
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrene-based ammonium bromides combined with g-C3N4 for the synergistically enhanced fixation reaction of CO2 and epoxides†
Tao Chang ab,
Xiaopeng Lia,
Yongjing Haoa,
Lianwei Kanga,
Tian Tian*a,
Xiying Fua,
Zheng Zhu*a,
Balaji Panchala and
Shenjun Qin*a
ab,
Xiaopeng Lia,
Yongjing Haoa,
Lianwei Kanga,
Tian Tian*a,
Xiying Fua,
Zheng Zhu*a,
Balaji Panchala and
Shenjun Qin*a
aKey Laboratory of CO2 Utilization of Handan City, College of Material Science and Engineering, Hebei University of Engineering, Handan 056038, Hebei, China. E-mail: tiantian19860102@126.com; zhuzheng@hebeu.edu.cn; sjqin528@hebeu.edu.cn
bKey Laboratory of Heterocyclic Compounds of Hebei Province, Handan College, Handan 056005, Hebei, China
First published on 10th September 2021
Abstract
A new type of pyrene-based ammonium bromides (PABs) was synthesized via the reaction of bromomethyl pyrene and tertiary amines with different alkyl chains combined with graphitic carbon nitride (g-C3N4) through π–π stacking interactions. The new pyrene-based ammonium bromides were investigated both in homogenous phase and heterogeneous phase combining with g-C3N4 for the CO2 fixation reaction of epoxides under mild conditions. Obviously, the combination was proved to be an efficient system for the conversion of epoxides. The interaction between g-C3N4 and PABs was confirmed by quantum chemical calculations. g-C3N4/Py-C12 exhibited an excellent yield of cyclic carbonates (above 93%) at 80 °C, atmospheric pressure and solvent-free conditions. A preliminary kinetic study was performed using g-C3N4/Py-C12 and the activation energy was calculated to be 61.5 kJ mol−1.
Introduction
The anthropogenic carbon dioxide emission in atmosphere has far-reaching effects on sustainable society, and is considered as a major component of global greenhouse gas, leading to global warming and extreme weather events.1 However, the effective conversion of CO2 into high value-added chemicals has attracting widespread attention to scientists, because of which it has been regarded as an alternative, nontoxic, and sustainable C1 resource.2–7 Among these conversions, the generation of cyclic carbonates, a popular group of chemicals, is recognized as an effective route to convert inert CO2 with epoxides.8–16 In addition, cyclic carbonates are established as not only valuable materials with various industrial applications for polymer synthesis, aprotic polar solvents, fuel additives and electrolyte solvents in lithium-ion batteries but also crucial intermediates in the manufacturing of fine chemicals.17,18In fact, there are quite a few materials on the utilization of CO2 fixation, including homogeneous and heterogenous catalysts, for example, ionic liquids,19,20 organocatalysts,21–23 Salen,24–26 porphyrins,27 alkali salts,28 covalent-organic frameworks (COFs),29 metal–organic frameworks (MOFs)30 and covalent organic polymers (COPs).9,31 More intriguingly, some quaternary ammonium salts have been selected as catalysts for CO2 fixation with epoxides, which are more sustainable than traditional Lewis acid (base) alternatives because of such suit of metal-free organocatalysts according to a green chemistry view point.32–37 Apparently, with regard to ammonium catalysts, hydrogen bonding activating strategy of epoxides is crucial for accomplishing CO2 fixation.38 In addition, the conversion of CO2 to cyclic carbonates under mild conditions, such as low reaction temperature, atmospheric pressure, metal-free and solvent-free, is still challenging.
Recently, the syntheses and application of 2D materials of graphite carbon nitride (g-C3N4) have attracted wide attention on CO2 absorption due to the large amount of basic groups of amine and guanidine as nucleophilic sites for CO2 activation.39 It is predicted that the as-prepared and adjusted g-C3N4 could be optimized as catalysts for the capture and conversion of CO2.40–54 The C3N4 materials could be selected independently as the catalysts for CO2 fixation with epoxides; however, these processes suffer from severe reaction conditions to obtain excellent conversion.41,46,56 Some strategies were developed to increase the activity of C3N4 materials, including adjusted acid-base duality with doping of metal atoms,40,49,54 alkalis,43 phosphorous,44 metal halides,45 and boric acid.50,51 Another is that co-catalysts, such as tetrabutyl ammonium bromide (TBAB),44,48,54 KI,50 and DMF,53 are introduced and combined with g-C3N4, which play a crucial role in accelerating the rate-limiting step of opening epoxides by the nucleophilicity of halide anions.28,31 Although significant advances have been accomplished in g-C3N4 syntheses, some protocols suffer from harsh conditions, such as high temperature, high pressure and the presence of metal salts and co-solvents. In addition, most of the optimized procedures focus on modulating the texture structure and surface properties of g-C3N4. Predictably, the development of novel catalysts is still a fascinated alternative, which could interact tightly and realize the reaction synergistically with g-C3N4.
In contrast, the non-covalent methodology is more convenient and effective for the catalyst construction without the chemical modification of homogeneous catalysts.55 The π–π stacking interactions are an available non-covalent immobilization, and g-C3N4 provides a significant alternative for non-covalent adjustment via π–π stacking interactions with polyaromatic molecules, such pyrene-based moiety.56 From the above point, the preparation of a pyrene-based non-covalent catalyst anchored on the g-C3N4 surface is a potentially valuable strategy of the heterogenization of homogeneous catalysis. Herein, quaternary ammonium salts connected with pyrene-based tag and different substituted alkyl chains, which possessed the properties of immobilization onto the g-C3N4 surface, are accordingly reported (Scheme 1).
 | ||
| Scheme 1 General synthesis strategy of pyrene-based quaternary ammonium salts. | ||
Results and discussion
In order to illustrate the potentiality for a catalytic reaction, the fixation of epichlorohydrin and CO2 as a model reaction was implemented in the presence of pyrene-based ionic liquids under mild conditions (Table 1, entries 1–5). Gratifyingly, all ionic liquids fabricated with mediated alkyl chains exhibited considered activities, which were superior to that of dodecyl trimethyl ammonium bromide (entry 6). Therefore, it is of significance to explore such catalysts deeply. As shown in Tables 1 and 2, when Py-Cn was used with different substitutes, there was only a slight change. The trend is that with the increase in the length of alkyl chains, the carbonate yield was ascending marginally. Predictably, the combination of g-C3N4 and Py-Cn was selected as the catalyst for CO2 conversion because a large number of amine and guanidine groups were grafted, which are beneficial to the absorption and activation of CO2. An increase in the fixation of CO2 was detected in the presence of g-C3N4 (Table 2, entries 1–5). Thus, it is certain that g-C3N4 plays a crucial role in realizing the CO2 fixation reaction. The results were better than that of DTAB combined with Py-Cn, which meant that Py-Cn could interact with g-C3N4. Therefore, a synergistic effect through π–π stacking interactions between Py-Cn and g-C3N4 was proposed, which is significant to realize the organic reaction.57,58| Entry | Py-Cn/C3N4 | Yield (%) |
|---|---|---|
| a Reaction conditions: ECH (10 mmol, 0.78 mL), Py-Cn (0.0375 mmol), g-C3N4 (12 mg), CO2 (0.1 MPa), 70 °C, 8 h.b In the absence of Py-Cn, no reaction. | ||
| 1 | Py-C4/g-C3N4 | 62.5 |
| 2 | Py-C8/g-C3N4 | 65.7 |
| 3 | Py-C12/g-C3N4 | 75.2 |
| 4 | Py-C16/g-C3N4 | 72.7 |
| 5 | Py-C18/g-C3N4 | 72.0 |
| 6 | DTAB/g-C3N4 | 42.6 |
| 7b | g-C3N4 | NR |
The reaction time was found to influence the CO2 fixation and be an important factor to realize the reaction completely. Then, the influence of the reaction time ranged from 4 to 24 h on the CO2 conversion was explored. As demonstrated in Fig. 1, the cyclic carbonates yield increased with prolonged time, and 75.2% yield was obtained after 8 h. From 8 h to 24 h, the well-distributed growth trend of the yield was detected. Therefore, the appropriate reaction time was selected as 16 h, beneficially, this time was not only effective, but also more economical.
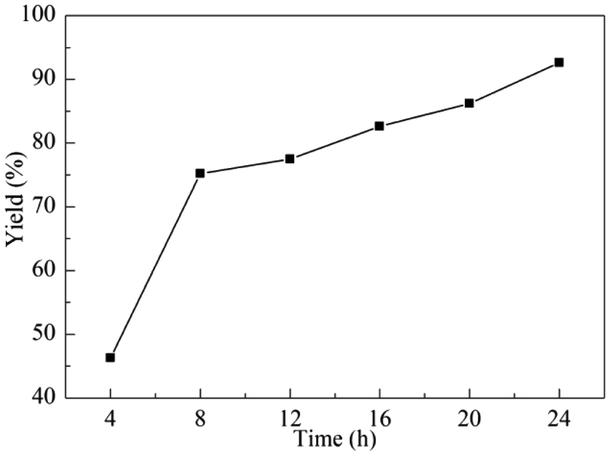 | ||
| Fig. 1 Effect of the reaction time. Reaction conditions: ECH (10 mmol, 0.78 mL), g-C3N4 (12 mg), Py-C12 (0.0375 mmol, 19 mg), CO2 (0.1 MPa), 70 °C. | ||
Temperature plays a crucial effect on organic reactions. Thus, the influence of temperature varied from 60 °C to 90 °C was investigated for the fixation reaction of ECH with CO2. As shown in Fig. 2, the yield improved rapidly when the reaction was carried out at 60 °C after 16 h, and a yield of 70.2% was detected. The varied yields of 82.6%, 90.1% and 95.2% were obtained when the temperature was risen from 70 °C to 90 °C. Although an excellent yield was explored at 90 °C, temperature of 80 °C was reasonable according to the energy cost.
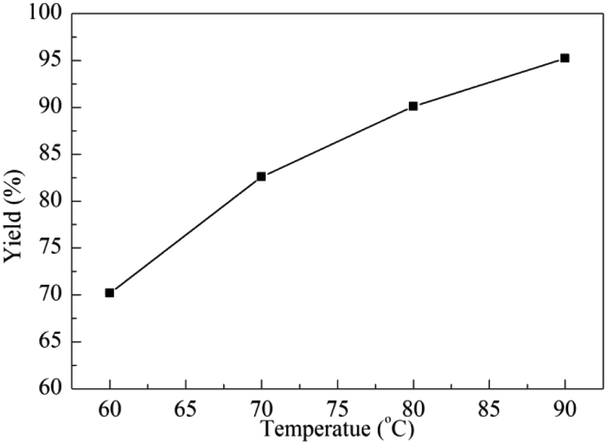 | ||
| Fig. 2 Effect of the reaction temperature. Reaction conditions: ECH (10 mmol, 0.78 mL), g-C3N4 (12 mg), Py-C12 (0.0375 mmol, 19 mg), CO2 (0.1 MPa), 16 h. | ||
The yield is strongly related to the amount of the combined catalyst in this reaction. The results were illustrated in Table 3. A yield of 80.1% was obtained at a low catalyst amount with a constant ratio of g-C3N4 to Py-C12 (entry 1) because lower catalytic concentration can not provide sufficient active sites to promote the reaction availably. Increasing the catalyst amount improved the activity of this combined system because of the enhancing synergistic interaction between the catalyst and substrates. A superior yield of 95.2% was observed, and a substantial increase was not obtained by increasing the catalytic amount to higher than 15 mg and 23.8 mg of g-C3N4 and Py-C12, respectively.
| Entry | g-C3N4 (mg) | Py-C12 (mg) | Yield (%) |
|---|---|---|---|
| a Reaction conditions: ECH (10 mmol, 0.78 mL), CO2 (0.1 MPa), 80 °C, 16 h. | |||
| 1 | 9 | 14.3 | 80.1 |
| 2 | 12 | 19 | 82.6 |
| 3 | 15 | 23.8 | 95.2 |
| 4 | 18 | 28.5 | 96.0 |
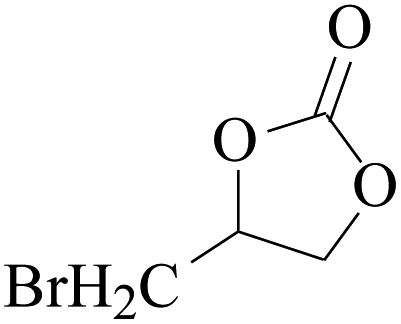
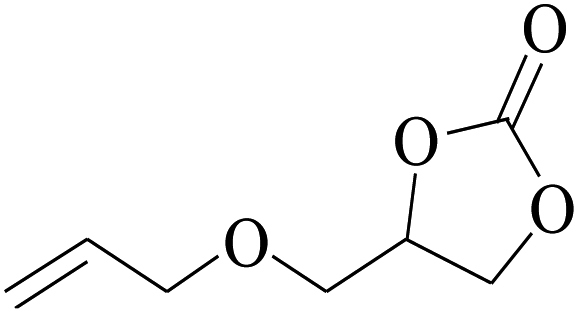
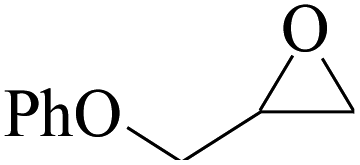
In order to expand the versatility of the present catalytic system, the catalyst of Py-C12 with g-C3N4 was evaluated for the CO2 fixation of various epoxides at optimum reaction conditions. All results are summarized in Table 4; as expected, excellent yields varied from 93% to 96% were observed for all epoxides tethered with diverse functional groups. Epoxides exhibited different conversion ability, and some epoxides such as ECH, epibromohydrin, glycidyl phenyl ether and allyl glycidyl ether were converted within 16 h (entries 1–4). In contrast, butyl glycidyl ether and styrene oxide needed a slightly longer time to obtain high yields (entries 5 and 6). In addition, glycol diglycidyl ether could also be considered as a candidate substrate and generated into corresponding cyclic carbonate with good yield at 48 h (entry 7).
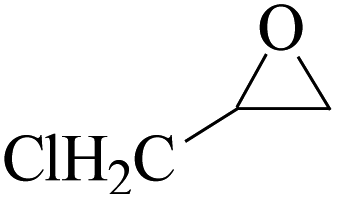
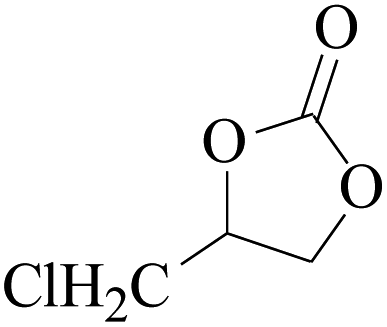
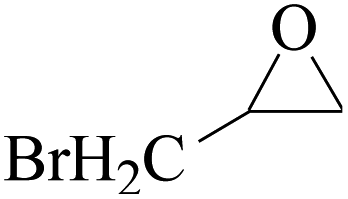

The catalytic activity of g-C3N4/Py-C12 was compared with other catalysts for the CO2 fixation reaction, as summarized in Table 5. In most studies, the CO2 fixation reactions were realized at high temperatures and pressures. Comparatively, the reactions were carried out at mild conditions and with lower catalyst amount in our study.
| Catalysts | Catalyst amount | Co-catalyst | Reaction conditions | Yield (%) | Reference |
|---|---|---|---|---|---|
| Zn-g-C3N4/SBA | 100 mg | — | 150 °C, 3.5 MPa, 1.5 h | 99 | 40 |
| u-g-C3N4 | 100 mg | — | 130 °C, 2 MPa, 24 h | 99 | 41 |
| ZnCl2/mp-C3N4 | 200 mg | — | 140 °C, 2.5 MPa, 6 h | 73 | 42 |
| g-C3N4-500-NaOH | 400 mg | — | 140 °C, 2 MPa, 6 h | 92 | 43 |
| P-g-C3N4 | 150 mg | TBAB | 100 °C, 2 MPa, 3 h | 91 | 44 |
| n-butlBr | 200 mg | — | 140 °C, 2.5 MPa, 6 h | 88 | 45 |
| u-g-C3N4 | 50 mg | — | 130 °C, 3.5 MPa, 2 h | 99 | 46 |
| g-C3N4 | 50 mg | TBAB | 105 °C, 1 bar, 20 h | >99 | 48 |
| Co@NxC | 50 mg | TBAB, CH3CN | 60 °C, 1 bar, 12 h | 84 | 49 |
| BGCN05 | 100 mg | KI | 60 °C, 1 bar, 60 h | 73 | 50 |
| BCN | 30 mg | — | 130 °C, 3.0 MPa, 24 h | 97 | 51 |
| u-C3N4 | 230 mg | — | 120 °C, 2.0 MPa, 2 h | 96 | 52 |
| GO | 2.5 mg | DMF | 100 °C, 1 bar, 12 h | 95 | 53 |
| 2.0Zn@g-C3N4-550 | 50 mg | TBAB | 90 °C, 1.5 MPa, 3 h | 99 | 54 |
| g-C3N4 | 15 mg | Py-C12 | 80 °C, 1 bar, 16 h | 95 | This work |
Clearly, the catalyst could catalyze the fixation reaction of CO2 by decreasing the activation energy (Ea), along with a high-speed reaction rate at a lower activation energy. In this regard, the capability of Py-C12 coordinated with g-C3N4 in lowering the activation energy of the fixation reaction was exactly examined at varied temperatures from 50 to 80 °C under the optimum conditions.
In general, the rate equation for the CO2 fixation was represented as eqn (1). Usually, eqn (1) could be simplified as eqn (2), because the reaction was implemented at constant catalyst concentration and excessively CO2 conditions. According to previous reports, the fixation reaction was described as pseudo first order reaction on the ECH concentration.59–62 Then, the equation was supported as eqn (3) and (4), where k is the rate constant, t is the reaction time and x is the conversion yield.
| r = dx/dt = k′′[ECH]α[CO2]β[cat.]γ | (1) |
| r = dx/dt = k′[ECH]α | (2) |
| r = dx/dt = k′[ECH] | (3) |
| ln(1 − x) = kt + C | (4) |
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k = Ea/RT + C k = Ea/RT + C
| (5) |
Therefore, the process kinetics and Ea of the production were investigated, and the results are demonstrated in Tables S1–S5†, Fig. 3 and 4. As shown in Fig. 3, a linear relationship between ln(1 − x) versus time was obtained, that meant that the hypothesis of pseudo first order reaction on the ECH concentration was rational. Thus, the k values described in Table S5† were calculated from curve of ln(1 − x) versus t at various temperatures. The Ea to cyclic carbonates could be obtained according to Arrhenius law (eqn (5)). The slope of lnk versus 1/T is the value of Ea/T, consequently, the Ea was calculated as 61.5 kJ mol−1 (Fig. 4).
 | ||
| Fig. 3 Logarithmic plots of (1 − x) versus time by Py-C12 in the presence of g-C3N4. | ||
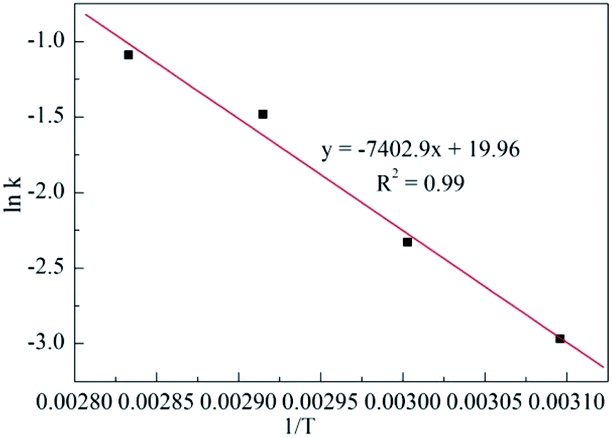 | ||
| Fig. 4 Arrhenius plots for CO2 fixation with ECH using Py-C12 in the presence of g-C3N4. | ||
In order to investigate the fixation reaction mechanism of CO2 and epoxides enhanced by pyrene-based ammonium bromides combined with g-C3N4, density functional theory (DFT) calculations were performed using a DMol3 module of Material Studio 2016. Molecular orbitals were used to investigate the type of bonding and the interactions between the new pyrene-based ammonium bromides and g-C3N4 during the catalytic process.63,64 The surfaces of the molecular orbitals were drawn at the 0.03 a. u. iso level, and they described the electron density in both the GS and the excited state in Fig. S1.† In case of g-C3N4/Py-C4 and g-C3N4/Py-C18, it was a π-type bond and the electron density was localized on g-C3N4 and the tertiary amine group (–(CH2)2–N–(CH3)2–) and Br−. The molecular orbitals that characterized this π-type bond were HOMO orbitals. The process included a slight delocalization of electron density from the g-C3N4 ring. On the basis of g-C3N4/Py-C8 and g-C3N4/Py-C16, the π-type bond exited between them, and the electron density was localized on the g-C3N4 and the pyrenephenyl group. The molecular orbitals that characterized this π-type bond were also HOMO orbitals. The π-extended electron density conjugation appeared as an effect of an intramolecular interaction between pyrenephenyl group and g-C3N4 ring. Based on g-C3N4/Py-C12, the stronger interaction was π–π conjugation effects, and the electron density was localized on the g-C3N4 and the tertiary amine group (–(CH2)2–N–(CH3)2–) and Br−. Meanwhile, the weaker interaction was π → π conjugation effects, and the electron density was localized on the g-C3N4 and pyrenephenyl group. Therefore, a series of varying alkyl chain-substituted pyrene-based ammonium bromides combining with g-C3N4 by π–π stacking interactions were exhibited to have predominant activity for CO2 fixation with various epoxides.
Therefore, based on the present experimental and previous references,31,41,44,50–52 the proposed mechanism derived from the concept of synergetic effect for CO2 fixation catalyzed by g-C3N4/Py-C12 is depicted in Scheme 2. First, the interaction of g-C3N4 with Py-C12 happens through π–π stacking certified by DFT and previous reports.57,58 Thus, the combined catalytic system is favorable for synergistic inteaction between catalytic sites of amino groups and the nucleophilic reagent (Scheme 2, intermediate I). Second, the amino groups in g-C3N4 play a significant role in activating the epoxides and carbon dioxide through hydrogen bonding. The activation of epoxides through hydrogen bonding with primary amino group and CO2 via secondary and tertiary amine possibly occurs simultaneously, which allows the coordination of intermediate II.26,41,44 As a result, the increasing electrophilicity of the terminal carbon of epoxides is attacked by the nucleophilic bromide ion from the pyrene-based ammonium interacting tightly with g-C3N4 to generate the negative bromo-alkoxide intermediate III, which is stabilized by hydrogen bonding. Consequently, the attack of activated CO2 with bromo-alkoxide and intermediate IV of linear bromo-carbonate are generated. Finally, the intramolecular ring-closing of intermediate IV occurs, and the generated cyclic carbonate regenerates the catalyst.
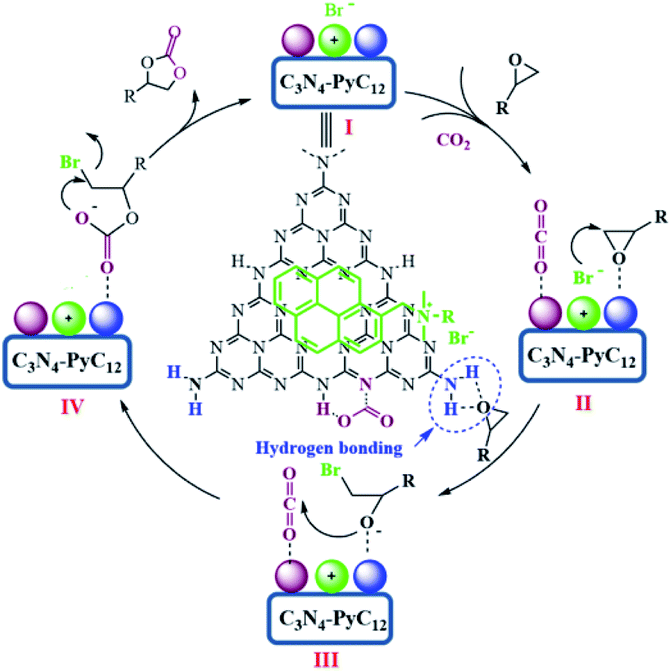 | ||
| Scheme 2 Probable mechanism for the fixation of CO2 to epoxides. | ||
Experimental section
Preparation of pyrene based quaternary ammonium salts and g-C3N4
Py-C4: 1H NMR (400 MHz, CD3OD) δ 8.82 (d, J = 9.2 Hz, 1H, ArH), 8.16–8.46 (m, 8H, ArH), 5.39 (s, 2H, ArCH2N), 3.54–3.58 (m, 2H, NCH2), 3.09 (s, 6H, NCH3), 1.84–1.88 (m, 2H, NCH2CH2), 1.32–1.37 (m, 2H CH2), 0.97 (t, J = 7.6 Hz, 3H, CH3); 13C NMR (100 MHz, CD3OD) δ 132.86. 132.81, 132.16, 131.11, 130.38, 129.38, 129.35, 127.71, 127.21, 126.79, 126.47, 125.23, 124.64, 124.03, 123.96, 122.09, 64.30, 63.92, 49.51, 24.40, 19.87, 14.02; ESI-MS: calcd for C23H26NBr, m/z [M − Br]+: 316.2, found: 316.1.
Py-C8: 1H NMR (400 MHz, CD3OD) δ 8.80 (d, J = 9.2 Hz, 1H, ArH), 8.16–8.46 (m, 8H, ArH), 5.38 (s, 2H, ArCH2N), 3.5–3.55 (m, 2H, NCH2), 3.08 (s, 6H, NCH3), 1.84 (s, 2H, NCH2CH2), 1.24–1.30 (m, 10H CH2), 0.85 (t, 3H, CH3); 13C NMR (100 MHz, CD3OD) δ 132.86. 132.77, 132.16, 131.12, 130.39, 129.39, 129.36, 127.71, 127.22, 126.80, 126.47, 125.23, 124.64, 124.03, 123.93, 122.11, 64.47, 63.98, 49.54, 31.61, 28.91, 28.88, 26.41, 22.49, 22.42, 14.42; ESI-MS: calcd for C27H34NBr, m/z [M − Br]+: 372.3, found: 372.2.
Py-C12: 1H NMR (400 MHz, CDCl3) δ 8.79 (d, J = 9.32 Hz, 1H, ArH), 8.26 (d, J = 7.88 Hz, 1H, ArH), 7.98–8.08 (m, 3H, ArH), 7.78–7.91 (m, 3H, ArH), 7.64 (d, J = 8.84 Hz, 1H, ArH), 6.04 (s, 2H, ArCH2N), 3.78 (t, J = 7.64 Hz, 2H, NCH2), 3.30 (s, 6H, NCH3), 1.71 (s, 2H, NCH2CH2), 0.97–1.24 (m, 18H, CH2), 0.85 (t, J = 6.8 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 132.50, 132.33, 131.85, 130.61, 129.96, 129.54, 128.78, 126.59, 126.20, 126.00, 125.83, 124.24, 123.63, 123.47, 120.07, 65.20, 64.19, 49.36, 31.86, 29.49, 29.45, 29.32, 29.28, 29.25, 26.33, 23.10, 22.68, 14.16; ESI-MS: calcd for C31H42NBr, m/z [M − Br]+: 428.3, found: 428.2.
Py-C16: 1H NMR (400 MHz, CDCl3) δ 8.80 (d, J = 9.28 Hz, 1H, ArH), 8.27 (d, J = 7.88 Hz, 1H, ArH), 7.99–8.09 (m, 3H, ArH), 7.79–7.92 (m, 3H, ArH), 7.65 (d, J = 8.84 Hz, 1H, ArH), 6.06 (s, 2H, ArCH2N), 3.79 (t, J = 7.56 Hz, 2H, NCH2), 3.31 (s, 6H, NCH3), 1.72 (s, 2H, NCH2CH2), 0.97–1.24 (m, 26H, CH2), 0.87 (t, J = 6.5 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 132.51, 132.34, 131.87, 130.62, 129.98, 129.57, 128.79, 126.59, 126.20, 126.01, 125.84, 124.25, 123.64, 123.48, 120.06, 65.22, 64.20, 49.37, 31.94, 29.72, 29.68, 29.65, 29.57, 29.48, 29.39, 29.35, 29.26, 26.33, 23.11, 22.71, 14.17; ESI-MS: calcd for C35H50NBr, m/z [M − Br]+: 484.4, found: 484.3.
Py-C18: 1H NMR (400 MHz, CDCl3) δ 8.77 (d, J = 8.92 Hz, 1H, ArH), 8.24 (d, J = 7.4 Hz, 1H, ArH), 7.93–8.03 (m, 3H, ArH), 7.76–7.86 (m, 3H, ArH), 7.61 (d, J = 8.56 Hz, 1H, ArH), 7.10–7.19 (m, 3H), 5.98 (s, 2H, ArCH2N), 3.77 (s, 2H, NCH2), 3.25 (s, 6H, NCH3), 2.29 (s, 2H, NCH2CH2), 1.68 (s, 2H, NCH2CH2), 0.84–1.21 (m, 30H, CH2); 13C NMR (100 MHz, CDCl3) δ 137.80, 132.48, 132.30, 131.83, 130.59, 129.93, 129.46, 128.99, 128.75, 128.18, 126.61, 126.17, 125.98, 125.81, 125.26, 124.23, 123.62, 123.49, 120.15, 65.09, 64.19, 49.32, 31.92, 29.70, 29.67, 29.64, 29.56, 29.48, 29.37, 29.29, 29.24, 26.35, 23.10, 22.70, 21.44, 14.16; ESI-MS: calcd for C37H54NBr, m/z [M − Br]+: 512.4, found: 512.4.
Procedure of CO2 fixation with epoxides
At room temperature, Py-Cn, g-C3N4 and epoxides were charged into a Schlenk tube, respectively. To enable the catalysts interact completely, the mixture was sonicated for 20 min. Then, CO2 was injected into the tube through a balloon, which was placed into a heater and stirred at a fixed temperature. After the reaction, the mixture was cooled at room temperature, and the excess CO2 was vented. After centrifugation, a small amount of clearly crude product was separated and detected by 1H NMR to identify the conversion.DFT calculations
The DFT calculations were performed using a DMol3 module of Material Studio 2016. During optimization, all atoms of the paddlewheel cluster and adsorbing molecules, except terminating atoms, could relax. The generalized gradient approximation (GGA) method with the Perdew–Burke–Ernzerhof (PBE) function was employed to describe the interactions between the core and electrons. An energy cutoff of 450 eV was used for the plane wave expansion of the electronic wave function. The force and energy convergence criteria were set to 0.03 eV Å−1 and 10−5 eV, respectively.Conclusions
A series of ranged alkyl chain-substituted pyrene-based ammonium bromides combining with g-C3N4 by π–π stacking interactions exhibited a predominant activity towards CO2 fixation with various epoxides, and above 93% yields of the corresponding cyclic carbonates were detected at 80 °C, 0.1 MPa CO2 pressure under solvent-free conditions. It could be predicted that the effectiveness of such a combination system originated from the synergetic interactions between π–π stacking, amino group and bromide ion nucleophile. This method provides a new strategy that the simple immobilized g-C3N4 can be used as a catalyst for the fixation reaction of CO2 under mild conditions.Author contributions
Tao Chang: Data curation, formal analysis, methodology, writing – original draft, review & editing. Xiaopeng Li: data curation, formal analysis. Yongjing Hao: data curation, formal analysis. Lianwei Kang: software, visualization. Tian Tian: data curation, methodology, software, supervision. Xiying Fu: data curation, software. Zheng Zhu: formal analysis, supervision. Balaji Panchal: writing – review & editing. Shenjun Qin: methodology, supervision, project administration, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) program (21902041), the National Science Foundation of Hebei province (B2019402020 B2020402002 E2021402017) and the Science and Technology Project of Hebei Education Department (ZD2019002, ZD2019055, QN2020165).Notes and references
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. B. Yi, D. Y. Zhao, K. Albahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumil, Green Chem., 2020, 22, 4002–4033 RSC.
- P. Sreejyothi and K. M. Swadhin, Chem. Sci., 2020, 11, 10571–10593 RSC.
- Z. E. Zhang, S. Y. Pan, H. Li, J. C. Cai, A. G. Olabi, E. J. Anthony and V. Manovic, Renewable Sustainable Energy Rev., 2020, 125, 109799 CrossRef CAS.
- M. Aresta, F. Nocito and A. Dibenedetto, Adv. Catal., 2018, 62, 49–111 CAS.
- X. L. Liu, J. L. Li, N. Li, B. Y. Li and X. H. Bu, Chin. J. Chem., 2021, 39, 440–462 CrossRef CAS.
- C. Claver, M. B. Yeamin, M. Reguero and A. M. Masdeu-Bultó, Green Chem., 2020, 22, 7665–7706 RSC.
- A. A. Marciniak, K. J. Lamb, L. P. Ozorio, C. J. A. Mota and M. North, Curr. Opin. Green Sustain. Chem., 2020, 26, 100365 CrossRef.
- F. D. Monica and A. W. Kleij, Catal. Sci. Technol., 2020, 10, 3483–3501 RSC.
- C. K. Ran, X. W. Chen, Y. Y. Gui, J. Liu, L. Song, K. Ren and D. G. Du, Sci. China: Chem., 2020, 63, 1336–1351 CrossRef CAS.
- B. Limburg, A. Cristofol, F. D. Monica and A. J. Kleij, ChemSuschem, 2020, 13, 6056–6065 CAS.
- S. L. Hou, J. Dong and B. Zhao, Adv. Mater., 2019, 32, 1806163 CrossRef PubMed.
- F. D. Monica, A. Buonerba and C. Capacchione, Adv. Synth. Catal., 2020, 361, 265–282 CrossRef.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- K. Huang, J. Y. Zhang, F. J. Liu and S. Dai, ACS Catal., 2018, 8, 9079–9102 CrossRef CAS.
- B. Schäffer, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- N. Yadav, F. Ssidi, D. Crespy and V. Dielia, ChemSusChem, 2019, 12, 724–754 CrossRef CAS PubMed.
- J. H. Wu, B. H. Lv, X. M. Wu, Z. M. Zhou and G. Jing, ACS Sustainable Chem. Eng., 2019, 7, 7312–7323 CrossRef CAS.
- W. Sun, M. Wang, Y. Zhang, W. Ding, F. Huo, L. Wei and H. He, Green Energy Environ., 2020, 5, 183–194 CrossRef.
- S. Suleman, H. A. Younus, Z. A. K. Khattak, H. Ullah, M. Elkadi and F. Verpoort, Mol. Catal., 2020, 493, 111071 CrossRef CAS.
- C. Sperandio, J. Rodriguez and A. Quintard, Org. Biomol. Chem., 2020, 18, 2637–2640 RSC.
- K. Naveen, H. Ji, T. S. Kim, D. Kim and D. H. Cho, Appl. Catal., B, 2021, 280, 119395 CrossRef CAS.
- Z. X. Yue, M. Pudukudy, S. Y. Chen, W. B. Zhao, J. Y. Wang, S. Y. Shan and Q. M. Jia, Appl. Catal., A, 2020, 601, 117646 CrossRef CAS.
- J. Liu, G. Q. Yang, Y. Liu, D. J. Zhang, X. B. Hu and Z. B. Zhang, Green Chem., 2020, 22, 4509–4515 RSC.
- X. Xin, H. W. Shan, T. Tian, Y. R. Wang, D. Yuan, H. P. You and Y. M. Yao, ACS Sustainable Chem. Eng., 2020, 8, 13185–13194 CrossRef CAS.
- Q. J. Wu, M. J. Mao, J. X. Chen, Y. B. Huang and R. Cao, Catal. Sci. Technol., 2020, 10, 8026–8033 RSC.
- Y. Hao, T. Tian, Y. Kang, T. Chang, X. Fu, Z. Zhu, X. Meng, B. Panchala and S. Qin, New J. Chem., 2020, 44, 15811–15815 RSC.
- H. Y. Cheng and T. Wang, Adv. Synth. Catal., 2021, 363, 144–193 CrossRef CAS.
- M. L. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- Y. J. Hao, X. L. Yan, Z. Zhu, T. Chang, X. C. Meng, X. Y. Fu, B. Panchal, L. W. Kang and S. J. Qin, Sustainable Energy Fuels, 2021, 5, 2943–2951 RSC.
- T. Ema, K. Fukuhara, T. Sakai, M. Ohbo, F. Q. Bai and J. Y. Hasegawa, Catal. Sci. Technol., 2015, 5, 2314–2321 RSC.
- J. P. Dong, C. Zhao, J. J. Qiu and C. M. Liu, J. Ind. Eng. Chem., 2020, 90, 95–104 CrossRef CAS.
- R. Yan, K. Chen, Z. J. Li, Y. Y. Qu, L. Y. Guo, H. Y. Tong, Y. Q. Li, J. Li, Y. Z. Hu and K. Guo, ChemSusChem, 2021, 14, 738–744 CrossRef CAS PubMed.
- H. Q. Yang, D. N. Zheng, J. S. Zhang, K. Chen, J. F. Li, L. Wang, J. L. Zhang, H. Y. He and S. J. Zhang, Chem. Ind. Eng. Prog., 2018, 57, 7121–7129 CAS.
- T. Chang, X. Gao, L. Bian, X. Fu, M. Yuan and H. Jing, Chin. J. Catal., 2015, 36, 408–413 CrossRef CAS.
- X. Fu, P. Xie, Y. Lian, L. He, W. Zhao, T. Chang and S. Qin, Chin. J. Catal., 2018, 39, 1854–1860 CrossRef CAS.
- M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev., 2018, 61, 214–267 CrossRef.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Z. J. Huang, F. B. Li, B. F. Chen, T. Lu, Y. Yuan and G. Q. Yuan, Appl. Catal., A, 2013, 136–137, 269–277 CrossRef CAS.
- Q. Su, J. Sun, J. Q. Wang, Z. F. Yang, W. G. Cheng and S. J. Zhang, Catal. Sci. Technol., 2014, 4, 1556–1662 RSC.
- J. Xu, F. Wu, Q. jiang, J. K. Shang and Y. X. Li, J. Mol. Catal. A: Chem., 2015, 403, 77–83 CrossRef CAS.
- J. Xu, J. K. Shang, Q. Jiang, Y. Wang and Y. X. Li, RSC Adv., 2016, 6, 55382–55392 RSC.
- D. H. Lan, H. T. Wang, L. Chen, C. T. Au and S. F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS.
- J. Xu, F. Wu, Q. Jiang and Y. X. Li, Catal. Sci. Technol., 2015, 5, 447–454 RSC.
- Z. J. Huang, F. B. Li, B. F. Chen and G. Q. Yuan, Catal. Sci. Technol., 2016, 6, 2942–2948 RSC.
- Y. Oh, V. D. Le, U. N. Maiti, J. O. Hwang, W. J. Park, J. Lim, K. E. Lee, Y. S. Bae, Y. H. Kim and S. O. Kim, ACS Nano, 2015, 9, 9148–9157 CrossRef CAS PubMed.
- T. Biswas and V. Mahalingam, New J. Chem., 2018, 41, 14839–14842 RSC.
- Y. Y. Yang, H. Li, S. P. Pei, F. Liu, W. Feng and Y. M. Zhang, RSC Adv., 2020, 10, 42408–42412 RSC.
- H. Chand, P. Choudhary, A. Kumar, A. Kumar and V. Krishnan, J. CO2 Util., 2021, 51, 101646 CrossRef CAS.
- J. Zhu, T. Diao, W. Wang, X. Xu, X. Sun, S. A. Carabineiro and Z. Zhao, Appl. Catal., B, 2017, 219, 92–100 CrossRef CAS.
- X. H. Song, Y. F. Wu, D. H. Pan, F. F. Cai and G. M. Xiao, Mol. Catal., 2017, 436, 228–236 CrossRef CAS.
- S. Zhang, H. hang, F. X. Cao, Y. Y. Ma and Y. Q. Qu, ACS Sustainable Chem. Eng., 2018, 6, 4204–4211 CrossRef CAS.
- Y. Q. Gu, R. Ping, F. S. Liu, G. Y. Zhang, M. S. Liu and J. M. Sun, Ind. Eng. Chem. Res., 2021, 60, 5723–5732 CrossRef CAS.
- E. Peris, Chem. Commun., 2016, 52, 5777–5787 RSC.
- M. Majdoub and Z. Anfar, ACS Nano, 2020, 14, 12390–12469 CrossRef CAS PubMed.
- X. J. Zhang, B. Y. Wang, Y. M. Lu, C. G. Xia and J. H. Liu, Mol. Catal., 2021, 504, 111452 CrossRef CAS.
- X. Zhang, C. C. Jia, Y. H. Xue and P. Yang, RSC Adv., 2017, 7, 43888–43893 RSC.
- J. Zhang, X. Li, Z. Zhu, T. Chang, X. Fu, Y. Hao, X. Meng, B. Panchal and S. Qin, Adv. Sustainable Syst., 2021, 5, 2000133 CrossRef CAS.
- X. Jiang, F. L. Gou, F. Chen and H. W. Jing, Green Chem., 2016, 18, 3567–3576 RSC.
- J. Y. Wang, Y. T. Liang, D. G. Zhou, J. P. Ma and H. W. Jing, Org. Chem. Front., 2018, 5, 741–748 RSC.
- Y. D. Zhang, G. J. Chen, L. Wu, K. Liu, H. Zhong, Z. Y. Long, M. M. Tong, Z. Z. Yang and S. Dai, Chem. Commun., 2020, 56, 3309–3312 RSC.
- K. Pramoda, U. Gupta, M. Chhetri, A. Bandyopadhyay, S. K. Pati and C. N. R. Rao, ACS Appl. Mater. Interfaces, 2017, 9, 10664–10672 CrossRef CAS PubMed.
- F. L. Lai, Y. Wang, D. D. Li, X. S. Sun, J. Peng, X. D. Zhang, Y. P. Tian and T. X. Liu, Nano Res., 2018, 11, 1099–1108 CrossRef CAS.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05328k |
| This journal is © The Royal Society of Chemistry 2021 |