
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide-directed Pd-decorated Au and PdAu nanocatalysts for degradation of nitrite in water†
Imann Mosleh and
Alireza Abbaspourrad*
and
Alireza Abbaspourrad*
Department of Food Science, College of Agricultural and Life Sciences, Cornell University, Stocking Hall, Ithaca, NY 14853, USA. E-mail: alireza@cornell.edu; Tel: +1 607 255 2923
First published on 5th October 2021
Abstract
In this work, a palladium binding peptide, Pd4, has been used for the synthesis of catalytically active palladium-decorated gold (Pd-on-Au) nanoparticles (NPs) and palladium–gold (PdxAu100−x) alloy NPs exhibiting high nitrite degradation efficiency. Pd-on-Au NPs with 20% to 300% surface coverage (sc%) of Au showed catalytic activity commensurate with sc%. Additionally, the catalytic activity of PdxAu100−x alloy NPs varied based on palladium composition (x = 6–59). The maximum nitrite removal efficiency of Pd-on-Au and PdxAu100−x alloy NPs was obtained at sc 100% and x = 59, respectively. The synthesized peptide-directed Pd-on-Au catalysts showed an increase in nitrite reduction three and a half times better than monometallic Pd and two and a half times better than PdxAu100−x NPs under comparable conditions. Furthermore, peptide-directed NPs showed high activity after five reuse cycles. Pd-on-Au NPs with more available activated palladium atoms showed high selectivity (98%) toward nitrogen gas production over ammonia.
Introduction
Water pollution has threatened clean water accessibility, and has become a major environmental issue with high water consumption common in energy production, agriculture, and domestic households.1 Unfortunately, water resources including streams, rivers, and groundwater have been found to contain various levels of nitrate, nitrite, phosphate, chlorate, and other ions.2–5 An ubiquitous contaminant present in more than two-thirds of water resource samples is nitrate (NO3−) coming from anthropogenic sources such as fertilizer run-off.6,7 Nitrate can be readily converted to nitrite (NO2−), and nitrite is known to lead to birth defects, hypertension, blue baby syndrome, and various forms of cancer.8–11 Several different treatments have been developed to eliminate NO3− and NO2− from drinking water including ion exchange, reverse osmosis, and electrodialysis.12–15 However, the main roadblock associated with membrane-based techniques has been the formation of secondary waste containing intact ions in the system. This leads to the reintroduction of NO3− and NO2− ions into the environment after completion of the process leading to additional operation steps for their removal. Another option is biological denitrification treatments, but long start-up times for biomass growth may hinder the elimination process of nitrite and nitrate.16 Therefore, to overcome these issues, heterogenous catalytic treatment, a low waste production technique with quick start-up time, has shown promise as an alternative approach for NO3− and NO2− elimination.In conventional catalytic reduction of nitrate, NO3− primarily reduces to NO2−. Subsequently, nitrite can be reduced and either NH3 or N2 produced as end products. Palladium-based catalysts such as Pd–Cu, Pd–In and Pd–Sn, were the first heterogenous catalytic systems explored for NO3− and NO2− reduction. Initially NO3− is reduced to NO2− after chemisorption onto the copper, indium or tin.17,18 This is followed by the reduction of NO2− to NH3 or N2 by the Pd sites. In addition to reduction by Pd, other metal catalysts have been explored for NO2− reduction via hydrogenation such as ruthenium (Ru), platinum (Pt), rhodium (Rh) as well as a mixed metal/main group catalyst, dinickel phosphide (Ni2P).19 However, these catalysts have shown low activity toward NO2− conversion, the need for highly acidic conditions, and high NH3 and N2H4 production all of which have made them less favorable for drinking water treatment when compared to Pd-based catalysts.20–22 In recent years, supported and unsupported Pd-based bimetallic alloy catalysts such as PdAu, PdAg, Pd-decorated Au (Pd-on-Au), and In-on-Pd have been explored for nitrite elimination and have shown that the combination of Pd with a secondary metal highly increases the catalytic activity and improves the tendency to form N2 as final product.23–26 Nevertheless, current nanocatalyst fabrication methods require a multi-steps process, a wide variety of chemicals, harsh reaction conditions, and prolonged reaction times. Therefore, introducing of a one-pot, straightforward, expeditious and practical procedure allowing the rational design of the catalysts under ambient conditions would be well received.
Recently, biomimetic structures such as peptides have been used as alternatives to traditional synthetic approaches creating functional structures with unique properties.27–30 Peptides composed of 7 to 12 amino acids, and engineered combinations of them, were used to generate different inorganic nanoparticles and bimetallic alloy NPs in aqueous solution under ambient conditions.31–35 Previous studies have shown that the Pd4 enables the formation of uniform sized nanoparticles, without it, uniform nanoparticles do not form.30,36
Herein we present an efficient, prompt, scalable, and bio-based technique using biocombinatorially derived Pd4 peptide (TSNAVHPTLRHL) for the preparation of reusable bimetallic nanocatalysts containing Pd and Au for selective nitrite reduction into N2. Two different bimetallic NP preparation approaches were applied for aqueous phase NO2− hydrogenation. In the first approach, Pd was decorated on the surface of pre-synthesized peptide-directed Au NPs. In the second one, Pd2+ and Au3+ ions were simultaneously reduced in the presence of Pd4 peptide to form alloy NPs. To better understand the impact of these routes and to identify the most suitable technique, the Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio was kept constant for both synthetic approaches. The nanocatalyst compositions and surface structures were identified using UV-Vis spectroscopy, high resolution transmission electron microscopy (HRTEM), energy dispersive X-ray analysis (EDX), and X-ray photoelectron spectroscopy (XPS). Subsequently, catalyst activity and selectivity toward NO2− reduction were correlated with accessible Pd content in bimetallic NPs.
:Au ratio was kept constant for both synthetic approaches. The nanocatalyst compositions and surface structures were identified using UV-Vis spectroscopy, high resolution transmission electron microscopy (HRTEM), energy dispersive X-ray analysis (EDX), and X-ray photoelectron spectroscopy (XPS). Subsequently, catalyst activity and selectivity toward NO2− reduction were correlated with accessible Pd content in bimetallic NPs.
Previously, the use of Pd nanocatalysts for organic transformations using fusion Pd4 peptide were reported.37–39 In this study, Pd4 peptide was used to prepare robust nitrite reduction bimetallic nanocatalysts in water. Nanoparticles with different Pd and Au ratios were prepared according two different routes. In the first route, the surface of a pre-synthesized peptide-directed Au NP was decorated with Pd atoms at different surface coverages. Based on the Au magic cluster model, Au NP concentration, total number of atoms, number of atoms in each shell, Au NP diameter, and Pd surface coverages were estimated.40 Subsequently, peptide-directed nanocatalysts with different Pd surface coverage and composition were fabricated. In the second pathway, peptide-based synthesis of bimetallic NPs was performed in water containing both Pd2+ and Au3+ ions simultaneously. Consequently, bimetallic NPs with different Pd to Au ratios were obtained (Table S1†).
Experimental section
Materials
Tetrachloroauric(III) acid (HAuCl4·3H2O, 99.99%), palladium(II) chloride (K2PdCl4, 99.99%), sodium borohydride (NaBH4, 98.0%), sodium nitrite (NaNO2, 99.7%), N-(1-naphthyl)ethyl-enediamine dihydrochloride (98.0%), and antifoam SE-15 were acquired from Sigma-Aldrich. Sulfanilamide, phosphoric acid (H3PO4), and Nessler's reagent (K2HgI4) were purchased from LabChem. Pd4 peptide (95.0%) was obtained from Bio-Synthesis, Inc., hydrogen gas (99.99%) was purchased from Airgas, and Milli-Q water (18 mΩ cm) was used in all experiments. Griess reagent stock solution was prepared by dissolving 1.0 g (5.8 mmol) of sulfanilamide, 0.1 g (0.54 mmol) of N-(1-naphthyl)ethyl-enediamine dihydrochloride, and 2.94 mL of H3PO4 (85 wt% in water) (0.066 mmol) in 100 mL of Milli-Q water. The final concentrations of sulfanilamide, N-(1-naphthyl)ethyl-enediamine dihydrochloride, and H3PO4 were 1, 0.1, and 5 wt%, respectively.Catalytic nanoparticle (NP) synthesis
:3.:3.To measure the concentration of nitrite and ammonia in the mixture, Griess and Nessler's reagents were used, respectively.24 Typically, to analyze the concentration of nitrite, 1 mL aqueous solution containing 0.1 mL of reaction mixture, 0.1 mL of Griess reagent, and 0.8 mL of DI water was prepared, mixed, and allowed to stand for 10 min. The absorbance at 540 nm measured using UV-Vis spectroscopy indicated the nitrite concentration of the mixture. To measure the ammonia presence, similar technique was followed except using Nessler's reagent instead of Griess reagent and the absorbance at 420 nm provided the ammonia concentration in the solution.
By calculating the H2 in excess, the nitrite reduction reaction was found to follow pseudo-first-order kinetics, following:
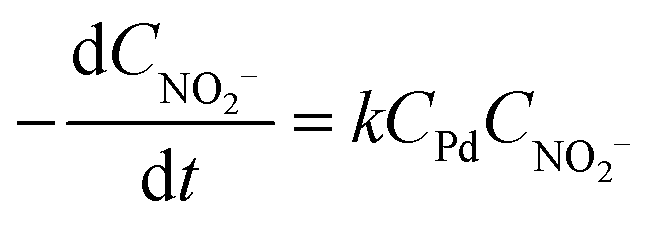 | (1) |
The intrinsic catalytic activity, turnover frequency (TOF), is calculated from the number of converted reactant molecules made per catalytic active site per unit time using:
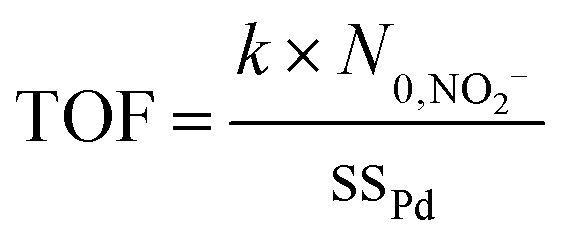 | (2) |
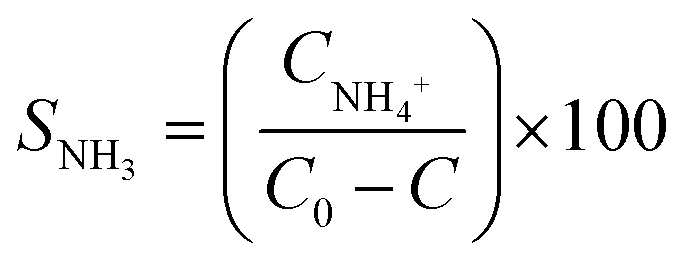 | (3) |
| SN2 = 100% − SNH3 | (4) |
For the reusability study of the catalysts, after completion of each cycle, the medium was bubbled with carbon dioxide and hydrogen gas for 20 min. Subsequently, NaNO2 solution was re-injected into the solution so that the NO2− concentration of solution was 40 mg L−1 before each cycle.
Results and discussion
NPs structure and properties
UV-Vis absorption spectra of prepared peptide-directed NPs with different Pd:Au ratios following surface coverage and bimetallic PdxAu100−x pathways are shown in Fig. 1. The surface plasmon resonance (SPR) band of Au NPs occurs around 550 nm in visible region while Pd NP pattern does not show any bands.44 By increasing the surface coverage of Pd-on-Au, the SPR band of Au broadened with the increasing amount of Pd atoms on the Au NP surface (Fig. 1a). Conversely, by increasing the concentration of Pd in bimetallic PdxAu100−x NPs, the SPR band gradually disappears. Once the surface was covered with 26% palladium atoms, Pd26Au74, no band was detected, and it did not return with higher amounts of palladium (Fig. 1b). The disappearance of the Au band is attributed to intermixing of Au and Pd leading to the formation of bimetallic PdxAu100−x NPs.
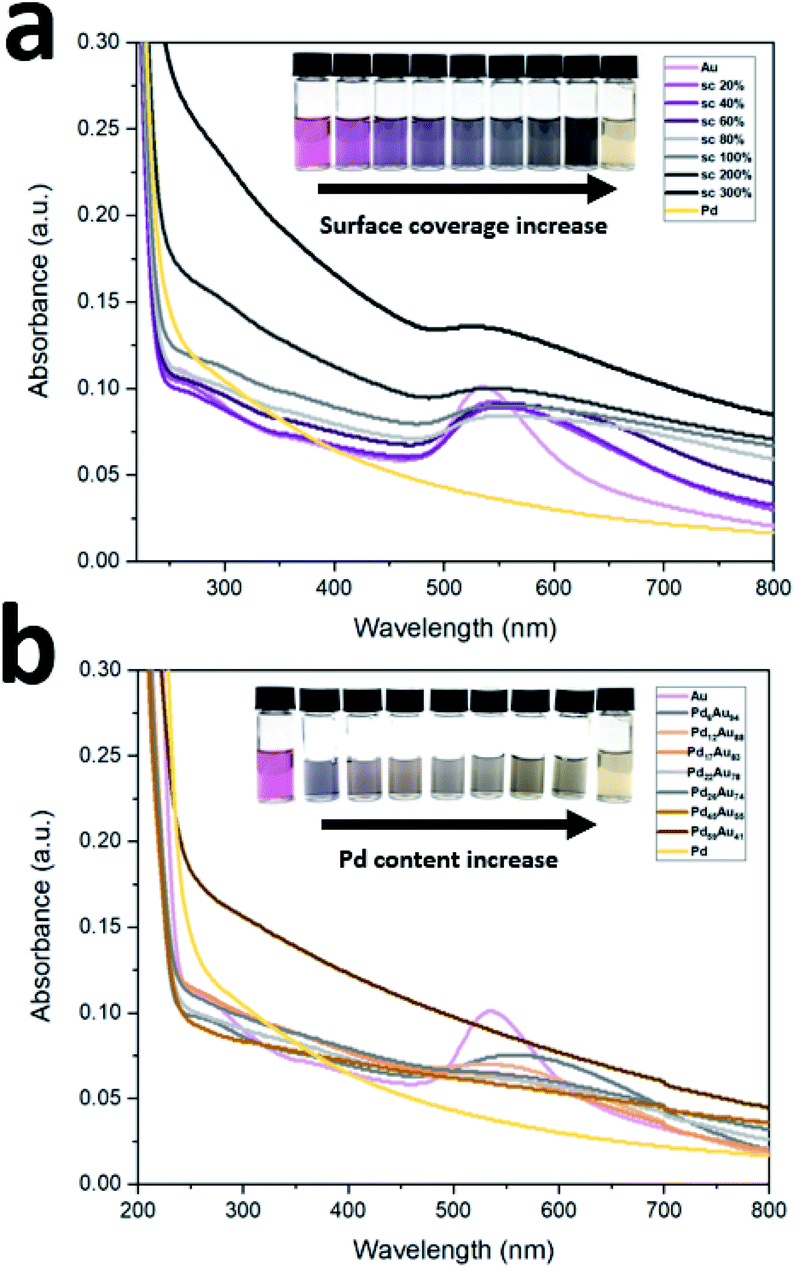 | ||
| Fig. 1 UV-Vis spectra of prepared NPs following (a) surface coverage and (b) bimetallic PdxAu100−x pathways. | ||
To confirm the morphology, size, and structure of peptide-directed Pd-on-Au and PdxAu100−x NPs, TEM analysis of prepared NPs was performed. Discrete and almost spherical Au NPs (aspect ratio close to unity) with average diameter of 4.0 ± 0.9 nm in the absence of large aggregates were initially prepared (Fig. 2a). The TEM analysis of 100 sc% Pd-on-Au NPs indicates that the average diameter size of NPs was 4.52 ± 1.20 nm (Fig. 2b). Comparing to average diameter size of as-synthesized Au NPs and magic cluster model calculations,40 the increase in the average NP size can be attributed to the formation of Pd layer on Au NPs. For bimetallic PdxAu100−x NPs, Pd26Au74 NPs, with the same overall Pd content as 100 sc% Pd-on-Au, were characterized. TEM analysis of Pd26Au74 NPs confirms the presence of spherical NPs with low amount of aggregation (Fig. 2c). The average diameter size of fabricated NPs was 4.63 ± 1.42 nm, with the majority of NPs in the range of 3–5 nm. Although the average diameter size of both NPs were comparable, the increase in the average diameter size of Pd26Au74 NPs compared to that of Au NP was attributed to the formation of an alloy structure through intermixing of Au and Pd.45 In addition, EDX analysis was used for assessing the composition of the synthesized NPs. The presence of Au and Pd were confirmed as peaks attributed to both elements were detected (Fig. 2d). The actual percentage of Pd was determined to be 27.0% and 26.2% in 100 sc% Pd-on-Au NP and Pd26Au74 NP, respectively, indicating that the composition of prepared peptide-directed NPs was dictated by the molar ratio of the precursors.
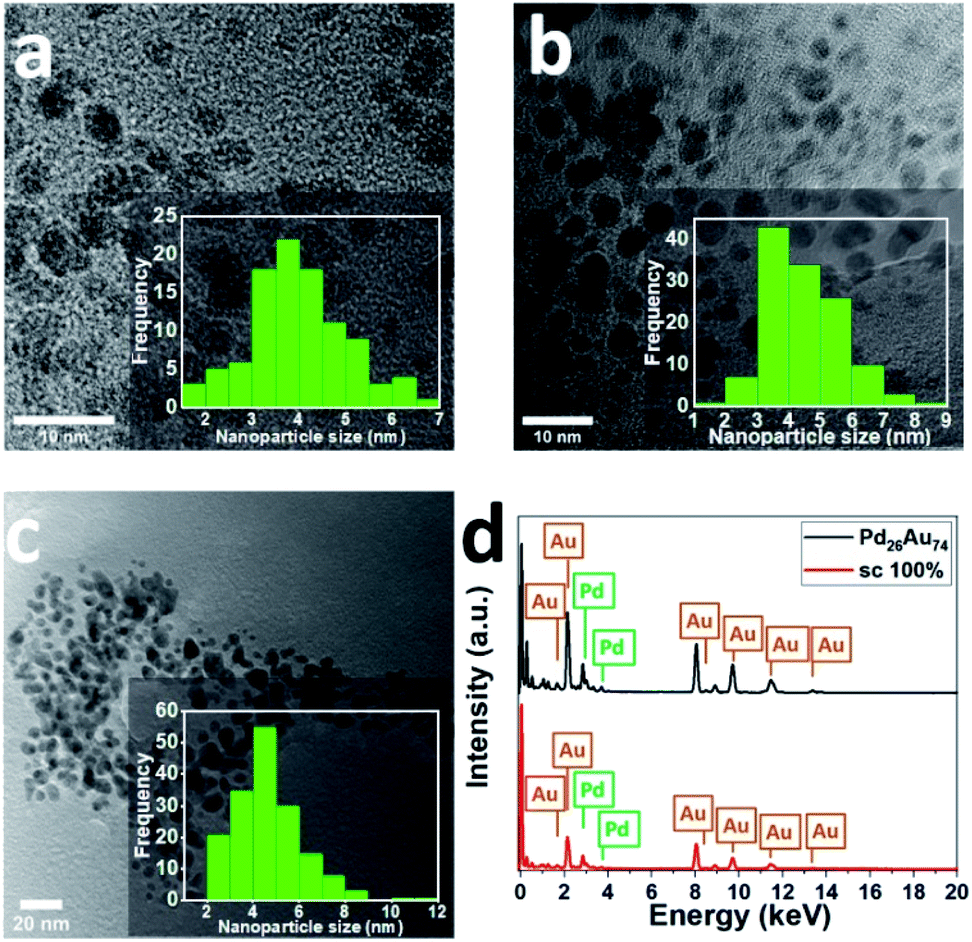 | ||
| Fig. 2 TEM image and size distribution of (a) Au NPs, (b) 100 sc% Pd-on-Au NPs, and (c) Pd26Au74 NPs. (d) EDX analysis of prepared bimetallic NPs. | ||
The formation of 100 sc% Pd-on-Au NPs and bimetallic alloy Pd26Au74 NPs was further confirmed by the d-spacing measurement of the lattice using high-resolution transmission electron microscopy (HRTEM) images. The analysis of Pd-on-Au NPs showed d-spacing of 0.236 nm which is in close agreement with theoretical d-spacing of 〈111〉 planes for pure face centered cubic (FCC) Au confirming the presence of Au as the core NP (Fig. 3a and Table S2†). The monolayer of Pd was formed on the surface of Au NP as lattices with d-spacing of 0.225 nm and 0.117 nm correspond to the 〈111〉 and 〈311〉 planes, respectively (Table S2†). Pd26Au74 NPs lattice d-spacing measurements showed a value of 0.233 nm for 〈111〉 FCC planes closely matching the calculated spacing of 0.232 nm (Fig. 3b and Table S2†). The d-spacing measurement of Pd26Au74 confirms the intermixing of Au and Pd where the d-spacing measurement is higher than that of pure Pd and lower than that of pure Au. There is, however, a slight Au bias due to the lower percentage of Pd in its structure.
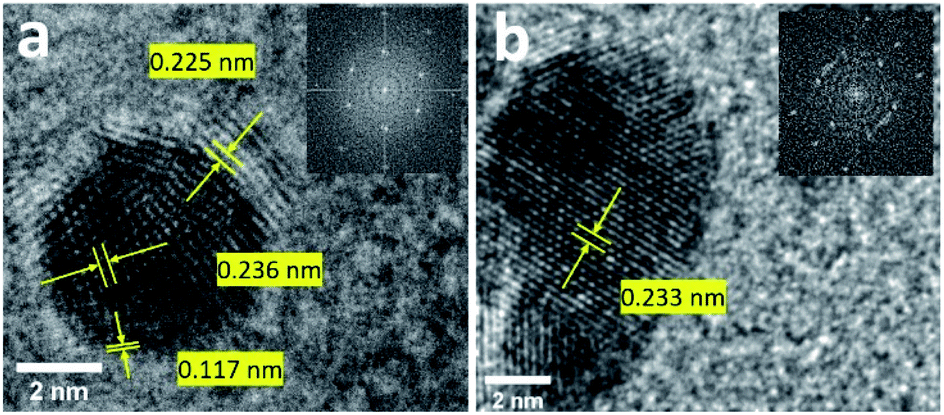 | ||
| Fig. 3 HRTEM image for 100 sc% Pd-on-Au (a) and (b) Pd26Au74 NPs. Fast Fourier transform images are shown as insets. | ||
Moreover, the oxidation state distribution of surface atoms in 100 sc% Pd-on-Au and Pd26Au74 NPs was analyzed through XPS measurements. Due to coordination with inner metal atoms, and not exposure to air, the valence for subsurface atoms were expected to be zero toward oxidation.45 In 100 sc% Pd-on-Au NP, as expected, only Pd signals were obtained while Au was not detected (Fig. S2†). The absence of Au signals could also be attributed to near perfect coverage of Pd on Au surface as Au signals can be detected when more than 20% of NP surface is possessed by Au atoms.25 In bimetallic Pd26Au74 NPs, Pd signals as well as high intensity signals pertaining to Au were detected confirming the presence of the alloy structure on the surface (Fig. S3†).
Catalytic nitrite reduction
After optimizing the nitrite reduction conditions, the activity of the prepared peptide-directed NPs were further evaluated for nitrite reduction (Table S3†). Catalytic nitrite reduction assessments for Pd4 peptide, Au NP, Pd NP, Pd26Au74 NP, and 100 sc% Pd-on-Au NP in which the Pd concentration for Pd, Pd26Au74, and 100 sc% Pd-on-Au NPs were the same (40 mg L−1) were conducted (Fig. 4). No progress was observed when Pd4 peptide and Au NP were used, emphasizing the need for Pd atoms for the hydrogenation of nitrite. The cooperation of Au led to enhanced catalytic activity in which higher yields were obtained when Pd26Au74 was used. The highest nitrite reduction was obtained using 100 sc% Pd-on-Au NP as catalyst further confirming that the coated Pd were activated toward reduction. The major product of nitrite hydrogenation process was dinitrogen, with 98% and 96% selectivity when 100 sc% Pd-on-Au, or Pd26Au74, were used as catalysts, respectively (Fig. S4†). Such a high selectivity towards dinitrogen makes these designed NPs ideal candidates for water purification.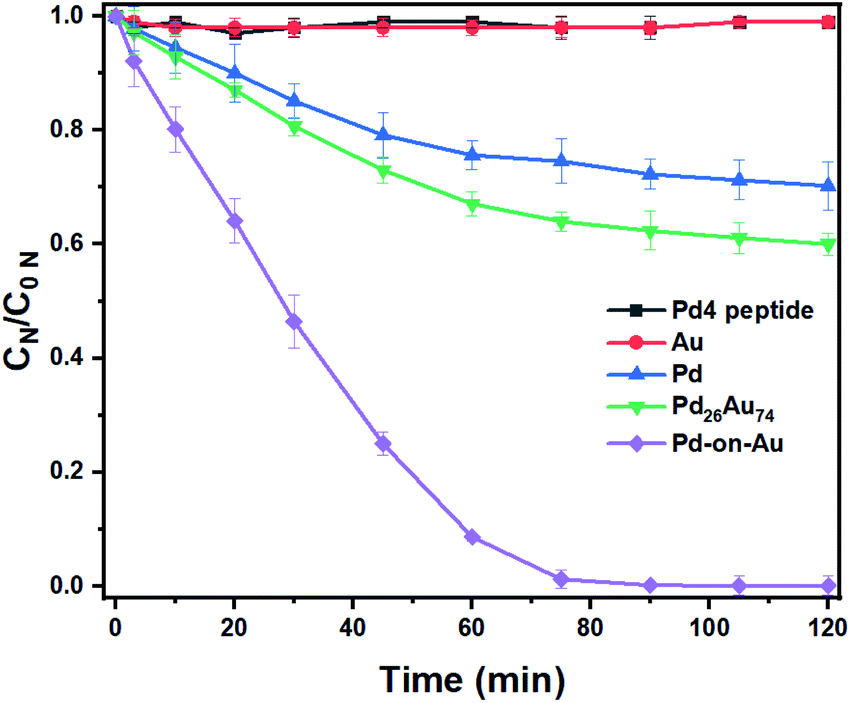 | ||
| Fig. 4 Nitrite time evolution for Pd4 peptide, Au, Pd, Pd26Au74, and 100 sc% Pd-on-Au catalyst. Reaction condition: NaNO2 (40 mg L−1), H2 (100 mL min−1), CO2 (100 mL min−1), catalyst (40 mg L−1 of Pd in Pd, Pd26Au74, and 100 sc% Pd-on-Au), H2O (20 mL), 600 rpm, 1 atm pressure, 120 min. Yields were determined by UV-Vis spectroscopy with Griess reagent as a nitrite concentration indicator. | ||
The activity of peptide-based Pd-on-Au in nitrite reduction reaction varied with Pd sc%. Although the use of NPs containing only Pd as catalyst led to nitrite reduction, Pd-on-Au catalysts showed enhanced activity. We attribute this increase in activity to the gold core of the Pd-on-Au catalyst which creates a more open packing structure for the palladium on its surface (Fig. 5 and Table S1†). The nitrite reduction activity steadily increased with increasing sc% starting from 20% Pd-on-Au to 100% Pd-on-Au where a monolayer of Pd was formed uniformly onto the peptide-capped Au NP. The obtained turnover frequency (TOF) of the peptide-directed 100 sc% Pd-on-Au catalyst was determined to be 5.33 (min−1) which is 3.6 times greater than that of monometallic Pd NPs, similar to a previous report.46 To evaluate the activity of NPs prepared using the Pd4 peptide, a review of recent reports indicated that our 100 sc% Pd-on-Au NP is comparable to some reports and better than others (Table S4†). However, increasing the amount of Pd loading above 100 sc%, the efficiency of catalyst decreased such that 200 sc% Pd-on-Au showed lower activity compared and further increasing Pd content to obtain 300 sc% Pd-on-Au had led to an even less active catalyst. Thus, supporting the need for a direct interaction between Pd and Au atoms for higher catalytic activity (Fig. 5 and Table S1†).
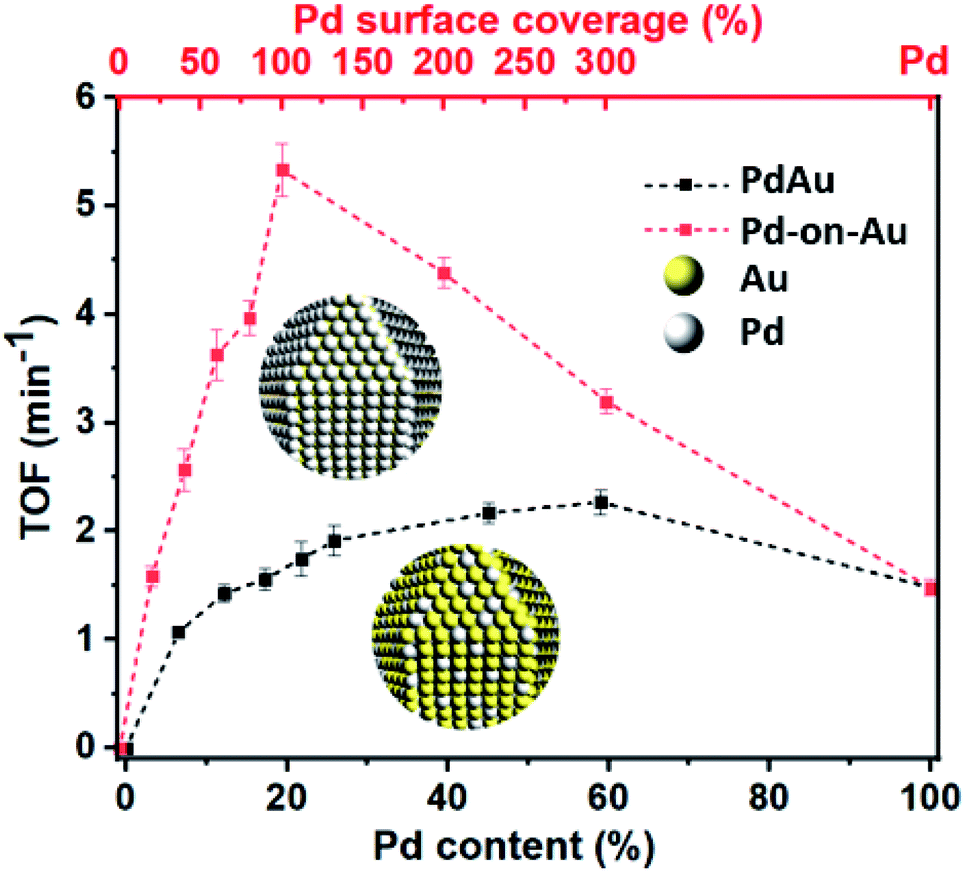 | ||
| Fig. 5 The experimentally obtained TOF for Pd-on-Au and PdAu catalysts as a function of Pd sc% and Pd content. | ||
Similar to the catalysts prepared with different surface coverages of Pd-on-Au, by increasing the amount of Pd content, a monotonical increase toward nitrite reduction activity was observed when peptide-based PdxAu100−x NPs were used as catalysts. However, in these reactions, the calculated TOF never exceeded that of reactions performed with Pd-on-Au catalysts with the same Pd loading. We attribute this to our peptide-based synthesis producing bimetallic PdAu alloy NPs with Pd atoms on the surface that are less active and because we are keeping the palladium content the same, some Pd atoms below the surface remain inaccessible to nitrite. Therefore, these NPs resulted in lower TOFs as compared to their counterparts that were prepared by the surface coverage technique.
In comparison, the PdAu NPs showed lower TOF overall and never achieved the TOF of the 100 sc% Pd-on-Au. Although the design of catalyst was to have the same accessible amount of Pd sites, the amount of active Pd sites were found to be more in 100 sc% Pd-on-Au. It was previously reported that the Pd atoms were reduced in the form of 2-D and 3-D islands on the Au NP surface instead of making a smooth layer of Pd. We can likely attribute the higher activity of 100 sc% Pd-on-Au catalyst to the formation of 2-D and 3-D islands. The geometric and electronic effects risen from such structure caused enhancement in the catalytic activity of the catalyst.47 Based on these results, we propose that the nitrite reduction reaction would take place on Pd active sites located on the surface of nanocatalysts. The NO2− ions were first adsorbed onto the Pd sites from solution, then in the presence of dissociated H* from H2 over Pd (0), NO2− ions were reduced to NO and N2O intermediate, which were then consumed and N2 was produced as the final product (Fig. 6).
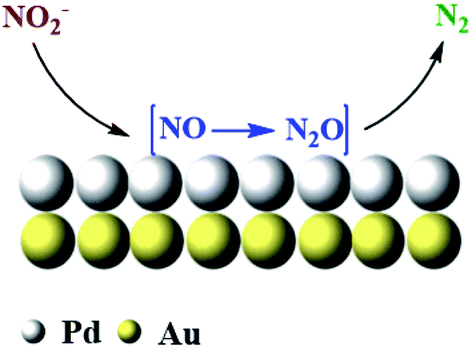 | ||
| Fig. 6 Degradation of nitrite reaction using 100 sc% Pd-on-Au nanocatalyst. | ||
Reusability of NPs
The reusability of 100 sc% Pd-on-Au and bimetallic alloy Pd26Au74 catalysts for NO2− reduction was also investigated and it was found that the NPs were catalytically active after five cycles (Fig. 7). The activity decreased by 22% and 19% for 100 sc% and 19% Pd26Au74 NPs after five runs, respectively. Furthermore, NPs remained discrete without significant size or morphology changes and no noticeable aggregation after five cycles.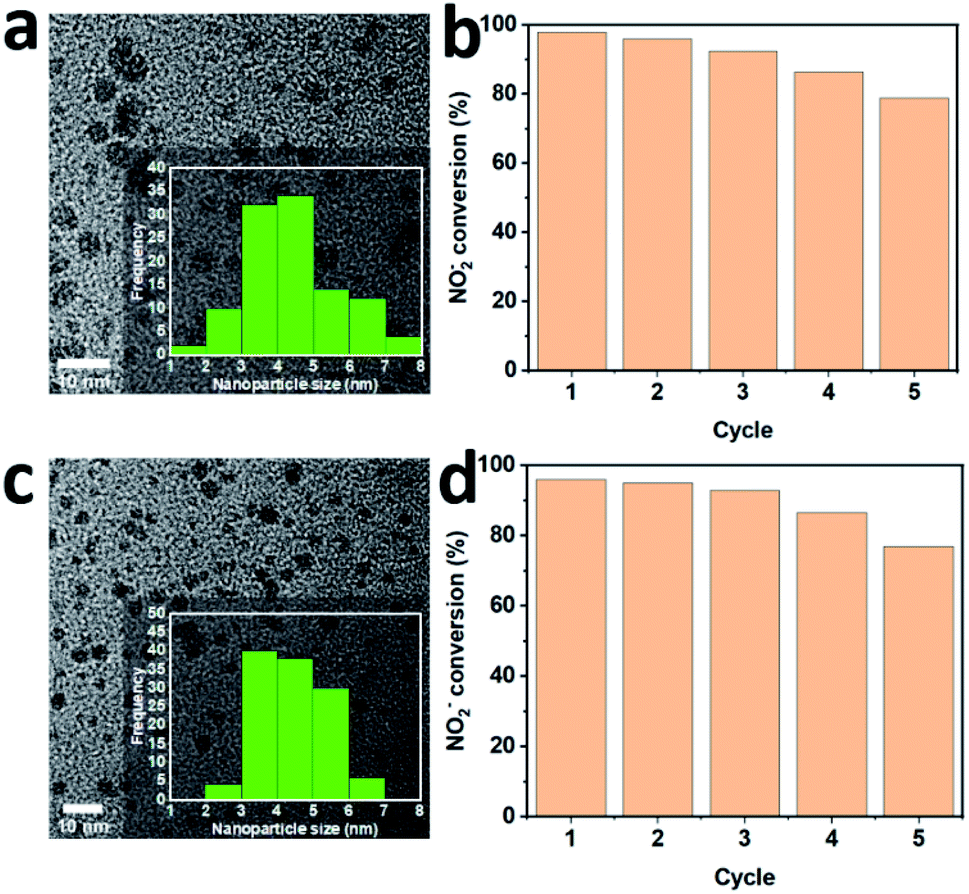 | ||
| Fig. 7 (a) TEM image of peptide-directed Pd-on-Au NP 100 sc% after five catalytic cycles. (b) Nitrite conversion for Pd-on-Au NP in five cycles. (c) TEM image of peptide-directed Pd26Au74 NP after five catalytic cycles. (d) Nitrite conversion for Pd26Au74 NP in five cycles (inset is the size distribution plot). Reaction condition: NaNO2 (40 mg L−1), H2 and CO2 (50 mL min−1), catalyst (400 mg L−1 of Pd), H2O (100 mL), 600 rpm, 1 atm pressure, 75 min. | ||
Conclusions
In this investigation, we have developed a convenient one-pot synthesis of Pd-decorated Au and PdxAu100−x NPs in water under ambient conditions. Stable Pd4-directed NPs with preserved morphologies and sizes in the absence of aggregated particles were obtained. Using the peptide-directed synthetic approach, a high-level of control over compositions and structures of Pd-on-Au and PdxAu100−x was achieved. Utilizing these synthesized bimetallic NPs as robust nanocatalysts for reduction of nitrite in the aqueous phase, high product selectivity toward nitrogen gas was also obtained. Other potential applications of fabricated nanocatalysts are currently being investigated in our lab.Author contributions
I. M. and A. A. developed the idea. I. M. performed the study and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the use of TEM and XPS facilities at the Cornell Center for Materials Research (CCMR) supported by the National Science Foundation Materials Research Science and Engineering Center (NSF MRSEC) program (DMR-1719875).Notes and references
- R. P. Schwarzenbach, T. Egli, T. B. Hofstetter, U. von Gunten and B. Wehrli, Annu. Rev. Environ. Resour., 2010, 35, 109–136 CrossRef.
- C.-M. Lee, S.-Y. Hamm, J.-Y. Cheong, K. Kim, H. Yoon, M. Kim and J. Kim, Environ. Res., 2020, 184, 109313 CrossRef CAS PubMed.
- J. Elser and E. Bennett, Nature, 2011, 478, 29–31 CrossRef CAS PubMed.
- P. F. Hudak, J. Hydrol., 2000, 228, 37–47 CrossRef CAS.
- P. Westerhoff, A. Atkinson, J. Fortner, M. S. Wong, J. Zimmerman, J. Gardea-Torresdey, J. Ranville and P. Herckes, Nat. Nanotechnol., 2018, 13, 661–669 CrossRef CAS PubMed.
- A. Liu, J. Ming and R. O. Ankumah, Sci. Total Environ., 2005, 346, 112–120 CrossRef CAS PubMed.
- B. T. Nolan and J. D. Stoner, Environ. Sci. Technol., 2000, 34, 1156–1165 CrossRef CAS.
- J. D. Brender, P. J. Weyer, P. A. Romitti, B. P. Mohanty, M. U. Shinde, A. M. Vuong, J. R. Sharkey, D. Dwivedi, S. A. Horel, J. Kantamneni, J. C. Huber, Q. Zheng, M. M. Werler, K. E. Kelley, J. S. Griesenbeck, F. B. Zhan, P. H. Langlois, L. Suarez and M. A. Canfield, Environ. Health Perspect., 2013, 121, 1083–1089 CrossRef PubMed.
- B. A. Kilfoy, Y. Zhang, Y. Park, T. R. Holford, A. Schatzkin, A. Hollenbeck and M. H. Ward, Int. J. Cancer, 2011, 129, 160–172 CrossRef CAS PubMed.
- R. R. Jones, P. J. Weyer, C. T. DellaValle, M. Inoue-Choi, K. E. Anderson, K. P. Cantor, S. Krasner, K. Robien, L. E. B. Freeman, D. T. Silverman and M. H. Ward, Environ. Health Perspect., 2016, 124, 1751–1758 CrossRef CAS PubMed.
- A. Temkin, S. Evans, T. Manidis, C. Campbell and O. V. Naidenko, Environ. Res., 2019, 176, 108442 CrossRef CAS PubMed.
- D. Cliford and X. Liu, J.–Am. Water Works Assoc., 1993, 85, 135–143 CrossRef.
- R. Epsztein, O. Nir, O. Lahav and M. Green, Chem. Eng. J., 2015, 279, 372–378 CrossRef CAS.
- S. Duan, T. Tong, S. Zheng, X. Zhang and S. Li, Water Res., 2020, 173, 115571 CrossRef CAS PubMed.
- A. El Midaoui, F. Elhannouni, M. Taky, L. Chay, M. A. Menkouchi Sahli, L. Echihabi and M. Hafsi, Sep. Purif. Technol., 2002, 29, 235–244 CrossRef CAS.
- S. G. Lehman, M. Badruzzaman, S. Adham, D. J. Roberts and D. A. Clifford, Water Res., 2008, 42, 969–976 CrossRef CAS PubMed.
- N. Barrabés and J. Sá, Appl. Catal., B, 2011, 104, 1–5 CrossRef.
- K. A. Guy, H. Xu, J. C. Yang, C. J. Werth and J. R. Shapley, J. Phys. Chem. C, 2009, 113, 8177–8185 CrossRef CAS.
- S. Hörold, K.-D. Vorlop, T. Tacke and M. Sell, Catal. Today, 1993, 17, 21–30 CrossRef.
- X. Huo, D. J. Van Hoomissen, J. Liu, S. Vyas and T. J. Strathmann, Appl. Catal., B, 2017, 211, 188–198 CrossRef CAS.
- X. Chen, X. Huo, J. Liu, Y. Wang, C. J. Werth and T. J. Strathmann, Chem. Eng. J., 2017, 313, 745–752 CrossRef CAS.
- C. A. Clark, C. P. Reddy, H. Xu, K. N. Heck, G. Luo, T. P. Senftle and M. S. Wong, ACS Catal., 2020, 10, 494–509 CrossRef CAS.
- S. Seraj, P. Kunal, H. Li, G. Henkelman, S. M. Humphrey and C. J. Werth, ACS Catal., 2017, 7, 3268–3276 CrossRef CAS.
- H. Li, S. Guo, K. Shin, M. S. Wong and G. Henkelman, ACS Catal., 2019, 9, 7957–7966 CrossRef CAS.
- J. P. Troutman, H. Li, A. M. Haddix, B. A. Kienzle, G. Henkelman, S. M. Humphrey and C. J. Werth, ACS Catal., 2020, 10, 7979–7989 CrossRef CAS.
- S. Guo, K. Heck, S. Kasiraju, H. Qian, Z. Zhao, L. C. Grabow, J. T. Miller and M. S. Wong, ACS Catal., 2018, 8, 503–515 CrossRef CAS.
- W. Wang, C. F. Anderson, Z. Wang, W. Wu, H. Cui and C.-J. Liu, Chem. Sci., 2017, 8, 3310–3324 RSC.
- J. T. Wagner, A. K. Long, M. B. Sumner, I. Cockman, M. Smith and B. B. Penland, J. Phys. Chem. C, 2020, 124, 27743–27753 CrossRef CAS.
- E. Pazos, E. Sleep, C. M. Rubert Pérez, S. S. Lee, F. Tantakitti and S. I. Stupp, J. Am. Chem. Soc., 2016, 138, 5507–5510 CrossRef CAS PubMed.
- T. R. Walsh and M. R. Knecht, Chem. Rev., 2017, 117, 12641–12704 CrossRef CAS PubMed.
- N. M. Bedford, Z. E. Hughes, Z. Tang, Y. Li, B. D. Briggs, Y. Ren, M. T. Swihart, V. G. Petkov, R. R. Naik, M. R. Knecht and T. R. Walsh, J. Am. Chem. Soc., 2016, 138, 540–548 CrossRef CAS PubMed.
- A. D. Parab, J. M. Slocik, R. R. Naik and M. R. Knecht, ACS Appl. Nano Mater., 2018, 1, 7149–7158 CrossRef CAS.
- N. M. Bedford, A. R. Showalter, T. J. Woehl, Z. E. Hughes, S. Lee, B. Reinhart, S. P. Ertem, E. B. Coughlin, Y. Ren, T. R. Walsh and B. A. Bunker, ACS Nano, 2016, 10, 8645–8659 CrossRef CAS PubMed.
- N. A. Merrill, E. M. McKee, K. C. Merino, L. F. Drummy, S. Lee, B. Reinhart, Y. Ren, A. I. Frenkel, R. R. Naik, N. M. Bedford and M. R. Knecht, ACS Nano, 2015, 9, 11968–11979 CrossRef CAS PubMed.
- D. Li, Z. Tang, S. Chen, Y. Tian and X. Wang, ChemCatChem, 2017, 9, 2980–2987 CrossRef CAS.
- A. Polasa, I. Mosleh, J. Losey, A. Abbaspourrad, R. Beitle and M. Moradi, bioRxiv, 2021, 2021.03.11.432607 Search PubMed.
- I. Mosleh, H. R. Shahsavari, R. Beitle and M. H. Beyzavi, ChemCatChem, 2020, 12, 2942–2946 CrossRef CAS.
- I. Mosleh, M. Benamara, L. Greenlee, M. H. Beyzavi and R. Beitle, Mater. Lett., 2019, 252, 68–71 CrossRef CAS.
- R. Tejada-Vaprio, I. Mosleh, R. P. Mukherjee, H. Aljewari, M. Fruchtl, A. Elmasheiti, N. Bedford, L. Greenlee, M. H. Beyzavi and R. Beitle, Biotechnol. Prog., 2020, 36, e2956 CrossRef CAS PubMed.
- M. O. Nutt, K. N. Heck, P. Alvarez and M. S. Wong, Appl. Catal., B, 2006, 69, 115–125 CrossRef CAS.
- H. Shin, S. Jung, S. Bae, W. Lee and H. Kim, Environ. Sci. Technol., 2014, 48, 12768–12774 CrossRef CAS PubMed.
- J. Sá and J. A. Anderson, Appl. Catal., B, 2008, 77, 409–417 CrossRef.
- S. Jung, S. Bae and W. Lee, Environ. Sci. Technol., 2014, 48, 9651–9658 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- P. Kunal, H. Li, B. L. Dewing, L. Zhang, K. Jarvis, G. Henkelman and S. M. Humphrey, ACS Catal., 2016, 6, 4882–4893 CrossRef CAS.
- H. Qian, Z. Zhao, J. C. Velazquez, L. A. Pretzer, K. N. Heck and M. S. Wong, Nanoscale, 2014, 6, 358–364 RSC.
- K. N. Heck, S. Garcia-Segura, P. Westerhoff and M. S. Wong, Acc. Chem. Res., 2019, 52, 906–915 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed characterization data (TEM, and XPS for compounds), catalysts activity comparison table, and references are provided. See DOI: 10.1039/d1ra05304c |
| This journal is © The Royal Society of Chemistry 2021 |