
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and mechanistic study of new benzenesulfonamide derivatives as anticancer and antimicrobial agents via carbonic anhydrase IX inhibition†
Mohamed T. M. Nemra,
Asmaa M. AboulMagd b,
Hossam M. Hassan*cd,
Ahmed A. Hamede,
Mohamed I. A. Hamedf and
Mohamed T. Elsaadigh
b,
Hossam M. Hassan*cd,
Ahmed A. Hamede,
Mohamed I. A. Hamedf and
Mohamed T. Elsaadigh
aPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Cairo University, Kasr El-Eini street 11562, Cairo, Egypt
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Nahda University, Beni-Suef 62513, Egypt
cPharmacognosy Department, Faculty of Pharmacy, Beni-Suef University, Beni-Suef 62513, Egypt. E-mail: hossam.mokhtar@nub.edu.eg
dPharmacognosy Department, Faculty of Pharmacy, Nahda University, Beni-Suef 62513, Egypt
eMicrobial Chemistry Department, National Research Center, Giza, Egypt
fOrganic and Medicinal Chemistry Department, Faculty of Pharmacy, Fayoum University, Fayoum 63514, Egypt
gPharmaceutical Chemistry Department, Faculty of Pharmacy, Beni-Suef University, Beni-Suef 62513, Egypt
hPharmaceutical Chemistry Department, Faculty of Pharmacy, Sinai University, Egypt
First published on 1st August 2021
Abstract
Changes in gene expression cause uncontrolled cell proliferation and consequently tumor hypoxia. The tumor cells shift their metabolism to anaerobic glycolysis with a significant modification in pH. Therefore, an over expression of carbonic anhydrase IX (CA IX) genes was detected in many solid tumors. Accordingly, selective inhibition of CA IX can be a useful target for discovering novel antiproliferative agents. The present study described the synthesis of new aryl thiazolone–benzenesulfonamides 4a–j as well as their carbonic anhydrase IX inhibitory effect. All the designed derivatives were evaluated for their anti-proliferative activity against triple-negative breast cancer cell line (as MDA-MB-231) and another breast cancer cell line (MCF-7) in addition to normal breast cell line MCF-10A. Compounds 4b–c, 4e, 4g–h showed significant inhibitory effect against both cancer cell lines at concentration ranges from 1.52–6.31 μM, with a high selectivity against breast cancer cell lines ranges from 5.5 to 17.5 times. Moreover, three sulfonamides derivatives 4e, 4g and 4h showed excellent enzyme inhibition against CA IX with IC50 10.93–25.06 nM and against CA II with IC50 1.55–3.92 μM that revealed their remarkable selectivity for CA IX over CA II. Additionally, 4e was able to induce apoptosis in MDA-MB-231 with a significant increase in the annexin V-FITC percent by 22 fold as compared with control. Cellular uptake on MDA-MB-231 cell lines were carried out using HPLC method on the three active compounds (4e, 4g and 4h). On the other hand inhibition of one or more CAs present in bacteria was reported to interfere with bacterial growth. So, the new benzenesulfonamides were evaluated against their antibacterial and anti-biofilm activities. Analogues 4e, 4g and 4h exhibited significant inhibition at 50 μg mL−1 concentration with 80.69%, 69.74% and 68.30% against S. aureus compared to the positive control CIP which was 99.2%, while compounds 4g and 4h showed potential anti-biofilm inhibition 79.46% and 77.52% against K. pneumonia. Furthermore, the designed compounds were docked into CA IX (human) protein (PDB ID: 5FL6) and molecular modeling studies revealed favorable binding interactions for the active inhibitors. Finally, the predictive ADMET studies showed that, compounds 4e, 4g and 4h possessed promising pharmacokinetic properties.
1 Introduction
Cancer remains one of the leading causes of death worldwide, thus necessitating the development of novel and effective therapies. Although modern therapeutic agents have been developed over the past 100 years, the successful treatment of cancer appears to be a powerful challenge at the start of the present century. This challenge lies in the difficulty to discover novel selective agents that inhibit the proliferation of tumor cells without being toxic to normal cells.1–4 According to GLOBOCAN, 2020 estimation, about 19.3 million cancer cases and 10 million cancer deaths are estimated to occur worldwide in 2020. Breast cancer (BC) is one of the most commonly diagnosed and the second leading cause of cancer related deaths in females. Also, BC represents 1 in 4 cancer cases diagnosed among women globally.5 It is often characterized by hypoxia regions due to poor vascularization.6 Moreover, BC cells that survive in this adverse media are found to be resistant to radiotherapy and chemotherapy.7 Among the genes that contributes to hypoxic condition production and the adaptation of hypoxic tumor cells are carbonic anhydrase IX (CA IX) and XII (CA XII).8 CA IX is reported to be overexpressed in various tumor entities, especially in those with invasive BC, immunohistochemical CA IX over expression is consistent with worse relapse-free and overall survival.9 Knowing the topology of carbonic anhydrase active site offers a promising strategy for developing selective inhibitors. This mammalian CAs active site is characterized by a cone-shaped cavity with two preserved environments, known as hydrophobic and hydrophilic walls, and a Zn2+ ion at the bottom. This Zn2+ ion is tetrahedrally conserved with three characteristic histidine residues namely (His94, His96, and His119) and a solvent molecule; which binds to the hydroxide ion that reacted with carbon dioxide to yield bicarbonate.10,11 As a result, CA inhibitors should have a specific zinc binding group (ZBG) that can coordinate efficiently with the Zn2+ ion at the binding site. Actually, ZBG binds with different chemical moieties that called the compound “tail” that enables the interactions with certain residues in the active site. One of the important mechanisms of anticancer activity of sulfonamides is the inhibition of carbonic anhydrase isozymes.12–15 The amino thiazole based coumarin benzenesulfonamide compound I (Fig. 1) showed potent inhibitory activity against hCA IX with a Ki value of 25.04 nM as compared to the reference compound acetazolamide II which had a Ki value of 25 nM, in addition, compound I showed potent inhibitory activity against hCA XII when compared with AZA, with Ki values of 3.94 and 5.7 nM respectively.16 A series of diamide-based benzenesulfonamides were synthesized and evaluated as inhibitors of the metalloenzyme CA IX, compound III (Fig. 1) achieved the best CA IX/I and IX/II selectivity with selective indexes (SIs) of 985 and 13.8, respectively.17 Recently a CA IX inhibitor, SLC-0111 (IVa, Fig. 1), entered in Phase I clinical trials by potentiating the cytotoxic activity of the traditional chemotherapeutic agents against MCF-7 breast cancer cells, and HCT-116 colorectal cancer cells. SLC-0111 and some of its analogues IVa–d are among isoform-selective sulfonamides that are discovered as CAIs in the last period.18–20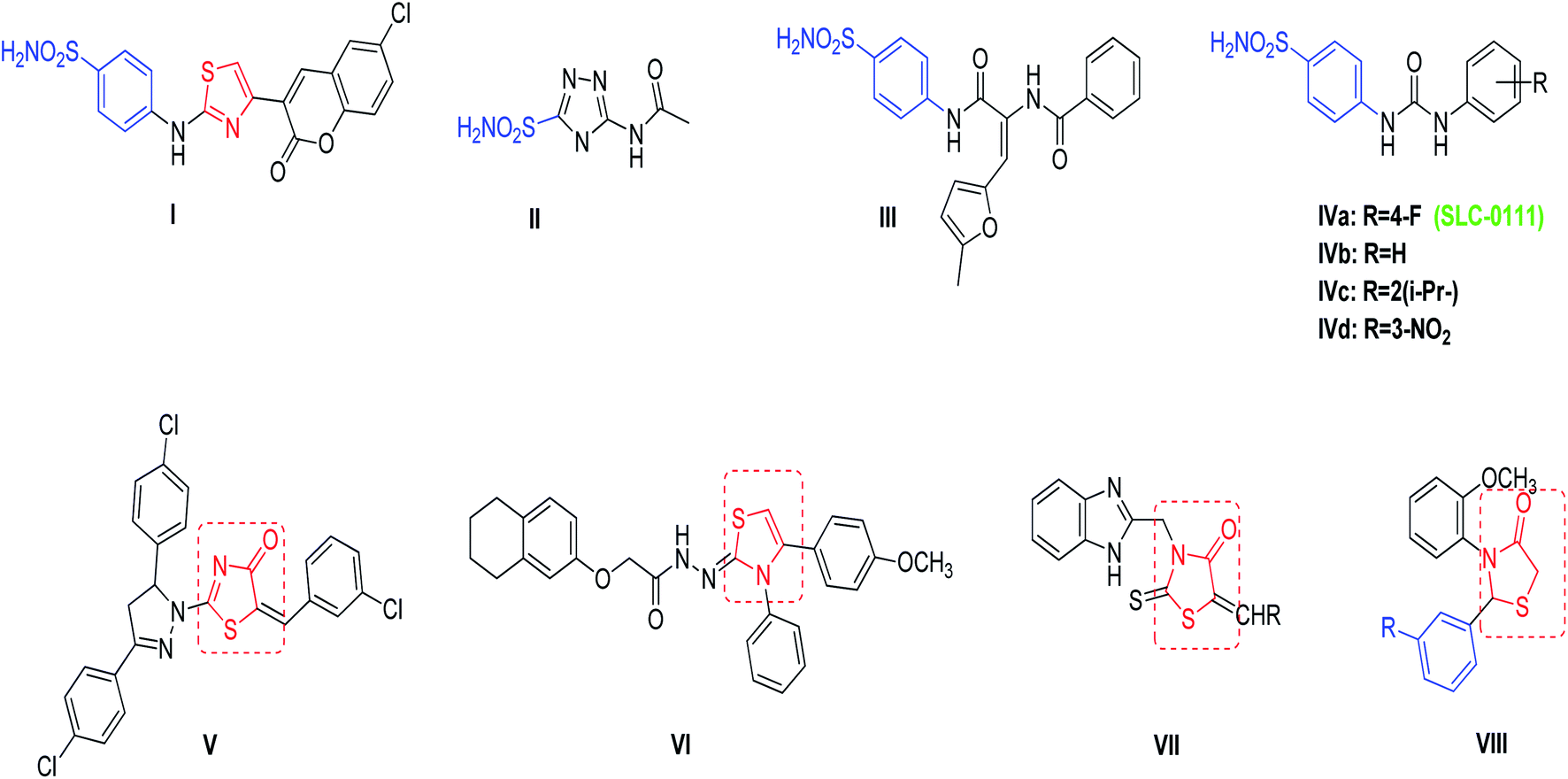 | ||
| Fig. 1 Reported benzenesulfonamides-based CAIs (I–IV), thiazolone and thiazolidine derivatives (V–VIII) as anticancer agents. | ||
4-Thiazolone and 4-thiazolidinones derivatives are important heterocyclic class possess a wide range of biological activities such as anticancer, anti-diabetic, anti-microbial, anti-viral, anti-inflammatory, and anti-convulsant.21–23 Compounds V–VIII (Fig. 1) have been reported to demonstrate significant antitumor activity against the human BC cell line (MCF-7).24–27
From another point of view, it has shown that interference with the growth and/or pathogenicity of many microorganisms is affected by inhibition of CAs.28–30 This can be explained by the participation of CAs enzymes in metabolic process control, or in the pH homeostasis, that are highly relevant in the bacteria.31,32 Chohan, et al.33 introduced a series of 4-hydroxycoumarin sulfonamides derivatives and studied their in vitro anti-bacterial activities for different Gram-positive (S. aureus, B. subtilis) and Gram-negative (S. flexneri, S. typhi, E. coli, P. aeruginosa) bacteria. The amino substituted ethyl coumarin benzenesulfonamide derivative IX (Fig. 2) exhibited the best activity against E. coli and S. flexenari with a zone inhibition (ZI) value of 10 and 09 mm, respectively, as compared to the standard reference drug imipenem which had a ZI values of 30 and 27 mm, respectively. Moreover, a series of thiazole derivatives were synthesized and evaluated for their antimicrobial activity and cytotoxicity. Compound Xa (Fig. 2) exhibited potent cytotoxicity more effective than cisplatin (IC50 = 13.7 ± 1.2 mg mL−1) and compound Xb was found to be the most promising antibacterial agent against Pseudomonas aeruginosa.34
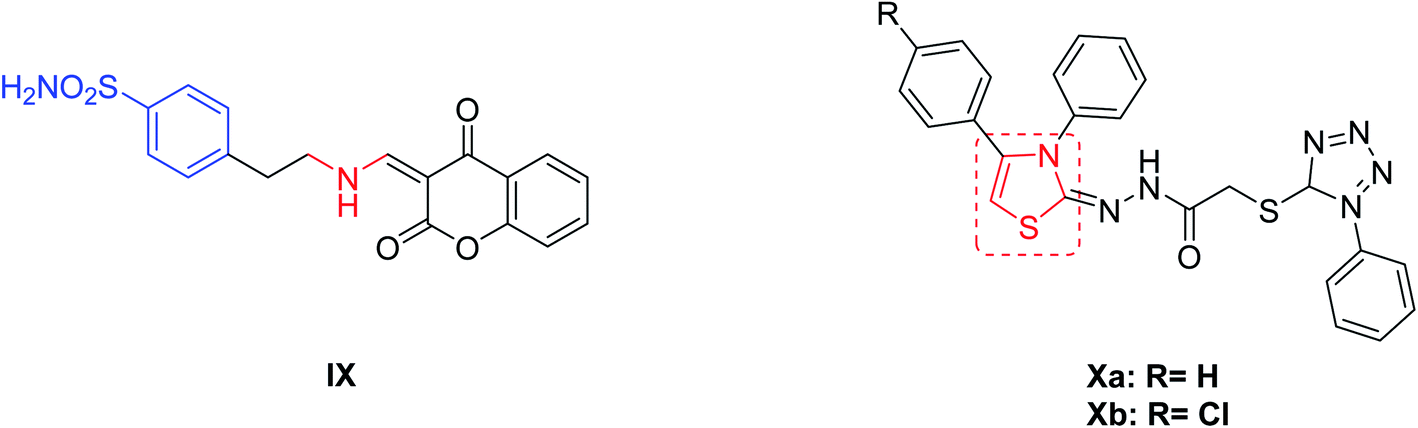 | ||
| Fig. 2 Reported benzenesulfonamides-based CAIs (IX) and thiazole derivatives (Xa,b) as antibacterial agents. | ||
From previously mentioned, subsequently, we represent the synthesis, and biological evaluation of a new series of 4-thiazolone-based benzenesulfonamides 4a–j with the “tail approach” (Fig. 3). It is based on appending “tails”, with a different chemical nature, to the aromatic or heterocyclic ring owning the zinc binding sulfonamide group, to interact with the middle and the rim parts of the active site cavity. Firstly, the bioisosteric replacement tactic was selected to replace the urea linker (compounds SLC-0111, Fig. 3) with the hetero 4-thiazolone that was expected to bind firmly with the active site's “spacer” region, also structural rigidification was employed to prevent the rotation and attains CA selectivity. Moreover, bioisosteric replacement was applied by using benzylidene tails incorporating different substituents to ensure appropriate SAR exploration concerning the hydrophobic region of the binding cleft.
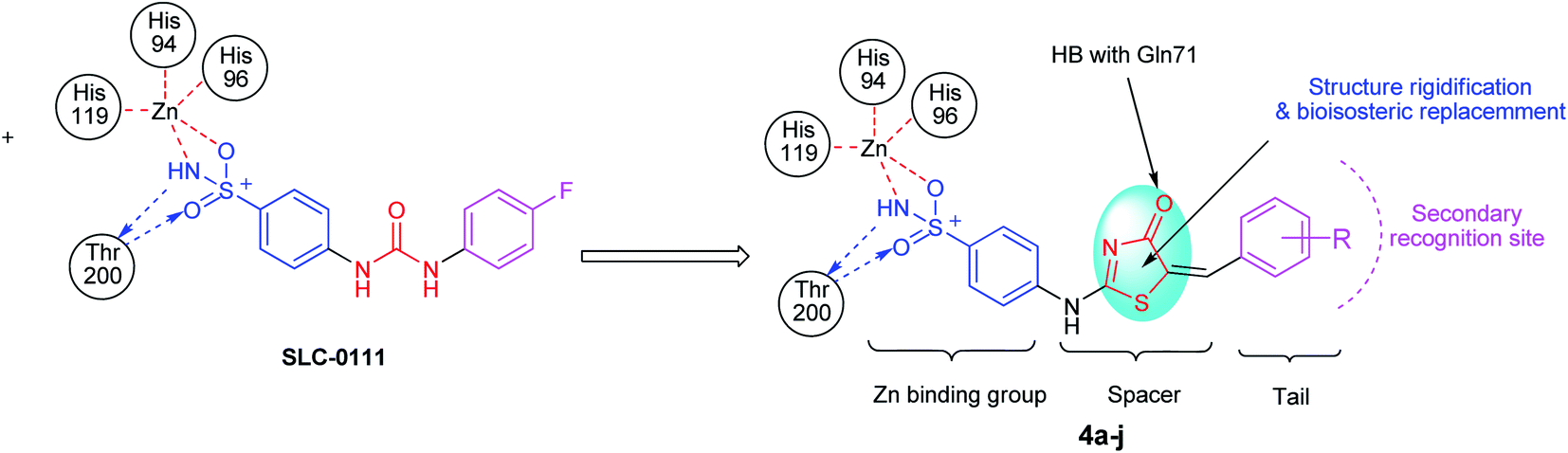 | ||
| Fig. 3 The design strategy for the new thiazolone–sulfonamide conjugates as selective hCA IX inhibitors. | ||
All the newly-synthesized 4-thiazolone-based benzenesulfonamides 4a–j were characterized and biologically for their anti-proliferative activity against breast cancer cell lines; MDA-MB-231 and MCF-7 in addition to normal breast cell line MCF-10A. Moreover, all the newly synthesized compounds were evaluated for their selective CA IX inhibitory effect to CA II, to gain insight and further explain the mechanism of the dual activity of the prepared hybrid structure as anticancer and antimicrobial agents. The most active derivative 5e was further examined for its apoptosis induction potential in MDA-MB-231 cells. Finally, docking study was performed to explore the affinity of the synthesized derivatives against CA IX in addition to ADMET profile to predict their pharmacokinetic properties.
2 Experimental
2.1 Chemistry
Melting points are uncorrected and have been measured on a Stuart melting point apparatus (Stuart Scientific, Redhill, UK). On the Shimadzu IR 435 spectrophotometer, the IR spectra (KBr) were calculated and values are shown in cm−1. Using a Varian Gemini 300-BB spectrophotometer, the 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were carried out (Bruker, Munich, Germany), using tetramethylsilane TMS as internal standard. Splitting patterns have been designated as follows: s: singlet; d: doublet; t: triplet; m: multiplet and chemical shift values are reported in ppm on δ scale. Progress of the reactions was tracked by TLC using TLC sheets precoated with Merck 60 F 254 UV florescent silica gel and visualized them using UV lamps. The chemicals used are supplied by Across from (New Jersey, USA). At the Microanalytical Center, Al-Azhar University, elemental analyses were performed.Yield 70%, m.p. 250–252 °C as reported.
2.1.3.1 (E)-4-(5-Benzylidene-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide 4a. Creamy solid: yield, 84%; m.p. 326 ± 2 °C; IR (KBr) νmax: 3313, 3284 (NH2), 3201 (NH), 2978 (CH aliph.), 1678 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) cm−1, 1H-NMR (DMSO-d6) δ: 7.02 (d, J = 8.0 Hz, 1H, Ar-H), 7.35 (s, 1H, CH), 7.42–7.64 (m, 5H, Ar-H), 7.68 (d, J = 8.0 Hz, 2H, Ar-H), 7.83 (d, J = 8.0 Hz, 1H, Ar-H), 8.00 (s, 2H, NH2), 12.35 (s, 1H, NH) ppm. MS (Mw: 359.04): m/z 360.94 (M + 2, 66.32%), 359.18 (43.74%), 329.61 (86.59%), 267.29 (100%); anal. calcd. For C16H13N3O3S2: C, 53.47; H, 3.65; N, 11.69. Found: C, 53.69; H, 3.91; N, 11.28% as reported.37
O) cm−1, 1H-NMR (DMSO-d6) δ: 7.02 (d, J = 8.0 Hz, 1H, Ar-H), 7.35 (s, 1H, CH), 7.42–7.64 (m, 5H, Ar-H), 7.68 (d, J = 8.0 Hz, 2H, Ar-H), 7.83 (d, J = 8.0 Hz, 1H, Ar-H), 8.00 (s, 2H, NH2), 12.35 (s, 1H, NH) ppm. MS (Mw: 359.04): m/z 360.94 (M + 2, 66.32%), 359.18 (43.74%), 329.61 (86.59%), 267.29 (100%); anal. calcd. For C16H13N3O3S2: C, 53.47; H, 3.65; N, 11.69. Found: C, 53.69; H, 3.91; N, 11.28% as reported.37
2.1.3.2 (E)-4-(5-(4-Hydroxybenzylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide 4b. Yellow solid: yield, 87%; m.p. 234 ± 2 °C; IR (KBr) νmax: 3441 (OH), 3348, 3255 (NH2), 3201 (NH), 2951 (CH aliph.), 1670 (C
O) cm−1, 1H-NMR (DMSO-d6) δ: 6.87 (d, J = 8.0 Hz, 1H, Ar-H), 6.95 (d, J = 8.0 Hz, 1H, Ar-H), 7.35 (s, 1H, CH), 7.49–7.58 (m, 3H, Ar-H), 7.65 (d, J = 8.0 Hz, 1H, Ar-H), 7.84 (d, J = 8.0 Hz, 1H, Ar-H), 7.91 (s, 1H, NH), 7.96 (d, J = 8.0 Hz, 1H, Ar-H), 10.41 (s, 2H, NH2), 12.17 (s, 1H, OH) ppm. 13C-NMR (DMSO-d6) δ: 116.8, 117.0, 117.1, 122.0, 124.4, 127.0, 127.7, 129.2, 132.5, 133.2, 134.4, 140.4, 144.9, 160.0, 165.6, 167.4 ppm. MS (Mw: 375.03): m/z 375.47 (M+, 23.67%), 344.80 (66.80%), 314.23 (49.20%), 300.56 (100%); anal. calcd. For C16H13N3O4S2: C, 51.19; H, 3.49; N, 11.19. Found: C, 50.97; H, 3.63; N, 11.45% as reported.37
2.1.3.3 (E)-4-(5-(4-Methoxybenzylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide 4c. Light brown solid: yield, 85%; m.p. 345 ± 2 °C; IR (KBr) νmax), 3329, 3251 (NH2), 3201 (NH), 2978 (CH aliph.), 1678 (C
O) cm−1, 1H-NMR (DMSO-d6) δ: 3.87 (s, 3H, OCH3), 7.05 (d, J = 8.0 Hz, 1H, Ar-H), 7.13 (d, J = 8.0 Hz, 1H, Ar-H), 7.18 (d, J = 8.0 Hz, 1H, Ar-H), 7.36 (s, 1H, CH), 7.46 (d, J = 8.0 Hz, 2H, Ar-H), 7.60 (d, J = 8.0 Hz, 2H, Ar-H), 7.84 (s, 2H, NH2), 7.95 (d, J = 8.0 Hz, 1H, Ar-H), 12.14 (s, 1H, NH) ppm. 13C-NMR (DMSO-d6) δ: 55.9, 115.4, 115.6, 120.8, 122.0, 125.9, 127.0, 127.1, 129.2, 132.2, 132.8, 133.9, 140.5, 144.9, 161.2, 161.8, 165.5 ppm. MS (Mw: 389.05): m/z 391.67 (M + 2, 16.26%), 389.82 (M+, 50.15%), 369.60 (81.39%), 283.86 (48.96%), 149.28 (100%); anal. calcd. For C17H15N3O4S2: C, 52.43; H, 3.88; N, 10.79. Found: C, 52.66; H, 4.05; N, 11.08%.
2.1.3.4 (E)-4-(5-(4-Chlorobenzylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide 4d. Yellow solid: yield, 81%; m.p. 314 ± 2 °C; IR (KBr) νmax), 3383, 3325 (NH2), 3251 (NH), 2978 (CH aliph.), 1674 (C
O) cm−1, 1H-NMR (DMSO-d6) δ: 7.19 (d, J = 8.0 Hz, 1H, Ar-H), 7.38 (s, 1H, CH), 7.51 (d, J = 8.0 Hz, 2H, Ar-H), 7.65 (d, J = 8.0 Hz, 1H, Ar-H), 7.70 (d, J = 8.0 Hz, 2H, Ar-H), 7.83 (d, J = 8.0 Hz, 1H, Ar-H), 7.95 (s, 2H, NH2), 8.00 (d, J = 8.0 Hz, 1H, Ar-H), 12.14 (s, 1H, NH) ppm. 13C-NMR (DMSO-d6) δ: 121.2, 122.0, 122.7, 127.0, 127.8, 129.2, 129.8, 130.0, 131.8, 132.3, 132.4, 135.0, 136.0, 140.9, 145.0, 167.2 ppm. MS (Mw: 393.00): m/z 394.78 (M + 2, 13.37%), 393.55 (M+, 39.40%), 389.85 (59.83%), 350.38 (62.60%), 236.22 (55.85%), 110.01 (100%); anal. calcd. For C16H12ClN3O3S2: C, 48.79; H, 3.07; N, 10.67. Found: C, 48.95; H, 3.20; N, 10.51%.
2.1.3.5 Synthesis of 5-allyl-2-hydroxy-3-methoxybenzaldehyde. POCl3 (0.1 mol) was added portion wise to DMF (10 mL) and stirred at room temperature for 30 minutes. Then, a solution of eugenol (0.1 mol) in DMF (1 mL) was added portion wise at room temperature. The whole mixture was stirred for 24 h and then poured onto ice (10 g). The product was neutralized with 40% NaOH and the resultant oil was separated and purified using column chromatography starting with petroleum to afford the 5-allyl-2-hydroxy-3-methoxybenzaldehyde.
2.1.3.6 (E)-4-(5-(5-Allyl-2-hydroxy-3-methoxybenzylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide 4e. Light brown solid: yield, 79%; m.p. 259 ± 2 °C; IR (KBr) νmax), 3523 (OH), 3360, 3271 (NH2), 3201 (NH), 2862 (CH aliph.), 1678 (C
O) cm−1, 1H-NMR (DMSO-d6) δ: 3.74 (d, J = 5.6 Hz, 2H, 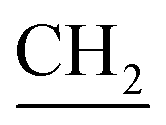 –CHCH2), 4.04 (s, 3H, OCH3), 5.08 (d, J = 8.2 Hz, 2H, CH2–CH
–CHCH2), 4.04 (s, 3H, OCH3), 5.08 (d, J = 8.2 Hz, 2H, CH2–CH ), 5.90 (m, 1H, CH2–
), 5.90 (m, 1H, CH2– CH2), 7.11 (d, J = 8.0 Hz, 1H, Ar-H), 7.22 (d, J = 8.0 Hz, 1H, Ar-H), 7.31 (s, 1H, CH), 7.46 (d, J = 8.0 Hz, 2H, Ar-H), 7.74 (d, J = 8.0 Hz, 2H, Ar-H), 7.80 (s, 2H, NH2), 7.95 (s, 1H, NH), 11.42 (s, 1H, OH) ppm. 13C-NMR (DMSO-d6) δ: 40.6, 58.4, 113.7, 112.7, 118.9, 121.8, 127.0, 127.5, 129.0, 130.2, 136.5, 140.1, 152.9, 157.1, 171.6, 172.4, 186.9 ppm. MS (Mw: 445.08): m/z 446.92 (M + 1, 9.33%), 413.56 (15.47%), 392.77 (61.79%), 388.09 (32.24%), 360.87 (17.87%), 326.78 (100%), 130.70 (51.36%); anal. calcd. For C20H19N3O5S2: C, 53.92; H, 4.30; N, 9.43. Found: C, 54.09; H, 3.87; N, 9.34%.
CH2), 7.11 (d, J = 8.0 Hz, 1H, Ar-H), 7.22 (d, J = 8.0 Hz, 1H, Ar-H), 7.31 (s, 1H, CH), 7.46 (d, J = 8.0 Hz, 2H, Ar-H), 7.74 (d, J = 8.0 Hz, 2H, Ar-H), 7.80 (s, 2H, NH2), 7.95 (s, 1H, NH), 11.42 (s, 1H, OH) ppm. 13C-NMR (DMSO-d6) δ: 40.6, 58.4, 113.7, 112.7, 118.9, 121.8, 127.0, 127.5, 129.0, 130.2, 136.5, 140.1, 152.9, 157.1, 171.6, 172.4, 186.9 ppm. MS (Mw: 445.08): m/z 446.92 (M + 1, 9.33%), 413.56 (15.47%), 392.77 (61.79%), 388.09 (32.24%), 360.87 (17.87%), 326.78 (100%), 130.70 (51.36%); anal. calcd. For C20H19N3O5S2: C, 53.92; H, 4.30; N, 9.43. Found: C, 54.09; H, 3.87; N, 9.34%.
2.1.3.7 (E)-4-((4-oxo-2-(4-Sulfamoylphenylamino)thiazol-5(4H)-ylidene)methyl)benzoic acid 4f. White solid: yield, 69%; m.p. 245 ± 2 °C; IR (KBr) νmax) 3518 (OH), 3344, 3255 (NH2), 3197 (NH), 2854 (CH aliph.), 1766, 1678 (2C
O) cm−1, 1H-NMR (DMSO-d6) δ: 5.52 (s, 1H, OH), 7.16 (d, J = 8.0 Hz, 1H, Ar-H), 7.32 (s, 1H, CH), 7.48 (m, 2H, Ar-H), 7.64 (d, J = 8.0 Hz, 1H, Ar-H), 7.82 (m, 3H, Ar-H), 7.86 (s, 2H, NH2), 7.96 (d, J = 8.0 Hz, 1H, Ar-H), 8.27 (s, 1H, NH) ppm. 13C-NMR (DMSO-d6) δ: 110.8, 122.0, 125.6, 127.7, 128.5, 130.0, 130.6, 131.4, 132.9, 140.5, 151.2, 168.2, 193.5 ppm. MS (Mw: 403.03): m/z 403.72 (M+, 23.08%), 394.62 (42.22%), 355.00 (67.74%), 320.27 (100%); anal. calcd. For C17H13N3O5S2: C, 50.61; H, 3.25; N, 10.42. Found: C, 50.85; H, 3.41; N, 10.33%.
2.1.3.8 (E)-4-(5-(4-Nitrobenzylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide 4g. Yellow solid: yield, 77%; m.p. 317 ± 2 °C; IR (KBr) νmax) 3417, 3314 (NH2), 3232 (NH), 2897 (CH aliph.), 1651 (C
O) cm−1, 1H-NMR (DMSO-d6) δ: 7.20 (d, J = 8 Hz, 1H, Ar-H), 7.35 (d, J = 8 Hz, 1H, Ar-H), 7.42 (s, 1H, CH), 7.51 (d, 2H, Ar-H), 7.77 (m, 4H, Ar-H), 7.98 (s, 2H, NH2), 8.02 (s, 1H, NH) ppm. MS (Mw: 404.02): m/z 404.38 (M+, 61.70%), 399.81 (24.26%), 351.23 (65.60%), 344.38 (67.88%), 218.86 (100%); anal. calcd. For C16H12N4O5S2: C, 47.52; H, 2.99; N, 13.85. Found: C, 47.68; H, 3.15; N, 14.02%.
2.1.3.9 (E)-4-(5-(4-Hydroxy-3-methoxybenzylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide 4h. Orange solid: yield, 82%; m.p. 263 ± 2 °C; IR (KBr) νmax) 3561 (OH), 3360, 3271 (NH2), 3201 (NH), 2862 (CH aliph.), 1678 (C
O) cm−1, 1H-NMR (DMSO-d6) δ: 4.04 (s, 3H, OCH3), 6.95 (d, J = 8.0 Hz, 1H, Ar-H), 7.35 (s, 1H, CH), 7.38 (m, 3H, Ar-H), 7.50 (s, 1H, OH), 7.55 (d, J = 8.0 Hz, 1H, Ar-H), 7.65 (d, J = 8.0 Hz, 1H, Ar-H), 7.84 (d, J = 8.0 Hz, 1H, Ar-H), 7.92 (s, 1H, NH), 7.99 (s, 2H, NH2) ppm. 13C-NMR (DMSO-d6) δ: 57.0, 116.8, 117.0, 117.2, 122.0, 124.4, 127.0, 127.5, 129.1, 132.5, 133.2, 134.5, 136.4, 140.3, 144.9, 160.0, 160.8 ppm. MS (Mw: 405.05): m/z 405.44 (M+, 58.86%), 476.91 (47.73%), 352.81 (96.34%), 329.33 (61.38%), 284.90 (100%); anal. calcd. For C17H15N3O5S2: C, 50.36; H, 3.73; N, 10.36. Found: C, 50.22; H, 3.94; N, 10.54%.
2.1.3.10 (E)-4-(4-oxo-5-(3,4,5-Trimethoxybenzylidene)-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide 4i. Yellow solid: yield, 71%; m.p. 275 ± 2 °C; IR (KBr) νmax) 3367, 3297 (NH2), 3255 (NH), 2839 (CH aliph.), 1681 (C
O) cm−1, 1H-NMR (DMSO-d6) δ: 3.72 (s, 3H, OCH3), 3.86 (s, 6H, OCH3), 7.05 (d, J = 8.0 Hz, 2H, Ar-H), 7.35 (s, 1H, CH), 7.50 (s, 1H, NH), 7.67 (d, J = 8.0 Hz, 1H, Ar-H), 7.85 (d, 2H, J = 8.0 Hz, Ar-H), 7.96 (d, J = 8.0 Hz, 1H, Ar-H), 8.00 (s, 2H, NH2) ppm. 13C-NMR (DMSO-d6) δ: 56.6, 60.7, 105.8, 108.2, 110.4, 112.6, 120.7, 127.1, 128.9, 129.2, 134.1, 136.2, 140.3, 145.0, 153.8, 159.5, 165.4, 167.2 ppm. MS (Mw: 449.07): m/z 449.36 (M+, 48.46%), 421.11 (30.76%), 295.15 (41.02%), 268.01 (40.96%), 183.35 (100%); anal. calcd. For C19H19N3O6S2: C, 50.77; H, 4.26; N, 9.35. Found: C, 51.03; H, 4.42; N, 9.47%.
2.1.3.11 (E)-4-(5-(4-(Dimethylamino)benzylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide 4j. Pale yellow solid: yield, 83%; m.p. 333 ± 2 °C; IR (KBr) νmax) 3356, 3305 (NH2), 3251 (NH), 2819 (CH aliph.), 1670 (C
O) cm−1, 1H-NMR (DMSO-d6) δ: 3.02 (s, 3H, CH3), 3.04 (s, 3H, CH3), 6.84 (d, J = 8.0 Hz, 1H, Ar-H), 7.34 (s, 1H, CH), 7.40 (m, 2H, Ar-H), 7.51 (d, J = 8.0 Hz, 2H, Ar-H), 7.63 (d, J = 8.0 Hz, 1H, Ar-H), 7.82 (s, 1H, NH), 7.86 (d, J = 8.0 Hz, 1H, Ar-H), 7.94 (d, J = 8.0 Hz, 1H, Ar-H), 7.99 (s, 2H, NH2) ppm. 13C-NMR (DMSO-d6) δ: 57.0, 116.8, 117.0, 117.2, 122.0, 124.4, 127.1, 127.5, 129.1, 132.5, 133.2, 134.5, 136.4, 140.3, 144.988, 160.02, 160.75 ppm. MS (Mw: 402.08): m/z 402.86 (M+, 72.86%), 354.24 (42.73%), 337.03 (61.34%), 323.27 (100%); anal. calcd. For C18H18N4O3S2: C, 53.71; H, 4.51; N, 13.92. Found: C, 53.60; H, 4.68; N, 14.08% as reported.37
2.2 Biological evaluation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well in a 96-well plate was evacuated to a centrifuge tube for 24 hours in a volume of 100 μL complete growth medium and 100 μL of the investigated substance per well before the MTT experiment. After that, the cell layer is rinsed with 0.25 percent (w/v) trypsin 0.53 mM EDTA solution to eliminate any traces of serum containing trypsin inhibitor. After adding 2.0 to 3.0 mL of trypsin EDTA solution to the flask, the cells were observed under an inverted microscope until the cell layer was dispersed, which normally took 5 to 15 minutes. A pipette was employed to gently aspirate cells after adding 6.0 to 8.0 mL of full growth media. The cell suspension was relocated to a another centrifuge tube containing the medium and cells from the first step to be centrifuged for 5 to 10 minutes at 125×g, with the supernatant discarded. In fresh growth medium, the cell pellet was resuspended. After that, the necessary aliquots of the cell suspension are added to fresh culture containers. After 24 hours of incubation at 37 °C, the cells were treated with serial quantities of the tested substance and incubated for 48 hours at 37 °C. After that, the plates were viewed under an inverted microscope and the MTT assay was carried out.38 For usage with −16 multiwall plates, the MTT technique of in vitro cytotoxicity control is well adapted. For best results, cells should be used in the log growth process, and the final cell density should not exceed 106 cells per cm2. Blanks should be comprised in each test to provide a full medium without cells. Take away the cultures from the incubator and place them in a sterile work environment, such as a laminar flow hood. To use, each vial of MTT [M-5655] was reconstituted with 3 mL of medium, excluding phenol red and serum. Reconstituted MTT was added to the culture medium in a volume equal to 10% of the total volume. Return the cultures to the incubator for another 2–4 hours, based on the cell type and density.
000 cells per well in a 96-well plate was evacuated to a centrifuge tube for 24 hours in a volume of 100 μL complete growth medium and 100 μL of the investigated substance per well before the MTT experiment. After that, the cell layer is rinsed with 0.25 percent (w/v) trypsin 0.53 mM EDTA solution to eliminate any traces of serum containing trypsin inhibitor. After adding 2.0 to 3.0 mL of trypsin EDTA solution to the flask, the cells were observed under an inverted microscope until the cell layer was dispersed, which normally took 5 to 15 minutes. A pipette was employed to gently aspirate cells after adding 6.0 to 8.0 mL of full growth media. The cell suspension was relocated to a another centrifuge tube containing the medium and cells from the first step to be centrifuged for 5 to 10 minutes at 125×g, with the supernatant discarded. In fresh growth medium, the cell pellet was resuspended. After that, the necessary aliquots of the cell suspension are added to fresh culture containers. After 24 hours of incubation at 37 °C, the cells were treated with serial quantities of the tested substance and incubated for 48 hours at 37 °C. After that, the plates were viewed under an inverted microscope and the MTT assay was carried out.38 For usage with −16 multiwall plates, the MTT technique of in vitro cytotoxicity control is well adapted. For best results, cells should be used in the log growth process, and the final cell density should not exceed 106 cells per cm2. Blanks should be comprised in each test to provide a full medium without cells. Take away the cultures from the incubator and place them in a sterile work environment, such as a laminar flow hood. To use, each vial of MTT [M-5655] was reconstituted with 3 mL of medium, excluding phenol red and serum. Reconstituted MTT was added to the culture medium in a volume equal to 10% of the total volume. Return the cultures to the incubator for another 2–4 hours, based on the cell type and density.When the incubation period had ended, crops were taken from the incubator then an amount of MTT Solubilization Solution [M-8910] equivalent to the original culture's were added to the formazan crystals. In a gyratory shaker, gentle mixing is recommended to improve dissolution. Trituration may be necessary on occasion, especially in thick cultures, to guarantee that the MTT formazan crystals are completely dissolved. The absorbance is measured at wavelength 570 nm with a spectrophotometer. Background absorbance of 16 multiwall plates was measured at 690 nm and was subtracted from the 570 nm reading.39 The concentration range used to determine IC50 was 100 μM, 25 μM, 6.25 μM, 1.56 μM, 0.39 μM.
000 rpm for 15 min. 100 μg of the cell extract was each acidified by 6N HCl (1:1 v/v) and vortexed for 30 s. 500 μL of the extracting buffer was then added to each of the acidified cell extracts, and samples were vortexed again and shaken in the Orbited shaker (at 100 rpm) for 15 min. The upper organic layer obtained after centrifugation at 18000 rpm for 20 min was filtered by a membrane filter. Finally this organic layer was transferred to a clean injection HPLC sample vials of about 100 μL for quantitative analysis using HPLC.
2.2.2.1 HPLC analysis of compounds 4e, 4g and 4h. The HPLC system used to carry out the HPLC analysis of compounds 4e, 4g and 4h was a Dionex Ultimate 3000 UHPLC equipped with auto-sampler, quaternary solvent delivery pump and diode array detector (Germany). The software used was Chromeleon software. The stationary phase used was ZORBAX Eclipse Plus® C18 column with dimensions of (250 × 4.6 mm, 5 μm) (California, USA). A mobile phase, which was composed of methanol
:water (90:10, v/v), was used, and the samples were each injected at an injection volume of 20 μL. Each sample was injected in triplicate and UV scanning was carried out at 254 nm.
| % Relative activity = ΔAb of S × 100 |
ΔAb of EC
| % Relative inhibition = ΔAb of EC − ΔAb of S × 100 |
ΔAb of EC
At 37 °C for 24 h, the plates were incubated overnight. The absorbance at 600 nm for bacteria and 340 nm for C. albicans was measured after incubation. Using a Spectrostar Nano Microplate Reader Albicans (BMG LABTECH GmbH, Allmendgrun, Germany). As a control, ciprofloxacin and nystatin were used.
3 Results and discussion
3.1 Chemistry
The design in this study is based in using SLC-0111 as a lead compound in which its ureido moiety is replaced by thiazol-4-one heterocyclic five membered ring that integrates the thioureido moiety involved in the reported lead compound. In this work, the design of synthesis of benzenesulfonamide derivatives containing thiazol-4-one scaffold was shown in Scheme 1. 4-((4-oxo-4,5-Dihydrothiazol-2-yl)amino)benzenesulfonamide 3 was prepared via intramolecular cyclization rearrangement reaction of previously prepared chloroacetamide derivative 2 when treated with ammonium thiocyanate in ethanol.23,47 Furthermore, intermediate 3 underwent coupling reaction with selected aromatic aldehydes on the active methylene group of compound 3 to afford 5-aryl-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamides 4a–j. The structure of the synthesized compounds 4a–j was confirmed by different spectral data. 1H-NMR spectrum of compounds 4a–j showed the disappearance of the characteristic dd signal corresponds to CH2 of the thiazol-4-one ring and the appearance of the characteristic singlet signal of CH proton at 7.31–7.38 ppm. Additionally, 1H-NMR spectrum revealed additional protons due to the presence of additional aromatic protons signals related to aromatic aldehydes. Moreover, 13C-NMR revealed the appearance of the characteristic signal of methine carbon at 145.0–161.2 ppm in addition to additional aromatic carbons which appeared in the aromatic region corresponding to substituted aromatic aldehyde moieties.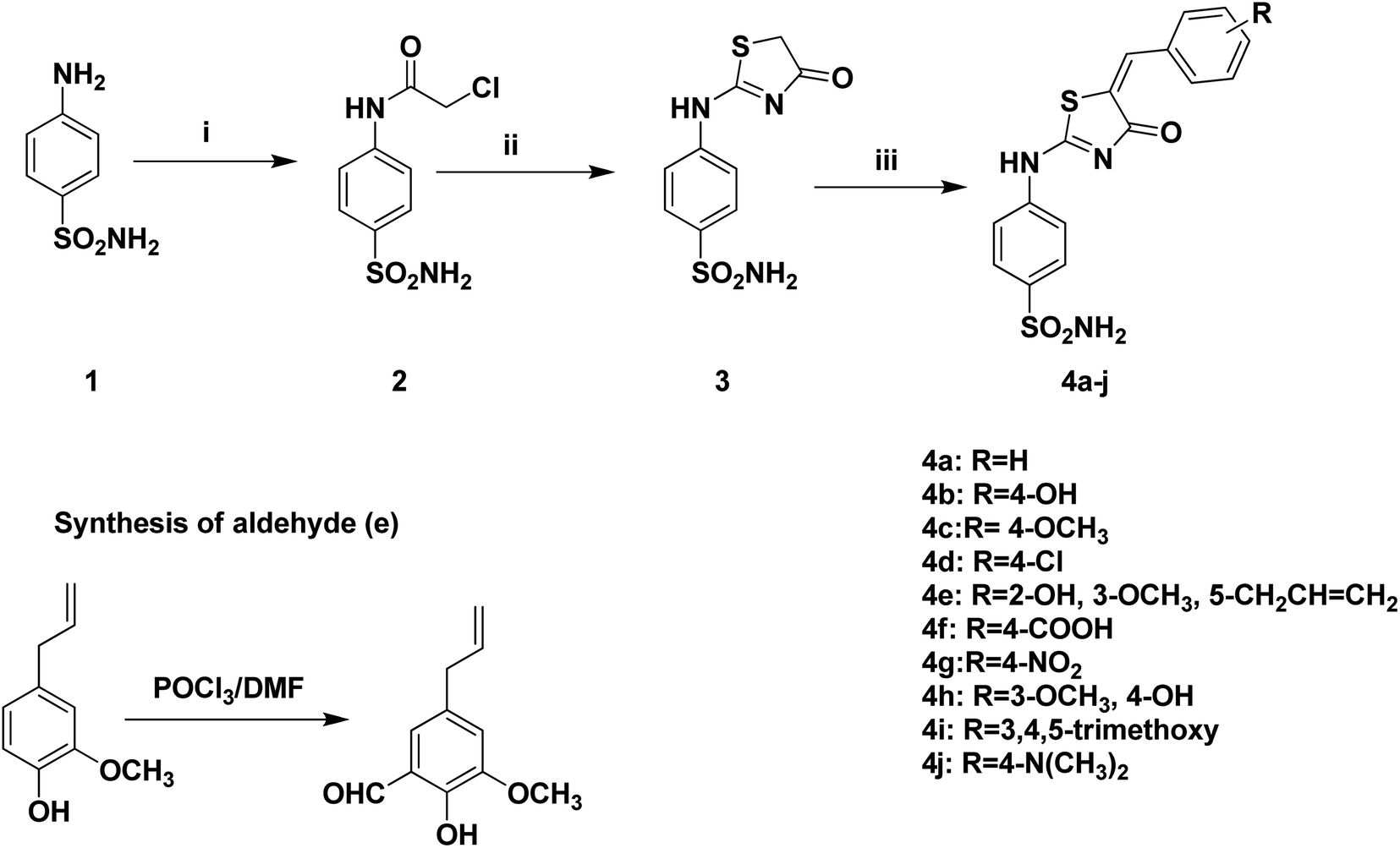 | ||
| Scheme 1 Synthesis of Benzenesulfonamides compounds 4a–j. (i) Chloroacetyl chloride, dry DMF, r.t., 2 h; (ii) ammonium thiocyanate, absolute ethanol, reflux, 1 h; (iii) appropriate aldehydes, sodium acetate, acetic acid, reflux, 24 h. | ||
3.2 Biological evaluation
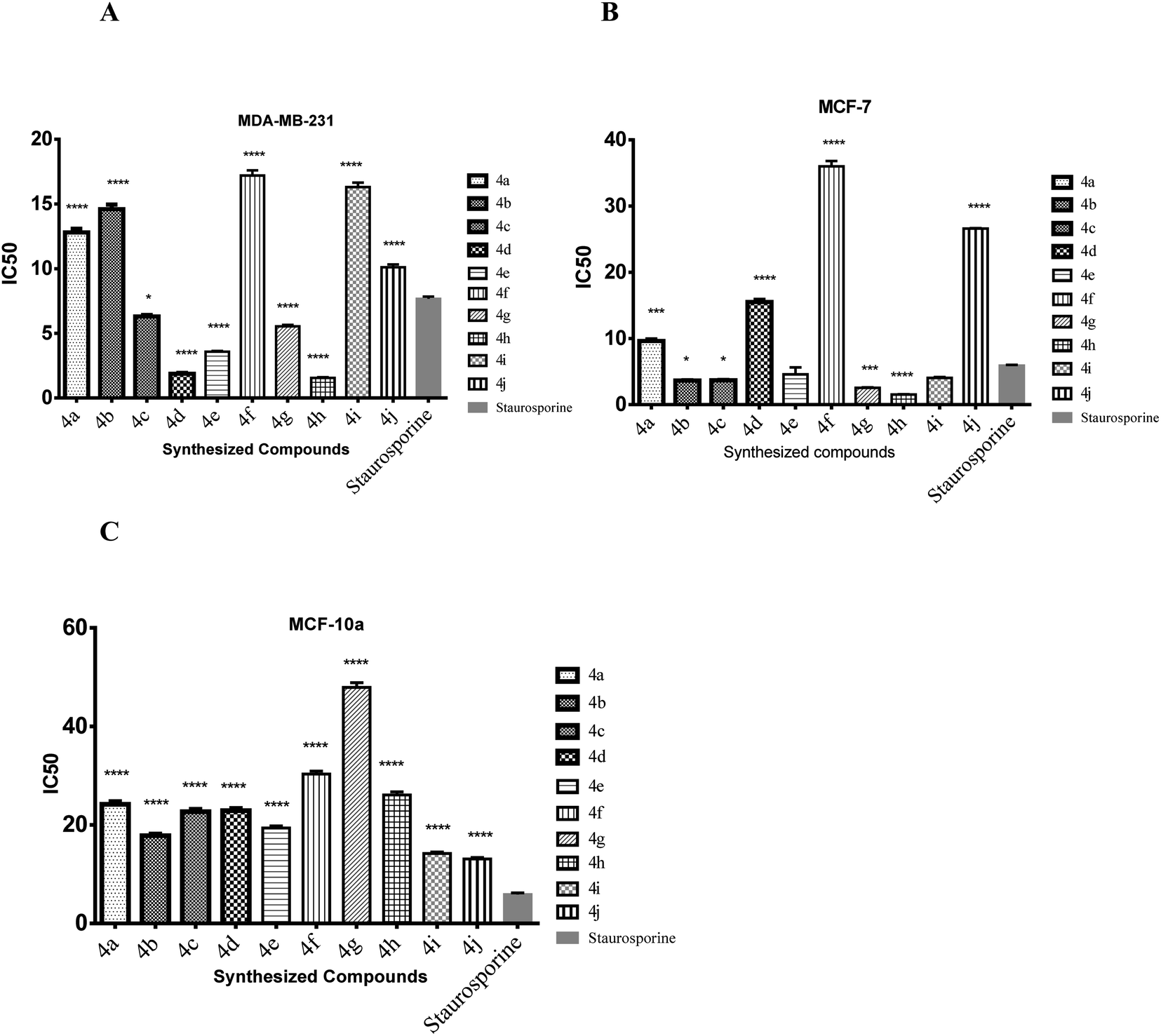 | ||
| Fig. 4 The bar graphs showing the IC50 of the target compounds ± standard error compared to staurosporine against MDA-MB-231 cell lines. ****High significantly different from staurosporine at p < 0.05, ***moderate significantly different from staurosporine at p < 0.05. *Mild significantly different from staurosporine at p < 0.05. | ||
| Compound no. | Cytotoxicity IC50 μM ± S.D. | ||
|---|---|---|---|
| MDA-MB-231 | MCF-7 | MCF 10A | |
| 4a | 12.8 ± 0.5 | 9.63 ± 0.4 | 24.2 ± 0.9 |
| 4b | 14.6 ± 0.6 | 3.63 ± 0.1 | 17.8 ± 0.6 |
| 4c | 6.31 ± 0.2 | 3.67 ± 0.1 | 22.7 ± 0.8 |
| 4d | 1.88 ± 0.1 | 15.5 ± 0.6 | 22.9 ± 0.8 |
| 4e | 3.58 ± 0.1 | 4.58 ± 1.8 | 19.4 ± 0.7 |
| 4f | 17.2 ± 0.7 | 36 ± 1.4 | 30.3 ± 1.1 |
| 4g | 5.54 ± 0.2 | 2.55 ± 0.1 | 47.9 ± 1.7 |
| 4h | 1.56 ± 0.1 | 1.52 ± 0.1 | 26.1 ± 1.1 |
| 4i | 16.3 ± 0.6 | 4.05 ± 0.2 | 14.2 ± 0.5 |
| 4j | 10.1 ± 0.4 | 26.6 ± 1.0 | 13.1 ± 0.5 |
| Staurosporine | 7.67 ± 0.3 | 5.89 ± 0.2 | 13.0 ± 0.5 |
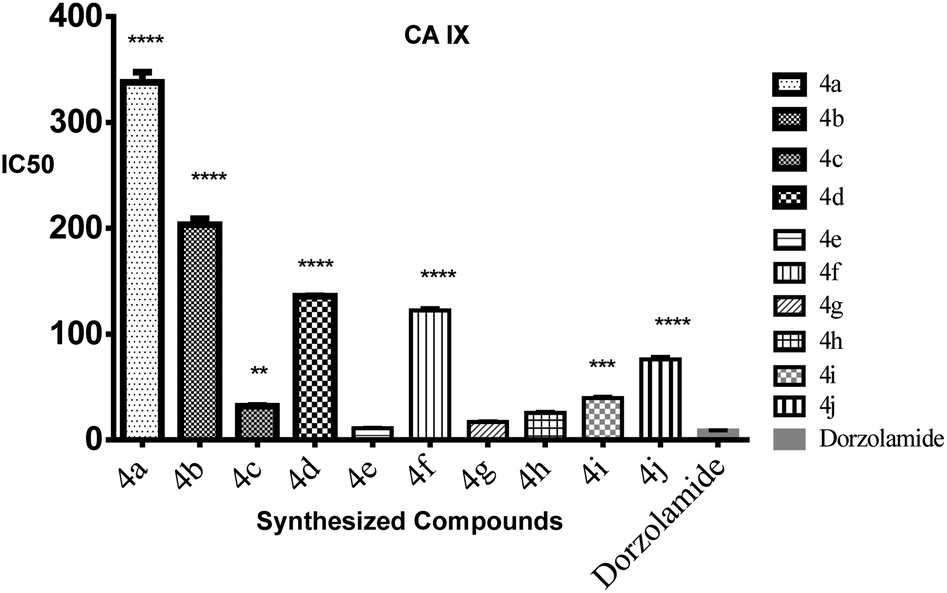 | ||
| Fig. 5 The bar graphs showing the IC50 of the target compounds ± standard error compared to Dorzolamide against CA IX cell lines. A: ****high significantly different from dorzolamide at p < 0.05, ***: moderate significantly different from dorzolamide at p < 0.05, **: mild significantly different from dorzolamide at p < 0.05. | ||
| Compound no. | CA IX IC50 (μM) | CA II IC50 (μM) |
|---|---|---|
| 4e | 0.011 | 3.92 |
| 4g | 0.017 | 1.55 |
| 4h | 0.026 | 2.19 |
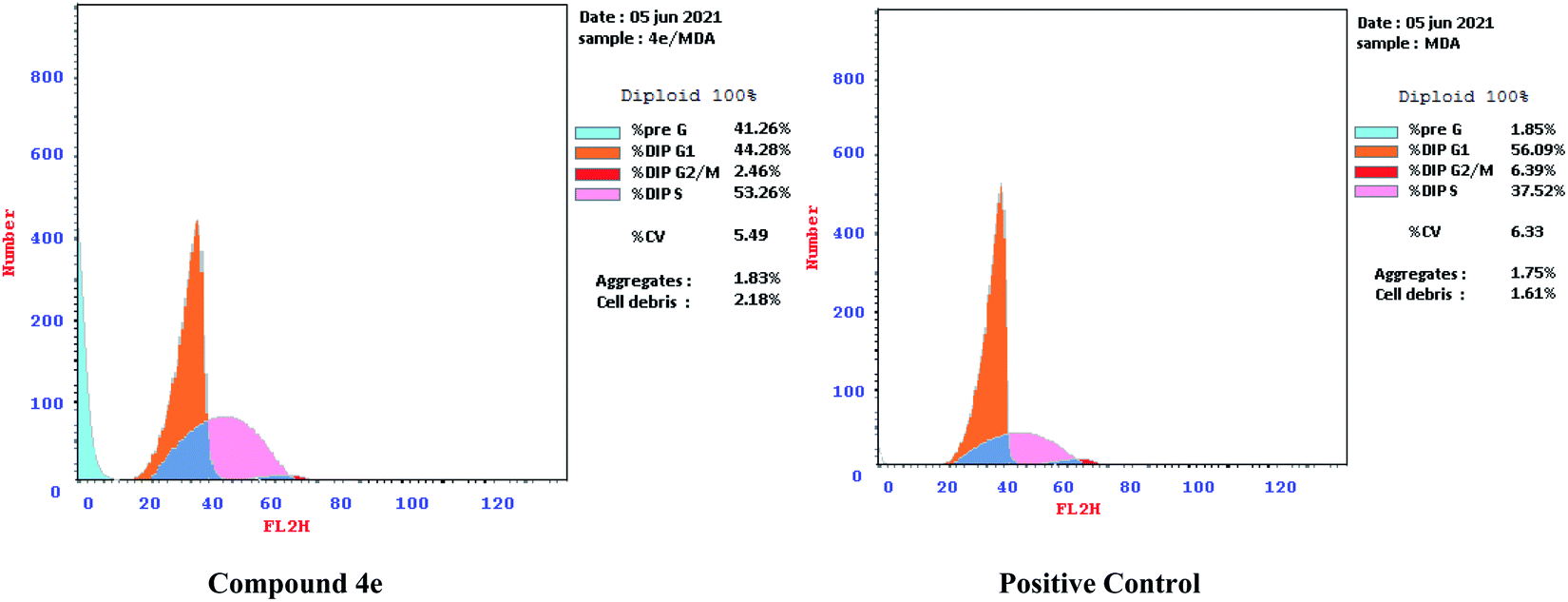 | ||
| Fig. 6 Effect of compound 4e on the phases of cell cycle of MDA-MB-231 cell lines. | ||
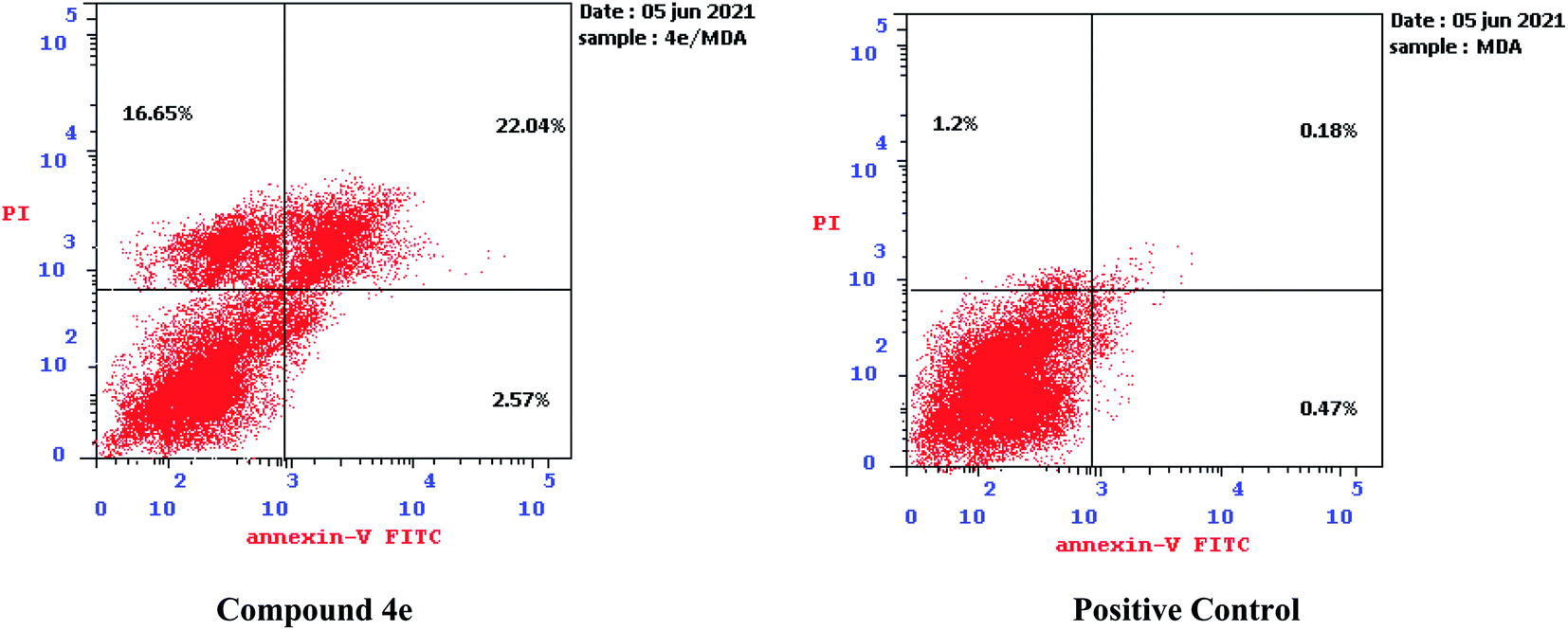 | ||
| Fig. 7 Effect of compound 4e on the percentage of annexin V-FITC-positive staining against MDA-MB-231. | ||
The outcomes showed that compound 4e had the ability to induce apoptosis against MDA-MB-231 cells that is evidenced by the considerable increase in the percent of annexin V-FITC-positive apoptotic cells, appearing in the late apoptotic phase (from 0.18% to 22.04%) in addition to necrosis phase (from 1.20% to 16.65%), which signifies about 22 fold total increase when compared to positive control.
| Test microbesa (inhibition ratio%) | |||||||
|---|---|---|---|---|---|---|---|
| Compound no. | K. pneumonia | E. coli | P. vulgaris | S. aureus | S. typhimurium | P. aeruginosa | C. albicans |
| a Cip: ciprofloxacin, Nys: nystatin, NA: not active. | |||||||
| 4a | NA | NA | NA | NA | NA | 15.33 | NA |
| 4b | NA | NA | NA | NA | NA | 18.61 | 8.87 |
| 4c | 14.43 | 25.68 | 0.00 | 15.58 | NA | 27.37 | NA |
| 4d | 59.28 | NA | 33.47 | 72.62 | 64.68 | 40.88 | NA |
| 4e | 62.89 | 60.66 | 41.95 | 80.69 | 75.09 | 32.12 | NA |
| 4f | 26.80 | 46.45 | 3.39 | 57.93 | 45.72 | NA | NA |
| 4g | NA | NA | 23.31 | 69.74 | 60.97 | 73.36 | NA |
| 4h | 33.51 | 29.51 | 19.07 | 68.30 | 59.11 | 27.01 | NA |
| 4i | NA | NA | NA | NA | NA | NA | NA |
| 4j | NA | NA | 5.08 | NA | NA | 37.59 | NA |
| Cip | 99.7 | 99.1 | 99.78 | 99.25 | 99.56 | 99.25 | — |
| Nys | — | — | — | — | — | — | 99.21 |
| Biofilm inhibition ratio% | ||||||
|---|---|---|---|---|---|---|
| Compound no. | K. pneumonia | E. coli | P. vulgaris | S. aureus | S. typhimurium | P. aeruginosa |
| 4a | NA | NA | 16.30 | NA | NA | NA |
| 4b | 78.29 | 18.78 | 16.85 | 53.06 | NA | 45.51 |
| 4c | 54.26 | NA | NA | NA | NA | NA |
| 4d | NA | NA | NA | NA | NA | NA |
| 4e | 18.60 | 15.47 | 61.41 | 45.92 | 63.43 | NA |
| 4f | 10.08 | 19.89 | 64.67 | 21.43 | 36.57 | NA |
| 4g | 79.46 | NA | NA | 35.24 | NA | NA |
| 4h | 77.52 | 19.89 | 50.54 | 19.39 | 66.86 | 35.96 |
| 4i | 5.43 | 49.72 | 7.61 | 22.55 | 22.86 | NA |
| 4j | NA | NA | NA | NA | NA | NA |
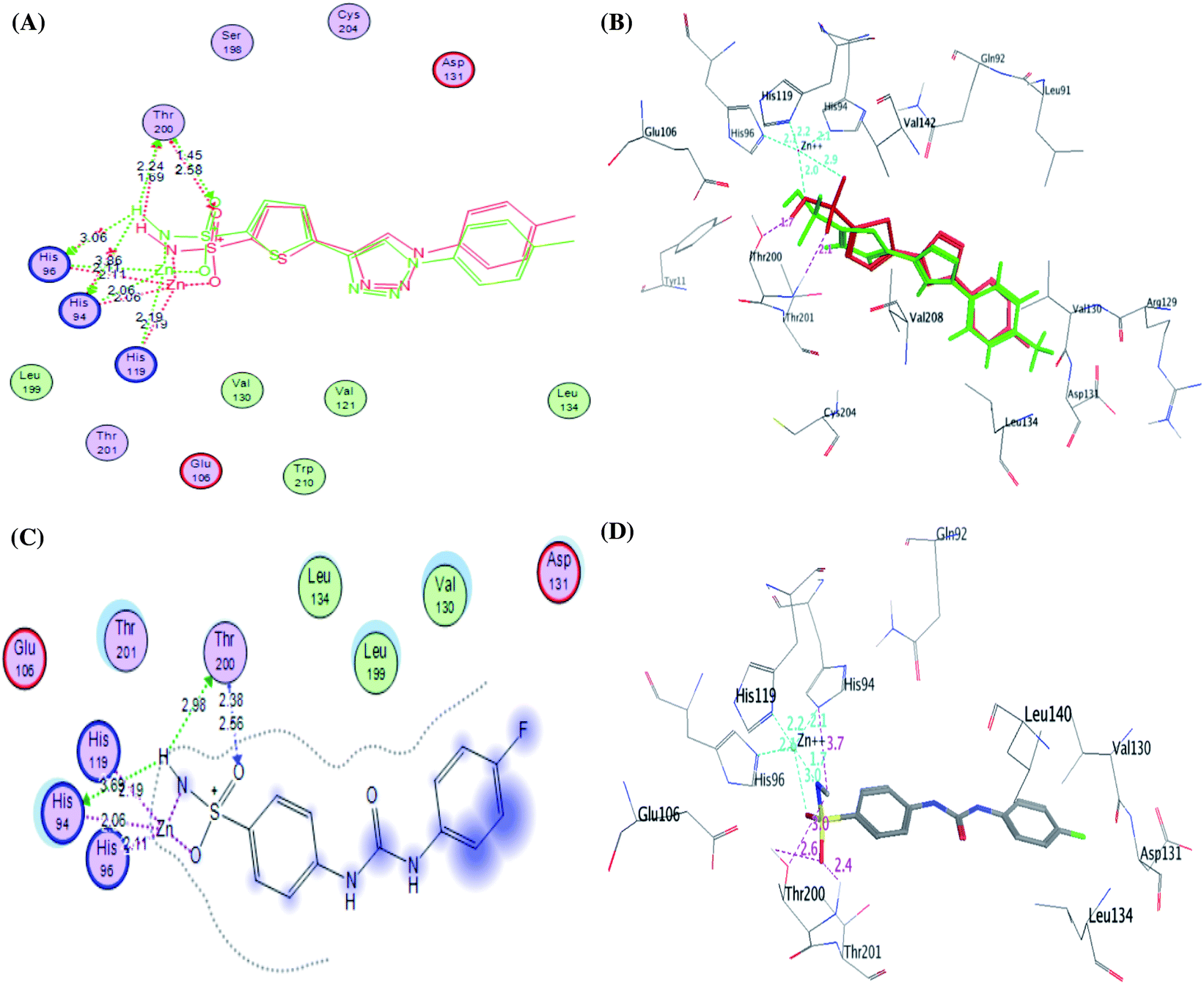 | ||
| Fig. 8 2D diagram (A) and 3D diagram (B) of the superimposed of the co-crystallized (red) and the docking pose (blue) of native ligand within active site of CA IX with RMSD of 1.41 Å. In addition, 2D diagram (C) and 3D representation (D) of compound SLC-0111 within the CA IX binding site. | ||
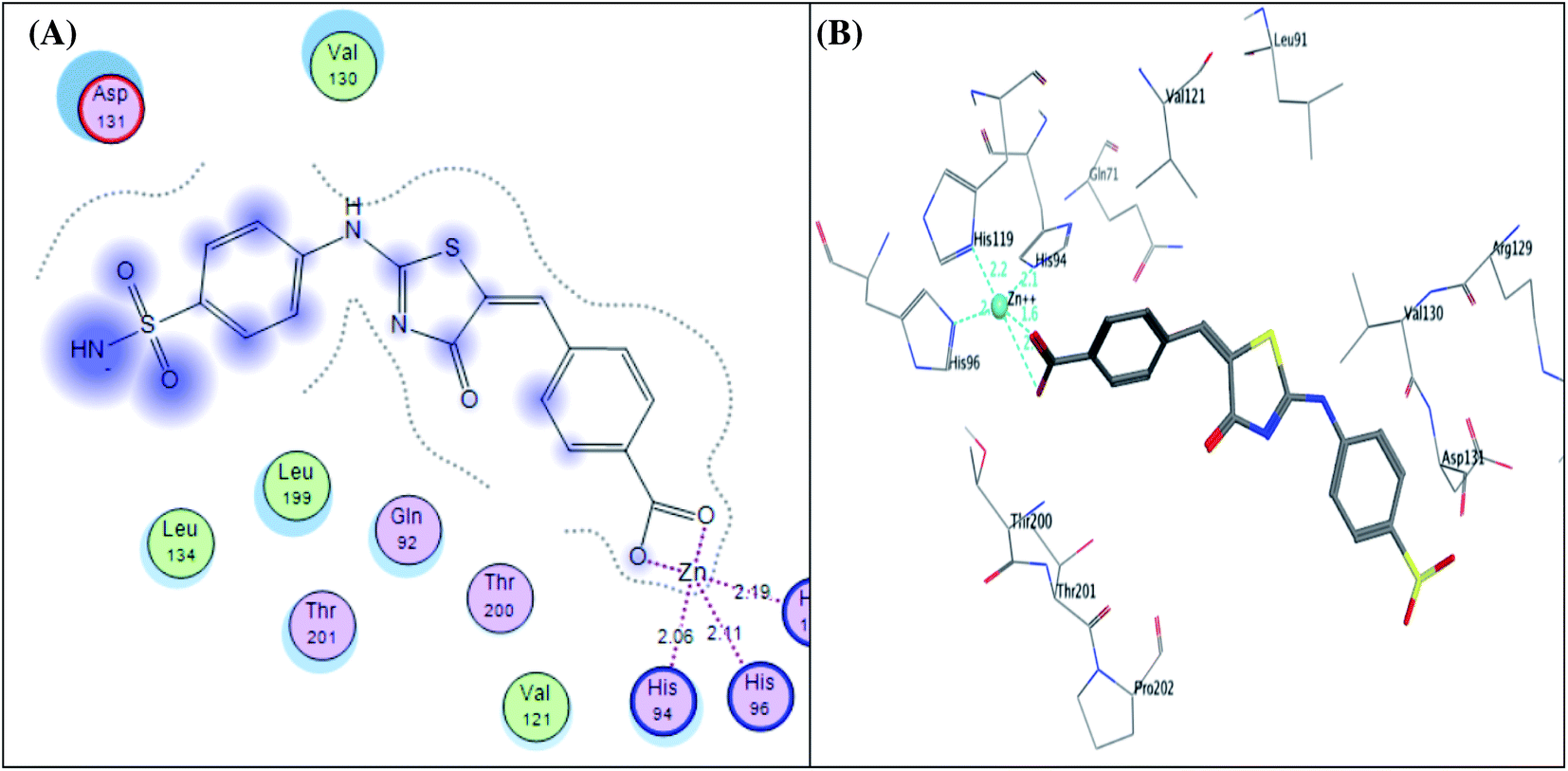
The CA IX catalytic pocket is reportedly as cone-shaped cavity, which is segregated into two characteristic conserved environments as mentioned before. So, docking results of the active analogues 4e, 4g and 4h suggested that they all fit accurately within the active site involving some interactions including coordination bonds and hydrogen bonds. Generally, it was recognized that the top docked poses have a common binding configuration and orientation that the phenyl-bearing sulfonamide moiety in the mentioned derivatives aligned towards His94, His96, His119, Thr200, Thr201, Tyr11 and Glu106 amino acids residues while the benzylidene moieties were extended towards Arg129, Val130, Asp131 and Leu134 residues forming some H bond interactions. Their remarkable potency against CA IX enzyme may be significantly due to the apparent similarity between the predicted binding patterns of these analogues and SLC-0111, the results of the docking studies are represented in Table 5.
| Cpd no. | S scorea kcal mol−1 | Amino acid/bond | Distance (Å) |
|---|---|---|---|
| a S: the score of placement of a compound into the binding pocket of protein using London dG scoring function. | |||
| 4e | −15.5765 | His94/Zn-coordination bond | 2.06 |
| His96/Zn-coordination bond | 2.11 | ||
| His119/Zn-coordination bond | 2.19 | ||
| His94/H-donor | 2.49 | ||
| Thr200/H-acceptor | 2.07 | ||
| Thr200/H-acceptor | 3.16 | ||
| Asp131/H-donor | 2.11 | ||
| Asp131/H-acceptor | 2.46 | ||
| 4f | −9.5118 | His94/Zn-coordination bond | 2.06 |
| His96/Zn-coordination bond | 2.11 | ||
| His119/Zn-coordination bond | 2.19 | ||
| 4g | −12.4361 | His94/Zn-coordination bond | 2.06 |
| His96/Zn-coordination bond | 2.11 | ||
| His119/Zn-coordination bond | 2.19 | ||
| His96/H-acceptor | 3.03 | ||
| Thr200/H-acceptor | 2.29 | ||
| Thr200/H-acceptor | 2.63 | ||
| Gln71/H-acceptor | 1.71 | ||
| 4h | −10.5835 | His94/Zn-coordination bond | 2.06 |
| His96/Zn-coordination bond | 2.11 | ||
| His119/Zn-coordination bond | 2.19 | ||
| Thr200/H-donor | 2.07 | ||
| Thr200/H-acceptor | 2.47 | ||
| Asp131/H-acceptor | 3.75 | ||
| SLC-0111 | −11.0025 | His94/Zn-coordination bond | 2.06 |
| His96/Zn-coordination bond | 2.11 | ||
| His119/Zn-coordination bond | 2.19 | ||
| His94/H-donor | 2.69 | ||
| Thr200/H-donor | 2.98 | ||
| Thr200/H-acceptor | 2.38 | ||
| Thr200/H-acceptor | 2.56 | ||
It was conceptualized that the type and number of these interactions could explain the variation between these ligands in their potency. The most active predicted eugenol derivative 4e (IC50 = 10.93 ± 0.53 nM) exhibited the highest number and types of interactions. It formed five different H-bonds; two H-bonds between the hydroxyl and methoxy groups of eugenol moiety and Asp131, one H-bond between NH group of sulphonamide and His94, two H-bonds between oxygen group of sulphonamide and Thr200. Furthermore, the sulfonamide group showed coordination bonds with Zn2+ ion and three histidine residues (His94, His96, and His119) as shown in Fig. 9A and B. Compared to eugenol derivative 4e, the decreased inhibition of p-nitrobenzylidene derivative 4g against CA IX (IC50 = 16.96 ± 0.83 nM) could be explained by the absence of some interactions such as two H-bonds with Asp131 indicating that the replacement of the EDG (OCH3 and OH) with EWG (P-NO2) resulted in the absence of an important H-bond with Asp131 amino acid in the active site of CA IX enzyme, on the other side, carbonyl group of thiazol-4-one moiety showed H-bond with Gln71 (Fig. 9C and D). However, 4-hydroxy-3-methoxybenzylidene derivative 4h showed the least activity (IC50 = 25.56 ± 1.25 nM) between the most active ones due to the reduction in the number of interactions despite of adopting a similar orientation (Fig. 9E and F).
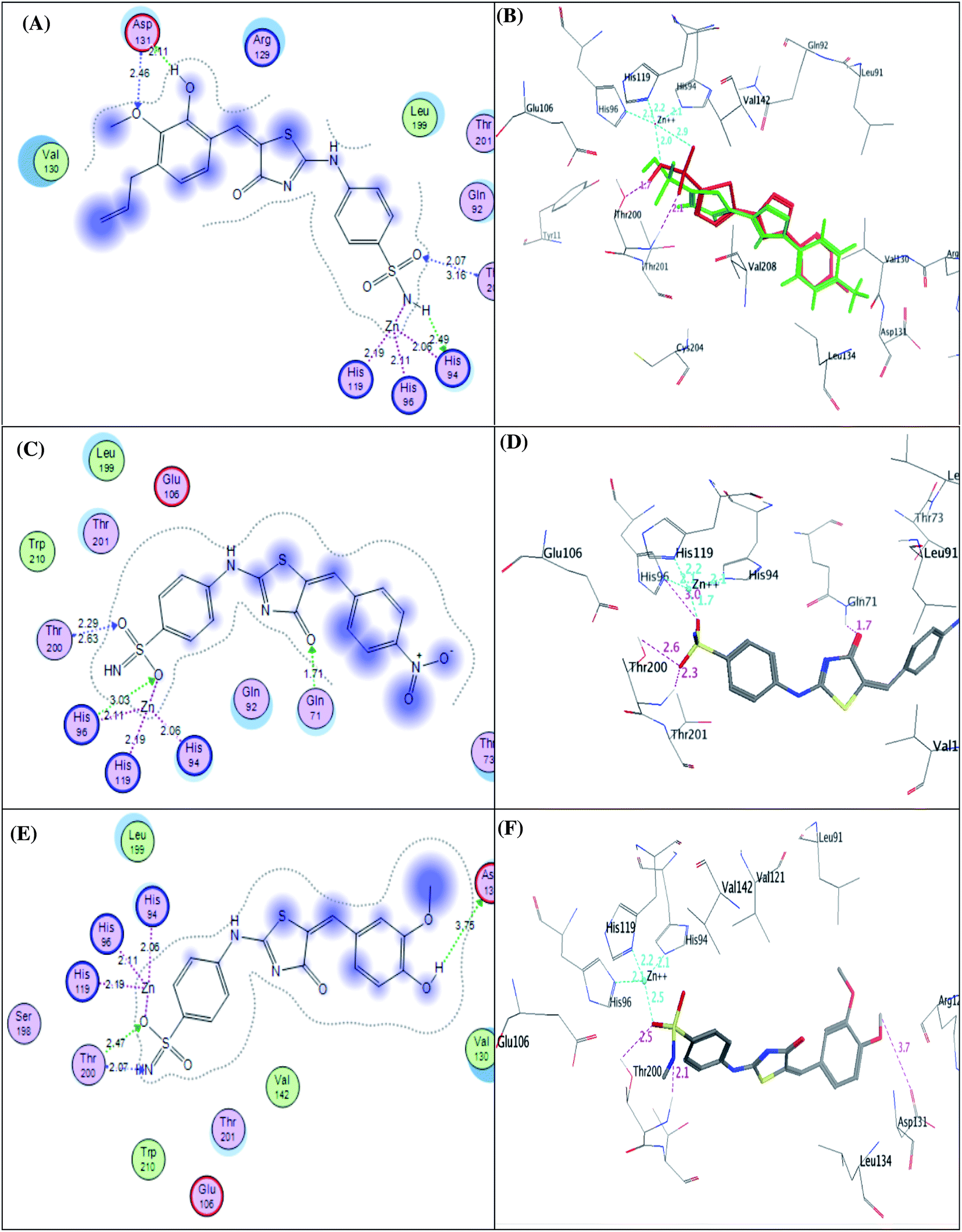 | ||
| Fig. 9 2D diagram (A) and 3D pose (B) of compound 4e, 2D diagram (C) and 3D pose (D) of compound 4g, and 2D diagram (E) and 3D pose (F) of compound 4h within binding site of CA IX binding site. | ||
Furthermore, the exploration of the docking outcomes of the least active compound 4f into the CA IX active pocket indicated that compound 4f possessed a totally opposing orientation and dispositioning compared with the previously docked active compounds (4e, 4g and 4h) as shown in Fig. 10. It was found that the benzoic acid group was located near His94, His96, His119, Thr200, Thr201, Tyr11 and Glu106 residues and exhibited coordination bonds with Zn2+ ion and three histidine residues (His94, His96, and His119), while the benzenesulfonamide core was extended towards Arg129, Val130, Asp131 and Leu134 residues and don't exhibited any interactions mirrored in a decrease in the CA IX inhibitory activity. As a result, docking study was coherent with the in vitro assay results, verifying that hybridization between the phenyl-bearing sulfonamide pharmacophoric and the thiazole-4-one CA IX moieties are promising leads for further optimization.
 | ||
| Fig. 10 2D diagram (A) and 3D pose (B) of compound 4f within the binding site of CA IX. | ||
P) revealed that all the studied analogues had a good lipophilicity as their logP values were less than 5 (ref. 51) (Table S2†). The predicted lipophilicities are negatively associated with their water solubility potentials but have a correlation with caco-2 permeability. Absorption is a very complex process that is defined as movement of a drug from an extravascular site of administration into the systemic circulation depending on permeability and solubility of the compound.52 For oral drugs, absorption occurs by either transcellular diffusion, paracellular diffusion, and transporter-mediated mechanisms and caco-2 cell lines53 was used to estimate the latter mechanism. Lagorce et al.54 reported that enzyme inhibitors showed lower caco-2 permeability than ion channels and nuclear receptors. Thus, this may explain the low absorption properties of the examined compounds. Furthermore, caco-2 permeability and human intestinal absorption have a role in determining the bioavailability of the drugs. From the results represented, all the examined derivatives illustrated an elevated cellular permeability prediction, particularly compound 4g that displayed 81.72% intestinal absorption (Table S2†). Skin permeability is a crucial consideration for the development of transdermal drug delivery. All the synthesized compounds showed good skin permeability. The three derivatives were found to be substrate of P-glycoprotein. Yet, neither of the evaluated derivatives were expected as P-glycoprotein II inhibitors. Additionally, the distribution of drug refers to its distribution throughout the body. Some parameters were investigated in silico regarding distribution that include blood brain barrier (BBB) permeability, and central nervous system (CNS) permeability. Also, the free (unbound) drug is also important to be monitored due to its availability to interact with the protein target.55 Prediction of distribution descriptors revealed that the synthesized analogues showed desirable theoretical dose related to uniform plasma distribution. Moreover, all the estimated benzenesulfonamides based derivatives expressed poor brain distribution but are capable of penetrating CNS (Table S2†). In the same context, metabolism is the biotransformation of the drug to facilitate tis excretion. Thus, metabolism prediction showed that all synthesized analogues showed low CYP promiscuity, however compound 4e can inhibit CYP2C19 and CYP2C9 while 4g analogue inhibit CYP3A4, however all compounds are CYP3A4 substrate. These findings indicated that the mentioned derivatives could have a role in drug–drug interactions and oxidative stress could be initiated. Analogue 4h is expected to be a Renal OCT2 substrate with respect to excretion prediction. Finally, toxicological predictions of the synthesized analogues were estimated using pkCSM software. Herein, via AMES toxicity testing, the bacterial mutagenic ability of the benzenesulfonamide derivatives showed that all analogues could be regarded as non-mutagenic agents. Nevertheless, the toxicity of all the analogues in T. pyriformis are high.Moreover, one of the significant parameters of toxicity, which is cardiotoxicity, was predicted for the investigated compounds; human ether-a-go-go-go-related gene I, II (hERG I, II). The predicted outcomes presented that the estimated compuds didn't inhibit hERG I, II, except for vanillin derivative 4e that inhibits hERG II, that may reflect normal cardiac rhythm.
4 Conclusion
In this work, a new series of sulfonamide derivatives containing thiazolone ring with different aryl moieties were designed and synthesized as potent carbonic anhydrase IX inhibitors. The synthesized derivatives were evaluated for their anticancer activity against two human BC cell lines (MDA-MB-231 and MCF-7) in addition to normal human cell line (MCF-10A). All the compounds were evaluated for their inhibitory activity against CA IX in a range with 10.93–25.06 nM. Three analogues 4e, 4g and 4h displayed significant selectivity towards tumor assisted CA IX over cytosolic CA II. From the results, we can conclude that the introduction of eugenol aldehyde or vanillin moieties (electron donating groups) are associated with enhanced anticancer activity (IC50 = 3.58 and 1.56 μM) against MDA-MB-231 and (IC50 = 4.58 and 1.52 μM) against MCF-7 as well as remarkable selectivity over normal breast cell line MCF-10A. This can be explained in accordance to docking studies due to their high binding affinities via forming high number and types of interactions. Additionally, compound 4e was able to induce apoptosis in MDA-MB-231 cells confirmed by the increase of annexinV – FITC percent by 22 fold. Moreover, the synthesized analogues were screened against antibacterial and anti-biofilm inhibition. The presence of electron withdrawing groups (as Cl and NO2) on the aryl moiety was shown to improve the antibacterial activity against S. aureus, and S. typhimurium while the presence of electron donating group enhanced the anti-biofilm effect towards Gram negative bacteria as K. pneumonia. Moreover, the poly substitution on the aryl ring (4e and 4h) enhanced the antibiofilm effect toward S. typhimurium.Compliance with ethical standards
Research involving human participants and/or animals
This work did not use human volunteers or animals.Ethics approval and consent to participate
Not applicable.Funding
This work is self-funded.Conflicts of interest
Authors declare that they have no conflict of interest.Acknowledgements
The authors express their appreciation and thanks to Dr Mohamed Abdel-Mohsen Elzayat, pharmacology department, NUB University to his kind assistance in performing ANOVA statistical analysis.References
- A. Kamal, K. S. Reddy, M. N. A. Khan, R. V. C. Shetti, M. J. Ramaiah, S. N. Pushpavalli, C. Srinivas, M. P. Bhadra, M. Chourasia, G. N. Sastry, A. Juvekar, S. Zingde and M. Barkume, Synthesis, DNA-binding ability and anticancer activity of benzothiazole/benzoxazole–pyrrolo[2,1-c][1,4]benzodiazepine conjugates, Bioorg. Med. Chem., 2010, 18, 4747–4761 CrossRef CAS PubMed.
- D. Verma, P. Kumar, B. Narasimhan, K. Ramasamy, V. Mani, R. K. Mishra and A. B. Abdul Majeed, Synthesis, antimicrobial, anticancer and QSAR studies of 1-[4-(substituted phenyl)-2-(substituted phenyl azomethyl)-benzo[b]-[1,4]diazepin-1-yl]-2-substituted phenylaminoethanones, Arabian J. Chem., 2019, 12, 2882–2896 CrossRef CAS.
- M. T. M. Nemr and A. M. AboulMagd, New fused pyrimidine derivatives with anticancer activity: synthesis, topoisomerase II inhibition, apoptotic inducing activity and molecular modeling study, Bioorg. Chem., 2020, 103, 104134 CrossRef CAS PubMed.
- M. T. M. Nemr, A. Sonousi and A. A. Marzouk, Design, synthesis and antiproliferative evaluation of new tricyclic fused thiazolopyrimidines targeting topoisomerase II: molecular docking and apoptosis inducing activity, Bioorg. Chem., 2020, 105, 104446 CrossRef CAS PubMed.
- https://www.uicc.org/news/globocan-2020-new-global-cancer-data.
- K. Ruan, G. Song and G. Ouyang, Role of hypoxia in the hallmarks of human cancer, J. Cell. Biochem., 2009, 107, 1053–1062 CrossRef CAS PubMed.
- J. P. Cosse and C. Michiels, Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression, Adv. Anticancer Agents Med. Chem., 2008, 8, 790–797 CrossRef CAS PubMed.
- U. Lendahl, K. L. Lee, H. Yang and L. Poellinger, Generating specificity and diversity in the transcriptional response to hypoxia, Nat. Rev. Genet., 2009, 10, 821–832 CrossRef CAS PubMed.
- A. Güttler, K. Theuerkorn, A. Riemann, H. Wichmann, J. Kessler, O. Thews, M. Bache and D. Vordermark, Cellular and radiobiological effects of carbonic anhydrase IX in human breast cancer cells, Oncol. Rep., 2019, 41, 2585–2594 Search PubMed.
- C. T. Supuran, Structure-based drug discovery of carbonic anhydrase inhibitors, J. Enzyme Inhib. Med. Chem., 2012, 27, 759–772 CrossRef CAS PubMed.
- C. T. Supuran, How many carbonic anhydrase inhibition mechanisms exist?, J. Enzyme Inhib. Med. Chem., 2016, 31, 345–360 CrossRef CAS PubMed.
- J. R. Casey, P. E. Morgan, D. Vullo, A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX, J. Med. Chem., 2004, 47, 2337–2347 CrossRef CAS PubMed.
- V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. D. Simone, Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms?, Chem. Rev., 2012, 112, 4421–4468 CrossRef CAS PubMed.
- E. Rosatelli, A. Carotti, M. Ceruso, C. T. Supuran and A. Gioiello, Flow synthesis and biological activity of aryl sulfonamides as selective carbonic anhydrase IX and XII inhibitors, Bioorg. Med. Chem. Lett., 2014, 24, 3422–3425 CrossRef CAS PubMed.
- B. P. Mahon, M. A. Pinard and R. McKenna, Targeting carbonic anhydrase IX activity and expression, Molecules, 2015, 20, 2323–2348 CrossRef PubMed.
- N. Chandak, M. Ceruso, T. C. Supuran and K. P. Sharma, Novel sulfonamide bearing coumarin scaffolds as selective inhibitors of tumor associated carbonic anhydrase isoforms IX and XII, Bioorg. Med. Chem., 2016, 2, 2882–2886 CrossRef PubMed.
- M. A. Abdelrahman, W. M. Eldehna, A. Nocentini, S. Bua, S. T. Al-Rashood, G. S. Hassan, A. Bonardi, A. A. Almehizia, H. M. Alkahtani, A. Alharbi, P. Gratteri and C. T. Supuran, Novel Diamide-Based Benzenesulfonamides as Selective Carbonic Anhydrase IX Inhibitors Endowed with Antitumor Activity: Synthesis, Biological Evaluation and In Silico Insights, Int. J. Mol. Sci., 2019, 20, 2484 CrossRef CAS PubMed.
- N. M. Abdel Gawad, N. H. Amin, M. T. Elsaadi, F. M. M. Mohamed, A. Angeli, V. D. Luca, C. Capasso and C. T. Supuran, Synthesis of 4-(thiazol-2-ylamino)-benzenesulfonamides with carbonic anhydrase I, II and IX inhibitory activity and cytotoxic effects against breast cancer cell lines, Bioorg. Med. Chem., 2016, 24, 3043–3051 CrossRef CAS PubMed.
- A. Grandane, M. Tanc, L. D. Mannelli, F. Carta, C. Ghelardini, R. Žalubovskis and C. T. Supuran, 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells, J. Med. Chem., 2015, 58, 3975–3983 CrossRef CAS PubMed.
- E. Andreucci, J. Ruzzolini, S. Peppicelli, F. Bianchini, A. Laurenzana, F. Carta, C. T. Supuran and L. Calorini, The carbonic anhydrase IX inhibitor SLC-0111 sensitises cancer cells to conventional chemotherapy, J. Enzyme Inhib. Med. Chem., 2019, 34, 117–123 CrossRef CAS PubMed.
- A. Insuasty, J. Ramírez, M. Raimondi, C. Echeverry, J. Quiroga, R. Abonia, M. Nogueras, J. Cobo, M. V. Rodrígue, S. A. Zacchino and B. Insuasty, Synthesis, antifungal and antitumor activity of novel (Z)-5-hetarylmethylidene-1,3-thiazol-4-ones and (Z)-5-ethylidene-1,3-thiazol-4-ones, Molecules, 2013, 18, 5482–5497 CrossRef CAS PubMed.
- D. Havrylyuk, B. Zimenkovsky, O. Vasylenko, L. Zaprutko, A. Gzella and R. Lesyk, Synthesis of novel thiazolone-based compounds containing pyrazoline moiety and evaluation of their anticancer activity, Eur. J. Med. Chem., 2009, 44, 1396–1404 CrossRef CAS PubMed.
- P. Vicini, A. Geronikaki, K. Anastasia, M. Incerti and F. Zani, Synthesis and antimicrobial activity of novel 2-thiazolylimino-5-arylidene-4-thiazolidinones, Bioorg. Med. Chem., 2006, 14, 3859–3864 CrossRef CAS PubMed.
- N. A. Khalil, E. M. Ahmed and H. B. El-Nassan, Synthesis, characterization, and biological evaluation of certain 1,3-thiazolone derivatives bearing pyrazoline moiety as potential anti-breast cancer agents, Med. Chem. Res., 2013, 22, 1021–1027 CrossRef CAS.
- G. Turan-Zitouni, L. Yurttas, A. Tabbi, G. A. Çiftçi, H. E. Temel and Z. A. Kaplancıklı, New Thiazoline-Tetralin Derivatives and Biological Activity Evaluation, Molecules, 2018, 23, 135 CrossRef PubMed.
- G. N. Masoud, A. M. Youssef, M. M. Abdel Khalek, A. E. Abdel Wahab, I. M. Labouta and A. A. B. Hazzaa, Design, synthesis, and biological evaluation of new 4-thiazolidinone derivatives substituted with benzimidazole ring as potential chemotherapeutic agents, Med. Chem. Res., 2013, 22, 707–725 CrossRef CAS.
- J. Wu, L. Yu, F. Yang, J. Li, P. Wang, W. Zhou, L. Qin, Y. Li, J. Luo, Z. Yi, M. Liu and Y. Chen, Optimization of 2-(3-(arylalkyl amino carbonyl)phenyl)-3-(2-methoxyphenyl)-4-thiazolidinone derivatives as potent antitumor growth and metastasis agents, Eur. J. Med. Chem., 2014, 80, 340–351 CrossRef CAS PubMed.
- C. T. Supuran, Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas, Expert Opin. Ther. Pat., 2018, 28, 709–712 CrossRef CAS PubMed.
- C. T. Supuran, Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases, Expert Opin. Ther. Pat., 2018, 28, 713–721 CrossRef CAS PubMed.
- C. T. Supuran, How many carbonic anhydrase inhibition mechanisms exist?, J. Enzyme Inhib. Med. Chem., 2015, 31, 345–360 CrossRef PubMed.
- T. V. Wani, S. Bua, P. S. Khude, A. H. Chowdhary, C. T. Supuran and M. P. Toraskar, Evaluation of sulphonamide derivatives acting as inhibitors of human carbonic anhydrase isoforms I, II and Mycobacterium tuberculosis b-class enzyme Rv3273, J. Enzyme Inhib. Med. Chem., 2018, 33, 962–971 CrossRef CAS PubMed.
- S. G. Alegaon and K. R. Alagawadi, New thiazolidinedione-5-acetic acid amide derivatives: synthesis, characterization and investigation of antimicrobial and cytotoxic properties, Med. Chem. Res., 2012, 21, 816–824 CrossRef CAS.
- H. Z. Chohan, U. A. Shaikh, A. Rauf and T. C. Supuran, Antibacterial, antifungal and cytotoxic properties of novel N-substituted sulfonamides from 4-hydroxycoumarin, J. Enzyme Inhib. Med. Chem., 2006, 21, 741–748 CrossRef PubMed.
- M. D. Altıntop, Z. A. Kaplancıklı, G. A. Çiftçi and R. Demirel, Synthesis and biological evaluation of thiazoline derivatives as new antimicrobial and anticancer agents, Eur. J. Med. Chem., 2014, 74, 264–277 CrossRef PubMed.
- A. M. Alafeefy, M. Ceruso, A. S. Al-Tamimi, S. Del Prete, C. Capasso and C. T. Supuran, Quinazoline–sulfonamides with potent inhibitory activity against the α-carbonic anhydrase from Vibrio cholerae, Bioorg. Med. Chem., 2014, 22, 5133–5140 CrossRef CAS PubMed.
- K. O. Mohamed, Y. M. Nissan, A. A. El-Malah, W. A. Ahmed, D. M. Ibrahim, T. M. Sakr and M. A. Motaleb, Design, synthesis and biological evaluation of some novel sulfonamide derivatives as apoptosis inducers, Eur. J. Med. Chem., 2017, 135, 424–433 CrossRef CAS PubMed.
- Y. M. Nissan, K. O. Mohamed, W. A. Ahmed, D. M. Ibrahim, M. M. Sharaky, T. M. Sakr, M. A. Motaleb, A. Maher and R. K. Arafa, New benzenesulfonamide scaffold-based cytotoxic agents: design, synthesis, cell viability, apoptotic activity and radioactive tracing studies, Bioorg. Chem., 2020, 96, 103577 CrossRef CAS PubMed.
- D. T. Vistica, P. Skehan, D. Scudiero, A. Monks, A. Pittman and M. R. Boyd, Tetrazolium-based assays for cellular viability: a critical examination of selected parameters affecting formazan production, Cancer Res., 1991, 51, 2515–2520 CAS.
- H. Takeuchi, M. Baba and S. Shigeta, An application of tetrazolium (MTT) colorimetric assay for the screening of anti-herpes simplex virus compounds, J. Virol. Methods, 1991, 33, 61–71 CrossRef CAS PubMed.
- W. M. Eldehna, D. H. El-Naggar, A. R. Hamed, H. S. Ibrahim, H. A. Ghabbour, H. A. Abdel-Aziz and H. A. Ghabbour, One-pot three-component synthesis of novel spirooxindoles with potential cytotoxic activity against triple-negative breast cancer MDAMB-231 cells, J. Enzyme Inhib. Med. Chem., 2018, 33, 309–318 CrossRef CAS PubMed.
- W. M. Eldehna, H. Almahli, G. H. Al-Ansary, H. A. Ghabbour, M. H. Aly, O. E. Ismael, A. Al-Dhfyan and H. A. Abdel-Aziz, Synthesis and in vitro anti-proliferative activity of some novel isatins conjugated with quinazoline/phthalazine hydrazines against triple-negative breast cancer MDA-MB-231 cells as apoptosis-inducing agents, J. Enzyme Inhib. Med. Chem., 2017, 32, 600–613 CrossRef CAS PubMed.
- R. A. Ingebrigtsen, E. Hansen, J. H. Andersen and H. C. Eilertsen, Light and temperature effects on bioactivity in diatoms., J. Appl. Phycol., 2016, 28, 939–950 CrossRef CAS PubMed.
- S. I. Elewa, A. O. Abdelhamid, A. A. Hamed and E. Mansour, Synthesis, characterization, antimicrobial activities, anticancer of some new pyridines from 2, 3-dihydro-2-oxo-4-phenyl-6-(thien-2-yl) pyridine-3-carbonitrile, Synth. Commun., 2021, 51, 151–161 CrossRef CAS.
- D. M. Eskander, S. M. M. Atalla, A. A. Hamed and E. D. A. El-Khrisy, Investigation of secondary metabolites and its bioactivity from Sarocladium kiliense SDA20 using shrimp shell wastes, Pharmacogn. J., 2020, 12, 636–644 CrossRef CAS.
- A. A. Hamed, H. Kabary, M. Khedr and A. N. Emam, Antibiofilm, antimicrobial and cytotoxic activity of extracellular green-synthesized silver nanoparticles by two marine-derived actinomycete, RSC Adv., 2020, 10, 10361–10367 RSC.
- Pharmacokinetic properties, http://biosig.unimelb.edu.au/pkcsm/prediction.
- S. Mahboobi, A. Sellmer, H. Hocher, E. Eichhorn, T. Bär, M. Schmidt, T. Maier, J. F. Stadlwieser and T. L. Beckers, [4-(Imidazol-1-yl)thiazol-2-yl]phenylamines. A novel class of highly potent colchicine site binding tubulin inhibitors: synthesis and cytotoxic activity on selected human cancer cell lines, J. Med. Chem., 2006, 49, 5769–5776 CrossRef CAS PubMed.
- A. M. Alafeefy, R. Ahmad, M. Abdulla, W. M. Eldehna, A. S. Al-Tamimi, H. A. Abdel-Aziz, O. Al-Obaid, F. Carta, A. A. Al-Kahtani and C. T. Supuran, Development of certain new 2-substituted-quinazolin-4-yl-aminobenzenesulfonamide as potential antitumor agents, Eur. J. Med. Chem., 2016, 109, 247–253 CrossRef CAS PubMed.
- J. Leitans, A. Kazaks, A. Balode, J. Ivanova, R. Zalubovskis, C. T. Supuran and K. Tars, Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX, J. Med. Chem., 2015, 58, 9004–9009 CrossRef CAS PubMed.
- https://www.rcsb.org/structure/5FL6.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- M. P. Gleeson, A. Hersey and S. Hannongbua, In-silico ADME models: a general assessment of their utility in drug discovery applications, Curr. Top. Med. Chem., 2011, 11, 358–381 CrossRef CAS PubMed.
- M. P. Gleeson, Generation of a set of simple, interpretable ADMET rules of thumb, J. Med. Chem., 2008, 51, 817–834 CrossRef CAS PubMed.
- D. Lagorce, D. Douguet, M. A. Miteva and B. O. Villoutreix, Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors, Sci. Rep., 2017, 7, 46277 CrossRef CAS PubMed.
- G. Moroy, V. Y. Martiny, P. Vayer, B. O. Villoutreix and M. A. Miteva, Toward in silico structure-based ADMET prediction in drug discovery, Drug Discovery Today, 2012, 17, 44–55 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05277b |
| This journal is © The Royal Society of Chemistry 2021 |