
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper(I)-catalyzed radical carboamination reaction of 8-aminoquinoline-oriented buteneamides with chloroform: synthesis of-β-lactams†
Zixu Gana,
Ke Zhanga,
Peng Shib,
Yingsheng Zhao *a and
Runsheng Zeng*a
*a and
Runsheng Zeng*a
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry Chemical Engineering and Materials Science, Soochow University, Suzhou, Jiangsu 215123, China. E-mail: yszhao@suda.edu.cn; zengrunsheng@suda.edu.cn
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg1, 52074 Aachen, Germany
First published on 19th August 2021
Abstract
A novel Cu(CH3CN)4PF6-catalyzed carboamination reaction of 8-aminoquinoline-oriented buteneamides with chloroform to afford 4-(2,2,2-trichloroethyl)-β-lactams is described. The reaction proceeded at 110 °C in air with di-t-butyl peroxide. Preliminary studies indicated that the reaction undergoes a free radical mechanism via a Cu(I)/Cu(II)/Cu(III) catalytic cycle.
Introduction
Nitrogen-containing heterocycles are notable compounds known for their bioactivity in nature.1 In particular, functionalized β-lactams are the core skeleton of many natural products and antibiotics drug molecules with specific effects, such as penicillins,2 carbapenems,3 cephalosporins4 and monocyclic β-lactams5 (Fig. 1).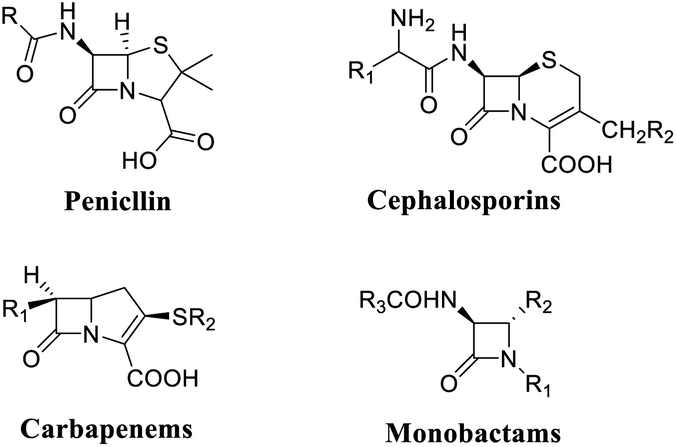 | ||
| Fig. 1 Natural products and antibiotics drug molecules. | ||
These β-lactams are known for their clinical use as antibiotics, which have high medicinal synthetic value due to their low toxicity, good bactericidal activity and wide indications.6 Therefore, during the past 2 decades, great efforts have been focused on the core skeleton construction of β-lactams in synthetic chemistry, such as the Staudinger ketene-imine [2 + 2] cycloaddition,7 Beckmann rearrangement,8 Kinugasa alkyne–nitrone cycloaddition,9 and Schmidt reactions,10 Until now, the C(sp3)–H bond activation,11 C–C,12 C–N,13 C–P,14 and C–S15 bond strategy have provided a straightforward pathway to synthesize β-lactams.
The research groups of Shi firstly used transition metal palladium-catalyzed intramolecular amination of aliphatic or amino acid derivatives with different guiding groups to generate α-amino-β-lactam.16 Afterwards, Wu group has also demonstrated that the propionamide-linked bidentate aminoquinoline (AQ or Q) directing group can facilitate the C(sp3)–H activation under Pd catalysis.17 The research groups of Ge,18 Kanai19 and Chantani20 used transition metal like nickel, cobalt and copper catalyst to generate α-amino-β-lactam by intramolecular amination of aliphatic or amino acid derivatives with different guiding groups (Scheme 1a). The C–H functionalization strategy is more efficient and convenient than classic intramolecular condensation and nucleophilic reactions. However, the difficulty of these methods was that it first need to obtain functionalized acids as the reaction substrates through monoarylation.
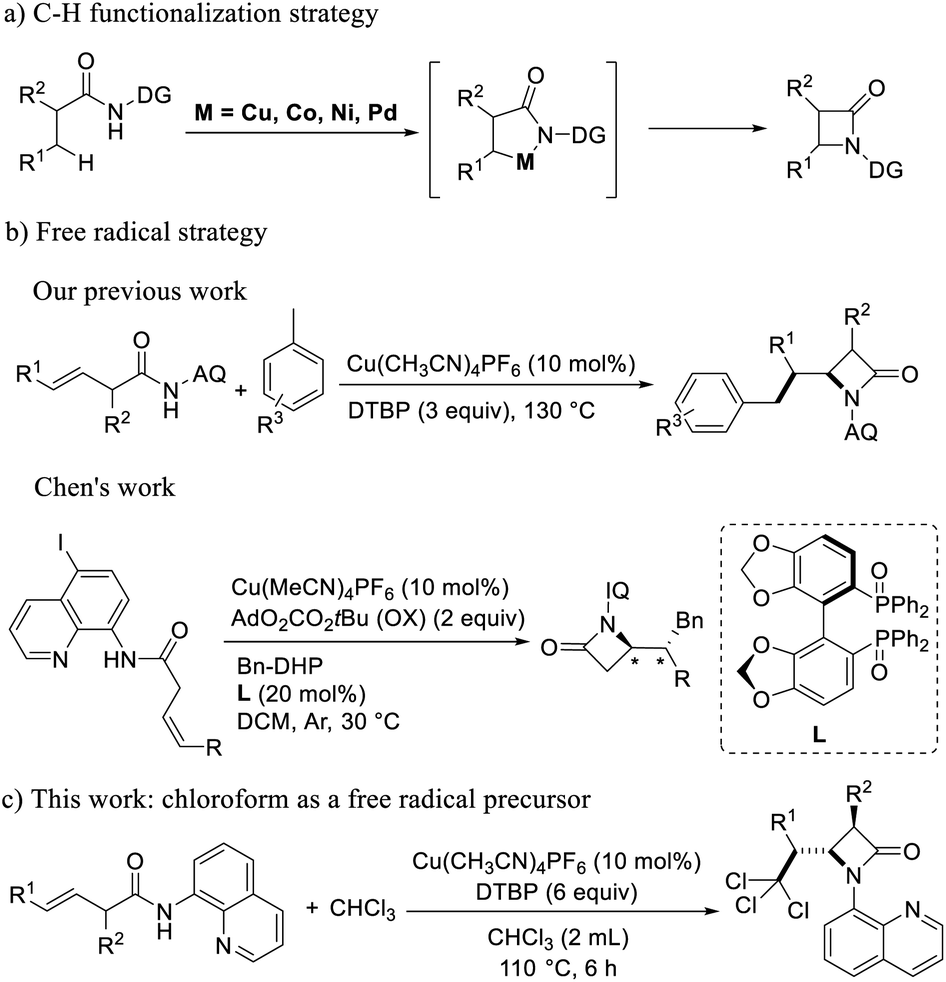 | ||
| Scheme 1 One step intramolecular amination reaction. | ||
Later, on the basis of the reaction of nucleophiles and 8-aminoquinoline-oriented buteneamide compounds in the Engle's research group,21 our group first used buteneamide compounds as substrates to oxidize toluene with DTBP to generate benzyl radicals, then benzyl radicals attacked inactive double bonds, and coordinated with copper to generate β-lactams successfully.22 The reaction mode is efficient and can enrich the preparation method of β-lactam. Subsequently, Chen23 group used cis-3-hexenamide compounds with 8-amino-5-iodoquinoline as the substrates, 4-benzyl Hantzsch esters as the alkyl radical precursor, and rarely used biaryl diphosphine oxide as a chiral ligand to synthesize a series of chiral β-lactam compounds (Scheme 1b). In order to verify the applicability of this method, we tried other free radicals. Doyle24 and Sheng25 used chloroform as a solvent and a source of free radicals to achieve the functionalization of unsaturated double bond. It can be seen that chloroform is a good source of polychloromethyl radicals,26 Therefore, we used chloroform as the free radical source and found that the chloroform was decomposed into trichloromethyl radicals triggered by DTBP, and then reacted with 8-aminoquinoline-oriented buteneamide compounds to achieve the 2,2,2-trichloroehtyl-β-lactams (Scheme 1c).
Results and discussion
When the model reaction of the directing group-protected butenoic acid derivative (1a) with chloroform was performed in the presence of oxidants such as tert-butyl hydroperoxide (TBHP), no desired products were obtained (Table 1, entries 7). After the addition of 10 mol% di-t-butyl peroxide (DTBP), the reaction proceeded smoothly to afford the desired product, 2,2,2-trichloroehtyl-β-lactam (2a).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (%) | Oxidant | Temperature | Base | Yieldb |
| a Reaction condition: 1a (0.2 mmol), DTBP (1 mmol), Cu(CH3CN)4PF6, (0.02 mmol), chloroform (2 ml), at 110 °C in air atmosphere, 6 h.b Yields are given for isolated products.c DTBP (1.2 mmol). | |||||
| 1 | CuBr | DTBP | 110 °C | 50% | |
| 2 | Cu(OAc)2 | DTBP | 110 °C | 45% | |
| 3 | Cu(OTf)2 | DTBP | 110 °C | 76% | |
| 4 | CuBr2 | DTBP | 110 °C | 62% | |
| 5 | Cu(acac)2 | DTBP | 110 °C | 61% | |
| 6 | Cu(CH3CN)4PF6 | DTBP | 110 °C | 88% | |
| 7 | Cu(CH3CN)4PF6 | TBHP | 110 °C | 0% | |
| c8 | Cu(CH3CN)4PF6 | DTBP | 110 °C | 92% | |
| c9 | Cu(CH3CN)4PF6 | DTBP | 120 °C | 41% | |
| c10 | Cu(CH3CN)4PF6 | DTBP | 100 °C | 83% | |
| c11 | Cu(CH3CN)4PF6 | DTBP | 90 °C | tr | |
| c12 | Cu(CH3CN)4PF6 | DTBP | 110 °C | Na2CO3 | 44% |
| c13 | Cu(CH3CN)4PF6 | DTBP | 110 °C | K2HPO4 | 53% |
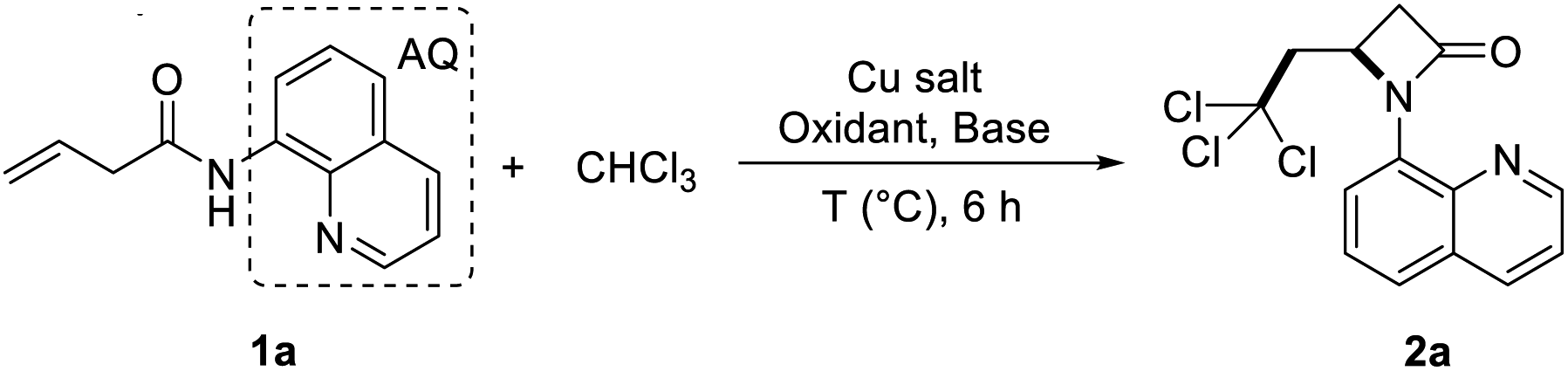
From the experiment results we can see that trichloromethyl free radical in the presence of copper(I) and DTBP carry out the cascade radical addition/intramolecular amination to perform 2,2,2-trichloroehtyl-β-lactam in a single step. When the crotonamide compound 1a with 8-aminoquinoline guiding group was reacted with copper acetate (10 mol%) and DTBP (5 equiv.) in chloroform at 110 °C for 6 h, the cyclized product 2a was obtained in 45% yield, and some of the starting material was recovered (Table 1, entry 2). Encouraged by this result, several Cu catalysts such as most commonly used CuBr, CuBr2, Cu(OTf)2, Cu(acac)2 and Cu(CH3CN)4PF6 were tested and the results showed that both Cu(I) and Cu(II) salt could get product 2a. Interestingly, Cu(CH3CN)4PF6 gave the best yield of 2a at 88% (Table 1, entry 6).
The control experiment also clearly showed that Cu(CH3CN)4PF6 and di-tert-butyl peroxide were indispensable for this reaction. Additionally, increasing the amount of DTBP to 6 equiv. gave a higher yield of 2a at 92% yield (Table 1, entry 8). However, as the reaction temperature rises to 120 °C, the yield of product 2a was reduced to 41% (Table 1, entry 9). The reaction could hardly go on when temperature was below 90 °C (Table 1, entry 11). To further improve the efficiency of the free radical reaction, we added several base to reaction system, but this measure has no effect (Table 1, entry 12-13).
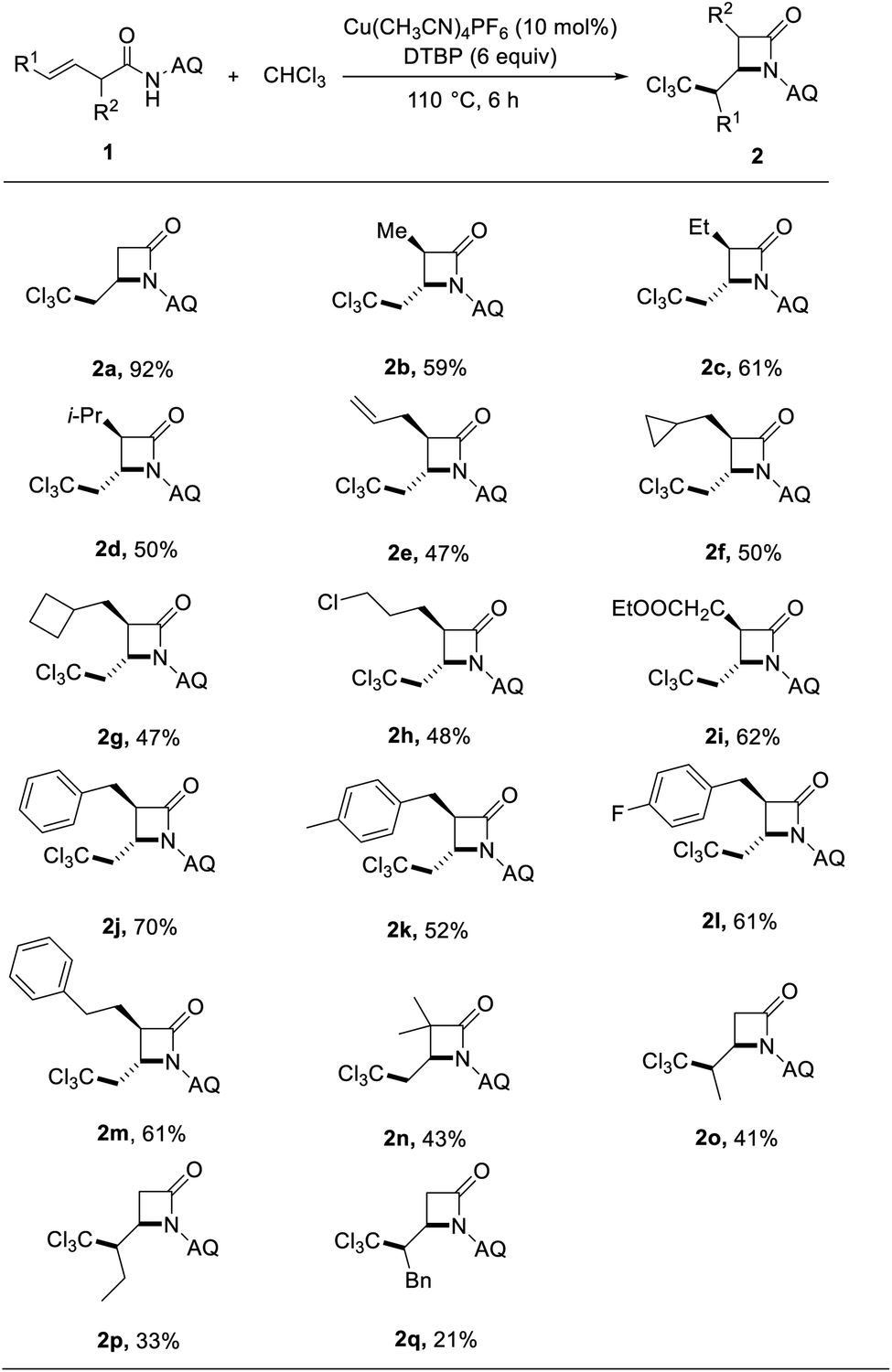
With the optimized reaction conditions established, we examined the substrate scope of N-(quinolin-8-yl)but-3-enamide derivatives under optimized conditions: DTBP as an oxidant, Cu(CH3CN)4PF6 as a catalyst at 110 °C, for 6 hours in air. As can be seen from the Table 2, the most α-substituted N-(quinolin-8-yl)but-3-enamide derivatives were well tolerated. Mono-substituted N-(quinolin-8-yl)but-3-enamide derivatives were all compatible and had less effect on the C–H activation reaction. Various functional groups, like methyl, ethyl, methylcyclopropyl and benzyl etc., gave the corresponding products (2b–2m) in moderate to good yields. Unfortunately, when there were two methyl groups at α position, the β-lactam product yield was only 43% (2n). When the γ-substituted N-(quinolin-8-yl)but-3-enamide derivatives were used as the substrates, the products were obtained in low yields (2o–2q), and a large amount of raw materials were not reacted completely. It may abate the activity of the reaction substrates due to steric hindrance.
a Reaction condition: 1a (0.2 mmol), DTBP (1.2 mmol), Cu(CH3CN)4PF6, (0.02 mmol), chloroform (2 ml), at 110 °C in air atmosphere, 6 h. Yield of isolated products are given. ‘dr’ decided by NMR is >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 if not stated otherwise. :1 if not stated otherwise. |
|---|
 |
To further demonstrate the synthetic utility of the reaction, the gram–scale reaction was further performed. We found that the corresponding β-lactam product 2a was acquired in 78% yield under the optimized reaction conditions (Scheme 2).
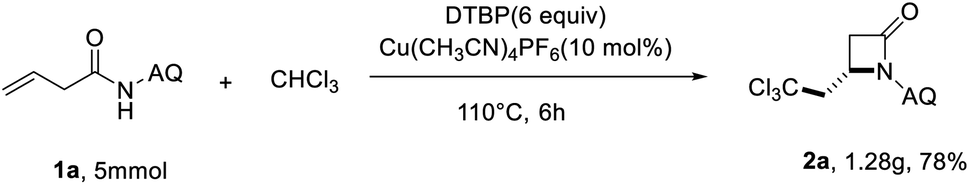 | ||
| Scheme 2 Gram-scale reaction. | ||
In order to understand the carboamination reaction mechanism better, we did the following control experiments (Scheme 3). First, we carried out the radical inhibition and capture experiment. When 2 equiv. of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) was added in the reaction system under standard conditions, the target product was not produced. It showed that the reaction may be a way of free radical reaction.
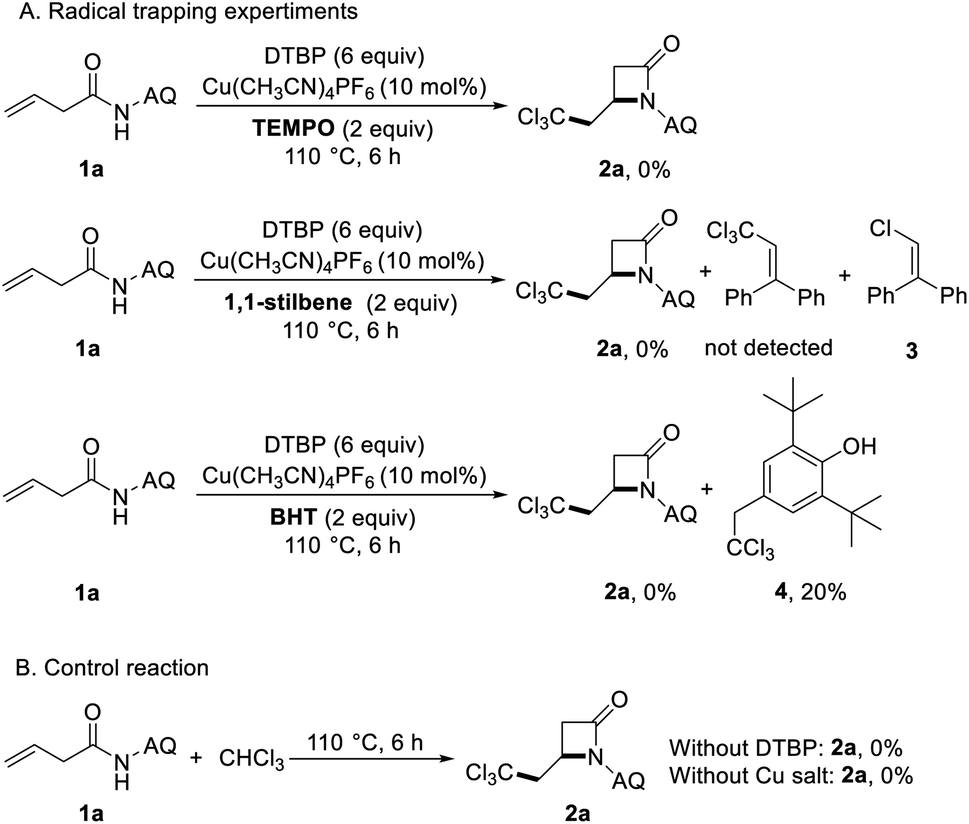 | ||
| Scheme 3 Control experiments. | ||
When using 1,1-stilbene to capture free radicals, we didn't detect the presence of trichloromethyl radical capture product, but obtained chlorine radical capture product 3. This may be due to the decomposition of trichloromethyl radicals into chlorine radicals and dichloromethyl carbene. Since Ghosh research group captured the tribromomethyl radical by using 2,6-di-tert-butyl-4-methylphenol (BHT) successfully,27 we try to captured the trichloromethyl radical by using the same method. It's exciting to obtain the BHT trapped trichloromethyl radical compound 4 (Scheme 3A). And then, when copper salt and DTBP were not added to the reaction system, the reaction did not proceed, which showed that copper salt and DTBP are very important in the reaction (Scheme 3B).
On the basis of the mechanistic studies and experimental results, a plausible mechanism is proposed in Scheme 4. Initially, the copper(I) catalyst reacted with di-tert-butyl peroxide, leading to the copper(II) catalyst, tert-butoxy radical and tert-butoxy anion. Thereafter, tert-butoxy radical reacted with chloroform to generate trichloromethyl radicals and tert-butoxy anion pulled out the hydrogen on the nitrogen of the reaction substrate (1a), the copper(II) catalyst was coordinated with nitrogen anion to form intermediate I. Finally, the intermediate I captured the trichloromethyl radical to form the intermediate II,28 which was reduced and eliminated to obtain the target product 2a and copper(I) catalyst. Preliminary studies indicated that the reaction undergoes a free radical mechanism via a Cu(I)/Cu(II)/Cu(III) catalytic cycle.
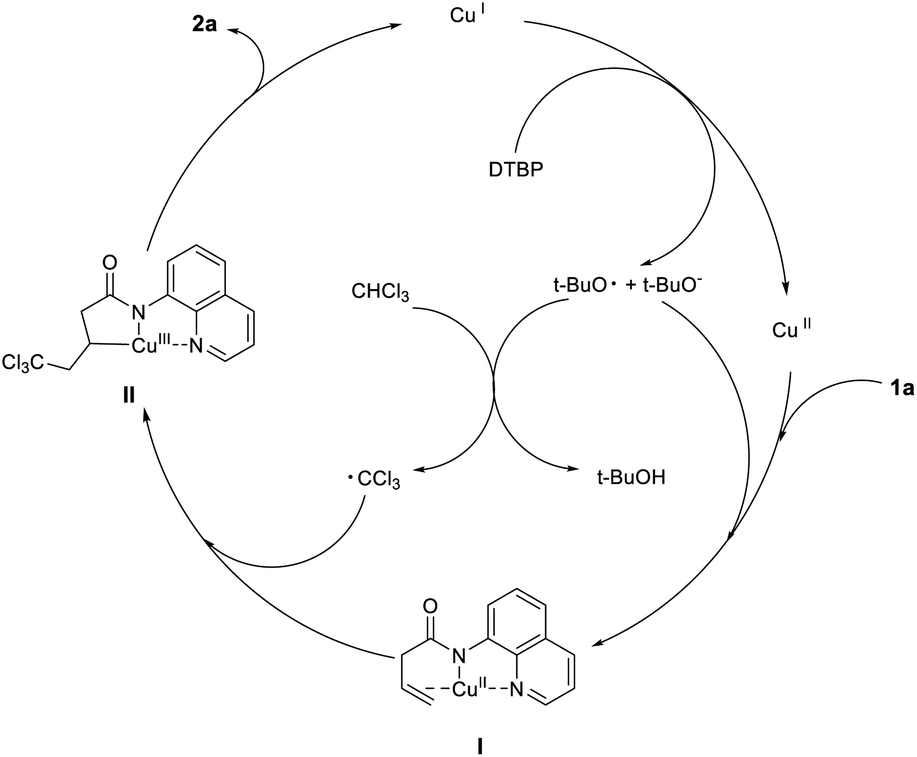 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In conclusion, a novel method for carbon–nitrogen bond formation was developed through the carboamination reaction of 8-aminoquinoline-oriented buteneamides with chloroform catalyzed by 10% Cu(CH3CN)4PF6. The reaction proceeded at 110 °C in air with di-t-butyl peroxide to afford 4-(2,2,2-trichloroethyl)-β-lactams from medium to good yields. Further we will find new free radicals and expand the applicability of the free radical reaction to form β-lactams.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Prospective Study Program of Jiangsu (BY2015039-08), the National Natural Science Foundation of China (No. 21572149), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- A. K. Mailyan, J. A. Eickhoff, A. S. Minakova, Z. H. Gu, P. Lu and A. Zakarian, Chem. Rev., 2016, 116, 4441 CrossRef CAS PubMed
.
- L. A. Mandell and R. G. Wunderink, Clin. Infect. Dis., 2012, 54, 1134 CrossRef CAS
- S. Oida, A. Yoshida and E. Ohki, Chem. Pharm. Bull., 1980, 28, 3494 CrossRef CAS
- W. A. Craig, Diagn. Microbiol. Infect. Dis., 1995, 22, 89 CrossRef CAS
- S. H. Lee, Bull. Korean Chem. Soc., 2014, 35, 2990 CrossRef CAS
- M. Chen, V. Buurma, M. Shah and G. Fahim, Am. J. Health-Syst. Pharm., 2019, 76, 1383 CrossRef
- S. France, A. Weatherwax, A. E. Taggi and T. Lectka, Acc. Chem. Res., 2004, 37, 592 CrossRef CAS PubMed
- P. S. Mahajan, V. T. Humne, S. D. Tanpure and S. B. Mhaske, Org. Lett., 2016, 18, 3450 CrossRef CAS
- J.-L. Qi, F. Wei, S. Huang, C.-H. Tung and Z.-H. Xu, Angew. Chem., Int. Ed., 2021, 60, 4561 CrossRef CAS PubMed
- W. Zhan, M. Tong, L. Ji, H. Zhang, Z.-M. Ge, X. Wang and R.-T. Li, Chin. Chem. Lett., 2019, 30, 973 CrossRef CAS
-
(a) C.-H. Shan, L. Zhu, L.-B. Qu, R.-P. Bai and Y. Lan, Chem. Soc. Rev., 2018, 47, 7552 RSC
-
(a) X. Lu, B. Xiao, Z.-Q. Zhang, T.-J. Gong, W. Su, J. Yi, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129 CrossRef
- J. A. Gurak, K. S. Yang, Z. Liu and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 5805 CrossRef CAS PubMed
- C. Liu, H. Zhang, W. Shi and A.-W. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed
- D.-J. Wang, K.-H. Zhou, J.-Y. Zhang and Y.-S. Zhao, Org. Chem. Front., 2020, 7, 3229 RSC
-
(a) Q. Zhang, K. Chen, W.-H. Rao, Y.-J. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588 CrossRef CAS
-
(a) W.-W. Sun, P. Cao, R.-Q. Mei, Y. Li, Y.-L. Ma and B. Wu, Org. Lett., 2014, 16, 480 CrossRef CAS PubMed
-
(a) X.-S. Wu, Y. Zhao, G.-W. Zhang and H.-B. Ge, Angew. Chem., Int. Ed., 2014, 53, 3706 CrossRef CAS
- Z. Wang, J.-Z. Ni, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 3496 CrossRef CAS
- Y. Aihara and N. Chatani, ACS Catal., 2016, 6, 4323 CrossRef CAS
-
(a) Z. Liu, T. Zeng, K. S. Yang and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 15122 CrossRef CAS
- P. Shi, J. Wang, Z.-X. Gan, R.-S. Zeng and Y.-S. Zhao, Chem. Commun., 2019, 55, 10523 RSC
-
(a) Z.-B. Bai, H. Zhang, H. Wang, H.-R. Yu, G. Chen and G. He, J. Am. Chem. Soc., 2021, 143, 1195 CrossRef CAS PubMed
- R. K. Neff, Y.-L. Su, S.-Q. Liu, M. Rosado, X.-H. Zhang and M. P. Doyle, J. Am. Chem. Soc., 2019, 141, 16643 CrossRef CAS PubMed
- H.-C. Ge, K.-Y. Du and W.-J. Sheng, Chin. J. Org. Chem., 2020, 40, 1625 CrossRef
-
(a) C. Chen, H. Tan, B.-F. Liu, C.-C. Yue and W.-B. Liu, Org. Chem. Front., 2018, 5, 3143 RSC
- T. Sahoo, C. Sen, H. Singh, E. Suresh and S. C. Ghosh, Adv. Synth. Catal., 2019, 361, 3950 CrossRef CAS
-
(a) H.-Y. Zhang, L.-L. Mao, B. Yang and S.-D. Yang, Chem. Commun., 2015, 51, 4101 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05233k |
| This journal is © The Royal Society of Chemistry 2021 |