
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew total synthesis and structure confirmation of putative (+)-hyacinthacine C3 and (+)-5-epi-hyacinthacine C3†
Lívia Dikošováa,
Barbora Otočkováa,
Tomáš Malatinskýa,
Jana Doháňošováb,
Mária Kopáčovác,
Anna Ďurinovád,
Lucie Smutnád,
František Trejtnar*d and
Róbert Fischer *a
*a
aInstitute of Organic Chemistry, Catalysis and Petrochemistry, Slovak University of Technology in Bratislava, Radlinského 9, 812 37 Bratislava, Slovak Republic. E-mail: robert.fischer@stuba.sk
bCentral Laboratories, Slovak University of Technology in Bratislava, Radlinského 9, 812 37 Bratislava, Slovak Republic
cInstitute of Chemistry, Slovak Academy of Sciences, Dúbravská cesta 9, 845 38 Bratislava, Slovak Republic
dCharles University, Faculty of Pharmacy in Hradec Kralove, Heyrovskeho 1203, 50005 Hradec Kralove, Czech Republic. E-mail: trejtnarf@faf.cuni.cz
First published on 24th September 2021
Abstract
A unique synthesis of polyhydroxylated pyrrolizidine alkaloids, namely (+)-hyacinthacine C3 and (+)-5-epi-hyacinthacine C3 is presented. The strategy relies on a 1,3-dipolar cycloaddition of an L-mannose derived nitrone, which owing to its great syn-stereoselectivity builds up the majority of the required stereocenters. The following key steps include Wittig olefination and iodine-mediated aminocyclisation, that provide two epimeric pyrrolizidines with the appropriate configuration. As a result, structure and steric arrangement of the first synthetically prepared (+)-hyacinthacine C3 are proved to be correct, clearly confirming the inconsistency with the stereochemistry assigned to the natural sample. With respect to the previously proven glycosidase inhibitory activities, the antiproliferative effect of (+)-hyacinthacine C3 and (+)-5-epi-hyacinthacine C3 was evaluated using several cell line models.
Introduction
Polyhydroxylated pyrrolizidine alkaloids are frequent candidates for the investigation of their potential antiviral properties and other medicinal applications such as treatment of diabetes, obesity, cancer, viral and bacterial infections.1 They exhibit powerful glycosidase inhibitory activities due to their structural similarity with the furanose ring found in the monosaccharides.2Hyacinthacines are naturally occurring polyhydroxylated pyrrolizidine alkaloids which have been isolated from plants of the Hyacinthaceae family. So far, more than 20 compounds belonging to this group have been isolated and described. Among them, hyacinthacines C2 (1) and C3 (2a) were isolated together with other hyacinthacines from the bulbs of Scilla socialis (Fig. 1).3 The glycosidase inhibitory studies revealed that hyacinthacine C2 exhibited a moderate inhibition of the human placenta α-L-fucosidase (IC50 = 17 μM). Hyacinthacine C3 was found to be a weak inhibitor of bovine liver β-galactosidase (IC50 = 52 μM). Both alkaloids inhibited C. saccharolyticum β-glucosidase with IC50 values of 13 and 25 μM, respectively.
 | ||
| Fig. 1 Examples of polyhydroxylated 3,5-bis(hydroxymethyl)pyrrolizidine alkaloids. | ||
Hyacinthacines C2 and C3, along with their unnatural 5-epimers, have been prepared synthetically only once.4 However, only the structure of 1 was identical with the natural sample indicating that the primarily assigned relative configuration of 2a could require a correction.
Due to the lack of absolute configuration information obtained from the X-ray structural analysis of hyacinthacines, there are currently many inaccuracies in their structures and configuration. In most cases, their structure and relative configuration have been determined only by 2D NMR experiments, based on the J(HH) coupling constants and the observed NOEs. For this reason, efforts toward the total syntheses of this group of alkaloids, to confirm or refute their relative and absolute configuration, are still attractive.5
Our previous success with the synthesis of the unnatural enantiomer of (+)-hyacinthacine B2 (3), belonging to the class of structurally intriguing 3,5-bis(hydroxymethyl)pyrrolizidine alkaloids (Fig. 1),6 inspired us to prepare the (+)-hyacinthacine C3 (2a) and its C-5 epimer 2b. Comparison of our results to the already reported data would bring definite clarity into the discussion about their structure, which would also create basis for their expected biological properties. Since it is well known that these compounds exhibit inherent glycosidase inhibitory activities,5 our aim was to evaluate the antiproliferative effects of the synthesized alkaloids. To the best of our knowledge, this would be for the first time when such polyhydroxylated pyrrolizidines would be tested against human cancer cell lines.
The essence of the new synthetic route is contained in three fundamental steps (Scheme 1), including highly syn-stereoselective 1,3-dipolar cycloaddition of L-mannose derived cyclic nitrone 7 which provides (3,3a-trans,3a,4-cis)-isoxazolidine exo-syn-6 ensuring the 1,7a-cis, as well as 7,7a-trans configuration in the desired pyrrolizidine. The conversion of the dioxolane sidechain into a protected hydroxymethyl substituent (acidic hydrolysis, oxidative cleavage, reduction, silylation) and acidic hydrolysis of the carbonate is followed by a very straightforward extension of the isoxazolidine diol 5 backbone by Wittig olefination. The introduced alkene moiety in pyrrolidine derivative 4 is a vital part of the structure, as it serves in the iodine-mediated aminocyclisation reaction, that offers two C-5-epimeric 6-iodo-pyrrolizidine structures. Their dehalogenation and final deprotection generates (+)-hyacinthacine C3 (2a) and (+)-5-epi-hyacinthacine C3 (2b).
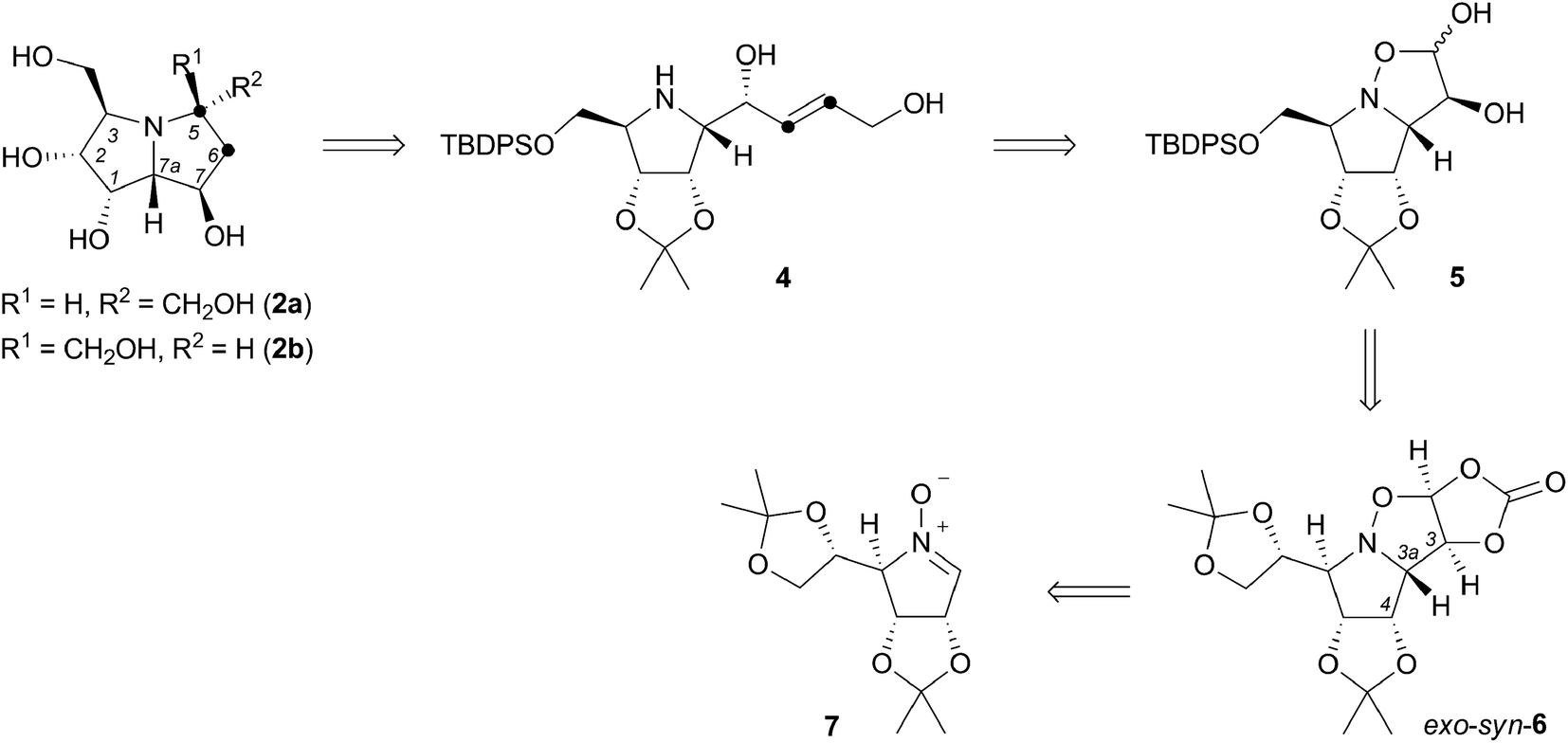 | ||
| Scheme 1 Retrosynthesis of (+)-hyacinthacine C3 (2a) and (+)-5-epi-hyacinthacine C3 (2b) starting from L-mannose derived nitrone 7. | ||
Results and discussion
Synthesis of isoxazolidine diol
The essential building block of the presented synthesis, L-mannose-derived nitrone 7, was prepared by a slightly modified procedure described in the literature in total yield of 54% in six steps.7,8 Its highly syn-stereoslective 1,3-dipolar cycloaddition with vinylene carbonate resulted in a formation of cycloadduct exo-syn-6 as a major isomer (exo-syn-6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :exo-anti-6, 90:10 ratio based on 1H NMR spectrum of the crude product) in 67% yield (ESI†).9 Prior to carbonate hydrolysis, we have decided to convert the acetonide side chain of 6 into a protected hydroxymethyl substituent (Scheme 2). The vicinal diol 8 was obtained in a 91% yield by a selective acid-catalysed alcoholysis with p-TSA·H2O in ethanol. The oxidative cleavage of the diol with sodium periodate followed by reduction of the obtained aldehyde with sodium borohydride led to formation of alcohol 9 in 80% over two steps.10 The reaction of 9 with TBDPSCl afforded the protected alcohol 10 in 92% yield. The desired isoxazolidine diol 5 was provided by a base-catalysed hydrolysis with Et3N in aqueous methanol as a mixture of two undefined anomers in an anomeric ratio 90:10.
:exo-anti-6, 90:10 ratio based on 1H NMR spectrum of the crude product) in 67% yield (ESI†).9 Prior to carbonate hydrolysis, we have decided to convert the acetonide side chain of 6 into a protected hydroxymethyl substituent (Scheme 2). The vicinal diol 8 was obtained in a 91% yield by a selective acid-catalysed alcoholysis with p-TSA·H2O in ethanol. The oxidative cleavage of the diol with sodium periodate followed by reduction of the obtained aldehyde with sodium borohydride led to formation of alcohol 9 in 80% over two steps.10 The reaction of 9 with TBDPSCl afforded the protected alcohol 10 in 92% yield. The desired isoxazolidine diol 5 was provided by a base-catalysed hydrolysis with Et3N in aqueous methanol as a mixture of two undefined anomers in an anomeric ratio 90:10.
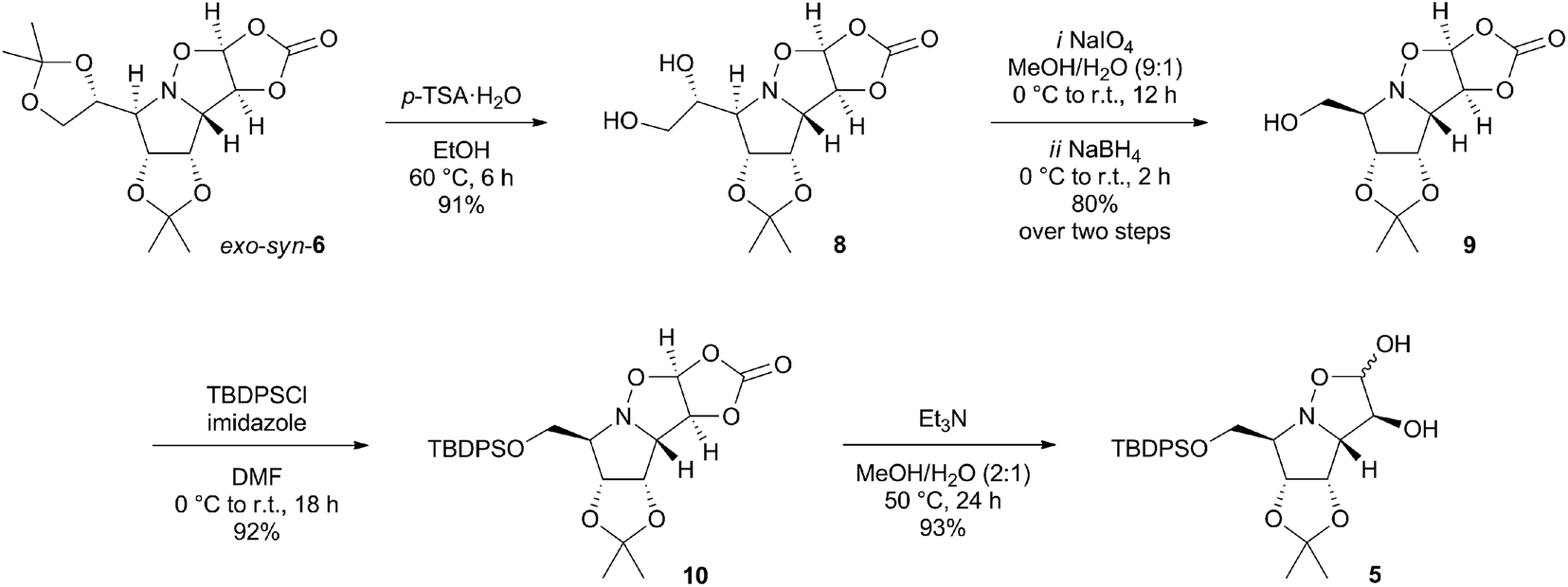 | ||
| Scheme 2 Synthesis of isoxazolidine diol 5. | ||
Synthesis of the pyrrolidine intermediate via Wittig olefination
With the starting diol 5 in hand, we were ready to examine the possibility of application of the Wittig olefination in the synthesis of pyrrolizidines.11 Accordingly, the isoxazolidine diol 5 was left to react with commercially available stable phosphorous ylide 11 at room temperature in dichloromethane for 48 h (Scheme 3) leading to the formation of α,β-unsaturated ester 12 in very good 89% isolated yield as almost exclusively (E)-isomer with large coupling constant JCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH = 15.5 Hz (E/Z, 95:5). The structure of the minor (Z)-isomer was predicted to be formed based on another double bond signals detected in 1H NMR spectrum of the crude reaction mixture, however, the compound was never isolated in a pure form due to its low stability. It is noteworthy that no spontaneous intramolecular oxa-Michael addition of hydroxylamine nucleophile in 12 occurred, that would eventually lead to formation of new bicyclic isoxazolidine. Subsequently, ester 12 was reduced with DIBAL-H in anhydrous CH2Cl2 at −20 °C to afford alkenol 13 in 78% yield (Scheme 3). Up to four equivalents of DIBAL-H were necessary for the complete consumption of the starting material. THF was also examined as a possible solvent, however, this reaction was not brought to completion even with additional amount of DIBAL-H. Finally, the reductive cleavage of the N–O bond of the hydroxylamine group in 13 was carried out with zinc powder in aqueous acetic acid.12 The reaction proceeded smoothly at room temperature, affording desired pyrrolidine 4 in an excellent 81% yield already after 30 minutes.
CH = 15.5 Hz (E/Z, 95:5). The structure of the minor (Z)-isomer was predicted to be formed based on another double bond signals detected in 1H NMR spectrum of the crude reaction mixture, however, the compound was never isolated in a pure form due to its low stability. It is noteworthy that no spontaneous intramolecular oxa-Michael addition of hydroxylamine nucleophile in 12 occurred, that would eventually lead to formation of new bicyclic isoxazolidine. Subsequently, ester 12 was reduced with DIBAL-H in anhydrous CH2Cl2 at −20 °C to afford alkenol 13 in 78% yield (Scheme 3). Up to four equivalents of DIBAL-H were necessary for the complete consumption of the starting material. THF was also examined as a possible solvent, however, this reaction was not brought to completion even with additional amount of DIBAL-H. Finally, the reductive cleavage of the N–O bond of the hydroxylamine group in 13 was carried out with zinc powder in aqueous acetic acid.12 The reaction proceeded smoothly at room temperature, affording desired pyrrolidine 4 in an excellent 81% yield already after 30 minutes.
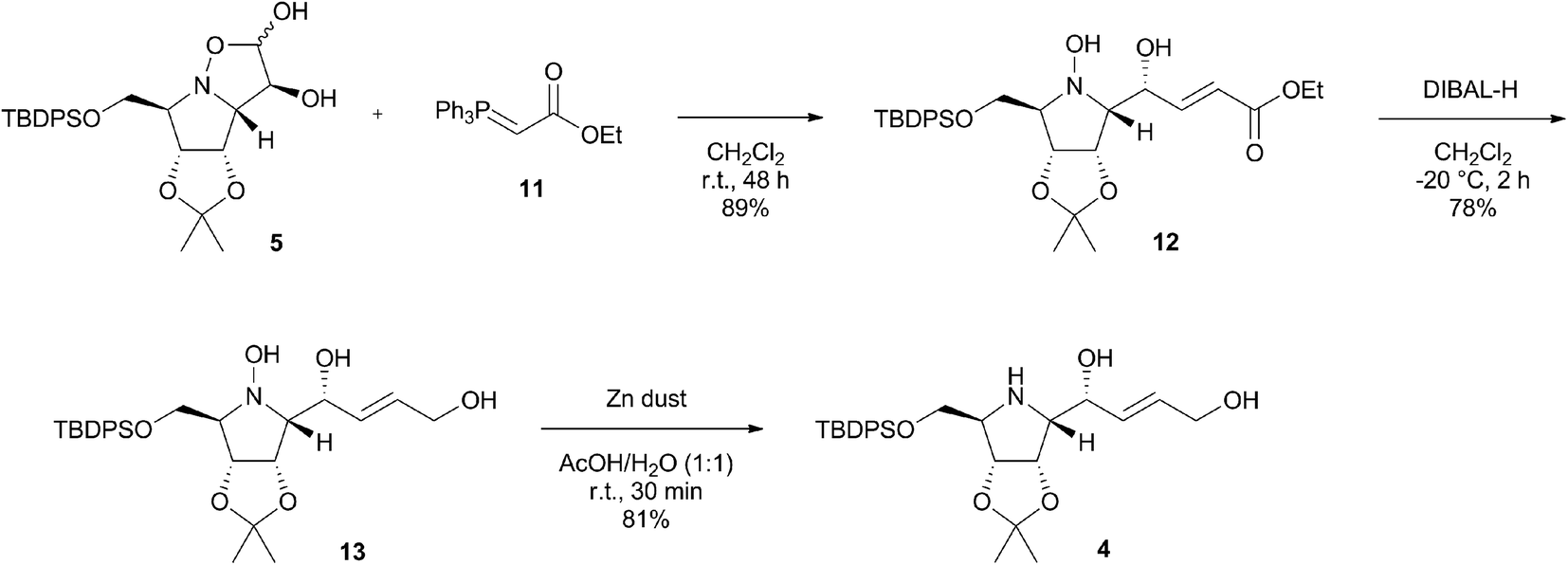 | ||
| Scheme 3 Synthesis of pyrrolidine intermediate 4 via Wittig olefination of 5 with stable phosphorous ylide 11. | ||
Synthesis of (+)-hyacinthacine C3 and its C-5 epimer
In order to prepare the desired pyrrolizidines, we were ready to examine the proposed iodine-mediated amino cyclisation reaction of 4.13 Its treatment with iodine in ethyl acetate in the presence of potassium carbonate at −20 °C afforded the 5-endo-trig cyclisation products as a mixture of two diastereomers 14 and 15 in a 55:45 ratio (Scheme 4), as determined by 1H NMR of the crude reaction mixture. To avoid problems associated with chromatographic separation of each individual isomer, that has emerged during this process, the isomers were isolated as product mixture and used directly in the next reaction. To determine the stereochemistry of the iodo-pyrrolizidines, a sample of the mixture was subjected to repeated preparative thin-layer chromatography to obtain both isomers in pure form. Their respective structures as well as the relative configurations were determined based on 1H and 13C NMR spectra, including COSY, HSQC and 1D NOESY experiments (ESI†). The NOE interactions between H-6 and H-7 protons in 14 were considerably stronger (2.9%) compared to the same interactions in 15 (0.4%). Very similar weak interactions were observed for H-5 and H-6 protons in both diastereomers 14 (0.3%) and 15 (0.3%). This corresponded with their expected trans orientation that arises from the mechanism of the intramolecular iodoamination. For the minor 5,6-trans-6,7-trans isomer 15, several other interactions were detected, especially between H-3 and H-5 protons (2.4%), H-6 and H-7a protons (1.1%) and between H-5 and H-7 (1.0%). Next, the dehalogenation of pyrrolizidines 14 and 15 was achieved under conditions adapted from literature for related pyrrolidine derivatives,14 where InCl3 together with a suitable hydride acts as a reducing agent. In our case, the use of InCl3 (2 equiv.) and DIBAL-H (10 equiv.) in THF at 0 °C resulted in formation of the desired C-6 unsubstituted products 16 and 17 in 30 and 25% yields, respectively, after two steps (Scheme 4). Strictly anhydrous conditions in an argon atmosphere were required to avoid the formation of C-6 hydroxylated by-products. It is worth noting that the typical radical dehalogenation with Bu3Sn catalysed by AIBN failed.
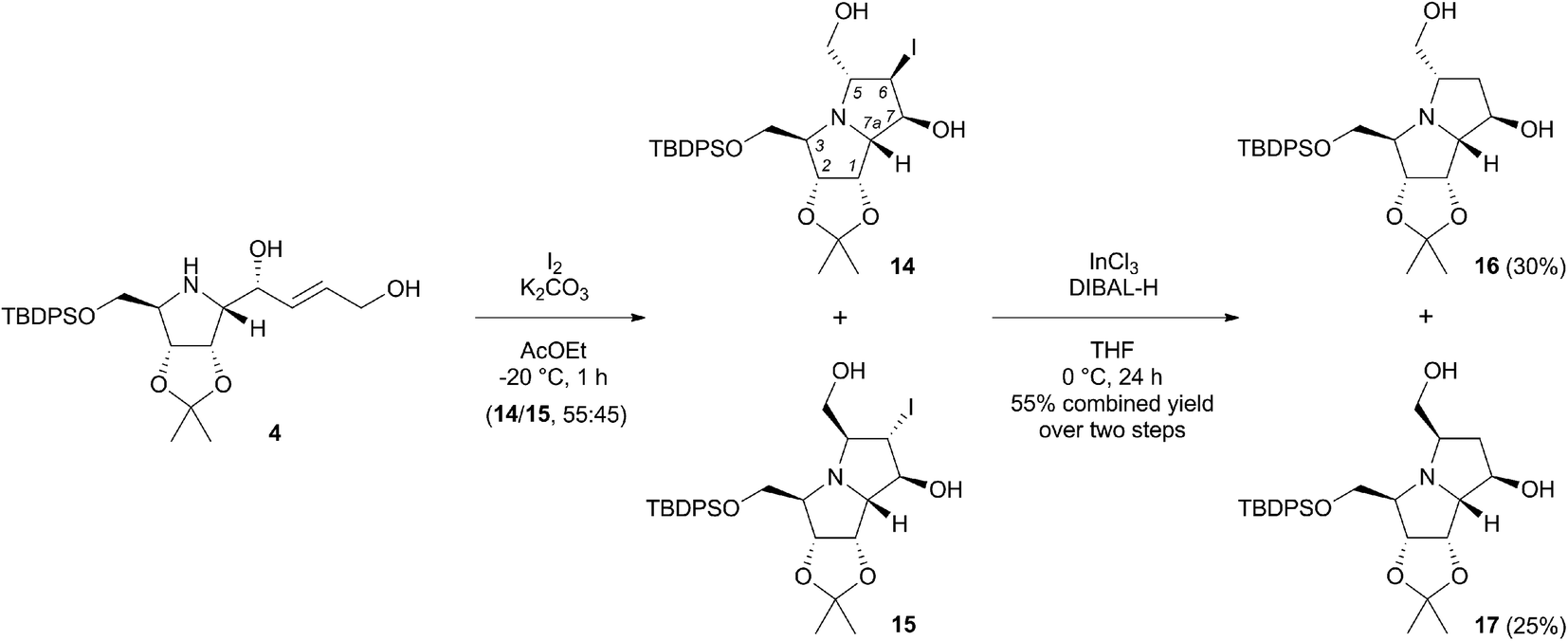 | ||
| Scheme 4 Intramolecular iodoamination of pyrrolidine 4 followed by InCl3 mediated reductive dehalogenation. | ||
Pyrrolizidines 16 and 17 were next used individually in the fluoride-mediated desilylation (Scheme 5). The reaction was carried out with CsF (5 equiv.) in MeOH at 50 °C, and the desilylated products 18 and 19 were obtained in very good 85 and 95% yields, respectively. The primary screening of deprotection agents has shown, that the most common reagent TBAF is not suitable for this transformation, as it contains trace amounts of Bu3N that contaminated the final product irretrievably.15 Eventually, hydrolysis of the acetonide protecting group with acidic ion exchange resin Dowex 50WX-8 in MeOH was carried out.16 After washing the resin with aqueous ammonia, target (+)-hyacinthacine C3 (2a) and (+)-5-epi-hyacinthacine C3 (2b) were obtained in almost quantitative 94 and 95% isolated yields, respectively. Their structures were assigned based on 1H, 13C NMR, COSY and HSQC spectra, as well as extensive 1D NOESY experiments (ESI†). For 2a, strong NOE interactions between H-2 and H-1, H-1 and H-7a, H-7 and H-6b, H-6a and H-5 were observed. No interaction between the H-5 and H-3 protons was detected, which supports its 3,5-trans configuration.
 | ||
| Scheme 5 Synthesis of (+)-hyacinthacine C3 (2a) and (+)-5-epi-hyacinthacine C3 (2b). | ||
On the other hand, strong 4.2% interaction was identified between H-5 and H-3 in case of 2b, thus proving the cis orientation of the two hydroxymethyl substituents. The obtained 1H and 13C NMR spectra were compared to the previously gathered data for synthetic (+)-hyacinthacine C3 and its C-5 epimer by Yoda et al., 2009,4 as well as with the spectroscopic data of the natural sample (Kato et al., 2007).3 While our data were in good agreement with those described by Yoda for both of the compounds (with a little exception for the H-3, H-5 and H-7a protons, whose chemical shift may be affected by a different chemical environment during the process of isolating and purifying),17 they were evidently inconsistent (in particular, 3JH–H coupling constants and 13C chemical shifts) with the characteristics of the natural sample of the compound isolated and identified as (+)-hyacinthacine C3 by Kato et al. (ESI, Tables S1 and S2†). To our delight, the optical rotation value for 2a {[α]22D = +9.3 (c 0.3, H2O)} closely resembled the value of the optical rotation for the (+)-hyacinthacine C3 {[α]22D = +8.8 (c 0.3, H2O)}, previously prepared by Yoda et al.4 Similarly, the value of optical rotation for the 5-epi-isomer 2b {[α]22D = +13.8 (c 0.3, H2O)} was in agreement with the published data for (+)-5-epi-hyacinthacine C3 (+)-hyacinthacine C3 {[α]22D = +14.8 (c 0.3, H2O)}. The above-mentioned evidence supports the argument, that the original structure proposed for (+)-hyacinthacine C3, isolated from natural source, is incorrect.
Evaluation of antiproliferative effect on human cancer cells
Previously published experimental data showed significant inhibitory effect of hyacinthacines towards many types of glycosidases.5b Since glycosidase inhibitors were established as possible treatments for tumor metastasis,5b the prepared synthetic hyacinthacine C3 (2a) and its C-5 epimer 2b were evaluated for its potential antiproliferative effect. The effect was examined on three different cancer cell lines U87-MG, HepG2 and JEG-3 as well as immortalized proximal tubular cells HK2. IC50 values of all tested compounds exceeded maximal value of the tested concentration range 1–1000 μM (ESI, Table S3†). We did not confirm any significant antiproliferative activity in the tested concentrations. The finding may suggest that the proved antiglycosidase potency of the tested hyacithacines is not so strong to be manifested in very vital cancer cells. Moreover, their low antiproliferative activity may be associated with the high level of hydrophility of the polyhydroxylated pyrrolizidines. It is known that derivatization of similar hydroxylated pyrrolidines with lipophilic groups enhances their ability to penetrate cell membranes, and thus increase their efficiency in human tumor cell inhibition.18 The lack of a lipophilic unit in the structure of the reviewed compounds may explain the obtained limited results. This fact implies that synthetized substances are not suitable for development of potential cytostatics. Due to antiglycosidase activity, hyacinthacines may exert a potency against viral infection and various lysosomal storage disorders.5b Therefore, the found low cytotoxicity can be a positive impulse for considerations of further studies on utilization of these potential effects.Conclusions
In summary, we have designed a new synthetic pathway towards structurally interesting polyhydroxylated pyrrolizidine alkaloids bearing two hydroxymethyl substituents at C-3 and C-5 and a hydroxy substituent at C-7 position. (+)-Hyacinthacine C3 and (+)-5-epi-hyacinthacine C3 were prepared from cyclic L-mannose derived nitrone, which underwent a highly stereoselective 1,3-dipolar cycloaddition, building up majority of the desired stereocenters. The following key steps included Wittig olefination that provided a necessary alkene basis for iodocyclisation, that afforded two pyrrolizidine isomers, that were eventually transformed into the desired compounds. (+)-Hyacinthacine C3 and (+)-5-epi-hyacinthacine C3 were prepared in 13 steps starting from the sugar derived nitrone in 6% and 5% yield, respectively. This total synthesis supports previously synthetically acquired data for these compounds, while disproving the structural proposal for the naturally occurring sample. Although the hyacinthacine C-type alkaloids exhibit inherent glycosidase inhibitory activities, synthetic hyacinthacine C3 and its C-5 epimer showed no antiproliferative activity against selected human cancer cell lines in vitro. On the other hand, the insignificant cytotoxicity forms a good base for employment of other biological effects of the tested compounds.Experimental section
General methods
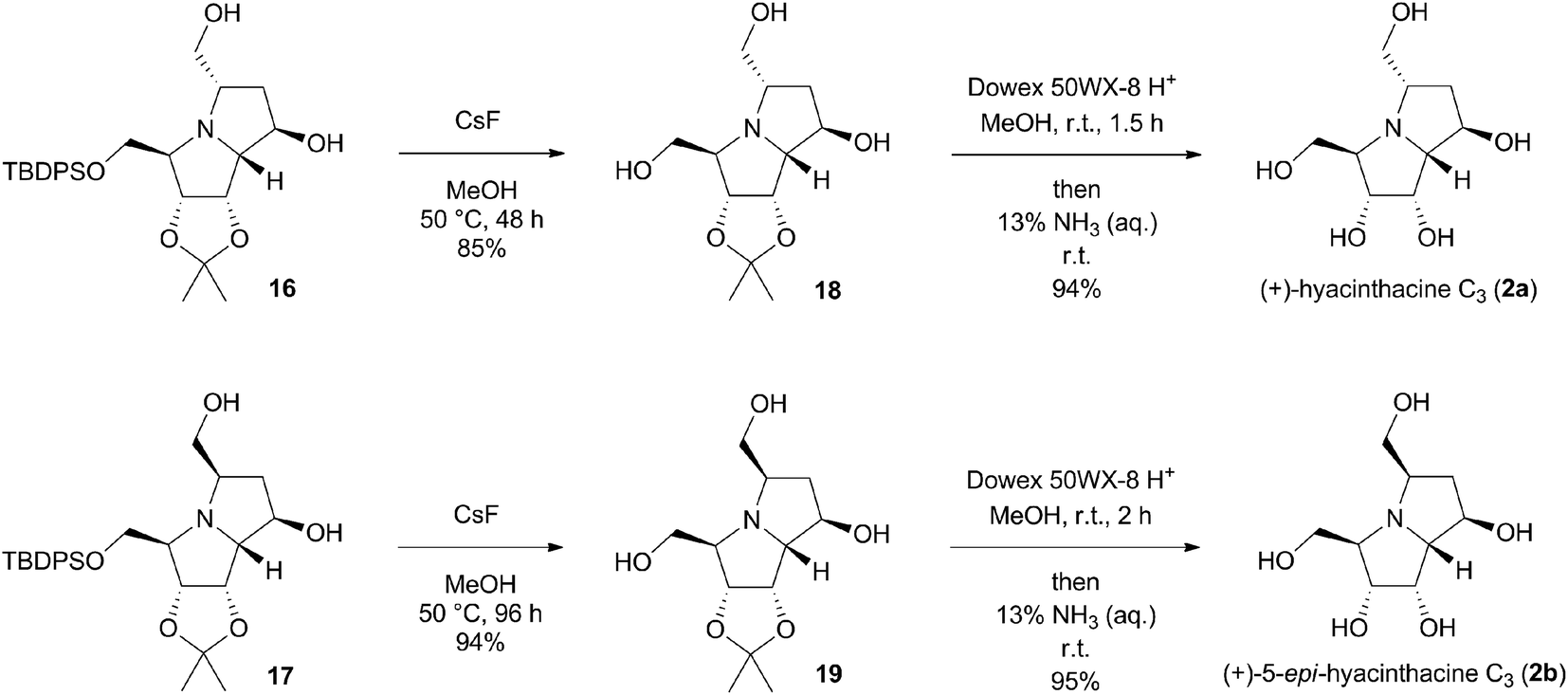
Melting points were measured with a Melting Point B-540 apparatus (Büchi). Flash column chromatography (FCC) was carried out with a Büchi system (Pump Manager C-615 and Fraction Collector C-660) using Normasil 60 silica gel (0.040–0.063 mm; VWR). Thin Layer Chromatography (TLC) analysis was carried out using TLC silica gel 60 F254 (aluminium sheets, Merck), and plates were visualized with UV light or by treatment with permanganate solution followed by heating. Optical rotations were measured with a JASCO P-2000 digital polarimeter with a Na-D lamp (10 cm cell length). Concentrations (c) are given in gram per 100 mL. Infrared (IR) spectra were recorded as neat samples with a Nicolet 5700 FTIR spectrometer with an ATR Smart Orbit Diamond adapter (Thermo Electron Corporation). NMR spectra were recorded with a Varian INOVA-300 spectrometer (1H, 299.95 MHz, and 13C, 75.42 MHz) and a Varian VNMRS-600 instrument (1H, 599.75 MHz, and 13C, 150.81 MHz) in CDCl3 (using tetramethylsilane as the internal standard), in CD3OD (residual [D3]methanol, δH = 3.31, 4.87 ppm, δC = 49.00 ppm), and in D2O (using TSP-d4 as the internal standard, δH,C = 0.00 ppm). Data are presented as follows: chemical shift, multiplicity, coupling constants and integration. 1H and 13C signals were assigned by 2D experiments (COSY and HSQC). HRMS analysis was carried out with an Orbitrap Velos Pro spectrometer (Thermo Fisher Scientific). Dichloromethane was distilled in the presence of calcium hydride. Tetrahydrofuran was distilled in the presence of sodium. Anhydrous ethyl acetate (Sigma-Aldrich), N,N-dimethylformamide (Acros Organics) and acetic acid (Acros Organics) were used as received. Simple distillation was used to purify other solvents prior to use if not stated otherwise. Phosphorus ylide 11 was commercially available. The pyrrolizidine ring protons in compounds 14–19 have been designated as follows: H-1, H-2, H-3, H-5, H-6, H-7, and H-7a, regardless of their naming.:9) to give diol 8 (2.27 g, 7.49 mmol, 91%) as a white solid.Rf = 0.26 (EtOAc), mp = 153–155 °C, [α]25D = +139.9 (c 1.02, MeOH). IR (ATR) νmax: 3477; 2937; 1789; 1377; 1190; 1043; 982; 864; 770; 722; 516 cm−1. 1H NMR (600 MHz, CD3OD) δ (ppm) 1.29 (s, 3H, CH3), 1.40 (s, 3H, CH3), 3.56 (dd, J = 6.2, 11.0 Hz, 1H, 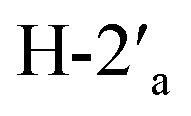 ), 3.69 (dd, J = 6.3, 11.0 Hz, 1H,
), 3.69 (dd, J = 6.3, 11.0 Hz, 1H, 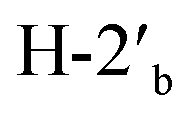 ), 3.77 (td, J = 3.0, 6.2 Hz, 1H, H-1′), 3.99 (d, J = 2.9 Hz, 1H, H-6), 4.05 (d, J = 5.3 Hz, 1H, H-3a), 4.89 (pseudo t, J = 5.6, 6.0 Hz, 1H, H-4), 4.98 (d, J = 6.3 Hz, 1H, H-5), 5.67 (d, J = 5.2 Hz, 1H, H-3), 6.13 (d, J = 5.2 Hz, 1H, H-2). 13C NMR (150 MHz, CD3OD) δ (ppm) 23.9 (CH3), 26.5 (CH3), 64.5 (C-5′), 73.4 (C-4′), 74.1 (C-6), 75.8 (C-3a), 83.2 (C-4), 87.0 (C-3), 88.6 (C-5), 103.0 (C-2), 114.0 [
), 3.77 (td, J = 3.0, 6.2 Hz, 1H, H-1′), 3.99 (d, J = 2.9 Hz, 1H, H-6), 4.05 (d, J = 5.3 Hz, 1H, H-3a), 4.89 (pseudo t, J = 5.6, 6.0 Hz, 1H, H-4), 4.98 (d, J = 6.3 Hz, 1H, H-5), 5.67 (d, J = 5.2 Hz, 1H, H-3), 6.13 (d, J = 5.2 Hz, 1H, H-2). 13C NMR (150 MHz, CD3OD) δ (ppm) 23.9 (CH3), 26.5 (CH3), 64.5 (C-5′), 73.4 (C-4′), 74.1 (C-6), 75.8 (C-3a), 83.2 (C-4), 87.0 (C-3), 88.6 (C-5), 103.0 (C-2), 114.0 [![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (CH3)2], 155.6 (CO). HRMS (ESI) m/z: for C12H17NNaO8 [M + Na]+, calcd: 326.0847; found: 326.0848.
(CH3)2], 155.6 (CO). HRMS (ESI) m/z: for C12H17NNaO8 [M + Na]+, calcd: 326.0847; found: 326.0848.
:1, 145 mL) and the mixture was cooled down to 0 °C in an ice/water bath. Afterwards, NaIO4 powder (2.33 g, 10.88 mmol, 1.5 equiv.) was slowly added and the reaction mixture was gradually warmed up to room temperature and stirred for 12 h. When TLC (CH2Cl2/MeOH 9:1) showed that the reaction was complete, the mixture was cooled down to 0 °C again and solid NaBH4 (550 mg, 14.5 mmol, 2 equiv.) was added. The reaction was stirred at room temperature for 2 h, until TLC (CH2Cl2/MeOH 9:1) showed complete disappearance of the aldehyde. After this time, the suspension was diluted with saturated aqueous solution of NH4Cl (20 mL), and methanol was partially removed under reduced pressure. The residue was diluted saturated aqueous solution of NaCl (100 mL) and extracted with EtOAc (4× 60 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by FCC (CH2Cl2/MeOH 85:15) to give alcohol 9 (1.58 g, 5.78 mmol, 80% over two steps) as a white solid.Rf = 0.46 (EtOAc), mp = 209–211 °C, [α]25D = −79.6 (c 0.97, MeOH). IR (ATR) νmax: 3272; 3002; 2928; 1823; 1383; 1208; 1157; 1063; 995; 850; 768; 519 cm−1. 1H NMR (600 MHz, CD3OD) δ (ppm) 1.29 (s, 3H, CH3), 1.40 (s, 3H, CH3), 3.62 (dd, J = 4.2, 11.3 Hz, 1H,  ), 3.67 (dd, J = 4.5, 11.3 Hz, 1H,
), 3.67 (dd, J = 4.5, 11.3 Hz, 1H, 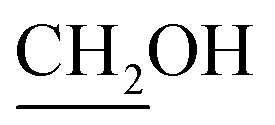 ), 3.86 (t, J = 4.3 Hz, 1H, H-6), 4.05 (d, J = 5.2 Hz, 1H, H-3a), 4.89 (pseudo t, J = 5.5, 6.0 Hz, 1H, H-4), 4,94 (d, J = 6.3 Hz, 1H, H-5), 5.68 (d, J = 5.2 Hz, 1H, H-3), 6.15 (d, J = 5.2 Hz, 1H, H-2). 13C NMR (150 MHz, CD3OD) δ (ppm) 23.9 (CH3), 26.4 (CH3), 62.5 (C-4′), 74.5 (C-6), 75.2 (C-3a), 83.3 (C-4), 86.8 (C-3), 87.1 (C-5), 103.1 (C-2), 114.1 [(CH3)2], 155.5 (CO). HRMS (ESI) m/z: for C11H15NNaO7 [M + Na]+, calcd: 296.0741; found: 296.0740.
), 3.86 (t, J = 4.3 Hz, 1H, H-6), 4.05 (d, J = 5.2 Hz, 1H, H-3a), 4.89 (pseudo t, J = 5.5, 6.0 Hz, 1H, H-4), 4,94 (d, J = 6.3 Hz, 1H, H-5), 5.68 (d, J = 5.2 Hz, 1H, H-3), 6.15 (d, J = 5.2 Hz, 1H, H-2). 13C NMR (150 MHz, CD3OD) δ (ppm) 23.9 (CH3), 26.4 (CH3), 62.5 (C-4′), 74.5 (C-6), 75.2 (C-3a), 83.3 (C-4), 86.8 (C-3), 87.1 (C-5), 103.1 (C-2), 114.1 [(CH3)2], 155.5 (CO). HRMS (ESI) m/z: for C11H15NNaO7 [M + Na]+, calcd: 296.0741; found: 296.0740.
:3) showed complete disappearance of the starting material, water (50 mL) was added, and the mixture was extracted with CH2Cl2 (3× 50 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by FCC (hexanes/EtOAc 80:20) to give isoxazolidine 10 (2.63 g, 5.14 mmol, 92%) as a white solid.Rf = 0.25 (n-hexane/EtOAc 3:1), mp = 145–148 °C, [α]25D = −44.2 (c 1.035, CHCl3). IR (ATR) νmax: 2931; 2860; 1812; 1373; 1107; 1062; 990; 742; 701; 615; 503 cm−1. 1H NMR (600 MHz, CDCl3) δ (ppm) 1.04 (s, 9H, Sit-Bu), 1.29 (s, 3H, CH3), 1.42 (s, 3H, CH3), 3.77 (dd, J = 3.4, 10.7 Hz, 1H, CH2OSi), 3.90 (dd, J = 2.5, 10.7 Hz, 1H, CH2OSi), 3.94 (t, J = 2.8 Hz, 1H, H-6), 4.15 (d, J = 5.3 Hz, 1H, H-3a), 4.88 (pseudo t, J = 5.6, 5.9 Hz, 1H, H-4), 4.98 (d, J = 6.2 Hz, 1H, H-5), 5.60 (d, J = 5.2 Hz, 1H, H-3), 6.02 (d, J = 5.2 Hz, 1H, H-2), 7.39–7.47 (m, 6H, H-Ph), 7.59–7.63 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3) δ (ppm) 19.2 [Si(CH3)3], 23.8 (CH3), 26.3 (CH3), 27.1 [SiC(H3)3], 64.5 (C-4′), 73.0 (C-6), 74.5 (C-3a), 82.3 (C-4). 85.2 (C-3), 86.2 (C-5), 101.1 (C-2), 113.3 [(CH3)2], 128.1 (CH, Ph), 128.2 (CH, Ph), 130.2 (CH, Ph), 130.3 (CH, Ph), 132.1 (C, Ph), 132.3 (C, Ph), 135.5 (CH, Ph), 135.7 (CH, Ph), 153.3 (CO). HRMS (ESI) m/z: for C27H33NNaO7Si [M + H]+, calcd: 534.1919; found: 534.1919).
:1, 50 mL), Et3N (6.9 mL, 49.5 mmol, 10 equiv.) was added at room temperature and the mixture was stirred for 24 h at 50 °C. The progress of the reaction was monitored by TLC (EtOAc). After the starting material was not detected in the reaction mixture anymore, MeOH was partially evaporated under reduced pressure. The residue was combined with saturated aqueous solution of NaCl (100 mL), and the mixture was extracted with CH2Cl2 (3× 100 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by FCC (hexanes/EtOAc 2:3) to give an anomeric mixture of isoxazolidine diol 5 (2.23 g, 4.59 mmol, 93%, anomeric ratio = 90:10) as a white solid foam.Rf = 0.50 (n-hexane/EtOAc 1:4). IR (ATR) νmax: 3406; 2931; 2856; 1427; 1372; 1104; 9933; 822; 740; 700; 501 cm−1. NMR data selected from the anomeric mixture after purification: 1H NMR (600 MHz, CDCl3, major anomer) δ (ppm) 1.05 (s, 9H, Sit-Bu), 1.28 (s, 3H, CH3), 1.42 (s, 3H, CH3), 3.41 (bs, 1H, 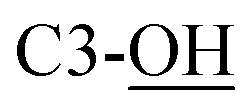 ), 3.74–3.77 (m, 2H, CH2OSi, H-6), 3.80–3.83 (m, 2H, CH2OSi, H-3a), 4.55 (bs, 1H,
), 3.74–3.77 (m, 2H, CH2OSi, H-6), 3.80–3.83 (m, 2H, CH2OSi, H-3a), 4.55 (bs, 1H, 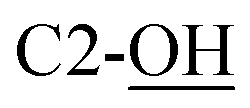 ), 4.82 (pseudo t, J = 5.7, 6.2 Hz, 1H, H-4), 4.86 (d, J = 6.4 Hz, 1H, H-5), 4.88 (d, J = 5.3 Hz, 1H, H-3), 5.36 (d, J = 5.3 Hz, 1H, H-2), 7.37–7.45 (m, 6H, H-Ph), 7.61–7.64 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3, major anomer) δ (ppm) 19.3 [Si(CH3)3], 24.1 (CH3), 26.4 (CH3), 27.1 [SiC(H3)3], 64.6 (C-4′), 72.4 (C-6), 76.6 (C-3), 77.4 (C-3a), 82.4 (C-5), 85.5 (C-4), 95.6 (C-2), 112.8 [(CH3)2], 127.9 (CH, Ph), 128.0 (CH, Ph), 130.1 (CH, Ph), 130.2 (CH, Ph), 132.6 (C, Ph), 132.8 (C, Ph), 135.8 (2× CH, Ph). HRMS (ESI + APCI) m/z: for C26H36NO6Si [M + H]+, calcd: 486.2307; found: 486.2.
), 4.82 (pseudo t, J = 5.7, 6.2 Hz, 1H, H-4), 4.86 (d, J = 6.4 Hz, 1H, H-5), 4.88 (d, J = 5.3 Hz, 1H, H-3), 5.36 (d, J = 5.3 Hz, 1H, H-2), 7.37–7.45 (m, 6H, H-Ph), 7.61–7.64 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3, major anomer) δ (ppm) 19.3 [Si(CH3)3], 24.1 (CH3), 26.4 (CH3), 27.1 [SiC(H3)3], 64.6 (C-4′), 72.4 (C-6), 76.6 (C-3), 77.4 (C-3a), 82.4 (C-5), 85.5 (C-4), 95.6 (C-2), 112.8 [(CH3)2], 127.9 (CH, Ph), 128.0 (CH, Ph), 130.1 (CH, Ph), 130.2 (CH, Ph), 132.6 (C, Ph), 132.8 (C, Ph), 135.8 (2× CH, Ph). HRMS (ESI + APCI) m/z: for C26H36NO6Si [M + H]+, calcd: 486.2307; found: 486.2.
:1) the solvent was removed under reduced pressure. The residue was purified by FCC (hexanes/EtOAc 3:1) to give ester 12 (1.48 g, 2.66 mmol, 89%) as a white solid foam.Rf = 0.25 (n-hexane/EtOAc 3:2), mp = 51–54 °C, [α]25D = −20.4 (c 1.01, CHCl3). IR (ATR) νmax: 3408; 2933; 1819; 1716; 1372; 1160; 1055; 973; 822; 701; 613; 503 cm−1. 1H NMR (600 MHz, CDCl3) δ (ppm) 1H NMR (600 MHz, CDCl3): 1.04 (s, 9H, Sit-Bu), 1.25 (s, 3H, CH3), 1.30 (t, J = 7.2 Hz, 1H, 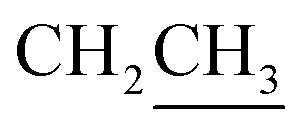 ), 1.41 (s, 3H, CH3), 3.48–3.51 (m, 1H, H-5′), 3.60–3.64 (m, 1H, H-2′), 3.68 (dd, J = 3.0, 11.0 Hz, 1H, CH2OSi), 3.84–3.97 (m, 1H,
), 1.41 (s, 3H, CH3), 3.48–3.51 (m, 1H, H-5′), 3.60–3.64 (m, 1H, H-2′), 3.68 (dd, J = 3.0, 11.0 Hz, 1H, CH2OSi), 3.84–3.97 (m, 1H, 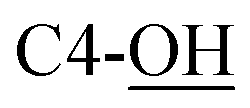 ), 4.17–4.25 (m, 2H,
), 4.17–4.25 (m, 2H, 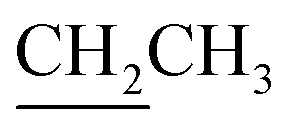 ), 4.32 (dd, J = 3.0, 11.0 Hz, 1H, CH2OSi), 4.67–4.70 (m, 2H, H-3′, H-4′), 4.79 (bs, 1H, H-4), 6.20 (dd, J = 2.0, 15.5 Hz, 1H, H-3), 7.10 (dd, J = 4.4, 15.5 Hz, 1H, H-2), 7.37–7.45 (m, 6H, H-Ph), 7.62–7.69 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3) δ (ppm) 14.4
), 4.32 (dd, J = 3.0, 11.0 Hz, 1H, CH2OSi), 4.67–4.70 (m, 2H, H-3′, H-4′), 4.79 (bs, 1H, H-4), 6.20 (dd, J = 2.0, 15.5 Hz, 1H, H-3), 7.10 (dd, J = 4.4, 15.5 Hz, 1H, H-2), 7.37–7.45 (m, 6H, H-Ph), 7.62–7.69 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3) δ (ppm) 14.4 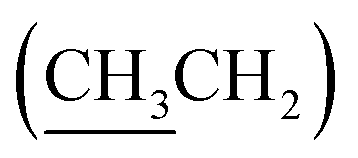 , 19.2 [Si(CH3)3], 23.9 (CH3), 25.8 (CH3), 27.0 [SiC(H3)3], 60.2 (C-6), 60.5
, 19.2 [Si(CH3)3], 23.9 (CH3), 25.8 (CH3), 27.0 [SiC(H3)3], 60.2 (C-6), 60.5 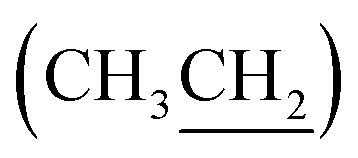 , 69.0 (C-5), 69.8 (C-2), 70.1 (C-4′), 78.4 (C-3), 79.4 (C-4), 111.3 [(CH3)2], 121.7 (C-3′), 127.9 (CH, Ph), 128.0 (CH, Ph), 130.0 (2× CH, Ph), 132.8 (C, Ph), 133.0 (C, Ph), 135.7 (CH, Ph), 135.8 (CH, Ph), 148.3 (C-2′), 166.6 (CO). HRMS (ESI) m/z: for C30H42NO7Si [M + H]+, calcd: 556.2726; found: 556.2728.
, 69.0 (C-5), 69.8 (C-2), 70.1 (C-4′), 78.4 (C-3), 79.4 (C-4), 111.3 [(CH3)2], 121.7 (C-3′), 127.9 (CH, Ph), 128.0 (CH, Ph), 130.0 (2× CH, Ph), 132.8 (C, Ph), 133.0 (C, Ph), 135.7 (CH, Ph), 135.8 (CH, Ph), 148.3 (C-2′), 166.6 (CO). HRMS (ESI) m/z: for C30H42NO7Si [M + H]+, calcd: 556.2726; found: 556.2728.
Rf = 0.27 (EtOAc), mp = 53–56 °C, [α]25D = −45.4 (c 1.02, CHCl3). IR (ATR) νmax: 3353; 2932; 2857; 1427; 1209; 1088; 972; 822; 740; 700; 613; 502 cm−1. 1H NMR (600 MHz, CDCl3) δ (ppm) 1.03 (s, 9H, Sit-Bu), 1.27 (s, 3H, CH3), 1.41 (s, 3H, CH3), 1.93 (bs, 1H, C4-OH), 3.48 (t, J = 3.1 Hz, 1H, H-5′), 3.54 (t, J = 4.9 Hz, 1H, H-2′), 3.58 (d, J = 8.1 Hz, 1H, 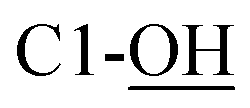 ), 3.68 (dd, J = 3.2, 11.0 Hz, 1H, CH2OSi), 4.16 (d, J = 4.9 Hz, 2H, H-4), 4.27 (dd, J = 3.5, 11.0 Hz, 1H, CH2OSi), 4.54–4,59 (m, 1H, H-1), 4.69 (d, J = 6.9 Hz, 1H, H-4′), 4.73 (dd, J = 5.2, 6.9 Hz, 1H, H-3′), 5.93 (tdd, J = 1.2, 5.9, 15.6 Hz, 1H, H-2), 5.99 (dt, J = 5.0, 15.6 Hz, 1H, H-3), 7.01 (s, 1H,
), 3.68 (dd, J = 3.2, 11.0 Hz, 1H, CH2OSi), 4.16 (d, J = 4.9 Hz, 2H, H-4), 4.27 (dd, J = 3.5, 11.0 Hz, 1H, CH2OSi), 4.54–4,59 (m, 1H, H-1), 4.69 (d, J = 6.9 Hz, 1H, H-4′), 4.73 (dd, J = 5.2, 6.9 Hz, 1H, H-3′), 5.93 (tdd, J = 1.2, 5.9, 15.6 Hz, 1H, H-2), 5.99 (dt, J = 5.0, 15.6 Hz, 1H, H-3), 7.01 (s, 1H, 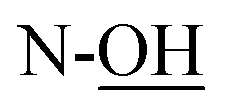 ), 7.37–7.44 (m, 6H, H-Ph), 7.62–7.69 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3) δ (ppm) 19.2 [Si(CH3)3], 24.0 (CH3), 25.8 (CH3), 27.0 [SiC(H3)3], 60.1 (C-1′), 63.2 (C-6), 69.3 (C-5), 70.7 (C-2), 70.9 (C-4′), 78.3 (C-3), 79.2 (C-4), 111.1 [(CH3)2], 127.8 (CH, Ph), 127.9 (CH, Ph), 129.9 (CH, Ph), 130.0 (CH, Ph), 131.2 (C-2′), 131.9 (C-3′), 132.9 (C, Ph), 133.0 (C, Ph), 135.7 (CH, Ph), 135.8 (CH, Ph). HRMS (ESI) m/z: for C28H40NO6Si [M + H]+, calcd: 514.2620; found: 514.2621.
), 7.37–7.44 (m, 6H, H-Ph), 7.62–7.69 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3) δ (ppm) 19.2 [Si(CH3)3], 24.0 (CH3), 25.8 (CH3), 27.0 [SiC(H3)3], 60.1 (C-1′), 63.2 (C-6), 69.3 (C-5), 70.7 (C-2), 70.9 (C-4′), 78.3 (C-3), 79.2 (C-4), 111.1 [(CH3)2], 127.8 (CH, Ph), 127.9 (CH, Ph), 129.9 (CH, Ph), 130.0 (CH, Ph), 131.2 (C-2′), 131.9 (C-3′), 132.9 (C, Ph), 133.0 (C, Ph), 135.7 (CH, Ph), 135.8 (CH, Ph). HRMS (ESI) m/z: for C28H40NO6Si [M + H]+, calcd: 514.2620; found: 514.2621.
:1). Zinc powder (2.16 g, 33 mmol, 20 equiv.) and the reaction mixture was vigorously stirred at room temperature for 30 min. The progress of the reaction was monitored by TLC (EtOAc). After observing the complete disappearance of the starting material, zinc was removed by filtration through filter paper under vacuum and washed with CH2Cl2 (200 mL). Saturated aqueous NaHCO3 was slowly added to the stirring solution until the pH was neutral. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3× 100 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by FCC (CH2Cl2/MeOH 97:3) to give alcohol 4 (665 mg, 1.34 mmol, 81%) as a yellowish oil.Rf = 0.27 (CH2Cl2/MeOH 95:5), [α]25D = −8.49 (c 1.07, CHCl3). IR (ATR) νmax: 3350; 2931; 2858; 1427; 1208; 1105; 1043; 742; 700; 613; 502 cm−1. 1H NMR (600 MHz, CDCl3) δ (ppm) 1. 05 (s, 9H, Sit-Bu), 1.31 (s, 3H, CH3), 1.50 (s, 3H, CH3), 2.35 (bs, 3H,  , NH), 3.10 (dd, J = 4.4, 5.8 Hz, 1H, H-2′), 3.36 (t, J = 5.9 Hz, 1H, H-5′), 3.59 (dd, J = 6.2, 10.5 Hz, 1H, CH2OSi), 3.64 (dd, J = 5.6, 10.5 Hz, 1H, CH2OSi), 4.15 (d, J = 5.2 Hz, 2H, H-4), 4.33 (t, J = 6.0 Hz, 1H, H-1), 4.67 (d, J = 5.8 Hz, 1H, H-4′), 4.70 (dd, J = 4.4, 5.8 Hz, 1H, H-3′), 5.84 (tdd, J = 1.5, 6.5, 15.6 Hz, 1H, H-2), 5.94 (dtd, J = 1.0, 5.2, 15.6 Hz, 1H, H-3), 7.37–7.45 (m, 6H, H-Ph), 7.62–7.66 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3) δ (ppm) 19.3 [Si(CH3)3], 23.9 (CH3), 26.1 (CH3), 27.0 [SiC(H3)3], 62.9 (C-1′), 64.7 (C-5), 64.9 (C-6), 65.1 (C-2), 72.1 (C-4′), 82.5 (C-3), 83.9 (C-4), 111.5 [(CH3)2], 127.9 (2× CH, Ph), 130.0 (2× CH, Ph), 131.5 (C-2′), 131.7 (C-3′), 133.2 (2× C, Ph), 135.7 (2× CH, Ph). HRMS (ESI) m/z: for C28H40NO5Si [M + H]+, calcd: 498.2671; found: 498.2676.
, NH), 3.10 (dd, J = 4.4, 5.8 Hz, 1H, H-2′), 3.36 (t, J = 5.9 Hz, 1H, H-5′), 3.59 (dd, J = 6.2, 10.5 Hz, 1H, CH2OSi), 3.64 (dd, J = 5.6, 10.5 Hz, 1H, CH2OSi), 4.15 (d, J = 5.2 Hz, 2H, H-4), 4.33 (t, J = 6.0 Hz, 1H, H-1), 4.67 (d, J = 5.8 Hz, 1H, H-4′), 4.70 (dd, J = 4.4, 5.8 Hz, 1H, H-3′), 5.84 (tdd, J = 1.5, 6.5, 15.6 Hz, 1H, H-2), 5.94 (dtd, J = 1.0, 5.2, 15.6 Hz, 1H, H-3), 7.37–7.45 (m, 6H, H-Ph), 7.62–7.66 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3) δ (ppm) 19.3 [Si(CH3)3], 23.9 (CH3), 26.1 (CH3), 27.0 [SiC(H3)3], 62.9 (C-1′), 64.7 (C-5), 64.9 (C-6), 65.1 (C-2), 72.1 (C-4′), 82.5 (C-3), 83.9 (C-4), 111.5 [(CH3)2], 127.9 (2× CH, Ph), 130.0 (2× CH, Ph), 131.5 (C-2′), 131.7 (C-3′), 133.2 (2× C, Ph), 135.7 (2× CH, Ph). HRMS (ESI) m/z: for C28H40NO5Si [M + H]+, calcd: 498.2671; found: 498.2676.
With the aim to completely characterize the new compounds, a sample of the mixture was subjected to preparative TLC (CH2Cl2/MeOH 9:1) to give two pure isomers 14 and 15 as yellowish solids.
Data for (6S,7S)-14. Rf = 0.21 (n-hexane/EtOAc 7
:3), mp = 51–54 °C, [α]25D = −124.7 (c 1.00, CHCl3). IR (ATR) νmax: 3435; 2930; 2856; 1427; 1207; 1111; 1083; 823; 740; 700; 613; 502 cm−1. 1H NMR (600 MHz, CDCl3) δ (ppm) 1.06 (s, 9H, Sit-Bu), 1.28 (s, 3H, CH3), 1.50 (s, 3H, CH3), 2.09 (bs, 1H, OH), 2.98 (bs, 1H, OH), 3.48–3.54 (m, 2H, H-3, H-8a), 3.56 (dd, J = 5.6, 10.1 Hz, 1H, H-8b), 3.64 (dd, J = 3.1, 9.6 Hz, 1H, H-5), 3.70 (d, J = 13.5 Hz, 1H, H-9a), 3.76 (d, J = 5.1 Hz, 1H, H-7a), 3.85 (d, J = 13.0 Hz, 1H, H-9b), 4.09 (d, J = 4.3 Hz, 1H, H-7), 4.40 (dd, J = 4.2, 9.7 Hz, 1H, H-6), 4.59 (t, J = 5.5 Hz, 1H, H-1), 4.66 (d, J = 5.8 Hz, 1H, H-2), 7.39–7.45 (m, 6H, H-Ph), 7.67–7.69 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3) δ (ppm) 19.2 [Si(CH3)3], 23.4 (CH3), 25.0 (CH3), 27.0 [SiC(H3)3], 35.6 (C-6), 58.1 (C-9), 64.7 (C-3), 66.1 (C-8), 68.5 (C-5), 71.5 (C-7a), 72.0 (C-7), 80.9 (C-1), 85.3 (C-2), 112.4 [(CH3)2], 128.0 (2× CH, Ph), 130.0 (2× CH, Ph), 133.2 (2× C, Ph), 135.8 (2× CH, Ph). HRMS (ESI) m/z: for C28H39INO5Si [M + H]+, calcd: 624.1637; found: 624.1640.
Data for (6R,7R)-15. Rf = 0.17 (n-hexane/EtOAc 7
:3), mp = 45–48 °C, [α]25D = −32.9 (c 1.01, CHCl3). IR (ATR) νmax: 3427; 2931; 2857; 1427; 1209; 1105; 1063; 823; 739; 700; 613; 501 cm−1. 1H NMR (600 MHz, CDCl3) δ (ppm) 1.06 (s, 9H, Sit-Bu), 1.30 (s, 3H, CH3), 1.52 (s, 3H, CH3), 2.33 (bs, 1H, OH), 3.29 (t, J = 5.6 Hz, 1H, H-3), 3.46–3.49 (m, 2H, H-8a,b), 3.51 (dt, J = 2.3, 10.2 Hz, 1H, H-5), 3.57–3.60 (m, 3H, H-7a, H-9a,b), 4.17 (dd, J = 8.3, 10.2 Hz, 1H, H-6), 4.59 (dd, J = 1.0, 5.7 Hz, 1H, H-2), 4.71 (t, J = 5.6 Hz, 1H, H-1), 4.86 (dd, J = 4.8, 8.3 Hz, 1H, H-7), 7.39–7.46 (m, 6H, H-Ph), 7.66–7.68 (m, 4H, H-Ph). 13C NMR (150 MHz, CDCl3) δ (ppm) 19.2 [Si(CH3)3], 24.0 (CH3), 26.8 (CH3), 27.0 [SiC(H3)3], 31.1 (C-6), 58.9 (C-9), 66.0 (C-8), 67.6 (C-3), 72.8 (C-5), 73.4 (C-7a), 79.7 (C-7), 81.7 (C-1), 85.3 (C-2), 112.6 [(CH3)2], 128.0 (2× CH, Ph), 130.0 (2× CH, Ph), 132.9 (2× C, Ph), 135.7 (2× CH, Ph). HRMS (ESI) m/z: for C28H39INO5Si [M + H]+, calcd: 624.1637; found: 624.1640.
:3) to give two pure pyrrolizidines 16 (195 mg, 0.39 mmol, 30% after two steps) and 17 (165 mg, 0.33 mmol, 25% after two steps) as yellowish oils.
Data for (6S)-16. Rf = 0.21 (n-hexane/EtOAc 1
:9), [α]25D = +1.04 (c 1.00, CHCl3). IR (ATR) νmax: 3402; 2928; 2854; 1430; 1380; 1206; 1106; 1047; 703; 617; 506 cm−1. 1H NMR (600 MHz, CD3OD) δ (ppm) 1.07 (s, 9H, Sit-Bu), 1.29 (s, 3H, CH3), 1.47 (s, 3H, CH3), 1.83–1.88 (m, 1H, H-6a), 1.92–1.98 (m, 1H, H-6b), 3.48–3.52 (m, 3H, H-5, H-3, H-9a), 3.55 (dd, J = 3.3, 5.2 Hz, 1H, H-7a), 3.57 (dd, J = 6.7, 10.2 Hz, 1H, H-8a), 3.60–3.64 (m, 1H, H-9b), 3.70 (dd, J = 4.9, 10.2 Hz, 1H, H-8b), 4.36 (ddd, J = 3.4, 4.2, 6.1 Hz, 1H, H-7), 4.59 (pseudo t, J = 5.4, 5.7 Hz, 1H, H-1), 4.70 (dd, J = 1.5, 5.9 Hz, 1H, H-2), 7.39–7.46 (m, 6H, H-Ph), 7.69–7.72 (m, 4H, H-Ph). 13C NMR (150 MHz, CD3OD) δ (ppm) 20.0 [Si(CH3)3], 24.3 (CH3), 26.1 (CH3), 27.4 [SiC(H3)3], 40.2 (C-6), 61.8 (C-7a), 64.0 (C-9), 64.8 (C-5), 66.6 (C-8), 70.6 (C-7), 76.9 (C-3), 81.2 (C-2), 87.3 (C-1), 113.1 [(CH3)2], 128.8 (CH, Ph), 128.9 (CH, Ph), 130.9 (CH, Ph), 131.0 (CH, Ph), 134.6 (2× C, Ph), 136.8 (2× CH, Ph). HRMS (ESI) m/z: for C28H40NO5Si [M + H]+, calcd: 498.2671; found: 498.2673.
Data for (6R)-17. Rf = 0.37 (n-hexane/EtOAc 1
:9), [α]25D = +2.13 (c 1.00, CHCl3). IR (ATR) νmax: 3399; 2934; 2858; 1427; 1374; 1206; 1112; 1041; 829; 741; 706; 618; 509 cm−1. 1H NMR (600 MHz, CD3OD) δ (ppm) 1.06 (s, 9H, Sit-Bu), 1.27 (s, 3H, CH3), 1.45 (s, 3H, CH3), 1.60 (ddd, J = 4.8, 7.4, 13.0 Hz, 1H, H-6a), 2.44 (dt, J = 7.4, 13.0 Hz, 1H, H-6b), 3.32–3.44 (m, 4H, H-3, H-5, H-9a,b), 3.51–3.54 (m, 2H, H-7a, H-8a), 3.64 (dd, J = 5.7, 10.4 Hz, 1H, H-8b), 4.56 (ddd, J = 2.4, 4.8, 7.2 Hz, 1H, H-7), 4.63 (pseudo t, J = 5.1, 5.7 Hz, 1H, H-1), 4.68 (dd, J = 1.3, 5.7 Hz, 1H, H-2), 7.40–7.46 (m, 6H, H-Ph), 7.70–7.73 (m, 4H, H-Ph). 13C NMR (150 MHz, CD3OD) δ (ppm) 19.9 [Si(CH3)3], 23.9 (CH3), 26.9 (CH3), 27.4 [SiC(H3)3], 40.3 (C-6), 67.1 (C-8), 67.2 (C-9), 68.2 (C-5), 71.9 (C-3), 72.0 (C-7), 77.7 (C-7a), 83.3 (C-2), 87.1 (C-1), 113.0 [(CH3)2], 128.8 (CH, Ph), 128.9 (CH, Ph), 130.9 (CH, Ph), 131.0 (CH, Ph), 134.5 (2× C, Ph), 137.8 (2× CH, Ph). HRMS (ESI) m/z: for C28H40NO5Si [M + H]+, calcd: 498.2671; found: 498.2672.
:1). After full disappearance of the starting pyrrolizidine MeOH was removed under reduced pressure. The residue was purified by FCC (CH2Cl2/MeOH 4:1) to give pyrrolizidine 18 (85 mg, 0.33 mmol, 85%) as a yellowish oil.Rf = 0.16 (CH2Cl2/MeOH 4:1), [α]25D = +206.3 (c 0.90, CHCl3). IR (ATR) νmax: 3311; 2981; 2937; 2360; 1383; 1206; 1165; 1024; 859; 512 cm−1. 1H NMR (600 MHz, CD3OD) δ (ppm) 1.32 (s, 3H, CH3), 1.49 (s, 3H, CH3), 1.90 (ddd, J = 2.8, 5.8, 12.9 Hz, 1H, H-6a), 2.00 (ddd, J = 5.3, 9.2, 12.9 Hz, 1H, H-6b), 3.60–3.65 (m, 2H, H-9a,b), 3.72–3.81 (m, 5H, H-3, H-5, H-7a, H-8a,b), 4.44 (dt, J = 2.6, 5.3 Hz, 1H, H-7), 4.72 (dd, J = 2.2, 6.0 Hz, 1H, H-2), 4.76 (t, J = 5.8 Hz, 1H, H-1). 13C NMR (150 MHz, CD3OD) δ (ppm) 24.3 (CH3), 26.2 (CH3), 39.1 (C-6), 62.7 (C-8), 63.2 (C-9), 64.2 (C-3), 66.1 (C-5), 70.8 (C-5), 78.2 (C-7a), 80.7 (C-1), 86.2 (C-2), 113.4 [(CH3)2]. HRMS (ESI) m/z: for C12H22NO5 [M + H]+, calcd: 260.1493; found: 260.1489.
:1). After full disappearance of the starting pyrrolizidine MeOH was removed under reduced pressure. The residue was purified by FCC (CH2Cl2/MeOH 4:1) to give pyrrolizidine 19 (80 mg, 0.31 mmol, 94%) as a yellowish oil.Rf = 0.34 (CH2Cl2/MeOH 4:1), [α]25D = +1.78 (c 1.00, CHCl3). IR (ATR) νmax: 3344; 2931; 2860; 1430; 1212; 1109; 1074; 1032; 700; 618; 505 cm−1. 1H NMR (600 MHz, CD3OD) δ (ppm) 1.30 (s, 3H, CH3), 1.46 (s, 3H, CH3), 1.63 (ddd, J = 4.6, 7.3, 13.1 Hz, 1H, H-6a), 2.45 (dt, J = 7.6, 13.1 Hz, 1H, H-6b), 3.28–3.30 (m, 1H, H-3), 3.39 (tt, J = 5.3, 7.7 Hz, 1H, H-5), 3.43 (dd, J = 7.2, 11.0 Hz, 1H, H-8a), 3.48 (dd, J = 5.8, 11.0 Hz, 1H, H-8b), 3.50 (d, J = 5.3 Hz, 2H, H-9a,b), 3.55 (dd, J = 2.3, 4.8 Hz, 1H, H-7a), 4.57 (ddd, J = 2.4, 4.6, 7.1 Hz, 1H, H-7), 4.60 (dd, J = 1.7, 5.7 Hz, 1H, H-2), 4.70 (pseudo t, J = 5.2, 5.4 Hz, 1H, H-1). 13C NMR (150 MHz, CD3OD) δ (ppm) 24.0 (CH3), 26.9 (CH3), 39.9 (C-6), 63.9 (C-8), 66.6 (C-9), 68.5 (C-5), 72.0 (C-7), 72.6 (C-3), 77.2 (C-7a), 82.8 (C-1), 86.5 (C-2), 113.2 [(CH3)2]. HRMS (ESI) m/z: for C12H22NO5 [M + H]+, calcd: 260.1493; found: 260.1491.
:3). Afterwards, the resin was well washed in the flask with 13% aqueous NH3 (26% NH3/H2O 1:1, 5× 2 mL), and the combined liquids were concentrated under reduced pressure to give (+)-hyacinthacine C3 (2a, 68 mg, 0.31 mmol, 94%) as a yellowish oil.Rf = 0.14 (CH2Cl2/MeOH/NH3 60:35:5), [α]22D = +9.30 (c 0.3, H2O), [ref. 4: [α]22D = +8.80 (c 0.3, H2O), ref. 3: [α]D = +3.5 (c 0.23, H2O)]. IR (ATR) νmax: 3282; 2931; 2878; 1409; 1342; 1124; 1024; 600; 547; 477 cm−1. 1H NMR (600 MHz, D2O, TSP-d4) δ (ppm) 1.99 (dt, J = 6.0, 13.1 Hz, 1H, H-6a), 2.16 (dt, J = 6.6, 13.2 Hz, 1H, H-6b), 3.33 (dt, J = 4.7, 8.3 Hz, 1H, H-3), 3.54 (t, J = 4.3 Hz, 1H, H-7a), 3.60–3.64 (m, 1H, H-5), 3.69 (dd, J = 5.3, 11.8 Hz, 1H, H-8a), 3.81 (dd, J = 6.0, 12.5 Hz, 1H, H-9a); 3.83 (dd, J = 4.2, 11.8 Hz, 1H, H-8b), 3.86 (dd, J = 4.4, 12.5 Hz, 1H, H-9b), 4.05 (dd, J = 4.1, 8.3 Hz, 1H, H-2), 4.23 (t, J = 4.3 Hz, 1H, H-1), 4.63 (td, J = 4.2, 5.8 Hz, 1H, H-7). 13C NMR (150 MHz, D2O) δ (ppm) 40.1 (C-6), 63.1 (C-9), 64.4 (C-8), 64.5 (C-3), 64.8 (C-5), 71.2 (C-7), 72.7 (C-1), 77.1 (C-2), 77.3 (C-7a). HRMS (ESI) m/z: for C9H18NO5 [M + H]+, calcd: 220.1180; found: 220.1180.
:3). Afterwards, the resin was well washed in the flask with 13% aqueous NH3 (26% NH3/H2O 1:1, 5× 2 mL), and the combined liquids were concentrated under reduced pressure to give (+)-5-epi-hyacinthacine C3 (2b, 65 mg, 0.29 mmol, 95%) as a yellowish oil.Rf = 0.25 (CH2Cl2/MeOH/NH3 60:35:5), [α]22D = +13.78 (c 0.3, H2O), [ref. 4: [α]22D = +14.80 (c 0.3, H2O)]. IR (ATR) νmax: 3193; 2917; 2880; 1422; 1333; 1112; 1022; 1000; 819; 737; 635; 561; 416 cm−1. 1H NMR (600 MHz, D2O, TSP-d4) δ (ppm) 1.66 (ddd, J = 6.9, 8.3, 13.0 Hz, 1H, H-6a), 2.41 (dt, J = 6.7, 13.0 Hz, 1H, H-6b), 2.93 (ddd, J = 3.8, 6.1, 9.1 Hz, 1H, H-3), 3.14–3.19 (m, 1H, H-5), 3.41 (t, J = 4.3 Hz, 1H, H-7a), 3.57 (dd, J = 5.4, 11.3 Hz, 1H, H-9a), 3.66 (dd, J = 6.1, 11.7 Hz, 1H, H-8a), 3.68 (dd, J = 6.4, 11.3 Hz, 1H, H-9b), 3.78 (dd, J = 3.8, 11.7 Hz, 1H, H-8b), 3.97 (dd, J = 4.1, 9.1 Hz, 1H, H-2), 4.16 (t, J = 4.0 Hz, 1H, H-1), 4.63 (td, J = 4.6, 6.9 Hz, 1H, H-7). 13C NMR (150 MHz, D2O) δ (ppm) 40.9 (C-6), 65.6 (C-9), 67.6 (C-8), 71.1 (C-5), 72.1 (C-7), 72.4 (C-3), 73.6 (C-1), 76.4 (C-7a), 77.6 (C-2). HRMS (ESI) m/z: for C9H18NO5 [M + H]+, calcd: 220.1180; found: 220.1178.
Cell culture and cultivation conditions
All the cell passage numbers were lower than 25. Cells were maintained under standard cultivation conditions: 37 °C, humidified atmosphere containing 5% CO2.The human glioblastoma astrocytoma cancer cell line U84-MG was purchased from ATCC, USA. Cells were cultivated in Dulbecco's Modified Eagle's Medium (DMEM) high glucose supplemented with L-glutamine 2 mM, sodium pyruvate 1 mM, 1% non-essential amino acids (NAA) and 10% fetal bovine serum (FBS).
The HK-2 (purchased from ATCC, USA), an immortalized proximal tubular cell line was cultivated in DMEM high glucose medium supplemented with 2 mM L-glutamine and 10% FBS.
The hepatocellular carcinoma cell line HepG2 (ECACC, UK) was cultivated in DMEM high glucose medium containing L-glutamine 2 mM, 10% FBS, 1% NAA.
The human placental choriocarcinoma cells JEG-3 (ECACC, UK) were cultivated in Minimum Essential Medium Eagle (MEM) containing 1% NAA, sodium pyruvate 1 mM and 10% FBS.
DMEM, MEM and all used supplements were purchased from Sigma-Aldrich, USA except for FBS, which was purchased from Biosera, South America.
Cell viability testing
All the tested cells were seeded in a 96-well plate (TPP, Switzerland) at low density 4 × 103 cells per well. Cells were treated 24 hours after seeding with medium containing tested substance at indicated concentrations, 1% DMSO (non-toxic control, 100% viability) or 10% DMSO (toxic control, 100% mortality). Stock solution of tested compounds were prepared by dissolution in DMSO (Sigma-Aldrich, USA) and dilution in cultivation medium to set wanted concentration. Amount of solvent was 1% (V/V) in all cases except toxic control (10%). The cells were incubated with compounds for 24, 48 and 72 hours, respectively, under standard culture conditions. Viability of cells was determined using CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, USA) according to the manufacturer's instructions.IC50 values were determined using the GraphPad Prism 8.3.1 software.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Slovak Grant Agency for Science VEGA (project no. 1/0552/18), and Charles University Research Programme Progres (grant no. Q42). The authors are sincerely grateful to Ing. Miroslav Koóš, DrSc. from the Slovak Academy of Sciences for the generous gift of L-mannose which was used as the starting material in the presented total synthesis.Notes and references
- (a) R. J. Nash, in Bioactive Natural Products, ed S. M. Colegate and R. J. Molyneux, CRC Press, Boca Raton, 2nd edn, 2008, pp. 407–420 Search PubMed; (b) T. D. Butters, R. A. Dwek and F. M. Platt, Chem. Rev., 2000, 100, 4683 CrossRef CAS PubMed; (c) I. Robina, A. J. Moreno-Vargas, A. T. Carmona and P. Vogel, Curr. Drug Metab., 2004, 5, 329 CrossRef CAS PubMed; (d) P.-H. Liang, W.-C. Cheng, Y.-L. Lee, H.-P. Yu, Y.-T. Wu, Y.-L. Lin and C.-H. Wong, ChemBioChem, 2006, 7, 165 CrossRef CAS PubMed; (e) V. H. Lillelund, H. H. Jensen, X. F. Liang and M. Bols, Chem. Rev., 2002, 102, 515 CrossRef CAS PubMed; (f) E. Borges de Melo, A. da Silva Gomes and I. Carvalho, Tetrahedron, 2006, 62, 10277 CrossRef CAS; (g) S. Gerber-Lemaire and L. Juillerat-Jeanneret, Mini-Rev. Med. Chem., 2006, 6, 1043 CrossRef CAS PubMed.
- (a) N. Asano, R. J. Nash, R. J. Molyneux and G. W. J. Fleet, Tetrahedron: Asymmetry, 2000, 11, 1645 CrossRef CAS; (b) N. Asano, J. Enzyme Inhib., 2000, 15, 215 CrossRef CAS PubMed; (c) A. A. Watson, G. W. J. Fleet, N. Asano, R. J. Molyneux and R. J. Nash, Phytochemistry, 2001, 56, 265 CrossRef CAS PubMed; (d) R. J. Nash, A. Kato, C.-Y. Yu and G. W. J. Fleet, Future Med. Chem., 2011, 3, 1513 CrossRef CAS PubMed.
- A. Kato, N. Kato, I. Adachi, J. Hollinshead, G. W. J. Fleet, C. Kuriyama, K. Ikeda, N. Asano and R. J. Nash, J. Nat. Prod., 2007, 70, 993 CrossRef CAS PubMed.
- T. Sengoku, Y. Satoh, M. Takahashi and H. Yoda, Tetrahedron Lett., 2009, 50, 4937 CrossRef CAS.
- (a) A. W. Carroll, A. C. Willis, M. Hoshino, A. Kato and S. G. Pyne, J. Nat. Prod., 2019, 82, 358 CrossRef CAS PubMed; (b) A. W. Carroll and S. G. Pyne, Curr. Org. Synth., 2019, 16, 498 CrossRef CAS PubMed; (c) T. Ritthiwigrom and S. G. Pyne, Stud. Nat. Prod. Chem., 2012, 36, 1 CAS; (d) R. Lahiri, A. A. Ansari and Y. D. Vankar, Chem. Soc. Rev., 2013, 42, 5102 RSC; (e) J. Robertson and K. Stevens, Nat. Prod. Rep., 2014, 31, 1721 RSC; (f) V. Desvergnes and Y. Landais, Stud. Nat. Prod. Chem., 2014, 42, 373 CAS; (g) S. T. Martinez, C. Belouezzane, A. C. Pinto and T. Glasnov, Org. Prep. Proced. Int., 2016, 48, 223 CrossRef CAS; (h) J. Robertson and K. Stevens, Nat. Prod. Rep., 2017, 34, 62 RSC.
- T. Malatinský, B. Otočková, L. Dikošová and R. Fischer, ChemistrySelect, 2019, 4, 4233 CrossRef.
- O. Tamura, A. Toyao and H. Ishibashi, Synlett, 2002, 1344 CrossRef CAS.
- (a) A. Peer and A. Vasella, Helv. Chim. Acta, 1999, 82, 1044 CrossRef CAS; (b) S. Desvergnes, S. Py and Y. Vallée, J. Org. Chem., 2005, 70, 1459 CrossRef CAS PubMed.
- (a) T. Malatinský, M. Spišáková, M. Babjak, J. Doháňošová, J. Marek, J. Moncol and R. Fischer, Eur. J. Org. Chem., 2017, 1086 CrossRef; (b) T. Malatinský, Z. Puhová, M. Babjak, J. Doháňošová, J. Moncol, Š. Marchalín and R. Fischer, Tetrahedron Lett., 2018, 59, 3867 CrossRef.
- E. A. Brock, S. G. Davies, J. A. Lee, P. M. Roberts and J. E. Thomson, Org. Biomol. Chem., 2013, 11, 3187 RSC.
- J. A. Tamayo, F. Franco and F. Sánchez-Cantalejo, Eur. J. Org. Chem., 2011, 7182 CrossRef CAS ; and references cited therein..
- T. S. Cooper, P. Laurent, C. J. Moody and A. K. Takle, Org. Biomol. Chem., 2004, 2, 265 RSC.
- Y. Kondo, N. Suzuki, M. Takahashi, T. Kumamoto, H. Masu and T. Ishikawa, J. Org. Chem., 2012, 77, 7988 CrossRef CAS PubMed.
- K. Inoue, A. Sawada, I. Shibata and A. Baba, J. Am. Chem. Soc., 2002, 124, 906 CrossRef CAS PubMed.
- H. Sun and S. G. DiMagno, J. Am. Chem. Soc., 2005, 127, 2050 CrossRef CAS PubMed.
- M. Jasiński, E. Moreno-Clavijo and H.-U. Reissig, Eur. J. Org. Chem., 2014, 442 CrossRef.
- (a) W.-J. Liu, J.-L. Ye and P.-Q. Huang, Org. Biomol. Chem., 2010, 8, 2085 RSC; (b) M. R. Wormald, R. J. Nash, P. Hrnciar, J. D. White, R. J. Molyneux and G. W. J. Fleet, Tetrahedron: Asymmetry, 1998, 9, 2549 CrossRef CAS; (c) T. J. Donohoe, H. O. Sintim and J. Hollinshead, J. Org. Chem., 2005, 70, 7297 CrossRef CAS PubMed; (d) T. J. Donohoe, R. E. Thomas, M. D. Cheeseman, C. L. Rigby, G. Bhalay and I. D. Linney, Org. Lett., 2008, 10, 3615 CrossRef CAS PubMed.
- H. Fiaux, F. Popowycz, S. Favre, C. Schütz, P. Vogel, S. Gerber-Lemaire and L. Juillerat-Jeanneret, J. Med. Chem., 2005, 48, 4237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of isoxazolidine intermediate 6, 1H and 13C NMR spectra of 2a, 2b, 4–10, 12–19, the comparison tables of the 1H and 13C NMR data between the synthetic (+)-hyacinthacine (2a) and the natural sample, NOESY spectra of 14, 15, 2a, 2b, evaluation of antiproliferative effect of 2a and 2b. See DOI: 10.1039/d1ra06225e |
| This journal is © The Royal Society of Chemistry 2021 |