
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmergent antibacterial activity of N-(thiazol-2-yl)benzenesulfonamides in conjunction with cell-penetrating octaarginine†
Poonam Ratreya,
Amarjyoti Das Mahapatrab,
Shiny Panditc,
Murtuza Hadianawalab,
Sasmita Majhia,
Abhijit Mishra a and
Bhaskar Datta*bc
a and
Bhaskar Datta*bc
aDepartment of Materials Science and Engineering, Indian Institute of Technology, Gandhinagar, Gujarat, India
bDepartment of Chemistry, Indian Institute of Technology, Gandhinagar, Gujarat, India. E-mail: bdatta@iitgn.ac.in; Fax: +91-79-2397-2622; Tel: +91-79-2395-2073
cDepartment of Biological Engineering, Indian Institute of Technology, Gandhinagar, Gujarat, India
First published on 25th August 2021
Abstract
Hybrid antimicrobials that combine the effect of two or more agents represent a promising antibacterial therapeutic strategy. In this work, we have synthesized N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide derivatives that combine thiazole and sulfonamide, groups with known antibacterial activity. These molecules are investigated for their antibacterial activity, in isolation and in complex with the cell-penetrating peptide octaarginine. Several of the synthesized compounds display potent antibacterial activity against both Gram-negative and Gram-positive bacteria. Compounds with 4-tert-butyl and 4-isopropyl substitutions exhibit attractive antibacterial activity against multiple strains. The isopropyl substituted derivative displays low MIC of 3.9 μg mL−1 against S. aureus and A. xylosoxidans. The comparative antibacterial behaviour of drug–peptide complex, drug alone and peptide alone indicates a distinctive mode of action of the drug–peptide complex, that is not the simple sum total of its constituent components. Specificity of the drug–peptide complex is evident from comparison of antibacterial behaviour with a synthetic intermediate–peptide complex. The octaarginine–drug complex displays faster killing-kinetics towards bacterial cells, creates pores in the bacterial cell membranes and shows negligible haemolytic activity towards human RBCs. Our results demonstrate that mere attachment of a hydrophobic moiety to a cell penetrating peptide does not impart antibacterial activity to the resultant complex. Conversely, the work suggests distinctive modes of antibiotic activity of small molecules when used in conjunction with a cell penetrating peptide.
1. Introduction
Bacterial infectious diseases are a significant cause of morbidity and mortality worldwide. Methicillin-resistant Staphylococcus aureus (MRSA), the first superbug, is said to cause approximately 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths annually in the United States alone.1 Similarly, Helicobacter pylori (H. pylori) affects more than half of the global population.2 The multidrug resistance of ESKAPE nosocomial pathogens is now accepted.3,4 ESKAPE is an acronym for Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species, that are collectively known to cause multitude of pandemic infections. Such life-threatening infections include meningitis, bacteremia, tuberculosis, pneumonia, typhoid, urinary tract infections and gonorrhea.5–7 Bacterial resistance to antimicrobial agents is not new but attracts significantly more attention now than ever before owing to the growing human population and improvements in overall life-expectancy. There is perennial need for research into effective and novel antimicrobial agents as well as targeting and delivery strategies.
000 deaths annually in the United States alone.1 Similarly, Helicobacter pylori (H. pylori) affects more than half of the global population.2 The multidrug resistance of ESKAPE nosocomial pathogens is now accepted.3,4 ESKAPE is an acronym for Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species, that are collectively known to cause multitude of pandemic infections. Such life-threatening infections include meningitis, bacteremia, tuberculosis, pneumonia, typhoid, urinary tract infections and gonorrhea.5–7 Bacterial resistance to antimicrobial agents is not new but attracts significantly more attention now than ever before owing to the growing human population and improvements in overall life-expectancy. There is perennial need for research into effective and novel antimicrobial agents as well as targeting and delivery strategies.
Therapeutic approaches to mitigate the drug-resistance phenomena include reasonably straight-forward combination therapy and use of derivatives of established drugs. More sophisticated strategies such as use of hybrid-antimicrobials, therapeutic adjuvants and nanotechnology-based constructs have been the subject of intensive research in recent years.8–12 In combination therapy, two or more antimicrobial agents are concomitantly administered. Nanotechnology-based approaches have enabled antimicrobial interventions by offering a plethora of delivery options and mechanisms of drug release.13 Other notable materials that have emerged in the context of innovative antimicrobial agents include siderophore–antibiotic conjugates, antimicrobial peptides, peptoids, and antimicrobial polymers.14–18 Siderophores are the small-sized iron chelators secreted by bacteria to transport iron(III) across the cell membrane, and a uniquely built-in internalization pathway exists for iron-scavenging. Taking advantage of the inherent mode of entry of siderophore derivatives, recently, antibiotics have been covalently linked with them to directly deliver antibiotics to the cytoplasm to circumvent the membrane-mediated resistance.19 Antimicrobial peptides (AMPs) are short peptides with an ability to disrupt the membrane integrity or inhibit vital cellular functions. It is hypothesized that they create punctures or holes in bacterial cell membranes leading to cell death and hence the development of resistance is abated due to a novel mechanism of action.20
The mentioned strategies, while promising, are constrained by various limitations including in vivo instability, unsuitable production costs, cytotoxicity, emergence of new resistance mechanisms and unsatisfactory translation to in vivo contexts. Hybridization of antimicrobial agents with a second functional moiety, especially peptides and nucleic acids, displays significant promise as a next-generation strategy. In particular, the use of cell-penetrating peptides (CPPs) as the second functional entity in the hybrid antibacterials has garnered immense interest.9,21–24 AMPs and CPPs fall into the category of membrane-active peptides, where AMPs kill cells while CPPs carry cargos across lipid bilayers.25 Cell-penetrating peptides (CPPs) are short peptides comprising of 5–30 amino acids, usually cationic and hydrophilic, and possess a unique capability of translocating across the cellular-membrane barrier.26 CPPs have been used as a delivery vehicle for various drugs with intracellular targets as well as for intracellular imaging applications.27,28 A detailed survey of the literature shows that they have been majorly used for mammalian cells and not for bacterial cells. Further, their activity is consistently measured in the context of established drugs or natural products. In this work, we study the antimicrobial activity of a new series of N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide derivatives in conjunction with octaarginine CPP.
Substituted thiazole and benzothiazole derivatives have been previously reported as possessing antibacterial properties.29–31 Several sulfonamide derivatives have been extensively studied as antibacterial agents.32–35 Antibacterial sulfonamides have been suggested as competitive inhibitors of enzyme dihydropteroate synthetase (DHPS), whose activity plays an important role for the synthesis of folate necessary in cells for the synthesis of nucleic acids.36 Inhibition of DHPS does not allow bacterial cell division, which results in a bacteriostatic effect. In the present study we have designed and synthesized N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide scaffold by incorporating both thiazole and sulfonamide moieties in a single molecule (as shown in Fig. 1). While heterocyclic compounds possessing separate thiazole and sulfonamide moieties have been systematically studied for their antibacterial activity,37,38 there is a scarcity of reports on molecules combining both groups. Further, most small molecule antibacterial drugs are hydrophobic and display antibacterial behaviour upon dissolution in organic solvents. Clinically viable drugs require higher bioavailability and hence greater water solubility.
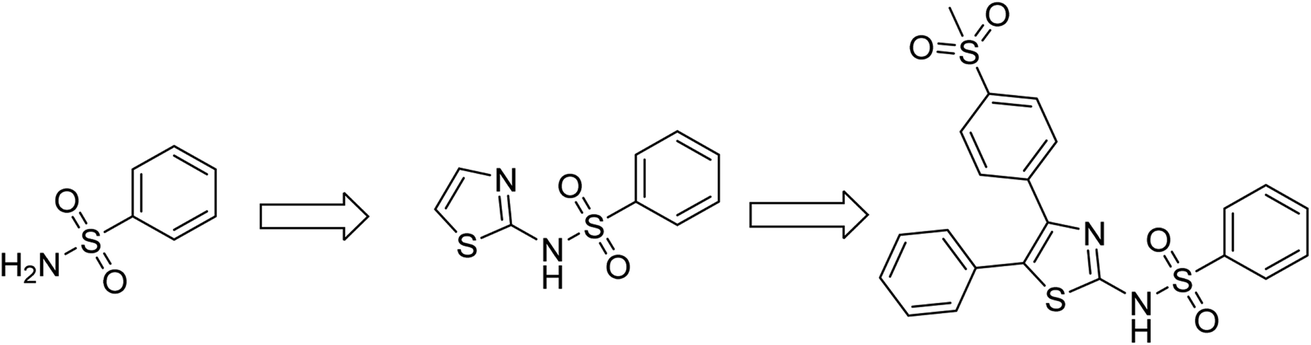 | ||
| Fig. 1 Thiazole and sulfonamide moieties in a single molecule. | ||
The present study employs a cell-penetrating peptide, an impressive but undervalued vector for bacterial cells, to form a sulfonamide based dual component molecular hybrid antibacterial. Interesting observations in terms of bioactivity of the drug–peptide complexes are presented, which augment the understanding of antibacterial drug-design harnessing combinatorial strategy. The work sheds light on antibacterial activity of hitherto untested small molecules alone and especially when used in conjunction with cell penetrating peptides.
2. Materials, cells and methods
Chemicals were procured from Sigma Aldrich or Merck or Alfa Aesar. Solvents were obtained from Merck Chemicals or SDFCL or FINAR and were used without further purification. Nuclear magnetic resonance (NMR) spectra were recorded on a 500 MHz Bruker Instrument using tetramethylsilane (TMS) as an internal reference. Chemical shifts were measured in ppm (δ) relative to TMS (0.00 ppm). Coupling constants (J) are reported in Hertz (Hz). Mass spectra were measured by LC-MS on a Waters SYNAPT-G2S-S using the electrospray ionization technique. Progress of the reactions were monitored by thin layer chromatography (TLC) analysis on silica gel plates. Octaarginine (R8) peptide was synthesized by standard fluorenylmethloxycarbonyl (FMOC) solid phase peptide synthesis method using rink amide resin and DIC (diisopropylcarbodiimide)-oxyma coupling reagents; characterized and purified by Mass spectrometry and preparative-HPLC respectively.Escherichia coli (ATCC 25922), Staphylococcus aureus (ATCC 25923), Bacillus subtilis (ATCC 6633), Staphylococcus epidermidis (MTCC 435), Paracoccus sp. (MTCC 11829), Achromobacter xylosoxidans (MTCC 1685), Escherichia coli K-12 (MTCC 1302) cells were collectively procured from National Collection of Industrial Microorganism (NCIM) and Microbial Type Culture Collection and Gene Bank (MTCC). Mueller Hintor agar and Mueller Hinton broth were obtained from Himedia, Mumbai India. Human erythrocytes or red blood cells (hRBCs) were procured from Unipath Specialty pathology laboratory Ahmedabad, India. RBCs from healthy persons of age group 22–30 years were obtained with their consent for research and experimental purpose. Cells were pelleted down and diluted with PBS (phosphate buffer saline) in the pathology lab. Haemolysis experiment was performed the same day without storage of the RBCs.
2.1 General procedure for the synthesis of 1-(4-(methylthio) phenyl)-2-phenylethan-1-one (1)
Thioanisole (3 mmol) was slowly added to a mixture of phenylacetyl chloride (3.25 mmol) and aluminium trichloride (3.85 mmol) in 5 mL of dry dichloromethane (DCM) under nitrogen gas atmosphere at 0–5 °C. Temperature of the reaction mixture was maintained for 2 h and then kept at room temperature for 2–3 h. Finally, the resulting reaction mixture was poured on crushed ice and acidified with diluted HCl (pH 5–6) followed by extraction with DCM. The organic extract was washed with saturated NaHCO3 solution followed by brine solution (saturated NaCl solution) and dried over anhydrous Na2SO4. Then organic layer was evaporated under vacuum and the resulting residue was finally purified by column chromatography to afford title compound 1 (80%). 1H NMR (CDCl3, 500 MHz): δ 7.92 (d, 2H, J = 8.5 Hz), 7.33–7.30 (m, 2H), 7.26–7.24 (m, 5H), 4.24 (s, 2H), 2.51 (s, 3H), 13C NMR (CDCl3, 125 MHz): 196.7, 146.1, 134.7, 132.9, 129.4, 129.1, 128.7, 126.9, 125.0, 45.4, 14.7 MS (ESI): m/z calculated [M + H]+: 243.0838 [M + H]+: 243.1024 (found).2.2 General procedure for the synthesis of 1-(4-(methylsulfonyl) phenyl)-2-phenylethan-1-one (2)
30% hydrogen peroxide (H2O2, 7 mmol) was slowly added to a mixture of compound 1 (2 mmol) in acetic acid (5 mL). The reaction mixture was then heated at 50 °C for 5–6 h. After completion of reaction, reaction mixture was cooled at room temperature and poured over crushed ice and extracted with DCM. Organic layer was then washed with saturated NaHCO3 solution, brine solution, dried over anhydrous Na2SO4 and evaporated under vacuum to obtain crude solid residue which was finally purified using column chromatography to get desire compound 2 (75%).1H NMR (CDCl3, 500 MHz): δ 8.17 (d, 2H, J = 8.4 Hz), 8.03 (d, 2H, J = 8.4 Hz), 7.36–7.33 (m, 2H), 7.29–7.26 (m, 3H), 4.32 (s, 2H), 3.07 (s, 3H), 13C NMR (CDCl3, 125 MHz): 196.4, 144.2, 140.5, 133.5, 129.5, 129.4, 128.9, 127.8, 127.3, 46.0, 44.3.
2.3 General procedure for synthesis of 2-bromo-1-(4-(methylsulfonyl)phenyl)-2-phenylethan-1-one (3)
To a solution of compound 2 (1.1 mmol) in DCM (5 mL) catalytic amount of HBr (0.1 mL) and a solution of Br2 in glacial acetic acid (1 mL, 7.5 mmol) were added with stirring and reaction mixture was refluxed for 5 h. Cooled reaction mixture was then poured over crushed ice and extracted with DCM. Organic layer was washed with saturated NaHCO3 solution, brine solution and dried over anhydrous Na2SO4. Solvent was evaporated under vacuum and the resulting crude residue was used for the next step without further purification.2.4 General procedure for the synthesis of 4-(4-(methylsulfonyl) phenyl)-5-phenylthiazol-2-amine (4)
Thiourea (0.39 mmol) and the crude residue obtained from the previous step was dissolved in ethanol (10 mL) and refluxed for 6 h. After completion of reaction, the reaction mixture was cooled at room temperature, poured in ice–cold water, and basified with saturated NaHCO3 solution (pH 8–9) followed by extraction with diethyl ether. The organic layer was washed with saturated NaHCO3 solution, brine solution and dried over anhydrous Na2SO4. The organic layer was removed under reduced pressure and the resulting crude residue was further purified using the column chromatography to obtain title compound 4 as a yellow colored solid (80%). 1H NMR (DMSO-d6, 500 MHz): δ 7.80 (d, 2H, J = 8.5 Hz), 7.61 (d, 2H, J = 8.5 Hz), 7.38–7.30 (m, 3H), 7.28–7.25 (m, 4H), 3.21 (s, 3H); 13C NMR (DMSO-d6, 125 MHz): 166.9, 143.4, 140.6, 139.6, 132.6, 129.7, 129.5, 129.4, 128.2, 127.2, 122.2, 43.9; MS (ESI): m/z calculated [M + H]+: 331.0569 [M + H]+: 331.0654 (found).2.5 General procedure for the synthesis of N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide (5a–5e)
We have recently reported the synthesis and SphK1 kinase inhibitory activity of phenylthiazolyl benzenesulfonamide derivatives.39 Suitable sulfonyl chloride (1.1 mmol) was added to a solution of compound 4 (0.6 mmol) in dry pyridine (7–8 mL) and the resultant mixture was stirred for 5 hours at room temperature. After the completion of the reaction, the reaction mixture was poured on crushed ice and acidified with diluted HCl (pH 5–6). The resulting solid was filtered and further purified using flash chromatography to obtain target compounds 5a–5e.2.6 4-Isopropyl-N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide (5a)
The title compound was synthesized as described above; yield 52%, 1H NMR (DMSO-d6, 500 MHz): δ 13.28 (brs, 1H), 7.90 (d, 2H, J = 8.0 Hz), 7.81 (d, 2H, J = 8.0 Hz), 7.60 (d, 2H, J = 8.0 Hz), 7.46 (d, 2H, J = 8.0 Hz), 7.38–7.27 (m, 5H), 3.26 (s, 3H), 3.00–2.95 (m, 1H), 1.22 (d, 6H, J = 6.5 Hz); 13C NMR (DMSO-d6, 125 MHz): 153.5, 151.7, 141.5, 140.2, 130.5, 130.3, 129.7, 129.4, 129.3, 127.6, 127.4, 126.6, 43.6, 33.8, 24.0, MS (ESI): m/z: calculated [M + H]+: 513.0971; found [M + H]+: 513.1001 [M + H]+.2.7 4-(tert-Butyl)-N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2yl)benzenesulfonamide (5b)
The title compound was synthesized as described above; yield 55%, 1H NMR (DMSO-d6, 500 MHz): δ 13.29 (brs, 1H), 7.91 (d, 2H, J = 8.5 Hz), 7.82 (d, 2H, J = 8.5 Hz), 7.62–7.59 (m, 4H), 7.39–7.38 (m, 3H), 7.28–7.26 (m, 2H), 3.26 (s, 3H), 1.31 (s, 9H); 13C NMR (DMSO-d6, 125 MHz): 166.4, 155.8, 141.7, 139.8, 134.5, 130.4, 130.3, 129.7, 129.4, 129.3, 127.6, 126.4, 126.3, 43.7, 35.3, 31.3; MS (ESI): m/z: calculated [M + H]+: 527.1127; found [M + H]+: 527.1125.2.8 4-Chloro-N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide (5c)
The title compound was synthesized as described above; yield 55%, 1H NMR (CDCl3, 500 MHz): δ 7.92 (d, 2H, J = 7.5 Hz), 7.84 (d, 2H, J = 8.0 Hz), 7.53 (d, 2H, J = 8.0 Hz), 7.45 (d, 2H, J = 8.0 Hz), 7.35–7.33 (m, 4H), 7.22 (d, 2H, J = 6.5 Hz), 3.06 (s, 3H); 13C NMR (CDCl3, 125 MHz): 167.2, 140.8, 140.6, 138.3, 134.4, 130.1, 129.9, 129.4, 129.2, 129.1, 129.1, 128.9, 127.9, 127.6, 122.4, 44.2; MS (ESI): m/z: calculated [M + H]+: 505.0112; [M + 2]+: 507.0180; found [M + H]+: 505.0101 [M + 2]+, 507.0057.2.9 4-Methyl-N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2yl)benzenesulfonamide (5d)
The title compound was synthesized as described above; yield 65%, 1H NMR (DMSO-d6, 500 MHz): δ 13.25 (brs, 1H), 7.91 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.0 Hz), 7.60 (d, 2H, J = 8.5 Hz), 7.40–7.39 (m, 5H), 7.27–7.27 (m, 2H), 3.26 (s, 3H), 2.39 (s, 3H); 13C NMR (DMSO-d6, 125 MHz): 143.1, 141.6, 139.7, 130.4, 130.3, 130.0, 129.7, 129.4, 129.3, 127.6, 126.5, 43.6, 21.4; MS (ESI): m/z: calculated [M + H]+: 485.0658; found [M + H]+: 485.0688.2.10 N-(4-(4-(Methylsulfonyl)phenyl)-5-phenylthiazol-2yl)benzenesulfonamide (5e)
The title compound was synthesized as described above; yield 58%, 1H NMR (CDCl3, 500 MHz): δ 7.95 (d, 2H, J = 7.5 Hz), 7.86 (d, 2H, J = 8.5 Hz), 7.56–7.50 (m, 3H), 7.46 (t, 2H, J = 8.0 Hz), 7.38–7.31 (m, 3H), 7.22 (d, 2H, J = 7.0 Hz), 3.04 (s, 3H); 13C NMR (CDCl3, 125 MHz): 166.4, 141.5, 141.1, 134.6, 132.5, 130.2, 129.5, 129.4, 129.4, 129.1, 128.9, 128.1, 126.6, 122.9, 44.3; MS (ESI): m/z: calculated [M + H]+: 471.0501; found [M + H]+: 471.0519.2.11 Complexation of peptide and drug
The peptide–drug complex was prepared by mixing aqueous peptide (1 mg) and ethanolic drug (0.5 mg) with stirring for 48 hours; ethanol was removed in vacuo followed by lyophilization to obtain a powder. The peptide to drug weight ratio was 2:1. This concentration was optimized such that there was no phase separation after ethanol removal and the complex is freeze-dried to obtain a homogenous single-phase product.
2.12 Antibacterial activity of the synthesized compounds by disk diffusion test
Antibacterial activity of all five N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide derivatives (5a–5e) and intermediate compound 4 were tested by disk diffusion method. Mid-log phase bacterial cultures, freshly prepared from full-grown overnight cultures, were streaked on Mueller Hinton agar plates. Sterile filter discs measuring 5.5 mm in diameter were mounted on agar plates followed by the addition of a known volume of test compounds into it. Chloramphenicol and DMSO were used as positive and negative controls, respectively. All potent compounds were tested for their antibacterial activity by disk/agar diffusion method using the drug's dilutions ranging from 8 mM to 1 mM in DMSO. After 18 hours of incubation at 37 °C, compounds were tested for their antibacterial activity by measuring the zone of inhibition (ZOI), expressed in millimeters (mm).2.13 Antibacterial activity by broth microdilution method
The bacterial cultures were grown in Mueller Hinton broth overnight till they reached the stationary phase. The stationary phase cultures were then diluted in broth and allowed to grow till the mid-log phase, for around two hours at 37 °C. Absorbance at 600 nm was read and optical density (OD) was adjusted to 0.5–0.7. A bacterial suspension of density 5 × 105 CFU per mL was prepared with mid-log phase bacteria by further diluting it with the broth.40 For minimum inhibitory concentration (MIC) determination, 10 μL of drug of varying concentrations, in two-fold dilutions were added, in quadruplets to 90 μL of the bacterial cell suspension and incubated for 16–18 hours at 37 °C. The data points in MIC plots represent mean ± SD for three independent experiments performed in quadruplets. Acetone was used to dissolve the bare sulfonamide derivatives as they were not soluble in water in its original form without peptide. Sterile Milli-Q water was used for peptide and peptide–drug complexes. Solvent without any drug added to the bacterial suspension serves as the positive control, while broth alone with the solvent is the negative control. Absorbance at 600 nm (OD600) was read, and the percentage of survival was calculated as below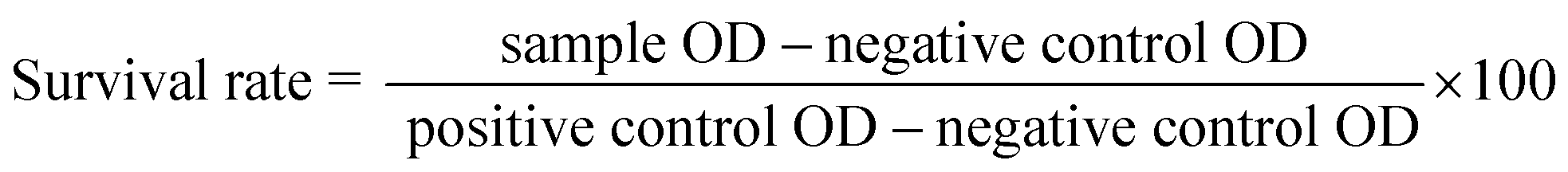
Checkerboard assay was performed by adding the combinations of R8 peptide and isopropyl substituted compound, 10 μL each to 80 μL volume of bacterial cells of density 5 × 105 CFU per mL. Peptide was diluted along the ordinate while isopropyl compound was diluted along the abscissa in a 96 microtiter well plate, and two-fold serial dilutions were prepared. 16× MIC initial concentration was used for the isopropyl drug while for R8 peptide 2× MIC was used as the starting concentration. After overnight incubation at 37 °C, OD600 was read where clear wells meant growth inhibition and turbidity signifies growth. This experiment was independently repeated thrice and positive and negative controls were kept similar to the MIC determination study. Fractional inhibitory concentration index (FICI) through the checkerboard assay was calculated using following relation:
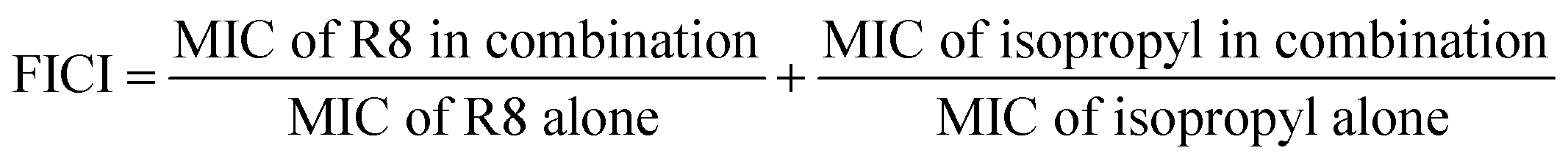
2.14 Killing-kinetics assay
Killing-kinetics or time-kill assay was conducted to compare the inhibitory activities of the peptide-based complex and the isopropyl compound with respect to time. A density of 5 × 105 CFU per mL bacterial cells was treated with 1× MIC and aliquots of the treated culture at specific time intervals were plated after dilution with sterile phosphate buffer saline of pH 7.4. The dilution factor was 50 and 20 μL volume was used for plating. The agar plates were incubated overnight at 37 °C and colonies were counted to find the reduction in viable cells. A plot of log10(CFU per mL) versus time points is plotted to assess the drug's kinetic performance of killing cells. The experiment was repeated thrice with a fixed initial inoculum size. A difference of only a few colonies was observed in between the trials which becomes negligible when logarithm is taken.2.15 Preparation of cells for scanning electron microscopy
Mid-logarithmic cells were treated with 1× MIC concentrations of the drugs for a period of 6 hours and cells were pelleted down and resuspended in 2.5% glutaraldehyde in PBS for fixation. After overnight fixation, cells were washed thrice with autoclaved Milli-Q water and lyophilized to obtain dry powder. A thin layer of cells was spread with clean brush onto the carbon tape glued over metallic stub and coated with platinum plasma under argon gas environment in a sputter coater. Scanning Electron Microscope (SEM) images were captured in JEOL 7600F Field Emission Scanning Electron Microscope at the Central Instrumentation Facility of IIT Gandhinagar.2.16 Assessment of haemolytic activity
Human RBCs diluted with 10 mM PBS to a final concentration of 2% was used for the haemolysis toxicity assay. Small modifications were made to the previous studies while designing this experiment.41,42 To 90 μL of RBCs suspension, 10 μL of drugs were added. Here we tested isopropyl–R8 complex in water, R8 peptide alone in water and isopropyl compound in acetone for their haemolytic activity in 1× MIC, 2× MIC and 4× MIC concentrations. Sterile Eppendorf tubes of volume 0.5 mL were used for this experiment. Compounds with RBCs were incubated for 1 hour at 37 °C with gentle shaking intermittently. Post-incubation, intact RBCs were pelleted down at 1100 rpm in a benchtop mini-centrifuge and 50 μL supernatant was collected, transferred to a 96 well plate and diluted with equal volume of PBS. 1% Triton-X served as positive control while respective solvents of drugs as negative controls. The assay was done in triplicates and independent experiments were done thrice, to calculate the error with standard deviation. Absorbance at 405 nm is read in a microplate reader (BioTek 800 TS) and % haemolytic activity is calculated as below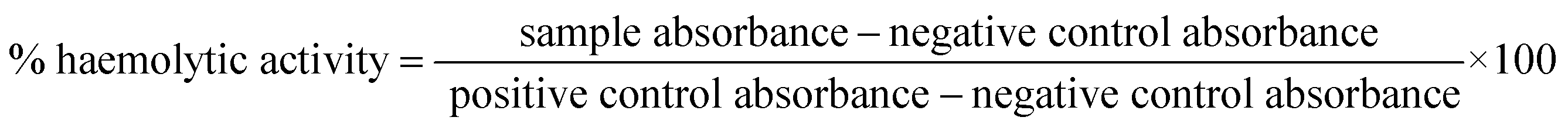
3. Results & discussion
3.1 Synthesis of N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide
In the present work, five derivatives of N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide with varying substitutions at para position of benzenesulfonamide containing phenyl ring, were synthesized according to the reported methods.39 Friedel Craft acylation of phenyl acetyl chloride with thioanisole in presence of aluminium chloride (AlCl3) and dichloromethane (DCM) as solvent was used for preparing compound 1, which on oxidation by hydrogen peroxide (H2O2) provided compound 2.43 Treatment of compound 2 with bromine in acetic acid and in the presence of hydrobromic acid (HBr) as a catalyst gave compound 3. A mixture of compound 3 and thiourea in ethanol was refluxed for preparing compound 4.44 It is a perfect example of Hantzsch's thiazole condensation reaction where an acyclic intermediate is formed by nucleophilic attack by sulfur on the carbon atom bearing the bromine atom, followed by cyclization and removal of water molecule lead the formation of thiazole derivatives. Target compounds 5a–5e were finally obtained by reaction of intermediate compound 4 with different sulfonyl chlorides in pyridine as shown in Scheme 1. All the synthesized molecules were characterized by different spectroscopic techniques such as 1H NMR, 13C NMR, and LC-MS mass spectrometry (ESI).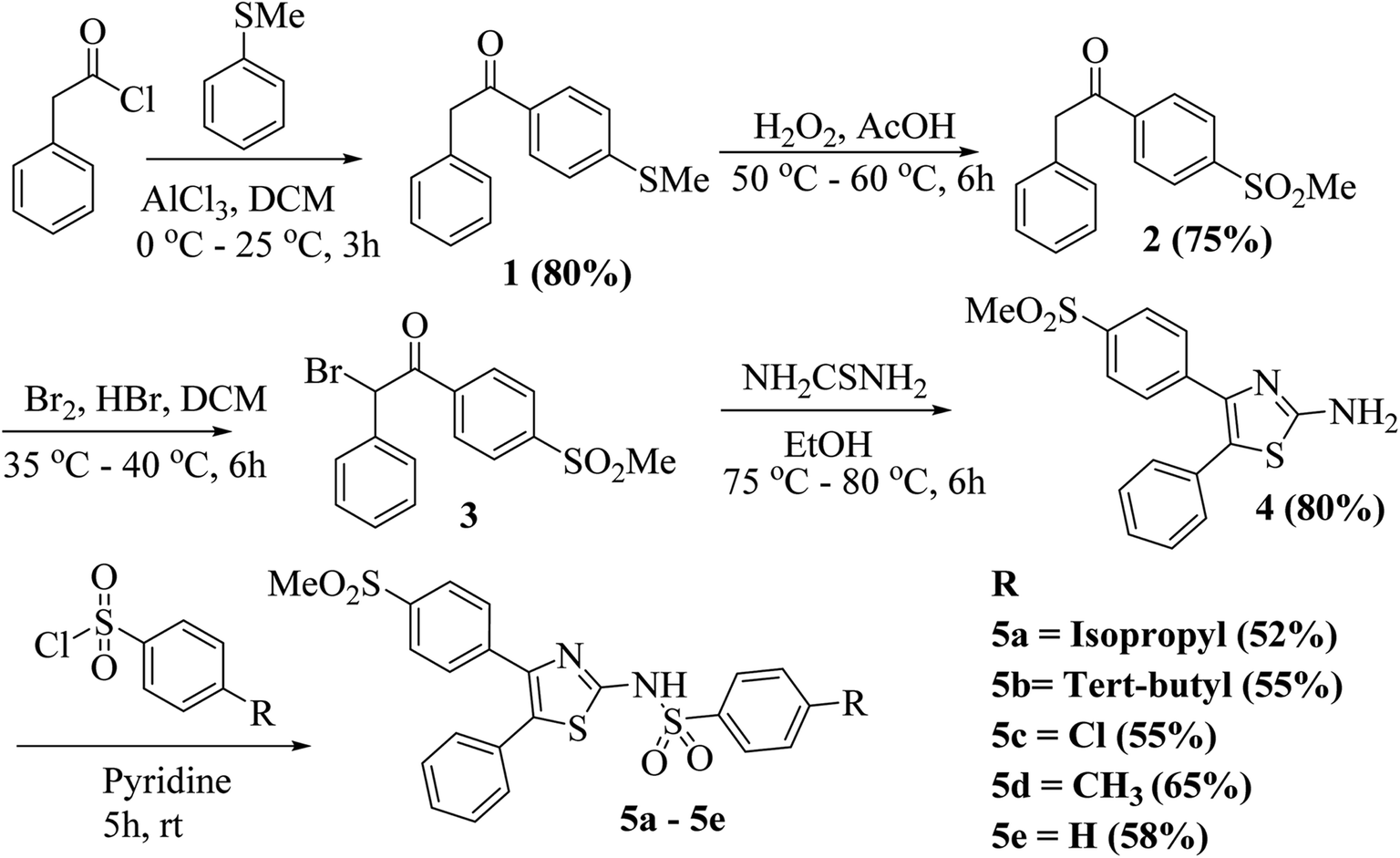 | ||
| Scheme 1 General synthetic scheme for the synthesis of N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide (5a–5e). | ||
3.2 Antibacterial activity of the synthesized compounds by disk diffusion test
Antibacterial activity for 4 and 5a–5e as assessed by the disk diffusion test are listed in Table 1 (refer to ESI† for the pictures of agar plates). Zone of inhibition implies antibacterial activity of the drugs while the hyphen marks in Table 1 represent no detectable ZOI. Screening results revealed all compounds, with the exception of intermediate 4, display antibacterial activity against E. coli while compounds 5a–5c were found to be active against S. aureus. Furthermore, the compounds 5a and 5b displayed antibacterial activity against B. subtilis and S. epidermidis. Among the synthesized compounds 5a and 5b exhibited attractive antibacterial activity, comparable to the standard chloramphenicol. Notably, the small molecule 5b bearing tert-butyl substituent displayed superior antibacterial activity in contrast to the alkyl derivatives. The availability of hydrophobic chain in this compound may be facilitating easier interaction and movement through the bacterial membrane. Such lipophilic basis of action may also justify the antibacterial activity of halide bearing compound 5c.| Compound name | Concentration (in mM) | Zone of inhibition (ZOI) in millimeters | |||
|---|---|---|---|---|---|
| E. coli | S. aureus | B. subtilis | S. epidermidis | ||
| –Isopropyl (5a) | 8 | 8 | 9 | 6 | 10.5 |
| 4 | 7.5 | 8 | — | 7 | |
| 2 | 7.5 | 6 | — | — | |
| 1 | 7 | — | — | — | |
| –tert-Butyl (5b) | 8 | 10.5 | 12 | 7 | 11 |
| 4 | 10 | 11 | — | 9.5 | |
| 2 | 9 | 10.5 | — | 8 | |
| 1 | 8 | 10 | — | — | |
| –Cl (5c) | 8 | 10 | 9 | — | — |
| 4 | 8.5 | 8 | — | — | |
| 2 | 7 | 6.5 | — | — | |
| 1 | — | — | — | — | |
| –CH3 (5d) | 8 | 9 | — | — | — |
| 4 | 8.5 | — | — | — | |
| 2 | 7 | — | — | — | |
| 1 | — | — | — | — | |
| –H (5e) | 8 | 8 | — | — | — |
| 4 | 7 | — | — | — | |
| 2 | — | — | — | — | |
| 1 | — | — | — | — | |
| Intermediate (4) | 8 | — | — | — | — |
| 4 | — | — | — | — | |
| 2 | — | — | — | — | |
| 1 | — | — | — | — | |
| Chloramphenicol | 8 | 19 | 16 | 25.5 | 23 |
| (Standard drug) | 4 | 15 | 13 | 23 | 21 |
| 2 | 10 | 10 | 19 | 20 | |
| 1 | 8 | 8 | 15 | 17 | |
3.3 Antibacterial activity of neat 5a by broth microdilution
Considering the broad activity observed for the isopropyl derivative (5a), MIC (minimum inhibitory concentration) values were analyzed by the method of broth microdilution. 5a and 5b emerged as the most active derivatives with the ZOIs (zones of inhibitions) of 5b pointing to a substantial antibacterial activity. We sought to select a scaffold that displayed broad activity without masking effect of peptide conjugation through an excessive antibacterial effect. Hence, 5a was chosen for physical conjugation/complexation experiments. The broth microdilution assay, also termed as MIC assay, is preferred in much lower concentration regimens, which cannot be effectively evaluated by the disk diffusion or ZOI assay.As shown in Fig. 2, 5a displayed potent antibacterial activity with concentration of 3.9 μg mL−1 inhibiting more than 90% bacterial growth. The drug is active against bacteria with rod and round or spherical-shaped morphology possessing Gram-positive as well as Gram-negative character. Results of the broth microdilution assay are summarized in Table 2. These results clearly indicate that 5a is active notwithstanding the curvature and chemical composition of bacterial membranes. Interestingly, 5a exerts absolutely no activity against E. coli at lower micromolar concentrations, used for the broth-dilution method, as shown in Fig. 2.
 | ||
| Fig. 2 Antibacterial activity of 5a against different bacteria in acetone solvent. | ||
| Sl. no. | Bacterial species | Activity of drug/MIC90 | Gram stain | Morphology |
|---|---|---|---|---|
| 1 | Staphylococcus aureus | Active (3.9 μg mL−1) | Positive | Spherical |
| 2 | Paracoccus sp. | Active (7.8 μg mL−1) | Negative | Rod (round in stationary phase) |
| 3 | Achromobacter xylosoxidans | Active (3.9 μg mL−1) | Negative | Rod shape |
| 4 | Escherichia coli ATCC 25922 | Not active | Negative | Rod shape |
| 5 | Escherichia coli K-12 | Not active | Negative | Rod shape |
3.4 Antibacterial studies with drug–peptide complexes
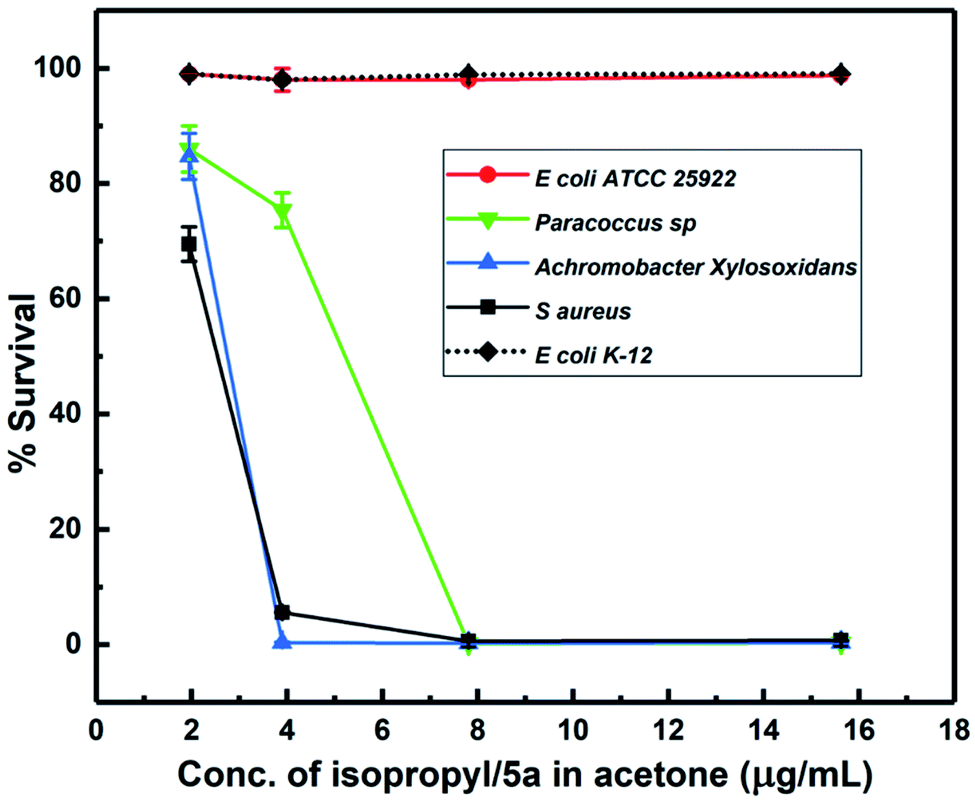
The anti-bacterial behavior of 5a provided us with a suitable candidate for testing in combination with a cell-penetrating agent. In particular, given the potent behavior of 5a against some bacterial strains while being inactive against others, we were interested in emergent properties of the drug when deployed in conjunction with a hydrophilic cell-penetrating peptide. Our working hypothesis relied on the hydrophilic cell-penetrating peptide rendering greater aqueous solubility to the drug while also actively enabling passage inside cells. We used a hydrophilic cell-penetrating peptide octaarginine (R8) to modify drug 5a non-covalently by physical complexation and subsequently studied antibacterial activity of the construct in aqueous media. The enhanced interaction of 5a–R8 conjugate with bacteria is expected based on increased intracellular delivery by crossing the cell membrane barrier or improved cell-membrane targeting. The antibacterial activity of the 5a–R8 complex was tested against S. aureus and E. coli. MIC assays showed that the complex almost completely inhibits the growth of S. aureus, while the neat 5a in water, without peptide complexation does not display inhibitory activity (Fig. 3(a)). The antibacterial activity of the peptide R8 is shown in Fig. 3(b), suggesting inhibition of bacteria only at high concentration ranges. In the complex form, we adjusted the dilution such that the peptide concentration was 100 μg mL−1 and lower.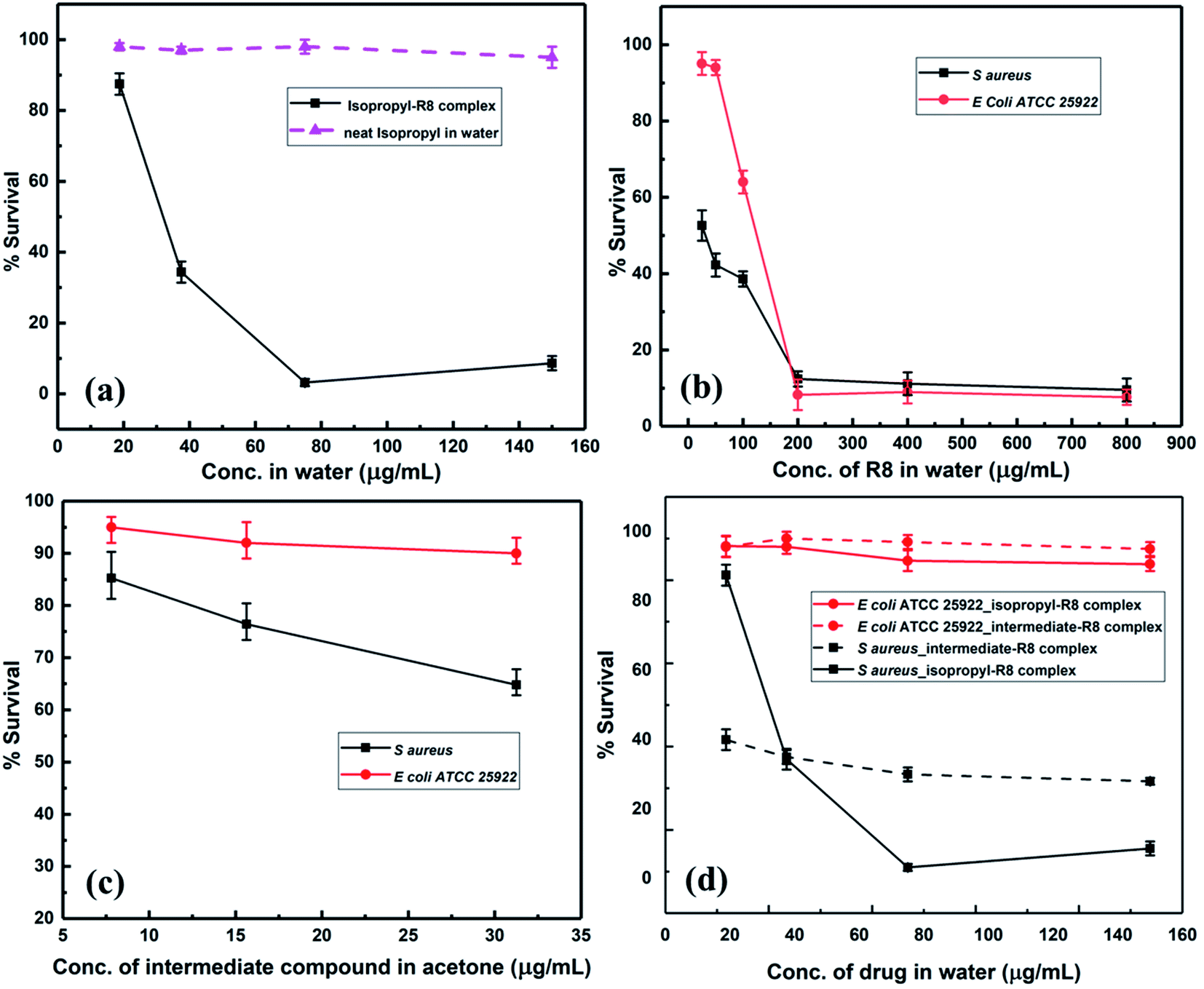 | ||
| Fig. 3 (a) Antibacterial activity of compound 5a in complex and neat form against S aureus. (b) Antibacterial activity of bare peptide in water. (c) Antibacterial activity of intermediate compound 4 in acetone. (d) Antibacterial activity of the complex of the peptide with 5a/isopropyl and 4/intermediate compounds in water. | ||
To obtain greater context for the performance of 5a–R8, we tested the synthetic intermediate 4 along similar lines. While intermediate compound 4 did not display any zone of inhibition when tested by disk diffusion method, this could be attributed to the limited sensitivity of the method as compared to the broth microdilution assay, as well as to the modest inhibition efficiency of the intermediate compound. As shown in Fig. 3(c), the compound 4 displayed modest activity against S. aureus when tested by broth microdilution. In the complex form, 4–R8 complex showed slightly enhanced activity against S. aureus while no activity against E. coli (Fig. 3(d)). Similarly, 5a–R8 complex displays absolutely no activity against E. coli cells (Fig. 3(d)). This behaviour follows the same pattern as bare 5a and the bare intermediate compound 4 (Fig. 2 and 3(c)). This indicates that the complex exerts antibacterial activity only when the drug itself is active against bacteria in its un-complexed form while the peptide functions as a carrier vehicle. From the comparative behavior of the 5a–R8 conjugate, 5a alone and R8 alone against S. aureus and E. coli, it is clear that the drug-cell penetrating peptide complex exerts a unique effect that is not a simple sum total effect of constituent components. Comparison of 4–R8 and 5a–R8 for their anti-bacterial action highlights the role of the chemical scaffold being used and confirms the selectivity of the peptide modified 5a drug complex. Nevertheless, from the cell penetrating peptide's perspective, the results suggest that mere attachment of a hydrophobic moiety is insufficient in imparting or enhancing antibacterial activity of the bare peptide. Taken together, our results illustrate the promising character of drug–peptide complexes with respect to antibacterial activity and by extension for targeted delivery.
The combination of a novel set of compounds with a CPP is likely to elicit distinctive mechanisms of action on bacteria. To this end, we modified the potent isopropyl derivative (5a) with octaarginine (R8) CPP and studied its antibacterial activity in water. An exciting feature of the N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide derivatives is that they are not active against E. coli bacterial cells at micromolar concentrations. The reason for this inactivity could be the presence of sulfonamide-resistant dihydropteroate synthase (DHPS).45 This feature was utilized to study the effect of the peptide-complexation on the overall activity of the complex against E. coli cells. The antibacterial activity observed for our N-(thiazol-2-yl)benzenesulfonamide–octaarginine CPP conjugate is attributed to a syncretic contribution from both constituents.
3.5 Checkerboard assay and FIC determination
The effect of the combinations of R8 peptide and isopropyl compound against S. aureus and E. coli was ascertained using the checkerboard assay, and the fractional inhibitory concentration index was calculated. Synergy measurement and FIC index (FICI) determination was conducted as per recent reports.46,47 We obtained the FIC index value of 0.3125 for S. aureus cells and 0.5 for E. coli (ATCC 25922) cells (see Table 3). These indices signify a synergistic interaction between R8 and isopropyl compound when individual drugs are directly mixed in solution. As per the checkerboard assay, the presence of R8 peptide potentiates the antibacterial activity of isopropyl compound against E. coli cells. This finding along with a null antibacterial activity observed for the isopropyl–R8 complex towards E. coli (Fig. 3(c)), further supports the emergent antibacterial behaviour of the isopropyl–R8 complex, distinct from the individual components. A FICI of 0.3125 against S. aureus cells signifies the greater sensitivity of the drug's combination towards this bacterial strain.| Bacterial cell | FIC index | FICR8 | FICisopropyl | Interaction |
|---|---|---|---|---|
| S. aureus | 0.3125 | 0.25 | 0.0625 | Synergy |
| E. coli | 0.5 | 0.5 | 0.0001 | Synergy |
3.6 Killing-kinetics assay
Killing-kinetics or time-kill assay is a colony-counting based method which provides information about how fast a drug eradicates viable counts and the bacterial load. Greater than 3log reduction in bacterial colonies points to a bactericidal mode of action. We found that the isopropyl compound alone at its MIC concentration, 3.9 μg mL−1, kills and removes all the S. aureus colonies within 4 hours (Fig. 4). This killing-time was reduced by half i.e., 2 hours, when isopropyl–R8 complex was fed to bacterial cells (refer to ESI† for agar plates showing colonies). In our previous report, we observed an analogous faster killing-kinetics of antibacterial agent when aided by R8 peptide through complexation.48
 | ||
| Fig. 4 Killing-kinetics assay of isopropyl–R8 complex and isopropyl alone against S. aureus cells. | ||
3.7 Preliminary investigation of mechanism of action
In order to gain a preliminary understanding of the mechanism of antibacterial action of the isopropyl–R8 complex, we observed the complex treated, isopropyl treated and control cells by scanning electron microscopy (SEM). As shown in Fig. 5, we found that the isopropyl–R8 complex caused damage in the form of pores, to the plasma membrane of S. aureus bacterial cells. Membrane damage can lead to leakage of cytoplasmic contents and eventual death of cells.49,50 Cells treated with only isopropyl drug does not display membrane damage pointing towards its intracellular targeting in bacterial cell. Interestingly, we could not find pores or membrane damage in case of complex-treated E. coli, possibly supporting the inactivity of the complex towards E. coli cells. We could however observe irregular membrane surface (membrane-ruffling) of the E. coli cells treated with isopropyl–R8 complex. These results further highlight the membrane-impinging strain-selective antibacterial activity of the isopropyl–R8 complex. | ||
| Fig. 5 Scanning electron micrographs of bacterial cells treated with isopropyl–R8 complex (a and d), isopropyl alone (b, c, e and f) are control group of cells. Top panel shows S. aureus cells while bottom panel images are of E. coli cells. (a) Pores are visible on the membrane surface of S. aureus cells treated with the complex, inset shows a magnified image. (d) Membrane ruffling on E. coli membrane surface on treatment with complex. | ||
3.8 Assessment of haemolytic activity
In an attempt to asses and compare the toxicity of R8 peptide, isopropyl compound and the isopropyl–R8 complex, their haemolytic activities were studied. We find that the isopropyl–R8 complex leads to only 5% haemolysis on average and does not cause significant toxicity to hRBCs even at 4× MIC concentration (Fig. 6). The MIC concentration of complex, which inhibits more than 90% bacterial cells growth was taken as reference for these studies. We observed that the isopropyl compound alone in high concentration results in higher haemolytic activity with close to 100% hemolysis at 16× MIC (see ESI, Fig. S7†). The reason behind haemolysis at high concentration of the hydrophobic isopropyl derivative could be due to the damage caused to erythrocyte membrane. Lower drug concentrations are likely accompanied by simple adsorption of drug molecules onto the membrane surface, as studied and described in detail in a recent report by Tacheva et al.51 Aligned to our observations, previous reports have described lower RBC-toxicity of octaarginine or R8 modification of drugs.52,53 The low toxicity of isopropyl–R8 complex towards red blood cells is an attractive feature when its clinical application is considered.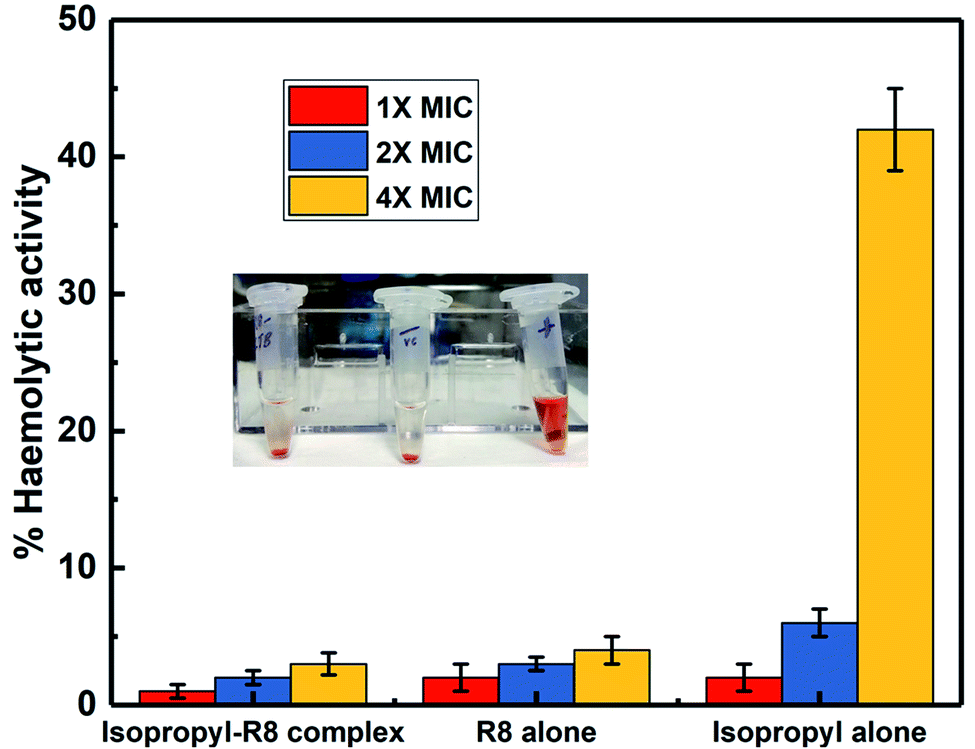 | ||
| Fig. 6 Comparison of haemolytic activity of the R8 peptide, isopropyl drug and isopropyl–R8 complex. Inset shows intact RBC pellet when treated with the complex, negative control and lysed RBCs upon Triton-X addition. | ||
4. Conclusions
A series of N-(thiazol-2-yl)benzenesulfonamides were synthesized and evaluated for antibacterial activity, based on the convergent presence of thiazole and sulfonamide moieties. Molecules bearing alkyl chains (methyl, isopropyl and butyl) exhibited the best antibacterial activity. The compound 5a is very potent as it inhibits nearly 100% bacterial cell growth at a low concentration of 3.9 μg mL−1 in an organic solvent. It is also active against a broad set of bacteria of different shapes and Gram character. The complex of compound 5a and octaarginine peptide is antibacterial towards S. aureus cells in aqueous medium and kills them rapidly without causing damage to human RBCs, thus providing scope for clinical studies in future. Interestingly, the complex of 5a with octaarginine does not inhibit the growth of E. coli cells, a pattern that matches the behaviour of bare compound 5a. These observations suggest the selective character of 5a–R8 in terms of targeting S. aureus as opposed to E. coli. Another way of stating this is that the cell penetrating peptide is unable to elicit sufficient antibiotic activity of 5a against E. coli pointing to the lack of cognate targets in the bacterial strain for the drug. While the peptide component in the complex is expected to play the role of a carrier to carry the N-(4-(4-(methylsulfonyl)phenyl)-5-phenylthiazol-2-yl)benzenesulfonamide derivative to the interior of the cell, additional modes may be at play.Owing to the cationic peptide component with guanidinium-ion rich species, it is possible that the complex exerts a membrane-active behaviour and causes punctures or dents in the membrane, as evident from the scanning electron microscope images of the complex treated bacterial cells. The complex may then enter the bacterial cell to exhibit intracellular toxic effects. In conclusion, there may be a combination of modes of action of the complex due to the multi functionalities of the individual units. Relatively little has been reported with respect to the antibacterial behaviour of untested molecules when used in conjunction with cell penetrating peptides. The present study addresses this question and highlights the distinctive underlying strategy that can be exploited to develop superior antibacterial agents. Detailed study of the mechanism of activity and enhancement of potency of such complexes is currently underway in our laboratories.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. D. is grateful to CSIR for financial support through grant no. 02(0342)/18/EMR-II. We thank Dr Mohan Galande from Unipath Specialty Pathology lab, Ahmedabad, India for providing red blood cell samples.References
- Y. Wu, et al., Optimization of biaryloxazolidinone as promising antibacterial agents against antibiotic-susceptible and antibiotic-resistant gram-positive bacteria, Eur. J. Med. Chem., 2020, 185, 111781 CrossRef CAS PubMed.
- P. Apostolis, et al., Helicobacter pylori eradication regimens in an antibiotic high-resistance European area: a cost-effectiveness analysis, Helicobacter, 2020, 25, e12666 Search PubMed.
- M. S. Mulani, E. E. Kamble, S. N. Kumkar, M. S. Tawre and K. R. Pardesi, Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: a review, Front. Microbiol., 2019, 10, 539 CrossRef PubMed.
- B. Deslouches, et al., Engineered cationic antimicrobial peptides to overcome multidrug resistance by ESKAPE pathogens, Antimicrob. Agents Chemother., 2015, 59, 1329–1333 CrossRef CAS PubMed.
- S. Wagner, et al., Novel strategies for the treatment of Pseudomonas aeruginosa infections, J. Med. Chem., 2016, 59, 5929–5969 CrossRef CAS PubMed.
- C. L. Ventola, The antibiotic resistance crisis Part 1: causes and threats, Pharmacy and therapeutics, 2015, 40, 277 Search PubMed.
- J. Li, et al., Gold nanoparticles cure bacterial infection with benefit to intestinal microflora, ACS Nano, 2019, 13, 5002–5014 CrossRef CAS.
- M. B. Calvert, V. R. Jumde and A. Titz, Pathoblockers or antivirulence drugs as a new option for the treatment of bacterial infections, Beilstein J. Org. Chem., 2018, 14, 2607–2617 CrossRef CAS PubMed.
- P. Klahn and M. Bronstrup, Bifunctional antimicrobial conjugates and hybrid antimicrobials, Nat. Prod. Rep., 2017, 34, 832–885 RSC.
- L.-Y. Guo, et al., Evaluation of hypocrellin A-loaded lipase sensitive polymer micelles for intervening methicillin-resistant Staphylococcus aureus antibiotic-resistant bacterial infection, Mater. Sci. Eng., C, 2020, 106, 110230 CrossRef CAS.
- H. Douafer, V. Andrieu, O. Phanstiel IV and J. M. Brunel, Antibiotic adjuvants: make antibiotics great again!, J. Med. Chem., 2019, 62, 8665–8681 CrossRef CAS.
- P. V. Baptista, et al., Nano-strategies to fight multidrug resistant bacteria – “A Battle of the Titans”, Front. Microbiol., 2018, 9, 1441 CrossRef PubMed.
- A. Gupta, S. Mumtaz, C.-H. Li, I. Hussain and V. M. Rotello, Combating antibiotic-resistant bacteria using nanomaterials, Chem. Soc. Rev., 2019, 48, 415–427 RSC.
- G. L. A. Mislin and I. J. Schalk, Siderophore-dependent iron uptake systems as gates for antibiotic Trojan horse strategies against Pseudomonas aeruginosa, Metallomics, 2014, 6, 408–420 CrossRef CAS PubMed.
- Y. J. Gordon and E. G. Romanowski, A Review of Antimicrobial Peptides and Their Therapeutic Potential as Anti-Infective Drugs, Curr. Eye Res., 2005, 30, 505–515 CrossRef CAS PubMed.
- F. Yuan, et al., Evaluation of topologically distinct constrained antimicrobial peptides with broad-spectrum antimicrobial activity, Org. Biomol. Chem., 2018, 16, 5764–5770 RSC.
- J. A. Patch and A. E. Barron, Helical Peptoid Mimics of Magainin-2 Amide, J. Am. Chem. Soc., 2003, 125, 12092–12093 CrossRef CAS PubMed.
- K. Chindera, et al., The antimicrobial polymer PHMB enters cells and selectively condenses bacterial chromosomes, Sci. Rep., 2016, 6, 23121 CrossRef CAS PubMed.
- Q. Laurent, L. K. Batchelor and P. J. Dyson, Applying a Trojan Horse strategy to ruthenium complexes in the pursuit of novel antibacterial agents, Organometallics, 2018, 37, 915–923 CrossRef CAS.
- B. Betchinger and S.-U. Gorr, Antimicrobial peptides: mechanisms of action and resistance, J. Dent. Res., 2017, 96, 254–260 CrossRef PubMed.
- H. Lee, et al., Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity, ACS Omega, 2019, 4, 15694–15701 CrossRef CAS PubMed.
- J. Ruczynski, et al., Transportan 10 improves the pharmacokinetics and pharmacodynamics of vancomycin, Sci. Rep., 2019, 9, 1–15 Search PubMed.
- M. Gomarasca, et al., Bacterium-derived cell-penetrating peptides deliver gentamicin to kill intracellular pathogens, Antimicrob. Agents Chemother., 2017, 61(4), 1–20 CrossRef PubMed.
- G. Barkowsky, et al., Influence of different cell-penetrating peptides on the antimicrobial efficiency of PNAs in Streptococcus pyogenes, Mol. Ther.–Nucleic Acids, 2019, 18, 444–454 CrossRef CAS PubMed.
- S. T. Henriques, M. N. Melo and M. A. R. B. Castanho, Cell-penetrating peptides and antimicrobial peptides: how different are they?, Biochem. J., 2006, 399, 1–7 CrossRef CAS.
- N. Schmidt, A. Mishra, G. H. Lai and G. C. L. Wong, Arginine-rich cell-penetrating peptides, FEBS Lett., 2010, 584, 1806–1813 CrossRef CAS PubMed.
- S. Futaki, et al., Arginine-rich peptide: An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS PubMed.
- T. Jiang, et al., Tumor imaging by means of proteolytic activation of cell-penetrating peptides, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17867–17872 CrossRef CAS.
- B. K. Sarojini, B. G. Krishna, C. G. Darshanraj, B. R. Bharath and H. Manjunatha, Synthesis, characterization, in vitro and molecular docking studies of new 2,5-dichloro thienyl substituted thiazole derivatives for antimicrobial properties, Eur. J. Med. Chem., 2010, 45, 3490–3496 CrossRef CAS.
- H. Mohammad, et al., Antibacterial Characterization of Novel Synthetic Thiazole Compounds against Methicillin-Resistant Staphyloccus pseudointermedius, PLoS One, 2015, 10, e0130385 CrossRef PubMed.
- S. Bondock, W. Fadaly and M. A. Metwally, Synthesis and antimicrobial activity of some new thiazole, thiophene and pyrazole derivatives containing benzothiazole moiety, Eur. J. Med. Chem., 2010, 45, 3692–3701 CrossRef CAS PubMed.
- Y. Genc, R. Ozkanca and Y. Bekdemir, Antimicrobial activity of some sulfonamide derivatives on clinical isolates of Staphyloccus aureus, Ann. Clin. Microbiol. Antimicrob., 2008, 7, 17 CrossRef PubMed.
- A. K. Gadad, C. S. Mahajanshetti, S. Nimbalkar and A. Raichurkar, Synthesis and Antibacterial Activity of Some 5-guanylhydrazone/thiocyanato-6-arylimidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide Derivatives, Eur. J. Med. Chem., 2000, 35, 853–857 CrossRef CAS.
- W. Boufas, et al., Synthesis and antibacterial activity of sulfonamides. SAR and DFT studies, J. Mol. Struct., 2014, 1074, 180–185 CrossRef CAS.
- A. R. Massah, et al., Synthesis, in vitro antibacterial and carbonic anhydrase II inhibitory activities of N-acylsulfonamides using silica sulfuric acid as an, 2008 Search PubMed.
- G. H. Miller, P. H. Doukas and J. K. Seydel, Sulfonamide structure–activity relation in a cell-free system. Correlation of inhibition of folate synthesis with antibacterial activity and physicochemical parameters, J. Med. Chem., 1972, 15, 700–706 CrossRef CAS PubMed.
- F. Naaz, et al., Molecular modeling, synthesis, antibacterial and cytotoxicity evaluation of sulfonamide derivatives of benzimidazole, indazole, benzothiazole and thiazole, Bioorg. Med. Chem., 2018, 26, 3414–3428 CrossRef CAS PubMed.
- E. S. Darwish, A. M. Abdel Fattah, F. A. Attaby and O. N. Al-Shayea, Synthesis and antimicrobial evaluation of some novel thiazole, pyridone, pyrazole, chromene, hydrazone derivatives bearing a biologically active sulfonamide, Int. J. Mol. Sci., 2014, 15, 1237–1254 CrossRef PubMed.
- S. Roy, et al., Design and Development of Novel Urea, Sulfonyltriurea, and Sulfonamide Derivatives as Potential Inhibitors of Sphingosine Kinase 1, Pharmaceuticals, 2020, 13, 118 CrossRef CAS.
- J. B. Patel et al., Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard-Tenth Edition, 2015, CLSI Document M07-A10 Search PubMed.
- H. Gong, et al., How do Self-Assembling Antimicrobial Lipopeptides Kill Bacteria?, ACS Appl. Mater. Interfaces, 2020, 12, 55675–55687 CrossRef CAS.
- C. Ergene and E. F. Palermo, Self-immolative polymers with potent and selective antibacterial activity by hydrophilic side chain grafting, J. Mater. Chem. B, 2018, 6, 7217–7229 RSC.
- S. K. Singh, et al., Synthesis and biological evaluation of 2,3-diarylpyrazines and quinoxalines as selective COX-2 inhibitors, Bioorg. Med. Chem., 2004, 12, 1881–1893 CrossRef CAS PubMed.
- A. H. Abdelazeem, M. Habash, I. A. Maghrabi and M. O. Taha, Synthesis and evaluation of novel diphenylthiazole derivatives as potential anti-inflammatory agents, Med. Chem. Res., 2015, 24, 3681–3695 CrossRef CAS.
- E. M. Wise and M. M. Abu Donia, Sulfonamide Resistance Mechanism in Escherichia coli: R Plasmids can determine Sulfonamide Resistant Dihydropteroate Synthases, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2621–2625 CrossRef CAS PubMed.
- P. Lekshmipriya, et al., Synergistic Effect of Frog Skin Antimicrobial Peptides in Combination with Antibiotics Against Multi Host Gram-Negative Pathogens, Int. J. Pept. Res. Ther., 2021, 27, 1529–1540 CrossRef CAS.
- A. Gonzalez, et al., Small Molecule Inhibitors of the Response Regulator Arsr Exhibit Bactericidal Activity against Helicobacter pylori, Microorganisms, 2020, 8(4), 1–16 CrossRef PubMed.
- P. Ratrey, S. V. Dalvi and A. Mishra, Enhancing Aqueous Solubility and Antibacterial Activity of Curcumin by Complexing with Cell-Penetrating Octaarginine, ACS Omega, 2020, 5, 19004–19013 CrossRef CAS PubMed.
- M. Yasir, D. Dutta and M. D. P. Willcox, Mode of action of the antimicrobial peptide Mel4 is independent of Staphylococcus aureus cell membrane permeability, PLoS One, 2019, 14, e0215703 CrossRef CAS.
- Y. Wu, et al., Antibacterial Activity and Membrane-Disruptive Mechanism of 3-p-trans-Coumaroyl-2-hydroxyquinic Acid, a Novel Phenolic Compound from Pine Needles of Cedrus deodara, against Staphylococcus aureus, Molecules, 2016, 21, 1084 CrossRef PubMed.
- B. Tacheva, et al., Drug Exchange between Albumin Nanoparticles and Erythrocyte Membranes, Nanomaterials, 2019, 9, 47 CrossRef PubMed.
- S. Pujals, et al., Curvature Engineering: Positive Membrane Curvature Induced by Epsin N-Terminal Peptide Boosts Internalization of Octaarginine, ACS Chem. Biol., 2013, 8, 1894–1899 CrossRef CAS.
- S. V. K. Rompicharla, P. Kumari, B. Ghosh and S. Biswas, Octa-Arginine Modified Poly (Amidoamine) Dendrimers for Improved Delivery and Cytotoxic Effect of Paclitaxel in Cancer, Artif. Cells, Nanomed., Biotechnol., 2018, 46, S847–S859 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03882f |
| This journal is © The Royal Society of Chemistry 2021 |