
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrostructure and electrochemical properties of high performance graphene/manganese oxide hybrid electrodes†
Fatima Hamade ,
Emmy Radich‡
and
Virginia A. Davis*
,
Emmy Radich‡
and
Virginia A. Davis*
Department of Chemical Engineering, Auburn University, Auburn, AL, USA. E-mail: davisva@auburn.edu
First published on 24th September 2021
Abstract
Hybrids consisting of 2D ultra-large reduced graphene oxide (RGO) sheets (∼30 μm long) and 1D α-phase manganese oxide (MnO2) nanowires were fabricated through a versatile synthesis technique that results in electrostatic binding of the nanowires and sheets. Two different hybrid (RGO/MnO2) compositions had remarkable features and performance: 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MnO2/RGO (75/25 wt%) denoted as 3H and 10:1 MnO2/RGO (90/10 wt%) denoted as 10H. Characterization using spectroscopy, microscopy, and thermal analysis provided insights into the microstructure and behavior of the individual components and hybrids. Both hybrids exhibited higher specific capacitance than their individual components. 3H demonstrated excellent overall electrochemical performance with specific capacitance of 225 F g−1, pseudocapacitive and electrochemical double-layer capacitance (EDLC) contributions, charge-transfer resistance <1 Ω, and 97.8% capacitive retention after 1000 cycles. These properties were better than those of 10H; this was attributed 3H's more uniform distribution of nanowires enabling more effective electronic transport. Thermal annealing was used to produce reduced graphene oxide (RGO) that exhibited significant removal of oxygen functionality with a resulting interlayer spacing of 0.391 nm, higher D/G ratio, higher specific capacitance, and electrochemical properties representing more ideal capacitive behavior than GO. Integrating ultra-large RGO with very high surface area and MnO2 nanowires enables chemical interactions that may improve processability into complex architectures and electrochemical performance of electrodes for applications in electronics, sensors, catalysis, and deionization.
:1 MnO2/RGO (75/25 wt%) denoted as 3H and 10:1 MnO2/RGO (90/10 wt%) denoted as 10H. Characterization using spectroscopy, microscopy, and thermal analysis provided insights into the microstructure and behavior of the individual components and hybrids. Both hybrids exhibited higher specific capacitance than their individual components. 3H demonstrated excellent overall electrochemical performance with specific capacitance of 225 F g−1, pseudocapacitive and electrochemical double-layer capacitance (EDLC) contributions, charge-transfer resistance <1 Ω, and 97.8% capacitive retention after 1000 cycles. These properties were better than those of 10H; this was attributed 3H's more uniform distribution of nanowires enabling more effective electronic transport. Thermal annealing was used to produce reduced graphene oxide (RGO) that exhibited significant removal of oxygen functionality with a resulting interlayer spacing of 0.391 nm, higher D/G ratio, higher specific capacitance, and electrochemical properties representing more ideal capacitive behavior than GO. Integrating ultra-large RGO with very high surface area and MnO2 nanowires enables chemical interactions that may improve processability into complex architectures and electrochemical performance of electrodes for applications in electronics, sensors, catalysis, and deionization.
Introduction
Development of energy storage devices with higher charge storage capabilities has received considerable attention in the past decade as a result of increased environmental concerns.1 Supercapacitors offer advantages over traditional electrical double-layer capacitors and batteries due to their higher power densities. Advancements in nanomaterial research have revealed unique combinations of material properties and behaviours that are desirable for many applications. Fundamental understanding of the effects of tailoring initial dispersion parameters on final properties provides a basis for developing structure-processing-property relationships. These relationships provide insight into the material interactions and microstructures that enable fluid-phase processing and fabrication into complex architectures.Nanomaterials such as single-walled carbon nanotubes (SWNTs),2–4 activated carbon,5,6 reduced graphene oxide (RGO),7–11 and titanium carbide (Ti3C2),12–15 possess exceptional electrical, mechanical, thermal, and optical properties enabling their applications in flexible electronics, catalysts, actuators, deionization, and capacitors. They store charge by electric double-layer capacitance (EDLC), which occurs at the interface between the conductive electrode surface and electrolyte.
Graphene is a 2D single-layer exfoliated sheet of graphite with sp2 hybridized carbon in a honeycomb lattice structure. Graphene's exceptional properties as a carbon-based nanomaterial include high theoretical specific surface area, Young's modulus, intrinsic phonon mobility, electrical conductivity, and optical transmittance.16 These properties have led to its use in applications such as transparent conductive films, sensors, rechargeable lithium ion batteries, and electrochemical double layer capacitors (EDLCs).17–19 Graphene has a robust and flexible nature and can be modified or functionalized in many ways to change its electronic structure and yield different surface properties.20,21 Graphene oxide (GO) is produced by chemical exfoliation and oxidation of graphite to separate the graphene layers and introduce oxygen functional groups. Throughout the widely accepted synthesis by modified Hummer's method, the graphite is sonicated to ensure dispersibility and produces carboxyl groups along the edges and hydroxyl, carbonyl, and epoxy groups along the basal planes of GO.16,22 This results in a system with monolayer to few-layer sheets, or flakes. GO is desirable for its ability to form multicomponent functional materials due to its hydrophilicity owing to its dispersibility in water and polar solvents and long-term stability.23,24
Ultra-large GO sheets are an order of magnitude larger in aspect ratio (length/diameter) than small GO sheets (∼25000 compared to ∼2000). In contrast to the synthesis of traditional GO, ultra-large GO sheets are prepared from thermally expandable graphite25 by increasing the interlayer spacing prior to oxidation. They are not subjected to any sonication due to their higher vulnerability to breakage under shear.26,27 Ultra-large GO has been less studied than its smaller counterparts, but previous studies on the phase behaviour and rheology of ultra-large GO revealed the formation of aligned liquid crystalline regions at a dispersion concentration three orders of magnitude lower than for small GO sheets.28,29 This can enable easier processability into aligned architectures and desirable properties for 3D printing, fiber spinning, electrochemical measurements, or as fillers for nanocomposites.24,30–34
To produce a more conductive material, GO can be reduced by chemical, thermal, or electrochemical methods. While the reduction techniques explored for small GO sheets have been previously investigated in great detail,9,35–37 reduction of ultra-large GO alone and as a component in multicomponent systems has only been studied by few reports.38–40 In this work, RGO was produced by thermal annealing ultra-large GO.
Manganese oxide (MnO2) has been studied for various applications including catalysts, absorbents, biosensors, and cathodes for alkaline batteries.41–44 MnO2 is a transition metal oxide that is environmentally friendly, cost-effective, and favorable for use in neutral electrolytes compared to RuO2, IrO2, and NiO metal oxides.1,45–47 MnO2 exhibits high specific capacitance by storing charge as a pseudocapacitor, where faradaic redox reactions with charge-transfer occur on the surface or by intercalation to store charge electrochemically.48 A significant advantage of MnO2-based electrodes is their ability to function in neutral electrolytes, which is a promising characteristic for large-scale applications.44 Furthermore, previous reports have shown that transition metal oxides, including MnO2, are valuable for increasing conductivity and preventing restacking, or agglomeration, of multilayer 2D nanosheets due to their electrostatic binding interactions.49–51
MnO2 exhibits a tetragonal structure that allows intercalation of species through its tunnelling configuration. The MnO2 crystal structure consists of MnO6 octahedron units that connect to produce different crystallographic forms resulting in distinctive morphologies (shapes, phases, aspect ratios, properties).52–54 MnO2 nanomaterials can be produced in a range of shapes and sizes using a variety of synthesis methods including hydrothermal,1,55 redox,54 and sol–gel56 reactions. However, the main disadvantages of these methods include introducing cross-linking agents, long reaction times, and expensive equipment. An alternative synthesis protocol using a precipitation method previously developed for MnO2 with a double-solvent system that is versatile, low cost, safe-handling, and short reaction time was used in this work.52 Nanowires (L/D ∼60) of α-type MnO2 were synthesized in the presence of ultra-large GO sheets. MnO2 can also form lower aspect ratio needles, rods, or spindles52 with aspect ratios ranging from 2 to 10. The geometry is determined by the rate of solvent addition and reaction time.53 The needle-like (L/D ∼10) morphologies form when deionized water in the potassium permanganate (KMnO4) solution is injected quickly into the boiling isopropanol and allowed to react for 10 min.52 Rod-like morphologies (L/D ∼2) form during very slow addition of KMnO4 solution during a 50 min reaction time. Spindle-like morphologies (L/D ∼2) form when four times as much water is rapidly added and the reaction is allowed to proceed for 10 min.52 In this work, longer reaction times of 30 min were used to obtain nanowires (L/D ∼60) electrostatically bound to the ultra-large sheets. The higher aspect ratio was desirable for both helping prevent sheet restacking and enhancing transport within the electrode structure.
Combining carbon-based nanomaterials with transition metals to form multicomponent structures enables interesting electrochemical properties that cannot be achieved than with either component alone. Studies have shown that the carbon support enhances the electrochemical performance and the geometry of 1D nanomaterials facilitate electron transport.48,57 One study described the synthesis and characterization of hollow cobalt oxide (Co3O4) attached onto holey graphene for use in lithium ion battery applications.58 Interestingly, the electrochemical performance demonstrated charge–discharge capacities that were faster for the in situ holey graphene–Co3O4 than either the normally holey graphene–Co3O4 or non-holey graphene–Co3O4 electrodes, which shorten diffusion lengths for enhancing lithium ion transport.58 In this work, we investigated the combination of ultra-large reduced graphene oxide (RGO) as a support with MnO2 nanowires attached onto the RGO sheets. The synthesis routes for attachment and formation of the nanostructures along the carbon-based materials enabled formation of novel architectures with interactions governing mass transport across the electrode interface.
Integrating small RGO sheets with MnO2 nanowires has previously shown enhanced cycling behaviour and capacity as electrodes tested in lithium ion cells,59 improved electrochemical stability in a three-electrode setup,60 and improved colloidal stability that promotes nanowire alignment.61 This work explored the use of MnO2 nanowires as guest materials to form a sandwich structure with ultra-large GO sheets. The nanowire-sheet interactions occur via electrostatic binding60 that occurs by nanowire nucleation and growth53,62 along the ultra-large GO sheets with simultaneous reduction to form a hybrid material (RGO/MnO2). To the best of the authors' knowledge, only a few studies have prepared multicomponent systems with ultra-large GO. The scope of this work included characterizing the individual and multicomponent, or hybrid, materials by spectroscopy, microscopy, and thermal analysis techniques to gain insights into the microstructures and probing the electrochemical responses of as-prepared electrodes. Results from this work provide understanding of the interactions of the ultra-large RGO sheets and MnO2 nanowires as well as the effects of combining 2D/1D geometries with very different aspect ratios.
Results and discussion
The individual materials expandable graphite, ultra-large graphene oxide (GO), partially reduced graphene oxide (P-RGO), reduced graphene oxide (RGO), and manganese oxide (MnO2) were prepared as described in the experimental section. Furthermore, hybrids (RGO/MnO2) were synthesized with addition of GO to the MnO2 synthesis enabling electrostatic binding of the MnO2 nanowires (NWs) onto the GO sheets. Two different hybrid compositions were studied for comparison with MnO2/GO ratios of (1) 75/25 wt% designated as 3:1 hybrid, or 3H, and (2) 90/10 wt% designated as 10:1 hybrid, or 10H. All the materials were then characterized by multiple spectroscopy techniques, microscopy and thermal analysis to understand the material properties including functionality, sizes, crystal structure, thermal stability and transitions, and graphitic domains.
Material sizes & morphology
:1 hybrid (3H) show a uniform distribution of nanowires along the GO sheets and 10:1 hybrid (10H) show regions of more concentrated bundles or clusters of MnO2 that do not have similar 1D structures to that of 3H (Fig. 1c and d). Fig. S1† demonstrates an AFM image of another 3H hybrid along with the z-height profile, showing the sheet thickness to be ∼1 nm and nanowire thickness to be ∼9 nm.
 | ||
| Fig. 1 AFM images of (a) GO with an average sheet size of 40 μm long and ∼1 nm thick, (b) MnO2 nanowires indicating network formation, and hybrids (c) 3H that represents uniform distribution of nanowires along the GO sheets and (d) 10H that represents clusters of MnO2 attached to RGO sheets. | ||
| Material | Length – L (nm) | Diameter (thickness) – D (nm) | Aspect ratio (L/D) |
|---|---|---|---|
| GO sheets | 30000 ± 4500 |
1.3 ± 0.5 | ∼25000 |
| MnO2 NWs | 850 ± 80 | 15 ± 10 | ∼60 |
 | ||
| Fig. 2 SEM images of dried dispersions of (a) GO, (b) MnO2 and (c) 3H hybrid. The morphology of the MnO2 on the hybrid illustrates lower image contrast in areas where the nanowires are more closely bound to the RGO sheets. | ||
Thermal analysis
Thermal degradation of the materials was investigated by thermogravimetric analysis (TGA). Fig. 3 shows corresponding TGA curves for GO, MnO2, and both hybrids (3H and 10H) in argon after a 30 min isothermal hold at 120 °C to remove most of the residual moisture. GO exhibits two thermal transitions, one with an onset temperature at 150 °C indicative of the decomposition of the oxygen functional groups and the other at 450 °C indicative of breaking the carbon backbone.68,69 Each transition corresponds to approximately half of the initial mass loss. The MnO2 (enlarged in Fig. S2 in the ESI†) exhibits three thermal transitions and is more thermally stable than GO. At 200 °C, MnO2 loses adsorbed water in the amount of ∼3 wt%. Next, at 425 °C, MnO2 is converted into Mn2O3 by the reaction 4MnO2 → 2Mn2O3 + O2 that contributes to ∼5 wt% mass loss. Then at 610 °C, Mn2O3 is converted into Mn3O4 by the reaction 6Mn2O3 → 4Mn3O4 + O2, which results in ∼4 wt% mass loss. These results are consistent with previous studies of MnO2 degradation.60,70,71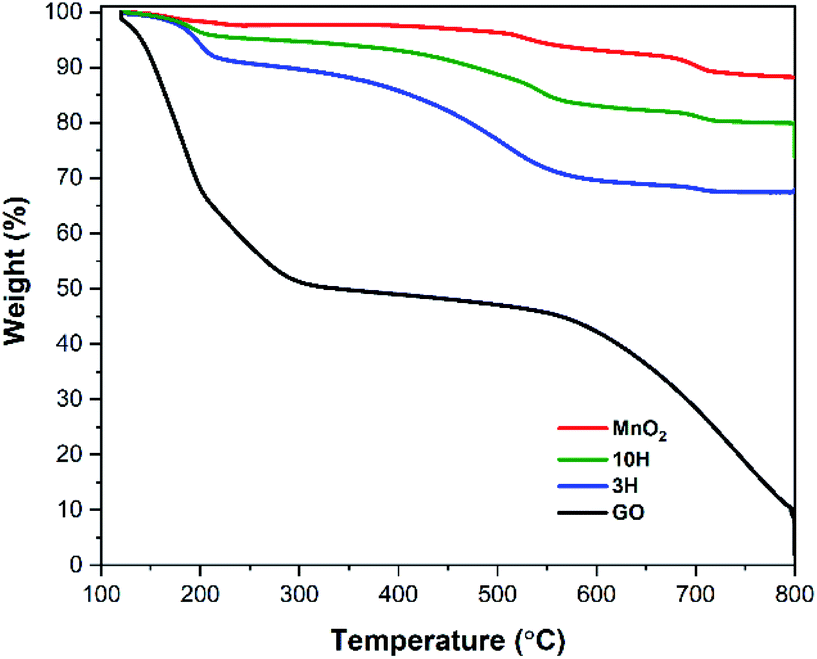 | ||
| Fig. 3 TGA performed under argon atmosphere at a rate of 5 °C min−1. GO exhibits two thermal transitions upon decomposition – loss of oxygen functionality and break carbon backbone. MnO2 is much more thermally stable than GO and is converted to Mn2O3 and then Mn3O4. The hybrid exhibits transitions consisting of both individual components. | ||
Throughout the hybrid syntheses, MnO2 nanowires are electrostatically bound to the GO sheets as illustrated from the AFM and SEM results. Therefore, the thermal degradation of the hybrids undergo the transitions of both individual components (Fig. 3). The 3H and 10H hybrids exhibited a thermal transition starting at 160 °C that resulted in amass loss of ∼10 wt% for 3H and ∼5% wt% for 10H. This was due to the combination of removing oxygen functionality from GO and adsorbed water from MnO2. The next transition occurred from 300–600 °C with a mass loss of 20 wt% for 3H and ∼11 wt% for 10H; this transition is attributed to conversion of MnO2 → Mn2O3 and possible partial destruction of the carbon skeleton. The onset of carbon breakdown at a lower temperature in the hybrids than GO (550–800 °C) may have occurred because of the presence of MnO2 and its phase changes, weaken the RGO sheets. The mass ratio of MnO2/RGO in the hybrids was calculated by taking the final mass after complete degradation at 800 °C divided by the initial mass at 120 °C. This showed the 3H hybrid consisted of a MnO2/RGO ratio of 3:1 (75% MnO2/25% RGO by wt), while 10H consisted of 10:1 (90% MnO2/10% RGO by wt). The residual mass of the hybrids being greater than that of the GO was due to the presence of the more thermally stable MnO2.
Spectroscopy study
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and a shoulder at 300 nm indicative of the n → π* transition of electrons in CO bonds connected to the CC bond in the GO plane.23 The shoulder in GO is a signature of the presence of oxygen functional groups in the material. The MnO2 spectra exhibits a d–d transition at a broad absorption band between 350 and 500 nm with the peak position at 410 nm of the manganese (Mn) ions from lower to higher d-orbital energy state in the MnO2 crystal.52 The UV-vis spectrum of the 3H hybrid shows a blue-shift in the conjugated carbon peak to 215 nm corresponding to GO sheet exfoliation60 and a broad Mn peak at 410 nm. Interestingly, 10H primarily exhibited a sharp Mn–O transition at 410 nm and much higher absorption values for the whole spectrum than GO and 3H, this demonstrates some restoration of the sp2 carbon framework.9,35 A control experiment was performed with only GO and the manganese salt as precursors (excluding KMnO4 addition) to determine the extent of GO reduction through the hybrid synthesis. The UV-vis spectra showed the electronic transitions present in the individual materials as well as some reduction of GO in both hybrid materials. These results indicate that the GO was not fully reduced by chemical reduction during the hybrid synthesis but formed a reduced graphene oxide (RGO) material (Fig. S3a in the ESI†).
C, and a shoulder at 300 nm indicative of the n → π* transition of electrons in CO bonds connected to the CC bond in the GO plane.23 The shoulder in GO is a signature of the presence of oxygen functional groups in the material. The MnO2 spectra exhibits a d–d transition at a broad absorption band between 350 and 500 nm with the peak position at 410 nm of the manganese (Mn) ions from lower to higher d-orbital energy state in the MnO2 crystal.52 The UV-vis spectrum of the 3H hybrid shows a blue-shift in the conjugated carbon peak to 215 nm corresponding to GO sheet exfoliation60 and a broad Mn peak at 410 nm. Interestingly, 10H primarily exhibited a sharp Mn–O transition at 410 nm and much higher absorption values for the whole spectrum than GO and 3H, this demonstrates some restoration of the sp2 carbon framework.9,35 A control experiment was performed with only GO and the manganese salt as precursors (excluding KMnO4 addition) to determine the extent of GO reduction through the hybrid synthesis. The UV-vis spectra showed the electronic transitions present in the individual materials as well as some reduction of GO in both hybrid materials. These results indicate that the GO was not fully reduced by chemical reduction during the hybrid synthesis but formed a reduced graphene oxide (RGO) material (Fig. S3a in the ESI†).
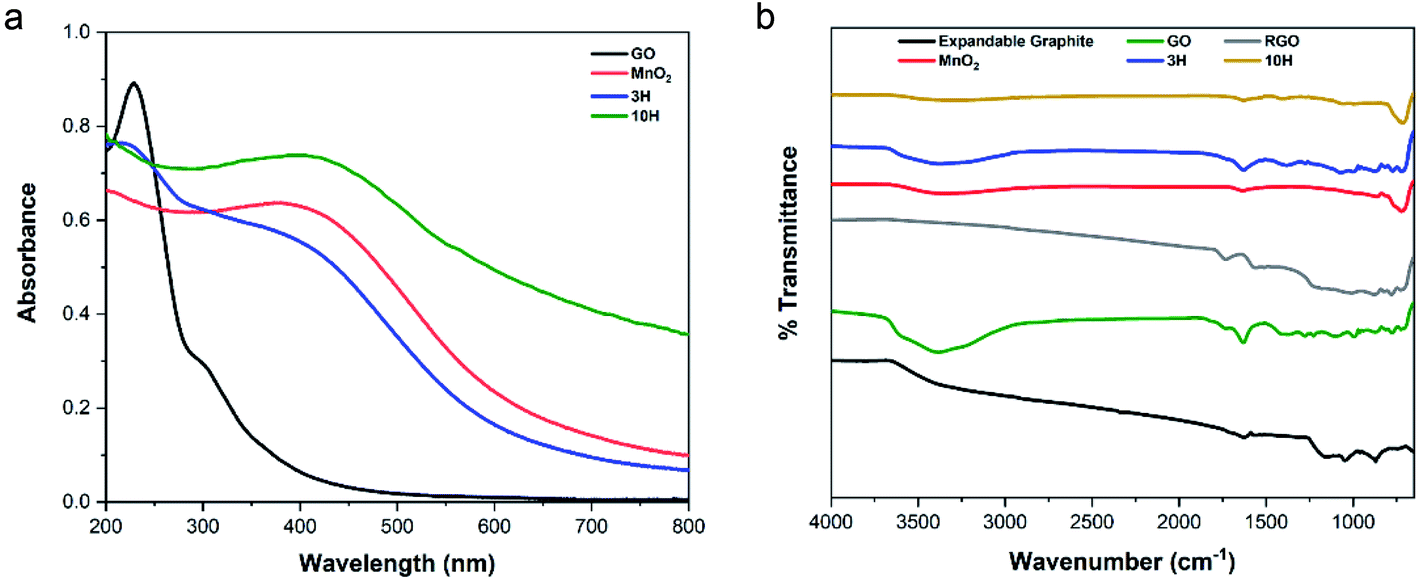 | ||
| Fig. 4 (a) UV-vis spectra indicating electronic transitions of ultra-large GO, MnO2, and both hybrids 3H (75% MnO2) and 10H (90% MnO2). Presence of oxygen functional groups in GO occurs at the shoulder of 300 nm. The 3H hybrid spectrum consists of the conjugated carbon peak blue-shifted to 215 nm ascribed to GO exfoliation and indication of the Mn–O transition with a broad peak at 410 nm. The higher MnO2 content of 10H primarily indicates the Mn–O transition. (b) FTIR spectra of expandable graphite, GO, RGO, and hybrids reveal the presence of oxygen functionality (hydroxyl, carboxyl, carbonyl, and epoxy) for GO, 3H, and 10H with 10H exhibiting the least intensities. FTIR spectra of MnO2 indicates hydroxyl functionality in its crystal lattice and contributes to both hybrid spectra. The RGO produced by thermal annealing of GO demonstrates no evident oxygen functionality similar to the expandable graphite (large flakes) used as a precursor to synthesizing GO and RGO in this work, indicating successful reduction. | ||
C) present in the expandable graphite, GO, RGO, and both hybrids. The oxygen functional groups occur at the following peaks: 3387 cm−1 (O–H stretch), 1730 cm−1 (CO, carboxyl), 1374 cm−1 (C–O, carbonyl), and 1227 cm−1 (C–O–C, epoxy). These peaks are clearly shown in the GO spectra but had lower intensities for 3H, and even lower intensities for 10H, due to the higher MnO2/RGO ratio. The MnO2 spectra indicate the presence of O–H groups in the crystal lattice via a broad stretching vibration at 3340 cm−1 and bending vibration at 1633 cm−1 (Fig. 4b). The P-RGO (Fig. S3b in the ESI†) spectra also included the conjugated carbon peak at 1630 cm−1 as well as low intensity peaks for the oxygen functionalities. This further confirms the UV-vis results which indicated that the GO was not completely reduced during the hybrid syntheses. For comparision, RGO was prepared by thermal annealing the ultra-large GO sheets. The RGO FTIR spectrum indicated complete removal of the hydroxyl peak and no other evident functionality.sinθ, and utilizing a Cu kα source with λ = 1.54 Å = 0.154 nm, the d-spacing of expandable graphite was 0.342 nm. GO exhibits an intense peak at 2θ = 9.9°, which corresponds to the (001) reflection indicative of oxygen functionality. The interlayer spacing of ultra-large GO was 0.892 nm as a result of the formation of functional groups resulting from oxidation. These values are in agreement with the literature.29 The XRD spectrum of the thermally annealed RGO produced in this work is shown in Fig. 5b. It exhibits loss of the (001) peak and the presence of a (002) peak at 2θ = 22.7°. The interlayer spacing calculated for RGO was 0.391 nm further confirming that the thermal annealing procedure successfully removed the majority of oxygen functionality from the GO and resulted in RGO.
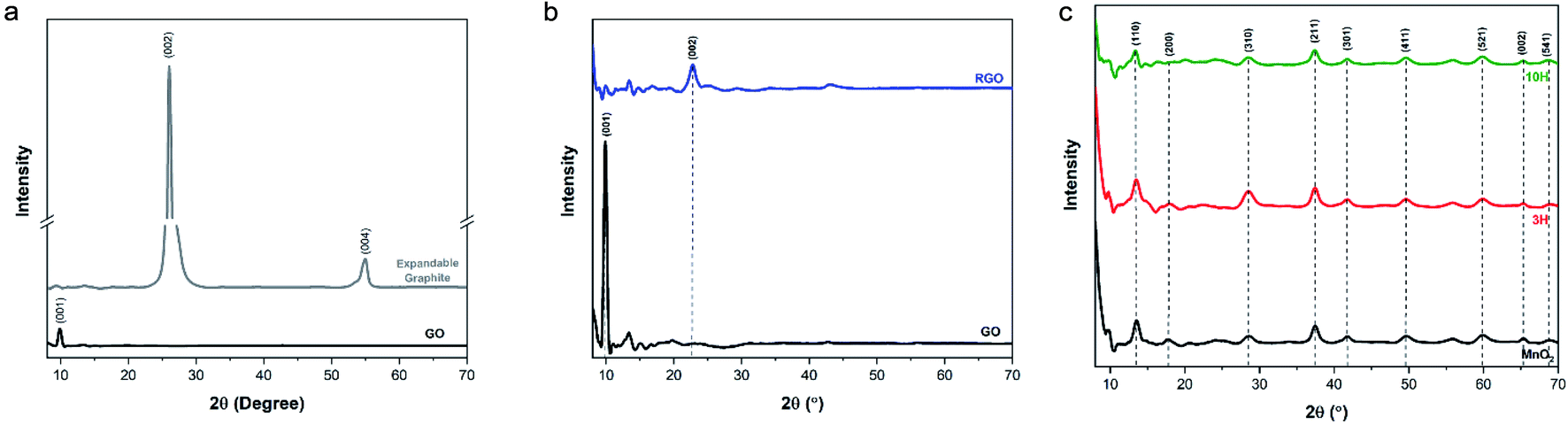 | ||
| Fig. 5 XRD patterns of the materials at slow scan rate of 0.01° s−1. (a) The expandable graphite exhibits two characteristic peaks that correspond to a d-spacing of 0.342 nm and GO exhibits an intense peak at 2θ = 9.9° with calculated d-spacing of 0.892 nm indicating oxidation of the sheets. (b) RGO spectrum exhibits a (002) peak at 2θ = 22.7° with calculated d-spacing of 0.391 nm, conveying successful reduction of graphene oxide. (c) The spectrum of MnO2 confirms formation of the α-phase crystal structure and both hybrids (3H and 10H) display a similar pattern. | ||
The diffraction spectrum of the produced MnO2 nanowires had similar peak identifications as those shown in previous reports52,54 and corresponded to the powder diffraction file from JCPDS 44-0141 (Fig. 5c). This confirms formation of the tetragonal α-phase structure of MnO2. The diffraction peaks of both hybrids (Fig. 5c) were almost identical to those of the MnO2 nanowires; the peak intensity of the (001) reflection of GO diminished significantly (see Fig. S4† showing a closer look at lower 2θ angles). This has also been previously observed with synthesizing RGO/MnO2 with small sheets.60
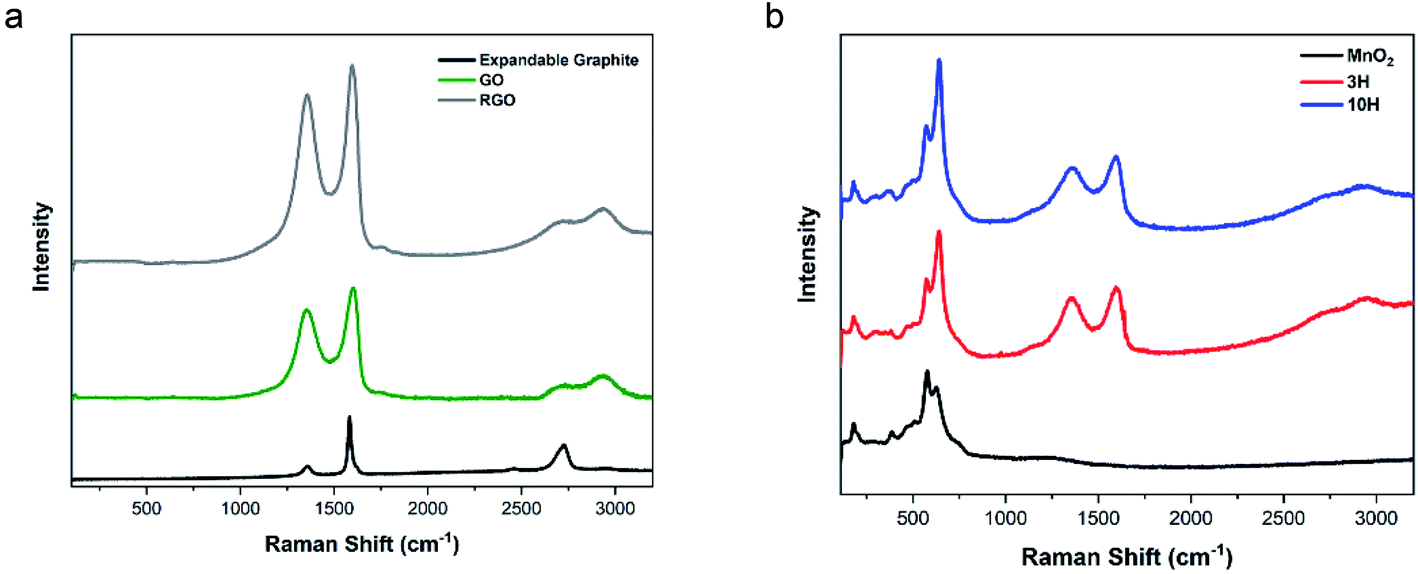 | ||
| Fig. 6 Raman spectra performed using a 514 nm laser. (a) Expandable graphite, ultra-large GO, and RGO result in D/G ratios of 0.22, 1.46 and 1.72, respectively. (b) The spectrum of MnO2 confirms α-phase morphology produced from potassium-based reactive species. This is also evident in both hybrid spectra. The 3H and 10H hybrid materials exhibit D/G ratios of 1.65 and 2.20, respectively. The ratio of Mn–O to G-peak is higher for 10H than 3H, further revealing the presence of more MnO2 content. | ||
The Raman spectrum of MnO2 consists of multiple peaks in the region of 100–1000 cm−1 (Fig. 6b). Magnification of this low wavenumber region is presented in Fig. S5.† In agreement with the literature,70 the Mn–O lattice vibrations result in more intense peaks at 180, 382, 579, and 629 cm−1 and five weak peaks at 281, 322, 463, 504, and 745 cm−1. These Raman shifts correspond to translational motion of the MnO6 octahedra framework at 180 cm−1, Mn–O bending vibrations at 380 cm−1, tunneling species at 579 and 629 cm−1, and antisymmetric Mn–O stretching at 745 cm−1.82 These results combined with the XRD show that the hybrids contained α-phase MnO2.
The hybrids showed peaks for both MnO2 and RGO. The values of D/G for 3H and 10H are 1.65 and 2.20, respectively. The initial GO has oxygen functionality that act as a support or scaffold for facilitating the reaction with the manganese ions from MnCl2 and KMnO4 to produce MnO2 attached to the RGO sheets. The 10H exhibited a higher ratio of the Mn–O to G-peak than 3H, further confirming the AFM and TGA data. The higher MnO2 in the 10H could have caused some clusters of MnO2 to form along the RGO sheets and prevented the longitudinal growth into nanowires and resulted in the clusters shown in Fig. 1D. The higher D/G ratio of 10H compared with 3H indicates more disorder in the carbon structure and an increase in restoration of the aromatic ring by forming many smaller sp2 domains.39,73,78 Furthermore, the content of MnO2 can be estimated by comparing the ratio of MnO2/RGO in the hybrid spectra.60,83,84
In summary, the microscopy, thermal, and spectroscopy analyses performed on the individual (expandable graphite, GO, RGO, P-RGO, and MnO2) and multi-component materials (3H and 10H) show that for the first time, intercalated graphite can be used to produce α-phase MnO2 attached to RGO sheets.
Electrochemical capacitive behavior
GO, RGO, MnO2 and 3H and 10H hybrids were fabricated into supercapacitor electrodes and characterized by cyclic voltammetry (CV) at varying scan rates to determine their electrochemical response and explore potential applications in energy storage. The electrochemical properties were measured using a three-electrode configuration in 1 M Na2SO4 electrolyte. Cyclic voltammetry was performed in the potential range of −0.2–0.8 V at scan rates of 5, 10, 20, 50, and 100 mV s−1.Fig. 7 shows the CV curves for all materials at the different scan rates. GO is a supercapacitive material that exhibits electrochemical double layer capacitance (EDLC), where non-faradaic reactions take place at the electrode surface. GO is not conductive; therefore, it has very low specific capacitance. However, RGO demonstrated higher capacitance than GO (Fig. 7b). In contrast to GO, manganese oxide (MnO2) is a transition metal oxide that exhibits a faradaic process along the surface of the electrode material, owing to its pseudocapacitive nature. Fig. 7c illustrates the quasi-rectangular shapes of the MnO2 CV curves, representing pseudocapacitive behavior. The combination of RGO and MnO2 in the hybrid materials enabled contributions of both pseudocapacitive and EDLC behavior as shown in the CV curves of Fig. 7d and e. The 10H CV curves look similar to the MnO2 curves, indicating more pseudocapacitive effect from the higher MnO2 content in 10H than 3H. An overlay of all the materials at the lowest scan rate of 5 mV s−1 is shown in Fig. 7f. This captures the shapes of the curves together with no obvious oxidation/reduction peaks.
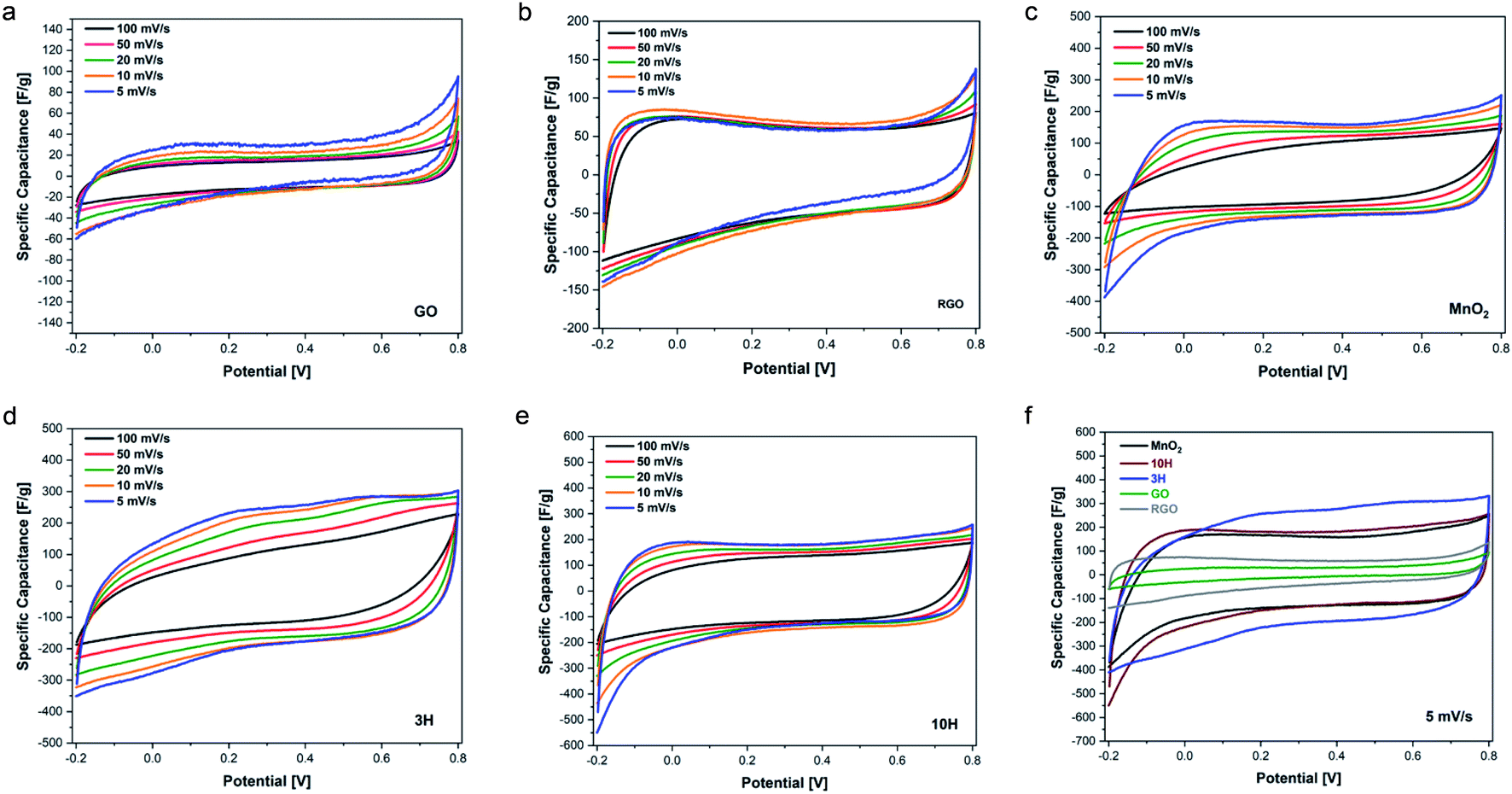 | ||
| Fig. 7 Cyclic voltammetry curves at different scan rates of (a) GO, (b) RGO, (c) MnO2, (d) 3H (75% MnO2/25% RGO by wt), and (e) 10H (90% MnO2/10% RGO by wt). (f) An overlay of the CV curves of all materials at 5 mV s−1 scan rate. The data is representative of ten cycles performed on two electrodes of similar mass with <10% difference in capacitance. | ||
The rate capability for the materials is shown in Fig. 8a. The RGO prepared in this work from ultra-large GO via thermal annealing at 300 °C demonstrates higher specific capacitance than GO. The higher RGO performance confirms the previously described physical characterization data showing that thermal annealing was successful in removing oxygen functional groups. These results suggest that increasing the annealing temperatures up to 1000 °C may result in enhanced electrical conductivity as has previously been found for small sheets of GO.9,78,79 The capacitive performance of MnO2 exceeds that of RGO. MnO2 consists of an octahedron configuration and is well-known for its exceptional performance due to its intricate tunnelling structure that enables more efficient ion transfer.
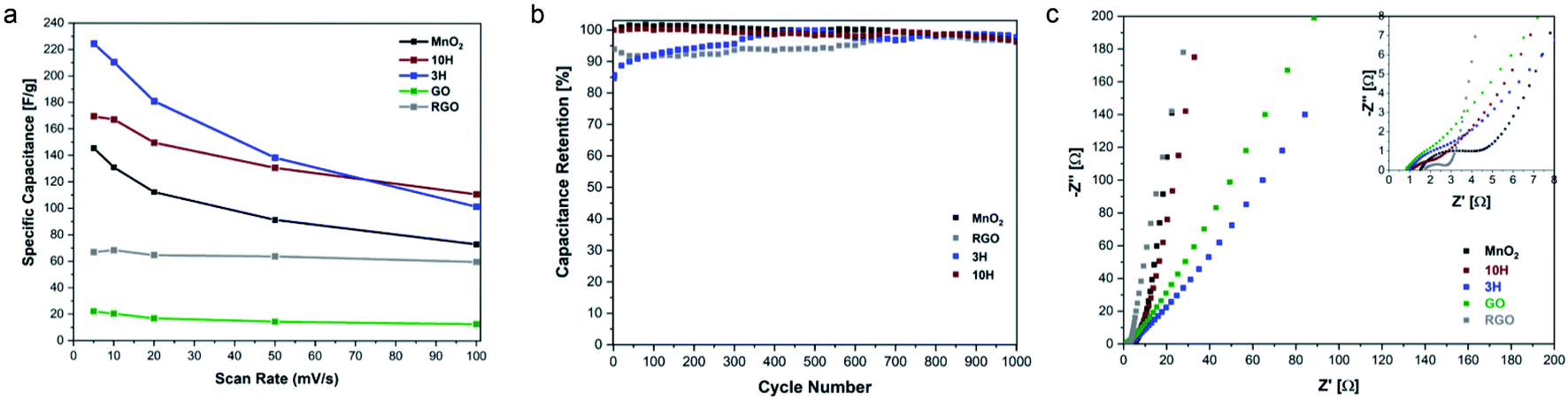 | ||
| Fig. 8 Electrochemical behavior with cyclability and EIS. (a) Comparison of specific capacitance as a function of scan rate for all materials. (b) Cycling behavior over a range of 1000 CV cycles performed at 20 mV s−1 scan rate. After 1000 cycles, the materials exhibit >95% capacitance retention. (c) Nyquist plot overlay of electrodes in 1 M Na2SO4 aqueous solution with inset showing high frequency region. | ||
Interestingly, the hybrids exhibit higher capacitance than MnO2 alone; this is attributed to more effective transport in the complex structure enabling faster charge transfer. Previous reports of combining carbon and transition metal systems, such as PdAgNTs57 and PdW/C,48 also convey superior electrochemical performance measurements compared to the individual components without the carbon support. This is due to the support providing high surface areas and active sites for electrode interactions. At the lowest scan rate studied, MnO2 has a specific capacitance of 145 F g−1, 3H exhibits a specific capacitance of ∼225 F g−1, and that of 10H is 170 F g−1. The lower capacitance of 10H compared to 3H may be due to the increased defects that are introduced during synthesis when more MnO2 is bound to the ultra-large sheets since defects and resulting morphology affect electronic transport properties. This was evident with the higher D/G ratio of 10H as demonstrated in the Raman spectroscopy results.
The cycling behavior for the individual and hybrid electrodes was also investigated to understand the electrochemical stability of the materials. Repeated CV cycles at 20 mV s−1 scan rate were performed for the conductive materials MnO2, RGO, 3H and 10H. The capacitance retention of these materials over the course of 1000 cycles is shown in Fig. 8b. The results from the cycling studies show that MnO2 retained 96.5% capacitance, while RGO retained 96.0%, 3H retains 97.8%, and 10H retained 96.4%. This work was inspired in part by few other studies that have investigated the electrochemical behavior of a hybrid of graphene oxide and manganese oxide materials. However, our work is unique in its use of ultra-large graphene oxide sheets (∼30 μm long) which produce larger active surface areas for ion and electron diffusion compared to the smaller graphene oxide sheets (∼2 μm long). Chen et al. reported a graphene–MnO2 supercapacitor exhibited 197 F g−1 specific capacitance and after 1000 cycles, the hybrid in their work retained 84% capacitance.60 In other work, an MnO2/RGO electrode produced via a hydrothermal method decorating MnO2 onto the graphene had pseudocapacitive and double-layer contributions with the nearly ideal rectangular CV shapes and 205 F g−1 specific capacitance at 5 mV s−1 scan rate.85 That work demonstrated ∼85% capacitance retention after 1000 charge/discharge cycles. Furthermore, the cycling stability of an asymmetric supercapacitor of MnO2/rGO hybrid after 1000 cycles remained near 80%.86 Interestingly, our hybrid electrodes demonstrated higher cycling stability which conveys longer lifetime with the 3H MnO2/RGO hybrid electrode also demonstrating a high specific capacitance of 225 F g−1.
The electrochemical performance of the MnO2 and hybrid electrodes was further investigated with an electrochemical impedance spectroscopy (EIS) study (Fig. 8c). The Nyquist profiles in the high frequency region indicate the solution resistance Rs as shown by the intercept of the real axis, which is related to the ionic resistance from the salt solution, contact resistance at the solution/electrode interface, and intrinsic resistance. Furthermore, the high frequency region illustrates a small quasi-semicircle, representing the charge-transfer resistance Rct. For GO, RGO, MnO2, 3H, and 10H, the values for Rs are 0.846, 1.75, 1.53, 1.01, and 1.15 Ω, respectively. The values for Rct can be observed from the inset in Fig. 8c where the straight line intersects the real axis. For all the materials studied, Rct is <3 Ω, which demonstrates excellent charge-transfer resistance. Rct for the hybrids is ∼1 Ω, illustrating higher electrical conductivity that enabled faster ion and electron diffusion than for the individual components which serves as an advantage of combining different morphologies. The Nyquist plots in the low-frequency region represent the diffusion resistance, which shows linear characteristics for the materials. The more vertical the diffusing line is along the imaginary axis, the more representative of ideal capacitive behavior. Comparing the GO and RGO results, it is evident that the RGO demonstrated electrochemical behavior closer to that of an ideal capacitor. The lower charge-transfer resistance and combination of pseudocapacitive and EDLC behavior of the hybrids provide advantages as electrode materials compared to their individual components.
EIS was performed after the 1000 CV cyclability study to determine the changes in resistivity (Fig. S6 in the ESI†). There was not a significant change in charge-transfer resistance for the hybrids. Interestingly, in the low-frequency region of the Nyquist profile, the linear characteristics of MnO2 and 10H exhibited a shift to a lower phase angle indicating a deviation from ideal capacitive performance. However, 3H exhibited a shift to a higher phase angle, closer to the imaginary axis, which can be attributed to the higher capacitance retention achieved than the other electrodes studied. The electrochemical behavior studies showed superior performance for the 3H hybrid electrodes, with 225 F g−1 specific capacitance, 97.8% capacitance retention after 1000 cycles, very low charge-transfer resistance, and contributions of pseudocapacitive and EDLC components.
Performance discussion
The 3H hybrid had excellent electrochemical performance compared to the 10H hybrid. In both cases, inclusion of MnO2 with ultra-large RGO results in the contributions of both materials to pseudocapacitance and EDLC thereby enhancing performance compared with the individual components. AFM (Fig. 1 and ES1†) and SEM images (Fig. 2) illustrate the morphology and microstructures of the materials with MnO2 attached to the sheets. The results indicate single to few-layer RGO sheets with bound MnO2, indicating successful synthesis of the hybrid with ultra-large sheets. The 3H had discrete nanowires on the sheets while the 10H had bundles of MnO2 on the sheets. TGA (Fig. 3) profiles quantified the higher MnO2 content in 10H as well as the possible breakdown of the carbon backbone at an earlier temperature compared to GO. This may be the result of the MnO2 causing the structure to weaken as oxygen reduction and MnO2 nucleation and growth occur through the hybrid synthesis. Next, the spectroscopy investigations provided insights to the functionality, crystal structure, reduction and defects for the materials (Fig. 4 and 5). Interestingly, 10H exhibited a much higher D/G ratio than 3H (Fig. 6), which suggests that the inclusion of a higher MnO2 content resulted in a more defective graphene structure as more MnO2 crowd the binding sites on the sheets. Therefore, the higher capacitive performance of 3H than 10H is attributed to the nanowire structure enabling faster electrode kinetics and diffusion compared to the clustered structure (Fig. 7 and 8).Conclusions
A novel hybrid material consisting of α-type manganese oxide (MnO2) nanowires anchored along ultra-large reduced graphene oxide (RGO) sheets, comprising of ∼30 μm long sheet size and higher specific surface areas than traditional RGO sheets, was successfully fabricated. Microscopy, spectroscopy, and thermal analysis characterizations of the individual components contributed to understanding the microstructural development and behavior of the hybrids. Electrochemical results showed that the 3H hybrid electrode consisting of MnO2/RGO (75/25 wt%) exhibited both electrochemical double-layer capacitance and pseudocapacitive contributions as well as low internal resistance (Rct ∼1 Ω). Furthermore, it demonstrated excellent specific capacitance of up to 225 F g−1 with 97.8% cycling stability after 1000 cycles. The outstanding electrochemical performance can be attributed to the uniform distribution of the nanowires along the sheets, suppressing restacking of the RGO sheets, as well as the ultra-large active surface area that reduced the ion and electron diffusion lengths and improved charge transfer. The combination of 2D and 1D nanomaterials from this work highlight that such hybrid materials are promising candidates for potential applications in electronic devices, catalysts, and sensors.Experimental
Ultra-large graphene oxide (GO) synthesis
Expandable graphite flakes from Asbury Graphite Mills USA (3772 grade) were placed in a cylindrical alumina crucible and microwaved (Panasonic, NE-12523) on high power for 30 seconds as a thermal treatment to increase the interlayer spacing of the layers within the graphite structure. Caution: only 50 mg expandable graphite was used at once in the microwave to prevent fire or explosion hazards and microwaving must be performed in the hood due to release of toxic fumes. This expansion was followed by using a modified Hummer's method to obtain GO.22,30 This resulted in very low density expanded graphite that was then transferred to an Erlenmeyer flask. Briefly, 100 mL of 93–98% sulfuric acid (H2SO4) was added to a flask containing 500 mg expanded graphite and stirred continuously at room temperature for three days. Next, 5 g of crystalline potassium permanganate (KMnO4) was added dropwise very slowly using a spatula while stirring the mixture. Caution: in the presence of strong acid, KMnO4 reacts to form dimanganeseheptoxide (Mn2O7) which is known to detonate at temperatures above 55 °C. Therefore, this step must be performed with very small amounts added to the mixture each time and the temperature monitored and kept at, or below, 22 °C to prevent a runaway reaction. After three days, the mixture became very viscous and dark green, indicating that most of the KMnO4 had reacted. The mixture was then placed in an ice bath with a thermometer to monitor reaction temperature upon water and hydrogen peroxide additions. The temperature was kept below 22 °C (68 °F) to prevent formation of a highly exothermic reaction during the addition of water to acid. 100 mL of deionized water was carefully added to the mixture using a pipet (about 1 mL min−1), thus changing the mixture color to dark red. Once the mixture was diluted with water, 25 mL of 30% by weight hydrogen peroxide (H2O2) solution was added slowly to react the leftover potassium permanganate and manganese oxide complexes that formed throughout the synthesis. This resulted in a light brown ultra-large GO mixture. The mixture was centrifuged with a hydrochloric acid solution (9:1 ratio water/hydrochloric acid by volume) and then multiple deionized water washes to achieve a pH value of ∼6.
Reduced graphene oxide (RGO) preparation
Concentrated dispersions (sludges) of GO were vacuum dried at ∼80 °C overnight. Dried samples were placed in aluminum foil boats. Samples were annealed in a tube furnace under nitrogen flow at a ramp rate of 5 °C min−1 from 25 °C to desired temperature 300 °C and then held at desired temperature for 2 h. Samples were then cooled before removing them from the tube furnace.Manganese oxide nanowires (MnO2 NWs) synthesis
The simple double-solvent reacting system previously reported by Chen et al.52 was used to produce the manganese oxide (MnO2) nanowires. Briefly, 450 mg of MnCl2·4 H2O was added to 80 mL of isopropanol (IPA) in a three-neck round-bottom flask and bath sonicated to dissolve the manganese salt. The mixture was then brought to reflux conditions (∼80 °C) while vigorously stirring and monitoring temperature with a thermocouple. 250 mg of crystalline potassium permanganate (KMnO4) was dissolved in 8 mL of deionized water and rapidly injected into the mixture. Caution: KMnO4 is highly reactive and should be handled very carefully when adding the solution to the syringe and injecting into the reaction mixture. The reaction proceeded for 30 min at a temperature of ∼83 °C. This resulted in a black precipitate of nanowires. Precise control of the shape and size of the MnO2 strongly depends on the reaction parameters, which affect the morphology of the product and its properties upon processing. A two-stage centrifugation technique was employed to purify as well as reduce aggregation in the final product. The precipitate was placed in conical centrifuge tubes and initially centrifuged at 4700 × g for 20 min. The bottoms were then dispersed in 200 proof ethanol and centrifuged at 47 × g for 3 min. The low centrifugation rate caused aggregates to settle to the bottom of the tubes, and allowed dispersed nanowires to remain in the supernatant. Polarized optical microscopy images of different parts of the tube were sampled to qualitatively determine removal of aggregates as previously reported by a study with silver nanowires.87 The supernatant was decanted into new centrifuge tubes and then centrifuged at 4700 × g for 20 min. This completed the first ethanol wash. The second ethanol wash was then performed by dispersing the bottoms of the tubes from the first wash and then centrifuging at 4700 × g for 20 min. A third ethanol wash was performed in the same manner as the second wash. The bottoms were then dispersed in water and washed multiple times at 4700 × g. The final product was then dispersed in water or dried in a vacuum oven for characterization.Hybrid (MnO2/RGO) synthesis
A hybrid of GO/MnO2 was synthesized by growth of the NWs along the ultra-large GO sheets. The hybrid synthesis method has been previously reported with small-sized GO sheets, but to the authors' knowledge had not previously been performed on ultra large sheets.59 100 mg of GO dispersed in 20 mL IPA was added to 450 mg manganese chloride salt dissolved in 60 mL IPA in a three-neck round-bottom flask. The previously described procedure for synthesizing and purifying the NWs alone was followed to form the final hybrid product, denoted as 3H for a 3:1 ratio of MnO2/RGO (75/25 wt%). Another composition of the hybrid was also synthesized for comparison, where the GO mass was reduced to achieve a 10:1 ratio of MnO2/RGO (90/10 wt%), denoted as 10H. As a control experiment to determine the extent of GO reduction in the hybrid, partially reduced graphene oxide (P-RGO) was also synthesized by taking 100 mg of GO dispersed in IPA and following the MnO2 synthesis excluding the injection of the KMnO4 solution (reactive species).
Electrodes
Dispersions of samples were dried under vacuum at 60 °C for 12 hours to remove residual adsorbed water. The electrodes were prepared by mixing 80 wt% samples, 15 wt% carbon black (CB), and 5 wt% polytetrafluoroethylene (PTFE) slurry. The mixtures were then pressed onto nickel foam with active surface area of 1 cm2 and dried before soaking for 1 day in 1 M Na2SO4 electrolyte.Materials characterization
Tapping-mode atomic force microscopy (AFM) was performed on an Anton Paar Tosca 400 to visualize nanomaterial sizes and morphology. AFM samples were prepared by drop-casting a very low dispersion concentration onto a silicon wafer with silicon oxide layer then heating for ∼1 min to quickly remove the solvent (water). The morphology and material architecture was also investigated by field emission scanning electron microscopy (FE-SEM) using a JEOL JSM-7000F. Dispersions drop-cast onto silicon wafers at ∼70 °C, which were then adhered to cylindrical SEM stubs using carbon tape. All SEM samples were sputtered with gold to increase conductivity for obtaining high resolution images. Ultraviolet-visible (UV-vis) spectroscopy measurements were performed using on a Cary 60 (Agilent Technologies) using 10 mL cuvettes. Fourier transform infrared radiation (FTIR) spectroscopy with attenuated total reflectance (ATR) on a Thermo Scientific Nicolet iS10 was performed on samples of dried dispersions on a germanium crystal. Thermogravimetric analysis (TGA) was performed on a TA Instruments Q50 to investigate thermal degradation profiles with at least 10 mg of solid material in a platinum pan under argon atmosphere. The degradation profile was measured by heating at a rate of 10 °C min−1 to 120 °C, holding isothermal for 30 min to remove residual adsorbed water, heating at 5 °C min−1 to 800 °C, and then holding isothermal for 45 min. Raman spectra were collected on a Renishaw inVia Raman microscope using a 514 nm laser. X-ray diffraction was performed on AXRD+ Powder Diffractometer (Proto Manufacturing) with a Cu Kα radiation (λ ≈ 1.54 Å) at a slow scan rate of 0.01° s−1 of diffraction angles 2θ from 5 to 70°.Electrochemical measurements
Once the materials were characterized, electrodes were fabricated for electrochemical measurements. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were carried out using a Gamry Interface 1000 E Potentiostat. The electrochemical experiments were performed in a three-electrode mode, including a silver/silver chloride (Ag/AgCl) reference electrode, platinum (Pt) wire counter electrode, and working electrode composed of sample/CB/PTFE (80/15/5% by wt) dried on nickel foam current collector. The electrolyte tested in this work is 1 M Na2SO4. The specific capacitance (F g−1) was calculated from the CV curves by the following equation:where I is current (A), ν is scan rate (V s−1), ΔV is potential window (V), and m is the mass (g).
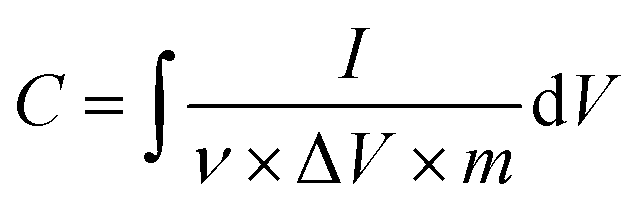
Author contributions
Davis and Radich contributed conceptualization, project administration, resources, and supervision. Hamade conducted the methodology and writing the original draft. All authors contributed to formal analysis, validation, reviewing and editing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The research project was financially supported by the U.S. Department of Education Graduate Assistance in Areas of National Need Award Number P200A180002. The authors would also like to acknowledge Dr Tae-Sik Oh from the Auburn University Department of Chemical Engineering for permission to use the XRD and Dr Majid Beidaghi from Auburn University's Materials Engineering Program in the Mechanical Engineering Department for useful discussions and assistance with the electrochemical characterization.Notes and references
- V. Subramanian, H. Zhu, R. Vajtai, P. M. Ajayan and B. Wei, J. Phys. Chem. B, 2005, 109, 20207–20214 CrossRef CAS PubMed.
- V. A. Davis, L. M. Ericson, A. N. G. Parra-Vasquez, H. Fan, Y. Wang, V. Prieto, J. A. Longoria, S. Ramesh, R. K. Saini, C. Kittrell, W. E. Billups, W. W. Adams, R. H. Hauge, R. E. Smalley and M. Pasquali, Macromolecules, 2004, 37, 154–160 CrossRef CAS.
- P. K. Rai, R. A. Pinnick, A. N. G. Parra-Vasquez, V. A. Davis, H. K. Schmidt, R. H. Hauge, R. E. Smalley and M. Pasquali, J. Am. Chem. Soc., 2006, 128, 591–595 CrossRef CAS PubMed.
- R. R. Kohlmeyer, M. Lor, J. Deng, H. Liu and J. Chen, Carbon, 2011, 49, 2352–2361 CrossRef CAS.
- Z. Chen, H. Zhang, C. Wu, L. Luo, C. Wang, S. Huang and H. Xu, Desalination, 2018, 433, 68–74 CrossRef CAS.
- F. Rodríguez-reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- B. Jia and L. Zou, Chem. Phys. Lett., 2012, 548, 23–28 CrossRef CAS.
- J. E. Kim, J. H. Oh, M. Kotal, N. Koratkar and I. K. Oh, Nano Today, 2017, 14, 100–123 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- M. Kotal, J. Kim, J. Oh and I.-K. Oh, Front. Mater., 2016, 3, 29 Search PubMed.
- X. Huang, C. L. Tan, Z. Y. Yin and H. Zhang, Adv. Mater., 2014, 26, 2185–2204 CrossRef CAS PubMed.
- K. Maleski, V. N. Mochalin and Y. Gogotsi, Chem. Mater., 2017, 29, 1632–1640 CrossRef CAS.
- J. Orangi, F. Hamade, V. A. Davis and M. Beidaghi, ACS Nano, 2020, 14, 640–650 CrossRef CAS PubMed.
- P. Sobolciak, A. Ali, M. K. Hassan, M. I. Helal, A. Tanvir, A. Popelka, M. A. Al-Maadeed, I. Krupa and K. A. Mahmoud, PLoS One, 2017, 12, 18 CrossRef PubMed.
- M.-Q. Zhao, M. Torelli, C. E. Ren, M. Ghidiu, Z. Ling, B. Anasori, M. W. Barsoum and Y. Gogotsi, Nano Energy, 2016, 30, 603–613 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- E. Mitchell, J. Candler, F. De Souza, R. K. Gupta, B. K. Gupta and L. F. Dong, Synth. Met., 2015, 199, 214–218 CrossRef CAS.
- F. Bonaccorso, A. Bartolotta, J. N. Coleman and C. Backes, Adv. Mater., 2016, 28, 6136–6166 CrossRef CAS PubMed.
- S. K. Deng and V. Berry, Mater. Today, 2016, 19, 197–212 CrossRef CAS.
- P. Poulin, R. Jalili, W. Neri, F. Nallet, T. Divoux, A. Colin, S. H. Aboutalebi, G. Wallace and C. Zakri, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11088–11093 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, K. Konstantinov, S. E. Moulton, J. M. Razal and G. G. Wallace, ACS Nano, 2013, 7, 3981–3990 CrossRef CAS PubMed.
- A. Yakovlev, A. Finaenov, S. Zabud’kov and E. Yakovleva, Russ. J. Appl. Chem., 2006, 79, 1741–1751 CrossRef CAS.
- Y. Geng, S. J. Wang and J.-K. Kim, J. Colloid Interface Sci., 2009, 336, 592–598 CrossRef CAS PubMed.
- M. M. Gudarzi and F. Sharif, J. Colloid Interface Sci., 2010, 349, 63–69 CrossRef CAS PubMed.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, K. Konstantinov, J. M. Razal, S. E. Moultona and G. G. Wallace, Mater. Horiz., 2014, 1, 87–91 RSC.
- S. H. Aboutalebi, M. M. Gudarzi, Q. B. Zheng and J. K. Kim, Adv. Funct. Mater., 2011, 21, 2978–2988 CrossRef CAS.
- S. Naficy, R. Jalili, S. H. Aboutalebi, R. A. Gorkin, K. Konstantinov, P. C. Innis, G. M. Spinks, P. Poulin and G. G. Wallace, Mater. Horiz., 2014, 1, 326–331 RSC.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, R. L. Shepherd, J. Chen, S. Aminorroaya-Yamini, K. Konstantinov, A. I. Minett, J. M. Razal and G. G. Wallace, Adv. Funct. Mater., 2013, 23, 5345–5354 CrossRef CAS.
- Y. Tan, Y. Song and Q. Zheng, Nanoscale, 2012, 4, 6997–7005 RSC.
- X. Y. Lin, X. Shen, Q. B. Zheng, N. Yousefi, L. Ye, Y. W. Mai and J. K. Kim, ACS Nano, 2012, 6, 10708–10719 CrossRef CAS PubMed.
- X. Zhou and Z. Liu, Chem. Commun., 2010, 46, 2611–2613 RSC.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- B. Dehghanzad, M. K. Razavi Aghjeh, O. Rafeie, A. Tavakoli and A. Jameie Oskooie, RSC Adv., 2016, 6, 3578–3585 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- S. Madani, C. Falamaki, H. Alimadadi and S. H. Aboutalebi, J. Energy Storage, 2019, 21, 310–320 CrossRef.
- C.-Y. Su, Y. Xu, W. Zhang, J. Zhao, X. Tang, C.-H. Tsai and L.-J. Li, Chem. Mater., 2009, 21, 5674–5680 CrossRef CAS.
- W. J. Basirun, M. Sookhakian, S. Baradaran, M. R. Mahmoudian and M. Ebadi, Nanoscale Res. Lett., 2013, 8, 397 CrossRef PubMed.
- X. Yang, W. Tang, Q. Feng and K. Ooi, Cryst. Growth Des., 2003, 3, 409–415 CrossRef CAS.
- D. Zheng, Z. Yin, W. Zhang, X. Tan and S. Sun, Cryst. Growth Des., 2006, 6, 1733–1735 CrossRef CAS.
- X.-L. Shi, M.-S. Cao, X.-Y. Fang, J. Yuan, Y.-Q. Kang and W.-L. Song, Appl. Phys. Lett., 2008, 93, 223112 CrossRef.
- K. B. Hatzell, L. Fan, M. Beidaghi, M. Boota, E. Pomerantseva, E. C. Kumbur and Y. Gogotsi, ACS Appl. Mater. Interfaces, 2014, 6, 8886–8893 CrossRef CAS PubMed.
- K.-C. Liu, J. Electrochem. Soc., 1996, 143, 124 CrossRef CAS.
- Y. S. Yoon, W. I. Cho, J. H. Lim and D. J. Choi, J. Power Sources, 2001, 101, 126–129 CrossRef CAS.
- V. I. Birss, B. Conway and J. Wojtowicz, J. Power Sources, 1997, 66, 1–14 CrossRef.
- F. Wang, S. Wang, D. Wu, H. Huang, W. Yuan and L. Y. Zhang, Appl. Surf. Sci., 2021, 537, 147860 CrossRef CAS.
- J. Zhang, J. Jiang and X. S. Zhao, J. Phys. Chem. C, 2011, 115, 6448–6454 CrossRef CAS.
- X. Feng, N. Chen, Y. Zhang, Z. Yan, X. Liu, Y. Ma, Q. Shen, L. Wang and W. Huang, J. Mater. Chem. A, 2014, 2, 9178–9184 RSC.
- J. Hao, Y. Zhong, Y. Liao, D. Shu, Z. Kang, X. Zou, C. He and S. Guo, Electrochim. Acta, 2015, 167, 412–420 CrossRef CAS.
- S. Chen, J. W. Zhu, Q. F. Han, Z. J. Zheng, Y. Yang and X. Wang, Cryst. Growth Des., 2009, 9, 4356–4361 CrossRef CAS.
- D. Portehault, S. Cassaignon, E. Baudrin and J.-P. Jolivet, Chem. Mater., 2007, 19, 5410–5417 CrossRef CAS.
- M. Musil, B. Choi and A. Tsutsumi, J. Electrochem. Soc., 2015, 162, A2058 CrossRef CAS.
- J.-L. Liu, L.-Z. Fan and X. Qu, Electrochim. Acta, 2012, 66, 302–305 CrossRef CAS.
- J. Liu, Y.-C. Son, J. Cai, X. Shen, S. L. Suib and M. Aindow, Chem. Mater., 2004, 16, 276–285 CrossRef CAS.
- Y. Ouyang, H. Cao, H. Wu, D. Wu, F. Wang, X. Fan, W. Yuan, M. He, L. Y. Zhang and C. M. Li, Appl. Catal., B, 2020, 265, 118606 CrossRef.
- D. Wu, C. Wang, H. Wu, S. Wang, F. Wang, Z. Chen, T. Zhao, Z. Zhang, L. Y. Zhang and C. M. Li, Carbon, 2020, 163, 137–144 CrossRef CAS.
- J. G. Radich and P. V. Kamat, ACS Catal., 2012, 2, 807–816 CrossRef.
- S. Chen, J. W. Zhu, X. D. Wu, Q. F. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS PubMed.
- Y. G. Li and Y. Y. Wu, J. Am. Chem. Soc., 2009, 131, 5851–5857 CrossRef CAS PubMed.
- J.-P. Jolivet, M. Henry and J. Livage, Metal oxide chemistry and synthesis: from solution to solid state, Wiley-Blackwell, 2000 Search PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394–3398 CrossRef CAS PubMed.
- F. Hamade, S. K. Amit, M. B. Woods and V. A. Davis, Crystals, 2020, 10(8), 715 CrossRef CAS.
- H. Lu, J. Zhang, J. Luo, W. Gong, C. Li, Q. Li, K. Zhang, M. Hu and Y. Yao, Composites, Part A, 2017, 102, 1–8 CrossRef CAS.
- A. G. Ricciardulli, S. Yang, G.-J. A. H. Wetzelaer, X. Feng and P. W. M. Blom, Adv. Funct. Mater., 2018, 28, 1706010 CrossRef.
- H.-K. Jeong, Y. P. Lee, M. H. Jin, E. S. Kim, J. J. Bae and Y. H. Lee, Chem. Phys. Lett., 2009, 470, 255–258 CrossRef CAS.
- A. G. Bannov, A. A. Timofeeva, V. V. Shinkarev, K. D. Dyukova, A. V. Ukhina, E. A. Maksimovskii and S. I. Yusin, Prot. Met. Phys. Chem. Surf., 2014, 50, 183–190 CrossRef CAS.
- T. Gao, M. Glerup, F. Krumeich, R. Nesper, H. Fjellvåg and P. Norby, J. Phys. Chem. C, 2008, 112, 13134–13140 CrossRef CAS.
- Z. Ai, L. Zhang, F. Kong, H. Liu, W. Xing and J. Qiu, Mater. Chem. Phys., 2008, 111, 162–167 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B, 2000, 61, 14095–14107 CrossRef CAS.
- M. M. Lucchese, F. Stavale, E. H. M. Ferreira, C. Vilani, M. V. O. Moutinho, R. B. Capaz, C. A. Achete and A. Jorio, Carbon, 2010, 48, 1592–1597 CrossRef CAS.
- V. López, R. S. Sundaram, C. Gómez-Navarro, D. Olea, M. Burghard, J. Gómez-Herrero, F. Zamora and K. Kern, Adv. Mater., 2009, 21, 4683–4686 CrossRef.
- C. Gómez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef PubMed.
- D. Luo, G. Zhang, J. Liu and X. Sun, J. Phys. Chem. C, 2011, 115, 11327–11335 CrossRef CAS.
- Y. Liu, X. Xu, T. Lu, Z. Sun, D. H. C. Chua and L. Pan, RSC Adv., 2015, 5, 34117–34124 RSC.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- A. Ambrosi, A. Bonanni, Z. Sofer, J. S. Cross and M. Pumera, Chem.–Eur. J., 2011, 17, 10763–10770 CrossRef CAS PubMed.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- T. Gao, H. Fjellvåg and P. Norby, Anal. Chim. Acta, 2009, 648, 235–239 CrossRef CAS PubMed.
- X. Zhao, Y. Hou, Y. Wang, L. Yang, L. Zhu, R. Cao and Z. Sha, RSC Adv., 2017, 7, 40286–40294 RSC.
- L. Zhang, H. Liu, H. Ruan, Y. Su, R. Hu, L. Tian, Z. Hu and J. Li, Int. J. Electrochem. Sci., 2016, 11, 10815–10826 CrossRef CAS.
- X. Qiu, D. Xu, L. Ma and Y. Wang, Int. J. Electrochem. Sci., 2017, 12, 2173–2183 CrossRef CAS.
- X. Zhu, P. Zhang, S. Xu, X. Yan and Q. Xue, ACS Appl. Mater. Interfaces, 2014, 6, 11665–11674 CrossRef CAS PubMed.
- S. Murali, T. Xu, B. D. Marshall, M. J. Kayatin, K. Pizarro, V. K. Radhakrishnan, D. Nepal and V. A. Davis, Langmuir, 2010, 26, 11176–11183 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05323j |
| ‡ Current address: Infiltrator Water Technologies. |
| This journal is © The Royal Society of Chemistry 2021 |