
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-dimensional porous carbon derived from different organic acid salts for application in electrochemical sensing†
Xiaoyue Yue abc,
Yan Lia,
Min Lia,
Xiaoyu Luoa and
Yanhong Bai*abc
abc,
Yan Lia,
Min Lia,
Xiaoyu Luoa and
Yanhong Bai*abc
aCollege of Food and Bioengineering, Zhengzhou University of Light Industry, Zhengzhou 450001, PR China. E-mail: baiyanhong212@163.com
bHenan Key Laboratory of Cold Chain Food Quality and Safety Control, Zhengzhou 450001, PR China
cHenan Collaborative Innovation Center of Food Production and Safety, Zhengzhou 450001, PR China
First published on 27th September 2021
Abstract
Three-dimensional porous carbon materials were synthesized by the one-step pyrolysis of organic salts with different numbers of hydroxyl groups on the side chain (sodium tartrate, sodium malate and sodium succinate). Further, the formation of these porous carbon materials was explored. And then, three kinds of carbon materials were used for constructing electrochemical sensors for nitrite detection, respectively. Porous carbon derived from sodium tartrate (PCST) showed the highest electrocatalytic ability for nitrite oxidation among all three materials. The PCST-based sensors allow for rapid detection of nitrite in a wide linear range of 0.1–100 μM with a low detection limit of 0.043 μM. The sensor was applied to detect nitrite in meat samples and the results tested by the developed sensor were consistent with the results obtained by HPLC. We envision that PCST-based electrochemical sensor is promising as an alternative choice for the development of electrochemical analysis.
1. Introduction
Electrode modified material is a decisive factor for constructing an electrochemical sensors and biosensors. To obtain desired electrochemical performance, various materials are currently being explored, thereinto, carbon materials are some of the most popular modified materials. Due to the advantages of high surface area, outstanding electrical conductivity, excellent structure stability and ease of synthesis, porous carbon is thought to be one of the most attractive materials in the fields of electrochemical energy storage1–4 and electrochemical sensing.5,6 As one kind of efficient electrode material for fabricating sensors, porous carbon has been widely researched.For instance, Xiang et al. prepared three-dimensional interconnected porous carbon (3DIPC) material via direct pyrolysis of sodium citrate.7 3DIPC material was successfully synthesized through an easy and one-step pyrolysis reaction without templates and substrates. The sodium citrate was carbonized in a ceramic crucible to high temperatures 700 °C under a N2 (99.999%) atmosphere with a ramp of 2 °C min−1. Each sample was held at the setting temperature for 2 h. The prepared material has a discrete fourier transform (DFT) surface area of 546.2 m2 g−1 and total volume of 1.67 cm3 g−1. Under the optimal condition, 3DIPC-based sensor allows for simultaneous determination of hydroquinone (HQ) and catechol (CA). For HQ detection, the developed sensor showed high sensitivity of 15.42 AM−1 cm−2 and a low detection limit of 2.1 × 10−8 M (S/N = 3). For CA detection, the proposed sensor showed good sensitivity of 13.58 AM−1 cm−2 and a low detection limit of 3.7 × 10−8 M (S/N = 3). By using formaldehyde and resorcinol as carbon precursors, Veerakumar et al. synthesized carbon porous materials (CPMs) by a facile microwave-assisted hydrothermal route.8 Specifically, the precursor precipitated mixture was subjected to microwave irradiation, followed by curing, washing and drying of the polymeric gel. And then, the dried sample was heated under a N2 environment at a rate 10 °C min−1 until reaching a temperature of 900 °C, followed by switching to a CO2 gas stream at a designated temperature and then being kept for 1–2 h. The prepared CPMs possess a high surface area up to 2660 m2 g−1, microwave-assisted route. And then CPMs were applied to the electrochemical detection of dopamine. The CPM-based sensor exhibited excellent selectivity, a desirable detection limit (2.9 nM), and extraordinary sensitivity (2.56 mA mM−1 cm−2) for detection of dopamine. Furthermore, porous carbon materials were also used to construct sensors for chloramphenicol detection,9 heavy metal ions determination,10 pressure and temperature sensing11 based on their different electrochemical behaviors. However, each study usually involved only one type of porous carbon material, which mainly derived from one carbon source. And, there was short of systematic research on the effect of porous carbon derived from multiple carbon sources on electrocatalytic performance. Therefore, the assessment of porous carbon materials derived from different carbon sources on electrochemical behavior is critical for developing sensitive sensors.
Nitrite, as a common food additive in meat food, functions as a colorant, is used for color protection and inhibits the breeding of clostridium botulinum and toxin secretion. Excessive nitrite can lead to abnormal methemoglobin levels.12 In the digestive system, nitrite is converted to nitrosamines, which are carcinogenic. Nitrite has teratogenic effects on the fetus. Therefore, it's necessary to establish a rapid and efficient method for nitrite detection. Electrochemical methods have attracted extensive attention due to their advantages of rapidness, high sensitivity and selectivity in quantitative analysis.13 To the best our knowledge, this kind of electrochemical sensor which was constructed based on different three-dimensional porous carbon derived from sodium tartrate, sodium malate and sodium succinate, were rarely reported. Otherwise, the morphologies of electrode materials have great influence on the analytical performance of electrochemical sensors.14 For the morphologies of porous carbon materials, the raw materials and preparing process play key roles.2 The above investigations encouraged us to explore the relationship between three-dimensional porous carbon derived from different organic salt precursors and the electrochemical performance for nitrite sensing.
In this work, we researched the effect of porous carbon materials derived from three different carbon sources on nitrite sensing, from the aspect of the construction of sensors, detection mechanism, detection performance. In terms of the material synthesis, the effects of different carbon sources on the formation of porous carbon structures were revealed. Specifically, the porous carbon materials were prepared by a one-step pyrolysis of organic salts (sodium tartrate (ST), sodium malate (SM) and sodium succinate (SS)) that consisted of similar carbon atoms and sodium ions but with different numbers of hydroxyl groups. Organic salts with different hydroxyl groups produced different porous carbon textural structures and expressed high electrocatalytic activities to nitrite. Thereinto, PCST-based sensor showed the optimal electrocatalytic ability for nitrite oxidization. The fabricated sensor based on PCST allows sensitive detection of nitrite with satisfactory analytical performance.
2. Experimental
2.1 Reagents and instruments
Sodium succinate(C4H4Na2O4, ≥98%), sodium tartrate (C4H4Na2O6, 99%), sodium malate (C4H4Na2O5, ≥95%) and other reagents were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). DMF (purity ≥99.5) used in the experiment was purchased from rom Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Thermogravimetric analysis (TGA) was performed with a PerkinElmer Diamond TG/DTA apparatus. The textural structure and micromorphology were examined by scanning electron microscopy (SEM; JSM-7001F, JEOL Ltd., Tokyo, Japan) and transmission electron microscopy (TEM; JEM-2100, JEOL Ltd., Tokyo, Japan). X-ray diffraction (XRD) spectra were recorded on a D8 ADVANCE X-ray diffractometer (Bruker Corp., Germany). Raman spectra were recorded with a Horiba LabRAM HR Evolution spectrometer. The N2 adsorption isotherms were measured using a micromeritics ASAP2020 analyzer. Brunauer–Emmett–Teller (BET) analysis and density functional theory (DFT) were utilized to obtain the surface area and pore size distribution, respectively. XPS spectra were measured on an ESCALAB 250Xi photoelectron spectrometer. All electrochemical tests were carried out on a CHI 660E electrochemical workstation.2.2 Materials synthesis
The preparation of the carbon electrodes from the different chemical structures was displayed in Scheme 1, specifically. Five grams each of sodium succinate, sodium malate and sodium tartrate were weighed and placed in three alumina crucibles, individually. The alumina crucibles were then transformed to a tubular furnace. Then, the furnace was heated at a heating rate of 3 °C min−1 once 800 degrees was reached and then the temperature was held at 800 degrees for 1 hour. 800 °C, as the carbonization temperature, referred to previous reports.15–17 By literature consulting, it is believed that 700–900 °C is a reasonable pyrolysis temperature. Increasing pyrolysis temperature (700 °C) can promote the formation of an ordered graphitic structure, but higher pyrolysis temperature would produce irregular carbon nanostructures. However, when the pyrolysis temperature is higher than 900 °C, no or very few products exist. Hence, we choose 800 °C as the pyrolysis temperature to synthesize porous carbon materials. Afterwards, the carbonized products were washed with 1 mol L−1 HCl to remove the alkaline sodium salt, and then washed with distilled water until the pH value of the eluent was neutral. Finally, the mushy materials were then freeze dried for 48 h to remove any trace of the solvent. The three carbon materials were named as PCSS (porous carbon derived from sodium succinate carbon source), PCSM (porous carbon derived from sodium malate) and PCST (porous carbon derived from sodium tartrate), respectively. | ||
| Scheme 1 The scheme for preparation of the carbon electrodes from the different chemical structures. | ||
2.3 Electrochemical measurements
Electrochemical analysis of nitrite was analyzed by three-electrode electrochemical system. A bare or modified glassy carbon electrode was used as the working electrode. The modified electrode was fabricated by dropping 6 μL of suspension on the surface of bare GCE, and air dried at room temperature. The dispersion solution was prepared by mixing N, N-dimethylformamide (DMF) and distilled water in proper ratio. Because porous carbon material has the physical property of easy agglomeration, it is difficult to disperse evenly with water or ethanol. Therefore, N,N-dimethylformamide (DMF) was chosen as one of important solvent. To obtain the optimal electrochemical performance, the ratio of DMF to water was optimized. As shown in Fig. S1,† the volume fraction of DMF was 50%, PCST/GCE-based sensor displayed a maximum current intensity. Therefore, 50% DMF was chosen as the dispersing agent of the porous carbon material during the subsequent experiments. 1.0 mg mL−1 porous carbon dispersion solution was obtained by ultrasonic dispersion. An Ag/AgCl electrode with saturated KCl solution and a platinum electrode were used as the reference electrode and the auxiliary electrode, respectively.2.4 Electrochemical detection of nitrite in actual sample
The pretreatment of actual sample referred to previous report.18 The liquid supernatant would be obtained after sample pretreatment. The actual samples were detected by amperometric measurements. For amperometric measurements, the potential parameter was set as 0.9 V. A magnetic stirring was conducted in the electrolytic cell until the electrochemical signal keep stable. And then added 5 μL supernatant to the electrolytic cell. Thus, the value of the increased current intensity was obtained. The data were substituted into the standard curve to calculate the concentration of NO2−in the real samples.3. Results and discussions
3.1 Physical characterization
To explain the formation of three-dimensional porous carbon, the unwashed and washed pyrolysis products derived from different carbon sources were characterized by SEM and TEM. As shown in Fig. 1(A, C and E), the unwashed samples showed encased granular structures. The formation of encased granular structures was due to that Na2CO3 crystals formed during calcining and were covered by a carbon sheet. After the removal of Na2CO3 crystal, the carbon nanosheets collapsed to form a superimposed morphology of large nanosheets, as shown in Fig. 1(B, D and F). The phenomenon also signified that Na2CO3 particles played a key template role in the formation of the porous network structure. As displayed in Fig. 1A and B, PCSS displayed a bulk and conglomerate morphology. Instead of the bulk and conglomerate structure, PCSM (Fig. 1D) retained a complete macroporous structure with sharp edges and smooth walls. Fig. 1D and S2C† showed that PCST possessed a three-dimensional network structure with many pores of different diameters linked to each other, thus increasing the surface area.19 The interconnected 3D structure may provide ion transport channels, which will improve the ability for electron transfer. The pore walls of PCST were broken and co-existed with some carbon nanoparticles attached to the edge of the broken pores. These macropores were beneficial to the diffusion of electrolyte ions. Compared with the other two materials, the PCST had few macroporous pore with the diameter of about 100–200 nm and a few nanoparticles with the size of about a few tens of nanometer. This may be because of the differences in the number of initial nucleation. The structural characteristics of the material were further studied by TEM. The PCSS displayed a large independent lamellar structure and visible disordered pores of 50–100 nm on the carbon sheet (Fig. S2A†). The PCSM was a network structure of carbon sheet accumulation with a relatively dense pore structure on the sheets, as exhibited in Fig. S2B.† Fig. S2C† showed the PCST was composed of many carbon nanosheets, and these carbon nanosheets formed a three-dimensional network structure. Besides, more lamellar edges were exposed and some broken carbon particles adhered to the carbon sheets. The lamellar edges and carbon particles could provide more active sites for the catalytic reaction. Importantly, HRTEM images (Fig. S2D–F†) showed that the prepared carbon material possessed mesopore structure, which would offer fast ion transport channels. | ||
| Fig. 1 SEM images of unwashed (A, C and E) and washed (B, D and F) pyrolysis products prepared from different carbon sources: sodium succinate (A and B), sodium malate (C and D) and sodium tartrate (E and F). | ||
Nitrogen physisorption measurements were carried out to analyze the specific surface area and pore size distribution of the obtained materials. As shown in Fig. S3A,† the absorption volume increased sharply at a lower relative pressure (P/P0 <0.01), implying the presence of a large number of micropores. The continuous increase of the absorption volume at a relative pressure of 0.1–0.5, and the large hysteresis loop at a high relative pressure (P/P0 > 0.5) signified the existence of abundant mesopores. Furthermore, the pore size distribution curves of PCSS, PCSM and PCST were displayed in Fig. S3B.† It shows seven sharp peaks which are centered at 2.74, 1.30 and 1.25 nm, respectively. The DFT pore size distribution curves are an integration of type I and type IV, which were similar to those reported in previous literature.17 The BET surface areas of PCST, PCSM and PCSS were calculated to be 380.62 m2 g−1, 658.04 m2 g−1 and 197.41 m2 g−1 with the pore volumes of 0.77, 1.13 and 0.28 cm3 g−1, respectively. The porosity of porous carbon materials was mainly made up of micropores, mesopores structures and few macropores structures. The prepared porous carbon material contains abundant micropores structure, mesopore structures, macropores structures and carbon particles. The three kinds of structures all play its role in improving the electrochemical performance, which could be ascribed to the follow aspects: (i) micropores structure could make the carbon sufficiently contact with the electrolyte and provide high surface area, ensuring a large specific capacitance and high energy density. (ii) The mesopores structure could provide fast ion transport channels and decrease the diffusion resistance; (iii) a certain amount of macropores and effective carbon particles (>50 nm) are helpful to shorten the transmission distance by providing ion-buffering reservoirs. The results indicated that the number of hydroxyl groups influenced the specific areas and the total pore volumes to a certain extent. The synthesized porous carbon exhibited an appropriate porous structure with abundant micropores and a part of mesopores. The micropore structure could make the electrode material expose more active sites.11
The thermodynamic behaviors of sodium succinate, sodium malate and sodium tartrate were examined at 50–850 °C under nitrogen flow. As shown in Fig. 2A, the TGA curve of PCST indicated a two-step mass loss. The mass loss steps for 0–200 °C corresponded to crystal water elimination. The mass loss at 250–300 °C was associated with sodium tartrate decomposition. Each unit of sodium tartrate produces two units of NaHCO3, and further decomposing into a molecule of H2O and a molecule of CO2. The theoretical mass loss of this reaction (∼32% = 18 + 44/194) was almost identical to the actual mass loss (∼33%), which meant that all Na of the sodium tartrate was converted from organic-Na to inorganic-Na. The three carbon sources exhibited different degrees of mass loss for 300–500 °C, which may be due to the differences in –COONa contents. Two units of –COONa removed a molecule of CO2 to produce a unit of Na2CO3 at ∼450 °C.
 | ||
| Fig. 2 (A) TGA thermograms of the sodium succinate, sodium malate and sodium tartrate; (B) XRD patterns; (C) Raman spectra of the porous carbon material derived from sodium succinate, sodium malate and sodium tartrate. (D) the corresponding fitted Raman spectrum of the porous carbon material derived from sodium succinate (D), sodium malate (E) and sodium tartrate (F). | ||
Three carbon material mass loss types occurred in reaction 1. The escape of CO2 accelerated the generation of a certain number of holes. Based on the higher number of microporous and the lower sp3 carbon content of the sodium malate, the extra mass loss of the sodium malate participated in reaction 2. The fine Na2CO3 grains that anchored inside the carbon sheet had a high reactivity because of their small size. As the reaction product, Na2O existed at a small size and with a high reactivity, which could react with active C (sp3) of the carbon sheets. Thus, additional micropores and mesoporous were produced.
| Na2CO3 → Na2O + CO2 | (1) |
| Na2O + C(sp3) → 2Na + CO | (2) |
The XRD patterns of porous carbon material in Fig. 2B displayed two broad peaks, which were typical carbonaceous materials with an amorphous-like structure.20 The diffraction peaks at ∼25° and 43° may be attributed to (002) and (100) index planes, respectively,21 indicating that PCSM had a higher degree of graphitization, which was in favour of obtaining a higher electrical conductivity.8 X-ray photoelectron spectroscopy (XPS) was used to study the distribution of elements on the surface of porous carbon materials. The surface element contents of the three porous carbon materials are shown in Table S1.† Three kinds of porous carbon materials were mainly consisted of C, O and Na. The content of oxygen atomic was 11.95 in PCST material, which may improve the dispersion of PCST material in the dispersing agent (50% DMF).
Moreover, degree of graphitization of the prepared porous carbon materials was examined by Raman spectroscopy. The Raman spectra of PCST, PCSM and PCSS were compared in Fig. 2C. The characteristic peaks of the typical carbon materials were the D-band (1350 cm−1) and G-band (1600 cm−1).22 Previous studies have shown that the two peaks can be fitted into four separate peaks at ∼1194 (peak 1), 1347 (peak 2), 1510 (peak 3) and 1585 cm−1 (peak 4). Peaks 1 and 3 resulted from a sp3-type diamond-like structure, and peaks 2 and 4 resulted from an sp2-type graphite-like structure. The integrated intensity ratio Isp2/Isp3 could be used to characterize the degree of carbon graphitization, where Isp2 and Isp3 were the total areas of (peak 2 + peak 4) and (peak 1 + peak 3), respectively.23 According to the fitting peaks of Fig. 2D–F, the Isp2/Isp3 values of PCST, PCSM and PCSS were calculated to be 1.72, 2.74 and 2.08, suggesting that PCSM exhibited a higher degree of graphitization. The results value is consistent with the XRD results.
3.2 Electrochemical behaviors
To explore the reasons for improving electrochemical performance, the interfacial properties of the electrode materials were studied by Electrochemical Impedance Spectroscopy (EIS). As shown in Fig. 3A, the Nyquist impedance plot comprised of a semicircle and a linear part. The semicircle part in the high-frequency region corresponds to an electron-transfer-limited process and the linear part in the low-frequency region represents a diffusion-limited process. A smaller semicircle means a low electrode charge-transfer resistance, suggesting a high charge-transfer rate.24 Therefore, porous carbon modified electrode had a high charge-transfer rate. Conversely, the bare GCE had a relatively low charge-transfer rate. The enlarged EIS curves with fitting curves were provided in Fig. S4.† The fitted values of the Rct for bare GCE, PCST/GCE, PCSM/GCE and PCSS/GCE were 166.7 Ω, 13.13 Ω, 0.01 Ω and 16.08 Ω, respectively, showing that porous carbon material greatly enhanced the charge-transfer efficiency. The cyclic voltammetric curves (Fig. 3B) of nitrite display obvious oxidation peaks at the modified electrodes. Compared with bare GCE, the capacitance and peak currents of the PCST/GCE increased apparently, which may be ascribed to that porous carbon material accelerate the electron transfer at the electrode surface. To provide an estimate of precision, repeated CV experiments were carried out. The mean value of peak current obtained by PCST/GCE, PCSM/GCE, PCSS/GCE and GCE was 94.01 μA, 57.87 μA, 66.77 μA and 40.45 μA with the standard deviation of 3.18, 4.06, 3.90 and 0.80169, respectively, which indicates a good precision of CV characterisation. To further evaluate the electrochemical performance of different electrodes, we carried out the cyclic voltammetry experiments in 5 mM [Fe(CN)6]3−/4− containing 0.1 M KCl. As shown in Fig. S5F,† compared with the other electrodes, PCST/GCE displayed superior electrochemical performance. | ||
| Fig. 3 (A) Electrochemical impedance spectroscopy of different modified electrodes (PCSS/GCE, PCSM/GCE and PCST/GCE and bare GCE) Inset: the Randles equivalent circuit. (B) Voltammetric behaviors of different electrodes in 1 mM nitrite (pH 3.0, 0.1 M PBS buffer) at the scan rate of 100 mV s−1. | ||
And the electrochemical active surface areas of four different electrodes were calculated according to the cyclic voltammograms (CVs) in Fig. S5.† Based on the Randles–Sevcik equation: Ip = (2.69 × 105)n3/2AD1/2v1/2C, where A is the geometric area of the electrode, C is the bulk reactant concentration, D is the reactant diffusion coefficient, v is the voltage scan rate and α is the transfer coefficient. n is the number of electrons F, R and T have their usual meanings, the effective surface areas (ECSAs)of the PCST/GCE, PCSM/GCE, PCSS/GCE and GCE were calculated to be 0.055, 0.043, 0.031 and 0.022 cm2, respectively. The results verify that porous carbon materials could upgrade the electrochemical active surface area and enhance the capacity of electron transfer, thus causing the improvements of electrocatalytic performance. Thereinto, PCST/GCE exhibited the best electrocatalytic performance.
Compared to the other types porous carbon derived from sodium tartrate (PCST)-based sensor showed the best catalytic activity, which may be ascribed to the several aspects: (1) PCST possessed a high graphitization degree, which is favorable for electron mobility and stabilization of the reactive intermediates in the radical-involved catalytic systems; (2) PCST/GCE displayed larger effective surface areas, which could enhance the capacity of electron transfer, thus causing the improvements of electrocatalytic performance. In conclusion, PCST/GCE exhibited the best electrocatalytic performance. (3) PCST had good mesoporous structures and abundant sp3 C, providing sufficient active sites for electrocatalytic reaction.
Our initial thought was that some relationship may exist between the contents of hydroxyl groups on the side chain in organic salt precursors and the morphology of porous carbon, which may result in regular electrochemical catalytic activities. After lots of experiment, it seems that hydroxyl groups on the side chain in organic salt precursors has some influence on the morphology of porous carbon, however, the effects on electrochemical performance were not as much as we thought. The phenomenon may be due to that there are many influencing factors on electrochemical catalytic activity, such as the type of material, geometry, surface factor, energy factor and so on. The factors influenced electrochemical catalytic activity require further study. This is the future trend and the direction of our efforts.
To investigate the reaction kinetics, the effect of scan rate on the electrocatalytic oxidation of nitrite at PCST/GCE was studied. As shown in Fig. 4A, the nitrite oxidation peak current (Ipa) showed a good linear dependence with the scan rate square root (v1/2). As shown in the inset of Fig. 4B, this linear dependence could be expressed by a linear equation, Ipa (μA) = 420.8v1/2 − 26.67 with a satisfactory correlation coefficient of 0.992, which represented a diffusion-controlled electrocatalytic process for 10–100 mV s−1.25 To further confirm the presence/absence of adsorption effects, we have plotted to higher scan rates against scan rates (100–500 mV s−1). However, the coefficient of determination was 0.956, which is lower than the coefficient of determination (0.992) obtained by plotting higher scan rates against square root scan rates, as exhabited in Fig. 4C and D. These results indicated that the oxidation reaction of nitrite at PCST/GCE were diffusion controlled-controlled processes. For a typical irreversible diffusion-controlled reaction, the electron transfer could be evaluated from the eqn (3) and the linear equation in Fig. 4B.26 The electrochemical oxidation of nitrite at the PCST/GCE was calculated to be a two-electron transfer process.
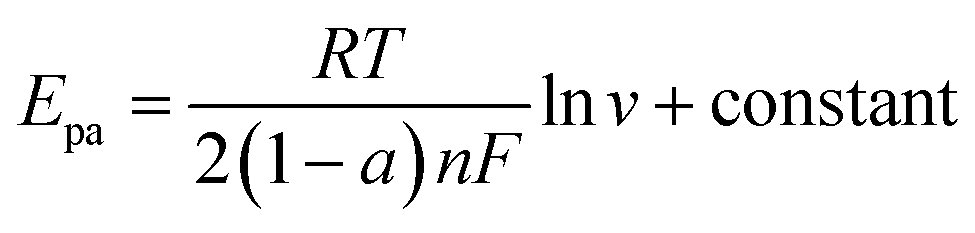 | (3) |
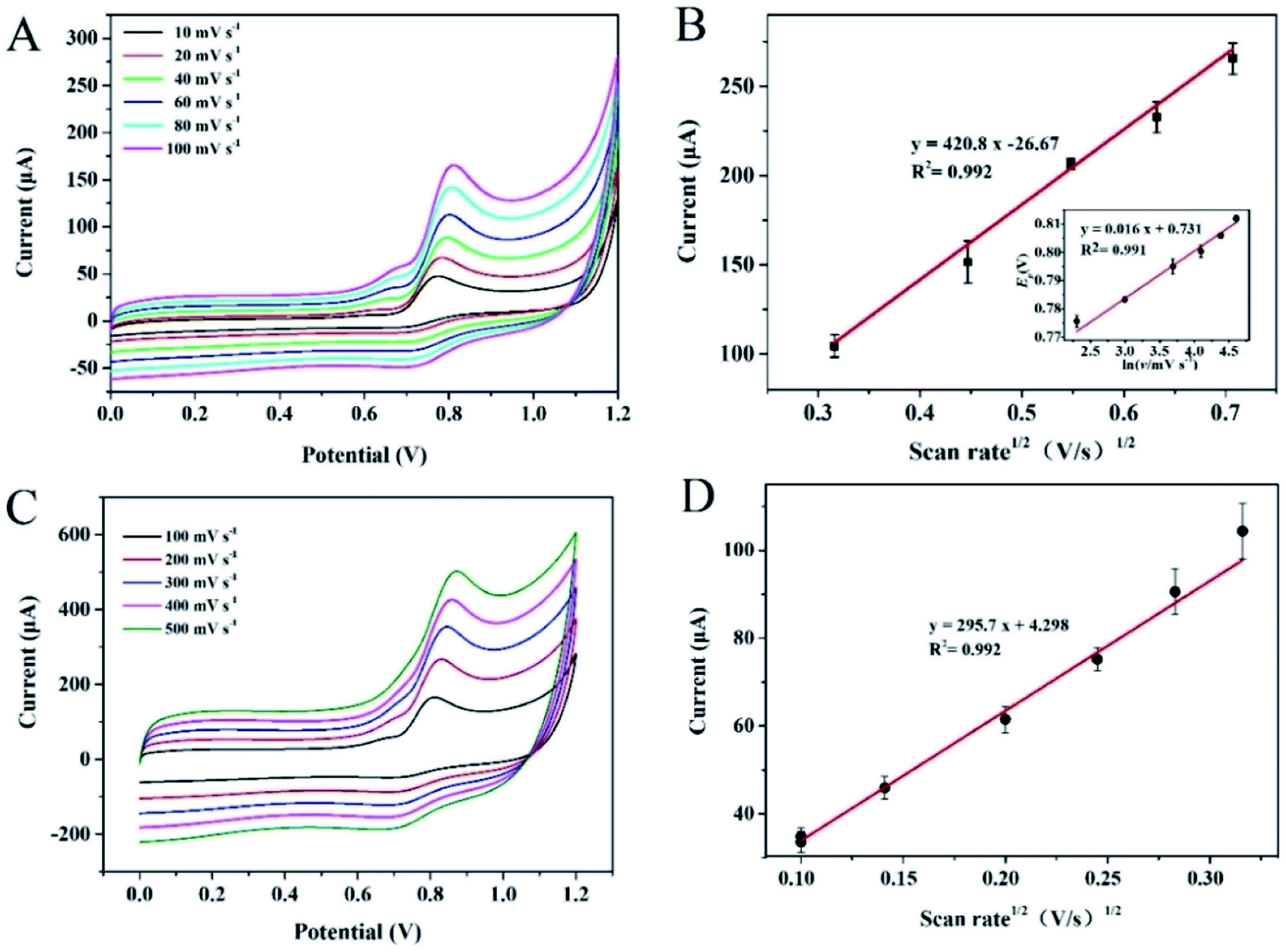 | ||
| Fig. 4 (A) Cyclic voltammograms of different electrodes recorded at different scan rates of 10–100 mV s−1 in 1 mM nitrite (pH 3.0, 0.1 M PBS buffer); (B) the corresponding Linear relationship between anodic peak currents (Ipa) vs. the square root of scan rates; Inset: the linear relationship between Ep versus the scan rates. (C) CV curves recorded at different scan rates in the range of 100–500 mV s−1 in 1 mM nitrite (pH 3.0, 0.1 M PBS buffer); (D) the corresponding linear relationship between anodic peak currents (Ipa) vs. the square root of scan rates. | ||
3.3 Electrochemical performance evaluation of the developed sensor
The linear range, detection limit, sensitivity and anti-interference are important factors to evaluate the electrochemical sensing ability. To study the analytical performance of a PCST-based sensor, the amperometric i–t curve was recorded by adding nitrite continuously at 50 s intervals (Fig. 5A). During testing, the electrolyte kept stirring at a speed of 500 rpm constantly to avoid the heterogeneous distribution of nitrite. The current intensity increased linearly with the increasing nitrite concentration in a wide linear range of 0.1–100 μM, as exhibited in Fig. 5B. The linear equation is y(I) = 0.1175x(c) + 0.04796 (R2 = 0.986). The detection limit is calculated to be as low as 0.043 μM, which is far below the maximum residue limit in ham sausage (30 mg kg−1) regulated by the Chinese standard. In other words, the proposed method completely meets the requirements of nitrite detection and shows better electrochemical performance than the electrochemical methods reported by our previous report.18 Otherwise, as displayed in Table 1, compared with these methods, the proposed method displayed comparable or even superior analytical performance. Although some previous methods have lower detection limits, the proposed method possess its unique novelties and advantages, specifically. As far as the preparation of electrode material, PCST was synthesized by one-step pyrolysis method. Besides, carbon material has the advantages of low cost and abundant raw materials. Conclusively, the proposed method allowed sensitive and selective detection of nitrite with satisfactory analytical performance. | ||
| Fig. 5 (A) Amperometric i–t curve of PCST/GCE to successive addition of different concentration of nitrite (pH 3.0, 0.1 M PBS buffer); (B) linear calibration plot of response current vs. the concentration of nitrite (pH 3.0, 0.1 M PBS buffer); (C) i–t response of nitrite in the presence of KCl, CO32−, HCO3−, Cu2+, ascorbic acid, glucose, sodium citrate and nitrate (pH 3.0, 0.1 M PBS buffer). | ||
| Electrode materials | Linear range | LOD | Ref. |
|---|---|---|---|
| Pt/CoO/GCE | 0.2 μM to 3670 mM; 3.67–23.7 mM; | 0.067 μM | 26 |
| Ag/Cu/MWNTs/GCE | 1.0 μM to 1.0 mM | 0.2 μM | 27 |
| Ag–Cu@ZnO/GCE | 0–1500 μM | 17 μM | 28 |
| P-3ABA/GCE | 10–140 μM | 0.15 μM | 29 |
| PANI–CNT/GCEs | 20 μM to 1.8 mM | 7.8 μM | 30 |
| Au NPs/Ti3C2TX/GCE | 1.0 μM to 4581.1 μM | 0.14 μM | 31 |
| AuNPs/CS@N,S–MWCNTS/GCE | 1–7000 μM | 0.2 μM | 32 |
| polyNiCo/GCE | 2.5 μM to 1.73 mM | 0.45 μM | 33 |
| AgNS/GCE | 0.1–8 μM | 0.031 μM | 34 |
| PCST/GCE | 0.1–100 | 0.043 μM | This work |
Selectivity plays a key role in evaluating the analytical performance of the constructed sensor. The electrochemical response of possible interferents, including nitrate, potassium chloride, carbonate, bicarbonate, glucose, cupric ion, ascorbic acid and sodium citrate, was inspected (Fig. 5C). The current almost unchanged after the addition of these possible interferents, however, the current increased obviously with the injection of NaNO2, suggesting that the constructed PCST-based sensor possessed a good selectivity. At the potential of 0.9 V, the sensor displayed relatively good selectivity for nitrite. Different substances possess different electrochemical active, resulting that they have different redox potentials. Nitrite oxidation potential is about 0.9 V. Therefore, nitrite shows good selectivity at this potential. However, if a substance has a similar oxidation potential to nitrite, and the substance may have some influence on the detection of nitrite. To investigate the stability of the constructed sensor, cyclic voltammograms of NaNO2 were tested once a day for a week. The oxidation peak current of 1.0 mM NaNO2 decreased by ∼3.83% after one week storage at 4 °C in air in fridge in absence of NaNO2, which indicated the excellent stability of the constructed sensor.
3.4 Real sample analysis
To confirm the practicability of the developed sensor, the electrochemical sensor was used to detect nitrite in meat samples. Meanwhile, ion chromatography was applied to validate the results that were obtained by the electrochemical sensor. Table 2 shows that the nitrite concentrations tested by the proposed sensor were consistent with the results obtained by ion chromatography. As summarized in Table 2, the relative standard deviation (RSD) of the repeated measurements for the eight samples ranged from 5.07% to 8.71%, which suggested the favorable precision of this developed sensor. In conclusion, the proposed sensor possessed good accuracy and reliability and could be further applied to determine nitrite in real samples.| Samples | Ion chromatography | Electrochemical method | Electrochemical method (mean ± SD) | RSD/% | ||
|---|---|---|---|---|---|---|
| N1 | N2 | N3 | ||||
| a Note: N1, N2 and N3 represents parallel number of measurements. N1 corresponds the value obtained by first detection. N2 corresponds the value obtained by second detection. N3 corresponds the value obtained by third detection. | ||||||
| Meat 1 | 28.08 | 23.1 | 26.29 | 27.39 | 25.59 ± 2.23 | 8.71 |
| Meat 2 | 31.3 | 31.43 | 32.98 | 35.01 | 33.14 ± 1.80 | 5.43 |
| Meat 3 | 46.04 | 41.87 | 44.58 | 47.33 | 44.59 ± 2.73 | 6.12 |
| Meat 4 | 66.4 | 66.45 | 71.39 | 73.38 | 70.41 ± 3.57 | 5.07 |
4. Conclusions
Porous carbon material was synthesized by the one-step pyrolysis of organic salts with different numbers of hydroxyl groups on the side chain (sodium tartrate, sodium malate and sodium succinate). In addition, the effects of different carbon sources on the formation of porous carbon structures were revealed and then, the electrochemical sensor constructed by different three-dimensional porous carbon was applied in electrochemical sensing of nitrite. PCST-based sensor presented the best electrochemical analytical performance with a wide linear range and a low detection limit. Meaningfully, the fabricated sensor could be applied to detect nitrite in meat samples with a good stability and reproducibility. This electrochemical analysis method provided technical support for relevant departments to monitor nitrite abuse by applying this electrochemical sensor to nitrite detection in processed meat products.Conflicts of interest
The authors declare that there is no conflict of interests regarding the publication of this paper.Acknowledgements
This research was financed by the National Natural Science Foundation of China (Project No. 31801630), the Foundation of Henan Educational Committee (19A550012), the Postgraduate Education Reform Project of Henan Province (2019SJGLX018Y) and the Key Scientific and Technological Project of Henan Province (No: 212102310493).References
- H. Wei, K. Liao, P. Shi, J. Fan, Q. Xu and Y. Min, Nanoscale, 2018, 10, 15842–15853 RSC.
- M. Song, Y. Zhou, X. Ren, J. Wan, Y. Du, G. Wu and F. Ma, J. Colloid Interface Sci., 2019, 535, 276–286 CrossRef CAS PubMed.
- M. Xu, Q. Yu, Z. Liu, J. Lv, S. Lian, B. Hu, L. Mai and L. Zhou, Nanoscale, 2018, 10, 21604–21616 RSC.
- R. Balgis, S. Sago, G. M. Anilkumar, T. Ogi and K. Okuyama, ACS Appl. Mater. Interfaces, 2013, 5, 11944–11950 CrossRef CAS PubMed.
- M. H. Sun, S. Z. Huang, L. H. Chen, Y. Li, X. Y. Yang, Z. Y. Yuan and B. L. Su, Chem. Soc. Rev., 2016, 45, 3479–3563 RSC.
- N. Lu, T. Zhang, X. Yan, Y. Gu, H. Liu, Z. Xu, H. Xu, X. Li, Z. Zhang and M. Yang, Nanoscale, 2018, 10, 14923–14930 RSC.
- L. l. Yunhui Xiang, H. liu, Z. Shi, Y. Tan, C. Wu, Y. Liu, J. Wang and S. Zhang, Sens. Actuators, B, 2018, 267, 302–311 CrossRef.
- R. M. Pitchaimani Veerakumar, S.-M. Chen, C.-T. Hung, P.-H. Tang, C.-B. Wang and S.-B. Liu, Analyst, 2014, 139, 4994–5000 RSC.
- R. X. Lili Xiao, Q. Yuan and F. Wang, Talanta, 2017, 167, 39–43 CrossRef PubMed.
- S. W. Xingxing Wu, Y. Li, C. Han, Q. Yuan and W. Gan, Anal. Chim. Acta, 2019, 1046, 115–122 CrossRef PubMed.
- X. H. Zhao, S. N. Ma, H. Long, H. Yuan, C. Y. Tang, P. K. Cheng and Y. H. Tsang, ACS Appl. Mater. Interfaces, 2018, 10, 3986–3993 CrossRef CAS PubMed.
- Y. Zhang, W. Zhu, Y. Wang, Y. Ma, J. Sun, T. Li, J. Wang, X. Yue, S. Ouyang and Y. Ji, Inorg. Chem. Front., 2019, 6, 1501–1506 RSC.
- L. Huang, D. W. Sun, H. Pu and Q. Wei, Compr. Rev. Food Sci. Food Saf., 2019, 18, 1496–1513 CrossRef PubMed.
- M. S. a. A. B. Fuertes, J. Mater. Chem. A, 2013, 1, 13738–13741 RSC.
- X. Yu, J. Zhao, R. Lv, Q. Liang, B. Yu, Z. Huang, W. Shen and F. Kang, RSC Adv., 2015, 5, 75403–75410 RSC.
- Z. Jia, D. Liu and D. Wang, Carbon, 2016, 100, 664–677 CrossRef.
- A. B. Fuertes and M. Sevilla, ACS Appl. Mater. Interfaces, 2015, 7, 4344–4353 CrossRef CAS PubMed.
- X. Yue, X. Luo, Z. Zhou, Y. Wu and Y. Bai, New J. Chem., 2019, 43, 4947–4958 RSC.
- H. Zhang, Y. Ling, Y. Peng, J. Zhang and S. Guan, Inorg. Chem. Commun., 2020, 115, 107856 CrossRef CAS.
- M. S. a. A. B. Fuertes, ACS Nano, 2014, 8, 5069–5078 CrossRef PubMed.
- W. Zhang, Y. Bao and A. Bao, J. Environ. Chem. Eng., 2020, 8, 103732 CrossRef CAS.
- H. Liang, R. Sun, B. Song, Q. Sun, P. Peng and D. She, J. Hazard. Mater., 2020, 387, 121987 CrossRef CAS PubMed.
- Y. Jin, C. P. Yang, X. H. Rui, T. Cheng and C. H. Chen, J. Power Sources, 2011, 196, 5623–5630 CrossRef CAS.
- Y. Ma, X. Song, X. Ge, H. Zhang, G. Wang, Y. Zhang and H. Zhao, J. Mater. Chem. A, 2017, 5, 4726–4736 RSC.
- S. Kang, H. Zhang, G. Wang, Y. Zhang, H. Zhao, H. Zhou and W. Cai, Inorg. Chem. Front., 2019, 6, 1432–1441 RSC.
- L. Lu, Sens. Actuators, B, 2019, 281, 182–190 CrossRef CAS.
- Y. Zhang, J. Nie, H. Wei, H. Xu, Q. Wang, Y. Cong, J. Tao, Y. Zhang, L. Chu, Y. Zhou and X. Wu, Sens. Actuators, B, 2018, 258, 1107–1116 CrossRef CAS.
- G. Manjari, S. Saran, S. Radhakrishanan, P. Rameshkumar, A. Pandikumar and S. P. Devipriya, J. Environ. Manage., 2020, 262, 110282 CrossRef CAS PubMed.
- W. A. A. Abdullah, M. Asiri, H. M. Marwani and M. M. Rahman, New J. Chem., 2020, 44, 2022–2032 RSC.
- F. Gao, H. Teng, J. Song, G. Xu and X. Luo, Anal. Methods, 2020, 12, 604–610 RSC.
- H. Zou, F. Zhang, H. Wang, J. Xia, L. Gao and Z. Wang, New J. Chem., 2019, 43, 2464–2470 RSC.
- H. Rao, Y. Liu, J. Zhong, Z. Zhang, X. Zhao, X. Liu, Y. Jiang, P. Zou, X. Wang and Y. Wang, ACS Sustainable Chem. Eng., 2017, 5, 10926–10939 CrossRef CAS.
- T. Islam, M. M. Hasan, S. S. Akter, N. H. Alharthi, M. R. Karim, M. A. Aziz, M. D. Hossain and A. J. S. Ahammad, ACS Appl. Polym. Mater., 2019, 2, 273–284 CrossRef.
- M. Shivakumar, K. L. Nagashree, S. Manjappa and M. S. Dharmaprakash, Electroanalysis, 2017, 29, 1–10 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05105a |
| This journal is © The Royal Society of Chemistry 2021 |