
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational methods directed towards drug repurposing for COVID-19: advantages and limitations†
Prem Prakash Sharma‡
a,
Meenakshi Bansal‡ a,
Aaftaab Sethi‡b,
Poonamc,
Lindomar Penad,
Vijay Kumar Goele,
Maria Grishinaf,
Shubhra Chaturvedig,
Dhruv Kumar*h and
Brijesh Rathi*a
a,
Aaftaab Sethi‡b,
Poonamc,
Lindomar Penad,
Vijay Kumar Goele,
Maria Grishinaf,
Shubhra Chaturvedig,
Dhruv Kumar*h and
Brijesh Rathi*a
aLaboratory For Translational Chemistry and Drug Discovery, Department of Chemistry, Hansraj College, University of Delhi, Delhi, 110007, India. E-mail: brijeshrathi@hrc.du.ac.in
bDepartment of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Hyderabad, India
cDepartment of Chemistry, Miranda House, University of Delhi, Delhi, 110007 India
dDepartment of Virology, Aggeu Magalhaes, Institute (IAM), Oswaldo Cruz Foundation (Fiocruz), Recife 50670-420, Pernambuco, Brazil
eSchool of Physical Sciences, Jawaharlal Nehru University, New Delhi, 110067, India
fSouth Ural State University, Laboratory of Computational Modelling of Drugs, Pr. Lenina, 76 454080, Russia
gDivision of Cyclotron and Radiopharmaceutical Sciences, Institute of Nuclear Medicine and Allied Sciences, New Delhi 110054, India
hAmity Institute of Molecular Medicine & Stem Cell Research (AIMMSCR), Amity University Uttar Pradesh, Noida 201313, India
First published on 10th November 2021
Abstract
Novel coronavirus disease 2019 (COVID-19) has significantly altered the socio-economic status of countries. Although vaccines are now available against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a causative agent for COVID-19, it continues to transmit and newer variants of concern have been consistently emerging world-wide. Computational strategies involving drug repurposing offer a viable opportunity to choose a medication from a rundown of affirmed drugs against distinct diseases including COVID-19. While pandemics impede the healthcare systems, drug repurposing or repositioning represents a hopeful approach in which existing drugs can be remodeled and employed to treat newer diseases. In this review, we summarize the diverse computational approaches attempted for developing drugs through drug repurposing or repositioning against COVID-19 and discuss their advantages and limitations. To this end, we have outlined studies that utilized computational techniques such as molecular docking, molecular dynamic simulation, disease–disease association, drug–drug interaction, integrated biological network, artificial intelligence, machine learning and network medicine to accelerate creation of smart and safe drugs against COVID-19.
1. Introduction
The mean investment required to bring a new therapeutic agent to the market has been estimated to be ∼$1.3 billion, accounting for the costs of failed clinical trials.1 Apart from the cost, the time involved in a conventional drug discovery process needs to be substantially reduced especially when response to a novel disease must be rapid, as is the case with the COVID-19 pandemic. It is an essential prerequisite that any new chemical entity being explored as a drug, must be safe and non-toxic.2 However, investigating the safety profile of any new chemical agent is a long and tedious procedure, therefore numerous attempts have made to discover anti-SARS-CoV-2 inhibitors from existing drugs.3,4Drug repurposing or repositioning refers to the usage of pre-existing drugs to tackle a disease other than the one that the drug was originally designed to target. It has several advantages such as reduction of time, risk and cost associated with the development of a new drug molecule.2,5,6 The benefit of drug repurposing is also highlighted by the fact that nearly 30% of new market entrants are derived from pre-existing drugs.7 There are several reasons for which a drug repurposing strategy can be used, some of them include, (a) reduction in time and cost of clinical trials, as evaluation in phase I and II might not be required; (b) already established pharmaceutical supply chains for distribution; (c) the additive or symbiotic effects of combinatorial therapy with other drugs in treatments in contrast to monotherapy; and (d) may explore new mechanisms of action for old drugs.8 The drug repurposing strategy has some limitations such as patent barriers. However, other factors such as the complexity of regulatory pathways, absence of funding opportunities, greater access to data from other industry-sponsored clinical trials, and the heterogeneity of the population for new clinical studies, have been instrumental in evolving drug repurposing as one of the most promising approaches for the drug development with unprecedented swiftness. Drug repurposing is also sometimes referred as drug redirecting, drug repositioning, drug reprofiling, drug retasking, and therapeutic switching.9
Drug repurposing has resulted in the successful development of various medicines. Several examples of drugs repositioned to treat diseases or disorders have come to the market, which include minoxidil, originally developed for hypertension and later used for hair loss; sildenafil, identified to treat erectile dysfunction as a result of a clinical trial for angina; and thalidomide, originally for morning sickness and now used to treat symptoms of leprosy.10 Several other medicines were repurposed and are being used to treat a multitude of ailments.11 A report published in July 2020 has shown that 8565 existing drugs are being repositioned for COVID-19.12
The conventional drug development process involves de novo detection and identification of new molecular entities, including the five phases, (a) discovery and preclinical; (b) safety review; (c) clinical research; (d) FDA review; and (e) FDA post-market safety monitoring. It is a time-consuming and expensive procedure with a high risk of failure as highlighted briefly before, while on the other hand, the drug repositioning has only four steps, including (a) compound identification; (b) compound acquisition; (c) development; and (d) FDA post-market safety monitoring.13 Several reports state that drug repurposing requires clearance from the FDA and/or the European Medicines Agency (EMA) for approval, but it approximately takes anywhere between 3–12 years and at a decreased cost of 50–60 percent.13 Drug repurposing has great significance in the current COVID-19 pandemic scenario where no standard and safe therapy is available yet against COVID-19.
Bioinformatics tool and other recently evolved computational methods based on Artificial Intelligence (AI), Machine Learning (ML) and network medicine are promising and on many occasions have been found to be robust in identifying new indications of existing drugs.14,15 The present article aims to provide highlights on computational approaches in drug development through drug repurposing for COVID-19, where each method is categorized into its core methodology, advantages and limitations. There are many studies published on a day-to-day basis describing a computational approach to identify drugs to treat SARS-CoV-2. This article is not a comprehensive overview of all those approaches as discussing the complete list of published studies would sound like a mere regurgitation and not be of any significant impact. Herein, we have tried to make an effort to provide relevant snippets of information from a huge volume of studies. There are several reviews published on this topic, but these articles have either focused extensively on drug repurposing approaches in general but not with respect to COVID-19 or a discussion on computational approaches has been limited to only a few popular techniques.6,16–18 We believe that the collective information on a wide variety of computational drug repurposing methods discussed in this article alongside with their critical analysis and scrutiny would be of great interest to a wide range of researchers and scientists.
2. Drug repurposing by virtual screening and molecular dynamics (MD) simulations
A large number of in silico studies have been carried out during the period of global lockdown. Structure-based drug design (SBDD) is a popular method in computational drug design in which X-ray crystallographic or NMR based 3D structures of proteins/targets are utilized, and small molecule are generally studied to evaluate the nature of interaction between protein–ligand.16,19,20 It also explains the conformational fitting of ligand within the binding pocket of protein. Therefore, the whole point of exploiting this technique is to find “binders” not just “fitters”.21 Ligands are small molecules that can act as activators or inhibitors of a protein by binding to it.22,23 Scoring functions are very useful and practical to score and rank all the screened ligands in the binding pocket based on the conformations generated, which leads to estimation of “binding affinity”, where the potential binders are referred to as “Hits”.21 There are several softwares available that can be utilized for molecular docking, a few are open-access, such as AutoDock Vina while others are commercial suites like Glide, GOLD, etc.Docking results must be validated with MD simulation to understand the interactions between receptor–ligand and their stability. It helps in estimation of the accurate thermodynamics and kinetics associated with drug–target recognition and binding.24 MD simulation are generally also helpful in separating true positives from false positives. There are several programs, e.g., Desmond and GROMACS among others that can be implemented for MD simulation on GUI interface or command line.
2.1. Methods
Approaches implemented for drug repurposing virtual screening and MD simulations have been described in Fig. 1, and include the following key steps.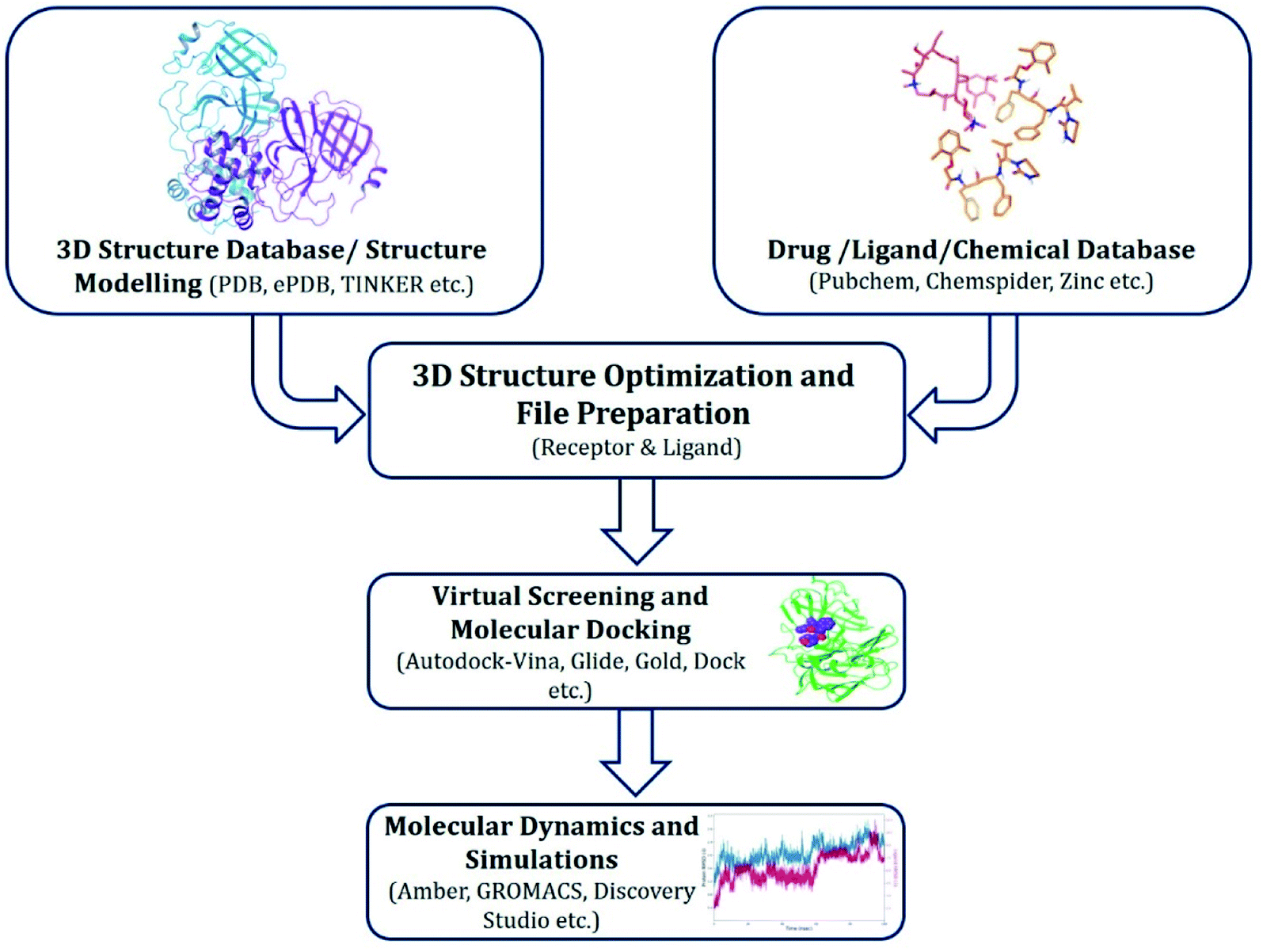 | ||
| Fig. 1 Workflow for structure-based drug design. | ||
(a) Retrieving experimentally determined crystal or cryo-EM protein structures from public databases. If unavailable, utilizing homology modelled protein structures or retrieving predicted structures from sources like AlphaFold (https://alphafold.ebi.ac.uk/);
(b) Protein and ligand preparation prior to docking;
(c) Screening of molecules that can bind to the target proteins;
(d) Selection of hit compounds based on respective scoring function algorithm; and
(e) Validation of most promising molecules by MD simulations.
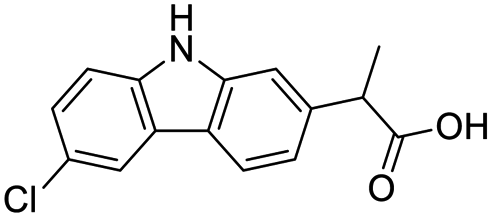
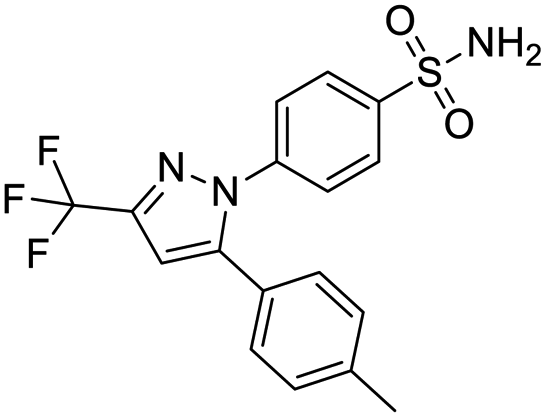
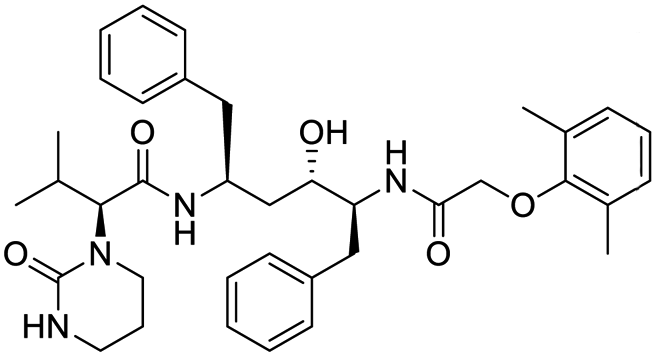
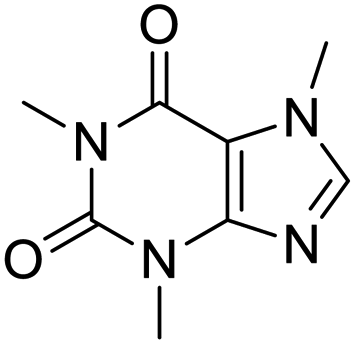
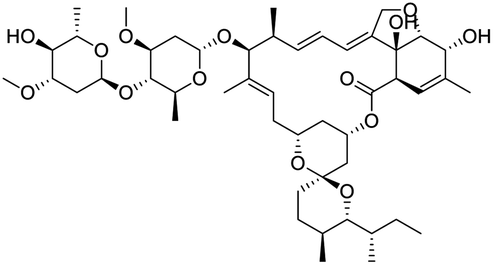
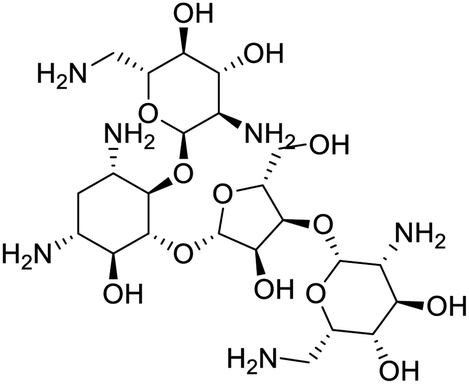
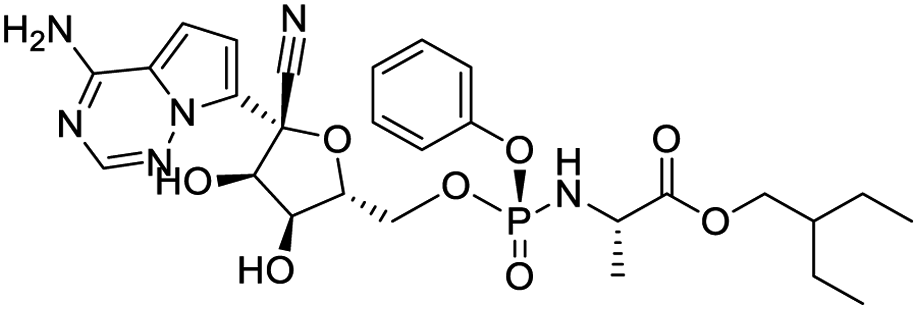
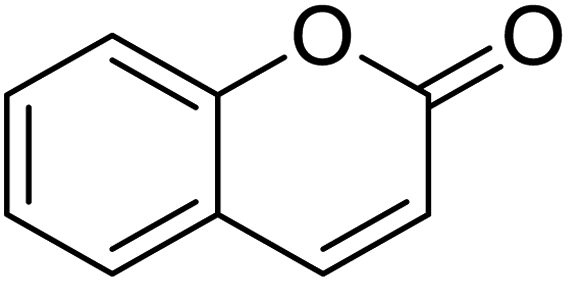
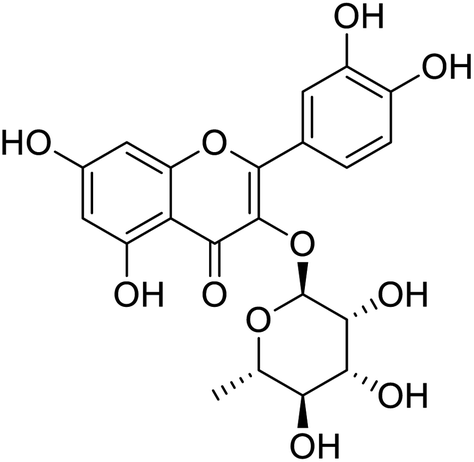
The structures of drug molecules described in this article are provided in Table 1. Recently, Gimeno et al.25 integrated the predictions of three molecular docking softwares (Glide, FRED, and AutoDock Vina) to repurpose drugs against the main protease (Mpro) (PDB: 6LU7) of SARS-CoV-2. The H41 and C145 are catalytic residues responsible for Mpro functionality. Given its pivotal role in mediating the replication and transcription of SARS-CoV-2, antiviral therapies targeting Mpro have the potential to disrupt critical steps in the viral life cycle. The carprofen interacted with both catalytic residues while celecoxib did not. In vitro study of carprofen and celecoxib showed inhibition of Mpro by 3.97 and 11.90% at 50 μM, respectively.25 Wang et al. proposed the potential of lopinavir along with other four drugs, namely, carfilzomib, eravacycline, valrubicin, and elbasvir, as promising molecules to inhibit Mpro (PDB: 6LU7) activity which were also validated by 100 ns MD simulation.26 These molecules interacted to hot spot residues H41, M49, N142, H164, M165, E166, and Q189. The combination of lopinavir and ritonavir showed a potential to better inhibit Mpro than lopinavir and ritonavir alone.27 For decades, caffeine has been utilized clinically to treat apnea in preterm infants and to enhance respiratory function because of its potential role in reducing inflammation, immune modulation, antiviral and improving respiratory symptoms.28 A computational study proposed a promising inhibiting nature of caffeine and linagliptin against Mpro (PDB: 6Y2E).29 Both caffeine and linagliptin interacted with the catalytic residues but caffeine interacted via H-bond and van der Waals interactions while linagliptin interacted through van der Waals interaction alone. Koulgi et al., proposed ensemble docking for in silico drug repurposing against Mpro (PDB: 6LU7) by screening two databases (DB), FDA approved drug-DB and SWEETLEAD-DB.30 Authors found that indinavir, ceftin, ivermectin, neomycin, vasopressin-tennate, and amikacin as top ranked molecules against Mpro. Indinavir, ceftin and ivermectin were observed to show interactions with T24, L27, V42, T45, R60, K61, N142, E166 and Q189. Neomycin, vasopressin-tennate, and amikacin interacted to either H41 or C145. In another study, Khan et al., reported that remdesivir, saquinavir, darunavir and natural products such as flavones and coumarin derivatives as promising hits for Mpro (PDB: 6LU7) inhibition.31 Both molecule interacted to C145, Q189, E166, and T190. Cherrak et al., proposed glycosylated flavonoid quercetin-3-O-rhamnoside as a promising Mpro (PDB: 6LU7) inhibitor.32 The quercetin-3-O-rhamnoside interacted to residues T26, F140, and L141 via H-bond while interacted to residues H163, G143, C145, M49, and R188 via other non-covalent interactions. Upadhyay et al., proposed a few plant extracts including green tea, black tea and haritaki as alternative therapeutic candidates for SARS-CoV-2 infection. These plant products have the polymeric polyphenols thearubigin, quercetin-3-O-rutinoside, and hesperidin which can potentially inhibit Mpro (PDB: 6LU7).33 The thearubigin interacted to catalytic residue, C145 while quercetin-3-O-rutinoside interacted to L141, N142, G143, and E166. Hesperidin hydrophobically interacted to M165 and Q189 while H-bond interacted to N142, and G146 residues.
 |
 |
 |
 |
| Carprofen | Celecoxib | Lopinavir | Carfilzomib |
 |
 |
 |
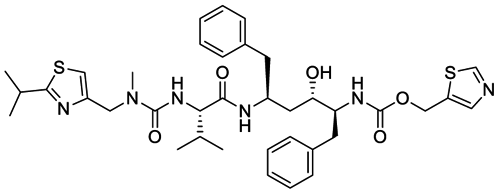 |
| Eravacycline | Valrubicin | Elbasvir | Ritonavir |
 |
 |
 |
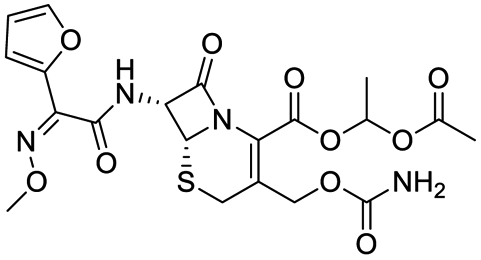 |
| Caffeine | Linagliptin | Indinavir | Ceftin |
 |
 |
 |
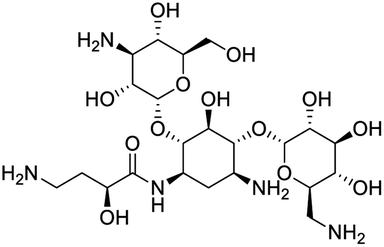 |
| Ivermectin | Neomycin | Vasopressin-tennate | Amikacin |
 |
 |
 |
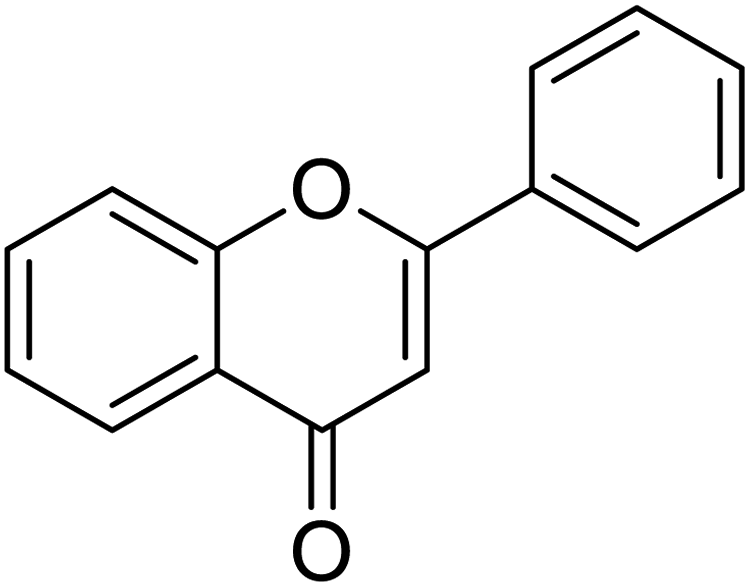 |
| Remdesivir | Saquinavir | Darunavir | Flavone |
 |
 |
 |
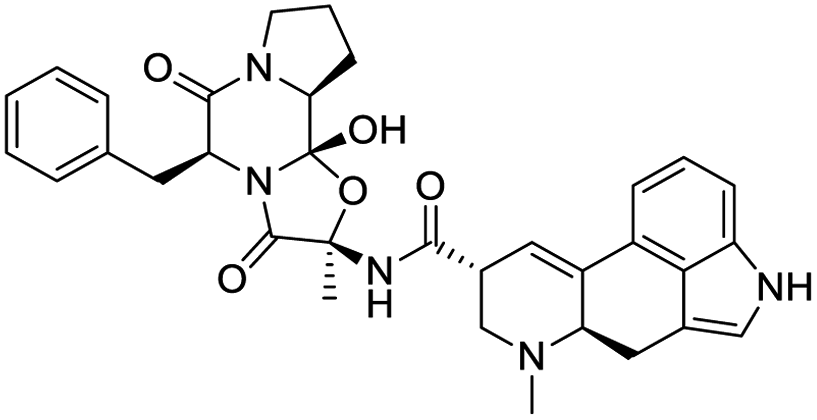 |
| Coumarin | Quercetin-3-O-rhamnoside | Thearubigin | Ergotamine |
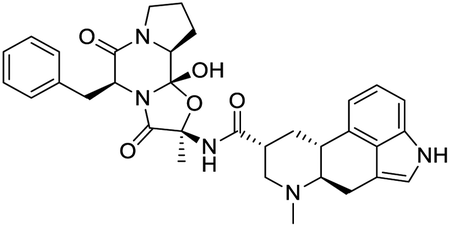 |
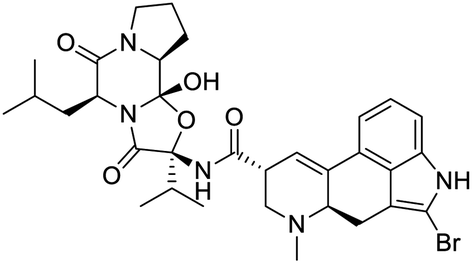 |
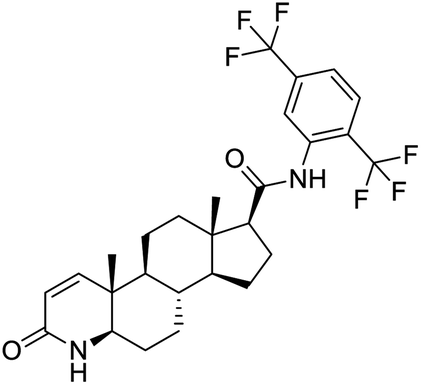 |
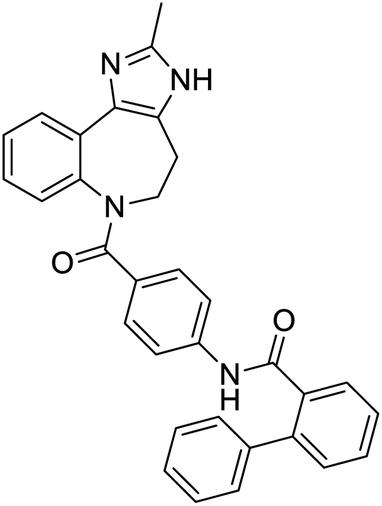 |
| Dihydroergotamine | Bromocriptine | Dutasteride | Conivaptan |
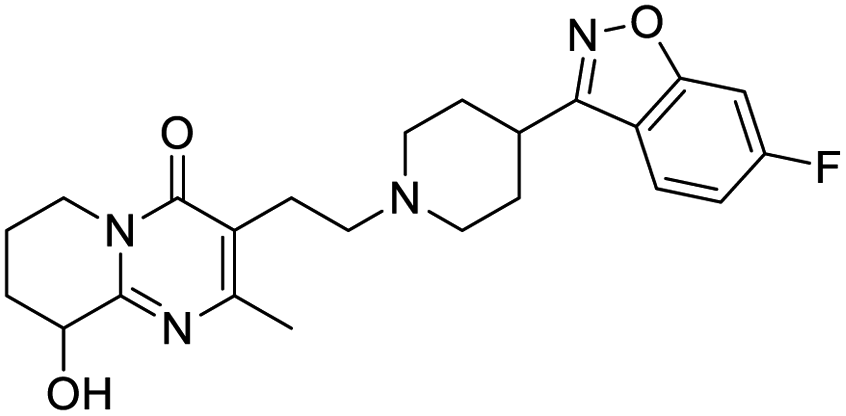 |
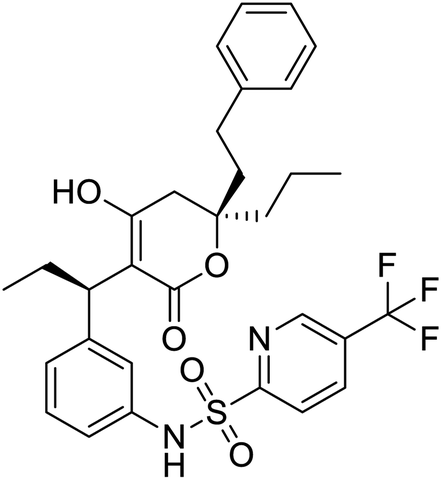 |
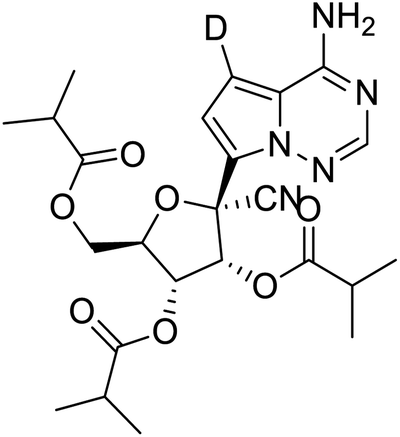 |
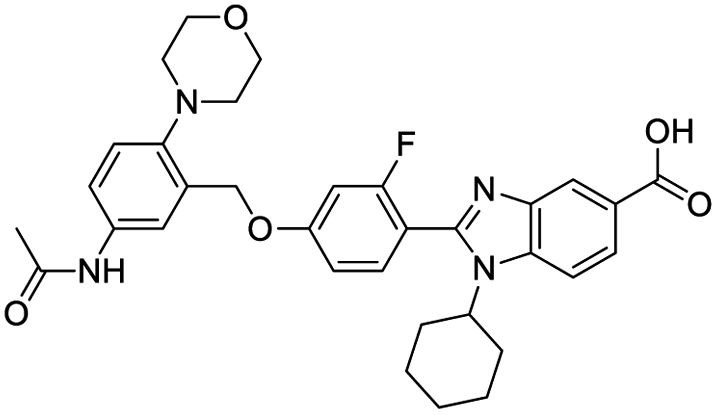 |
| Paliperidone | Tipranavir | Remdesivir derivative | IN-6 |
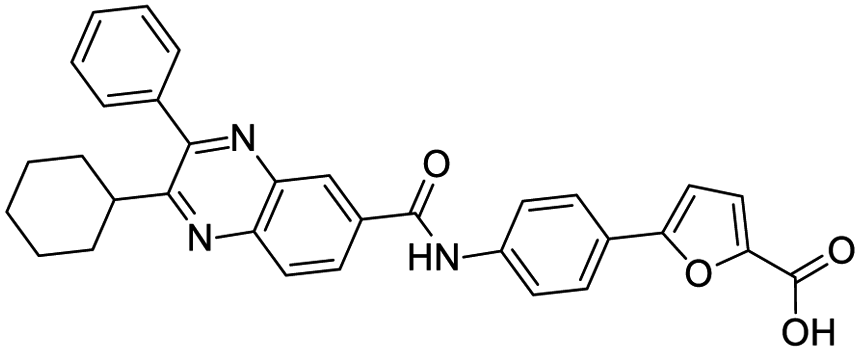 |
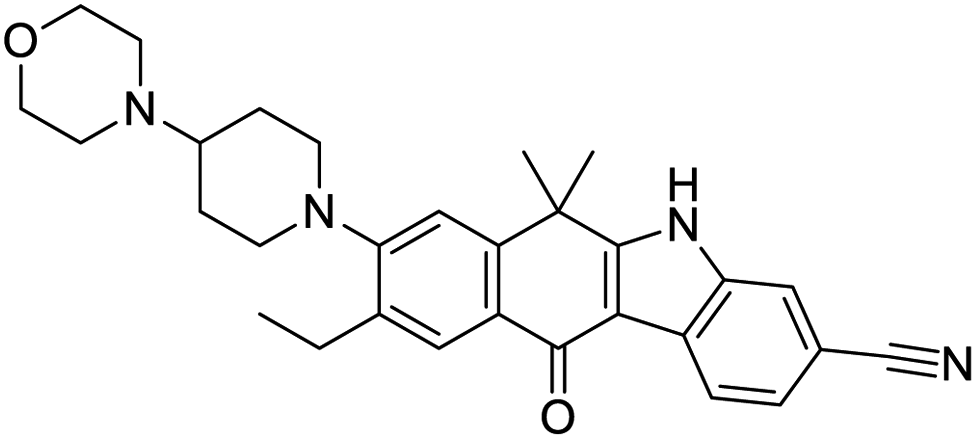 |
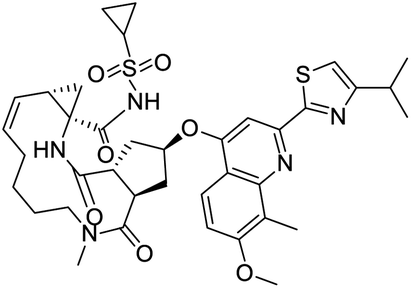 |
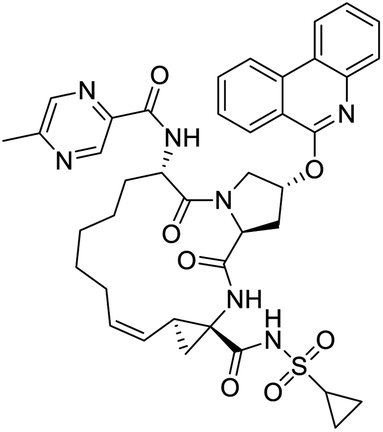 |
| IN-17 | Alectinib | Simeprevir | Paritaprevir |
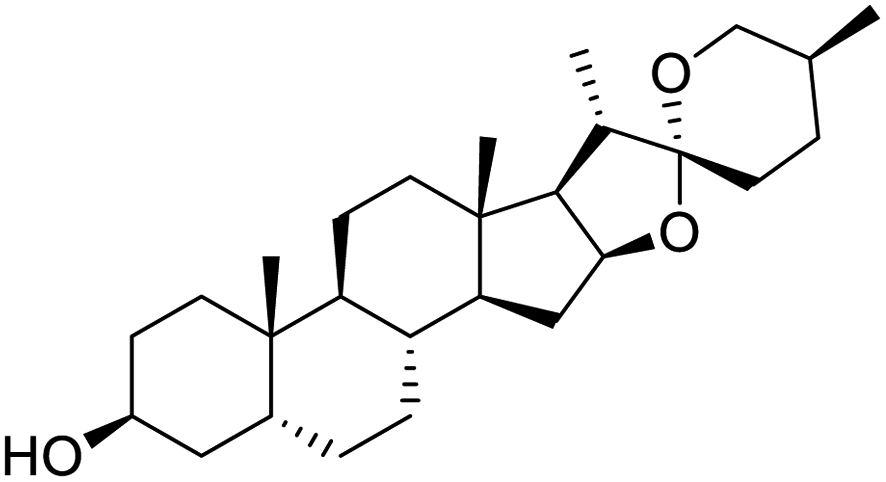 |
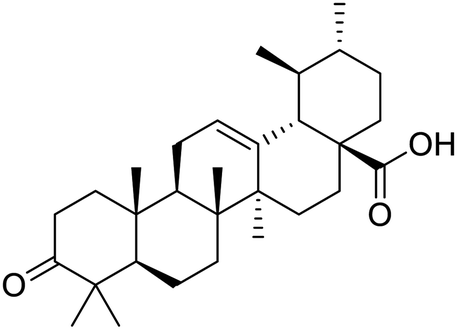 |
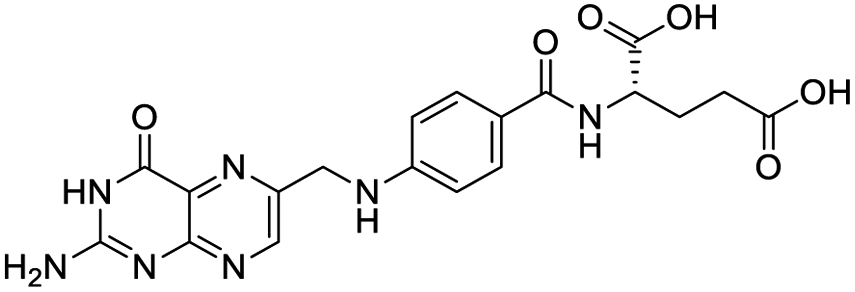 |
 |
| Sarsasapogenin | Ursolic acid | Folic acid | Telmisartan |
 |
 |
 |
 |
| Methotrexate | Bosentan | Lapatinib | Gefitinib |
 |
 |
 |
 |
| Ketoconazole | Carvedilol | Glyburide | Avanafil |
 |
 |
 |
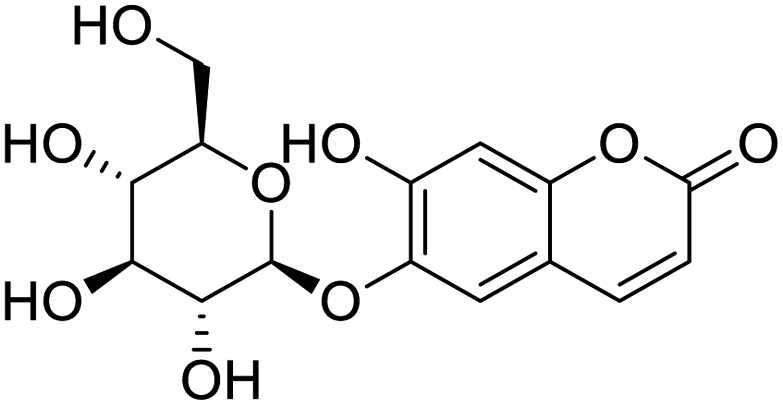 |
| Lumacaftor | Cepharanthine | Edoxudine | Esculin |
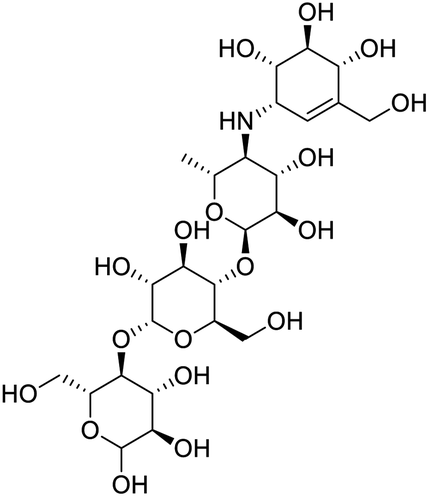 |
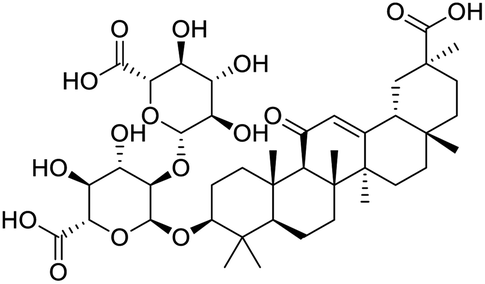 |
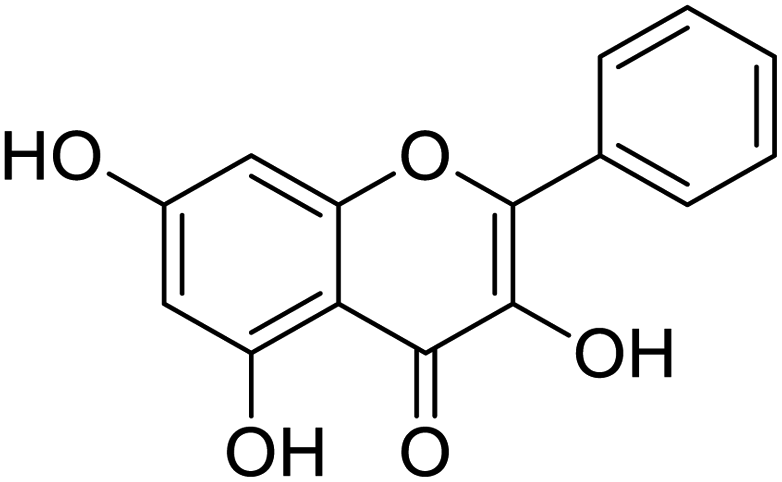 |
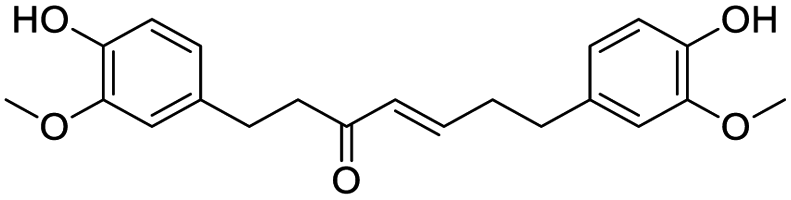 |
| Acarbose | Glycyrrhizic acid | Galangin | Gingerenone A |
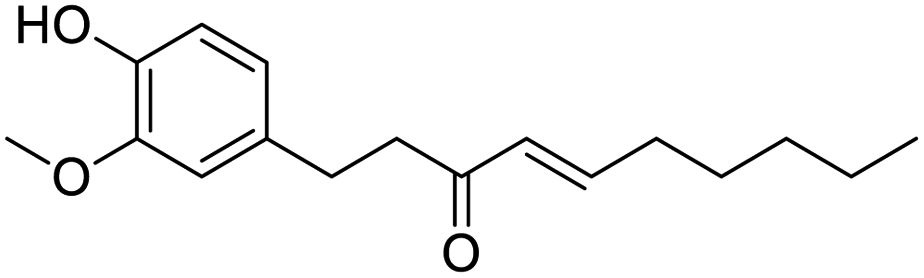 |
 |
 |
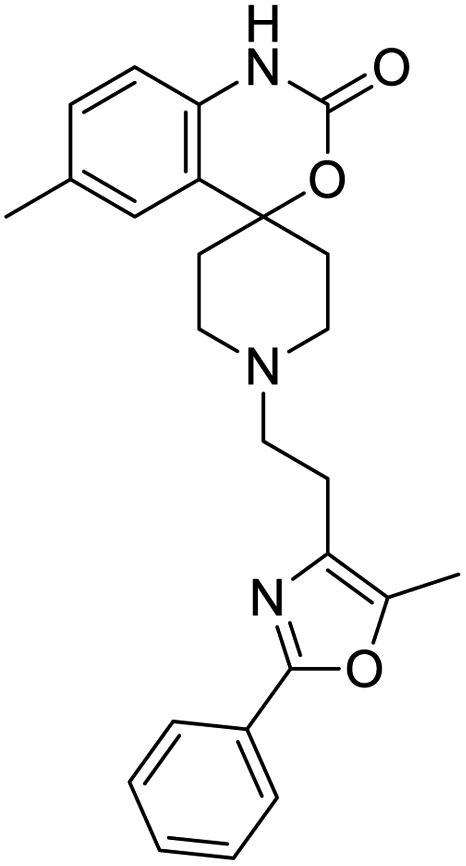 |
| Shogaol | GR 127935 | GNF5 | RS504393 |
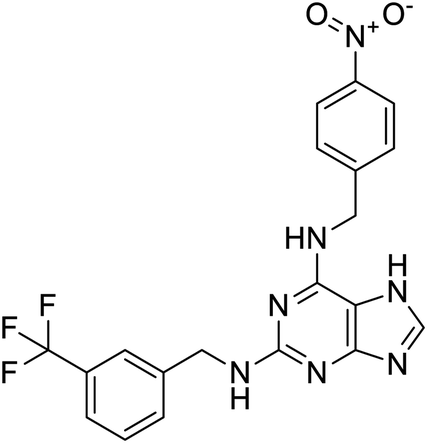 |
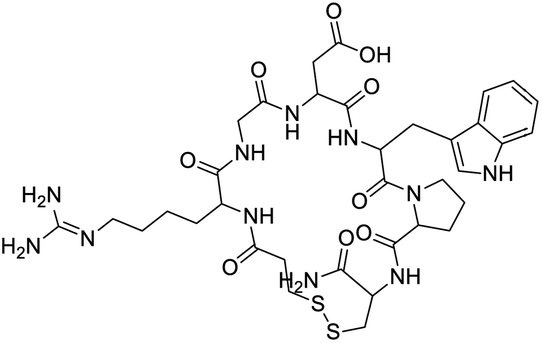 |
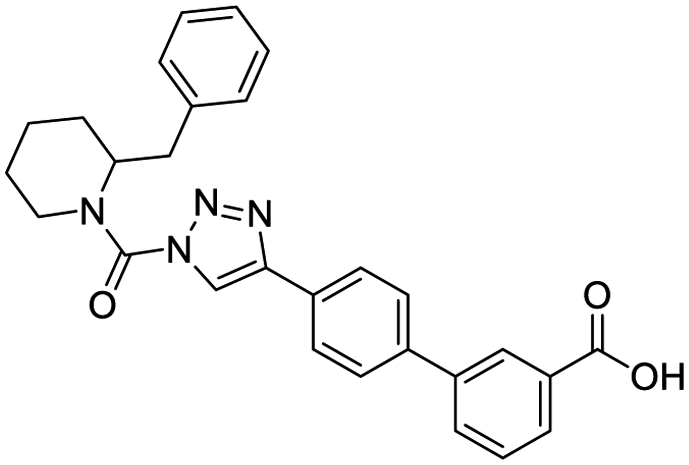 |
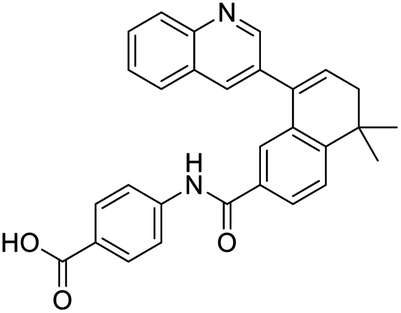 |
| TNP | Eptifibatide acetate | KT203 | BMS195614 |
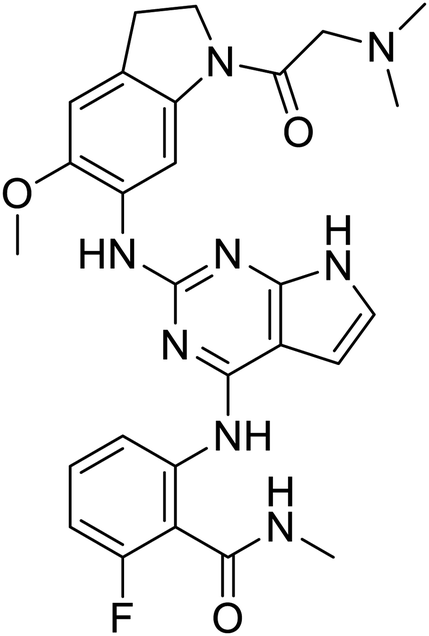 |
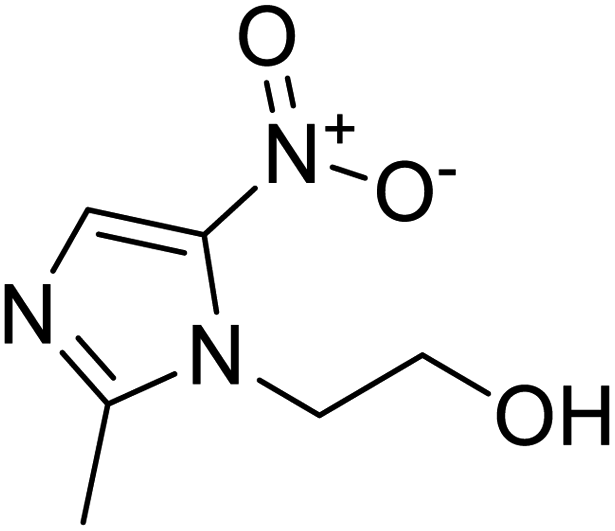 |
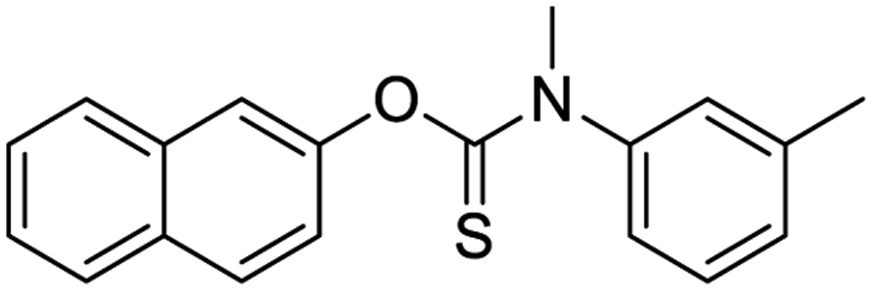 |
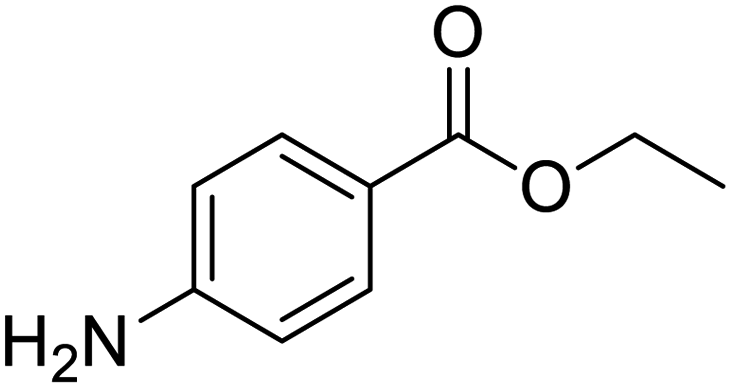 |
| GSK1838705A | Metronidazole | Tolnaftate | Benzocaine |
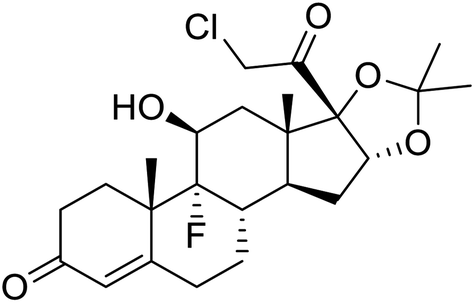 |
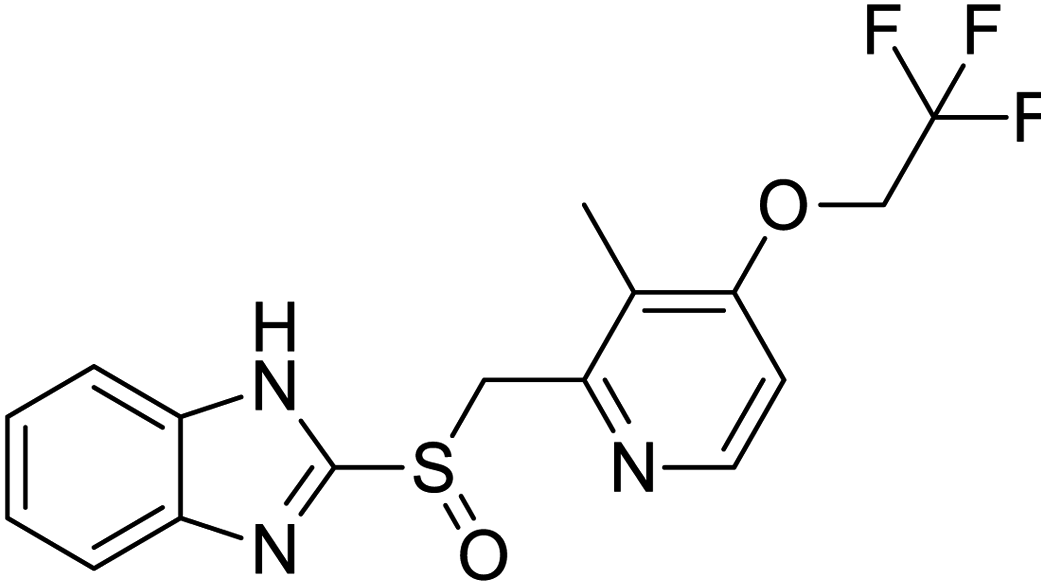 |
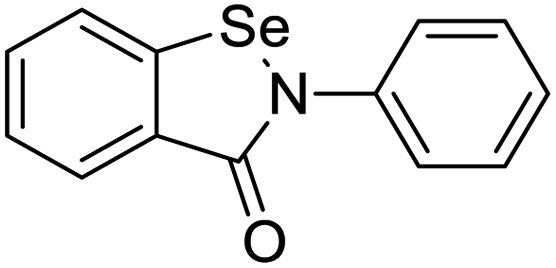 |
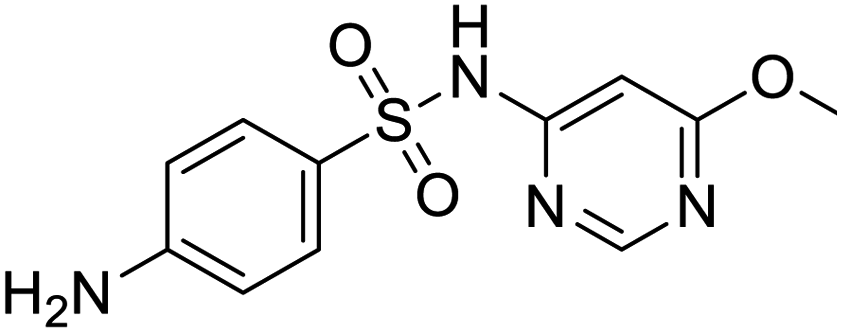 |
| Halcinonide | Lansoprazole | Ebselen | Sulfamonomethoxine |
 |
 |
 |
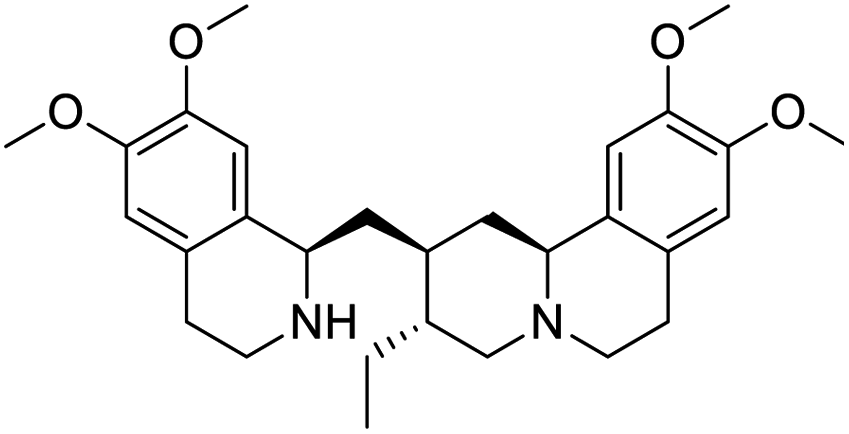 |
| Lidocaine | Histone deacetylase butyrate (phenylbutyric acid) | HSP 90 inhibitor (radicicol) | Emetine |
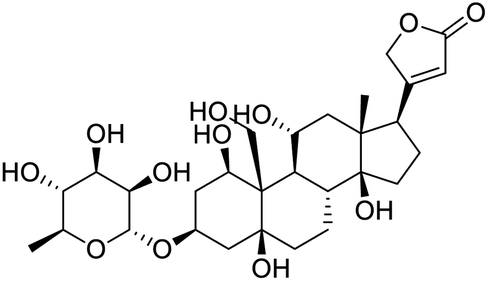 |
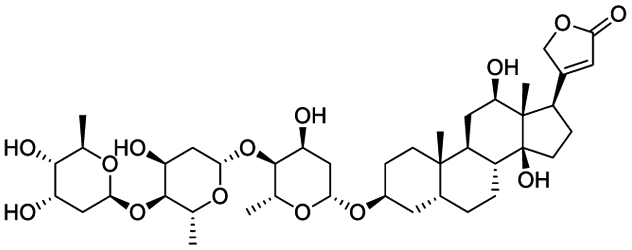 |
 |
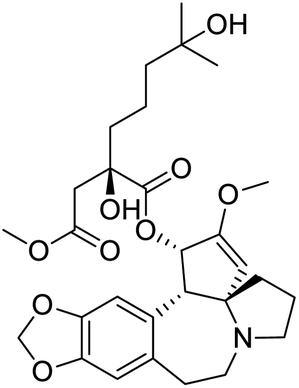 |
| Ouabain | Digoxin | Niclosamide | Homoharringtonine |
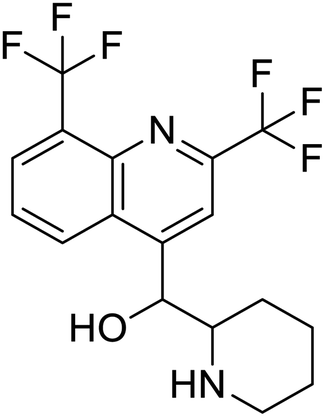 |
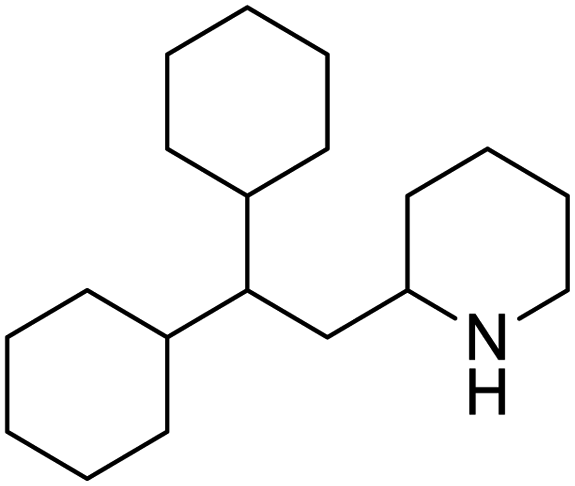 |
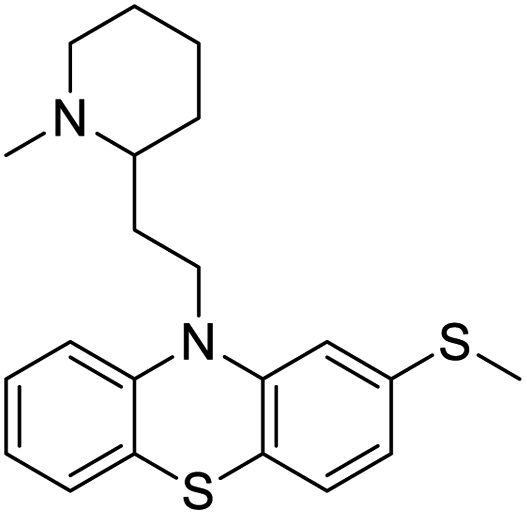 |
 |
| Mefloquine | Perhexiline | Thioridazine | Thapsigargin |
 |
|||
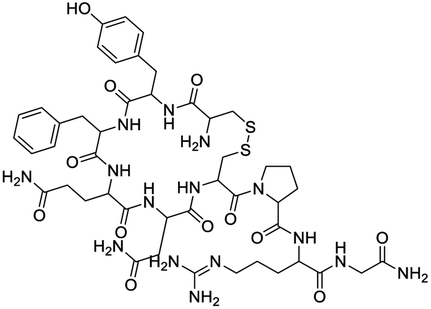
| Azithromycin |
Multi-targeted drugs such as ergotamine, dihydroergotamine, bromocriptine, dutasteride, conivaptan, paliperidone, and tipranavir were reported as potential promising inhibitors of both Mpro (PDB: 6Y84) and RNA-dependent RNA polymerase (RdRp) (PDB: 6NUR).34 The RdRp is essential for SARS-CoV-2 replication and has been considered as a major drug target.14 Remdesivir is an FDA-approved adenosine analogue, which has an EC90 value of 1.76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μM against SARS-CoV-2 in Vero E6 cell and even displayed activity against the virus in human liver cancer Huh-7 cells.35 Yin et al., developed remdesivir derivative, which was five-fold potent than remdesivir in inhibition of viral replication.14 Singh et al., screened >350 potential RdRp (PDB: 7BV2) inhibitors from the BRENDA library and proposed IN-6 and IN-17 as two promising hits compounds with the potential to inhibit RdRp with enhanced potency than remdesivir (Table 1).36 These two molecules maintained interaction with key residues K551, R553, R555, and T556. Alectinib is also proposed as a promising RdRp inhibitor, which was first used for non-small cell lung cancer (NSCLC) treatment.37
μM against SARS-CoV-2 in Vero E6 cell and even displayed activity against the virus in human liver cancer Huh-7 cells.35 Yin et al., developed remdesivir derivative, which was five-fold potent than remdesivir in inhibition of viral replication.14 Singh et al., screened >350 potential RdRp (PDB: 7BV2) inhibitors from the BRENDA library and proposed IN-6 and IN-17 as two promising hits compounds with the potential to inhibit RdRp with enhanced potency than remdesivir (Table 1).36 These two molecules maintained interaction with key residues K551, R553, R555, and T556. Alectinib is also proposed as a promising RdRp inhibitor, which was first used for non-small cell lung cancer (NSCLC) treatment.37
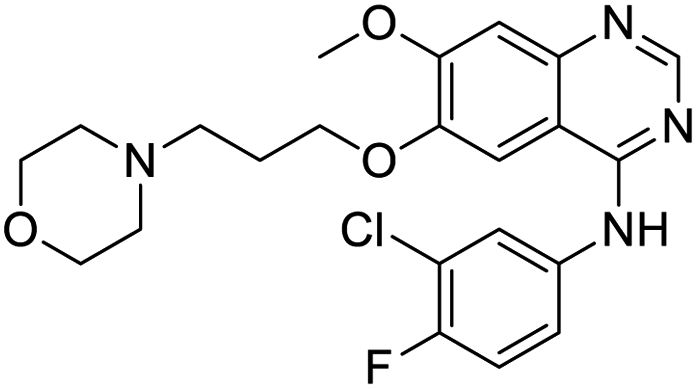
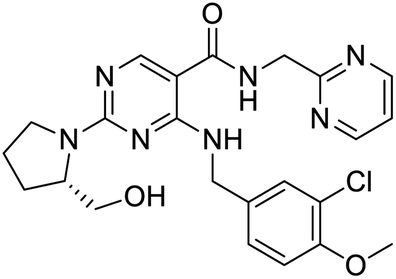
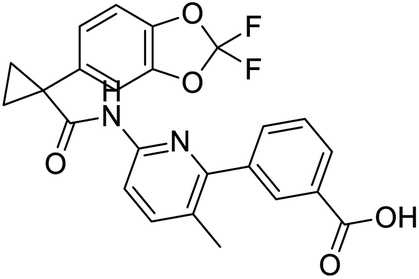
Nonstructural protein (NSP) 15, also referred as uridylate-specific endoribonuclease, primarily cleaves uridine at 3′ end and plays an important role in host cells for viral replication. Studies have shown that viral replication can be delayed after inhibition of NSP15.38 Khan et al., proposed simeprevir and paritaprevir, antivirals originally developed against hepatitis C infection as potential inhibitors of SARS-CoV-2 NSP15 (PDB: 6W01).39 The simeprevir and paritaprevir interacted with residues via H-bond (H235, K290), pi-interaction (A232, H235), and van der Waals interaction (E234). Kumar et al., found computationally two phytochemicals sarsasapogenin and ursolic acid that may be potent inhibitors of NSP15 (PDB: 6VWW).38 The ursolic acid interacted to residues K290, G248, T341, H250, and H235 while sarsasapogenin interacted to K290. Jung et al., proposed FDA approved compounds such as folic acid, telmisartan, methotrexate, bosentan, lapatinib, gefitinib, ketoconazole, carvedilol, glyburide and avanafil as potential NSP3 (PDB: 6W0J) inhibitor, which also hold the promise of potential activity against SARS-CoV-2 with mutations in the spike protein.40
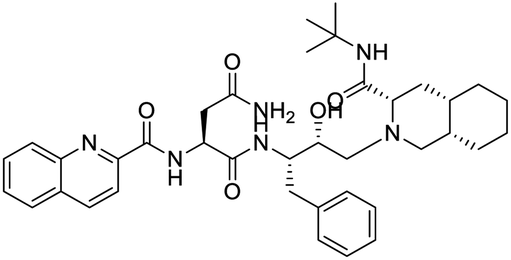
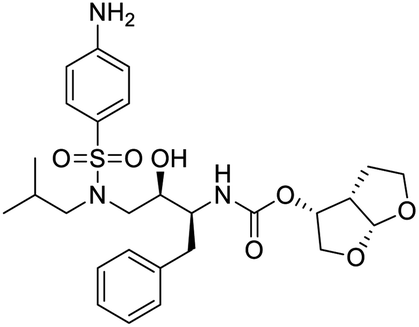
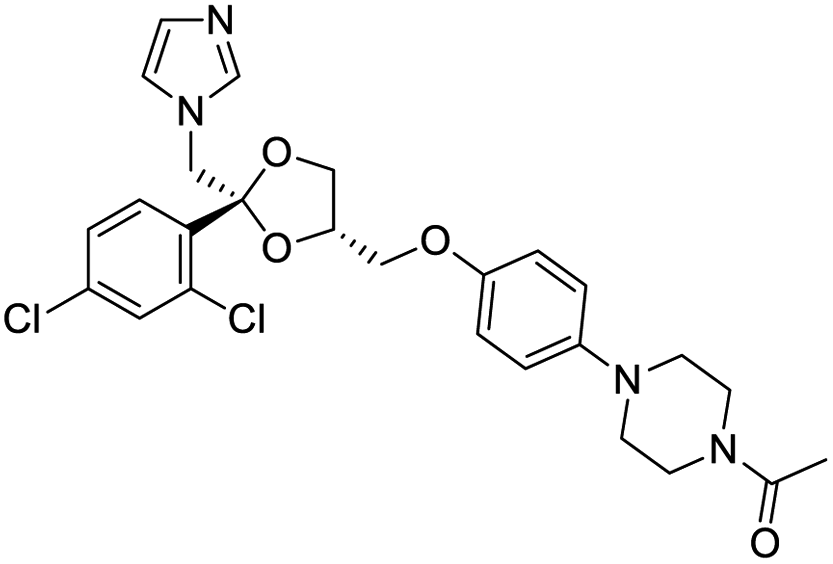
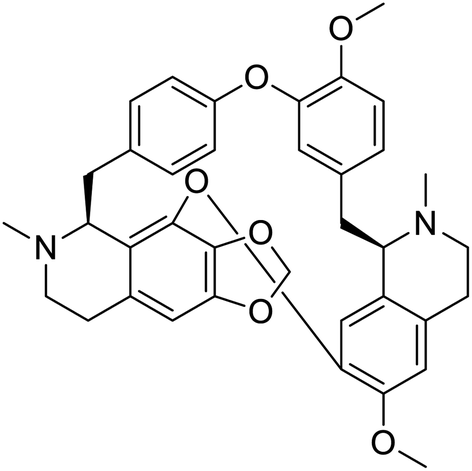
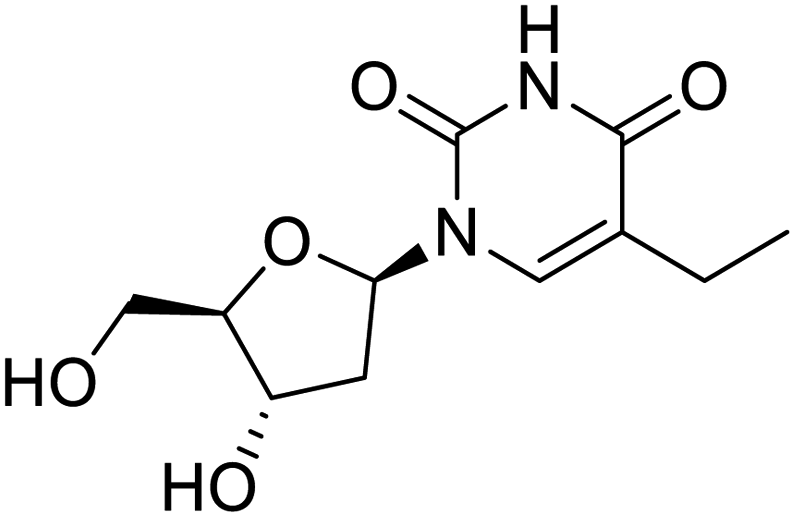
NSP13 helicase is a crucial component of viral replication among the 16 known coronavirus NSP proteins and shares the highest sequence conservation within the coronavirus family, highlighting its significance for viral viability.41 White et al., proposed lumacaftor and cepharanthine inhibiting NSP-13 ATPase activity after screening ∼970000 compounds from two libraries (the enamine libraries (AC and PC) and ZINC-in-trials library) with estimated IC50 values of 0.3 and 0.4 mM, respectively.41 Based on sequence similarity of SARS-CoV-2 NSP 1 protein to the NSP 1 protein of SARS-CoV (PDB: 2HSX), Sharma et al., hypothesized that NSP 1 inhibits host cell expression and inhibition of NSP 1 may result into SARS-CoV-2 attenuated form.42 Authors proposed that a few FDA approved compounds like remdesivir, edoxudine, esculin, acarbose, glycyrrhizic acid, galangin, gingerenone, and shogaol may interact with NSP-1.42 The esculin interacted with residues (R62, S63, A68, F72, and M74) via H-bond. The cidofovir, edoxudine, brivudine, remdesivir, SN00003849, and SN00003832 interacted to R62, R66, and M74 via H-bond. There were also H-bond interactions involved in stabilization of compounds iopromide (H2, V75, R113, N115), a cabose (R32, E76, L77, N115, G116, K114) and SN00216190 (R66, H72, M74, A65). It is suspected that the binding of SARS-CoV-2 spikes with human angiotensin-converting enzyme 2 (ACE2) protein promotes the entry of SARS-CoV-2 into human cells, making this mechanism a successful target.43 Choudhary et al., discovered that FDA-approved molecules (GR 127935 hydrochloride hydrate, GNF-5, RS504393, TNP, and eptifibatide acetate) bind effectively to ACE2 receptor virus binding motifs (PDB: 2AJF), whereas molecules KT203, BMS195614, KT185, RS504393, and GSK1838705A were identified to bind with the viral S-protein (homology model, template PDB: 6VSB) virus binding location.43 The ACE2 receptor has K31 and K353 residues, which are reported as two main sites of virus binding to SARS-CoV. It was observed that GR 127935 hydrochloride hydrate, and GNF-5 interacted to K353 via hydrophobic interactions. The receptor-binding domain of S –protein interacted with ACE2 protein through residues L455, F486, Q493, N487, and S494. The residues L455, F486, Q493 interacted to K31 residues while N487, and S494 interacted to K353 residues of ACE2. The molecules KT203, BMS195614, KT185, RS504393, and GSK1838705A interacted to residues of receptor-binding domain majorly by hydrophobic interaction except BMS195614 (H-bond N487, and S494), and RS504393 (H-bond N487). Sethi et al., proposed TD-139, a galectin inhibitor currently in phase IIb clinical trials to treat idiopathic pulmonary fibrosis, to be able to disrupt the SARS-CoV-2 spike protein and ACE2 interaction (PDB: 6M0J).44 It was shown in several prior studies that the coronaviruses have a galectin fold within their spike protein and additionally that glycosylated ligands have the potential to bind to spike protein. This led the authors to investigate galectin inhibitors with the potential to disrupt SARS-CoV-2:ACE2 interaction. The TD-139 interacted with residues E471, I472, and Q474 through H-bond. TD-139 is now being assessed in phase III clinical trials as a rapid experimental medicine to treat COVID-19.45
These pre-developed and pre-evaluated molecules hold the potential to assist in the control of COVID-19 pandemic by not only inhibiting the entry or replication of virus but also serving as anti-inflammatory agents that may offer relief from the pulmonary inflammation.
3. Diseases–disease association
Different diseases may have overlapping molecular mechanisms underlying their pathobiology, which is known as disease–disease association. Molecular relationships between distinct disease pathologies need to be explored for drug repurposing, even if there is no apparent similarity at the phenotypic or clinical level.9 There are a few methods of drug repurposing based on only disease–disease relationships. In these computational methods, the transcriptional signatures of the drugs and targets, the pathways perturbed as well as the diseases and side effects serve as multifarious modes for carrying out the drug repurposing. Transcriptional signatures of molecules could be used to explore therapeutic relationships between known drugs and new indications.47 It is based on a similarity between the transcriptomic signature of a particular disease and the specific signature of a drug, resulting in a connectivity map (CMap) being developed.483.1 Methodology
Disease–disease associations-based drug repurposing strategies use the DESeq2 algorithms,49 which show the strength of associations in p-values as shown in Fig. 2. | ||
| Fig. 2 Drug repurposing by disease–disease association method. | ||
The key steps involved in disease–disease association are illustrated below.
(a) Download microarray dataset from Gene Expression Omnibus (GEO) datasets of National Center for Biotechnology Information (NCBI);
(b) Normalize datasets i.e., the value distributions are median-centered across samples;50
(c) Determine differential gene expression using DESeq2;
(d) Analyse gene expression based on p-value results.
There are some other platforms like DisGeNET that may also be explored for drug repurposing against COVID-19.47 In order to find alternative therapies for COVID-19, Mousavi and his team members conducted drug repurposing analysis focused on the CMap concept.48 They studied differentially expressed genes (DEGs) in NHBE, a primary human lung epithelial cell line and A549, a transformed lung alveolar cell line infected with SARS-CoV-2. Then, the identified DEGs were analyzed by connectivity map (CMap) with the goal of identifying similarly acting drug candidates.
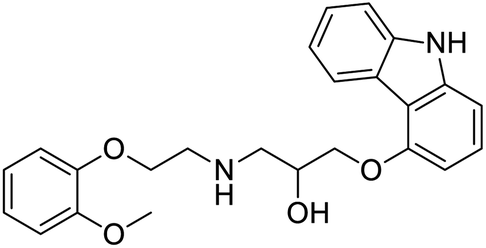
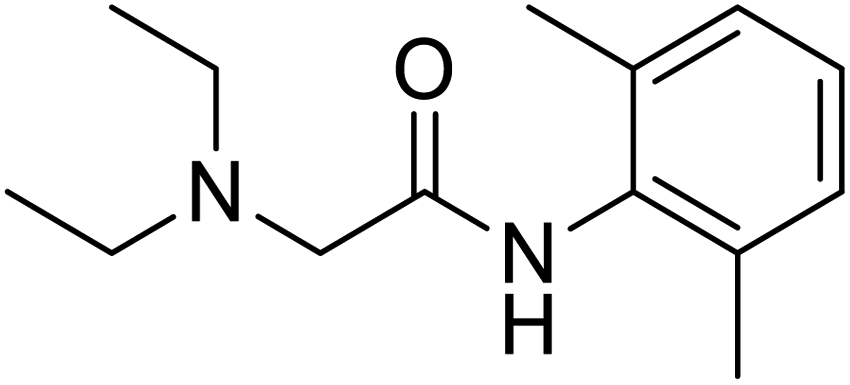
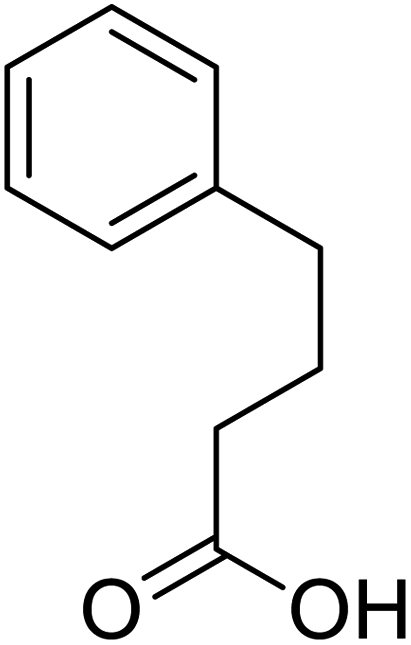
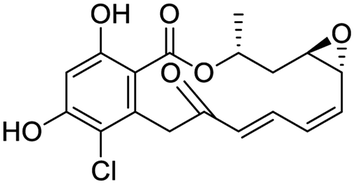
Several drugs demonstrated positive effects in reversing cell gene expression profiles in SARS-CoV-2 infected cells, including lansoprazole, folic acid, sulfamonomethoxine, tolnaftate, diclofenamide, halcinonide, saquinavir, metronidazole, ebselen, lidocaine and benzocaine, histone deacetylase (HDAC) inhibitors, and heat shock protein 90 (HSP 90) inhibitors (Table 1).48 Killick et al. used the transcriptional response of lung epithelial cells following infection by the original Severe Acute Respiratory Syndrome coronavirus (SARS) to identify repurposable drugs against SARS-CoV-2.51 The authors combined CMap and LINCS repositories of the transcriptomic effects of approved drugs, which provide signature data enrichment. They found positive correlation of the SARS signature and the striking result that half of the high scoring drugs have reported anti-viral activities (23/45 in CMap and 22/45 in LINCS). Among the drugs are emetine, ouabain, digoxin, niclosamide, homoharringtonine, mefloquine, perhexiline, thioridazine, and thapsigargine shown to have the potential to inhibit SARS-CoV-2.
4. Drug–drug interaction (DDI)
This section has been included even though drug–drug interaction does not directly aid in drug repurposing, because it is a crucial component to keep in mind when repurposing drugs. The adverse events due to DDI can be avoided with a prior knowledge. Many commercial DDI knowledge bases are widely used in hospitals and pharmacies such as Micromedex, Multum, Medispan, First Databank (FDB) etc.52 We discuss IBM Micromedex in detail.4.1. Methods
Drug–drug interaction method includes the following steps53(a) Search for potential DDIs between diseases on IBM Micromedex;
(b) Select medication for diseases from the search field and add to the ‘drugs to check’ in the interaction tool;
(c) Collect information on DDIs manually using Micromedex. Micromedex grades the severity of DDIs as contraindicated, significant, mild, uncertain; and
(d) Additionally, micromedex also offers an overview of all DDIs and clinical management data on some cases.
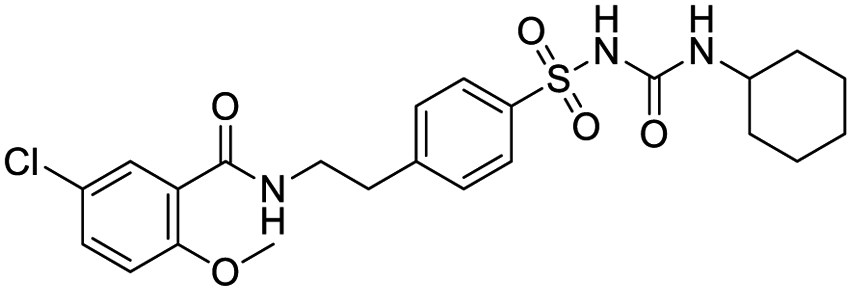
With IBM Micromedex's drug interaction checker, Baburaj and his team members performed a test for possible DDIs between lung cancer treatments and repurposed COVID-19 products.53 A total of 61 potential DDIs between COVID-19 repurposed drugs and lung cancer medications along with their severity were identified from Micromedex. Mechanism of DDI of repurposed COVID-19 drugs with lung cancer pharmacotherapies includes several factors such as pharmacodynamic interactions, QT prolongation, pharmacokinetic interactions, drug absorption, drug metabolism, and drug transportation. Authors found a high chance for the occurrence of major DDIs associated with the concomitant use of COVID-19 repurposed treatments with lung cancer medications, with QT prolongation being the most commonly identified DDI.53 The QT prolongation, measured on an electrocardiogram (ECG), represents the duration of the ventricular electrical systole results onto cardiac arrhythmias.54 In COVID-19 patients, the addition of azithromycin to hydroxychloroquine resulted in superior viral clearance compared with hydroxychloroquine alone.55 But chloroquine and hydroxychloroquine are associated with QT prolongation while azithromycin exposure has been reported to cause ventricular arrhythmia, and sudden cardiac arrest. Hence, concomitant use of azithromycin with hydroxychloroquine can cause a greater change in the QT prolongation than hydroxychloroquine alone.53 This raises alert for clinicians and pharmacists for developing novel strategies for screening, identifying, reporting, and managing potential DDIs in lung cancer patients co-infected with COVID-19 infection. Administration of ritonavir along with oral prednisolone may increase systemic corticosteroid exposure and result in the development of Cushing syndrome.56 Therefore, concomitant use of these drugs with powerful inhibitors of CYP3A, such as lopinavir and ritonavir, can raise the risk of adverse reactions and toxicity. Thus, possible medication interactions between cancer supportive care medications and repurposed COVID-19 drugs are necessary to consider.53
5. Integrating biological networks in drug repositioning
Protein–protein interaction (PPI) data represent the proteins, which can physically interact, but it does not suggest any information about how an organism will react to the stimuli. Gene expression data, however may display how an organism reacts to stimuli regarding the amount of RNA produced, but does not provide information on the physical mechanisms by which changes in the actions of the organism are manifested. Every type of link in a network represents specific information about an organism. Therefore, the integration of distinct types and ESI† would seem necessary.9 Drug repositioning semantic mining (DReSMin)57 as well as bipartite network projection and prioritization algorithms58 suggest integration of different biological networks to reposition drugs. In these approaches, each prediction is made separately based on distinct metrics, and then these metrics are aggregated to arrive at the final result.9 Zhang et al., proposed an approach that combines literature-based discovery (lbd) and knowledge graph completion for COVID-19 drug repurposing where the ABC model was considered for lbd in their study.595.1. Method
Semantic predications (SemMedDB and CORD-19 relations) having semantic relations between subject and object, automatically extracted from biomedical literature (PubMed and CORD-19) utilizing SemRep natural language processing tool (Fig. 3). SemMedDB has supported a wide range of computational applications, ranging from gene regulatory network inference60 to in silico screening for drug repurposing,61 medical reasoning,62 and has also found widespread applicability for the literature-based knowledge discovery and hypothesis generation.63 LitCovid, NCBI's bibliographic database for more than 83k COVID-19 literature (accessed on 27-12-2020).64 COVID-19 Open Research Dataset (CORD-19), which contains articles more than 200k articles65 but not all of them are included in PubMed.59 Both, PubMed and CORD-19 were used for adequate coverage of COVID-19 knowledge. Data were preprocessed before developing biomedical knowledge graph to remove some biases such as (i) eliminated relations involving generic biomedical concepts and relations with identical subject and object arguments; (ii) excluded a subset of predicate types that were not expected to be useful for drug repurposing; (iii) excluded the relations in which the subject or the object belongs to one of the following semantic groups: activities & behaviors, concepts & ideas, objects, occupations, organizations, and phenomena; and (iv) eliminated uninformative semantic relations. Normalization was also done to the range [0; 1] and summed them up into a final score.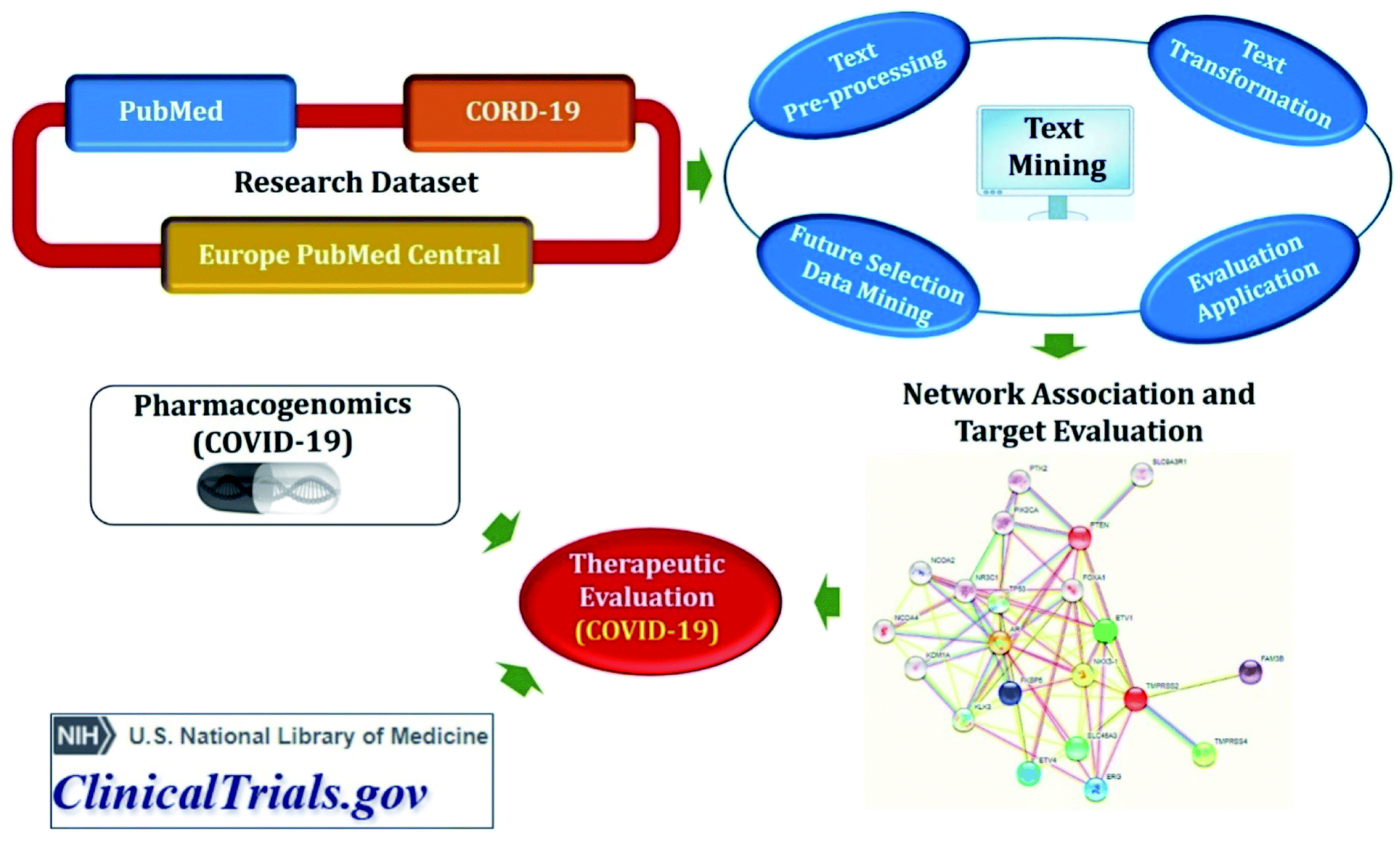 | ||
| Fig. 3 Drug repurposing by integrated biological network approach. | ||
Knowledge graph completion methods include multiple algorithms such as TransE66 and RotatE67 for translational models, DistMult68 and ComplEx69 for semantic matching models, and STELP70 for context-based encoding. The TransE has simplicity and good prediction performance but is able to model only one-to-one relations and fails to embed one-to-many, many-to-one, and many-to-many relations. Therefore, RotatE is considered to counter TrasnE limitations. DistMult is also the simplest approach among semantic matching models but is limited only to symmetric relations. Hence, ComplEx extends DistMult to the complex domain. Semantic Triple Encoder for Link Prediction (STELP) is a context-based encoding approach to knowledge graph completion. At its core is a Siamese BERT model that leverages sharing one set of weights across two models to produce encoded, contextual representations of the predications that are then fed to either multi-layer perceptron (MLP) for classification or a similarity function for contrasting. By using these models, open discovery patterns can be explored to identify drugs that can be repurposed for COVID-19 as well as can propose plausible mechanisms for the drug identification.
6. Artificial intelligence (AI), machine learning (ML) and network medicine approaches
In recent years, the development of next generation computational methods using Artificial Intelligence (AI), Machine Learning (ML) and network medicine approaches have positively impacted the different stages of a drug development process (Fig. 4).72,73 AI and ML-based tools have been the main focus of the pharmaceutical industry which have significantly helped in property prediction and target proteins and small molecule search. These tools can support drug repurposing approaches by rapidly accelerating the pace of all the approaches discussed in previous sections. The first AI invented drug to enter clinical trials took 12 months as opposed to 5 years through a conventional method.74 Additionally, using the AI and ML approaches, researchers have found many similarities between the regulatory proteins of SARS-CoV-2 and SARS.75 Huge amount of COVID-19 data including clinical as well as basic research data is now available in public domain, which can be further used to train the AI and ML based model to repurpose smart drugs against SARS-CoV-2 infection.76 Several reviews have elegantly summarized the principles and approaches to drug discovery using ML/AI.77,78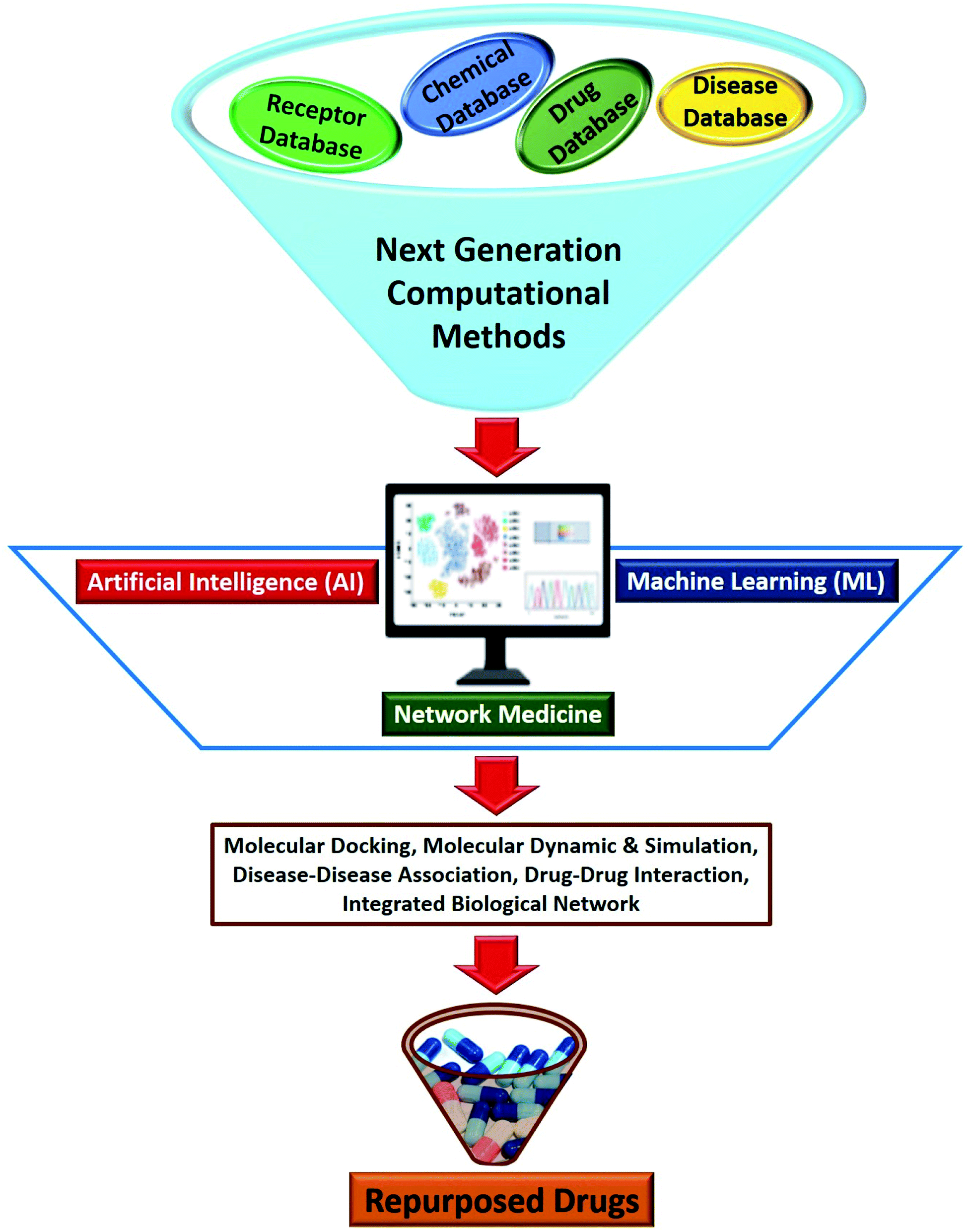 | ||
| Fig. 4 Next generation computational methods based on Artificial Intelligence (AI), Machine Learning (ML) and network medicine for drug repurposing. | ||
6.1. Methods
As summarized in the reviews,71,72 AI has found applications in different sectors of COVID management, (1) tracking through contact tracing; (2) diagnosis; (3) screening of potential drug candidates; (4) proteomics and genomics; and lastly (5) automation of procedures to reduce health worker exposure. The general approach has been to fuse the structure-based and ligand-based screening methods with AI algorithms to build prediction models. The AI and ML algorithms like deep learning, support vector machine (SVM), random forest (RF), Naive Bayesian, and neural networks have been used extensively for high throughput screening with thousand plus dataset of molecules. Specialized statistical parameters, namely root-mean-square error (RMSE), Z-score, area under the curve (AUC), mean absolute error (MAE), Cohen's K value, correlation coefficient (R2), etc. are used to evaluate the performance (precision + accuracy) of the models.7. Conclusion
Drug repurposing or repositioning is a technique in which existing drugs are remodeled to treat other diseases. In last few months, COVID-19 has greatly affected mankind. Though several vaccines have been developed recently, yet, no therapeutic drugs have been found to be effective. However, there are number of available drugs approved by the FDA for the treatment of other diseases, and these can be potentially repurposed for COVID-19. In recent years, the development of next generation computational methods including molecular docking, MD simulation, disease–disease association, drug–drug interaction, integrated biological network, artificial intelligence (AI), machine learning (ML) and network medicine have paved the way to repurpose drugs rapidly. Although drug repurposing has huge potential to decrease the time, resources and money required for a drug to reach the market compared to the conventional drug discovery process, several challenges still remain that need to be overcome. These challenges are twofold.One inherently associated with drug repurposing; the commercial and legal barriers. Repurposed drugs are still a high-risk investment. One may argue that a pre-approved drug has a suitable safety profile and thereby phase I and II studies could be avoided for repurposed drug. However, as regulatory science progresses so do its standards, and therefore, the previous data might no longer be applicable for the new indication. Even if the safety profile is found to meet present standards, the variation in benefit for the new indication might alter the overall benefit–risk profile of the drug. Even from the perspective of patentability, it faces certain hurdles. If there are studies in scientific literature/public domain that highlight the potential role of the preexisting drug for the new indication, this affects the novelty of the repurposed drug and thereby patentability. Thus, there is an urgent need to incentivize the repurposing of drugs as many believe that existing drugs would be beneficial in treating other ailments.
The second challenge is associated with the current pitfalls of the use of computational tools. The predictability of computational models has been time and again questioned. Specifically, the binding mode of interaction in molecular docking. Different software packages use different algorithms and scoring functions which leads to variability and inconsistency in data associated with docking. AI//ML based tools rely on large datasets, however, the lack of dissemination of clinical trial data by pharmaceutical companies could hamper predictability of AI/ML algorithms due to insufficient size of training data. Additionally, the data which is available is heterogenous, which makes integration of this data an extremely pain staking endeavor. Obtaining openly accessible standardized and structured data is crucial for progress of computational tools in drug repurposing. Every method has its own set of pitfalls, even in vitro data generated in anti-viral assays failed to translate in vivo due to phospholipidosis.79 Thus, results generated by every tool at our disposal need to be critically analyzed and scrutinized, to improve it further.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Department of Science & Department of Science & Technology (DST) under BRICS multilateral project (DST/INT/BRICS/COVID-19/2020). This study was funded by RFBR, DST, CNPq, SAMRC, project number 20-53-80002.References
- O. J. Wouters, M. McKee and J. Luyten, JAMA, J. Am. Med. Assoc., 2020, 323, 844 CrossRef PubMed.
- M. Placchi and R. J. Phillips, The Benefits and Pitfalls of Repurposing Drugs, https://oliver-dev.s3.amazonaws.com/ 2018/11/07/21/59/08/240/Benefits%20and%20Pitfalls% 20of%20Repurposing%20Drugs.pdf, accessed Febuary 26, 2021 Search PubMed.
- L. Riva, S. Yuan, X. Yin, L. Martin-Sancho, N. Matsunaga, L. Pache, S. Burgstaller-Muehlbacher, P. D. De Jesus, P. Teriete, M. V. Hull, M. W. Chang, J. F.-W. Chan, J. Cao, V. K.-M. Poon, K. M. Herbert, K. Cheng, T.-T. H. Nguyen, A. Rubanov, Y. Pu, C. Nguyen, A. Choi, R. Rathnasinghe, M. Schotsaert, L. Miorin, M. Dejosez, T. P. Zwaka, K.-Y. Sit, L. Martinez-Sobrido, W.-C. Liu, K. M. White, M. E. Chapman, E. K. Lendy, R. J. Glynne, R. Albrecht, E. Ruppin, A. D. Mesecar, J. R. Johnson, C. Benner, R. Sun, P. G. Schultz, A. I. Su, A. García-Sastre, A. K. Chatterjee, K.-Y. Yuen and S. K. Chanda, Nature, 2020, 586, 113 CrossRef CAS PubMed.
- L. Yang, R.-j. Pei, H. Li, X.-n. Ma, Y. Zhou, F.-h. Zhu, P.-l. He, W. Tang, Y.-c. Zhang, J. Xiong, S.-q. Xiao, X.-k. Tong, B. Zhang and J.-p. Zuo, Acta Pharmacol. Sin., 2020, 42, 1347 CrossRef PubMed.
- S. Naylor and J. M. Schonfeld, Therapeutic Drug Repurposing, Repositioning and Rescue Part I: Overview, https://www.ddw-online.com/therapeutic-drug-repurposing-repositioning-and-rescue-part-i-overview-1463-201412/, accessed March15, 2021 Search PubMed.
- G. Galindez, J. Matschinske, T. D. Rose, S. Sadegh, M. Salgado-Albarrán, J. Späth, J. Baumbach and J. K. Pauling, Nat. Comput. Sci., 2021, 1, 33 CrossRef.
- A. Persidis, The Benefits of Drug Repositioning, https://www.ddw-online.com/the-benefits-of-drug-repositioning-1779-201104/, accessed January 26, 2021 Search PubMed.
- S. G. V. Rosa and W. C. Santos, Revista Panamericana de Salud Publica, 2020, 44, e40 Search PubMed.
- M. Lotfi Shahreza, N. Ghadiri, S. R. Mousavi, J. Varshosaz and J. R. Green, Briefings Bioinf., 2017, 19, 878 CrossRef PubMed.
- R. A. Hodos, B. A. Kidd, K. Shameer, B. P. Readhead and J. T. Dudley, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2016, 8, 186 Search PubMed.
- P. Polamreddy and N. Gattu, Drug Discovery Today, 2019, 24, 789 CrossRef PubMed.
- K. Gao, D. D. Nguyen, J. Chen, R. Wang and G.-W. Wei, J. Phys. Chem. Lett., 2020, 11, 5373 CrossRef CAS PubMed.
- M. Rudrapal, S. Khairnar and A. Jadhav, in Drug Repurposing - Hypothesis, Molecular Aspects and Therapeutic Applications, ed. F. A. Badria, IntechOpen, 2020, DOI:10.5772/intechopen.93193.
- W. Yin, X. Luan, Z. Li, Y. Xie, Z. Zhou, J. Liu, M. Gao, X. Wang, F. Zhou, Q. Wang, Q. Wang, D. Shen, Y. Zhang, G. Tian, H. A. Aisa, T. Hu, D. Wei, Y. Jiang, G. Xiao, H. Jiang, L. Zhang, X. Yu, J. Shen, S. Zhang and H. E. Xu, bioRxiv, 2020 DOI:10.1101/2020.11.01.363812.
- A. Tuerkova and B. Zdrazil, J. Cheminf., 2020, 12, 71 CAS.
- A. B. Gurung, M. A. Ali, J. Lee, M. A. Farah and K. M. Al-Anazi, BioMed Res. Int., 2021, 2021, 8853056 Search PubMed.
- T. N. Jarada, J. G. Rokne and R. Alhajj, J. Cheminf., 2020, 12, 46 CAS.
- H. Tayara, I. Abdelbaky and K. To Chong, Brief. Bioinform., 2021 DOI:10.1093/bib/bbab339.
- E. Hernández-Domínguez, L. Castillo-Ortega, Y. García-Esquivel, V. Mandujano-González, G. Diaz-Godínez and J. Alvarez Cervantes, in Computational Biology and Chemistry, ed. P. Behzadi, IntechOpen, 2019. DOI: DOI:10.5772/intechopen.89594.
- R. Angles, M. Arenas-Salinas, R. García, J. A. Reyes-Suarez and E. Pohl, BMC Bioinf., 2020, 21, 85 CrossRef CAS PubMed.
- F. Sohraby, M. Bagheri and H. Aryapour, Methods Mol. Biol., 2019, 1903, 23 CrossRef CAS PubMed.
- G. Vettoretti, E. Moroni, S. Sattin, J. Tao, D. A. Agard, A. Bernardi and G. Colombo, Sci. Rep., 2016, 6, 23830 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289 CrossRef CAS PubMed.
- M. De Vivo, M. Masetti, G. Bottegoni and A. Cavalli, J. Med. Chem., 2016, 59, 4035 CrossRef CAS PubMed.
- A. Gimeno, J. Mestres-Truyol, M. J. Ojeda-Montes, G. Macip, B. Saldivar-Espinoza, A. Cereto-Massagué, G. Pujadas and S. Garcia-Vallvé, Int. J. Mol. Sci., 2020, 21, 3793 CrossRef CAS PubMed.
- J. Wang, J. Chem. Inf. Model., 2020, 60, 3277 CrossRef CAS PubMed.
- M. Mahdi, J. A. Mótyán, Z. I. Szojka, M. Golda, M. Miczi and J. Tőzsér, Virol. J., 2020, 17, 190 CrossRef CAS PubMed.
- F. Monji, A. Al-Mahmood Siddiquee and F. Hashemian, Eur. J. Pharmacol., 2020, 887, 173561 CrossRef CAS PubMed.
- A. O. Elzupir, J. Biomol. Struct. Dyn., 2020 DOI:10.1080/07391102.2020.1835732.
- S. Koulgi, V. Jani, M. Uppuladinne, U. Sonavane, A. K. Nath, H. Darbari and R. Joshi, J. Biomol. Struct. Dyn., 2020, 39, 5735 CrossRef PubMed.
- S. A. Khan, K. Zia, S. Ashraf, R. Uddin, Z. Ul-Haq and Z. Ul-Haq, J. Biomol. Struct. Dyn., 2020, 39, 2607 CrossRef PubMed.
- S. A. Cherrak, H. Merzouk and N. Mokhtari-Soulimane, PLoS One, 2020, 15, e0240653 CrossRef CAS PubMed.
- S. Upadhyay, P. K. Tripathi, S. Raghavendhar, M. Bhardwaj, A. K. Patel and M. Singh, Phytother. Res., 2020, 34, 3411 CrossRef CAS PubMed.
- S. Gul, I. H. Kavakli, O. Ozcan, I. Baris, I. H. Kavakli, S. Asar and A. Okyar, J. Biomol. Struct. Dyn., 2021, 39, 6772 CrossRef CAS PubMed.
- M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi and Z. Hu, Cell Res., 2020, 30, 269 CrossRef CAS PubMed.
- P. K. Singh, S. Pathania and R. K. Rawal, SAR QSAR Environ. Res., 2020, 31, 857 CrossRef CAS PubMed.
- S. Barage, A. Karthic, R. Bavi, N. Desai, R. Kumar, V. Kumar and K. W. Lee, J. Biomol. Struct. Dyn., 2020 DOI:10.1080/07391102.2020.1841026.
- S. Kumar, P. Kashyap, S. Chowdhury, S. Kumar, A. Panwar and A. Kumar, Phytomedicine, 2020, 85, 153317 CrossRef PubMed.
- R. J. Khan, R. K. Jha, E. Singh, M. Jain, G. M. Amera, R. P. Singh, J. Muthukumaran and A. K. Singh, J. Biomol. Struct. Dyn., 2020 DOI:10.1080/07391102.2020.1814870.
- L. S. Jung, T. M. Gund and M. Narayan, Protein J., 2020, 39, 619 CrossRef CAS PubMed.
- M. A. White, W. Lin and X. Cheng, J. Phys. Chem. Lett., 2020, 11, 9144 CrossRef CAS PubMed.
- A. Sharma, V. Tiwari and R. Sowdhamini, J. Biosci., 2020, 45, 100 CrossRef CAS PubMed.
- S. Choudhary, Y. S. Malik and S. Tomar, Front. Immunol., 2020, 11, 14 CrossRef PubMed.
- A. Sethi, S. Sanam, S. Munagalasetty, S. Jayanthi and M. Alvala, RSC Adv., 2020, 10, 29873 RSC.
- Rapid Experimental Medicine for COVID-19 (DEFINE), https://clinicaltrials.gov/ct2/show/NCT04473053, accessed September 26, 2021 Search PubMed.
- Q. Vanhaelen, P. Mamoshina, A. M. Aliper, A. Artemov, K. Lezhnina, I. Ozerov, I. Labat and A. Zhavoronkov, Drug Discovery Today, 2017, 22, 210 CrossRef CAS PubMed.
- P. Balasundaram, R. Kanagavelu, N. James, S. Maiti, S. Veerappapillai and R. Karuppaswamy, Methods Mol. Biol., 2019, 1903, 129 CrossRef CAS PubMed.
- S. Z. Mousavi, M. Rahmanian and A. Sami, Infect., Genet. Evol., 2020, 86, 104610 CrossRef CAS PubMed.
- M. I. Love, W. Huber and S. Anders, Genome Biol., 2014, 15, 550 CrossRef PubMed.
- T. Barrett, S. E. Wilhite, P. Ledoux, C. Evangelista, I. F. Kim, M. Tomashevsky, K. A. Marshall, K. H. Phillippy, P. M. Sherman, M. Holko, A. Yefanov, H. Lee, N. Zhang, C. L. Robertson, N. Serova, S. Davis and A. Soboleva, Nucleic Acids Res., 2012, 41, D991 CrossRef PubMed.
- R. Killick, C. Ballard, P. Doherty and G. Williams, Virus Res., 2020, 290, 198176 CrossRef CAS PubMed.
- K. W. Fung, J. Kapusnik-Uner, J. Cunningham, S. Higby-Baker and O. Bodenreider, J. Am. Med. Inform. Assoc., 2017, 24, 806 CrossRef PubMed.
- G. Baburaj, L. Thomas and M. Rao, Arch. Med. Res., 2020, 52, 261 CrossRef PubMed.
- L. Guglielmetti, S. Tiberi, M. Burman, H. Kunst, C. Wejse, T. Togonidze, G. Bothamley and C. Lange, Eur. Respir. J., 2018, 52, 1800537 CrossRef PubMed.
- P. Gautret, J.-C. Lagier, P. Parola, V. T. Hoang, L. Meddeb, M. Mailhe, B. Doudier, J. Courjon, V. Giordanengo, V. E. Vieira, H. Tissot Dupont, S. Honoré, P. Colson, E. Chabrière, B. La Scola, J.-M. Rolain, P. Brouqui and D. Raoult, Int. J. Antimicrob. Agents, 2020, 56, 105949 CrossRef CAS PubMed.
- N. E. Albert, S. Kazi, J. Santoro and R. Dougherty, Am. J. Med. Sci., 2012, 344, 72 CrossRef PubMed.
- J. Mullen, S. J. Cockell, H. Tipney, P. M. Woollard and A. Wipat, PeerJ, 2016, 4, e1558 CrossRef PubMed.
- A. J. Berenstein, M. P. Magariños, A. Chernomoretz and F. Agüero, PLoS Neglected Trop. Dis., 2016, 10, e0004300 CrossRef PubMed.
- R. Zhang, D. Hristovski, D. Schutte, A. Kastrin, M. Fiszman and H. Kilicoglu, J. Biomed. Inf., 2021, 115, 103696 CrossRef PubMed.
- G. Chen, M. J. Cairelli, H. Kilicoglu, D. Shin and T. C. Rindflesch, PLoS Comput. Biol., 2014, 10, e1003666 CrossRef PubMed.
- T. Cohen, D. Widdows, C. Stephan, R. Zinner, J. Kim, T. Rindflesch and P. Davies, CPT: Pharmacomet. Syst. Pharmacol., 2014, 3, e140 CAS.
- S. R. Sukumar, A Reasoning And Hypothesis-Generation Framework Based On Scalable Graph Analytics, Oak Ridge National Lab (ORNL), Oak Ridge, TN (United States), 2016 Search PubMed.
- G. Crichton, S. Baker, Y. Guo and A. Korhonen, PLoS One, 2020, 15, e0232891 CrossRef CAS PubMed.
- Q. Chen, A. Allot and Z. Lu, Nature, 2020, 579, 193 CrossRef CAS PubMed.
- L. L. Wang, K. Lo, Y. Chandrasekhar, R. Reas, J. Yang, D. Eide, K. Funk, R. M. Kinney, Z. Liu, W. Merrill, P. Mooney, D. Murdick, D. Rishi, J. Sheehan, Z. Shen, B. Stilson, A. D. Wade, K. Wang, C. Wilhelm, B. Xie, D. Raymond, D. S. Weld, O. Etzioni and S. Kohlmeier, 2020, ArXiv, https://arxiv.org/abs/2004.10706.
- A. Bordes, N. Usunier, A. García-Durán, J. Weston and O. Yakhnenko, NeurIPS, 2013, vol. 2, p. 2787 Search PubMed.
- Z. Sun, Z.-H. Deng, J.-Y. Nie and J. Tang, 2019, arXiv e-prints, arXiv:1902.10197.
- B. Yang, W.-t. Yih, X. He, J. Gao and L. Deng, 2014, arXiv e-prints, arXiv:1412.6575.
- T. Trouillon, J. Welbl, S. Riedel, É. Gaussier and G. Bouchard, 2016, arXiv e-prints, arXiv:1606.06357.
- B. Wang, T. Shen, G. Long, T. Zhou and Y. Chang, ArXiv, 2020, abs/2004.14781.
- D. Zhang, D. He, N. Zou, X. Zhou and F. Pei, 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 2018. DOI:10.1109/BIBM.2018.8621316.
- Y. Zhou, F. Wang, J. Tang, R. Nussinov and F. Cheng, The Lancet Digital Health, 2020, 2, e667 CrossRef PubMed.
- W. P. Walters and R. Barzilay, Expert Opin. Drug Discovery, 2021, 16, 1–11 CrossRef PubMed.
- J. Wakefield, Artificial intelligence-created medicine to be used on humans for first time, https://www.bbc.com/news/technology-51315462, accessed July 26, 2021 Search PubMed.
- H. S. Gns, S. Gr, M. Murahari and M. Krishnamurthy, Biomed. Pharmacother., 2019, 110, 700 CrossRef PubMed.
- K.-K. Mak and M. R. Pichika, Drug Discovery Today, 2019, 24, 773 CrossRef PubMed.
- A. Keshavarzi Arshadi, J. Webb, M. Salem, E. Cruz, S. Calad-Thomson, N. Ghadirian, J. Collins, E. Diez-Cecilia, B. Kelly, H. Goodarzi and J. S. Yuan, Front. Artif. Intell., 2020, 3, 1–13 CrossRef PubMed.
- Z. Tanoli, M. Vähä-Koskela and T. Aittokallio, Expert Opin. Drug Discovery, 2021, 16, 977 CrossRef CAS PubMed.
- T. A. Tummino, V. V. Rezelj, B. Fischer, A. Fischer, M. J. O'Meara, B. Monel, T. Vallet, K. M. White, Z. Zhang, A. Alon, H. Schadt, H. R. O'Donnell, J. Lyu, R. Rosales, B. L. McGovern, R. Rathnasinghe, S. Jangra, M. Schotsaert, J.-R. Galarneau, N. J. Krogan, L. Urban, K. M. Shokat, A. C. Kruse, A. García-Sastre, O. Schwartz, F. Moretti, M. Vignuzzi, F. Pognan and B. K. Shoichet, Science, 2021, 373, eabi4708 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05320e |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |