
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pH-sensitive and sustained-release oral drug delivery system: the synthesis, characterization, adsorption and release of the xanthan gum-graft-poly(acrylic acid)/GO–DCFP composite hydrogel†
Li Li ab,
Xiyan Zhenga,
Chunjiao Pana,
Hao Pana,
Zhongqiu Guoa,
Bingmi Liuac and
Yu Liu*ab
ab,
Xiyan Zhenga,
Chunjiao Pana,
Hao Pana,
Zhongqiu Guoa,
Bingmi Liuac and
Yu Liu*ab
aSchool of Pharmacy, Liaoning University, Shenyang, 110036, China. E-mail: lnuliuyu@163.com
bLiaoning Key Laboratory of New Drug Research & Development, Shenyang, 110036, China
cLiaoning Pharmaceutical Engineering Research Center for Natural Medicine, Shenyang, 110036, China
First published on 30th July 2021
Abstract
In this study, graphene oxide (GO) was successfully prepared using the improved Hummers method, and the prepared GO powder was dissolved in distilled water and subjected to ultrasonic stripping. Diclofenac potassium (DCFP) was selected as a model drug to systematically evaluate the adsorption mechanism of DCFP by GO. Different reaction models were constructed to fit the adsorption kinetics and adsorption isotherms of DCFP on GO, in order to further explore the underlying adsorption mechanism. The results demonstrated that the pseudo-second-order kinetic model and Freundlich model could better delineate the adsorption process of DCFP by GO. Both π–π stacking and hydrophobic interaction were mainly involved in the adsorption process, and there were electrostatic interaction and hydrogen bonding at the same time. Then, the xanthan gum-graft-poly(acrylic acid)/GO (XG-g-PAA/GO) composite hydrogel was synthesized by in situ polymerization as a slow-release drug carrier. For this reason, a XG-g-PAA/GO–DCFP composite hydrogel was synthesized, and its in vitro drug release and pharmacokinetic data were assessed. The results showed that the synthesized XG-g-PAA/GO composite hydrogel had a certain mechanical strength and uniform color, indicating that GO is evenly distributed in this composite hydrogel. Moreover, the results of a swelling ratio test demonstrated that the swelling ratios of the XG-g-PAA/GO composite hydrogel were significantly increased with increasing pH values, implying that this material is sensitive to pH. The in vitro drug release experiment showed that the cumulative release of DCFP after 96 h was significantly higher in artificial intestinal fluid than in artificial gastric fluid. These findings indicate that the XG-g-PAA/GO–DCFP composite hydrogel exhibits pH sensitivity under physiological conditions. Besides, the results of in vivo pharmacokinetic analysis revealed that the t1/2 of DCFP group was 2.03 ± 0.35 h, while that of the XG-g-PAA/GO–DCFP composite hydrogel group was 10.71 ± 2.04 h, indicating that the synthesized hydrogel could effectively prolong the drug action time. Furthermore, the AUC(0–t) of the DCFP group was 53.99 ± 3.18 mg L−1 h−1, while that of the XG-g-PAA/GO–DCFP composite hydrogel group was 116.79 ± 14.72 mg L−1 h−1, suggesting that the bioavailability of DCFP is greatly enhanced by this composite hydrogel. In conclusion, this study highlights that the XG-g-PAA/GO–DCFP composite hydrogel can be applied as a sustained-release drug carrier.
1. Introduction
Graphene oxide (GO) is a highly oxidized form of graphene made from natural graphite. Due to its unique structure and excellent properties, it has received widespread attention. GO is a two-dimensional sheet material composed of a typical graphite planar structure, which has a large specific surface area. Such a huge specific surface area may contribute to its high drug-loading capacity.1 Compared with the hydrophobic graphene that easily causes irreversible agglomeration,2 GO contains both hydrophobic graphene domains and hydrophilic edges, thus providing excellent amphiphilic properties for this material.3 The GO plane mainly includes hydroxyl and epoxy groups, and there are carboxyl groups on the edge of the sheet structure.4 The presence of oxygen-containing functional groups allows GO to be easily peeled off into a single-layer sheet, with good water dispersibility and excellent ability to penetrate cell membranes. At the same time, these groups allow GO to be functionalized,5,6 which in turn improves the performance of GO.7,8 The functional groups of GO can also capture, anchor or immobilize polymers and biomolecules, such as nucleic acids, drugs, etc.9 Therefore, GO is suitable for delivering bioactive molecules with antiviral, antibacterial or anticancer activities.10 With the continuous in-depth study of their unique structure and properties, GO and its derivatives have gradually become the focus of attention in the field of pharmaceutical medicine.Traditional hydrogels have relatively poor mechanical properties and limited functional characteristics; thus, they are often subjected to certain restrictions during their application.11 Several strategies, such as cold treatment,12 macroporous or nanoporous structures13 and hydrophobic crosslinking agents,14 have been widely used for improving the hydrogel. Rehman et al. developed super-absorbent hydrogels with extraordinarily high swelling and mechanical behaviors by fabricating different amounts of pH-sensitive monomer 2-acrylamido-2-methylpropane sulphonic acid (AMPS) in an acrylamide/sodium alginate hydrogel.15 Khan et al. synthesized physically cross-linked hydrogels using supramolecular interactions between the metal ions (Fe3+) and the deprotonated functional groups of acrylic acid and non-modified gum arabic, which was successfully confirmed by the FTIR spectra.16 They also physically cross-linked the synthesized hydrogel composites using a one-step method based on acrylic acid and gum arabic. The results revealed that the mechanical properties of the hydrogels could be altered by different amounts of TiO2.17 Shah et al. reported that the encapsulation of GO particles within the hydrogel network provided better mechanical properties and the presence of GO could improve the surface adsorption of the material via changing its pH sensitivity.18 By introducing nano-materials to modify the hydrogel, the performance of the polymer network itself is not destroyed, and the application limitations of the hydrogel have been greatly broken through. Among many nanomaterials, GO is a nanomaterial with many excellent features19 such as large specific surface area, good solubility, cost-effective and can be easily combined with the hydrogel through simple chemical modification, thus making it an attractive material for large-scale applications. In addition, GO has a large number of oxygen-containing functional groups, which can enhance its swelling performance and mechanical properties. GO composite hydrogel has gradually become a research hotspot in recent years.
Xanthan Gum (XG) is an extracellular polysaccharide generated by the microorganism Xanthomonas campestris. The main structure is a repeating pentasaccharide unit that consists of 2 glucose units, 2 mannose units, and 1 glucuronide.20 XG can be dissolved in either cold water or hot water, and low-concentration XG has high viscosity, which can be used as a dispersing agent in thicken aqueous solutions and as a stabilizer in both emulsions and suspensions. XG is non-toxic, does not cause skin or eye irritation, and has been widely used in pharmaceutical preparations, cosmetics and agricultural products. For example, it serves as a suspending agent and stabilizer in topical and oral formulations as well as a sustained-release agent in hydrophilic matrix tablets and pills.21,22
Diclofenac potassium (DCFP) is a new type of non-steroidal anti-inflammatory drug,23,24 and its mechanism of action is mainly to inhibit the synthesis of prostaglandins, thereby exerting anti-inflammatory and analgesic effects. Compared with diclofenac sodium, DCFP has better solubility in water and is rapidly absorbed. DCFP formulation can be used to treat acute and chronic pain, and it is particularly suitable for the diseases that require immediate treatment, such as migraine.25,26 However, like other non-steroidal anti-inflammatory drugs, oral administration of DCFP has a stimulating effect on the gastrointestinal tract.27 Moreover, the half-life of DCFP is short, and it is absorbed quickly after oral administration. Therefore, it is necessary to develop an oral sustained-release preparation of DCFP to reduce its side effects on the stomach, while prolonging its oral administration time and reducing the number of medications.
In this work, DCFP was selected as the model drug to investigate the adsorption mechanism of GO as a drug carrier. Through the use of XG and GO as raw materials, acrylic acid as graft monomer, N,N′-methylenebisacrylamide and ammonium persulfate as reaction crosslinking agents and initiator, XG-g-PAA/GO composite hydrogel was successfully prepared and employed as a carrier for controlled drug release. By taking DCFP as a model drug, the drug loading of XG-g-PAA/GO–DCFP composite hydrogel was investigated to determine the best preparation method for this composite hydrogel. The in vitro drug release of XG-g-PAA/GO–DCFP composite hydrogel was also tested at different pH values, by using artificial gastric juice and artificial intestinal juice as the release medium. Furthermore, DCFP and XG-g-PAA/GO–DCFP composite hydrogel were administered orally into rats, and the pharmacokinetic characteristics of the drug were evaluated in vivo.
2. Experimental details
2.1 Materials and animals
Graphite powder was obtained from Tianjin Bonop Co., Ltd. DCFP, ketoprofen and pancreatin were purchased from Dalian Meilun Biology Technology Co., Ltd. Sulphuric acid (98%), hydrogen peroxide, hydrochloric acid and phosphate-buffered saline were purchased from Tianjin Laibo Chemical Co., Ltd. Pepsin was supplied by Biotopped. Potassium permanganate and sodium nitrate were obtained from Shandong Yu Wang Industrial Co., Ltd. XG was purchased from Maya Reagent. Acrylic acid was purchased from Shanghai Macklin Biochemical Co., Ltd. N,N′-Methylenebisacrylamide and ammonium persulfate were supplied by Tianjin Damao Chemical Reagent Factory. Ordinary small animal capsules were purchased from Guangdong Qiangji Pharmaceutical Co., Ltd. SPF-grade Sprague-Dawley male rats were obtained from Liaoning Changsheng Biotechnology Co., Ltd.2.2 Preparation of GO
The modified Hummers method was referred for the preparation of GO. First, 2.5 g of graphite powder and 2 g of sodium nitrate were added to a three-necked flask containing 84.4 mL of concentrated sulfuric acid. Then, under stirring, 11.25 g of potassium permanganate was added in the batches within 1 h. The mixture was transferred from three-necked round bottom flask to an oil bath with magnetic heating. The temperature of the magnetic stir was adjusted as follows: 35 °C for 30 min, 65 °C for 30 min, and 85 °C for 30 min. The product was allowed to stand at room temperature for one week, and then transferred to a beaker containing hot water. Hydrogen peroxide (30%) was added dropwise until the color became bright yellow. The suspension was centrifuged, washed and freeze-dried to a constant weight, in order to obtain a flake-shaped solid GO. An appropriate amount of GO was accurately weighed and placed into a beaker, followed by the addition of distilled water. After sonication, the solution was continuously stirred until it became brownish yellow. Finally, the solution was stripped, centrifuged and washed to achieve a stable GO aqueous solution.2.3 Characterization of GO
The Fourier transform infrared (FT-IR) spectrum was measured using an IRAffinity-1 spectrometer (Shimadzu, Japan). The microstructure of GO was analyzed using a JEM-2100 transmission electron microscope (TEM; JEOL, Japan). The ultraviolet-visible spectrum was performed on an UV-2550 spectrometer (Shimadzu, Japan). The thermal stability and other quantitative parameters were characterized by means of thermogravimetric analysis under a nitrogen atmosphere at 20–400 °C. The particle size, ZP and polydispersity coefficient (PDI) value of GO solution were measured using a Malvern Zetasizer nanometer instrument (Nano-ZS 90, Malvern, UK). X-ray diffraction (XRD) analysis of GO was conducted using a Bruker D8 ADVANCE X-ray powder diffractometer (Bruker, Germany) with CuKα radiation and 2θ angle range (10–80°).2.4 Synthesis of GO–DCFP
GO was weighed by the mass M1 (mg) and dissolved in an appropriate amount of distilled water. The resulting GO aqueous solution was ultrasonically stripped in a beaker at a concentration of C0 (mg mL−1), which was fixed on a magnetic stirrer at a constant temperature. DCFP was precisely weighed by the mass M2 (mg) and dissolved in an appropriate amount of distilled water. Under stirring conditions, the completely dissolved DCFP aqueous solution was gradually added dropwise to the GO aqueous solution. The reaction took place in the dark under continuous stirring. After completion of the reaction, the solution was centrifuged, and the obtained precipitate was rinsed and freeze-dried to obtain the final product GO–DCFP.2.5 Adsorption experiments
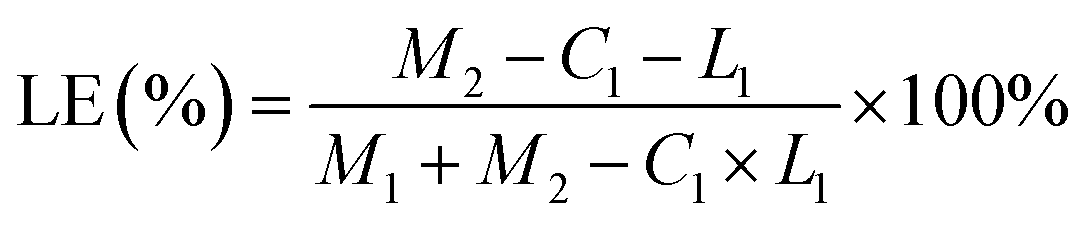
M1: GO quality; M2: total dose; C1: free drug concentration; L1: solution volume.
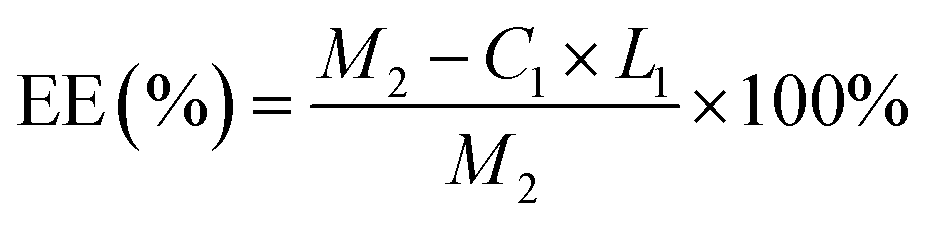
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GO were maintained as 1:5, 1:2, 1:1.5, 1:1 and 1.5:1. The loading efficiency and encapsulation efficiency were calculated at different drug and carrier feeding ratios.
:GO were maintained as 1:5, 1:2, 1:1.5, 1:1 and 1.5:1. The loading efficiency and encapsulation efficiency were calculated at different drug and carrier feeding ratios.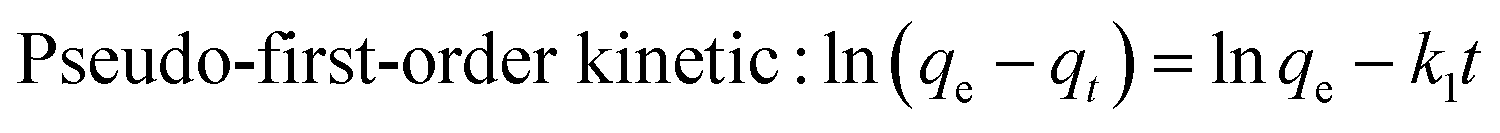

qe: balanced adsorption capacity (mg g−1); qt: adsorption capacity at time t (mg g−1); t: reaction time (min); k1: pseudo-first-order kinetic rate constant (min−1); k2: pseudo-second-order kinetic (g mg−1 min−1).
Ce: equilibrium concentration of the adsorbate (mg L−1); qe: equilibrium adsorption amount (mg g−1); qm: theoretical maximum adsorption capacity of monolayer (mg g−1); KL: Langmuir adsorption constant (L mg−1).
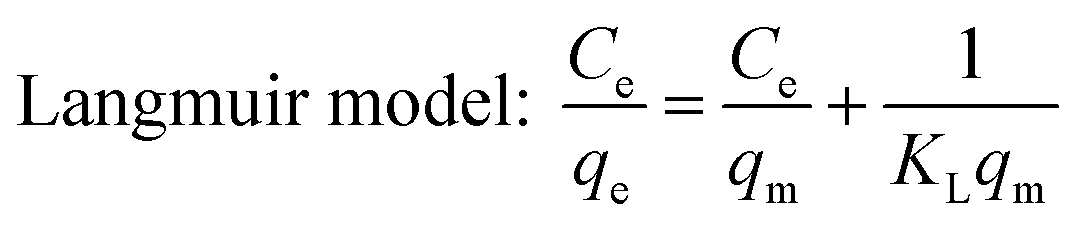
The Freundlich model presumes that the adsorption phenomenon takes place on a heterogeneous surface, that is, the energy at the surface adsorption sites is not uniform, and the adsorption is multi-layered.24 The general expression is as follows:
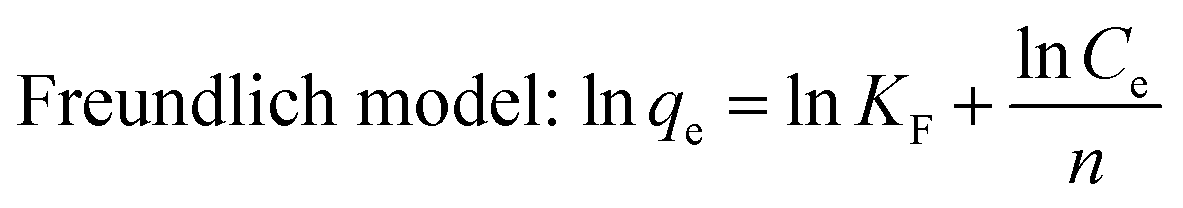
2.6 Synthesis of XG-g-PAA/GO and XG-g-PAA/GO–DCFP
First, GO was weighed into a beaker, added with distilled water, and ultrasonically peeled to obtain a yellow-brown GO aqueous solution. Under magnetic stirring, a certain amount of XG was completely dissolved into the GO solution, and the temperature was adjusted to 60 °C. A particular amount of ammonium persulfate was then mixed with the solution. After stirring for 10 min, specific amounts of acrylic acid and N,N′-methylenebisacrylamide were gradually added to the reaction solution until reaching the final volume of 30 mL. To form a composite hydrogel, the reaction was continuously heated at 60 °C. After completion of the reaction, the resulting product was cooled to room temperature, cut into smaller pieces and washed repeatedly with distilled water, in order to eliminate the residual reactants. The XG-g-PAA/GO composite hydrogel was finally obtained after freeze-drying in a vacuum for 24 h.To synthesize XG-g-PAA/GO–DCFP, the as-prepared XG-g-PAA/GO composite hydrogel was mixed with DCFP solution and then magnetically stirred at room temperature for 1 h. After magnetic stirring, other reactants were added according to the above-mentioned method, in order to obtain the final XG-g-PAA/GO–DCFP composite hydrogel.
2.7 Determination of swelling ratio
The as-prepared XG-g-PAA/GO composite hydrogel was dried to a constant weight and accurately weighed, and the mass of the dried gel was recorded as Wd. The xerogel was then submerged in distilled water, and a swelling test was performed. The surface moisture was absorbed with clean filter paper, and the sample was weighed until reaching a constant weight and swelling balance. The mass at this time point is recorded as Ws. The swelling ratio can be calculated by the following formula:SR: swelling ratio of the hydrogel (%); Ws: mass of the gel (mg) when swelling equilibrium is reached; Wd: mass of dry gel (mg).
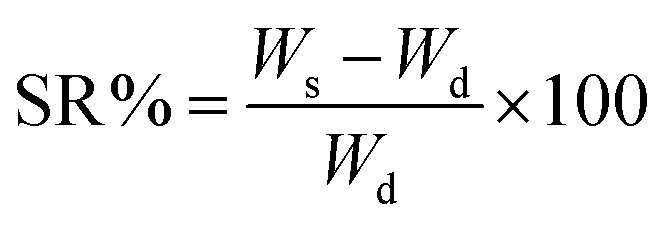
2.8 Characterization of XG-g-PAA/GO
2.9 XG-g-PAA/GO–DCFP drug loading
The concentration of DCFP was detected by high-performance liquid chromatography, and the loading efficiency and encapsulation efficiency of XG-g-PAA/GO composite hydrogel were calculated. The calculation formulas of loading efficiency (LE%) and encapsulation efficiency (EE%) are as follows:| LE (%) = M1/M2 × 100% |
| EE (%) = M1/M3 × 100% |
2.10 In vitro drug release testing
The in vitro release of XG-g-PAA/GO–DCFP composite hydrogel was assessed using the small cup method. The release media are artificial gastric juice and artificial intestinal fluid, respectively. First, 15 mg of the prepared XG-g-PAA/GO–DCFP composite hydrogel was accurately weighed, transferred into small capsules, and placed into artificial intestinal juice and artificial gastric juice at 37 ± 5 °C. The time points were set at 0, 5, 10, 20, 30 min, 1, 1.5, 2, 2.5, 3, 4, 8, 12, 24, 48, and 72 h. After centrifugation at 5000 rpm for 5 min at room temperature, the supernatant was subjected to pharmacokinetic study.2.11 Pharmacokinetic study and data analysis
The rats were randomly divided into two groups. DCFP and XG-g-PAA/GO–DCFP composite hydrogel were orally administered to the rats in the two groups, respectively, at a dosage of 25 mg kg−1. Blood samples were collected from the orbit at 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 48, and 72 h after oral administration. After centrifugation at 12000 rpm for 5 min, the upper plasma was subjected to liquid phase analysis.
3. Results and discussion
3.1 Characterization of GO
The PSD and ZP of GO are shown in Fig. 1. The mean particle size, PDI and ZP values of the prepared GO were approximately 336.9 nm, 0.183 and −39.6 mV, respectively. The PDI value of less than 0.3 indicates a good PSD; while the negative ZP suggests the presence of oxygen-containing functional groups in GO. The X-ray spectrum (Fig. 2) showed that the peaks at 10° and 26° were the characteristic peaks of GO, confirming the successful synthesis of GO. In addition, the absolute value of ZP was greater than 30 mV, indicating that the electrostatic repulsion between the particles of GO sheet is large. Thus, it is not easy to cause aggregation or precipitation, and the stability of GO solution is good.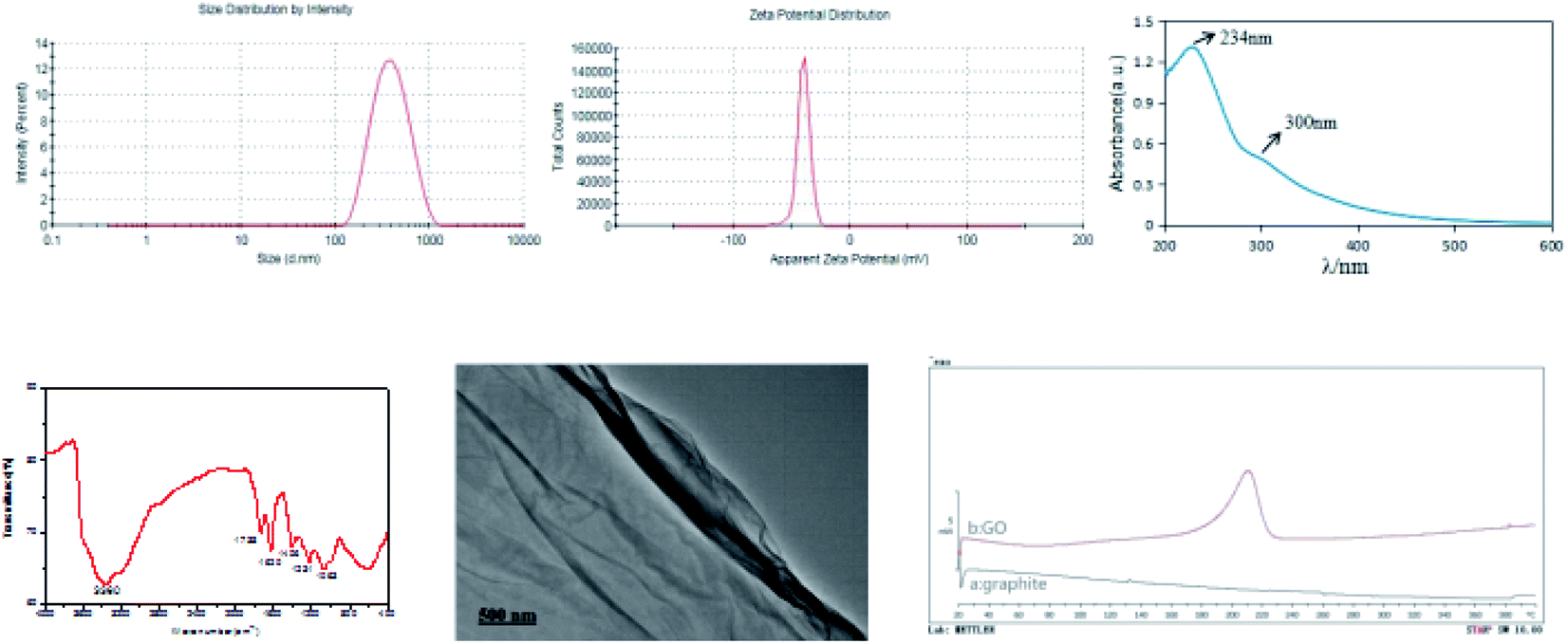 | ||
| Fig. 1 Particle size distribution, zeta potential, UV spectrum, infrared spectrum, TEM image, and differential scanning calorimetric curves of graphite (a) and GO (b). | ||
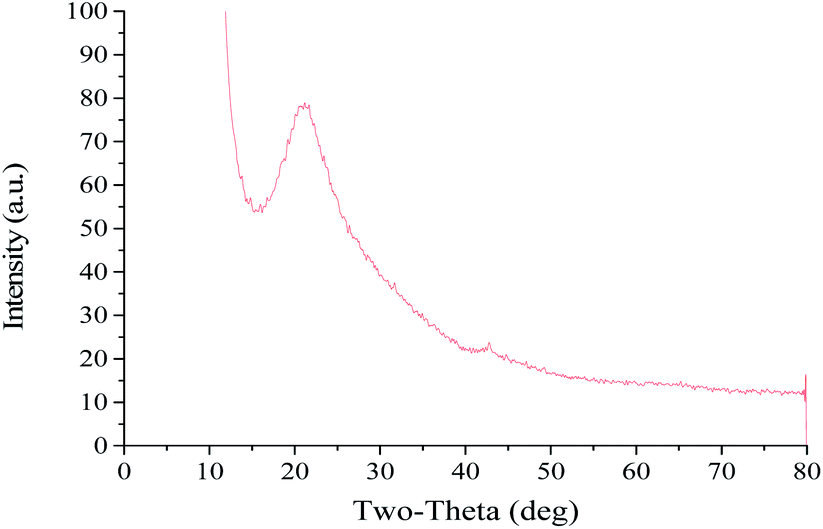 | ||
| Fig. 2 XRD pattern of GO. | ||
The UV spectral data of GO aqueous solution are presented in Fig. 1. Notably, the GO UV spectrum exhibited two features that could be used for identification. One was the maximum absorption wavelength at 234 nm, which corresponded to the π electron transition of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in GO structure. Another feature was the shoulder peak at 300 nm, which could be ascribed to the n → π* transition of CO. These results demonstrate that GO with higher oxidation degree has been successfully synthesized.
C bond in GO structure. Another feature was the shoulder peak at 300 nm, which could be ascribed to the n → π* transition of CO. These results demonstrate that GO with higher oxidation degree has been successfully synthesized.
The infrared spectrum of GO is shown in Fig. 1. Infrared spectroscopy revealed that –OH (3360 cm−1), CO (1738 cm−1) and CC (1620 cm−1) were existed in the synthesized GO, proving the successful introduction of oxygen-containing functional groups. In addition, there were C–OH stretching vibration peaks of carboxyl groups at 1406 cm−1, and absorption peaks of C–O groups at 1231 cm−1 and 1063 cm−1. These characteristic absorption peaks of GO infrared spectrum indicate that the graphite conjugate structure is destroyed and GO has been successfully synthesized.
TEM image of GO is displayed in Fig. 1. It was found that GO had a lamellar structure with wrinkles on the surface and edges. GO can be existed in the form of single layer or multiple layers, and the single-layer sheet structure is prone to folding and curling in space, which explains the slightly wrinkled sheet structure of GO.
The DSC spectra of graphite powder and GO are shown in Fig. 1. The results showed that there was a clear difference in DSC curves between graphite powder and GO. Graphite powder had no obvious melting point peak in the detected temperature range, and the synthesized GO sample exhibited a melting point peak at around 200 °C, which corresponded to the decomposition of unstable oxygen-containing functional groups. These findings also confirm the successful synthesis of GO.
3.2 Adsorption experiments
Based on the results, the loading effect of DCFP/GO was found to be most ideal at the DCFP:GO feeding ratio of 1:1.5. At this ratio, the loading efficiency and entrapment efficiency of DCFP/GO were 36.0% and 84.36%, respectively. Loading efficiency refers to the ratio of the amount of drug loaded into DCFP/GO to the total weight. Entrapment efficiency refers to the ratio of the amount of drug loaded into DCFP/GO to the amount of drug administered. The loading efficiency and entrapment efficiency of DCFP/GO are shown in Fig. 3 and 4.
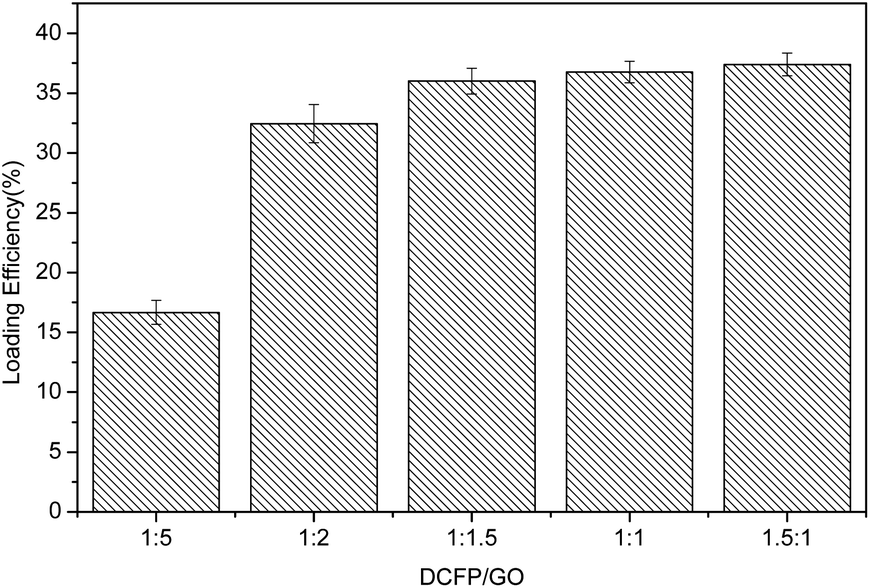 | ||
| Fig. 3 Loading efficiency of DCFP/GO. | ||
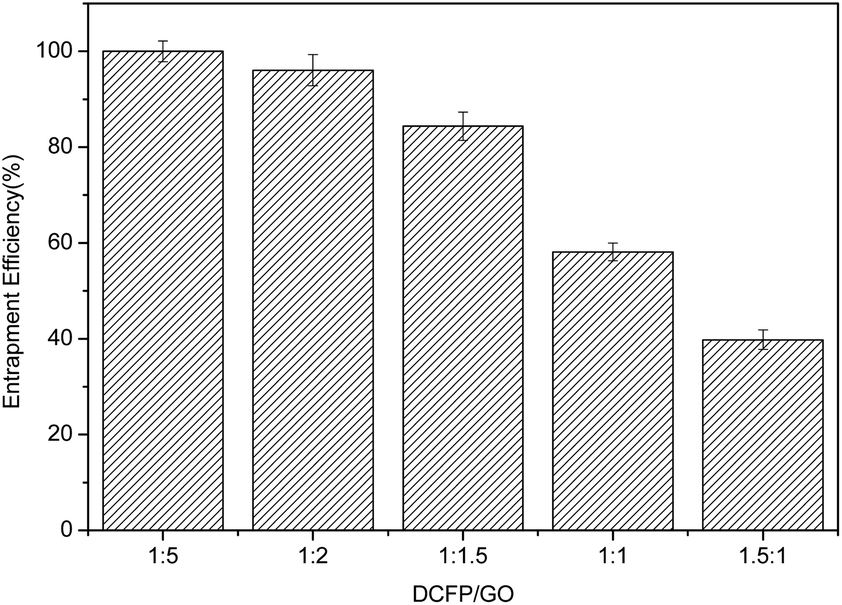 | ||
| Fig. 4 Entrapment efficiency of DCFP/GO. | ||
The effects of reaction time on DCFP-loaded GO were determined under different temperature conditions (293, 303, and 313 K). As demonstrated in Fig. 5, the initial reaction rate was relatively large. This may be due to the fact that the concentration of DCFP in the solution is high at the beginning of the reaction. Moreover, there are many unoccupied adsorption sites on the surface of GO, which can enhance its adsorption rate. As the reaction progressed, DCFP was gradually consumed in the solution and the adsorption sites on GO surface were gradually occupied, which in turn led to a gradual decrease in reaction rate until reaching equilibrium. The reaction equilibrium time was about 30 minutes.
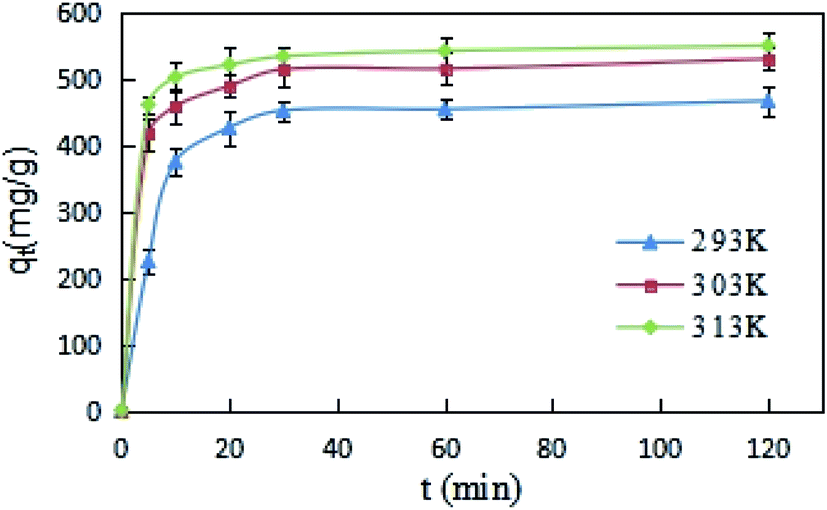 | ||
| Fig. 5 Adsorption kinetics curves of DCFP on GO. | ||
The impact of pH change on DCFP adsorption by GO was evaluated, different concentrations (200 and 250 μg mL−1) of DCFP were tested separately. As displayed in Fig. 6, the adsorption of GO on DCFP was inhibited with increasing pH values, and the equilibrium adsorption capacity of DCFP under alkaline conditions was very small. DCFP exists in a neutral form in a low pH environment (pH < pKa), and its pKa value is 4.0. When pH > pKa, DCFP gradually ionized, electrostatic repulsion occurred between the dissociated anions, and GO became negatively charged, thus leading to a decrease in the amount of DCFP adsorbed on GO. However, if the pH value was between 3 and 5, the adsorption capacity did not reduce significantly. This may be attributed to the fact that the electrostatic repulsion is weak at this time and has little effect on the adsorption results, which also implies that the electrostatic repulsion is not the only adsorption mechanism. The amino groups of DCFP and the oxygen-containing groups on GO surface can form hydrogen bonds, and the adsorption process is also affected by hydrogen bonds. The formation of hydrogen bonds under alkaline conditions is not conducive to the formation of hydrogen bonds, which may also lead to a decrease in adsorption capacity. It should be pointed out that GO has a hydrophobic surface and an aromatic ring structure. It can cause π–π stacking and hydrophobic interaction with DCFP, thus affecting the adsorption process. Overall, the adsorption of DCFP on GO can be affected through multiple mechanisms.
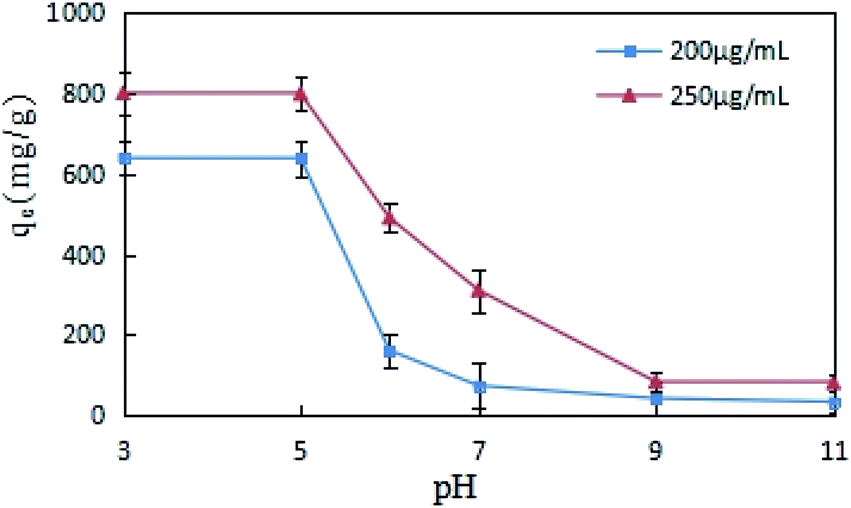 | ||
| Fig. 6 Effects of pH on the adsorption of DCFP on GO. | ||
Under the conditions of 293, 303 and 313 K, DCFP solutions with different initial concentrations were selected for this experiment, and the adsorption isotherms of the reaction between GO and DCFP were measured. As shown in Fig. 7, the amounts of DCFP absorbed on GO were increased with increasing equilibrium concentrations, and tended to be saturated when its equilibrium concentration reached a certain value. Similarly, the equilibrium adsorption capacity of GO increased with increasing reaction temperatures, indicating that a high temperature could exert profound effects on DCFP adsorption by GO.
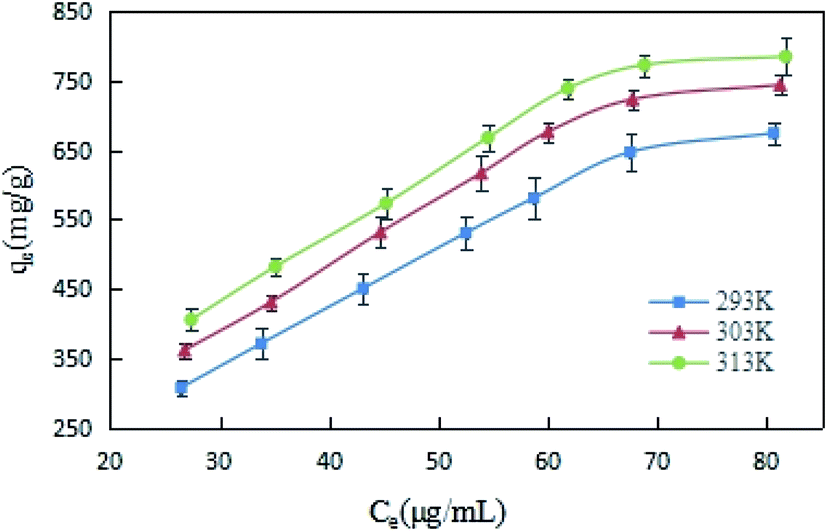 | ||
| Fig. 7 Adsorption isotherms of DCFP on GO. | ||
The fitting results of adsorption kinetics data are demonstrated in Fig. 8 and Table 1. The correlation coefficient R2 derived by fitting the two kinetic models was greater than 0.9, indicating that both pseudo-first-order and pseudo-second-order kinetic models could be employed to describe the kinetic process of DCFP adsorption on GO. Generally, pseudo-first-order kinetic model is only suitable for describing the adsorption characteristics of the initial adsorption stage, but not the whole adsorption process. By comparing the fitted data with the experimental data, it could be seen that the qe value of pseudo-second-order kinetic model was more comparable to that measured experimentally, and the R2 values of pseudo-first-order kinetic model obtained at three different temperatures were lesser than those of pseudo-second-order kinetic model, suggesting that the pseudo-second-order kinetic model is more appropriate to denote the adsorption process of DCFP on GO. The adsorption process is primarily regulated by the interaction between the adsorbate and the adsorbent. Based on the structural characteristics of GO and DCFP, it can be speculated that the interactions between the two are π–π stacking, hydrophobic interaction, electrostatic interaction and hydrogen bonding. Taken together, multiple mechanisms may play an essential role during the adsorption process of DCFP on GO.
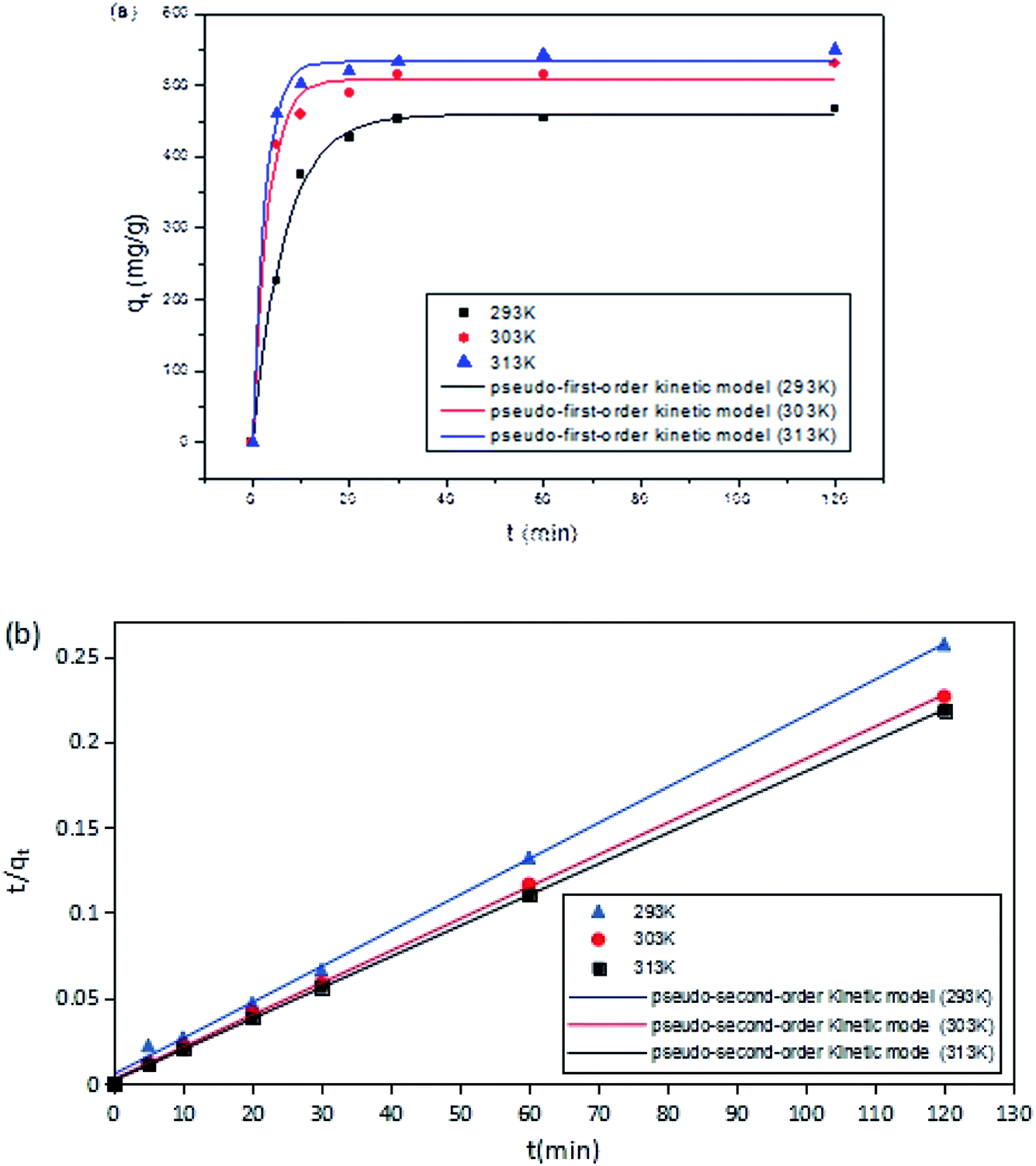 | ||
| Fig. 8 Adsorption kinetic model fitting. (a) Pseudo-first-order kinetic model. (b) Pseudo-second-order kinetic model. The point in the figure is the experimental value; while the line represents the calculated value of the model. | ||
| T (K) | qe, exp (mg g−1) | Pseudo-first-order kinetic model | Pseudo-second-order kinetic model | ||||
|---|---|---|---|---|---|---|---|
| qe, cal (mg g−1) | k1 (min−1) | R2 | qe, cal (mg g−1) | k2 (g mg−1 min−1) | R2 | ||
| 293 | 466.92 | 459.41 | 0.1485 | 0.9946 | 476.19 | 0.0008 | 0.9985 |
| 303 | 529.94 | 508.14 | 0.3181 | 0.9897 | 533.33 | 0.0015 | 0.9997 |
| 313 | 550.04 | 533.37 | 0.3841 | 0.9954 | 552.79 | 0.0021 | 0.9999 |
The adsorption isotherms of DCFP on GO were delineated by Langmuir model and Freundlich model. The fitting curves and data are presented in Fig. 9 and Table 2, respectively. By comparing the correlation coefficient R2 derived from the two models, it was observed that the Freundlich model could better describe the adsorption process. As mentioned above, there are multiple mechanisms involved during the adsorption process. The dominant mechanisms are π–π stacking and hydrophobic interaction, and these interactions tend to cause multi-layer adsorption. An elevated temperature can also affect the adsorption of DCFP by GO.
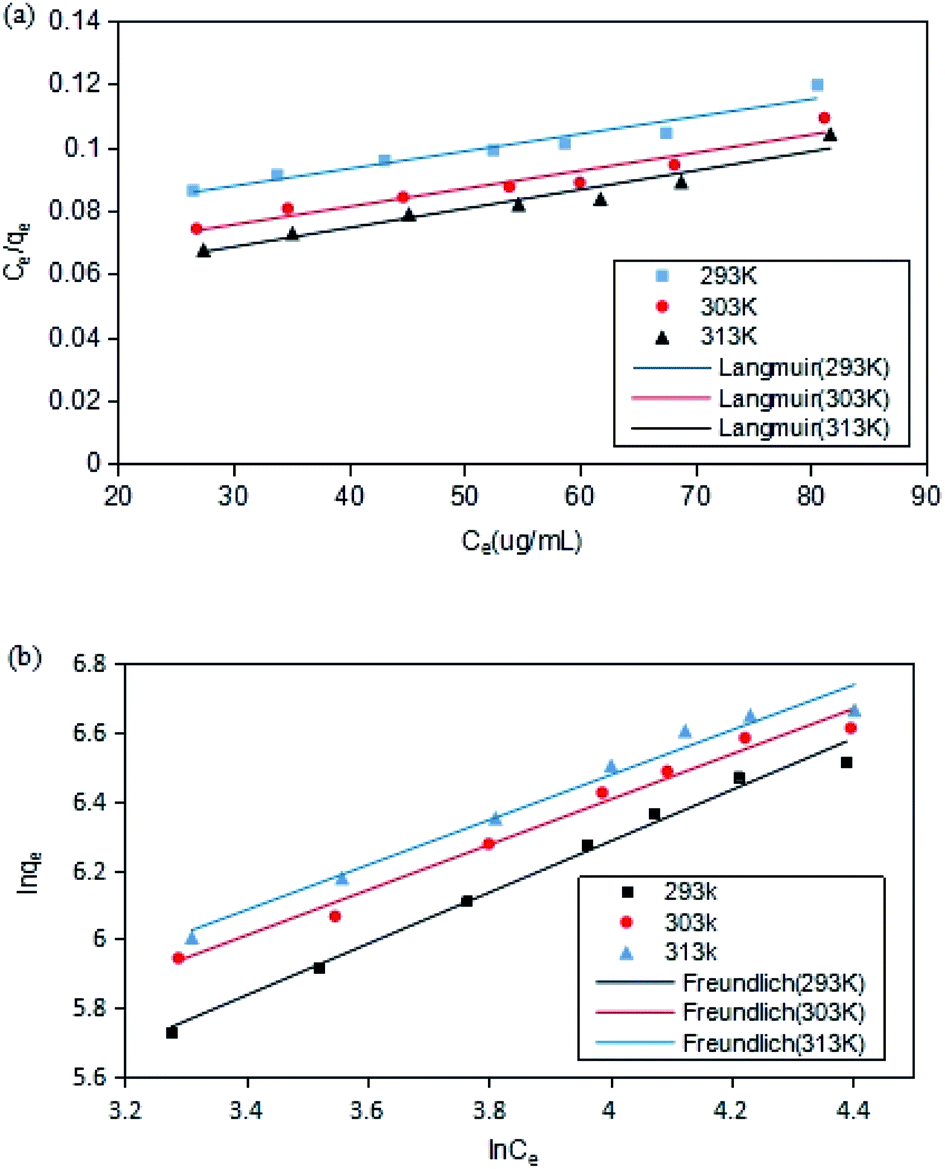 | ||
| Fig. 9 Adsorption isotherm model fitting. (a) Langmuir. (b) Freundlich. The point in the figure is the experimental value; while the line represents the calculated value of the model. | ||
| T (K) | Langmuir | Freundlich | ||||
|---|---|---|---|---|---|---|
| qm (mg g−1) | KL | R2 | KF | n | R2 | |
| 293 | 697 | 0.0201 | 0.9397 | 27.19 | 1.34 | 0.9883 |
| 303 | 776 | 0.0221 | 0.9302 | 43.54 | 1.52 | 0.9827 |
| 313 | 821 | 0.0242 | 0.9412 | 47.65 | 1.53 | 0.9756 |
3.3 Characterization of XG-g-PAA/GO composite hydrogel
As shown in Fig. 10, the prepared composite hydrogel was brown and uniform in color, and had certain mechanical strength. | ||
| Fig. 10 The appearance of XG-g-PAA/GO composite hydrogel. | ||
The SEM results of XG-g-PAA/GO composite hydrogel are shown in Fig. 11. It was found that the synthesized XG-g-PAA/GO composite hydrogel had a rough surface, multiple folds, and obvious porous structures. These features could enhance the absorption of other compounds more rapidly. The synthesis roadmap of the hydrogels is shown in Fig. 12.
 | ||
| Fig. 11 SEM image of XG-g-PAA/GO composite hydrogel. | ||
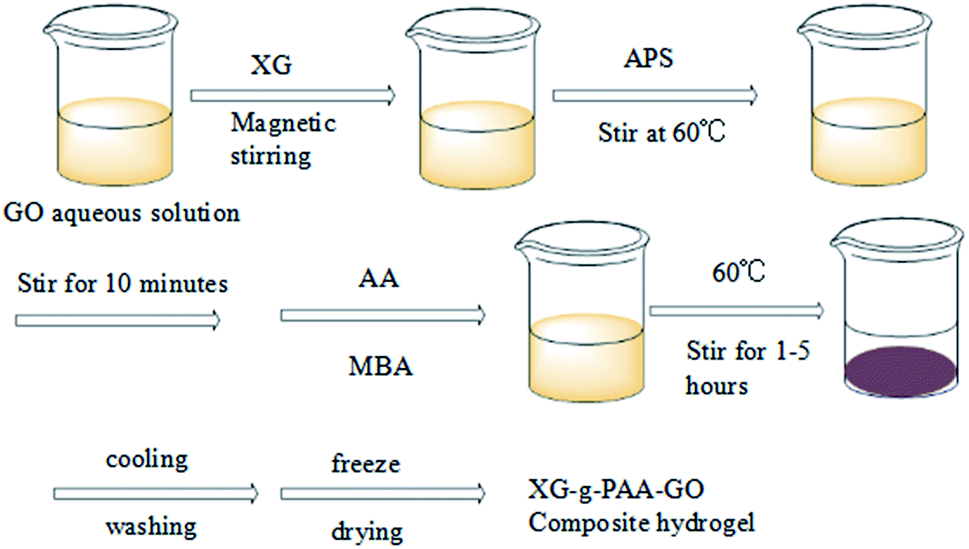 | ||
| Fig. 12 The synthesis roadmap of hydrogels. | ||
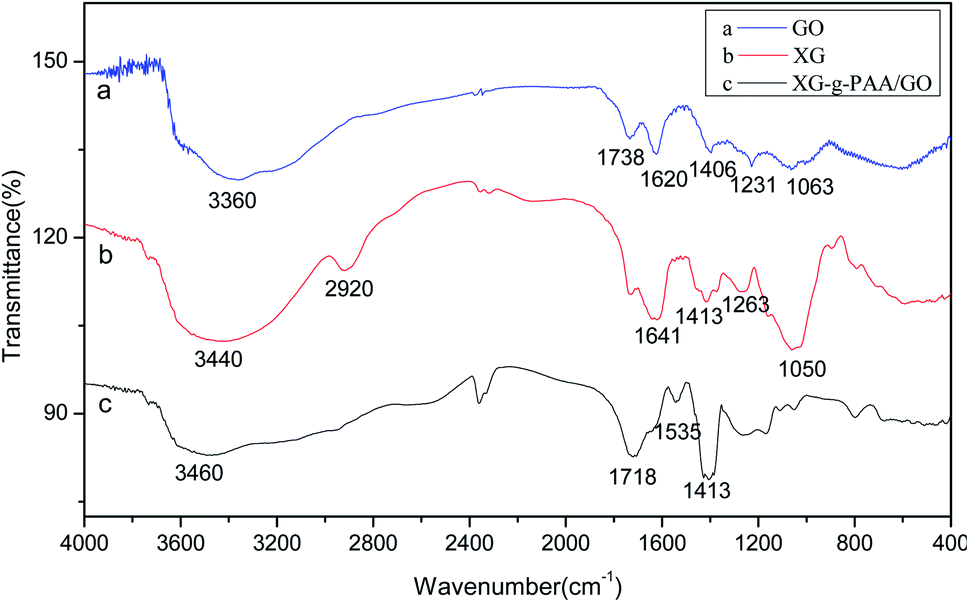
The infrared spectra of GO (a), XG (b) and XG-g-PAA/GO (c) are displayed in Fig. 13. Notably, the CO stretching peak of carboxyl group at 1738 cm−1, CC absorption peak at 1620 cm−1, and C–OH peak of carboxyl group at 1406 cm−1 were observed in the infrared spectrum of GO. These stretching vibration absorption peaks imply that GO has oxygen-containing groups. The absorption peaks of C–O group appeared at 1231 and 1063 cm−1. For the infrared spectrum of XG, the absorption peak at 3440 cm−1 was the primary and secondary –OH characteristic stretching vibrations. The absorption band at 2920 cm−1 could be attributed to the –CH stretching of alkyl group. The absorption peaks at 1641 and 1413 cm−1 were assigned to the asymmetric and symmetric COO-stretching vibrations of carboxylate group, respectively. The absorption peaks at 1263 and 1050 cm−1 were attributed to C–O bending vibration. Besides, the infrared spectrum of XG-g-PAA/GO composite hydrogel showed an increase in the peak intensity of the characteristic band at 1413 cm−1, which might be ascribed to the symmetric stretching mode of carboxylate group. These spectral data revealed the successful formation of XG-g-PAA/GO composite hydrogel. The peak at 1535 cm−1 was attributed to the CO asymmetric stretching of acrylic acid in the composite hydrogel. In addition, for XG, there was a broad absorption peak at 3440 cm−1, and after surface modification with acrylic acid, a new broad peak was emerged at 3200 cm−1. These results indicate that the copolymer of acrylic acid has been well grafted onto XG.
 | ||
| Fig. 13 The infrared spectra of GO (a), XG (b) and XG-g-PAA/GO (c). | ||
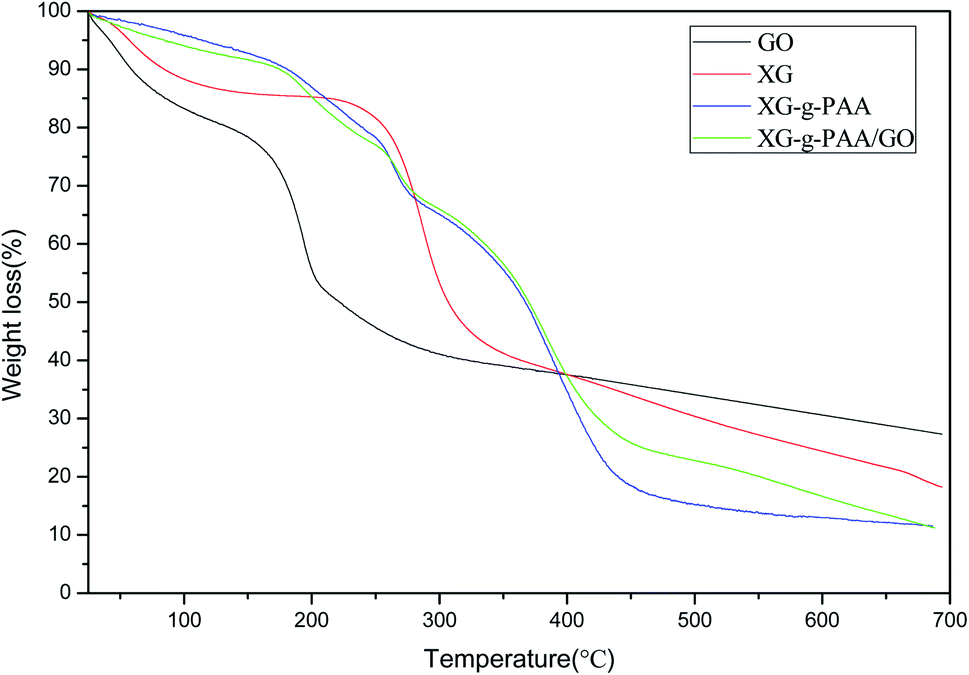
The results of thermogravimetric analysis (TGA) are revealed in Fig. 14. The TGA curve of GO demonstrated a weight loss in the range of 25–200 °C, probably due to the gradual pyrolysis of numerous oxygen-containing functional groups on GO surface, thereby producing CO2 and CO gases. The TGA curve of XG showed that the first degradation occurred in the range of 30–120 °C was associated with the endothermic peak, which might be due to the elimination of water. As the temperature increased to 200–300 °C, the weight loss rate also increased. The TGA curves of XG-g-PAA hydrogel and XG-g-PAA/GO composite hydrogel revealed similar trends of weight loss. The first decomposition stage was noted in the range of 250–300 °C. Combined with the TGA curve of XG, it can be speculated that the weight loss in this range is due to the decomposition of XG framework. In the curves of XG-g-PAA hydrogel and XG-g-PAA/GO composite hydrogel, the second weight loss between 375–450 °C was the consequence of residual polymer degradation. The TGA curve vividly indicated that the thermal stability of XG-g-PAA hydrogel and XG-g-PAA/GO composite hydrogel was higher than that of raw material XG at the temperature range of 25–400 °C. Thus, it can be inferred that the thermal stability of XG is increased after formation of the hydrogel.
 | ||
| Fig. 14 Thermogravimetric analysis curves of GO, XG, XG-g-PAA and XG-g-PAA/GO. | ||
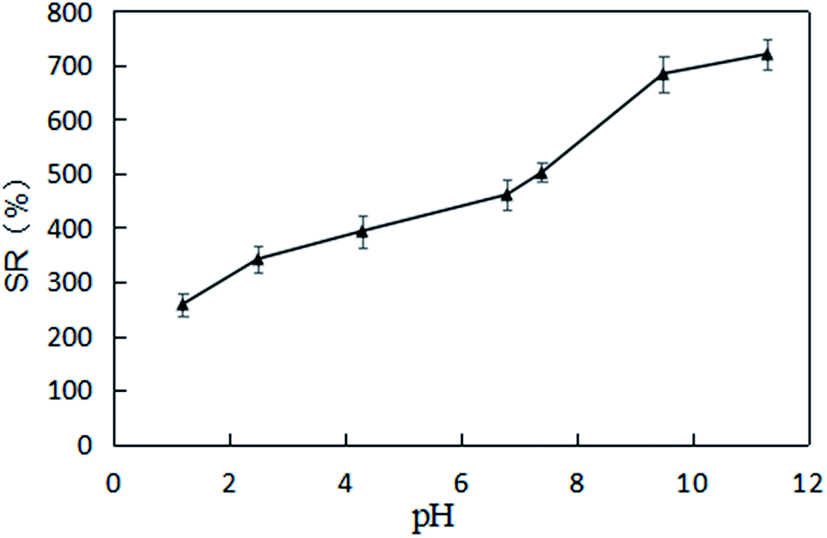
The experimental results for the effects of solution pH value on the swelling ratio of XG-g-PAA/GO composite hydrogel are presented in Fig. 15. Notably, the swelling ratios of XG-g-PAA/GO composite hydrogel were significantly increased with increasing pH values. The possible reason is that under a low pH environment, the carboxylate ions of XG-g-PAA/GO composite hydrogel are protonated, the electrostatic repulsion is weakened, and hydrogen bonds are formed between the carboxyl groups generated by protonation, which in turn leads to the formation of hydrogen bonds in the composite hydrogel. The network structure of XG-g-PAA/GO composite hydrogel is in a contracted state, the external liquid is not easy to enter the hydrogel, and the swelling ratio is relatively low. As the pH value continues to increase, the hydrogen bond is destroyed, the carboxyl group gradually dissociates into carboxylate ions, and the electrostatic repulsion is enhanced. Consequently, the network structure of the gel gradually expands to facilitate the external solution to enter the voids of the hydrogel, and thus, the swelling ratio is also increased.
 | ||
| Fig. 15 Curve of the swelling ratios of XG-g-PAA/GO composite hydrogel under different pH values. | ||
3.4 Drug loading of XG-g-PAA/GO composite hydrogel
The dose optimization results of the drug-loaded XG-g-PAA/GO composite hydrogel are summarized in Table 3. The XG-g-PAA/GO composite hydrogel was used as a carrier, the non-steroidal anti-inflammatory drug DCFP was used as a loading drug, and the loading efficiency was calculated to determine the best prescription dosage. The results showed that when the drug concentration was 10 mg mL−1, the drug loading capacity of the composite hydrogel was the largest (10.57%), with the encapsulation rate of 50.48%. The prescription with the largest drug loading was considered as the optimal prescribed dose. It can be seen from Table 3 that the drug loading capacity increased with increasing DCFP concentrations, but when the concentration was higher than 10 mg mL−1, the drug loading capacity decreased. Hence, the best prescription dose was determined to be 10 mg mL−1.| C (mg mL−1) | LE (%) | EE (%) |
|---|---|---|
| 1.25 | 1.88 | 44.15 |
| 2.5 | 3.49 | 53.12 |
| 5 | 8.49 | 57.32 |
| 10 | 10.57 | 50.48 |
| 15 | 10.31 | 50.70 |
3.5 In vitro DCFP release test
The in vitro drug release test results of XG-g-PAA/GO–DCFP composite hydrogel are summarized in Fig. 16. Under the conditions of simulated gastric and intestinal juice, the cumulative release of DCFP after 96 h was 34.57% and 68.41%, respectively. The drug embedded in the composite hydrogel was less released in the stomach, but was released well in the intestine. According to the test results of the swelling ratio of XG-g-PAA/GO composite hydrogel in different pH buffer solutions, it was found that the swelling ratios of the composite hydrogel elevated with increasing pH values. A high pH value is conducive to the entry of the external solution, so that the drug can be well released. In addition, the higher the pH values, the stronger the electrostatic repulsion between the anions ionized by DCFP and GO, which is also conducive to the release of DCFP. Therefore, the amount of drug released by the composite hydrogel is much higher in the artificial intestinal fluid at a high pH value, which in turn protects the drug from being released in the stomach and avoids the side effects of drug on the stomach.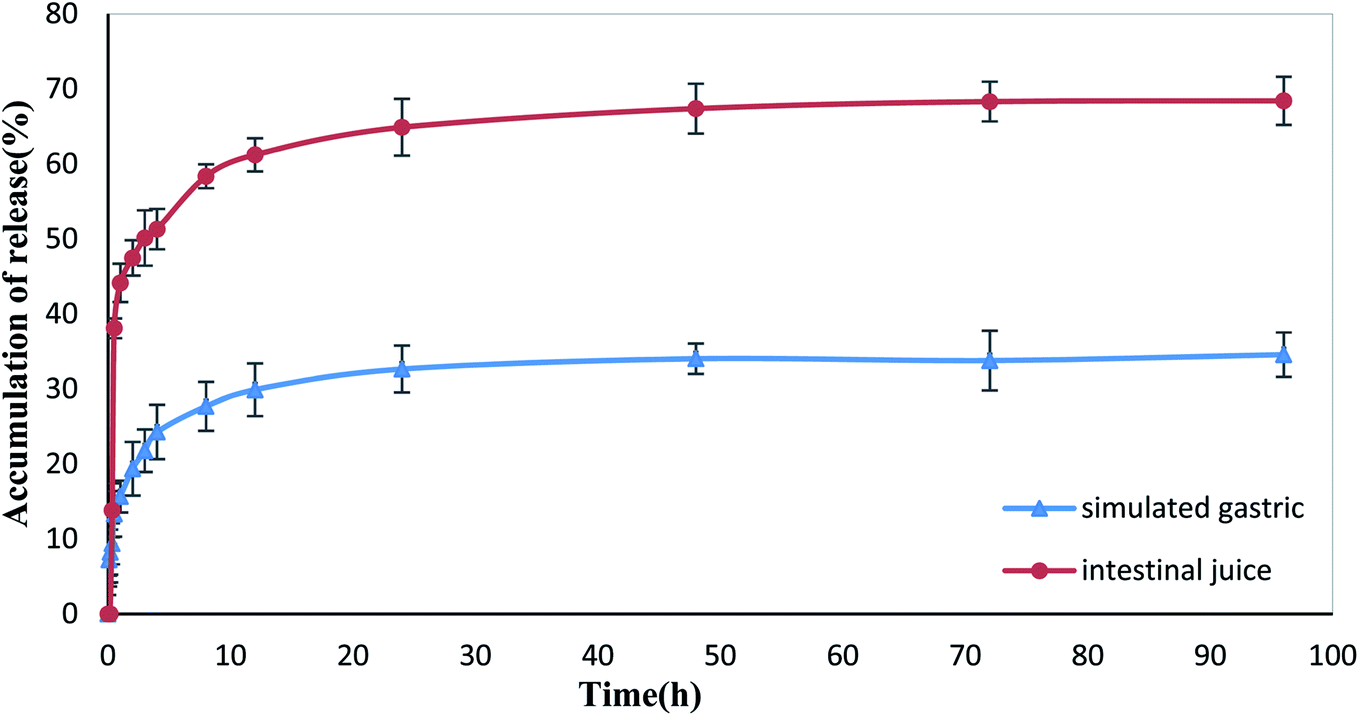 | ||
| Fig. 16 Drug release curves of XG-g-PAA/GO composite hydrogel in the simulated gastric and intestinal conditions. | ||
3.6 Pharmacokinetic studies
The plasma concentration–time curves obtained from the oral administration of DCFP and XG-g-PAA/GO–DCFP composite hydrogel to the rats are displayed in Fig. 17, and the basic pharmacokinetic parameters are listed in Table 4. From the figure, it can be seen that the blood drug concentration was not detectable in both DCFP and XG-g-PAA/GO–DCFP composite hydrogel groups within 1 h. This is because the rat enteric-coated capsules could not be degraded in the stomach. There is almost no release of the drug in the stomach. The t1/2 of DCFP group was 2.03 ± 0.35 h, and that of XG-g-PAA/GO–DCFP composite hydrogel group was 10.71 ± 2.04 h. The blood drug concentration of DCFP group was nearly undetectable at 10 h, while that of XG-g-PAA/GO–DCFP composite hydrogel group remained detectable at 24 and 48 h. This indicates that the elimination rate of DCFP is fast, and the composite hydrogel can slow down the elimination rate of the drug in the body, thus producing a sustained release effect. The AUC(0-t) of DCFP group was 53.99 ± 3.18 mg L−1 h−1, and that of XG-g-PAA/GO–DCFP composite hydrogel group was 116.79 ± 14.72 mg L−1 h−1, which was about 2.16 times higher than that of DCFP group. Therefore, it can be concluded that the bioavailability of DCFP in the rat body is low after oral administration, and the preparation of XG-g-PAA/GO–DCFP composite hydrogel can greatly improve its bioavailability and stability.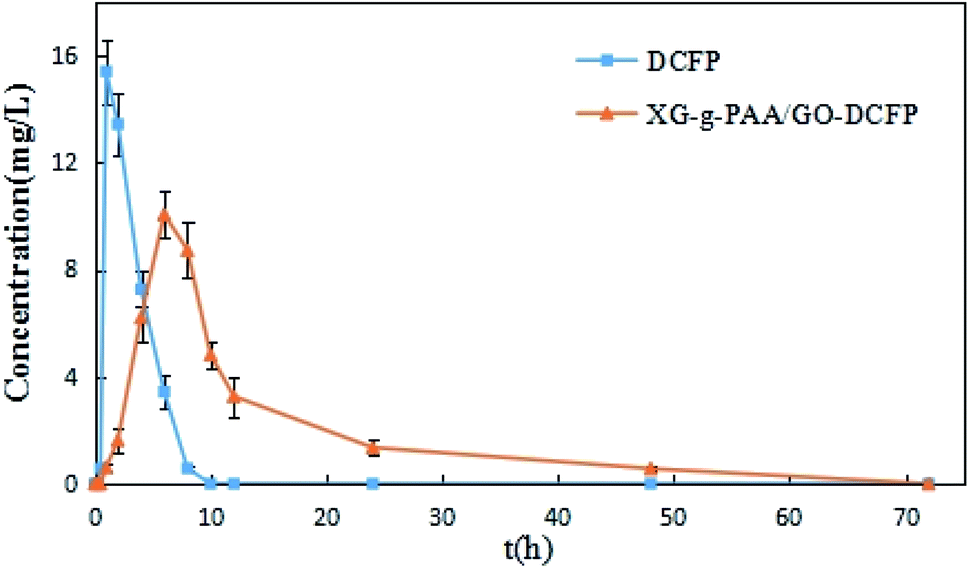 | ||
| Fig. 17 The in vivo plasma concentration versus time curves of DCFP and XG-g-PAA/GO–DCFP through oral administration. | ||
| Parameters | Unit | DCFP | XG-g-PAA/GO–DCFP |
|---|---|---|---|
| AUC(0–t) | mg L−1 h−1 | 53.99 ± 3.18 | 116.79 ± 14.72 |
| AUC(0–∞) | mg L−1 h−1 | 59.22 ± 5.23 | 123.90 ± 12.14 |
| t1/2 | h | 2.03 ± 0.35 | 10.71 ± 2.04 |
| Tmax | h | 1.33 ± 0.52 | 6.67 ± 1.03 |
| Cmax | mg L−1 | 15.52 ± 1.05 | 10.15 ± 0.90 |
4. Conclusion
First, we successfully synthesized nano-level GO using the improved Hummers method and ultrasonic peeling method. The non-steroidal anti-inflammatory drug DCFP was selected as the model drug to investigate the adsorption of GO to DCFP under different conditions, and different theoretical models were employed to fit the adsorption kinetics and adsorption isotherm model to further explore the adsorption mechanism of GO towards DCFP. The results showed that the adsorption capacity of GO on DCFP decreased with increasing pH values. By integrating the fitting results of adsorption kinetics and adsorption isotherm models, the pseudo-second-level kinetic model and Freundlich model could better delineate the adsorption process of DCFP on GO. It is speculated that a variety of adsorption mechanisms co-exist during the adsorption process. The most dominant mechanisms are π–π stacking and hydrophobic interaction, and to a lesser extent, hydrogen bonds and electrostatic repulsion. The pH-sensitive XG-g-PAA/GO composite hydrogel was successfully synthesized as a carrier, loaded with DCFP, and the loading efficiency and encapsulation efficiency for its optimal formulation were 10.57% and 50.48%, respectively. The results of in vitro release experiment demonstrated that the cumulative release of DCFP was higher in simulated intestinal fluid (68.41%) than in simulated gastric juice (34.57%) after 96 h. This implies that drug loading into a composite hydrogel can avoid the side effects of drugs on the stomach. Pharmacokinetic data revealed that XG-g-PAA/GO–DCFP composite hydrogel group had longer t1/2, and the AUC(0–t) was approximately 2.16 times higher than that of DCFP group. XG-g-PAA/GO–DCFP composite hydrogel enables DCFP to be gradually released in the body over time, thereby prolonging the action time of the drug in the body and greatly improving the bioavailability of DCFP in the body. In summary, the proposed XG-g-PAA/GO composite hydrogel has stable structure, uniform color, and pH sensitivity. The oral route of XG-g-PAA/GO–DCFP composite hydrogel can effectively prolong the action time of DCFP and enhance its bioavailability. Hence, this composite hydrogel can be applied as a sustained-release drug carrier for further research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their gratitude to EditSprings (https://www.editsprings.com/) for the expert linguistic services provided. The study was supported by the Natural Science Foundation of Liaoning province (20180510016, 2019-MS-153) and LiaoNing Revitalization Talents Program (XLYC2007054).References
- S. F. Kiew, L. V. Kiew, H. B. Lee, T. Imae and L. Y. Chung, J. Controlled Release, 2015, 217–228 Search PubMed.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061–1105 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- S. S. Nanda, G. C. Papaefthymiou and D. K. Yi, Crit. Rev. Solid State Mater. Sci., 2015, 40, 291–315 CrossRef CAS.
- M. de Sousa, H. Z. Martins Carlos and S. Franqui Lidiane, J. Mater. Chem. B, 2018, 6, 2803–2812 RSC.
- L. M. Veca, F. Lu, M. Meziani, L. Cao, P. G. Zhang and S. Y. P. Shrestha, Chem. Commun., 2009, 14, 2565–2567 RSC.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC.
- S. Bullo, K. Buskaran, R. Baby, D. Dorniani, S. Fakurazi and M. Z. Hussein, Pharm. Res., 2019, 36, 91 CrossRef PubMed.
- H. Tiwari, N. Karki, M. Pal, S. Basak, R. Kumar, V. Rajaram, B. Narain, D. Kandpal, G. Bisht and N. S. Gopal, Colloids Surf., B, 2019, 178, 452–459 CrossRef CAS PubMed.
- X. Q. Sun, D. Liu, X. Y. Xu, Y. F. Shen, Y. J. Huang, Z. S. Zeng, M. Xia and C. S. Zhao, Asian J. Pharm. Sci., 2020, 15, 713–727 CrossRef PubMed.
- P. Calvert, Adv. Mater., 2009, 21, 743–756 CrossRef CAS.
- T. Serizawa, K. Wakita and M. Akashi, Macromolecules, 2002, 35, 10–12 CrossRef CAS.
- A. B. Imran, T. Seki, K. Ito and Y. Takeoka, Macromolecules, 2010, 43, 1975–1980 CrossRef.
- T. U. Rehman, S. Bibi, M. Khan, I. Ali, L. A. Shah, A. Khan and M. Ateeq, RSC Adv., 2019, 9, 40051–40061 RSC.
- M. Khan, L. A. Shah, M. A. Khan, N. S. Khattak and H. B. Zhao, Mater. Sci. Eng., C, 2020, 116, 111278 CrossRef CAS PubMed.
- M. Khan, L. A. Shah, T. Rehman, A. Khan, A. Iqbal, M. Ullah and S. Alam, Iran. Polym. J., 2020, 29, 351–360 CrossRef CAS.
- L. A. Shah, T. U. Rehman and M. Khan, Polym. Bull., 2020, 77, 3921–3935 CrossRef CAS.
- F. Li, X. Jiang, J. Zhao and S. Zhang, Nano Energy, 2015, 16, 488–515 CrossRef CAS.
- F. García-Ochoa, V. E. Santos, J. A. Casas and E. Gómez, Biotechnol. Adv., 2000, 18, 549–579 CrossRef.
- H. Santos, F. Veiga, M. E. Pina and J. Sousa, Int. J. Pharm., 2005, 295, 17–27 CrossRef PubMed.
- J. Fan, K. Wang, M. Liu and Z. He, Carbohydr. Polym., 2008, 73, 241–247 CrossRef CAS.
- S. D. Jadhav, S. R. Butle, S. D. Patil and P. K. Jagtap, Arabian J. Chem., 2015, 8, 118–128 CrossRef CAS.
- J. R. Zuniga, H. Malmström, R. J. Noveck, J. H. Campbell, S. Christensen, R. S. Glickman, B. J. Tomasetti and S. E. Boesing, J. Oral Maxillofac. Surg., 2010, 68, 2735–2742 CrossRef PubMed.
- H. C. Diener, P. Montagna, G. Gács, P. Lyczak and K. Edson, Cephalalgia, 2010, 26, 537–547 CrossRef PubMed.
- W. McNeely and K. L. Goa, Drugs, 1999, 57, 991–1003 CrossRef CAS PubMed.
- S. A. Chime, A. A. Attama, P. F. Builders and G. C. Onunkwo, J. Microencapsulation, 2013, 30, 335–345 CrossRef CAS PubMed.
- H. Freundlich, Z. Phys. Chem., 1907, 57, 385–470 CrossRef CAS.
- M. Xu, Y. H. Mou, M. M. Hu, W. X. Dong, X. T. Su, R. X. Wu and P. Zhang, Asian J. Pharm. Sci., 2018, 13, 373–382 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01012c |
| This journal is © The Royal Society of Chemistry 2021 |