H, ArCH); δC (125 MHz, CDCl3) 9.4, 9.9, 13.9, 14.3, 18.7, 22.1, 35.9, 61.5, 113.0, 115.1, 120.7, 126.1, 126.7, 127.6, 128.4, 129.1, 131.5, 136.7, 146.4, 149.6, 150.3, 154.1, 161.5, 171.1; HRMS (ESI) calcd for C26H28O5 (M)+: m/z 420.1937, found 420.1940.O), 1721 (CO), 1597 (CC), 1117 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.87–0.98 (m, 6H, Me), 1.33–1.48 (m, 6H, CH2), 1.54–1.65 (m, 2H, CH2), 1.77–1.86 (m, 2H, CH2), 2.10 (s, 3H, Me), 2.21 (s, 3H, Me), 2.64 (t, J = 7.6 Hz, 2H, CH2), 2.70–2.76 (m, 2H, CH2), 3.46 (s, 3H, OMe), 6.63 (d, J = 16.5 Hz, 1H, CH), 7.30–7.44 (m, 4H, ArCH and CH), 7.50–7.57 (m, 2H, ArCH); δC (125 MHz, CDCl3) 9.4, 9.9, 14.0, 14.1, 22.4, 23.0, 24.8, 28.3, 31.5, 31.9, 34.1, 61.5, 113.0, 115.1, 120.7, 126.2, 126.5, 126.7, 128.3, 129.1, 131.6, 136.8, 146.4, 149.6, 150.3, 154.1, 161.6, 171.3; HRMS (ESI) calcd for C30H36O5 (M)+: m/z 476.2563, found 476.2565.O), 1712 (CO), 1595 (CC), 1120 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.77–0.93 (m, 6H, Me), 1.17–1.48 (m, 14H, CH2), 1.53–1.67 (m, 2H, CH2), 1.77–1.85 (m, 2H, CH2), 2.10 (s, 3H, Me), 2.22 (s, 3H, Me), 2.65 (t, J = 7.6 Hz, 2H, CH2), 2.69–2.75 (m, 2H, CH2), 3.46 (s, 3H, OMe), 6.62 (d, J = 16.5 Hz, 1H, CH), 7.30–7.44 (m, 4H, ArCH and CH), 7.50–7.57 (m, 2H, ArCH); δC (125 MHz, CDCl3) 9.5, 9.9, 14.2, 22.7, 22.8, 25.1, 28.5, 29.0, 29.3, 29.6, 29.7, 29.8, 31.7, 31.8, 34.1, 61.5, 113.0, 115.1, 120.7, 126.2, 126.5, 126.6, 128.3, 129.0, 131.4, 136.7, 146.5, 149.5, 150.2, 154.0, 161.7, 171.4; HRMS (ESI) calcd for C34H44O5 (M)+: m/z 532.3189, found 532.3185.O), 1712 (CO), 1595 (CC), 1121 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.82–0.92 (m, 6H, Me), 1.13–1.48 (m, 22H, CH2), 1.55–1.64 (m, 2H, CH2), 1.76–1.85 (m, 2H, CH2), 2.10 (s, 3H, Me), 2.21 (s, 3H, Me), 2.64 (t, J = 7.6 Hz, 2H, CH2), 2.69–2.75 (m, 2H, CH2), 3.46 (s, 3H, OMe), 6.62 (d, J = 16.5 Hz, 1H, CH), 7.31–7.43 (m, 4H, ArCH and CH), 7.51–7.55 (m, 2H, ArCH); δC (125 MHz, CDCl3) 9.4, 9.9, 14.2, 22.7, 22.8, 25.2, 28.5, 29.3, 29.4, 29.5, 29.6, 29.7, 29.8, 32.0, 34.1, 61.5, 113.0, 115.1, 120.7, 126.2, 126.6, 126.7, 128.3, 129.0, 131.6, 136.8, 146.4, 149.5, 150.3, 154.0, 161.7, 171.3; HRMS (ESI) calcd for C38H52O5 (M)+: m/z 588.3815, found 588.3810.O), 1712 (CO), 1595 (CC), 1120 (C–O–C) cm−1; δH (500 MHz, CDCl3) 0.84–0.92 (m, 6H, Me), 1.17–1.50 (m, 30H, CH2), 1.54–1.66 (m, 2H, CH2), 1.76–1.86 (m, 2H, CH2), 2.10 (s, 3H, Me), 2.21 (s, 3H, Me), 2.64 (t, J = 7.6 Hz, 2H, CH2), 2.68–2.75 (m, 2H, CH2), 3.46 (s, 3H, OMe), 6.62 (d, J = 16.5 Hz, 1H, CH), 7.29–7.43 (m, 4H, ArCH and CH), 7.51–7.55 (m, 2H, ArCH); δC (125 MHz, CDCl3) 9.4, 9.9, 14.3, 22.8, 25.2, 28.5, 29.3, 29.4, 29.5, 29.6, 29.6, 29.7, 29.8, 29.9, 32.1, 34.1, 61.5, 113.0, 115.1, 120.7, 126.2, 126.6, 126.7, 128.3, 129.1, 131.6, 136.8, 146.4, 149.6, 150.3, 154.1, 161.6, 171.3; HRMS (ESI) calcd for C42H60O5 (M)+: m/z 644.4441, found 644.4438.Geometry optimisation
The Gaussian 16 package was used to carry out the calculations. 3D structures and conformer distributions were established using Avogadro software46 using the Molecular Mechanics Force Field (MMFF). The lowest energy conformer was further performed by geometrical optimisation running on the Gaussian16 package47,48 using DFT/B3LYP as the function and 6-311++G (d,p) as the basis set for the calculation. Thermal free energy was employed to determine the relative free energy between conformers.In silico molecular docking study
Molecular docking was performed to predict the binding poses of the DMC derivatives on cyclin-dependent kinase 2 (CDK2) which plays an important role in cell division.49 AutoDock Vina™ was utilised in all docking experiments.50 All the calculations were performed on an Intel Xeon Silver 4114 CPU@2.20 GHz of HP Z8 G4 workstation, with 32 GB DDR4 Ram compiled under the Ubuntu18.04 operating system. The study was performed using human cyclin-dependent kinase 2 (CDK2) in complex with an inhibitor (PDB ID 4GCJ), which is available from the protein database http://www.rcsb.org.51 3D structures of the ligands were established using the Avogadro software and the energy further minimised by theoretical B3LYP/6-31G density functional theory (DFT) using the Gaussian16 package.47,48 The protein was prepared via BIOVIA discovery studio 2016 software52 using the protein preparation tool. All water molecules were removed, and standard protonation states were verified using the ProPka server.53,54 A co-crystallised inhibitor was used to determine the active site for docking. The docking protocol was validated by the redocking of the co-crystallised ligand to the protein active site obtaining a binding energy value of −9.7 kcal mol−1 and an RMSD value of 0.976 Å. The grid size was set to 30 × 30 × 30xyz points. The grid centre was designated at dimensions (x, y and z): −54.5170, 98.5972 and −59.1310. The best score of each ligand was chosen and the protein–ligand interactions visualised in 2D and 3D plots.Evaluation of anticancer activity
Anticancer activity was evaluated as the cytotoxicity of carcinoma cell lines. Slightly modified sulforhodamine B (SRB) was used as a standard method55 with an ellipticine as the positive control drug. The semi-synthetic DMC analogues were dissolved in 10% DMSO solution with concentration range 0.1–50 μM. Cell lines were grown at 104 cell per well and seeded onto a 96-well plate as follows: KKU-M213, FaDu, HT-29, MDA-MB-231, A-549, SH-SY5Y, and CL. To the cultures were added 10 μL of cytotoxic substance solution in various concentrations and then they were incubated at 37 °C in a humidified incubator with 5% CO2 atmosphere for 72 h. The cultures were fixed by 10% trichloroacetic acid (TCA) and washed with slow-running tap water followed by staining with 100 μL of 0.4% sulforhodamine B solution. The unbound dye was removed by washing with 1% acetic acid solution. Cellular bound dye was extracted and solubilised with 10 mM Trizma base solution (pH 10.5). The absorbance was measured at 510 nm using a Fluostar optima BMG plate reader. The cytotoxicity was expressed as IC50 (the concentration which indicated 50% cell-growth inhibition) in the micromolar unit of concentration. The selectivity index (SI) was calculated by the ratio of IC50 on a normal cell divided by that on a cancer cell.Antibacterial activity evaluation
The agar well diffusion method was used to investigate the antimicrobial activity of synthetic compounds, as described by L. M. Cintas et al.56 with a slight modification. Bacterial strains including E. coli (ATCC® 25922™), S. aureus (ATCC® 25923™), and P. aeruginosa (ATCC® 27853™) were grown on Trypticase soy agar (TSA) at 37 °C for 18 h and adjusted to achieve a turbidity comparable to 0.5 McFarland standard (1.5 × 108 CFU mL−1) with sterile 0.85% NaCl solution. Then, the tested strain was swabbed on the surface of Mueller–Hinton Agar (MHA) and wells were made using a sterile cork borer (6 mm in diameter). 20 μL of tested synthetic compounds which were previously dissolved in DMSO at 5 mg mL−1 starting concentration was added to the respective wells. The plates were placed in the refrigerator for 30 minutes to let the extracts diffuse well into the agar. Then, the plates were incubated at 37 °C for 16–20 h. Antimicrobial activity was expressed as a zone of inhibition (ZOI) in mm. DMSO at a concentration of 10% was employed as a negative control.Broth microdilution method
Broth microdilution method was achieved as described by M. Bolouiri et al.57 with some modifications. The inocula were prepared by adjusting the turbidity of the suspension to match the 0.5 McFarland standard and diluted to 1:100 for the broth microdilution procedure. Antibiotic samples were dissolved in DMSO at 5 mg mL−1 starting concentration. Dilutions ranging from 5 to 0.3125 mg mL−1 of the tested compounds were prepared in sterile 96-well microplates through a 2-fold serial dilution. To the well containing 1400 μL of Mueller–Hinton Broth (MHB) was added about 50 μL of tested compounds followed by 10 μL of bacterial strains to a final volume of 100 μL. The final inoculum was 5 × 104 CFU mL−1. DMSO was taken as a negative control. The inoculated plates were incubated in 37 °C for 16–20 hours. After incubation, 10 μL of resazurin was added to each well and further incubated at 37 °C for 30 minutes. The MIC is the lowest concentration of the compound at which the microorganism tested does not demonstrate visible growth. The MBC values were determined by sub-culturing 10 μL of the test dilutions onto a fresh MHA and incubated for a further 18–24 h. The highest dilution that yielded no bacterial growth was taken as the MBC. All experiments were done in triplicate.
Conclusions
In summary, DMC was isolated from a crude EtOAc extract of S. nervosum seeds. The DMC was further modified by acylation, alkylation, and sulfonylation accessing 4′-O-monosubstituted-DMCs (2a–2n), 7-O-acylated-4-hydroxycoumarins (3a–3e), stilbene–coumarins (4a–4e), 2′,4′-O-disubstituted-DMCs (5f–5n), and flavanone derivatives (6g and 6m). These compounds were tested for cytotoxicity and antimicrobial activity against six cancer cell lines and three bacterial strains. Most semi-synthetic DMC derivatives showed higher cytotoxicity and selectivity to cancer cell lines than DMC. 4′-O-Caproylated-DMC (2b) and 4′-O-methylated-DMC (2g) exhibited the strongest cytotoxicity against SH-SY5Y with IC50 values of 5.20 and 7.52 μM, respectively. 4′-O-Benzylated-DMC (2h) displayed the strongest cytotoxicity against A549 and FaDu cell lines with IC50 values of 9.99 and 13.98 μM, respectively. In silico molecular docking revealed the poses of DMC derivatives on cyclin-dependent kinase 2 (PDB ID 4GCJ). The results suggested that 4′-O-methylated-DMC (2g) and 4′-O-benzylated-DMC (2h) displayed lower binding energy than DMC, especially compound 2h, which indicated key interactions with amino acid residues of CDK2. Most derivatised DMC, on the other hand, lacked antimicrobial activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project was supported by the National Research Council of Thailand (NRCT) and TSRI. In addition, we would also like to thank the TA/RA Scholarship of Graduate School, the Center of Excellence for Innovation in Chemistry (PERCH-CIC), the Department of Chemistry, Faculty of Science, Chiang Mai University and the Graduate School, Chiang Mai University, Center of Excellence in Materials Science and Technology, Faculty of Science, Chiang Mai University for financial support.Notes and references
- A. L. Harvey, Drug Discovery Today, 2008, 13, 894–901 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- A. Bauer and M. Brönstrup, Nat. Prod. Rep., 2014, 31, 35–60 RSC.
- H. Yao, J. Liu, S. Xu, Z. Zhu and J. Xu, Expert Opin. Drug Discovery, 2017, 12, 121–140 CrossRef CAS PubMed.
- K. S. Parvathy, P. S. Negi and P. Srinivas, Food Chem., 2010, 120, 523–530 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- A. P. Subramanian, A. A. John, M. V. Vellayappan, A. Balaji, S. K. Jaganathan, E. Supriyanto and M. Yusof, RSC Adv., 2015, 5, 35608–35621 RSC.
- L. Falzone, S. Salomone and M. Libra, Front. Pharmacol., 2018, 9, 1300 CrossRef CAS PubMed.
- P. A. Cohen, A. Jhingran, A. Oaknin and L. Denny, Lancet, 2019, 393, 169–182 CrossRef.
- H. K. Wang, IDrugs, 1998, 1, 92–102 CAS.
- N. T. Dung, J. M. Kim and S. C. Kang, Food Chem. Toxicol., 2008, 46, 3632–3639 CrossRef CAS PubMed.
- C. Wang, P. Wu, S. Tian, J. Xue, L. Xu, H. Li and X. Wei, J. Nat. Prod., 2016, 79, 2912–2923 CrossRef CAS PubMed.
- P. M. Giang, V. T. Phuong and T. T. Chinh, Nat. Prod. Commun., 2016, 11, 29–30 CrossRef PubMed.
- B.-S. Min, C. V. Thu, N. T. Dat, N. H. Dang, H.-S. Jang and T. M. Hung, Chem. Pharm. Bull., 2008, 56, 1725–1728 CrossRef CAS PubMed.
- B.-S. Min, T. D. Cuong, J.-S. Lee, M.-H. Woo and T. M. Hung, Bull. Korean Chem. Soc., 2010, 31, 2392–2394 CrossRef CAS.
- B. S. Min, T. D. Cuong, J.-S. Lee, B.-S. Shin, M. H. Woo and T. M. Hung, Arch. Pharmacal Res., 2010, 33, 1665–1670 CrossRef CAS PubMed.
- C. L. Ye, Y. H. Lu, X. D. Li, D. Z. Wei, C. S. Whitehead and S. Afr, J. Bot., 2005, 71, 312–315 CAS.
- S. Charoensin, S. Taya, S. Wongpornchai and R. Wongpoomchai, Interdiscip. Toxicol., 2012, 5, 201–206 Search PubMed.
- J.-C. Su, S. Wang, W. Cheng, X.-J. Huang, M.-M. Li, R.-W. Jiang, Y.-L. Li, L. Wang, W.-C. Ye and Y. Wang, J. Org. Chem., 2018, 83, 8522–8532 CrossRef CAS PubMed.
- I. A. Ansari and M. S. Akhtar, in Natural Bio-active Compounds: Volume 2: Chemistry, Pharmacology and Health Care Practices, ed. M. K. Swamy and M. S. Akhtar, Springer Singapore, Singapore, 2019, pp. 53–80, DOI:10.1007/978-981-13-7205-6_3.
- D. M. Kopustinskiene, V. Jakstas, A. Savickas and J. Bernatoniene, Nutrients, 2020, 12, 457 CrossRef CAS PubMed.
- A. Rammohan, J. S. Reddy, G. Sravya, C. N. Rao and G. V. Zyryanov, Environ. Chem. Lett., 2020, 18, 433–458 CrossRef CAS.
- M. N. Gomes, E. N. Muratov, M. Pereira, J. C. Peixoto, L. P. Rosseto, P. V. L. Cravo, C. H. Andrade and B. J. Neves, Molecules, 2017, 22, 1210 CrossRef PubMed.
- Y.-L. Tang, X. Zheng, Y. Qi, X.-J. Pu, B. Liu, X. Zhang, X.-S. Li, W.-L. Xiao, C.-P. Wan and Z.-W. Mao, Bioorg. Chem., 2020, 98, 103748 CrossRef CAS PubMed.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS PubMed.
- P. Takac, M. Kello, M. B. Pilatova, Z. Kudlickova, M. Vilkova, P. Slepcikova, P. Petik and J. Mojzis, Chem.-Biol. Interact., 2018, 292, 37–49 CrossRef CAS PubMed.
- Y.-T. Wang, Y.-J. Qin, Y.-L. Zhang, Y.-J. Li, B. Rao, Y.-Q. Zhang, M.-R. Yang, A.-Q. Jiang, J.-L. Qi and H.-L. Zhu, RSC Adv., 2014, 4, 32263–32275 RSC.
- M. J. Mphahlele, M. M. Maluleka, N. Parbhoo and S. T. Malindisa, Int. J. Mol. Sci., 2018, 19, 2012 CrossRef PubMed.
- H. Zhu, L. Tang, C. Zhang, B. Wei, P. Yang, D. He, L. Zheng and Y. Zhang, Front. Pharmacol., 2019, 10, 1341 CrossRef CAS PubMed.
- V. R. Yadav, S. Prasad, B. Sung and B. B. Aggarwal, Int. Immunopharmacol., 2011, 11, 295–309 CrossRef CAS PubMed.
- C. Karthikeyan, N. S. Moorthy, S. Ramasamy, U. Vanam, E. Manivannan, D. Karunagaran and P. Trivedi, Recent Pat. Anti-Cancer Drug Discovery, 2015, 10, 97–115 CrossRef CAS PubMed.
- B. Kumar, S. Singh, I. Skvortsova and V. Kumar, Curr. Med. Chem., 2017, 24, 4729–4752 CAS.
- S. Gafner, J. L. Wolfender, S. Mavi and K. Hostettmann, Planta Med., 1996, 62, 67–69 CrossRef CAS PubMed.
- J. W. Choi, M. Kim, H. Song, C. S. Lee, W. K. Oh, I. Mook-Jung, S. S. Chung and K. S. Park, Metabolism, 2016, 65, 533–542 CrossRef CAS PubMed.
- T. K. Q. Ha, T. T. Dao, N. H. Nguyen, J. Kim, E. Kim, T. O. Cho and W. K. Oh, Fitoterapia, 2016, 110, 135–141 CrossRef CAS PubMed.
- T.-T. Dao, B.-T. Tung, P.-H. Nguyen, P.-T. Thuong, S.-S. Yoo, E.-H. Kim, S.-K. Kim and W.-K. Oh, J. Nat. Prod., 2010, 73, 1636–1642 CrossRef CAS PubMed.
- T. J. Anuruk Chailungka, W. Pompimon, N. Nuntasaen and P. Meepowpan, Maejo Int. J. Sci. Technol., 2017, 11, 58–67 Search PubMed.
- C.-L. Ye, F. Qian, D.-Z. Wei, Y.-H. Lu and J.-W. Liu, Leuk. Res., 2005, 29, 887–892 CrossRef CAS PubMed.
- H. N. Tuan, B. H. Minh, P. T. Tran, J. H. Lee, H. V. Oanh, Q. M. T. Ngo, Y. N. Nguyen, P. T. K. Lien and M. H. Tran, Molecules, 2019, 24, 2538 CrossRef CAS PubMed.
- C.-L. Ye, J.-W. Liu, D.-Z. Wei, Y.-H. Lu and F. Qian, Pharmacol. Res., 2004, 50, 505–510 CrossRef CAS PubMed.
- X. Wei, X. Mo, F. An, X. Ji and Y. Lu, Food Chem. Toxicol., 2018, 119, 252–259 CrossRef CAS PubMed.
- F. Qian, C. L. Ye, D. Z. Wei, Y. H. Lu and S. L. Yang, J. Chemother., 2005, 17, 309–314 CrossRef CAS PubMed.
- F. Belluti, G. Fontana, L. Dal Bo, N. Carenini, C. Giommarelli and F. Zunino, Bioorg. Med. Chem., 2010, 18, 3543–3550 CrossRef CAS PubMed.
- S. S. Abd El-Karim, Y. M. Syam, A. M. El Kerdawy and T. M. Abdelghany, Bioorg. Chem., 2019, 86, 80–96 CrossRef CAS PubMed.
- H.-C. Tang and C. Y.-C. Chen, BioMed Res. Int., 2014, 2014, 798742 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6.1, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- C. Benson, S. Kaye, P. Workman, M. Garrett, M. Walton and J. de Bono, Br. J. Cancer, 2005, 92, 7–12 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- E. Schonbrunn, S. Betzi, R. Alam, M. P. Martin, A. Becker, H. Han, R. Francis, R. Chakrasali, S. Jakkaraj, A. Kazi, S. M. Sebti, C. L. Cubitt, A. W. Gebhard, L. A. Hazlehurst, J. S. Tash and G. I. Georg, J. Med. Chem., 2013, 56, 3768–3782 CrossRef CAS PubMed.
- BIOVIA, Dassault Systèmes, BIOVIA Discovery Studio Visualizer, version 21.1.0.20298, San Diego, Dassault Systèmes, 2020 Search PubMed.
- C. R. Søndergaard, M. H. M. Olsson, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef PubMed.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- L. M. Cintas, J. M. Rodriguez, M. F. Fernandez, K. Sletten, I. F. Nes, P. E. Hernandez and H. Holo, Appl. Environ. Microbiol., 1995, 61, 2643–2648 CAS.
- M. Balouiri, M. Sadiki and S. K. Ibnsouda, J. Pharm. Anal., 2016, 6, 71–79 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data of anticancer and antimicrobial activities, molecular docking, 1H- and 13C-NMR spectra of synthetic compounds. See DOI: 10.1039/d1ra05445g |
| This journal is © The Royal Society of Chemistry 2021 |