
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of a novel bridged bis(β-cyclodextrin) chiral stationary phase by thiol–ene click chemistry for enhanced enantioseparation in HPLC†
Ning Zhang,
Siyu Guo and
Bolin Gong *
*
School of Chemistry and Chemical Engineering, North Minzu University, No. 204 Wenchang North Street, Xixia District, Yinchuan 750021, China. E-mail: gongbolin@163.com
First published on 5th November 2021
Abstract
A bridged bis(β-cyclodextrin) ligand was firstly synthesized via a thiol–ene click chemistry reaction between allyl-ureido-β-cyclodextrin and 4-4′-thiobisthiophenol, which was then bonded onto a 5 μm spherical silica gel to obtain a novel bridged bis(β-cyclodextrin) chiral stationary phase (HTCDP). The structures of HTCDP and the bridged bis(β-cyclodextrin) ligand were characterized by the 1H nuclear magnetic resonance (1H NMR), solid state 13C nuclear magnetic resonance (13C NMR) spectra spectrum, scanning electron microscope, elemental analysis, mass spectrometry, infrared spectrometry and thermogravimetric analysis. The performance of HTCDP in enantioseparation was systematically examined by separating 21 chiral compounds, including 8 flavanones, 8 triazole pesticides and 5 other common chiral drugs (benzoin, praziquantel, 1-1′-bi-2-naphthol, Tröger's base and bicalutamide) in the reversed-phase chromatographic mode. By optimizing the chromatographic conditions such as formic acid content, mobile phase composition, pH values and column temperature, 19 analytes were completely separated with high resolution (1.50–4.48), in which the enantiomeric resolution of silymarin, 4-hydroxyflavanone, 2-hydroxyflavanone and flavanone were up to 4.34, 4.48, 3.89 and 3.06 within 35 min, respectively. Compared to the native β-CD chiral stationary phase (CDCSP), HTCDP had superior enantiomer separation and chiral recognition abilities. For example, HTCDP completely separated 5 other common chiral drugs, 2 flavanones and 3 triazole pesticides that CDCSP failed to separate. Unlike CDCSP, which has a small cavity (0.65 nm), the two cavities in HTCDP joined by the aryl connector could synergistically accommodate relatively bulky chiral analytes. Thus, HTCDP may have a broader prospect in enantiomeric separation, analysis and detection.
1. Introduction
Many studies have shown that the enantiomers of chiral compounds tend to demonstrate obvious differences in biological activity, and pharmacological and toxicological effects,1–3 which makes enantioseparation a hot research topic in food chemistry, the pharmaceutical industry and environmental protection.4,5 So far, conventional enantiomer resolution methods include chemical resolution, enzyme kinetic resolution, high-performance liquid chromatography (HPLC),6 gas chromatography,7 and capillary electrochromatography.8 Among them, HPLC is widely used because of its high sensitivity, reproducibility, rapid separation speed, large column capacity, and broad applicability. At present, various chiral stationary phases (CSPs) combined with HPLC have been proved to be one of the most effective methods for direct separation of enantiomers.9 Therefore, the development of multifunctional and efficient CSPs is the key to improving the enantiomeric resolution. β-cyclodextrin (β-CD) is a mesa-shaped oligosaccharide formed by connecting 7 D-glucopyranose units via α-1,4 glycosidic bonds. The unique cavity structure of β-CD allows the formation of inclusion complexes with chiral analytes, which has been proved to be one of most important roles in driving enantioseparation.10 Additionally, the introduction of various substituents through the functionalization of the hydroxyl group on β-CD can also promote the separation of enantiomers. For example, many interactions including hydrogen bonding, π–π conjugation, dipole–dipole, steric repulsion, and static electricity can be introduced through functionalization of the hydroxyl group on β-CD.11–14 The introduction of these interactions enables β-CD CSP to enhance the identification and separation of chiral analytes. Therefore, various types of β-CD CSPs are widely prepared and used in chiral separations. Moreover, compared with vancomycin,15 teicoplanin,16 cellulose,17 amylos18 and polymer CSPs,19 such multi-functional enantiomeric separation mode is also an important reason why β-CD CSPs have received widespread attention and application.However, the separation ability and stability of β-CD CSPs are significantly affected by the functionalization of its hydroxyl groups and bond chemistry on silica gel.20,21 β-CD hydroxyl derivatization includes single,22 partial,11 and complete23 derivatization. Single-derivatized β-CD CSPs have a simple structure and stable chromatographic performance but limited ability to separate chiral enantiomers.17 By comparison, partially derivatized β-CD CSPs show improved enantioseparation but the number and position of the derived substituents are difficult to control, resulting in poor chromatographic reproducibility.24 Fully derivatized β-CD CSPs have more functional sites but too many substituents inevitably reduce the inclusion capacity of the β-CD cavity.25 Shuang et al.26 prepared a novel of single-derivatized β-CD CSP and separated 9 triazole pesticides. However, the resolution was not high, which may be caused by the low degree of derivatization. Armstrong research group27–29 prepared a series of fully derivatized β-CD CSPs. The enantioselectivity of β-CD CSPs was improved through the introduction of carbamate linkages, and successfully resolved chiral alcohols, amino acids and chiral amines. However, fully derivatized β-CD CSPs could not separate crown ether and dansyl amino acid enantiomers, which could be separated on natural β-CD CSPs. According to Stalcup and Chang,30 the fully derivatized β-CD CSPs may be due to the high degree of derivatization blocking the cavity of β-CD. Moreover, the separation of enantiomers is an extremely complex and delicate process that requires multiple recognition sites to cooperate with each other,31 with the inclusion effects and hydrogen bonding playing important roles.32 Therefore, it may not be sufficient to rely solely on the functionalization of the hydroxyl group on β-cyclodextrin to improve the enantiomeric selectivity. In recent years, the emergence of bridged β-CD CSPs is a very promising candidate.33
The Bridged bis(β-CD) is a supramolecular dimer connected by 2 single-derived CD units that has been widely used for molecular recognition and as a targeted drug carrier, photochemical, and asymmetric catalyst34–37 because of its greater capacity for synergistic inclusion and molecular recognition compared to single β-CD.38 In recent years, although the bridged β-CD has been widely used in many fields, the CSPs used for the separation of enantiomers are rarely reported. Wang's research group prepared a series of triazolyl-bridged bis(β-CD) CSPs and successfully separated enantiomers such as dansyl amino acids, aryl carboxylic acids and isoxazoline with high resolution.39,40 Yuan's research group prepared two bridged β-CD CSPs and separated 4 aromatic compound positional isomers and 5 amino acid enantiomers.41 Li's research group prepared a series of bridged bis(β-CD) CSPs and separated β-blockers, flavanones, triazoles, dansyl amino acids and other enantiomers with high resolution.42–44 Obviously, bridged β-CD CSPs have potential development prospects in improving the resolution of enantiomers. Liu and Chen45 also found that the bridged spacer arm of the bridged bis(β-CD) can be used as a pseudo-cavity to provide additional sites of action, thereby improving the ability of molecular recognition. Morever, thiol–ene click chemistry has been used to bridge spacer arms between mono-β-CD and silica gel due to its advantages of mild reaction conditions, fast reaction speed, and high yield.46
Herewith, we prepared a new bridged bis(β-cyclodextrin) chiral stationary phase (HTCDP) by thiol–ene click chemistry, applying the HTCDP's synergistic inclusion, hydrogen bonds and π–π interaction to the enantiomeric separation of chiral compounds at the same. The chromatographic performance of HTCDP was comprehensively evaluated by using 21 chiral drugs and pesticides as probes, and the chromatographic conditions affecting the enantioseparation were optimized. The results demonstrate that the enantioselectivity of HTCDP was significantly better than that of CDCSP.
2. Experimental
2.1. Materials
Mono-(6-amino-6-deoxy)-β-cyclodextrin (NH2-β-CD, purity ≥98%) was purchased from Shandong Binzhou Zhiyuan Biotechnology Co (Binzhou, China). 4,4′-Thiobisbenzenethiol (purity ≥98%), 2,2′-azobis(2-methylpropionitrile) (AIBN, purity ≥98%), 3-isocyanatopropyltriethoxysilane (purity ≥98%), formic acid (FA), acetic acid (HOAc), and triethylamine (TEA) were purchased from Shanghai Aladdin Bio-Chem Technology Co (Shanghai, China). Allyl isocyanate (purity ≥98%) was obtained from Alfa Aesar (Tewksbury, MA, USA). Pyridine and acetone were provided by Sinopharm Chemical Reagent Co (Shanghai, China). Methanol (MeOH) and acetonitrile (ACN) (HPLC grade) purchased from Tedia (Shanghai, China). Sephadex C-25 was provided by Rui Da Heng Hui Science Technology Development Co (Beijing, China). Triazole racemic standards (purity ≥98%) were purchased from Shanghai Pesticide Research Institution (Shanghai, China). Flavanones, benzoin, praziquantel, 1,1-bi-2-naphthol, Tröger's base, and bicalutamide racemic standards were obtained from Sigma-Aldrich (St. Louis, MO, USA). Spherical silica gel (5 μm, 25 nm) was provided by Lanzhou Institute of chemical physics (Lanzhou, China). Pyridine was treated with calcium hydride and distilled before use. The water was of ultrapure grade.2.2. Apparatus
All chiral analytes were separated on an LC-20AT high-performance liquid chromatograph (Shimadzu, Kyoto, Japan) equipped with an SPD-20A UV-VIS detector (Shimadzu). The 13C NMR spectra were collected on Bruker Advance III WB 400M (Bruker Daltonics, Bremen, Germany). The 1H NMR spectrum was collected on the AvanceII 400 MHz NMR spectrometer (Bruker, Switzerland). Scanning electron micrograph were obtained using Zeiss Sigma 300 field emission scanning electron microscopy (Zeiss, Germany). Thermogravimetric analysis was performed on an STA 449C synchronous thermogravimetric analyzer (Netzsch, Selb, Germany). Mass spectrometry (MS) analysis was performed on a 6540 TOF high-resolution mass spectrometer (Agilent, Santa Clara, CA, USA). C, H, N, and S contents were determined using a VarioEL III element analyzer (Elementar Analysensysteme GmbH, Langenselbold, Germany). IR analysis was performed on an FTIR-8400S Fourier Transform IR Spectrometer (Shimadzu). The synthesized bridged bis(β-CD) chiral ligand was packed using a CGY-100B chromatographic column packing machine (Beijing Fusiyuan Machinery Processing Department, Beijing, China). Ultrapure water was obtained using the VE-AS water purification system (Shenzhen Vamia Environmental Protection Co, Shenzhen, China).2.3. Preparation of HTCDP and CDCSP
The synthetic route of HTCDP was displayed in Fig. 1. NH2-β-CD (2 g, 1.76 mmol) was dissolved in 40 mL of anhydrous pyridine. The anhydrous pyridine solution of allyl isocyanate (10 mL anhydrous pyridine contained 0.234 g allyl isocyanate) was slowly dripped at ice bath temperature (0–5 °C) and stirred for 30 min, then the mixed solution was reacted at 80 °C for 24 h under the protection of nitrogen. At the end of the reaction, the unreacted anhydrous pyridine and allyl isocyanate were evaporated by vacuum distillation, and a white precipitate was obtained by adding acetone to the residual liquid. The precipitate was purified with a Sephadex C-25 column and dried under vacuum at 50 °C for 24 h to obtain 2.09 g of allyl-ureido-β-CD with the yield of 61%. ESI-MS (m/z), [M + H] + was calculated for 1218.48 found at 1218.41 exhibited in Fig. S1a.†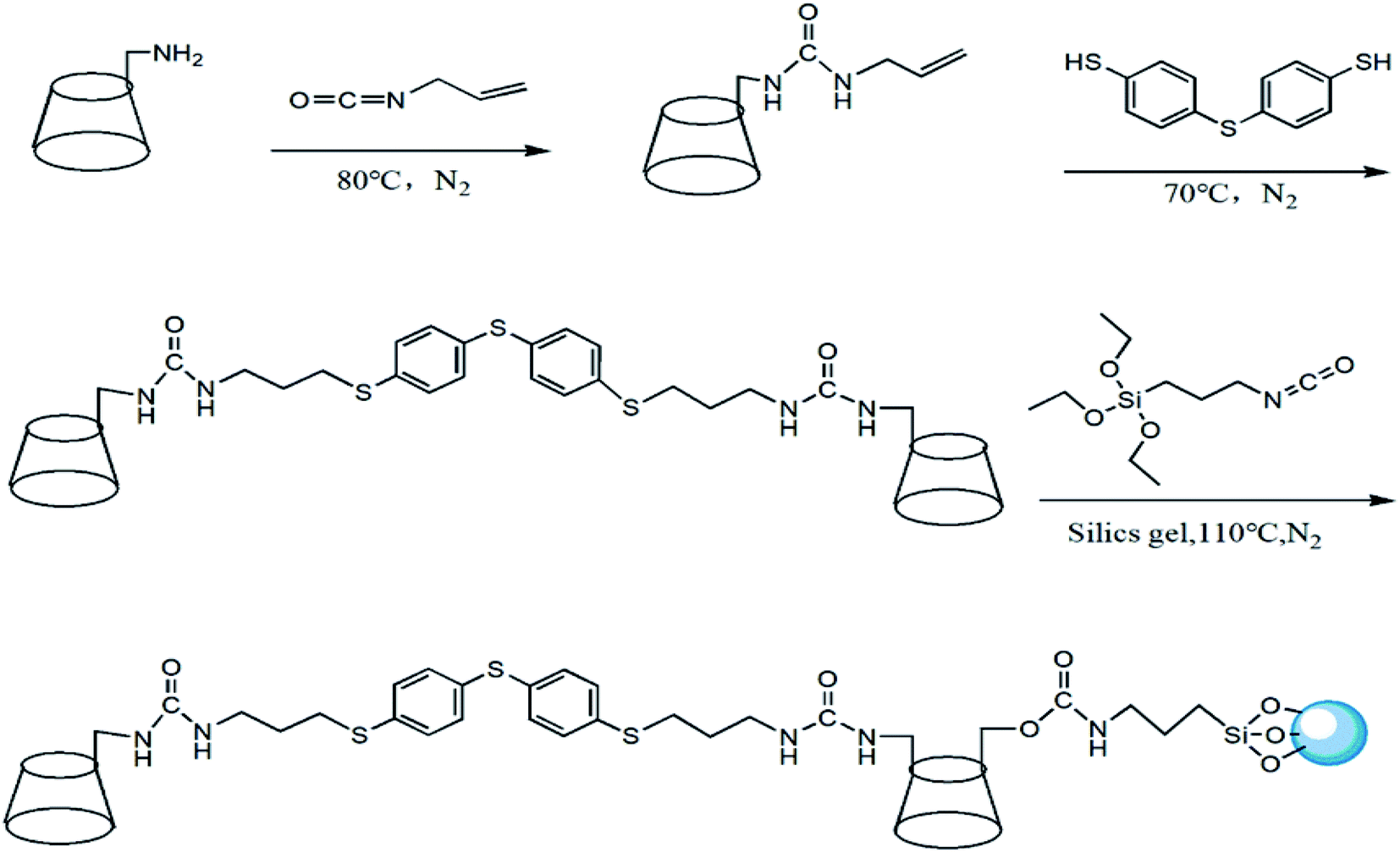 | ||
| Fig. 1 The synthetic route of HTCDP. | ||
Allyl-ureido-β-CD (2 g, 1.64 mmol), 4,4′-thiobisbenzenethiol (0.103 g), and AIBN (30 mg) were dissolved in 40 mL of anhydrous pyridine with stirring for 30 min, and reacted at 70 °C for 24 h under the protection of nitrogen. After the reaction, part of the anhydrous pyridine was evaporated by distillation under reduced pressure and acetone was added to the remaining liquid to obtain a pale yellow solid. The pale yellow solid was purified with a Sephadex C-25 column and dried under vacuum at 50 °C for 24 h to obtain 1.32 g of 4,4′-thiobisthiophenol bridged bis(β-CD) with the yield of about 60%. ESI-MS (m/z), [M + H] + was calculated for 2686.38 found at 2686.35 exhibited in Fig. S1b.† As shown in Fig. S2,† the 1HNMR results (DMSO-d6, 400 MHz, ppm) were found as: δ6.04 (2H, –NH–CO–NH), 5.87–5.50 (28H, OH, 2,3-position of CD), 5.29–4.87 (8H, phenyl), 4.83 (12H, 6-position of CD), 4.69–4.18 (14H, C (1)H of CD), 3.61 (56H, C (3,5,6) H of CD), 3.36–3.14 (28H, C (2,4) H of CD).
4,4′-Thiobisthiophenol bridged bis(β-CD) (1.2 g, 0.45 mmol) was dissolved in 40 mL of anhydrous pyridine, and 0.23 mL of 3-isocyanatopropyltriethoxysilane was added dropwise with stirring for 30 min. The temperature was increased to 80 °C and the solution was reacted for 12 h under nitrogen protection. After cooling to room temperature, silica gel (3 g) was added to the mixture, which was reacted at 115 °C for 24 h under nitrogen protection. After filtration, the solid product was successively washed several times with pyridine, MeOH, and acetone, and dried under vacuum at 50 °C for 24 h to obtain HTCDP.
CDCSP was prepared in a manner similar to HTCDP. β-CD (1 g, 0.88 mmol) was dissolved in 30 mL of anhydrous pyridine; 0.44 mL of 3-isocyanatopropyltriethoxysilane was added dropwise with stirring for 30 min, and the mixture was reacted at 80 °C and for 12 h under nitrogen protection. The temperature was lowered to room temperature and silica gel (3 g) was added to the mixture, which was reacted at 115 °C for 24 h under nitrogen protection to obtain CDCSP.
2.4. Packing of the chromatographic column
HTCDP and CDCSP were packed into stainless steel columns (150 × 4.6 mm inner diameter) using a standard slurry method. A 2.5 g of HTCDP or CDCSP was dispersed in 50 mL of MeOH and dispersed by ultrasound for 5 min, then poured into the homogenizing tank; with MeOH used as the displacement liquid, the tank was filled at 40 MPa pressure for 35 min. Finally, the pressure was gradually released to atmospheric level to complete the packing of the chromatographic column.2.5. Chromatographic methods
The mobile phase consisted of MeOH or ACN and water. According to the actual separation of analytes, a certain amount of FA or TEA and HOAc was added to the water. The 1% of triethylammonium acetate buffer (1% TEAA) was prepared from TEA and water in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, and the pH was adjusted with HOAc. All chiral drugs and pesticides were dissolved in MeOH to prepare standard solutions of 100–1000 μg mL−1. All standard analytes and mobile phases were passed through a 0.22 μm membrane filter and degassed by ultrasound before use. The detection wavelength was set to 214 nm. The flow rate was set to 0.8 mL min−1. The injection volume was 3–10 μL. The column temperature was adjusted according to the separation conditions of analytes. HPLC analysis was performed under isocratic conditions. The storage temperature of the analyzed sample was 4 °C.
:100, and the pH was adjusted with HOAc. All chiral drugs and pesticides were dissolved in MeOH to prepare standard solutions of 100–1000 μg mL−1. All standard analytes and mobile phases were passed through a 0.22 μm membrane filter and degassed by ultrasound before use. The detection wavelength was set to 214 nm. The flow rate was set to 0.8 mL min−1. The injection volume was 3–10 μL. The column temperature was adjusted according to the separation conditions of analytes. HPLC analysis was performed under isocratic conditions. The storage temperature of the analyzed sample was 4 °C.
According to the USP standard, three parameters for evaluating chromatographic performance were calculated. (1) Retention factor (k′) = (tR − t0)/t0, where t0 is the retention time of the first baseline interference of the solvent peak (unit: min), and tR is the retention time of each enantiomer (unit: min); (2) separation factor (α) = k2′/k2′, where k1′ and k2′ are the retention factors of the two enantiomers eluted before and after; (3) resolution (Rs) = 1.18(tR2 − tR1)/(W1 + W2), where tR1 and W1 are the retention time and half-width of the enantiomer that is eluted first, respectively; tR2 and W2 are the retention time and half-width of the enantiomer that is eluted later, respectively.
3. Results and discussion
3.1. Characterization of HTCDP
The prepared HTCDP was characterized by infrared (IR) spectroscopy, elemental analysis, thermogravimetric analysis, 13C NMR and scanning electron microscopy. The IR spectrum of the bridged bis(β-CD) ligand (Fig. S3a†) showed a strong peak at 3413.58 cm−1 corresponding to the stretching vibration peak of the residual hydroxyl (ν–OH) group; the peak at 2928.08 cm−1 was related to the stretching vibration of νC–H; peaks at 1638.35 and 1710.21 cm−1 were attributable to the carbonyl (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching vibration in the bridge arms; peak clusters at 1535.51 and 1411.39 cm−1 reflected tensile vibration of the benzene ring (νCC); and peaks at 1029.37 and 1238.37 cm−1 were related to the νC–O stretching vibration of the bridged bis(β-CD) ligand. These results indicate that the bridged bis(β-CD) ligand was successfully prepared. In the IR spectrum of HTCDP (Fig. S3b†), the strong peak at 3416.38 cm−1 corresponded to the stretching vibration of the residual hydroxyl groups (ν–OH) on the silica gel and bridged bis(β-CD) ligand; the peak at 1618.39 cm−1 was related to the stretching vibration of the carbonyl (νCO) on the bridged bis(β-CD) ligand; and the strong peak at 1114.93 cm−1 was attributable to the stretching vibration of Si–O–Si. This demonstrated that the bridging bis(β-CD) ligand was bonded to the silica gel.
O) stretching vibration in the bridge arms; peak clusters at 1535.51 and 1411.39 cm−1 reflected tensile vibration of the benzene ring (νCC); and peaks at 1029.37 and 1238.37 cm−1 were related to the νC–O stretching vibration of the bridged bis(β-CD) ligand. These results indicate that the bridged bis(β-CD) ligand was successfully prepared. In the IR spectrum of HTCDP (Fig. S3b†), the strong peak at 3416.38 cm−1 corresponded to the stretching vibration of the residual hydroxyl groups (ν–OH) on the silica gel and bridged bis(β-CD) ligand; the peak at 1618.39 cm−1 was related to the stretching vibration of the carbonyl (νCO) on the bridged bis(β-CD) ligand; and the strong peak at 1114.93 cm−1 was attributable to the stretching vibration of Si–O–Si. This demonstrated that the bridging bis(β-CD) ligand was bonded to the silica gel.
The results of the elemental analysis of HTCDP and CDCSP were depicted in Table S1.† HTCDP contained 3.23% C, 0.46% H, 0.24% N, and 0.12% S, whereas CDCSP contained 1.6% C, 0.28% H, and 0.38% N. The loads of HTCDP and CDCSP were calculated based on their carbon contents using the following formula:43 C%/(12 × Nc × S) × 106, where C% is the percentage of carbon in the CSP, 12 is the atomic mass of carbon, Nc (about 123) is the number of carbon atoms in each molecule of the CD unit, and S is the specific surface area of silica gel (60 m2 g−1). As a result, the loads of HTCDP and CDCSP were comparable at 0.45 and 0.51 μmol m−2, respectively.
The thermogravimetric analysis curve of HTCDP was exhibited in Fig. S4.† The weight loss rate of HTCDP was about 9.26% with a heating rate of 10 °C min−1 from 30 °C to 800 °C. HTCDP began to lose weight significantly at about 300 °C, indicating that HTCDP had good chemical and thermal stability.
The synthesized HTCDP was characterized by scanning electron microscope and solid state 13C NMR. The images were shown in Fig. S5 and S6,† respectively.
The above characterization results and excellent enantiomeric separation ability indicated that HTCDP and CDCSP were successfully prepared.
3.2. Performance of HTCDP in the chromatographic separation of enantiomers
In the study, 21 common chiral compounds with different chemical structures and properties were selected as analytes to examine the performance of HTCDP in enantioseparation, of which included 8 flavanones, 8 triazole pesticides and 5 other drugs. Morever, by separating 21 selected analytes under reversed-phase chromatographic mode, some related theories of enantiomeric separation were discussed preliminarily.| No. | Compounds | k1 | k2 | α | Rs | Mobile phases (v/v) | Temp. (°C) | CSPs |
|---|---|---|---|---|---|---|---|---|
| a k′, retention factor; α, separation factor; Rs, resolution; temp., temperature (°C); CSPs, chiral stationary phase; HTCDP, a novel bridged bis(β-cyclodextrin) chiral stationary phase; CDCSP, native β-cyclodextrin chiral stationary phase; —, no separation. | ||||||||
| 1 | Flavanone | 9.17 | 11.16 | 1.22 | 3.06 | ACN/0.1% FA (15/85) | 20 °C | HTCDP |
| 2.81 | — | — | — | ACN/0.1% FA (15/85) | 20 °C | CDCSP | ||
| 2 | 2-Hydroxy flavanone | 2.11 | 2.96 | 1.40 | 3.89 | ACN/0.1% FA (20/80) | 20 °C | HTCDP |
| 2.34 | 2.96 | 1.26 | 1.92 | ACN/0.1% FA (15/85) | 20 °C | CDCSP | ||
| 3 | 4-Hydroxy flavanone | 2.94 | 4.36 | 1.48 | 4.48 | ACN/0.1% FA (20/80) | 20 °C | HTCDP |
| 2.19 | — | — | — | ACN/0.1% FA (15/85) | 20 °C | CDCSP | ||
| 4 | 6-Hydroxy flavanone | 11.76 | 12.78 | 1.09 | 1.26 | MeOH/0.3% FA (20/80) | 30 °C | HTCDP |
| 1.96 | — | — | — | MeOH/0.3% FA (15/85) | 30 °C | CDCSP | ||
| 5 | 6-Methoxy flavanone | 10.77 | 11.79 | 1.09 | 1.19 | MeOH/0.3% FA (25/75) | 30 °C | HTCDP |
| 2.88 | — | — | — | MeOH/0.3% FA (15/85) | 30 °C | CDCSP | ||
| 6 | Naringin | 2.73 | 3.35 | 1.23 | 1.82 | MeOH/0.1% FA (15/85) | 20 °C | HTCDP |
| 2.40 | 2.76 | 1.15 | 1.19 | MeOH/0.1% FA (10/90) | 20 °C | CDCSP | ||
| 7 | Hesperidin | 4.06 | 4.66 | 1.14 | 1.50 | ACN/0.1% FA (20/80) | 20 °C | HTCDP |
| 1.59 | — | — | — | ACN/0.1% FA (15/85) | 20 °C | CDCSP | ||
| 8 | Silymarin | 1.50 | 4.73 | 3.15 | 4.34 | ACN/0.1% FA (15/85) | 20 °C | HTCDP |
| 0.40 | 0.70 | 1.75 | 1.47 | ACN/0.1% FA (15/85) | 20 °C | CDCSP | ||
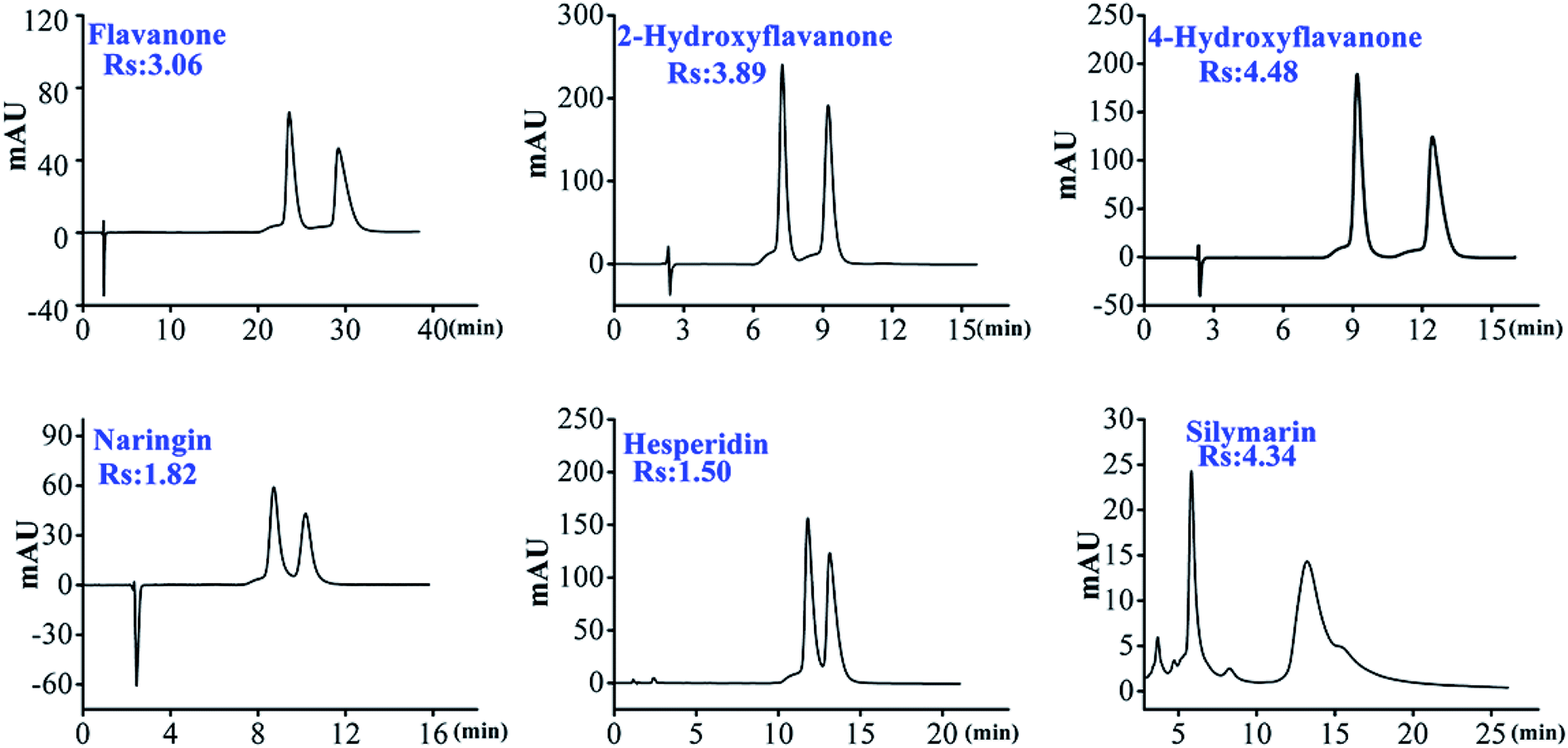 | ||
| Fig. 2 The representative chromatograms of flavanone enantiomers on HTCDP. | ||
As shown in Table 1, we can find that HTCDP separated all 8 flavanones, with 6 of these completely separated at high resolution (1.5–4.48). In contrast, CDCSP separated only 3 flavanones at a lower resolution and did not separate flavanone, 4-hydroxyflavanone, 6-hydroxyflavanone, 6-methoxyflavanone, and hesperidin. Morever, most derivatized mono-CD CSPs separate flavanones at a lower resolution than HTCDP,17,47 suggesting that the two bridging cavities of HTCDP promote the recognition of flavanone enantiomers. We also found that the resolution of flavanone, 4-hydroxyflavanone, 6-hydroxyflavanone, 6-methoxyflavanone, and hesperidin was significantly higher on HTCDP than on CDCSP without π-conjugated aryl-bridged bis(β-CD),42 indicating that the aryl bond arm of the bridged bis(β-CD) ligand was also important for the recognition of chirality.
On the other hand, hydrogen bonds also play a vital role in the separation of enantiomers. Both HTCDP and CDCSP were able to separate 2-hydroxyflavanone, naringenin, and silymarin. As shown in the Fig. S7,† the chiral carbons of the 3 analytes were directly connected to the hydroxyl-containing benzene ring and were potentially responsible for their separation, in line with the previous observation that hydrogen bonding contributes to chiral separation.32 HTCDP separated these 3 analytes at a higher resolution than CDCSP, possibly as a result of the introduction of urea groups into the bridged bonding arms of HTCDP that strengthened the hydrogen bonding. We also found that the resolution of 6-hydroxyflavanone and 6-methoxyflavanone separated by HTCDP was relatively low. This may be because the hydroxyl and methoxy groups at position 6 could not fully enter the hydrophobic cavity of CD, thereby weakening their inclusion.17 However, 6-hydroxyflavanone had a higher resolution than 6-methoxyflavanone, possibly because the former formed hydrogen bonds with the urea group on the spacer arm of HTCDP and thereby enhanced its chiral recognition.17 In summary, the two cavities of HTCDP provided synergistic inclusion while the underivatized hydroxyl group engaged in hydrogen bonding and the aromatic ring on the bridge engaged in π–π interactions; the combination of these interactions enhanced the chiral recognition ability of HTCDP, consistent with previous reports.43,45
3.2.1.1. Effect of mobile phase and column temperature conditions on the separation of flavanones. The composition of the mobile phase is a key factor affecting the chiral separation of β-CD CSPs. In this study, because most of the selected chiral flavanones contained acidic phenolic hydroxyl groups, FA was added to the mobile phase in order to inhibit the ionization of acidic phenolic hydroxyl groups.29 As shown in Table 1, all 8 flavanones could be separated on HTCDP using MeOH–FA or ACN–FA aqueous solution as the mobile phase. However, we found that MeOH–FA and ACN–FA aqueous solution respectively used as mobile phases caused a huge difference in the chiral resolution of HTCDP. For example, as shown in Table S2,† using ACN–formic acid aqueous solution as the mobile phase, the separation factor and resolution of 2-hydroxyflavanone were 1.40 and 3.89, respectively; the separation factor and resolution of 4-hydroxyflavanone were 1.48 and 4.48, respectively. Whereas using MeOH–formic acid aqueous solution as the mobile phase, the separation factor and resolution of 2-hydroxyflavanone were 1.29 and 2.06, respectively; the separation factor and resolution of 4-hydroxyflavanone are 1.31 and 2.68, respectively. This may be because MeOH competes with HTCDP for the hydrogen bonding sites of the analyte, which weakens the retention and separation factors of the analyte on HTCDP, thereby weakening the interaction of the latter.32 Interestingly, the opposite results were obtained in the separation of 6-hydroxyflavanone, naringenin, and 6-methoxyflavanone. Specifically, there was little separation of 6-hydroxyflavanone and 6-methoxyflavanone using ACN–FA aqueous solution as the mobile phase. This may be because MeOH is more polar than ACN and can increase the interaction time between the them and the β-CD hydrophobic cavity, thereby enhancing the inclusion interaction between the them and the cavity.17 Morever, we investigated the influence of MeOH content in the mobile phase on the separation of 6-methoxyflavanone. As shown in Fig. 3a, as the MeOH content increased from 20% to 35%, the resolution of 6-hydroxyflavanone increased before decreasing while the retention time also decreased. The resolution of 6-hydroxyflavanone was maximal (Rs: 1.19) at 25% MeOH. Thus, in order to improve the resolution of enantiomers, the proportioning and type of mobile phase need to be finely optimized.
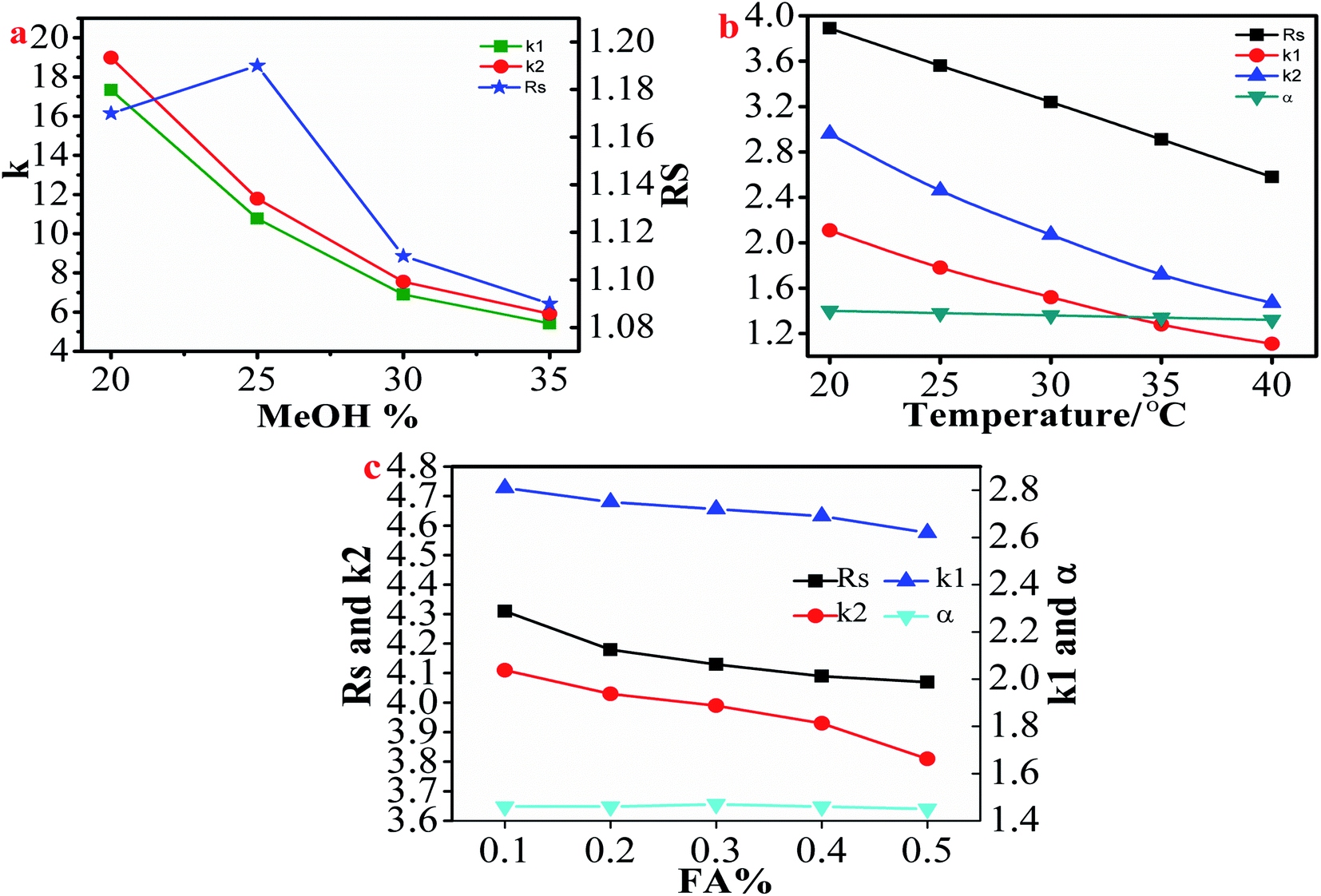 | ||
| Fig. 3 The effect of methanol content in the mobile phase on the resolution, and retention factor of 6-methoxyflavanone enantiomers (a); the effect of column temperature on the resolution, separation factor and retention factor of 2-hydroxyflavanone enantiomers (b) and the effect of formic acid content in the mobile phase on the resolution, separation factor and retention factor of 4-hydroxyflavanone enantiomers (c). | ||
Column temperature also plays an important role in the chromatographic separation of chiral compounds. Some enantiomers showed decreasing resolution with increasing column temperature while others showed the opposite trend.43,48 We examined the effect of column temperature on the resolution of 8 selected flavanones. Using 2-hydroxyflavanone as an example, as shown in Fig. 3b and Table S3,† the retention time (k′), separation factor (α), and resolution (Rs) of 2-hydroxyflavanone decreased with increasing column temperature. In order to improve the resolution of flavanones, we selected the lowest possible column temperature. However, the retention times of 6-hydroxyflavanone and 6-methoxyflavanone were longer at 20 °C. Given that raising the temperature can reduce retention time, they were separated at 30 °C.
3.2.1.2. Effect of FA content on the separation of flavanones. As we described earlier, due to most flavanones contain acidic phenolic hydroxyl groups that ionize when resolved by HTCDP. In order to prevent this process and maintain ionization equilibrium, FA was added to the mobile phase.32 We explored the effect of the ratio of FA to water on the resolution of flavanone compounds using 4-hydroxyflavanone as an example. As can be seen from Fig. 3c, the FA content had no effect on the separation factor of 4-hydroxyflavanone; however, as it increased from 0.1% to 0.5%, the resolution and retention factor of 4-hydroxyflavanone continued to decrease. Therefore, in order to achieve rapid separation and prevent the ionization of phenolic hydroxyl groups, an appropriate amount of FA should be added to the mobile phase. In this study, most selected flavanones were sufficient to achieve rapid and baseline separation with 0.1% FA content. However, given the long retention time of 6-hydroxyflavanone and 6-methoxyflavanone in HTCDP, we increased the FA concentration in the mobile phase from 0.1% to 0.3%, which enhanced the resolution of these 2 analytes.
| No. | Compounds | k1 | k2 | α | Rs | Mobile phases (v/v) | Temp. (°C) | CSPs |
|---|---|---|---|---|---|---|---|---|
| a k′, retention factor; α, separation factor; Rs, resolution; temp., temperature (°C); CSPs, chiral stationary phase; HTCDP, a novel bridged bis(β-cyclodextrin) chiral stationary phase; CDCSP, native β-cyclodextrin chiral stationary phase; —, no separation. | ||||||||
| 9 | Hexaconazole | 4.48 | 5.42 | 1.21 | 2.19 | ACN/H2O (20/80) | 20 °C | HTCDP |
| 3.02 | 3.50 | 1.16 | 1.32 | ACN/H2O (15/85) | 20 °C | CDCSP | ||
| 10 | Imazalil | 5.49 | 6.76 | 1.23 | 2.14 | ACN/1% TEAA (10/90) | 20 °C | HTCDP |
| 2.48 | — | — | — | ACN/1% TEAA (10/90) | 20 °C | CDCSP | ||
| 11 | Tebuconazole | 7.46 | 8.76 | 1.17 | 1.91 | ACN/H2O (20/80) | 20 °C | HTCDP |
| 9.30 | — | — | — | ACN/H2O (10/90) | 20 °C | CDCSP | ||
| 12 | Flutriafol | 4.80 | 5.57 | 1.16 | 1.84 | ACN/H2O (12/88) | 20 °C | HTCDP |
| 3.51 | 3.85 | 1.10 | 0.94 | ACN/H2O (12/88) | 20 °C | CDCSP | ||
| 13 | Paclobutrazol | 7.76 | 9.64 | 1.26 | 1.79 | ACN/H2O (28/72) | 20 °C | HTCDP |
| 5.33 | 5.75 | 1.08 | 0.71 | ACN/H2O (10/90) | 20 °C | CDCSP | ||
| 14 | Diniconazole | 6.02 | 7.04 | 1.17 | 1.71 | ACN/H2O (20/80) | 20 °C | HTCDP |
| 3.99 | 4.34 | 1.09 | 0.72 | ACN/H2O (10/90) | 20 °C | CDCSP | ||
| 15 | Uniconazole | 15.97 | 19.40 | 1.22 | 1.54 | ACN/H2O (25/75) | 20 °C | HTCDP |
| 3.93 | — | — | — | ACN/H2O (15/85) | 20 °C | CDCSP | ||
| 16 | Triticonazole | 8.98 | 10.60 | 1.18 | 1.96 | ACN/H2O (20/80) | 20 °C | HTCDP |
| 3.61 | 3.87 | 1.07 | 0.62 | ACN/H2O (15/85) | 20 °C | CDCSP | ||
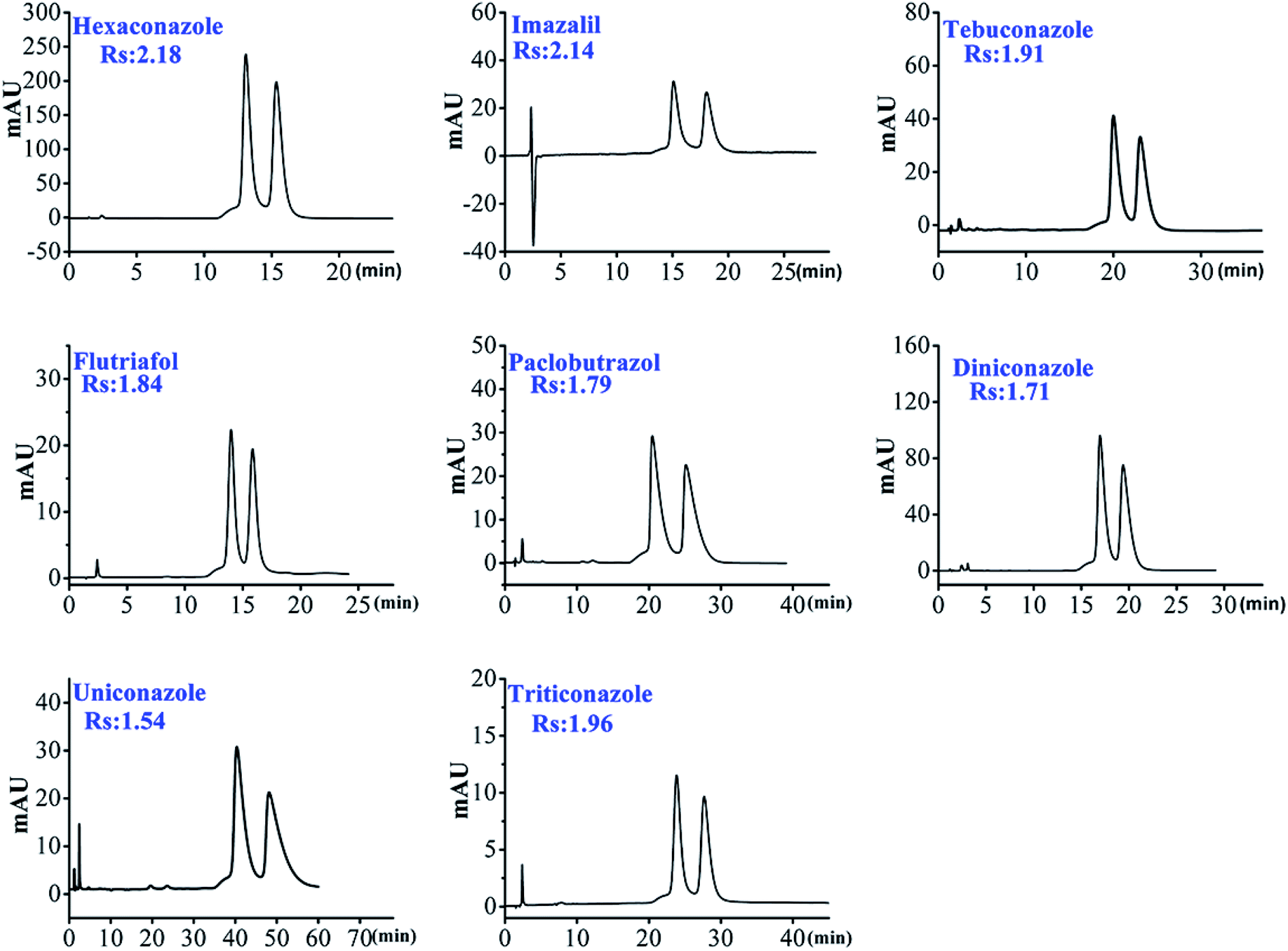 | ||
| Fig. 4 The chromatogram of 8 triazole pesticide enantiomers on HTCDP. | ||
As shown in Table 2, most of the selected chiral triazole analytes could be separated by HTCDP and CDCSP under simple ACN–H2O mobile phase conditions, demonstrating that the inclusion effects of HTCDP and CDCSP were the main driving force for their separation.49 However, the resolution of the 8 selected chiral triazole analytes on HTCDP and CDCSP differed markedly: HTCDP completely separated all 8 analytes at high resolution (Rs: 1.54–2.19) whereas CDCSP separated only 5 at a lower resolution (Rs: 0.62–1.32), failing to separate uniconazole, tebuconazole, and imazalil. As HTCDP and CDCSP are both underivatized β-CD CSPs, their different abilities to resolve the 8 analytes can be attributed to their distinct modes of chiral recognition. For example, the resolution of imazalil on HTCDP was as high as 2.14 whereas CDCSP did not separate this compound, possibly because of the lack of hydroxyl groups that could form hydrogen bonds on the chiral carbon of imazalil and the small size of the CDCSP cavity (0.65 nm). However, the two cavities in HTCDP linked via an aryl connector synergistically were able to accommodate the bulky imazalil molecule. Moreover, there was a π–π interaction between the aromatic ring of imazalil and the aryl connector of HTCDP. The combination of these two interactions enhanced the separation of imazalil by HTCDP, consistent with the previously reported mechanism.43,45
In addition to the inclusion effect and π–π interaction, hydrogen bonding plays an extremely important role in chiral separation. In addition to the 8 triazole pesticides, we also tried to separate myclobutanil, ornidazole, voriconazole, and lansoprazole (Fig. S9†) on HTCDP. All 4 compounds had a very short retention time and were not separated (Table S4†), unlike the 8 triazole pesticides (Table 1); this is likely because the chiral carbons of the latter compounds have hydroxyl groups that could form hydrogen bonds with HTCDP, thereby promoting their separation. This is consistent with the results previously reported.43,44
3.2.2.1. Effect of column temperature on separation. In order to improve the resolution of the selected triazole analytes, we next studied the effect of column temperature (ranging from 20 °C to 45 °C in increments of 5 °C) on the resolution of all 8 triazole chiral analytes. In the paper, we chose hexaconazole as the example for description. As shown in Fig. 5a and Table S5,† the retention factor, separation factor and resolution of hexaconazole decreased with the increase of column temperature. However, column temperature from 20 °C to 25 °C, the resolution, separation factor and retention factor of hexaconazole decreased rapidly. Therefore, in order to achieve a higher resolution, the separation of the selected triazole chiral analytes should be carried out at ≤20 °C.
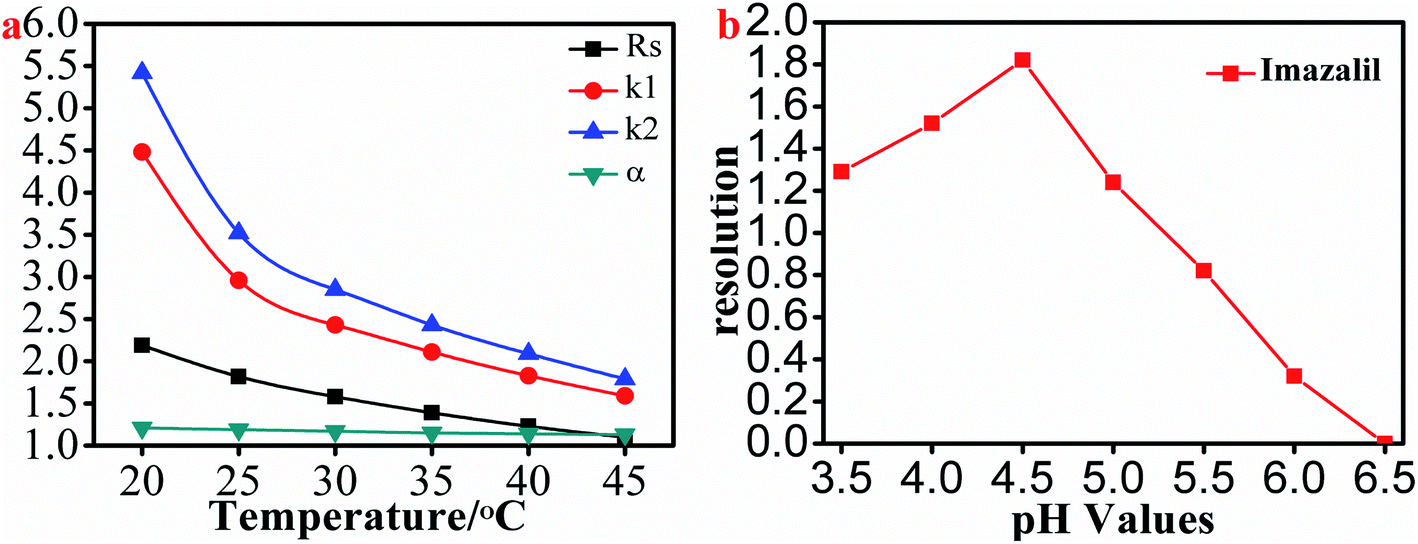 | ||
| Fig. 5 The effect of column temperature on the resolution, separation factor and retention factor of hexaconazole enantiomers (a) and the effect of pH values in mobile phase on the resolution of imazalil enantiomers (b). | ||
3.2.2.2. Effect of pH on the imazalil enantiomer resolution. We found that the above-mentioned other chiral triazole analytes could be separated with simple ACN–H2O as the mobile phase with the exception of imazalil. In order to achieve the separation of imazalil enantiomers, we used ACN–1% TEAA as the mobile phase to separate imazalil. Interestingly, imazalil was separated to a certain extent by adjusting the pH of the 1% TEAA solution. We evaluated the effect of pH on the resolution of imazalil and found that as the pH value decreased, the resolution first increased before decreasing (Fig. 5 b); the resolution was maximal (1.82) at pH 4.5 and was 1.52 at pH 4. This should be related to the change in charge and polarity of the imazalil at different pH values, which significantly affected the interaction and separation of the imazalil and HTCDP.38 Thus, the optimal pH to achieve baseline separation of imazalil is between 4 and 4.5. We also determined that imazalil was separated at the highest resolution (2.14) using HTCDP when the proportion of ACN was 10%.
| No. | Compounds | k1 | k2 | α | Rs | Mobile phases (v/v) | Temp. (°C) | CSPs |
|---|---|---|---|---|---|---|---|---|
| a k′, retention factor; α, separation factor; Rs, resolution; temp., temperature (°C); CSPs, chiral stationary phase; HTCDP, a novel bridged bis(β-cyclodextrin) chiral stationary phase; CDCSP, native β-cyclodextrin chiral stationary phase; —, no separation. | ||||||||
| 17 | Troger's base | 7.42 | 8.71 | 1.17 | 1.75 | ACN/H2O (17/83) | 20 °C | HTCDP |
| 3.67 | — | — | — | ACN/H2O (15/85) | 20 °C | CDCSP | ||
| 18 | 1-1′-Bi-2-naphthol | 7.24 | 9.01 | 1.24 | 1.72 | MeOH/1%TEAA (15/85) | 20 °C | HTCDP |
| 1.85 | — | — | — | MeOH/1%TEAA (15/85) | 20 °C | CDCSP | ||
| 19 | Bicalutamide | 4.71 | 5.45 | 1.16 | 1.62 | MeOH/H2O (15/85) | 20 °C | HTCDP |
| 2.07 | — | — | — | MeOH/H2O (15/85) | 20 °C | CDCSP | ||
| 20 | Praziquantel | 9.18 | 10.80 | 1.18 | 1.54 | ACN/H2O (15/85) | 20 °C | HTCDP |
| 4.10 | — | — | — | ACN/H2O (12/88) | 20 °C | CDCSP | ||
| 21 | Benzoin | 12.54 | 14.10 | 1.12 | 1.51 | ACN/1%TEAA (5/95) | 20 °C | HTCDP |
| 5.53 | — | — | — | ACN/1%TEAA (10/90) | 20 °C | CDCSP | ||
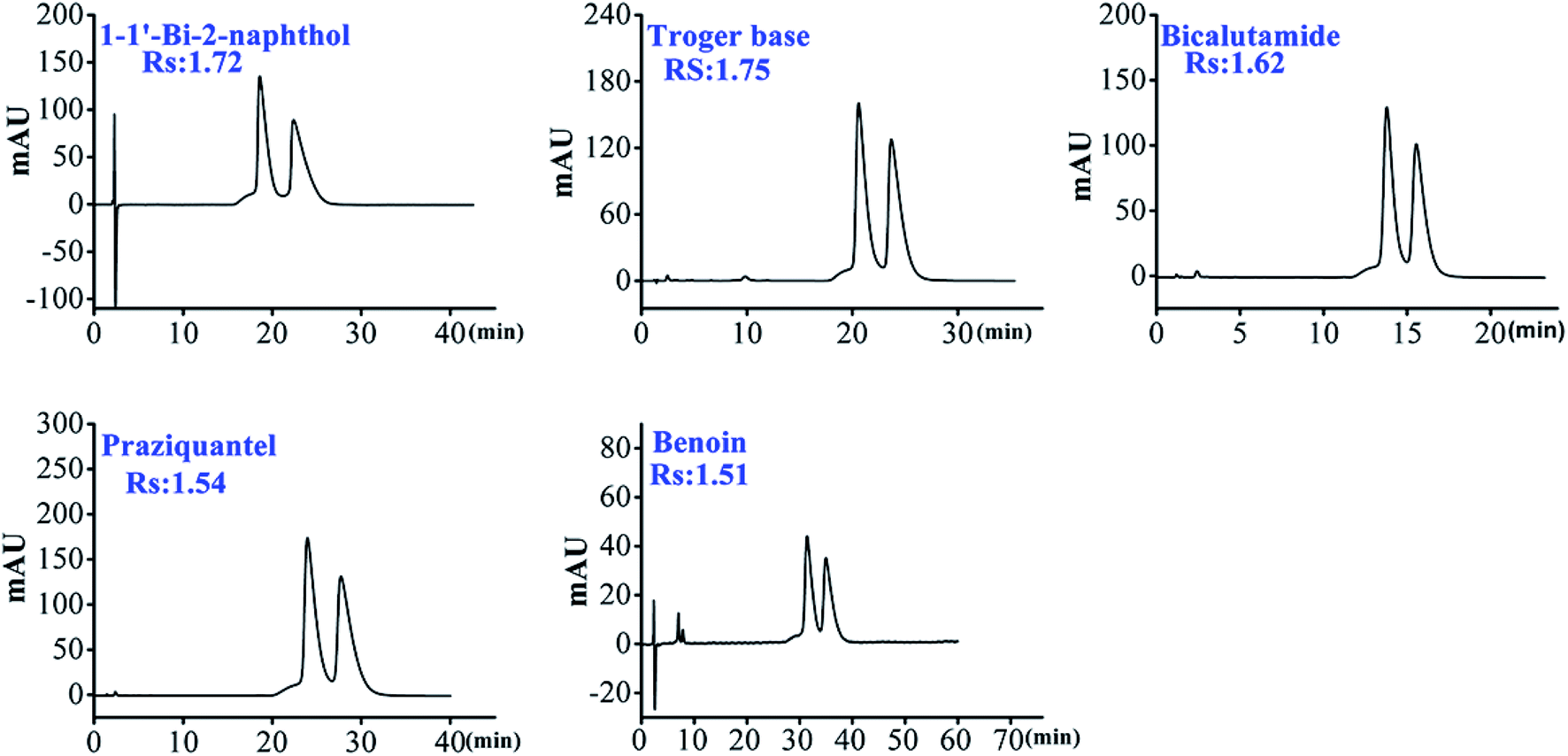 | ||
| Fig. 6 The chromatograms of 5 common enantiomers on HTCDP. | ||
As shown in Table 3, HTCDP completely separated all 5 analytes while none was separated by CDCSP. It is worth noting that the resolution of benzoin on HTCDP was higher than that on all reported bridged β-CD CSPs.39,40 The above results indicate that HTCDP exhibited high enantiomeric separation ability to the selected 5 analytes. As can be seen from Fig. S10,† the 5 selected analytes were relatively large molecules and most were composed solely of branched chains and benzene rings. As CDCSP is an underivatized β-CD CSP with a relatively small cavity (0.65 nm), there was no inclusion effect on the 5 analytes, which ultimately led to the failure of separation. However, all 5 compounds were separated by HTCDP at higher resolution; this may be because the analytes were included in the two cavities of HTCDP and engaged in π–π interaction with the benzene ring on HTCDP bridge arms.43,45
4. Conclusions
In this paper, a novel HTCDP was prepared through an efficient, simple and rapid thiol–ene click chemistry reaction. The chiral chromatographic performance of HTCDP was systematically studied and evaluated by separating 21 chiral triazole pesticides and drugs in reverse phase chromatographic mode. Morever, we also studied in detail the effects of formic acid content, column temperature, pH values, and mobile phase composition on enantiomeric resolution. The results showed that in addition to 6-hydroxyflavanone and 6-methoxyflavanone, HTCDP completely separated the other 19 selected analytes with relatively high resolution (1.50–4.48). By constructing a novel bridged bis(β-CD) ligand with a bridging linker (with a urea group and an aromatic ring) and two cavities, many interactions such as π–π, hydrogen bonds, and synergistic inclusion were introduced into the enantiomeric separation, thereby improving the enantiomeric separation and selection capabilities of HTCDP. Thus, HTCDP can potentially be used for quality and safety analyses of chiral drugs in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Key Research and Development Program of Ningxia (Special Project for Foreign Cooperation) (No. 2019BFH03004), Introduced Team Program of Ningxia (Rouxing) (No. 2019RXTD0004) and Graduate Innovation Project of North Minzu University (No. YCX20141).References
- Y. P. Xue, C. H. Cao and Y. G. Zheng, Chem. Soc. Rev., 2018, 47, 1142 RSC
.
- S. R. Crowell, W. M. Henderson, J. F. Kenneke and J. W. Fisher, Toxicol. Lett., 2011, 205, 154 CrossRef CAS PubMed
- L. Z. Chen, D. Q. Zhu and P. Xiang, Bioanalysis, 2021, 13, 493 CrossRef CAS PubMed
- B. Zhang, S. H. Yang, J. P. Deng and K. Pan, Adv. Sci., 2021, 8, 2003681 CrossRef PubMed
- A. Cavazzini, L. Pasti, A. Massi, N. Marchetti and F. Dondi, Anal. Chim. Acta, 2011, 706, 205 CrossRef CAS PubMed
- O. Horacek, M. Papajova-Janetkova, B. Gruner, L. Lochman, P. SterbovaKovarikova, R. Vespalec and R. Kucera, Talanta, 2021, 222, 121652 CrossRef CAS PubMed
- B. Tang, X. Zhang, L. N. Geng, L. Q. Sun and A. Q. Luo, J. Chromatogr. A, 2021, 1636, 461792 CrossRef CAS PubMed
- Z. T. Li, C. J. Hu, Y. K. Liu, Q. Y. Li, Y. Y. Fu and Z. L. Chen, J. Chromatogr. A, 2021, 1643, 462082 CrossRef CAS PubMed
- M. Lämmerhofer, J. Chromatogr. A, 2010, 1217, 814 CrossRef PubMed
- J. Voskuhl, K. Schaepe and B. J. Ravoo, Int. J. Mol. Sci., 2021, 12, 4637 CrossRef PubMed
- Y. F. Poon, I. W. Muderawan and I. W. Ng, J. Chromatogr. A, 2006, 1101, 185 CrossRef CAS PubMed
- X. B. Yao, T. T. Y. Tan and Y. Wang, J. Chromatogr. A, 2014, 1316, 80 CrossRef PubMed
- J. Zhou, B. Yang, J. Tang and W. H. Tang, J. Chromatogr. A, 2016, 1467, 169 CrossRef CAS PubMed
- R. Q. Wang, T. T. Ong and S. C. Ng, Tetrahedron Lett., 2012, 53, 2312 CrossRef CAS
- Z. A. AlOthman, A. G. Alanazi, M. Suhail and I. Ali, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1157, 122335 CrossRef CAS PubMed
- S. Ravichandran, J. R. Collins, N. Singh and I. W. Wainer, J. Chromatogr. A, 2012, 1269, 218 CrossRef CAS PubMed
- L. Li, H. Wang, Y. Z. Shuang and L. S. Li, Talanta, 2019, 202, 494 CrossRef CAS PubMed
- A. Rosetti, C. Villani, M. Pierini and R. Cirilli, Molecules, 2021, 26, 1747 CrossRef CAS PubMed
- S. H. Huang, Z. W. Bai, Q. Chuan, S. R. Li and Z. Q. Pan, Chirality, 2007, 19, 129 CrossRef CAS PubMed
- C. J. Easton and S. F. Lincoln, Chem. Soc. Rev., 1996, 25, 163 RSC
- X. X. Li, X. Jin, X. B. Yao, X. F. Ma and Y. Wang, J. Chromatogr. A, 2016, 1467, 279 CrossRef CAS PubMed
- L. Li, B. P. Chen, R. D. Zhou, Z. G. Cao, C. Zeng and L. S. Li, Talanta, 2017, 174, 179 CrossRef CAS PubMed
- J. Y. Sun, S. M. Ma, B. B. Liu, J. Yu and X. J. Guo, Talanta, 2019, 204, 817 CrossRef CAS PubMed
- A. M. Stalcup, S. C. Chang and D. W. Armstrong, J. Chromatogr. A, 1991, 540, 113 CrossRef CAS
- G. K. E. Scriba, J. Chromatogr. A, 2016, 1467, 56 CrossRef CAS PubMed
- Y. Z. Shuang, Z. G. Cao, T. C. Zhang and L. S. Li, Anal. Lett., 2020, 53(15), 2481 CrossRef CAS
- S. C. Chang, G. L. R. Iii, S. Chen, C. D. Chang and D. W. Armstrong, Anal. Chem., 1993, 12, 144 CAS
- P. Sun, C. L. Wang, Z. S. Breitbach, Y. Zhang and D. W. Armstrong, Anal. Chem., 2009, 81, 10215 CrossRef CAS PubMed
- Q. Q. Zhong, L. F. He, T. E. Beesley, W. S. Trahanovsky, P. Sun, C. L. Wang and D. W. Armstrong, J. Chromatogr. A, 2006, 1115, 19 CrossRef CAS PubMed
- A. M. Stalcup, S. C. Chang and D. W. Armstrong, J. Chromatogr. A, 1991, 540, 113 CrossRef CAS
- X. Y. Yu and Z. P. Yao, Anal. Chim. Acta, 2017, 968, 1 CrossRef CAS PubMed
- K. E. G. Scriba, J. Chromatogr. A, 2016, 1467, 56 CrossRef PubMed
- L. F. Zhang, J. Zhou and Y. Wang, Acta Chim. Sin., 2015, 73, 1182 CrossRef CAS
- M. Nakamura, T. Ikeda, A. Nakamura, H. Ikeda, A. Ueno and F. Toda, Chem. Lett., 1995, 5, 343 CrossRef
- A. Ruebner, J. G. Moser, D. Kirsch, B. Spengler, S. Andress and S. Roehrs, J. Inclusion Phenom. Macrocyclic Chem., 1996, 25, 35 CrossRef CAS
- D. Q. Yuan, J. Lu, M. Atsumi, J. M. Yan, M. Kai and K. Fujita, Org. Biomol. Chem., 2007, 5, 2932 RSC
- B. Zhang and R. Breslow, J. Am. Chem. Soc., 1997, 119, 1676 CrossRef CAS
- R. Breslow, N. Greenspoon, T. Guo and R. Zarzycki, J. Am. Chem. Soc., 1989, 111, 8296 CrossRef CAS
- J. Zhao, X. H. Lu, Y. Wang and J. Lv, J. Chromatogr. A, 2015, 1381, 253 CrossRef CAS PubMed
- J. Zhao, X. H. Lu, Y. Wang and T. T. Y. Tan, J. Chromatogr. A, 2014, 1343, 101 CrossRef CAS PubMed
- P. Ai, L. N. Han, M. Zi, L. Meng, F. T. Zi and L. M. Yuan, Chin. J. Anal. Chem., 2006, 34, 1459 CAS
- Y. Z. Shuang, Y. Q. Liao, T. C. Zhang and L. S. Li, J. Chromatogr. A, 2020, 1619, 460937 CrossRef CAS PubMed
- Y. Z. Shuang, T. C. Zhang and L. S. Li, J. Chromatogr. A, 2020, 1614, 460702 CrossRef CAS PubMed
- Y. Z. Shuang, Y. Q. Liao, H. Wang, Y. X. Wang and L. S. Li, Chirality, 2020, 32, 168 CrossRef CAS PubMed
- Y. Liu and Y. Chen, Acc. Chem. Res., 2006, 39, 681 CrossRef CAS PubMed
- M. Chen, X. L. Lu, X. F. Ma, Y. Xiao and Y. Wang, Analyst, 2021, 146, 3025 RSC
- T. C. Zhang, Y. Z. Shuang, H. Zhong, L. Li and L. S. Li, Anal. Sci., 2021, 37, 1095 CrossRef CAS PubMed
- E. Küsters, V. Loux, E. Schmid and P. Floersheim, J. Chromatogr. A, 1994, 666, 421 CrossRef
- P. Jandera, Anal. Chim. Acta, 2011, 692, 1 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04697g |
| This journal is © The Royal Society of Chemistry 2021 |