
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper(II)-catalyzed and acid-promoted highly regioselective oxidation of tautomerizable C(sp3)–H bonds adjacent to 3,4-dihydroisoquinolines using air (O2) as a clean oxidant†
Yun-Gang He,
Yong-Kang Huang,
Qi-Qi Fan,
Bo Zheng,
Yong-Qiang Luo,
Xing-Liang Zhu and
Xiao-Xin Shi *
*
Engineering Research Center of Pharmaceutical Process Chemistry of the Ministry of Education, School of Pharmacy, East China University of Science and Technology, Shanghai 200237, People's Republic of China. E-mail: xxshi@ecust.edu.cn
First published on 6th September 2021
Abstract
A mild, efficient and eco-friendly method for the oxidation of 1-Bn-DHIQs to 1-Bz-DHIQs without concomitant excessive oxidation of 1-Bz-DHIQs to 1-Bz-IQs is very important for the syntheses of 1-Bz-DHIQ alkaloids and analogues. In this article, we developed a novel Cu(II)-catalyzed and acid-promoted highly regioselective oxidation of tautomerizable C(sp3)–H bonds adjacent to the C-1 positions of various 1-Bn-DHIQs. It was observed that when 0.2 equiv. of Cu(OAc)2·2H2O was used as the catalyst, 3.0 equiv. of AcOH was used as the additive and air (O2) was used as a clean oxidant, various 1-Bn-DHIQs could be efficiently oxidized to corresponding 1-Bz-DHIQs at 25 °C in DMSO. Especially, almost no concomitant excessive oxidation of 1-Bz-DHIQs to 1-Bz-IQs was observed during the above reaction. In addition, this method was successfully applied in the first total synthesis of the alkaloid canelillinoxine.
Introduction
Oxidation of organic compounds is a kind of fundamental transformation in organic and medicinal chemistry. Development of novel efficient and highly selective oxidation of activated or non-activated C(sp3)–H bonds under mild reaction conditions is a challenging task for chemists due to its great importance, and it has become an exciting and promising research area in modern organic synthesis.1 Recently, significant efforts of chemists have been devoted to solving the issues of oxidation of C(sp3)–H bonds in a variety of substrates.2 Although many oxidants can be used in the above oxidation of C(sp3)–H bonds, air (O2) is certainly the best choice in the light of its abundance, easy availability, cheapness and eco-friendliness. On the other hand, copper is a low toxic and inexpensive transition metal. Therefore, an increasing amount of copper-catalyzed aerobic oxidations of various organic compounds have been recently developed.31-Benzoyl-3,4-dihydroisoquinolines (1-Bz-DHIQs) represent an important kind of isoquinoline alkaloids, which could be isolated from numerous plants.4 Since 1-Bz-DHIQ alkaloids and their synthetic analogues have exhibited various interesting biological activities,5 a mild, efficient and eco-friendly method for synthesis of 1-Bz-DHIQ alkaloids might be highly desirable for organic and medicinal chemists. 1-Bz-DHIQs usually can be synthesized via oxidation of 1-benzyl-3,4-dihydroisoquinolines (1-Bn-DHIQs), which could be readily prepared via the Bischler–Napieralski cyclization6 of corresponding amides. Some known methods for the oxidation of 1-Bn-DHIQs to 1-Bz-DHIQs have been reported.7–14 However, these known methods usually suffered from some serious drawbacks: (a) use of poisonous and hazardous strong oxidants such as CrO3,7 SeO2,8 MnO2,9 CAN [Ce(NH4)2(NO3)6],10 Pb(OAc)4,11 and singlet oxygen (1O2);8,12 (b) concomitant excessive oxidation of 1-Bz-DHIQs could not be avoided, significant amount of undesired 1-benzoyl-isoquinolines (1-Bz-IQs) were formed as by-products during the reaction;7–13 (c) yields of 1-Bz-DHIQs as the desired products are only moderate in most instances; (d) use of precious palladium catalyst (Pd/C) in Andreu's method.14 In order to overcome the above-mentioned drawbacks, development of a mild, efficient and eco-friendly method for the oxidation of 1-Bn-DHIQs to 1-Bz-DHIQs without concomitant excessive oxidation of 1-Bz-DHIQs to 1-Bz-IQs remains a challenge to organic chemists. Herein, we want to disclose a very mild and efficient copper(II)-catalyzed and acid-promoted highly regioselective oxidation of tautomerizable C(sp3)–H bonds adjacent to the C-1 positions of various 1-Bn-DHIQs using air (O2) as a clean oxidant.
Results and discussion
At first, we chose the oxidation of 1-Bn-DHIQ 1a in dimethyl sulfoxide (DMSO) as the model reaction, and tried this model reaction under various conditions, the results are summarized in Table 1. As can be seen from Table 1, the oxidation was sluggish in the absence of a catalyst (entry 1). The aerobic oxidation took place smoothly to give both of 1-Bz-DHIQ 2a and 1-Bz-IQ 3a as products in different ratios with a cupric salt as the catalyst (entries 2–22). Four cupric salts such as cupric chloride, cupric bromide, cupric sulfate and cupric acetate have been tested as the catalyst for the model reaction without additives (entries 2–8). Oxidation product 1-Bz-DHIQ 2a was formed as the major product, but undesired excessive oxidation product 1-Bz-IQ 3a was also formed in a significant amount.
|
||||
|---|---|---|---|---|
| En. | Catalyst (equiv.) | Additive (equiv.) | Time (h) | Yield %b (2a/3a) |
| a Reaction conditions: 1a (2 mmol), catalyst, additive, DMSO (4.0 mL), stirred at 25 °C under an air atmosphere.b Isolated yields.c 70% of 1a was recovered.d Trace amount of 1-Bz-IQ 3a (<0.5%) was detected.e DBU = 1,8-Diazabicyclo[5,4,0]undec-7-ene. | ||||
| 1 | None | None | 72 | 25/1c |
| 2 | CuCl2·2H2O (0.2) | None | 24 | 76/16 |
| 3 | CuBr2·2H2O (0.2) | None | 24 | 75/17 |
| 4 | CuSO4·2H2O (0.2) | None | 24 | 71/21 |
| 5 | Cu(OAc)2·2H2O (0.1) | None | 25 | 79/15 |
| 6 | Cu(OAc)2·2H2O (0.2) | None | 16 | 82/13 |
| 7 | Cu(OAc)2·2H2O (0.5) | None | 12 | 81/14 |
| 8 | Cu(OAc)2·2H2O (1.0) | None | 10 | 80/14 |
| 9 | Cu(OAc)2·2H2O (0.2) | HCl (1.0) | 15 | 83/1 |
| 10 | Cu(OAc)2·2H2O (0.2) | H2SO4 (1.0) | 18 | 82/1 |
| 11 | Cu(OAc)2·2H2O (0.2) | H3PO4 (1.0) | 14 | 86/1 |
| 12 | Cu(OAc)2·2H2O (0.2) | CF3CO2H (1.0) | 12 | 87/1 |
| 13 | Cu(OAc)2·2H2O (0.2) | AcOH (1.0) | 10 | 90/1 |
| 14 | Cu(OAc)2·2H2O (0.2) | AcOH (2.0) | 9 | 92/1 |
| 15 | Cu(OAc)2·2H2O (0.2) | AcOH (3.0) | 8 | 95/<0.5d |
| 16 | Cu(OAc)2·2H2O (0.2) | AcOH (4.0) | 8 | 93/<0.5d |
| 17 | Cu(OAc)2·2H2O (0.2) | AcOH (5.0) | 9 | 91/1 |
| 18 | Cu(OAc)2·2H2O (0.2) | DBUe (1.0) | 15 | <0.5/89 |
| 19 | Cu(OAc)2·2H2O (0.2) | Py (1.0) | 15 | 20/72 |
| 20 | Cu(OAc)2·2H2O (0.2) | Et3N (1.0) | 15 | 25/68 |
| 21 | Cu(OAc)2·2H2O (0.2) | K2CO3 (1.0) | 15 | 23/65 |
| 22 | Cu(OAc)2·2H2O (0.2) | Na2CO3 (1.0) | 15 | 23/64 |
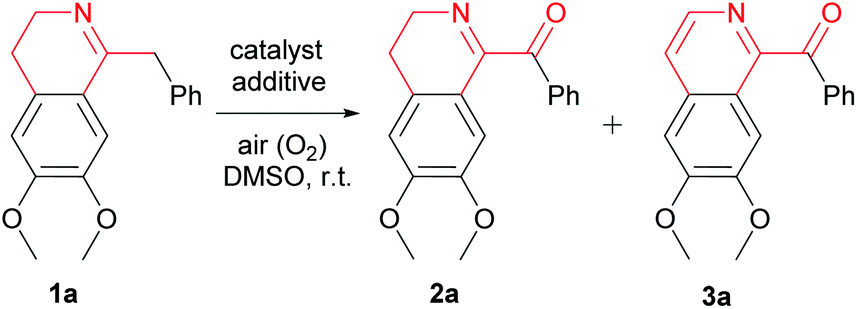
Subsequently, we tried effects of acid and base additives on the reaction, and found that acid and base additives dramatically changed the ratios of products 1-Bz-DHIQ 2a and 1-Bz-IQ 3a (entries 9–22). When acids were used as additives, 1-Bz-DHIQ 2a was formed as the major product (entries 9–17); when bases were used as additives, 1-Bz-IQ 3a was formed as the major product (entries 18–22).15 Especially, weak acid additive is better than strong acid (entry 13 versus entries 9–12), when 3.0 equiv. of acetic acid (AcOH) was used as the additive (entry 15), the desired product 1-Bz-DHIQ 2a was obtained in the best yield (95%), and only a trace amount (<0.5%) of undesired excessive oxidation product 1-Bz-IQ 3a was detected.
We have also tired the model reaction in different solvents, and found that the reaction is much faster in DMSO, and the yield of the desired product 2a is higher in DMSO than in several other solvents including N,N-dimethylforamide, acetonitrile, dichloromethane, chloroform, tetrahydrofuran, ethyl acetate, acetone, 1,4-dioxane, 1,2-dimethoxyethane, isopropanol and ethanol. Consequently, we concluded that the optimized reaction conditions for the above model reaction are as follows: DMSO is the solvent, cupric salt Cu(OAc)2·2H2O (0.2 equiv.) is the catalyst, acid AcOH (3.0 equiv.) is the additive, and the reaction were performed under an air (O2) atmosphere at room temperature (25 °C).
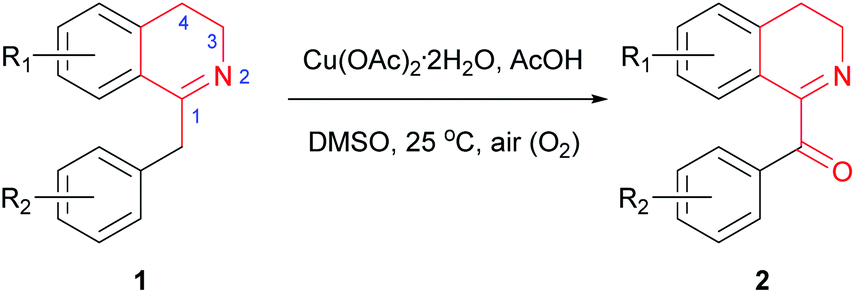
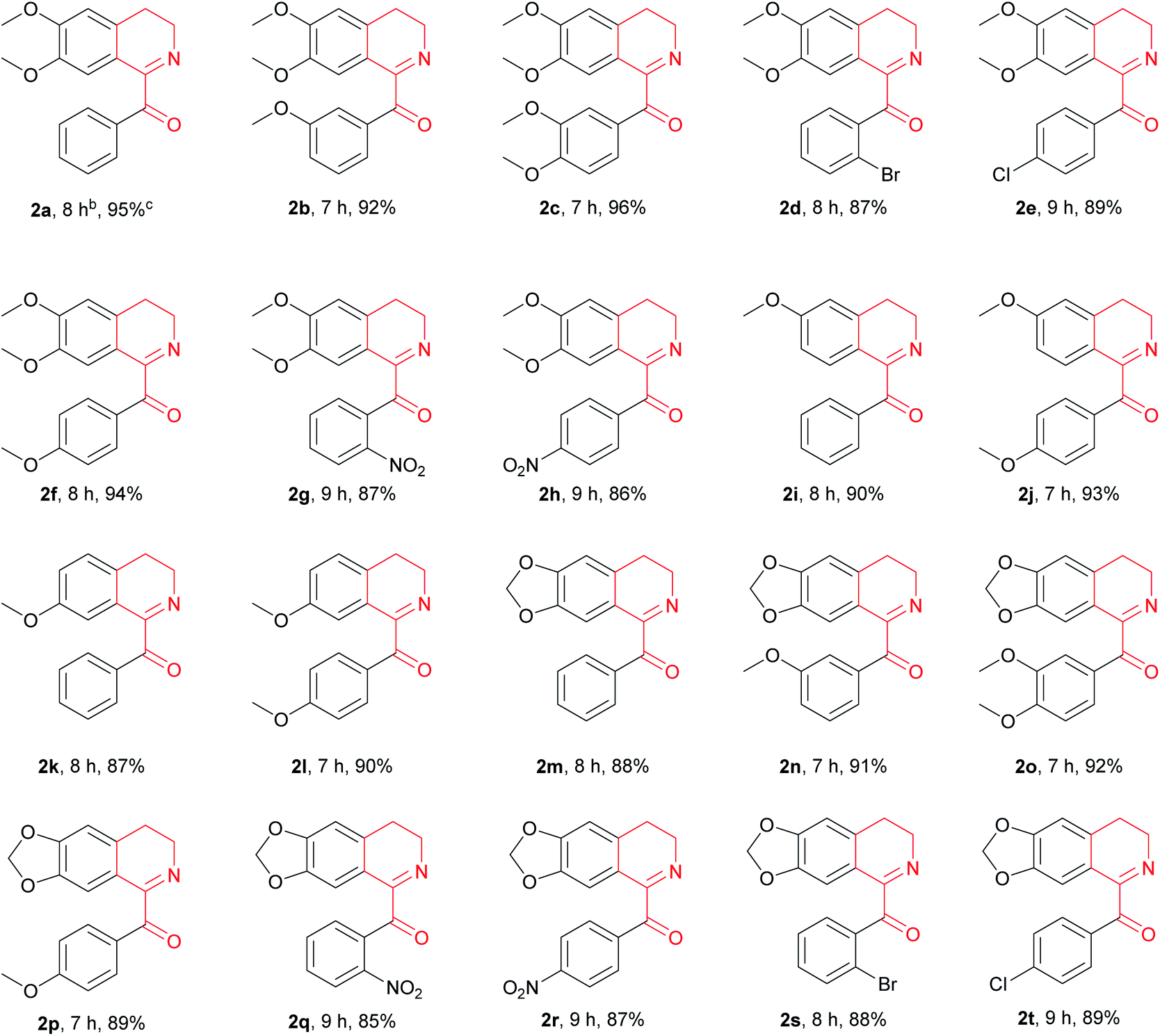
With the above optimized reaction conditions in hand, we then attempted Cu(OAc)2-catalyzed oxidation of variously substituted 1-Bn-DHIQs 1, which were prepared from corresponding amides according to the known method,15 under the above optimized conditions to prepare 1-Bz-DHIQs 2. As can be seen from Table 2, a total of twenty 1-Bn-DHIQs 1a–1t have been examined for the reaction, corresponding 1-BzDHIQs 2a–2t were thus obtained in high yields (85–96%). It is worth noting that the regioselectivity of oxidation of all tested substrates is extremely high, only tautomerizable C(sp3)–H bonds adjacent to the C-1 positions of 1-Bn-DHIQs were oxidized. Although C-4 is also a benzylic position, almost none of oxidation of C(sp3)–H bond at the C-4 position happened.
A possible mechanism for the above Cu(OAc)2-catalyzed and acid promoted aerobic oxidation of 1-Bn-DHIQs 1 was proposed in Scheme 1. 1-Bn-DHIQs 1 would first undergo acid-promoted tautomerization16 to form enamines 1′. Enamines 1′ would then coordinate with Cu(OAc)2 to produce Cu-complex I-A, which would further coordinate with molecule O2 (in air) to give more reactive Cu-complex I-B.17 Next, complex I-B would undergo intramolecular Jenkins-like oxidation18 to furnish an unstable perhydroxide compound I-C. Finally, decomposition19 of I-C would afford 1-Bz-DHIQs 2. Although acids can speed up tautomerization of imines 1 to enamines 1′, strong acids (HCl, H2SO4, etc.) would also react with imines 1 to form ammonium 1′-H+ reversibly, which could not coordinate with Cu(OAc)2 to form I-A, and that would retard the oxidation (Table 1, entries 9–12).
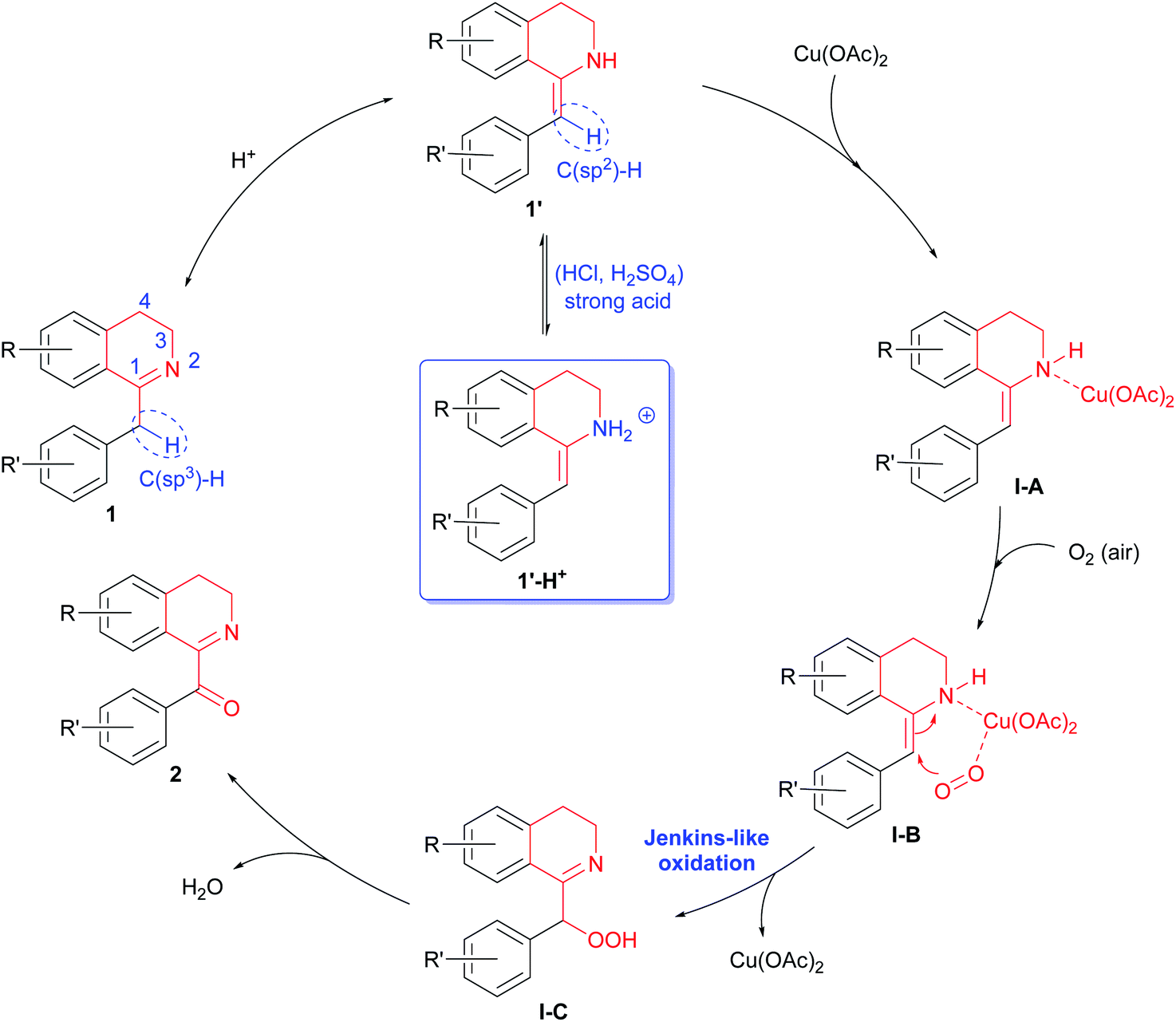 | ||
| Scheme 1 Possible mechanism for the Cu(OAc)2-catalyzed and acid-promoted aerobic oxidation of C(sp3)–H bonds adjacent to the C-1 positions of 1-Bn-DHIQs 1. | ||
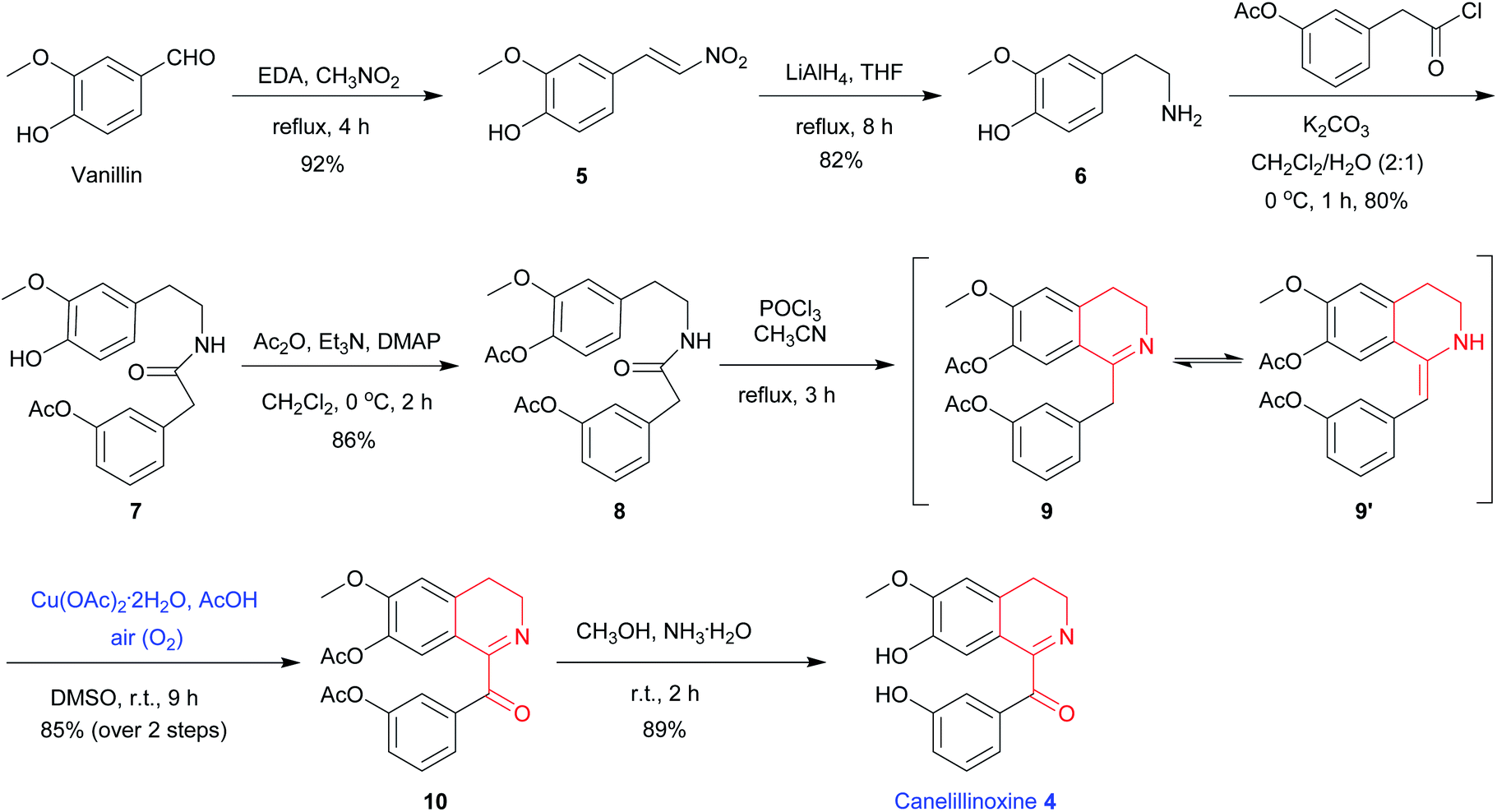
To demonstrate the utility of above-described methodology, we have applied the protocol to an efficient total synthesis of 1-Bz-DHIQ canelillinoxine 4. The alkaloid canelillinoxine 4 was isolated from the stem bark of Aniba canelilla H.B.K. (Lauraceae) in 1993.20 Herein we would like to report the first total synthesis of canelillinoxine 4 starting from vanillin by using the above mild Cu(OAc)2-catalyzed and acid-promoted oxidation of as the key step. As depicted in the Scheme 2, EDA-catalyzed condensation of vanillin with nitromethane produced nitroalkene 5 in 92% yield.21 Compound 5 was then treated with 5.0 equiv. of LiAlH4, simultaneous reduction of the double bond and nitro group took place to give amine 6 in 82% yield. When compound 6 was exposed to 1.1 equiv. of freshly prepared 2-(3-acetoxy-phenyl) acetyl chloride and 3.0 equiv. of K2CO3, amide 7 was obtained in 80% yield. Treatment of compound 7 with 3.0 equiv. of Ac2O, 3.0 equiv. of Et3N and 0.1 equiv. of N,N-dimethylamino-pyridine (DMAP) gave compound 8 in 86% yield. Next, exposure of amide 8 to 3.0 equiv. of POCl3, Bischler–Napieralski cyclization6 took place smoothly to produce the tautomeric mixture of imine 9 and enamine 9′, which were then treated with 0.2 equiv. of Cu(OAc)2·2H2O and 3.0 equiv. of AcOH in DMSO under an air atmosphere at 25 °C to furnish compound 10 in 85% yield (over 2 steps from 8). Finally, when compound 10 was treated with excessive NH3·H2O, protecting groups (two Ac groups) were removed to afford 1-Bz-DHIQ canelillinoxine 4 in 89% yield.
 | ||
| Scheme 2 The first total synthesis of canelillinoxine 4 from vanillin. | ||
Conclusions
In conclusion, we have studied the Cu(OAc)2-catalyzed and acid-promoted highly regioselective oxidation of tautomerizable C(sp3)–H bonds adjacent to C-1 positions of 1-Bn-DHIQs, and found that Cu(OAc)2·2H2O is the most effective catalyst, DMSO is the best solvent, weak acid AcOH is the best additive, and air (O2) is the suitable oxidant. The reaction is highly controlled by acid/base additives, acid additives led to formation of 1-Bz-DHIQ as the major product, while base additives led to formation of 1-Bz-IQ as the major product. The reaction is applicable for a variety of substituted 1-Bn-DHIQs to afford 1-Bz-DHIQs in high yields. The present method is superior to the known methods7–14 due to some advantages such as mildness, high regioselectivity, high yields, good practicability and eco-friendliness. In addition, the methodology was successfully applied to the first total synthesis of 1-Bz-DHIQ alkaloid canelillinoxine, it was synthesized from vanillin via 7 steps in 39% overall yield.Experimental
General methods
1H and 13C NMR spectra were acquired on a Bruker AM-400 magnetic resonance instrument. Chemical shifts were given on the delta scale as parts per million (ppm) with tetramethylsilane (TMS) as the internal standard. IR spectra were recorded on a Nicolet Magna IR-550 spectrometer. MS spectra were recorded on a Mariner mass spectrum equipment. Melting points were determined on a Mei-TEMP II apparatus. Column chromatography was performed on silica gel (Qingdao Chemical Factory). All chemicals are analytically pure, and were used as such as received from the chemical suppliers.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the solution was concentrated under vacuum to dryness, the residue was dissolved in CH2Cl2 (50 mL). An aqueous solution of K2CO3 (20 mL, 15% w/w) was added. After the mixture was vigorously stirred for 5 min, two phases were separated, and the aqueous phase was extracted twice with CH2Cl2 (2 × 25 mL). Organic extracts were combined, dried over anhydrous MgSO4, and then concentrated under vacuum to give crude product 1-Bn-DHIQ 1 in an almost quantitative yield, which was used as such for the next step.:1), a dilute ammonia aqueous solution (5% w/w, 20 mL) and EtOAc (20 mL) were added into the mixture. After the mixture was vigorously stirred for 5 min, two phases were separated. The aqueous phase was twice extracted with EtOAc (20 mL × 2). The organic extracts were combined, and dried over anhydrous MgSO4. Removal of solvent by vacuum distillation gave crude product, which was purified by flash chromatography (eluent: CH2Cl2/EtOAc = 20:1 to 5:1) to afford 1-Bz-DHIQ 2 in 85–96% yields. Characterization data of compounds 2a–t are as follows:
:1), the solution was concentrated under vacuum to dryness, the residue was dissolved in CH2Cl2 (50 mL). An aqueous solution of K2CO3 (20 mL, 15% w/w) was added. After the mixture was vigorously stirred for 5 min, two phases were separated, and the aqueous phase was extracted twice with CH2Cl2 (2 × 25 mL). Organic extracts were combined, dried over anhydrous MgSO4, and then concentrated under vacuum to give crude product 1-Bn-DHIQ 1 in an almost quantitative yield, which was used as such for the next step.:1), a dilute ammonia aqueous solution (5% w/w, 20 mL) and EtOAc (20 mL) were added into the mixture. After the mixture was vigorously stirred for 5 min, two phases were separated. The aqueous phase was twice extracted with EtOAc (20 mL × 2). The organic extracts were combined, and dried over anhydrous MgSO4. Removal of solvent by vacuum distillation gave crude product, which was purified by flash chromatography (eluent: CH2Cl2/EtOAc = 20:1 to 5:1) to afford 1-Bz-DHIQ 2 in 85–96% yields. Characterization data of compounds 2a–t are as follows:(6,7-Dimethoxy-3,4-dihydroisoquinolin-1-yl)(phenyl)methanone 2a: pale yellow crystals, m.p. 78–79 °C (lit.22 m.p. 78.8–79.4 °C). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 7.3 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.47 (dd, J1 = 7.3 Hz, J2 = 7.4 Hz, 2H), 6.95 (s, 1H), 6.76 (s, 1H), 3.99–3.87 (m, 2H), 3.92 (s, 3H), 3.78 (s, 3H), 2.88–2.77 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 193.97, 164.39, 151.72, 147.62, 135.57, 133.85, 131.17, 130.43, 128.54, 119.30, 110.57, 109.59, 56.07, 56.02, 47.31, 25.37. IR (KBr film) ν 3080, 2936, 2907, 2844, 1656, 1586, 1516, 1485, 1265, 1239, 1146, 1024, 929, 865, 752 cm−1.
(6,7-Dimethoxy-3,4-dihydroisoquinolin-1-yl)(3-methoxyphenyl)-methanone 2b: pale yellow crystals, m.p. 98–100 °C. 1H NMR (400 MHz, CDCl3) δ 7.59 (s, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.37 (dd, J1 = 7.8 Hz, J2 = 7.9 Hz, 1H), 7.15 (d, J = 7.9 Hz, 1H), 6.93 (s, 1H), 6.76 (s, 1H), 3.98–3.89 (m, 2H), 3.94 (s, 3H), 3.86 (s, 3H), 3.79 (s, 3H), 2.88–2.77 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 193.82, 164.54, 159.73, 151.77, 147.66, 136.85, 131.15, 129.58, 123.63, 120.63, 119.30, 113.96, 110.54, 109.58, 56.11, 56.05, 55.48, 47.21, 25.40. IR (KBr film) ν 3070, 2921, 2852, 1666, 1596, 1592, 1569, 1515, 1461, 1425, 1135, 1072, 1033, 935, 807, 754, 709 cm−1. HRMS (ESI) m/z calcd for C19H19NO4Na [M + Na]+: 348.1212, found: 348.1211.
(6,7-Dimethoxy-3,4-dihydroisoquinolin-1-yl)(3,4-dimethoxy-phenyl)methanone 2c: pale yellow crystals, m.p. 188–189 °C (lit.23 m.p. 188–190 °C). 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.61 (d, J = 8.4 Hz, 1H), 6.91 (s, 1H), 6.89 (d, J = 8.4 Hz, 1H), 6.76 (s, 1H), 3.96 (s, 3H), 3.95–3.88 (m, 2H), 3.95 (s, 3H), 3.94 (s, 3H), 3.79 (s, 3H), 2.89–2.76 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.76, 164.63, 154.16, 151.67, 149.16, 147.64, 131.08, 128.59, 126.61, 119.49, 111.21, 110.50, 110.02, 109.61, 56.16, 56.11, 56.04, 56.02, 47.25, 25.43. IR (KBr film) ν 3013, 2935, 2833, 2604, 1659, 1583, 1514, 1461, 1278, 1133, 1023, 866, 791, 755 cm−1.
(2-Bromophenyl)(6,7-dimethoxy-3,4-dihydroisoquinolin-1-yl)methanone 2d: pale yellow crystals, m.p. 159–161 °C. 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 7.8 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H),7.44 (dd, J1 = 7.8 Hz, J1 = 7.7 Hz, 1H), 7.36 (dd, J1 = 7.7 Hz, J2 = 7.7 Hz, 1H), 7.32 (s, 1H), 6.74 (s, 1H), 3.94 (s, 3H), 3.89 (s, 3H), 3.88–3.80 (m, 2H), 2.83–2.62 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 195.75, 163.67, 151.61, 147.54, 140.23, 133.14, 132.51, 131.68, 131.11, 127.51, 120.75, 119.16, 110.32, 110.28, 56.15, 56.02, 48.19, 25.12. IR (KBr film) ν 3054, 2942, 2899, 2844, 1681, 1587, 1499, 1478, 1460, 1381, 1315, 1266, 1032, 924, 868 cm−1. HRMS (ESI) m/z calcd for C18H16NO3Na [M + Na]+: 396.0211, found: 396.0215.
(4-Chlorophenyl)(6,7-dimethoxy-3,4-dihydroisoquinolin-1-yl)methanone 2e: white crystals, m.p. 132–133 °C (lit.24 m.p. 130–131 °C). 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.6 Hz, 2H), 7.45 (d, J = 8.6 Hz, 2H), 6.96 (s, 1H), 6.75 (s, 1H), 3.97–3.89 (m, 2H), 3.94 (s, 3H), 3.80 (s, 3H), 2.86–2.76 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.56, 163.93, 151.83, 147.69, 140.35, 134.01, 131.87, 131.22, 128.85, 119.17, 110.55, 109.61, 56.12, 56.06, 47.38, 25.38.
(6,7-Dimethoxy-3,4-dihydroisoquinolin-1-yl)(4-methoxyphenyl)-methanone 2f: pale yellow crystals, m.p. 92–93 °C (lit.25 m.p. 91–92 °C). 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.9 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 6.92 (s, 1H), 6.75 (s, 1H), 3.96–3.89 (m, 2H), 3.94 (s, 3H), 3.88 (s, 3H), 3.78 (s, 3H), 2.87–2.77 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.73, 164.67, 164.26, 151.61, 147.62, 132.91, 131.10, 128.50, 119.48, 113.87, 110.47, 109.61, 56.12, 55.56, 47.27, 29.72, 25.45.
(6,7-Dimethoxy-3,4-dihydroisoquinolin-1-yl)(2-nitrophenyl)-methanone 2g: pale yellow crystals, m.p. 98–100 °C. 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 7.8 Hz, 1H), 7.78 (dd, J1 = 7.8 Hz, J2 = 7.9 Hz, 1H), 7.71–7.61 (m, 3H), 6.70 (s, 1H), 3.97 (s, 3H), 3.94 (s, 3H), 3.74–3.62 (m, 2H), 2.70–2.56 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.66, 163.10, 151.69, 147.81, 147.56, 136.08, 134.17, 131.81, 131.04, 130.19, 123.57, 119.03, 110.97, 109.92, 56.17, 56.00, 47.84, 25.20. IR (KBr film) ν 3072, 2964, 2919, 2851, 1697, 1600, 1562, 1529, 1512, 1468, 1405, 1346, 1280, 1197, 1147, 1047, 906, 877, 800, 750, 707 cm−1. HRMS (ESI) m/z calcd for C18H17N2O5 [M + H]+: 341.1137, found: 341.1135.
(6,7-Dimethoxy-3,4-dihydroisoquinolin-1-yl)(4-nitrophenyl)-methanone 2h: white crystals, m.p. 160–161 °C. 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 8.8 Hz, 2H), 8.20 (d, J = 8.8 Hz, 2H), 7.07 (s, 1H), 6.77 (s, 1H), 4.00–3.92 (m, 2H), 3.95 (s, 3H), 3.84 (s, 3H), 2.89–2.76 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 191.71, 163.27, 152.06, 150.42, 147.75, 140.75, 131.53, 131.43, 123.49, 118.87, 110.58, 109.68, 56.16, 56.12, 47.57, 25.32. IR (KBr film) ν 3003, 2924, 2853, 1674, 1637, 1606, 1564, 1519, 1458, 1347, 1200, 1043, 906, 859, 800, 781 cm−1. HRMS (ESI) m/z calcd for C18H17N2O5 [M + H]+: 341.1137, found: 341.1143.
(6-Methoxy-3,4-dihydroisoquinolin-1-yl)(phenyl)methanone 2i: pale yellow crystals, m.p. 89–90 °C (lit.26 m.p. 88–90 °C). 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 7.4 Hz, 2H), 7.58 (t, J = 7.5 Hz, 1H), 7.45 (dd, J1 = 7.4 Hz, J2 = 7.5 Hz, 2H), 7.31 (d, J = 8.5 Hz, 1H), 6.76 (s, 1H), 6.73 (d, J = 8.5 Hz, 1H), 3.99–3.88 (m, 2H), 3.81 (s, 3H), 2.91–2.79 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 194.09, 164.86, 162.02, 139.57, 135.56, 133.86, 130.37, 128.56, 128.50, 120.16, 113.47, 112.13, 55.42, 47.13, 26.19.
(6-Methoxy-3,4-dihydroisoquinolin-1-yl)(4-methoxyphenyl)-methanone 2j: white crystals, m.p. 156–157 °C. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 9.0 Hz, 2H), 7.29 (d, J = 8.5 Hz, 1H), 6.94 (d, J = 9.0 Hz, 2H), 6.76 (s, 1H), 6.72 (d, J = 8.5 Hz, 1H), 3.97–3.87 (m, 2H), 3.85 (s, 3H), 3.82 (s, 3H), 2.90–2.80 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.81, 165.11, 164.24, 161.93, 139.53, 132.77, 128.53, 128.50, 120.28, 113.87, 113.40, 112.09, 55.56, 55.38, 47.05, 26.20. HRMS (ESI) m/z calcd for C18H18NO3 [M + H]+: 296.1287, found: 296.1292.
(7-Methoxy-3,4-dihydroisoquinolin-1-yl)(phenyl)methanone 2k: pale yellow crystals, m.p. 130–131 °C. 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 7.7 Hz, 2H), 7.60 (t, J = 7.8 Hz, 1H), 7.48 (dd, J1 = 7.7 Hz, J2 = 7.8 Hz, 2H), 7.17 (d, J = 8.2 Hz, 1H), 6.97 (d, J = 8.2 Hz, 1H), 6.92 (s, 1H), 4.03–3.89 (m, 2H), 3.73 (s, 3H), 2.87–2.76 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 193.80, 165.11, 158.55, 135.49, 133.94, 130.41, 129.19, 128.74, 128.60, 127.16, 117.68, 111.67, 55.49, 47.77, 24.78. IR (KBr film) ν 3058, 2973, 2946, 2836, 1664, 1598, 1575, 1498, 1475, 1448, 1353, 1311, 1259, 1222, 1199, 1112, 1031, 921, 879, 858, 740, 698 cm−1. HRMS (ESI) m/z calcd for C17H15NO2Na [M + Na]+: 288.1000, found: 288.1005.
(7-Methoxy-3,4-dihydroisoquinolin-1-yl)(4-methoxyphenyl)-methanone 2l: white crystals, m.p. 58–60 °C. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 9.0 Hz, 2H), 7.16 (d, J = 8.2 Hz, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.94 (d, J = 9.0 Hz, 2H), 6.90 (s, 1H), 3.98–3.91 (m, 2H), 3.87 (s, 3H), 3.73 (s, 3H), 2.86–2.76 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.53, 165.38, 164.32, 158.54, 132.82, 129.17, 128.69, 128.42, 117.60, 113.91, 113.76, 111.69, 55.57, 55.48, 47.67, 24.80. IR (KBr film) ν 3037, 2947, 2924, 2833, 1652, 1603, 1572, 1499, 1468, 1422, 1357, 1317, 1259, 1222, 1209, 1169, 1126, 1040, 1019, 948, 926, 881, 819, 767 cm−1. HRMS (ESI) m/z calcd for C18H17NO3Na [M + Na]+: 318.1106, found: 318.1102.
(7,8-Dihydro-[1,3]dioxolo[4,5-g]isoquinolin-5-yl)(phenyl)-methanone 2m: pale yellow crystals, m.p. 76–77 °C. 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.0 Hz, 2H), 7.50 (t, J = 8.1 Hz, 1H), 7.37 (dd, J1 = 8.0 Hz, J2 = 8.1 Hz, 2H), 6.76 (s, 1H), 6.62 (s, 1H), 5.85 (s, 2H), 3.88–3.74 (m, 2H), 2.74–2.63 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 193.80, 164.41, 149.91, 146.51, 135.42, 133.88, 132.99, 130.34, 128.55, 120.37, 108.29, 106.88, 101.48, 47.19, 25.85. IR (KBr film) ν 3054, 2921, 2846, 1664, 1621, 1581, 1502, 1483, 1461, 1058, 1037, 991, 931, 869, 730, 688 cm−1. HRMS (ESI) m/z calcd for C17H13NO3Na [M + Na]+: 302.0793, found: 302.0800.
(7,8-Dihydro-[1,3]dioxolo[4,5-g]isoquinolin-5-yl)(3-methoxy-phenyl)methanone 2n: pale yellow crystals, m.p. 70–72 °C. 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 1H), 7.55 (d, J = 7.9 Hz, 1H), 7.36 (dd, J1 = 7.9 Hz, J2 = 8.0 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 6.83 (s, 1H), 6.72 (s, 1H), 5.96 (s, 2H), 3.93–3.85 (m, 2H), 3.86 (s, 3H), 2.83–2.74 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 193.77, 164.53, 159.76, 149.94, 146.54, 136.71, 132.99, 129.59, 123.59, 120.71, 120.45, 113.82, 108.31, 106.98, 101.49, 55.51, 47.23, 25.92. IR (KBr film) ν 3077, 2941, 2903, 2837, 1677, 1592, 1568, 1481, 1459, 1429, 1375, 1318, 1264, 1191, 1104, 1038, 992, 935, 872, 751, 679 cm−1. HRMS (ESI) m/z calcd for C18H15NO4Na [M + Na]+: 332.0899, found: 332.0907.
(7,8-Dihydro-[1,3]dioxolo[4,5-g]isoquinolin-5-yl)(3,4-dimethoxy-phenyl)methanone 2o: pale yellow crystals, m.p. 154–155 °C (lit.5d m.p. 153–154 °C). 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 1H), 7.50 (d, J = 8.4 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.74 (s, 1H), 6.64 (s, 1H), 5.88 (s, 2H), 3.88 (s, 3H), 3.86 (s, 3H), 3.85–3.76 (m, 2H), 2.76–2.65 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.72, 164.68, 154.19, 149.87, 149.20, 146.52, 132.93, 128.47, 126.55, 120.64, 111.07, 110.01, 108.27, 107.04, 101.46, 56.18, 56.06, 47.18, 25.93.
(7,8-Dihydro-[1,3]dioxolo[4,5-g]isoquinolin-5-yl)(4-methoxy-phenyl)methanone 2p: pale yellow crystals, m.p. 131–132 °C. 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.9 Hz, 2H), 6.94 (d, J = 8.9 Hz, 2H), 6.83 (s, 1H), 6.71 (s, 1H), 5.95 (s, 2H), 3.94–3.82 (m, 2H), 3.86 (s, 3H), 2.84–2.73 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.60, 164.70, 164.27, 149.83, 146.49, 132.95, 132.80, 128.33, 120.57, 113.88, 108.26, 107.04, 101.45, 55.59, 47.17, 25.93. IR (KBr film) ν 3069, 2920, 2841, 1650, 1599, 1505, 1486, 1257, 1171, 1038, 932, 849, 768 cm−1. HRMS (ESI) m/z calcd for C18H16NO4 [M + H]+: 310.1079, found: 310.1073.
(7,8-Dihydro-[1,3]dioxolo[4,5-g]isoquinolin-5-yl)(2-nitrophenyl)-methanone 2q: pale yellow crystals, m.p. 180–181 °C (lit.7 m.p. 180–183 °C). 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 7.8 Hz, 1H), 7.70 (dd, J1 = 7.8 Hz, J2 = 7.9 Hz, 1H), 7.63–7.54 (m, 2H), 7.48 (s, 1H), 6.60 (s, 1H), 5.94 (s, 2H), 3.62–3.50 (m, 2H), 2.56–2.44 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.38, 163.19, 149.90, 147.77, 146.52, 136.00, 134.21, 133.78, 131.08, 130.26, 123.61, 120.01, 108.49, 107.70, 101.47, 47.78, 25.79.
(7,8-Dihydro-[1,3]dioxolo[4,5-g]isoquinolin-5-yl)(4-nitrophenyl)-methanone 2r: white crystals, m.p. 160–161 °C. 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 8.9 Hz, 2H), 8.18 (d, J = 8.9 Hz, 2H), 6.96 (s, 1H), 6.75 (s, 1H), 6.00 (s, 2H), 4.01–3.81 (m, 2H), 2.88–2.60 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 191.55, 163.31, 150.44, 150.25, 146.66, 140.56, 133.33, 131.46, 123.51, 119.95, 108.37, 106.99, 101.62, 47.47, 25.81. HRMS (ESI) m/z calcd for C17H13N2O5 [M + H]+: 325.0824, found: 325.0821.
(2-Bromophenyl)(7,8-dihydro-[1,3]dioxolo[4,5-g]isoquinolin-5-yl)methanone 2s: pale yellow crystals, m.p. 147–148 °C. 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.7 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.43 (dd, J1 = 7.7 Hz, J2 = 7.8 Hz, 1H), 7.35 (dd, J1 = 7.8 Hz, J2 = 7.9 Hz, 1H), 7.25 (s, 1H), 6.71 (s, 1H), 6.00 (s, 2H), 3.91–3.73 (m, 2H), 2.77–2.63 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 195.61, 163.63, 149.79, 146.43, 140.24, 133.04, 132.46, 131.05, 127.53, 120.66, 120.22, 108.02, 107.81, 101.46, 48.20, 25.70. HRMS (ESI) m/z calcd for C17H12BrNO3Na [M + Na]+: 379.9898, found: 379.9895.
(4-Chlorophenyl)(7,8-dihydro-[1,3]dioxolo[4,5-g]isoquinolin-5-yl)methanone 2t: pale yellow crystals, m.p. 133–134 °C (lit.5d m.p. 132–133 °C). 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.2 Hz, 2H), 7.45 (d, J = 8.2 Hz, 2H), 6.86 (s, 1H), 6.73 (s, 1H), 5.97 (s, 2H), 3.99–3.83 (m, 2H), 2.87–2.72 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 192.36, 164.05, 150.10, 146.60, 140.45, 133.82, 133.14, 131.79, 128.90, 120.24, 108.33, 107.01, 101.53, 47.20, 25.88. IR (KBr film) ν 3004, 2923, 2852, 1668, 1622, 1587, 1502, 1484, 1462, 1208, 1089, 1041, 936, 899, 821, 769 cm−1.
:2), the nitromethane was removed by vacuum distillation to give a crude yellowish solid product, which was then triturated in aqueous methanol (CH3OH/H2O = 2:1, 20 mL). Pale yellow crystals were collected on a Buchner funnel by suction, and rinsed twice with aqueous methanol (CH3OH/H2O = 1:1, 2 × 10 mL). After being dried overnight under a warm air, compound 5 (11.68 g, 59.84 mmol) was obtained as yellow crystals in 92% yield, m.p. 164–165 °C (lit.27 m.p. 162–164 °C). 1H NMR (400 MHz, acetone-d6) δ 8.58 (brs, 1H, OH), 8.02 (d, J = 13.5 Hz, 1H), 7.91 (d, J = 13.5 Hz, 1H), 7.49 (s, 1H), 7.32 (d, J = 8.2 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 3.94 (s, 3H). 13C NMR (100 MHz, acetone-d6) δ 151.85, 149.02, 140.44, 136.03, 126.41, 123.18, 116.47, 112.21, 56.42.:1) to afford pure compound 6 (4.115 g, 24.61 mmol) as white crystals in 82% yield, m.p. 158–160 °C (lit.28 m.p. 158.5–160.5 °C). 1H NMR (400 MHz, DMSO-d6) δ 6.83 (s, 1H), 6.76 (d, J = 8.0 Hz, 1H), 6.63 (d, J = 8.0 Hz, 1H), 3.76 (s, 3H), 3.02–2.91 (m, 2H), 2.81 (t, J = 7.9 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 147.51, 145.24, 128.02, 120.71, 115.47, 112.74, 55.52, 55.47, 32.61.:2) to afford pure compound 7 (4.945 g, 14.40 mmol) as white crystals in 80% yield, m.p. 108–110 °C. 1H NMR (400 MHz, CDCl3) δ 7.30 (dd, J1 = 7.9 Hz, J2 = 7.8 Hz, 1H), 7.02 (d, J = 7.8 Hz, 1H), 7.00 (d, J = 7.9 Hz, 1H), 6.94 (s, 1H), 6.77 (d, J = 8.0 Hz, 1H), 6.60 (s, 1H), 6.51 (d, J = 8.0 Hz, 1H), 5.70 (brs, 1H, NH), 3.79 (s, 3H), 3.49 (s, 2H), 3.47–3.36 (m, 2H), 2.66 (t, J = 6.9 Hz, 2H), 2.29 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.55, 169.52, 150.95, 146.76, 144.30, 136.41, 130.37, 129.88, 126.83, 122.60, 121.28, 120.51, 114.50, 111.23, 55.84, 43.37, 40.97, 35.02, 21.10. IR (KBr film) ν 3378, 3301, 3079, 2935, 2848, 1762, 1736, 1653, 1607, 1512, 1449, 1371, 1278, 1206, 1147, 1124, 1032, 963, 933, 861, 791, 693 cm−1. HRMS (ESI) m/z calcd for C19H21NO5Na [M + Na]+: 366.1317, found: 366.1313.:1), an aqueous solution of hydrochloric acid (1 N, 50 mL) was added. After the mixture was vigorously stirred for 5 min, two phases were separated, and organic layer was washed with an aqueous solution of potassium carbonate (15% w/w, 30 mL). The aqueous phase was extracted again with CH2Cl2 (30 mL). The organic extracts were combined, dried over anhydrous MgSO4, and then concentrated under vacuum to give crude product, which was purified by flash chromatography (eluent: EtOAc/hexane = 1:3) to afford pure compound 8 (3.866 g, 10.03 mmol) as white crystals in 86% yield, m.p. 95–97 °C. 1H NMR (400 MHz, CDCl3) δ 7.32 (dd, J1 = 7.8 Hz, J2 = 7.9 Hz, 1H), 7.04 (d, J = 7.8 Hz, 1H), 7.00 (d, J = 7.9 Hz, 1H), 6.96 (s, 1H), 6.89 (d, J = 8.0 Hz, 1H), 6.71 (s, 1H), 6.61 (d, J = 8.0 Hz, 1H), 5.71 (brs, 1H, NH), 3.76 (s, 3H), 3.50 (s, 2H), 3.48–3.40 (m, 2H), 2.72 (t, J = 6.9 Hz, 2H), 2.30 (s, 3H), 2.29 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.46, 169.46, 169.23, 151.01, 150.95, 138.25, 137.73, 136.44, 129.94, 126.82, 122.69, 122.60, 120.79, 120.51, 112.78, 55.85, 43.39, 40.69, 35.36, 21.13, 20.67. IR (KBr film) ν 3300, 3037, 2938, 2854, 1769, 1649, 1601, 1540, 1448, 1424, 1373, 1267, 1206, 1151, 1120, 1028, 967, 905, 807, 790, 690 cm−1. HRMS (ESI) m/z calcd for C21H23NO6Na [M + Na]+: 408.1423, found: 408.1425.The above tautomeric mixture of 3,4-dihydroisoquinoline 9 and enamine 9′ were dissolved in DMSO (15 mL). Cu(OAc)2·2H2O (339.0 mg, 1.557 mmol) and CH3COOH (1.400 g, 23.31 mmol) were added. The resulting solution was then stirred at room temperature for 9 h under an atmosphere of air. After the reaction was completed (checked by TLC, EtOAc/hexane = 1:1), a dilute ammonia aqueous solution (5% w/w, 60 mL) and EtOAc (60 mL) were added. After the mixture was vigorously stirred for 5 min, two phases were separated, and the aqueous phase was extracted again with EtOAc (50 mL). The organic extracts were combined, and dried over anhydrous MgSO4. Removal of solvent by vacuum distillation gave crude product, which was purified by flash chromatography (eluent: EtOAc/hexane = 1:3) to afford pure compound 10 (2.525 g, 6.621 mmol) as pale yellow crystals in 85% yield, m.p. 80–82 °C. 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.0 Hz, 1H), 7.78 (s, 1H), 7.47 (dd, J1 = 8.0 Hz, J2 = 7.9 Hz, 1H), 7.34 (d, J = 7.9 Hz, 1H), 7.16 (s, 1H), 6.83 (s, 1H), 4.01–3.94 (m, 2H), 3.88 (s, 3H), 2.92–2.79 (m, 2H), 2.31 (s, 3H), 2.27 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 192.12, 169.11, 168.90, 163.37, 153.78, 150.74, 138.18, 137.11, 136.82, 129.54, 128.14, 127.25, 123.21, 121.52, 119.38, 111.55, 56.10, 47.10, 25.71, 21.13, 20.54. IR (KBr film) ν 3072, 2923, 2849, 1763, 1673, 1608, 1585, 1563, 1512, 1438, 1367, 1282, 1200, 1127, 1068, 1045, 1008, 912, 886, 808, 756, 701 cm−1. HRMS (ESI) m/z calcd for C21H20NO6 [M + H]+: 382.1291, found: 382.1291.
:1), solvents were removed by vacuum distillation to give crude product, which was then purified by flash chromatography (eluent: CH2Cl2/CH3OH = 10:1) to afford pure compound 4 (696.0 mg, 2.341 mmol) as pale yellow crystals in 89% yield, m.p. 120–122 °C. 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.9 Hz, 1H), 7.20 (s, 1H), 7.17 (dd, J1 = 7.9 Hz, J2 = 8.0 Hz, 1H), 6.89 (s, 1H), 6.87 (d, J = 8.0 Hz, 1H), 6.70 (s, 1H), 3.93 (s, 3H), 3.87–3.80 (m, 2H), 2.81–2.73 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 194.03, 164.32, 157.60, 150.45, 144.86, 136.16, 129.99, 128.87, 121.44, 120.80, 118.63, 115.64, 112.52, 111.46, 55.63, 46.65, 24.46. IR (KBr film) ν 3391, 3072, 2928, 2847, 1672, 1600, 1585, 1513, 1452, 1374, 1286, 1133, 1048, 1024, 1000, 880, 826, 762 cm−1. HRMS (ESI) m/z calcd for C17H16NO4 [M + H]+: 298.1079, found: 298.1075.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 20972048) for the financial support of this work.Notes and references
- Recent reviews, see: (a) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed; (b) P. Wang and L. Deng, Chin. J. Chem., 2018, 36, 1222 CrossRef CAS; (c) Q. Zhang and B.-F. Shi, Chin. J. Chem., 2019, 37, 647 CrossRef CAS; (d) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC; (e) H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946 CrossRef CAS PubMed; (f) R. Wang, Y. Luan and M. Ye, Chin. J. Chem., 2019, 37, 720 CrossRef CAS; (g) M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988 CrossRef CAS PubMed; (h) X. Lu, B. Xiao, R. Shang and L. Liu, Chin. Chem. Lett., 2016, 27, 305 CrossRef CAS; (i) T. Aneeja, M. Neetha, C. M. A. Afsina and G. Anilkumar, RSC Adv., 2020, 10, 34429 RSC.
- Recent examples for the oxidation of C(sp3)–H bonds: (a) G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noel, Angew. Chem., Int. Ed., 2018, 57, 4078 CrossRef CAS PubMed; (b) Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293 CrossRef CAS PubMed; (c) J. Romero-Ibanez, S. Cruz-Gregorio, J. Sandoval-Lira, J. M. Hernandez-Perez, L. Quintero and F. Sartillo-Piscil, Angew. Chem., Int. Ed., 2019, 58, 8867 CrossRef CAS PubMed; (d) Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448 CrossRef CAS PubMed; (e) J. B. C. Mack, J. D. Gipson, J. D. Bois and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 9503 CrossRef CAS PubMed; (f) T. Nanjo, E. C. de Lucca Jr and M. C. White, J. Am. Chem. Soc., 2017, 139, 14586 CrossRef CAS PubMed; (g) M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 12796 CrossRef CAS PubMed; (h) M. N. Wenzel, P. K. Owens, J. T. W. Bray, J. M. Lynam, P. M. Aguiar, C. Reed, J. D. Lee, J. F. Hamilton, A. C. Whitwood and I. J. S. Fairlamb, J. Am. Chem. Soc., 2017, 139, 1177 CrossRef CAS PubMed; (i) T. K. Beng, V. Shearer, R. Davey and I. Redman, RSC Adv., 2020, 10, 20264 RSC; (j) R. Chen, B. Liu, W. Li, K.-K. Wang, C. Miao, Z. Li, Y. Lv and L. Liu, RSC Adv., 2021, 11, 8051 RSC.
- (a) S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756 CrossRef CAS PubMed; (b) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed; (c) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (d) T.-Z. Meng, J. Zheng, T. H. Trieu, B. Zheng, J.-J. Wu, Y. Zhang and X.-X. Shi, ACS Omega, 2018, 3, 544 CrossRef CAS PubMed; (e) B. Zheng, T. H. Trieu, F.-L. Li, X.-L. Zhu, Y.-G. He, Q.-Q. Fan and X.-X. Shi, ACS Omega, 2018, 3, 8243 CrossRef CAS PubMed; (f) B. Zheng, T. H. Trieu, T.-Z. Meng, X. Lu, J. Dong, Q. Zhang and X.-X. Shi, RSC Adv., 2018, 8, 6834 RSC.
- (a) A. F. Marinho, E. d. J. Oliveira, J. F. Tavares, R. B. Filho and J. M. Barbosa-Filho, Magn. Reson. Chem., 2013, 51, 312 CrossRef CAS PubMed; (b) S. N. Sulaiman, M. R. Mukhtar, A. H. A. Hadi, K. Awang, H. Hazni, A. Zahari, M. Litaudon, K. Zaima and H. Morita, Molecules, 2011, 16, 3119 CrossRef CAS PubMed; (c) Y. Dang, H. F. Gong, J. X. Liu and S. J. Yu, Chin. Chem. Lett., 2009, 20, 1218 CrossRef CAS; (d) X. H. Duan and J. Q. Jiang, Chin. Chem. Lett., 2008, 19, 308 CrossRef CAS; (e) L. Gan, X. Zhao, W. Yao, L. Wu, L. Li and C. J. Zhou, J. Chem. Res., 2008, 2008, 285 CrossRef; (f) Y. Nishiyama, M. Moriyasu, M. Ichimaru, K. Iwasa, A. Kato, S. G. Mathenge, P. B. C. Mutiso and F. D. Juma, Phytochemistry, 2006, 67, 2671 CrossRef CAS PubMed; (g) H. Zhang and J.-M. Yue, Nat. Prod. Res., 2006, 20, 553 CrossRef CAS PubMed; (h) B. Chen, C. Feng, B.-G. Li and G.-L. Zhang, Nat. Prod. Res., 2003, 17, 397 CrossRef CAS PubMed; (i) G. A. d. Lira, L. M. d. Andrade, K. C. Florencio, M. S. d. Silva, J. M. Barbosa-Filho and E. V. L. da-Cunha, Fitoterapia, 2002, 73, 356 CrossRef PubMed; (j) M. Lebceuf, A. Ranaivo, A. Cave and H. Moskowitz, J. Nat. Prod., 1989, 52, 516 CrossRef CAS.
- (a) A. A. Salim, N. Bidin, A. S. Lafi and F. Z. Huyop, Mater. Des., 2017, 132, 486 CrossRef CAS; (b) K. N. Yogendra, D. Dhokane, A. C. Kushalappa, F. Sarmiento, E. Rodriguez and T. Mosquera, Plant Sci., 2017, 256, 208 CrossRef CAS PubMed; (c) A. Bermejo, I. Andreu, F. Suvire, S. Leonce, D. H. Caignard, P. Renard, A. Pierre, R. D. Enriz, D. Cortes and N. Cabedo, J. Med. Chem., 2002, 45, 5058 CrossRef CAS PubMed; (d) J. A. Weisbach, J. L. Kirkpatrick, E. Macko and B. Douglas, J. Med. Chem., 1968, 11, 752 CrossRef CAS PubMed.
- (a) A. Bischler and B. Napieralski, Ber. Dstch. Chem. Ges., 1893, 26, 1903 CrossRef; (b) Z. M. A. Judeh, C. B. Ching, J. Bu and A. McCluskey, Tetrahedron Lett., 2002, 43, 5089 CrossRef CAS; (c) M. A. Epishina, A. S. Kulikov, M. I. Struchkova, N. V. Ignat’ev, M. Schulte and N. N. Makhova, Mendeleev Commun., 2012, 22, 267 CrossRef CAS ; and the references cited therein..
- W. I. Taylor, Tetrahedron, 1961, 14, 42 CrossRef CAS.
- N. H. Martin, S. L. Champion and P. B. Belt, Tetrahedron Lett., 1980, 21, 2613 CrossRef CAS.
- S. V. Kessar, T. Mohammad and Y. P. Gupta, Indian J. Chem., Sect. B, 1983, 22, 321 Search PubMed.
- R.-Y. Kuo, F.-R. Chang, C.-C. Wu, R. Patnam, W.-Y. Wang, Y.-C. Du and Y.-C. Wu, Bioorg. Med. Chem. Lett., 2003, 13, 2789 CrossRef CAS PubMed.
- G. R. Lenz and C. Costanza, J. Org. Chem., 1988, 53, 1176 CrossRef CAS.
- N. H. Martin and C. W. Jefford, Helv. Chim. Acta, 1982, 65, 762 CrossRef CAS.
- P. B. Wakchaure and N. P. Argade, Synthesis, 2008, 15, 2321 Search PubMed.
- I. Andreu, N. Cabedo, G. Atassi, A. Pierre, D. H. Caignard, P. Renard, D. Cortes and A. Bermejo, Tetrahedron Lett., 2002, 43, 757 CrossRef CAS.
- B. Zheng, H.-Y. Qu, T.-Z. Meng, X. Lu, J. Zheng, Y.-G. He, Q.-Q. Fan and X.-X. Shi, RSC Adv., 2018, 8, 28997 RSC.
- (a) R. A. Clark and D. C. Parker, J. Am. Chem. Soc., 1971, 93, 7257 CrossRef CAS; (b) S.-P. Lu and A. H. Lewin, Tetrahedron, 1998, 54, 15097 CrossRef CAS; (c) L. O. Davis, M. A. Putri, C. L. Meyer and C. P. Durant, Tetrahedron Lett., 2014, 55, 3100 CrossRef CAS.
- P. Gamez, P. G. Aubel, W. L. Driessen and J. Reedijk, Chem. Soc. Rev., 2001, 30, 376 RSC ; and the references cited therein..
- M. D. Garcia, A. J. Wilson, D. P. G. Emmerson and P. R. Jenkins, Chem. Commun., 2006, 2586–2588 RSC.
- (a) K. Maeda, M. Nakamura and M. J. Sakai, Chem. Soc. Perkin Trans., 1983, 1, 837 RSC; (b) Y. Mori and K. Maeda, Bull. Chem. Soc. Jpn., 1994, 67, 1204 CrossRef CAS.
- J.-M. Oger, A. Fardeau, P. Richomme, H. Guinaudeau and A. Fournet, Can. J. Chem., 1993, 71, 1128 CrossRef CAS.
- J.-X. Yang, J. Dong, X. Lu, Q. Zhang, W. Ding and X. Shi, Chin. J. Chem., 2012, 30, 2827 CrossRef CAS.
- M. D. Rozwadowska, M. Chrzanowska, A. Brossi, C. R. Creveling, M. E. Bembenek and C. W. Abell, Helv. Chim. Acta, 1988, 71, 1598 CrossRef CAS.
- R. Zhu, Z. Xu, W. Ding, S. Liu, X. Shi and X. Lu, Chin. J. Chem., 2014, 32, 1039 CrossRef CAS.
- S.-D. Cho, D.-H. Kweon, Y.-J. Kang, S.-G. Lee, W. S Lee and Y.-J. Yoon, J. Heterocycl. Chem., 1999, 36, 1151 CrossRef CAS.
- C. C. Silveira, C. R. Bernardi, A. L. Braga and T. S. Kaufman, Tetrahedron Lett., 2001, 42, 8947 CrossRef CAS.
- B.-X. Zhao, Y. Yu and S. Eguchi, Org. Prep. Proced. Int., 1997, 29, 185 CrossRef CAS.
- A. Fierroa, M. C. Rezendea, S. Sepúlveda-Bozab, M. Reyes-Paradab and B. K. Cassels, J. Chem. Res., 2001, 294 CrossRef.
- K. Rice and A. Brossi, J. Org. Chem., 1980, 45(4), 592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05671a |
| This journal is © The Royal Society of Chemistry 2021 |