DOI:
10.1039/D1RA04694B
(Paper)
RSC Adv., 2021,
11, 30132-30147
The SARS-CoV-2 B.1.618 variant slightly alters the spike RBD–ACE2 binding affinity and is an antibody escaping variant: a computational structural perspective
Received
17th June 2021
, Accepted 26th July 2021
First published on 9th September 2021
Abstract
Continuing reports of new SARS-CoV-2 variants have caused worldwide concern and created a challenging situation for clinicians. The recently reported variant B.1.618, which possesses the E484K mutation specific to the receptor-binding domain (RBD), as well as two deletions of Tyr145 and His146 at the N-terminal binding domain (NTD) of the spike protein, must be studied in depth to devise new therapeutic options. Structural variants reported in the RBD and NTD may play essential roles in the increased pathogenicity of this SARS-CoV-2 new variant. We explored the binding differences and structural-dynamic features of the B.1.618 variant using structural and biomolecular simulation approaches. Our results revealed that the E484K mutation in the RBD slightly altered the binding affinity through affecting the hydrogen bonding network. We also observed that the flexibility of three important loops in the RBD required for binding was increased, which may improve the conformational optimization and consequently binding of the new variant. Furthermore, we found that deletions of Tyr145 and His146 at the NTD reduced the binding affinity of the monoclonal antibody (mAb) 4A8, and that the hydrogen bonding network was significantly affected consequently. This data show that the new B.1.618 variant is an antibody-escaping variant with slightly altered ACE2–RBD affinity. Moreover, we provide insights into the binding and structural-dynamics changes resulting from novel mutations in the RBD and NTD. Our results suggest the need for further in vitro and in vivo studies that will facilitate the development of possible therapies for new variants such as B.1.618.
1. Introduction
Coronaviruses, which have affected the world with their continuous emergence at various intervals and their staggering distribution levels, are positioned within four genera, α, β, γ, and δ, of the subfamily Orthocoronavirinae in the family Coronaviridae.1,2 The epidemics of SARS, MERS, and SARS-CoV-2 in 2003, 2012, and 2019, respectively, which were caused by β coronaviruses, were responsible for serious negative effects on human health, society, and economics worldwide.3,4 These epidemics were associated with the ability of β coronaviruses to spread among humans.5–7 The recent pandemic caused by SARS-CoV-2 in late 2019 has continued (i.e., in the “third wave”) with the evolution of more contagious and virulent variants such as B.1.1.7 (United Kingdom), B.1.135, and P.1 (reported in South Africa and Brazil). These new variants have contributed to higher infectivity and contiguity, which has resulted in increased hospitalization.8 To date, the number of people affected globally is 171.90 million, with 3.57 million deaths recorded. The case fatality ratio (CFR) for SARS-CoV-2 is 3%, which is less than the 10% and 35% CFR induced by SARS and MERS, respectively.5,9 Nevertheless, the rapid expansion of SARS-CoV-2 and the emergence of new variants have presented increased risks to public health. Consequently, researchers have used various methods to reduce risks, including determining the evolutionary pattern between coronaviruses, developing novel vaccines, identifying the mutational landscape, drug repositioning, and developing novel drugs.10–17
Using multi-omics data to facilitate the control of COVID-19 primarily relies on understanding the SARS-CoV-2 proteome, which comprises sixteen nonstructural proteins (NSP1–NSP16) and four structural proteins S, E, N, and M.18 These structural proteins have an array of functions, including attachment to the host receptor, transcription, and replication.19 When attempting to control the spread of SARS-CoV-2, the spike protein and proteases are the main targets for the development of antiviral drugs and vaccines. These therapies either activate the innate immune response to cope with the pathogens or block the viral protein from binding with receptor proteins.20
The SARS-CoV-2 spike protein interacts with the host angiotensin-converting enzyme (ACE2), establishing a connection that helps the attachment to and invasion of the virus into the host cell.19 The transmission of SARS-CoV-2 is facilitated by two subunits (S1 and S2),21 which activate an immune response that triggers the host immune system upon infection and provides a primary route for the innate response.22 Thus, inhibition of the ACE2–receptor-binding domain (RBD) complex is crucial for controlling the infection caused by SARS-CoV-2.23 On the one hand neutralizing monoclonal antibodies (mAbs) that target the RBD site of the spike protein is gaining more attention; however it may suffer from resistance caused by the virus when used alone. Alternatively, antibodies that target the non-RBD site and that can be used as a cocktail may work more efficiently to neutralize the virus. A recent study also reported that a neutralizing antibody, 4A8, targets the NTD of the spike protein extracted from the convalescent plasma of COVID-19 patients. Among the 10 naturally produced antibodies, 4A8 has been reported to bind strongly to the NTD and effectively neutralize the viral infection.24 Therefore, the spike protein is an important and promising target for antiviral studies.25
Throughout the SARS-CoV-2 pandemic, which is currently in its third wave, numerous variants exhibiting various mutations have been reported. Several variants are reported to posses multiple mutations on the spike protein RBD domain. However, three main SARS-CoV-2 variants, namely B.1.1.7, B.1.351, and P.1, are recognized as variants of concern because of their increased transmissibility, severity of infection, and potential evasion of the host immune response.26
Recently, a new variant, B.1.618, has been reported in different countries around the world, including India, the UK, Pakistan, and Ireland; it has one mutation in the RBD (E484K) and two deletions (Tyr145 and His146) at the antibody binding site, i.e., the N-terminal binding domain (NTD) of the spike protein. Studies on the B.1.618 variant are needed to devise suitable therapeutic strategies. A detailed molecular-level investigation will be indispensable for revealing the role of the E484K substitution and Tyr145 and His146 deletions, and is required to decipher the structural and functional changes they produce. Consequently, in the present study, we employed consensus protein–protein interaction and biomolecular simulation methods to investigate the structural determinants that alter the binding affinity of ACE2–RBD and evaluate how the binding of mAb is impacted by the deletions. These analyses will help researchers to understand the structural and binding changes in the RBD and NTD domains and to assess the future repercussions of these changes.
2. Material and methods
2.1 Structure retrieval and mutant modelling
The primary amino acid sequences of the RBD and NTD of the SARS-CoV-2 spike protein were retrieved from UniProt using accession number P0DTC2. The RBD domain corresponds to positions 319–541, whereas the NTD corresponds to positions 13–303. In the RBD sequence, lysine was introduced at position 484 by replacing glutamic acid. However, tyrosine at position 145 and histidine at position 146 were deleted from the sequence, which was subjected to structural modelling. Chimera-embedded Modeller version 14.0 was used to model the 3D structures of RBD. Loop modelling of NTD missing from the crystal structure was also undertaken which is important for the interaction with mAb, and the modelling of residues 51–55 was performed. The modelled structures were subjected to structural refinement and validation.
2.2 Mutation-induced stability and function correlation
Mutation stability and function correlation were detected through computational algorithms deployed online. For this purpose, various online web servers, such as BeAtMuSiC,27 mCSM-PPI2,28 and DynaMut2,29 were used. To compute the stability changes associated with E484K replacement, we used BeAtMuSiC, which defines the backbone torsion angles, pairwise distances between residues, and solvent accessibilities, to estimate the mutational effect on stability and interface by applying statistical potentials. This method determines the folding and binding free energy changes of the two interacting protein partners as follows:
| ΔGbind = Gcomplex − GpartnerA − GpartnerB. |
Upon substitution, the binding free energy changes are calculated using the following equation:
| ΔΔGbind = Gmutantbind − Gwildtypebind. |
The equilibrated structure from the MD trajectory was utilized to estimate the binding energy differences between the wild-type and mutant complexes.
For interface analysis, mCSM-PPI2, which exercises graph kernels by combining evolutionary, network, and energetic terms to estimate the impact of a mutation on the inter-residue noncovalent interaction network, was used. Moreover, DynaMut2, which uses contingent graph-based signatures and normal mode analysis to determine the protein stability variation and dynamics upon mutation from Gibbs free energy of folding, was applied.
2.3 Consensus docking of wild-type and mutant spike RBD and ACE2
The MD relaxed sampled structure of E484K was subjected to interaction modelling via the HADDOCK (high ambiguity-driven protein–protein docking) algorithm30 to explore the binding differences between the wild-type and E484K variant. Restraint docking was performed by determining the interface residues at 21, 24, 27, 28, 30, 35, 38, 79, 80, 82, 83, and 353 for ACE2, and at 449, 453, 455, 456, 486, 487, 489, 493, 496, 498, 500, 501, 502, and 505 for RBD. The Guru interface was used to virtualize the docking interface, which uses all the available structural features for protein–protein or protein–DNA/RNA docking and is considered the best interface operated by the HADDOCK server. PDBsum was used for comprehension of hydrogen bonds, electrostatic, and salt bridge interaction patterns. For cross-validation, a hybrid algorithm that combined both template-based and free approaches to estimate the docking conformation and affinity was deployed as HDOCK.
2.4 Consensus docking of mAbs to wild-type and mutant NTD
Similarly, for mAbs docking against the wild-type and truncated (B.1.618) NTDs, the Guru interface of the HADDOCK webserver was utilized.30 Restraint docking was performed by determining the interface residues at 25–32, 51–58, and 100–116 for mAbs, and at 145–150 for NTD. PDBsum was used for comprehension of hydrogen bonds, electrostatic, and salt bridge interaction patterns, while HDOCK was used for cross-validation, as described in Section 2.3.
2.5 Dissociation constant (KD) determination
Protein networks and interactions are important for regulating cellular processes and functions. Determining the strength of protein associations helps researchers understand their biological significance and role in various diseases. We used an online webserver, PRODIGY (PROtein binDIng enerGY prediction), to provide insights into the binding strengths of wild-type-RBD–ACE2, E484K-RBD–ACE2, wild-type-NTD–mAbs, and truncated-(B.1.618)-NTD–mAbs complexes by employing an experimental and contact-based prediction model. The predicted KD values for wild-type-RBD–ACE2, E484K-RBD–ACE2, wild-type-NTD–mAbs, and truncated-(B.1.618)-NTD–mAbs provide further insights into these interactions.
2.6 Conformational dynamics of wild-type and mutant complexes
Solvated complexes of wild-type-RBD–ACE2, E484-RBD–ACE2, wild-type-NTD–mAbs, and truncated-(B.1.618)-NTD–mAbs neutralized by counter ions were subjected to all atomic molecular dynamics simulations using force field ff18 in the AMBER20 simulation program.31,32 Two steps, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 6000 conjugate gradient energy minimization cycles, were completed to relax the complexes and remove any bad clashes. With default heat parameters of 300 K for 200 ps, each complex was heated. For density equilibration, weak restraint was used for 2 ns at a constant pressure, and a GPU-accelerated 500 ns MD simulation of each complex was achieved using constant pressure. To control the temperature, a Langevin thermostat with 1 atm pressure and 300 K reading capacity was employed.33 The particle mesh Ewald algorithm was used to evaluate long-range interactions with a cutoff distance of 10 Å. To treat the covalent interactions involving hydrogen, the SHAKE algorithm was used.34
000 and 6000 conjugate gradient energy minimization cycles, were completed to relax the complexes and remove any bad clashes. With default heat parameters of 300 K for 200 ps, each complex was heated. For density equilibration, weak restraint was used for 2 ns at a constant pressure, and a GPU-accelerated 500 ns MD simulation of each complex was achieved using constant pressure. To control the temperature, a Langevin thermostat with 1 atm pressure and 300 K reading capacity was employed.33 The particle mesh Ewald algorithm was used to evaluate long-range interactions with a cutoff distance of 10 Å. To treat the covalent interactions involving hydrogen, the SHAKE algorithm was used.34
2.7 Post-simulation trajectory analysis
Structural-dynamics features were estimated using CPPTRAJ and PTRAJ35 to assess the influence of the newly evolved variant on stability, flexibility, compactness, hydrogen bonding, and protein motion. The dynamic stability of each complex was calculated as the root mean square deviation (RMSD), whereas the residual flexibility was evaluated as root mean square fluctuation (RMSF). Structural compactness was calculated as the radius of gyration (Rg) and hydrogen bonding over the simulation period.
2.8 Hydrogen bonding network analysis
The total number of hydrogen bonds and their sustainability during the MD simulation were calculated to reveal the critical interactions in the hydrogen bonding network.
2.9 Binding energy difference estimations
To compute the binding differences elicited by variations in the protein structure upon mutation, structural frames from the conformational dynamics were used for the estimation of binding free energy. The MM/GBSA method was employed to calculate the contributed van der Waals, electrostatic, and total binding energies of each complex.36 These methods are widely used and have been reported to strongly correlate with experimental results.15,25,37–41 Each energy term, including vdW, electrostatic, GB, and SA, was calculated as a part of the total binding energy.
For the free energy calculation, the following equation was used:
| ΔG(bind) = ΔG(complex) − [ΔG(receptor) + ΔG(ligand)]. |
Each component of the total free energy was estimated using the following equation:
| G = Gbond + Gele + GvdW + Gpol + Gnpol, |
where
Gbond,
Gele, and
GvdW denote the bonded, electrostatic, and van der Waals interactions, respectively. In contrast, polar and nonpolar are represented by
Gpol and
Gnpol.
3. Results and discussion
3.1 Structure modelling and analysis
The spike glycoprotein is the primary determinant of COVID-19 infection; it instigates pathogenesis by binding to the host ACE2 receptor through the receptor-binding motif rooted in the RBD domain. This trimeric multidomain protein is receptive to genetic variations, which helps the virus to evolve effectively and increases the frequency of infection accordingly. Contingent factors, i.e., ecological and epigenetics, contribute to the lethal evolution of the new strains of SARS-CoV-2. Recently reported strains of SARS-CoV-2 in the UK, South Africa, and Brazil are capable of causing infection that is more likely to be lethal and are 70% more transmissible than the Wuhan strain. The evolution of SARS-CoV-2 is ongoing; as such, the new B.1.618 variant has emerged with E484K in the RBD domain, D614G at the furin binding site, and deletions of Tyr145 and His146 from the NTD domain where the neutralizing antibodies bind and neutralize the virus.
A recent study reported that variant B.1.617 slightly alters the binding of S-protein to ACE2 and can evade neutralizing antibodies.42 However, a detailed study on the new variant is needed to understand the binding differences and explore the atomic-level interactions of the wild type and E484K mutant on the RBD domain. In addition, the effect of deletions on the NTD domain with regards to neutralizing antibodies must be determined. A previous structural study demonstrated how the new variants reported in the UK, South Africa, and Brazil interact with the host receptor and increase infectivity.38 These mutations, i.e. E484K, D614G, and the deletions that may be associated with functional complexity, could be used to counter infection in a specific manner. Given the importance of the SARS-CoV-2 spike protein RBD in the pathogenesis of the virus and the role of NTD in the attachment of neutralizing antibodies, we subjected the RBD and NTD domains to comparative binding and biophysical investigation upon their interaction with ACE2 and mAb.
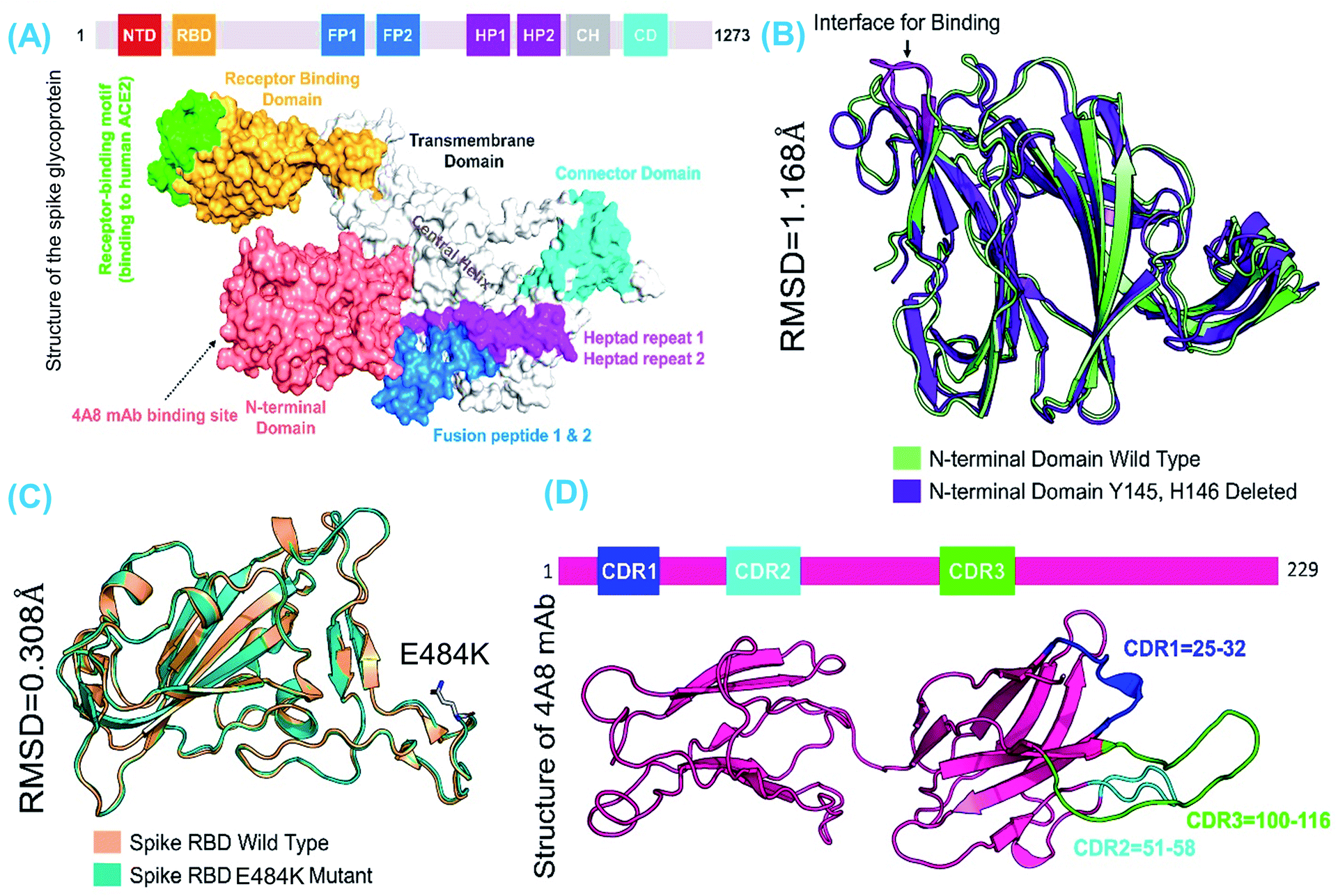
The structures of the wild-type SARS-CoV-2 spike protein RBD and mAb are available at (and were retrieved from) the RCSB website using the accession numbers PDB ID: 6M0J and 7C2L, respectively. Structural modelling of E484K-RBD and Y145–H146-deleted NTD was performed using Modeller integrated with the molecular visualization software Chimera (REF). The loop region (51–55) in the S-protein wild-type-NTD is missing; therefore, it was modelled using the loop modelling module in Modeller.43 The modelled structures were relaxed with a conjugate gradient energy-minimization algorithm and validated using a Ramachandran plot, which revealed that only 1.4% and 0.38% of the total residues lie in the outlier region while the rest of the structures are well conserved. Comparable secondary structural element distributions were observed among the wild-type and mutant modelled structures. RMSD differences were also calculated to understand the impact of mutations on structural deviation. The wild-type-RBD–ACE2 and E484K-RBD–ACE2 exhibited structural similarities with an RMSD difference of only 0.308 Å, whereas significant deviation was observed between the wild-type and truncated (B.1.618) NTDs. Specifically, the RMSD difference between the wild-type and truncated (B.1.618) NTDs was 1.108 Å. This indicates that the deletion has caused significant structural deviance, which may in turn affect mAb binding. Fig. 1 depicts the structure of the spike protein domain with the domain organization (A), the superimposed structures of wild-type and E484K RBD (B), the superimposed structure of the wild-type and truncated (B.1.618) NTDs (C), and the structure of mAb along with the distribution of three complementarity-determining regions (CDRs) (D).
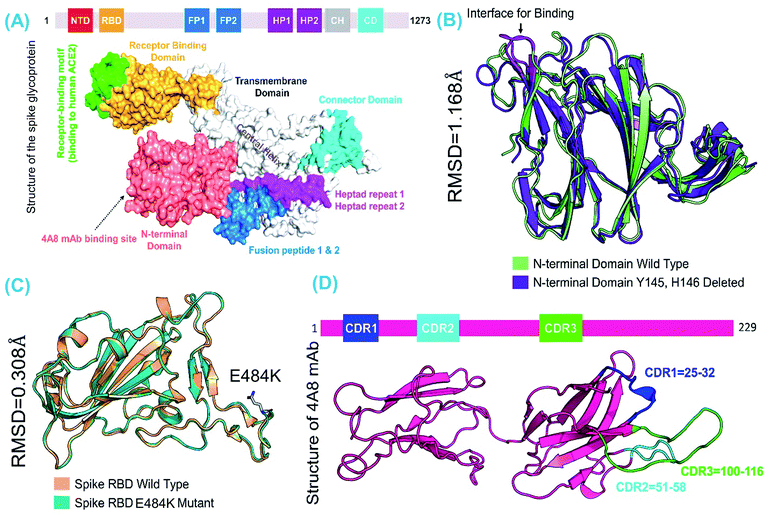 |
| | Fig. 1 The comparative structural evaluation of wild-type and mutant domains. (A) The complete structure of the spike trimeric protein with domain organization, (B) the superimposed structures of wild-type and B.1.618 RBDs, (C) the superimposed structures of wild-type and truncated (B.1.618) NTDs, and (D) the structure of mAb along with the distribution of three complementarity-determining regions (CDRs), which are compulsory for interaction with the NTD. | |
3.2 Mutation-induced stability and function correlation
In an analysis of the stability and function of E484K, results from BeAtMuSiC, DynaMut, and mCSM-PPI2 showed that the substitution produces a destabilizing effect but that the binding affinity remains comparable. An inter-residue interaction comparison revealed that the wild type is enriched with mixed vdW, hydrogen, polar, aromatic, and carbonyl bonds, whereas the E484K typed is enriched with two polar and a few mixed hydrophobic and carbonyl interactions. These results are consistent with previous findings, i.e., that enriched polar contacts increase stability and are strongly correlated with binding affinity.44 Hence, the polar contents are reduced in the E484K complex compared to the polar contents in the wild type. Moreover, the global stability trend of the RBD indicated that stabilizing mutation increases the binding affinity and eventually the infectivity.44 However, the effect of deletions cannot be determined using the aforementioned servers.
3.3 Spike RBD–ACE2 docking
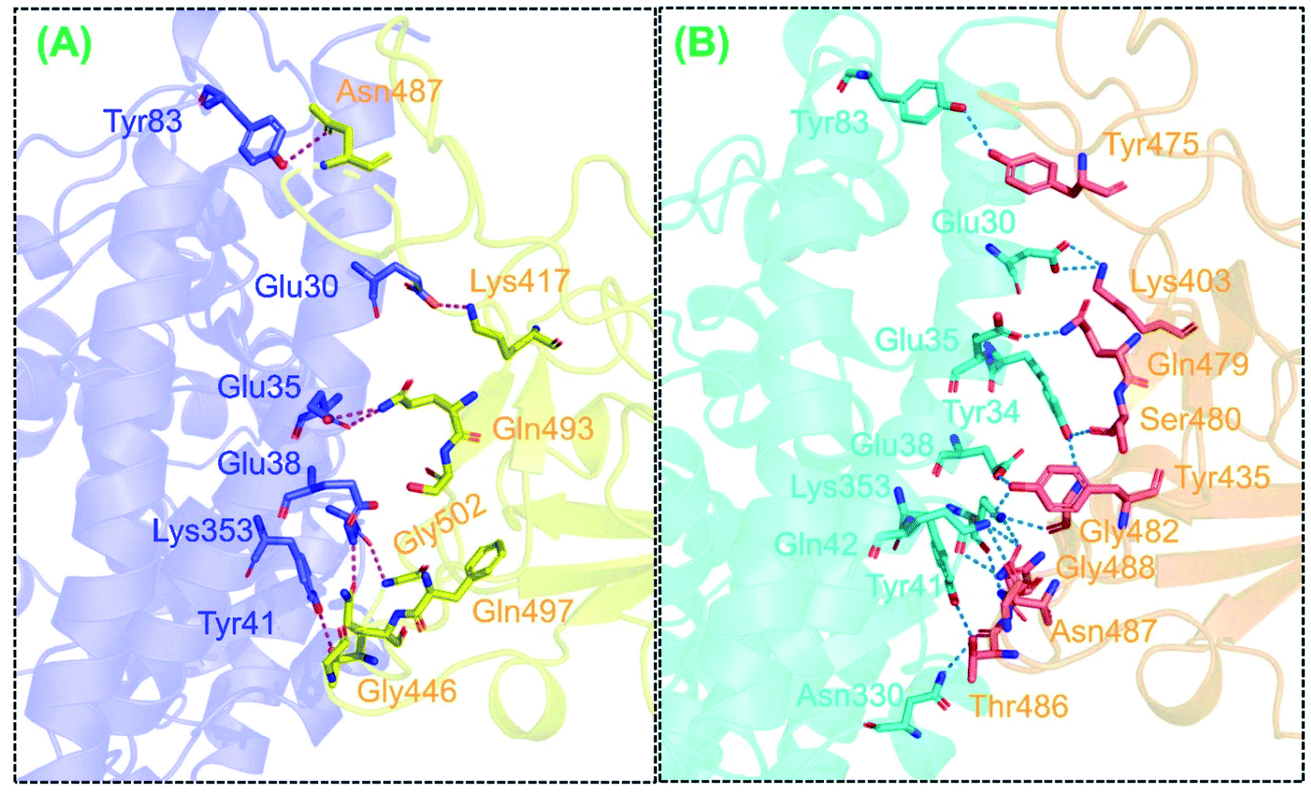
We also determined binding differences using a protein–protein docking approach. The association of biological macromolecules, i.e. proteins that regulate essential cellular process, plays an important role in maintaining normal cell functions. Any incongruity in protein–protein interactions may lead to biologically malfunctioning pathways, which may in turn trigger disease phenotypes. The association of proteins in cells is a complex phenomenon; in many diseases, the interacting interface is a key drug target when developing treatments for diseases. Any mutation at the interface or distinct site may directly affect binding and affect expression. Thus, identifying the binding of different proteins, particularly in SARS-CoV-2, helps to develop novel therapeutic strategies, especially given that the association of the RBD of the spike glycoprotein instigates COVID-19 infection upon binding to the host ACE2 receptor. Recently, variations reported in the UK, South Africa, and Brazil have been shown to cause substantial structural deviation and altered binding properties.38 However, the newly reported variant in India, i.e., that with an E484K mutation, has yet to be analyzed to determine whether the mutation is detrimental to ACE2 binding and therefore viral transmission. In the present study, using structural and biophysical investigations, we found that the E484K mutation did not alter the binding affinity significantly; however, the bonding pattern was changed. The docking scores and interactions between the wild-type and E484K complexes were comparable. In total, 11 hydrogen bonds and only one salt bridge were reported in the wild-type complex. The docking scores for the wild-type complex predicted by HADDOCK and HDOCK were −122.6 ± 0.7 and −302.12 kcal mol−1, respectively. With a salt bridge between Glu30 and Lys417, the hydrogen bonding interactors included Glu30–Lys417, Glu35–Gln493, Glu38–Tyr449, Glu38–Gly496, Tyr41–Thr500, Tyr41–Thr500, Gln42–Gln498, Asn330–Thr500, Lys353–Gly502, Lys353–Gly496, and Lys353–Gln498. These findings are consistent with previously reported ACE2–RBD interaction data.38
The docking score for the B.1.618-RBD–ACE2 complex was −123.6 ± 3.1 according to HADDOCK with a cluster size of 67. The HDOCK predicted docking score for the B.1.618-RBD–ACE2 complex was −311.03 kcal mol−1. The E484K-RBD complex resulted in one salt bridge (between Lys403 and Glu30), whereas 12 hydrogen bonds were shown to be involved between E484K-RBD and ACE2 residues: Lys403–Glu30, Gly432–Gln42, Tyr435–Glu38, Tyr435–Gln42, Gln479–Glu35, Ser480–Tyr34, Gly482–Lys353, Gln484–Lys353, Gln484–Gln42, Thr486–Tyr41, Thr486–Tyr41, and Gly488–Lys353. Results suggest that the mutation has changed the binding paradigm and that a key residue important for recognition, i.e., Lys353, formed multiple interactions with the RBD domain. Interestingly, some interactions were highly conserved between the wild-type and E484K-mutant complexes, particularly the interactions of Lys353 from ACE2 with the spike RBD. Previously reported MD-extracted coordinates indicate that this interaction is sustained.45,46 The binding patterns of the wild-type and E484K complexes are shown in Fig. 2A and B.
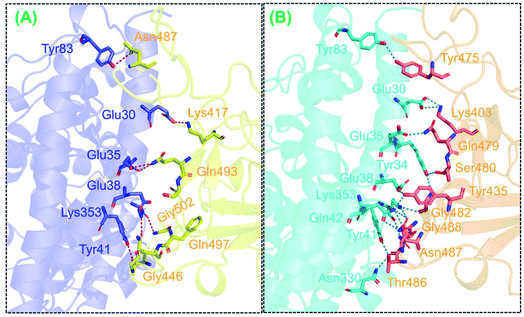 |
| | Fig. 2 The interaction pattern of the wild-type and B.1.618 variant (E484K) mutant docking complexes. (A) A stick representation of the wild-type-RBD and ACE2. Marine blue represents ACE2 while yellow represents the wild-type spike-RBD. (B) The binding interface of B.1.618 and ACE2 shown as sticks. The cyan colour represents ACE2 while the orange colour represents mutated RBD. | |
Considerable differences in the electrostatic energy between the wild-type and mutant complexes were observed. The electrostatic energy for the wild type was −223.0 ± 20.0, whereas that for the B.1.618 variant (E484K) mutant was −295.9 ± 12.7. This finding of greater electrostatic energy is supported by prior studies in which the binding of SARS-CoV-2 and SARS-CoV RBDs with ACE2 was evaluated; these studies revealed that more hydrogen bonds and electrostatic interactions existed in SARS-CoV-2.22,47 Thus, the results collectively indicate that the stronger binding of the mutant complexes is mainly due to electrostatic contributions. Moreover, these findings suggest that the key dissimilarities in interaction conformation are important for higher infectivity.
3.4 NTD–mAb docking
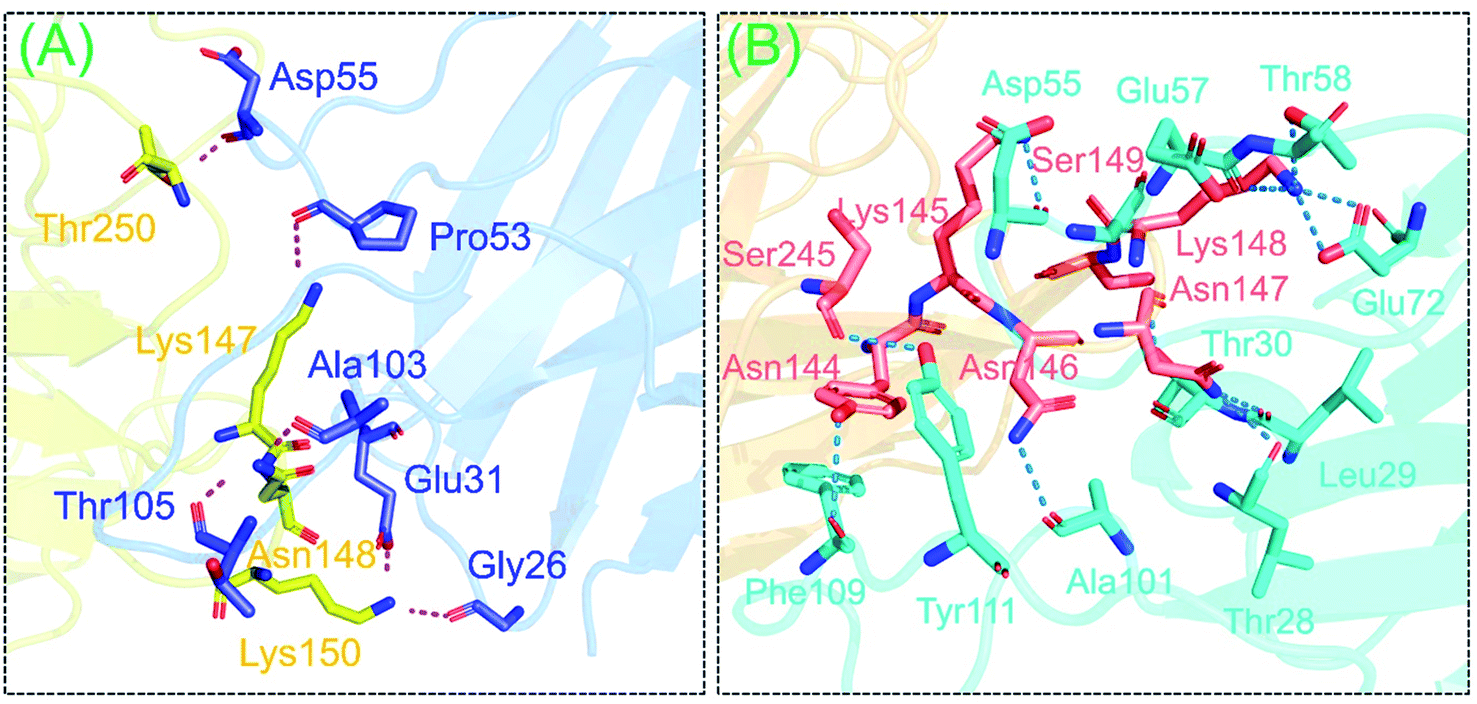
The binding pattern of the wild-type-NTD and truncated (B.1.618) NTD with mAb antibody was also evaluated here to assess the deletion-specific impact on the recognition and binding of mAb. Considerable differences in total binding energy, vdW, and electrostatic energies were observed. For the wild type, the total binding energy was −89.6 ± 6.3; for the truncated (B.1.618) NTD (Y145–H146 deletions) the total binding energy was −80.8 ± 2.9. In addition, the vdW for these complexes were −47.3 ± 4.9 and −39.0 ± 5.9, respectively, whereas the electrostatic energies were −223.0 ± 20.0 and −295.9 ± 12.7, respectively. The HDOCK docking scores were −339.26 kcal mol−1 for the wild-type and −283.11 kcal mol−1 for the truncated (B.1.618) NTD. Binding patterns revealed that the wild-type and truncated (B.1.618) NTDs possessed seven and five hydrogen bonds, respectively. In each complex, multiple salt bridges were also detected. The residues Tyr145 and His146 were previously reported to play an important role in the binding of neutralizing antibodies (mAb).24 Although the binding pattern showed that the mutant had more bonds than the wild type, this demonstrates that binding with the residues Tyr145 and His146 was more robust, which was also shown by the docking scores. Additionally, these two residues may alter the function of the spike protein upon binding with mAb. These results show that the new variant is an antibody-escaping variant and does not have substantially altered binding affinity. The binding patterns of the wild-type-NTD and truncated (B.1.618) NTD complexes are shown in Fig. 3A and B.
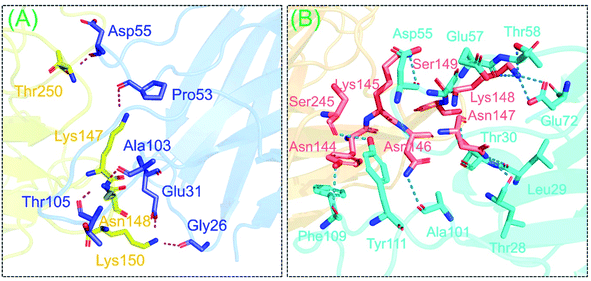 |
| | Fig. 3 The interaction patterns of the wild-type and B.1.618 variant (Y145–H146 deleted) NTD docking complexes. (A) A stick representation of the wild-type-NTD and mAb. (B) The binding interface of Y145–H146 deleted and mAb (as sticks). | |
3.5 Dissociation constant calculations
To more deeply assess the binding variations, we also calculated the KD (i.e., dissociation constant) of the wild-type-RBD–ACE2, E484K-mutant, wild-type-NTD, and truncated-(B.1.618)-NTD complexes. KD is frequently used to estimate and rank the strengths of biomolecular interfaces,48 and KD kinetics are commonly used to estimate the affinity of antigen–antibody, protein-small molecule, and large biological protein complexes. Stronger binding of the two interacting partners can be inferred from the lowest KD.49 This approach is most frequently employed to determine the binding strengths of different biomacromolecules.50 Here, the PRODIGY algorithm was employed to reveal the binding differences between wild-type and mutant complexes, i.e., wild-type-RBD–ACE2, B.1.618-RBD–ACE2, wild-type-NTD, and B.1.618-NTD. The results showed that the binding affinities of the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2 were tighter than that of B.1.618-RBD–ACE2. The binding affinities of the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2 were 5.2 × 10−10 and 4.7 × 10−10, respectively. Previously, lower KD values for B.1.1.7, B.1.351, and P.1 were reported, which eventually increase infectivity. These reports are in agreement with the significantly lower equilibrium KD values obtained in in vitro binding assays of SARS-CoV-2 compared with the KD values for SARS-CoV.51,52 KD was also determined for wild-type-NTD and B.1.618-NTD to evaluate the binding of mAb to the wild type and B.1.618. The KD values for wild-type-NTD (3.1 × 10−8) indicated stronger binding than was observed for B.1.618-NTD (with a KD of 9.2 × 10−8). These results are consistent with recent findings showing the reduced binding of an antibody with the NTD.53 All the parameters, including HADDOCK docking scores, cluster size, vdW energy, electrostatic energy, and KD, of all the complexes are given in Table 1.
Table 1 The HADDOCK predicted docking scores for all mutant complexes and additional parameters, including cluster size, vdW energy, electrostatic energy, and Z-score. The table also tabulates the KD value (dissociation constant) for each complex predicted using PRODIGY (PROtein binDIng enerGY prediction)
| Docking parameter |
Wild-type-NTD |
B.1.618-NTD |
Wild-type-RBD–ACE2 |
B.1.618-RBD–ACE2 |
| HADDOCK score |
−89.6 ± 6.3 |
−80.8 ± 2.9 |
−122.6 ± 0.7 |
−123.6 ± 3.1 |
| Cluster size |
61 |
22 |
64 |
67 |
| RMSD from the overall lowest-energy structure |
13.6 ± 0.5 |
10.5 ± 0.5 |
1.7 ± 1.0 |
1.2 ± 0.8 |
| van der Waals energy |
−47.3 ± 4.9 |
−39.0 ± 5.9 |
−59.6 ± 2.3 |
−55.5 ± 2.4 |
| Electrostatic energy |
−223.0 ± 20.0 |
−295.9 ± 12.7 |
−181.4 ± 15.5 |
−196.8 ± 16.6 |
| Desolvation energy |
−8.7 ± 3.1 |
−4.0 ± 3.1 |
−27.1 ± 3.4 |
−29.8 ± 2.0 |
| Restraint violation energy |
110.2 ± 15.5 |
213.8 ± 57.6 |
4.7 ± 3.8 |
31.0 ± 16.7 |
| Buried surface area |
1573.9 ± 125.4 |
1328.7 ± 39.4 |
1965.3 ± 120.6 |
1911.4 ± 26.2 |
| Z-score |
−1.2 |
−1.4 |
−1.9 |
−1.3 |
| HDOCK scores |
−339.26 |
−283.11 |
−302.12 |
−311.03 |
| KD (dissociation constant) |
3.1 × 10−8 |
9.2 × 10−8 |
5.2 × 10−10 |
4.7 × 10−10 |
3.6 Structural stability of RBD–ACE2 and NTD–mAb complexes
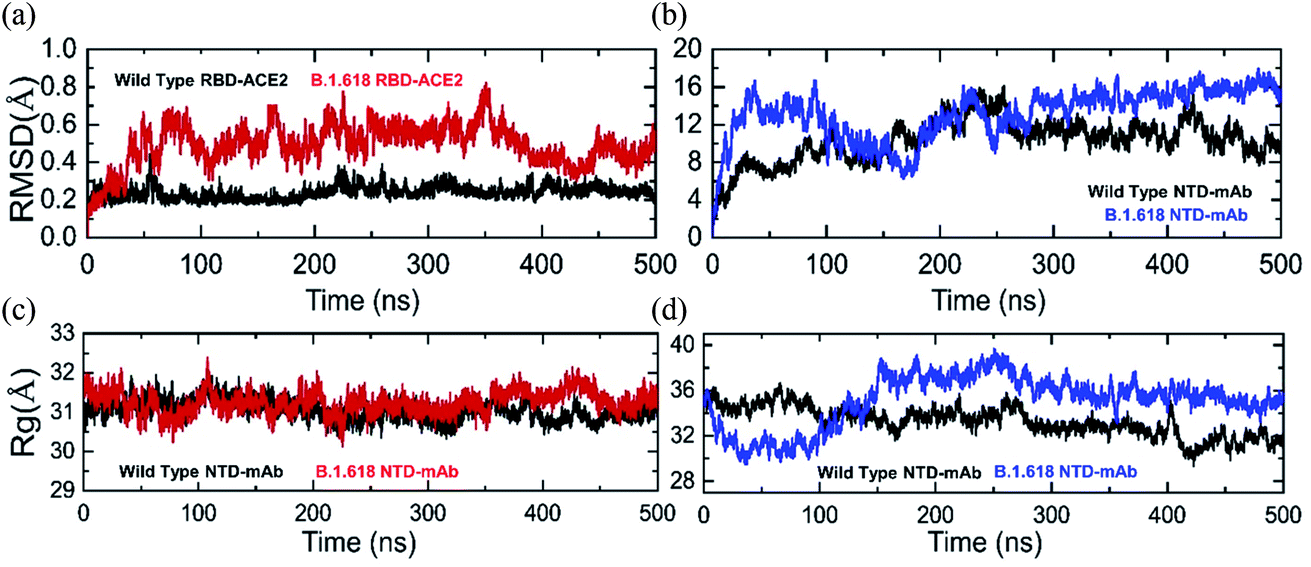
Structural deviation in the dynamic conditions was analyzed to evaluate variations in the stability of each complex as RMSD. Results of comparative stability analysis revealed that the wild-type-RBD–ACE2 complex was dynamically more stable and presented intermolecular constraint deviation relative to the B.1.618-RBD–ACE2 variant. Except for smaller deviations at 50–60, 230–260, and 380–400 ns, the RMSD trajectory of the wild type remained highly stable and reached an equilibrium state after 5 ns. The average RMSD value of the wild type was ∼2.0 Å, although the aforementioned sharp deviation reached 4.0 Å (Fig. 4a). In contrast, the B.1.618-RBD–ACE2 variant showed dynamically unstable behavior over the simulation period. The RMSD of B.1.618-RBD–ACE2 gradually increased at the start of the simulation and remained flattened until 35 ns; afterwards, significant structural perturbation was experienced over the remaining time. Specifically, significant deviation occurred between 36 and 350 ns. During this simulation period, the RMSD of the B.1.618-RBD–ACE2 complex was in continuous oscillation and faced substantial perturbations (Fig. 4a). This can be interpreted as a poorly docked intermolecular configuration with persistent formation and deformation of chemical interactions, which result from the effort to acquire the correct binding mode of the different molecules. During the simulation period (36–350 ns), the RMSD remained high at approximately 6.0 Å. The RMSD subsequently decreased and remained stable until 500 ns, which can be interpreted as the stability in the intermolecular docked pose finally having been achieved. The average RMSD over the last 150 ns was 4.0 Å. In summary, these results show that the wild-type complex is more behaviorally stable, although the B.1.618-RBD–ACE2 complex also reached stability at 350 ns. The behavior of the wild-type-RBD–ACE2 complex is consistent with the previous finding, including a similar RMSD trend. Furthermore, our conclusions are in agreement with the results of previous studies. First, global RBD stability leads to higher ACE2 binding affinity, as previously reported.44 Moreover, earlier studies have shown a close link between RBD stability and affinity, with mutations that maximize structural stability and inflexibility causing increases in binding affinity.54,55
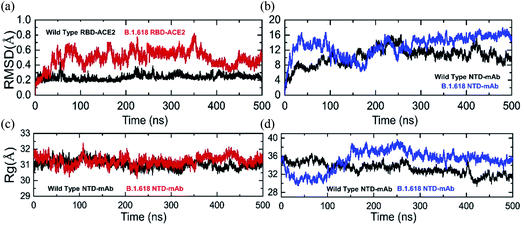 |
| | Fig. 4 The RMSD(s) and Rg(s) of all the complexes in different colours. (a) and (b) The RMSDs of the wild-type-RBD–ACE2 and wild-type-NTD are shown in black while the RMSDs of B.1.618-RBD–ACE2 and B.1.618-NTD are shown in red and blue, respectively. (c) and (d) The Rg(s) of wild-type-RBD–ACE2 and wild-type-NTD are shown in black while the Rg(s) of B.1.618-RBD–ACE2 and B.1.618-NTD are shown in red and blue, respectively. | |
In addition, we calculated the RMSDs of wild-type-NTD and B.1.618-NTD–mAb-bound complexes during the 500 ns simulation time. The RMSD trends for the wild-type and B.1.618 NTDs were comparable. Apparently, the RMSDs for both complexes increased initially up to 1.2 Å; the RMSD of the wild-type-NTD complex then increased to 0.8 Å and reached an equilibrium point at 50 ns, whereas the RMSD of the B.1.618-NTD complex increased to 1.6 Å during the first 100 ns. Thereafter, at 100–250 ns, inverse RMSD trends were observed; the RMSD of the wild type increased, whereas that of the B.1.618-NTD complex gradually decreased. During the last 250 ns, the RMSDs of the wild-type-NTD and B.1.618-NTDs were comparable. Intriguingly, the RMSD of the B.1.618-NTD complex remained higher than that of the wild type but was found to be more stable. However, the RMSD of the wild-type-NTD complex faced structural perturbation at different time intervals. Comparatively, the B.1.618-NTD–mAb-bound complex was more stable than the wild-type-NTD–mAb-bound complex. The RMSDs of both complexes are shown in Fig. 4b.
3.7 Structural compactness of RBD–ACE2 and NTD–mAb complexes
Structural compactness during the simulation may help to understand the packing of the protein complexes and reveal the binding and unbinding events that occur during the simulation. We evaluated the compactness of each complex as a function of time to understand the key differences in the binding between the wild-type and mutant complexes. As shown in Fig. 4c the structures of the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2 remained comparable; however, minor deviation was observed at different time intervals. The B.1.618-RBD–ACE2 had a more compact topology during the first 100 ns than did the wild type. Afterwards, the Rg values increased until 210 ns; for the B.1.618-RBD–ACE2, the compactness then increased for a short period (210–230 ns). Thereafter, the Rg values of the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2 remained similar in the 231–350 ns period. For a short period, i.e., 350–450 ns, the differences in the Rg values between the wild-type-NTD and B.1.618-NTD were obvious; however, at 500 ns the Rg pattern again aligned between the two. These data show that the two structures passed through several binding and unbinding events, particularly the wild type, whereas the compactness of the mutant (E484K) was observed to be conserved more during the MD simulation. The average Rg value for both complexes was 31.5 Å during the 500 ns simulation time.
Unlike the RBD complexes, the NTD complexes showed noteworthy variations in structural compactness. During the first 0–100 ns, the structure of the B.1.618-NTD remained more compact than that of the wild-type-NTD. During the first 100 ns, the average Rg values for the wild-type-NTD and B.1.618-NTD were 36.0 Å and 32.0 Å, respectively. Afterwards, B.1.618-NTD lost its structural compactness and reached 38.0 Å, and it remained at this higher level until 350 ns. The Rg then decreased and remained uniform during the last 150 ns. For the wild type, the average Rg value remained at 34.0 Å during the last 400 ns. An inverse trend was observed during the last 400 ns for the wild-type-NTD and B.1.618-NTD. In conclusion, these data show that, after 100 ns, mAb binding is destabilized by the deletions and then unbinding events are strongly favored. The Rg graphs of the wild-type-NTD and B.1.618-NTD are shown in Fig. 4d.
3.8 Residual flexibility of RBD–ACE2 and NTD–mAb complexes
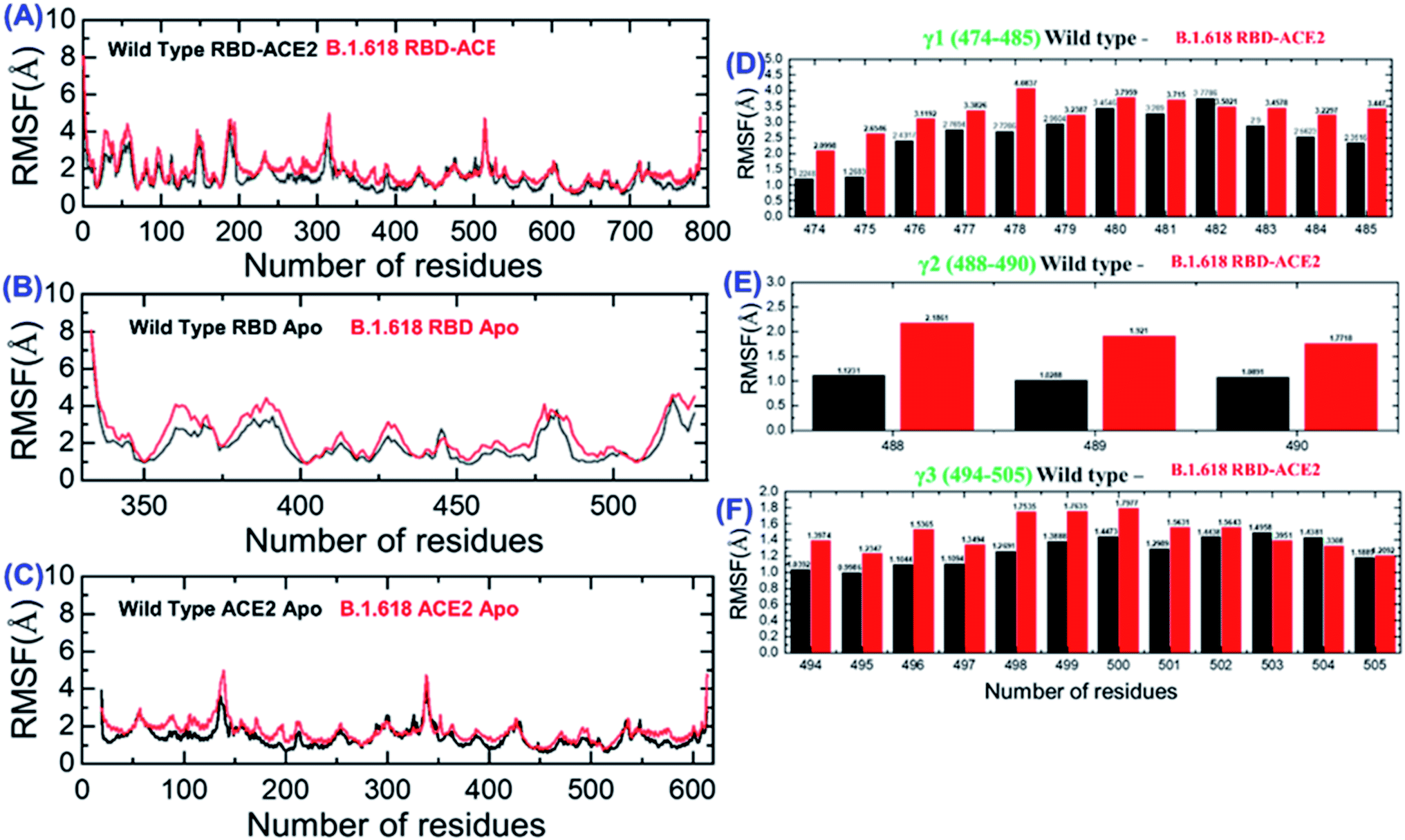
We also evaluated the residual flexibility of each residue in all the complexes with the aim of understanding the strength conferred by each residue to the intermolecular binding, the molecular recognition, and the possible impact on the global function of the biological macromolecules. To better explain the differences in flexibility at the residue level, we calculated RMSF as a complex, apo (RBD and ACE2), and for the three important loops, γ1 (474–485), γ2 (488–490), and γ3 (494–505), previously reported as crucial for binding.16,22,38 The RMSF of the RBD–ACE2 complex is given in Fig. 5A; it can be seen that the flexibility of some regions, particularly 350–400 and 450–526, was greatly increased (Fig. 5B). In addition, in ACE2, the region 19–200 displayed higher fluctuation (Fig. 5C). This shows that the mutation induces flexibility that results in better conformational optimization during the simulation time period and consequently alters the binding of RBD to the host ACE2. To provide further insights into the three aforementioned loops, per-residue RMSF was calculated (Fig. 5D–F). Results showed that the flexibility of these three loops was increased by the mutation as an allosteric effect, which improved the chances of bonding with nearby residues to connect and form a stable connection. Overall, these findings indicate that the spike protein undergoes structural adjustments to bind efficiently to the ACE2 receptor and, in turn, increases entry to the host cells.
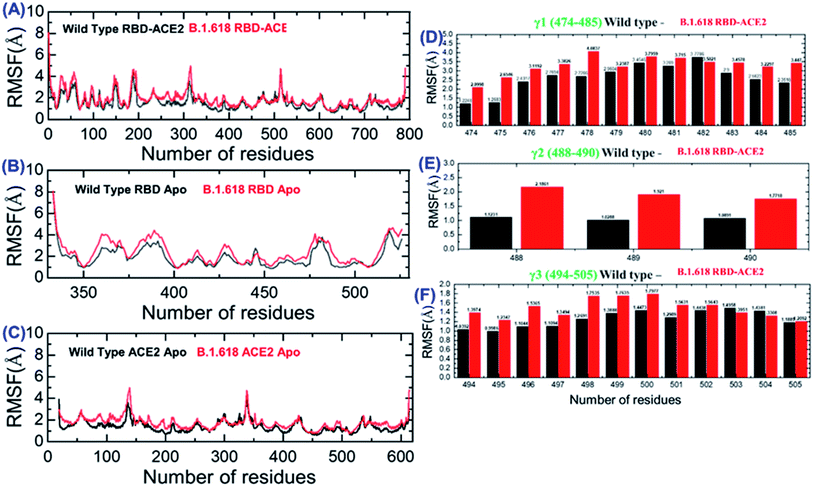 |
| | Fig. 5 Root mean square fluctuations to estimate the residual flexibility. (A) The RMSF for the wild-type and mutant complexes. (B) The RMSF for the wild-type and mutant RBD. (C) The RMSF for ACE2. (D–F) The RMSF values of the three important loops in the RBD required for binding. | |
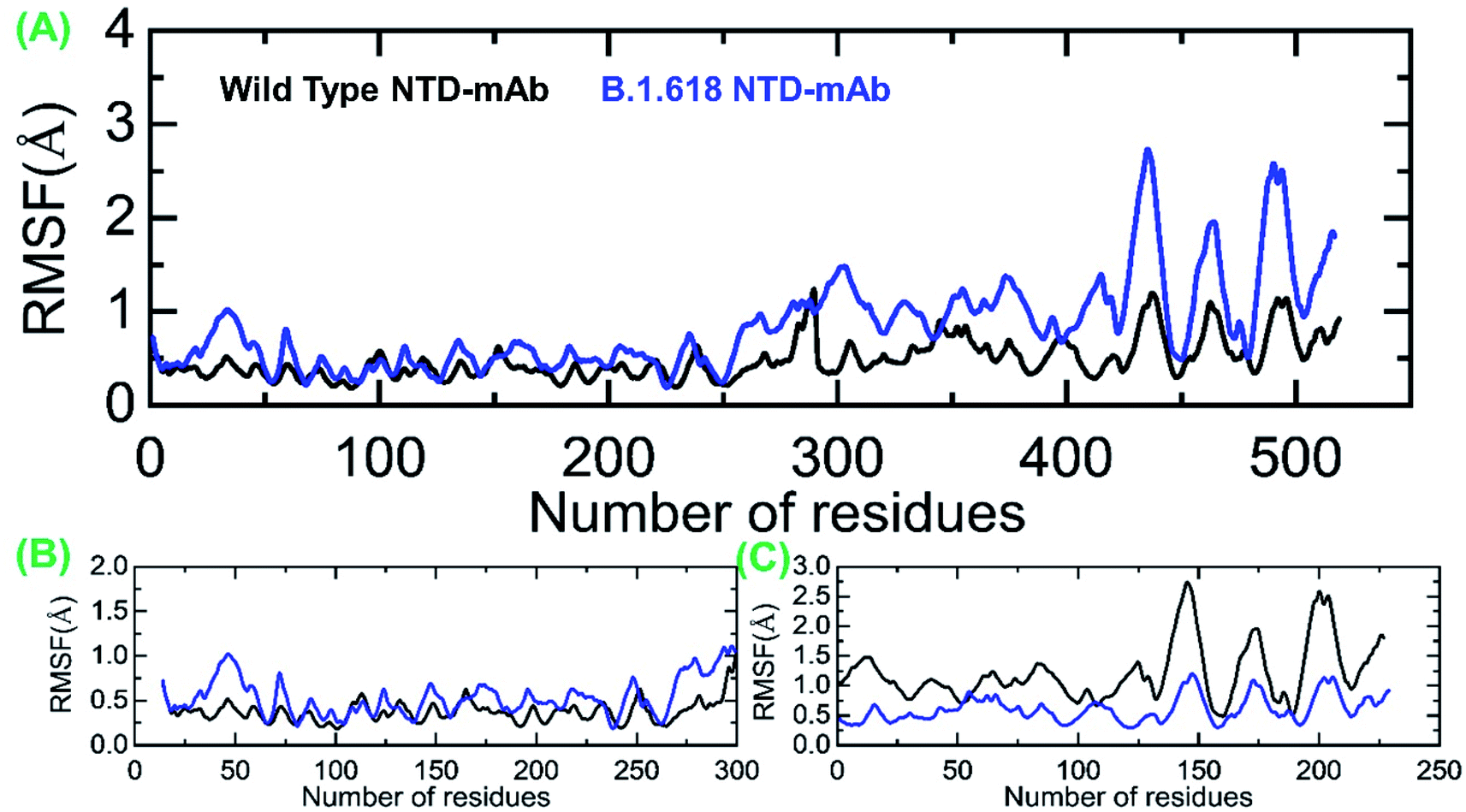
Moreover, we also estimated the RMSF for wild-type-NTD and B.1.618-NTD complexes to show the residual flexibility differences in complex and apo states. From Fig. 6A, it can be concluded that region 19–300 exhibits comparable fluctuation except in regions 19–75 and 250–300 in the B.1.618-NTD higher fluctuation, recorded in Fig. 6B. Significant differences in structural flexibility were seen in mAb (0–230) in Fig. 6C. Overall, the flexibility dynamics reveal that the deletions have caused the mAb to bind to the NTD weakly, and thus this variant potentially acts like a neutralizing-antibody-evading variant.
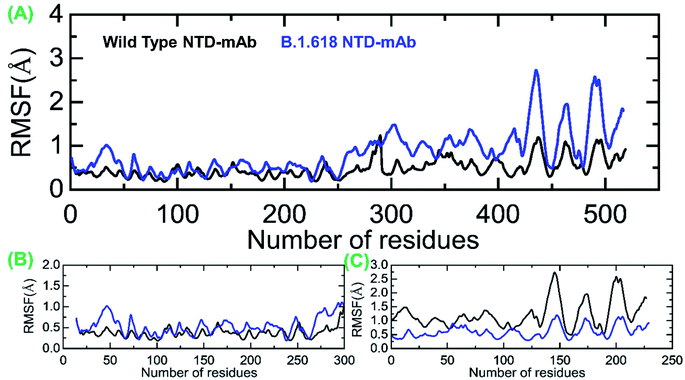 |
| | Fig. 6 Root mean square fluctuations to estimate the residual flexibility. (A) The RMSF for the wild-type and mutant complexes. (B) The RMSF for the wild-type and mutant NTD. (C) The RMSF for the mAb. | |
3.9 Hydrogen bond analysis
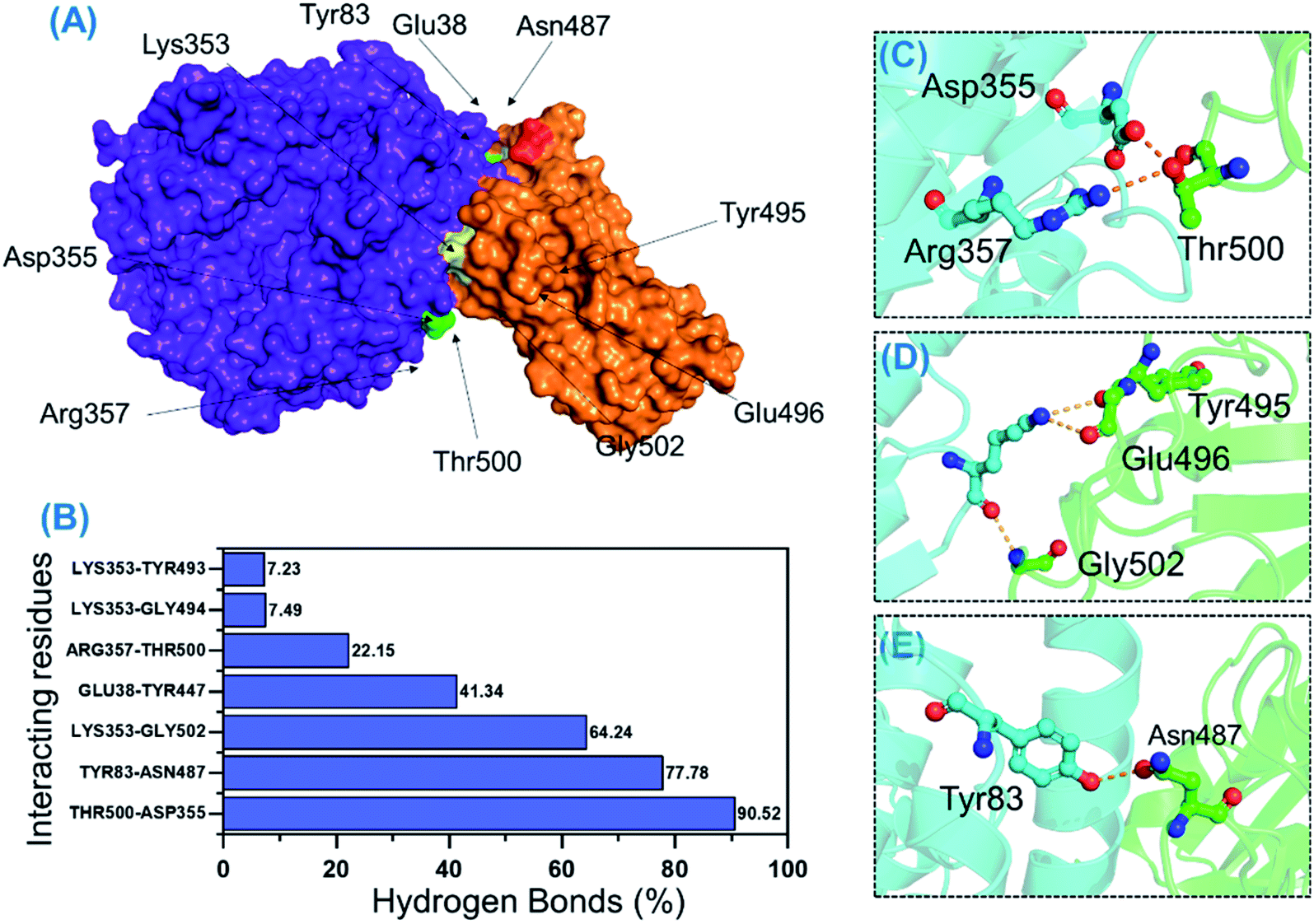
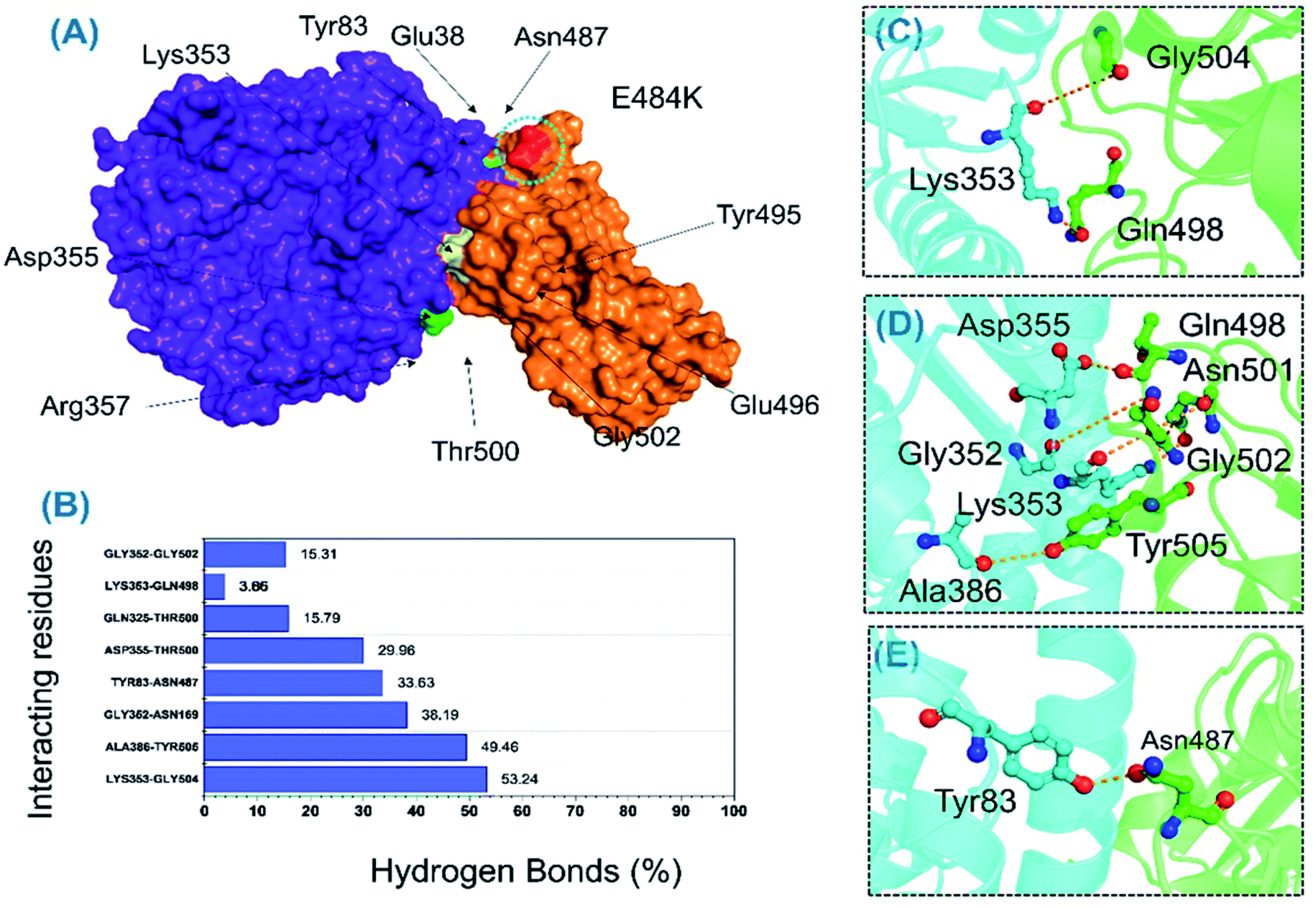
Protein–protein association is mainly guided by a variety of factors among which hydrogen bonds and hydrophobic interactions are key players. Interactions at protein interfaces are always conducted by water molecules which compete with the hydrogen bonds between residues.56 The processes behind protein–protein coupling, as well as the implications in which hydrogen bonds play a role, in this association are unknown.57 Whether hydrogen bonds govern protein–protein docking, in particular, is a long-standing concern with a poorly understood mechanism.58,59 Thus, to understand the bonding pattern between the wild-type-RBD–ACE2, B.1.618-RBD–ACE2, wild-type-NTD, and B.1.618-NTD complexes, post-simulation hydrogen bonding analysis was conducted to determine the binding specificity for ACE2 and mAb given biochemical events steered by hydrogen bonding. Essential equilibrated stable interactions with consistent contacts between different molecules over longer simulation periods that execute essential biological functions were monitored over 500 ns simulations for each complex. Interestingly, during interactions with ACE2, several hydrogen bonds were well preserved in both the wild-type-RBD–ACE2 and the B.1.618-RBD–ACE2 complexes. As shown in Table 2, in the wild type, the bond between Thr500 and Asp355 was well preserved in 90.52% (22630 frames) of 25000 structures; in the B.1.618-RBD–ACE2 complex, this interaction was sustained in only 29.96% of structural frames. An essential interaction, Tyr83–Asn487, which has previously been reported as a key interaction that differentiates SAR-CoV and SAR-CoV-2 binding in terms of binding energy,60 is reportedly responsible for the enhanced interaction between SARS-CoV-2 RBD and the host ACE2 receptor. In the wild-type complex, this bond was sustained in 77.78% (19446) of structural frames; in the B.1.618-RBD–ACE2 complex, it was detected in only 33.63% (8408) of structural frames. Lys353 is a key residue that may assist in the recognition and fixation of robust interactions between RBD and ACE2, and it is required for the entry of SARS-CoV-2 to the host cell.60 Indeed, this residue forms a cluster of interactions with Tyr495, Gly496, Gln498, and Gly502 to facilitate this entry.22,47 Interestingly, the interaction paradigm of Lys353 was well preserved while being altered in the wild type. In B.1.618-RBD–ACE2, the interaction of Lys353 with different residues was sustained in 53.24%, 3.85%, 3.65%, and 3.85% of structures, respectively. However, the interaction of Lys353 in the wild-type complex accounted for only 64% of the total frames. Moreover, only six key interactions in the wild-type B-RBD–ACE2 were involved and sustained during the simulation at different fractions, whereas ten key interactions had these qualities in B.1.618-RBD–ACE2. The role of interfacial water molecules between the RBD and ACE2 is also vital for the stronger affinity. A recent comparative study on SARS-CoV and SARS-CoV-2 reported that during the 100 ns the number of water molecules at the interface site was greater in SARS-CoV than in SARS-CoV-2.61 They reported that Gln493 is abridged with Glu35 by a hydrogen bond mediated by water molecules entering the interface during the simulation. Moreover, Arg403 stabilizes the interface via water-mediated interaction with Asn33, His34, Glu37, and Asp38 of ACE2 and thus contributes toward the higher affinities.62 Conclusively we speculated that the water molecules are also important for the higher infectivity because it mediates the interaction. All the interactions along with the frames and percentages of the total simulation are given in Table 2; the interactions obtained from the average equilibrated structures (wild-type-RBD–ACE2 and B.1.618-RBD–ACE2) are shown in Fig. 7 and 8.
Table 2 Hydrogen bond occupancy of the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2 complexes during the 500 ns simulation time
| ACE2 |
Wild-type-RBD–ACE2 |
Frames |
Percentage |
| Asp355@OD2 |
Thr500@HG1 |
22630 |
90.52 |
| Tyr83@HH |
Asn487@OD1 |
19446 |
77.78 |
| Lys353@O |
Gly502@H |
16059 |
64.24 |
| Glu38@OE1 |
Tyr447@HH |
10334 |
41.34 |
| Arg357@HH22 |
Thr500@OG1 |
5537 |
22.15 |
| Lys353@HZ3 |
Tyr493@O |
1807 |
7.23 |
| ACE2 |
B.1.618-RBD–ACE2 |
Frames |
Percentage |
| Lys353@O |
Gly504@H |
13309 |
53.24 |
| Ala386@O |
Tyr505@HH |
12365 |
49.46 |
| Gly352@O |
Asn169@HD22 |
9548 |
38.19 |
| Tyr83@HH |
Asn487@OD1 |
8408 |
33.63 |
| Asp355@OD1 |
Thr500@HG1 |
7490 |
29.96 |
| Gln325@HE22 |
Thr500@O |
3948 |
15.79 |
| Lys353@HZ1 |
Gln498@OE1 |
914 |
3.85 |
| Lys353@HZ3 |
Gln498@OE1 |
900 |
3.66 |
| Lys353@HZ1 |
Gln498@OE1 |
963 |
3.85 |
| Gly352@O |
Gly502@H |
3828 |
15.31 |
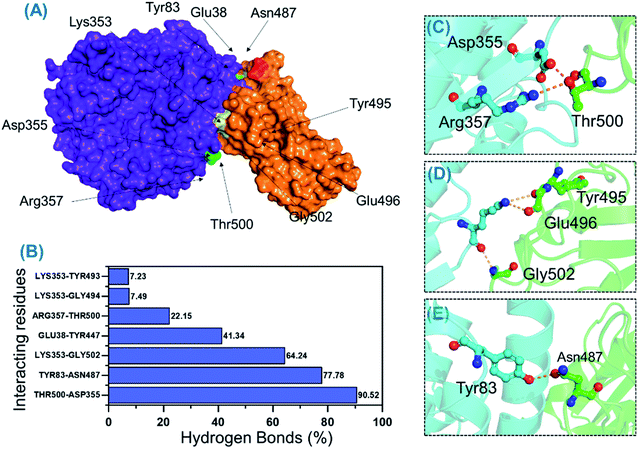 |
| | Fig. 7 H-bonding analysis of the 500 ns trajectory of wild-type-RBD–ACE2. (A) The interacting hotspots, (B) the hydrogen bond percentages, and (C–E) the key interactions observed to be sustained during the simulation. | |
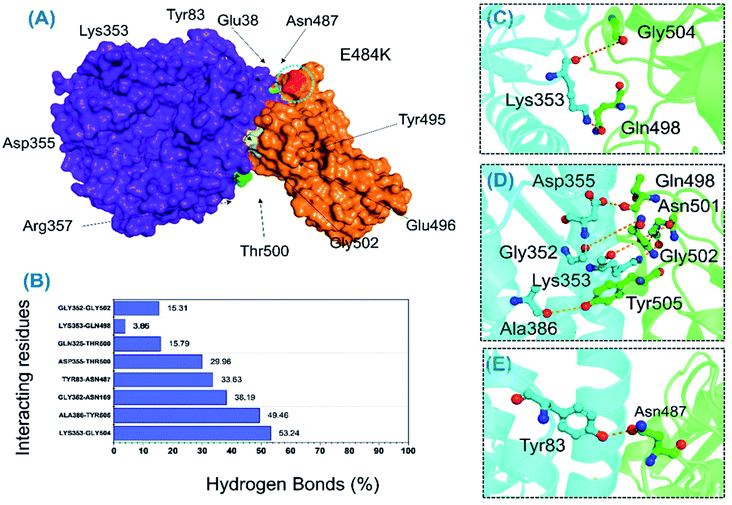 |
| | Fig. 8 H-bonding analysis of the 500 ns trajectory of B.1.618-RBD–ACE2. (A) The interacting hotspots. (B) The hydrogen bond percentages. (C–E) The key interactions observed to be sustained during the simulation. | |
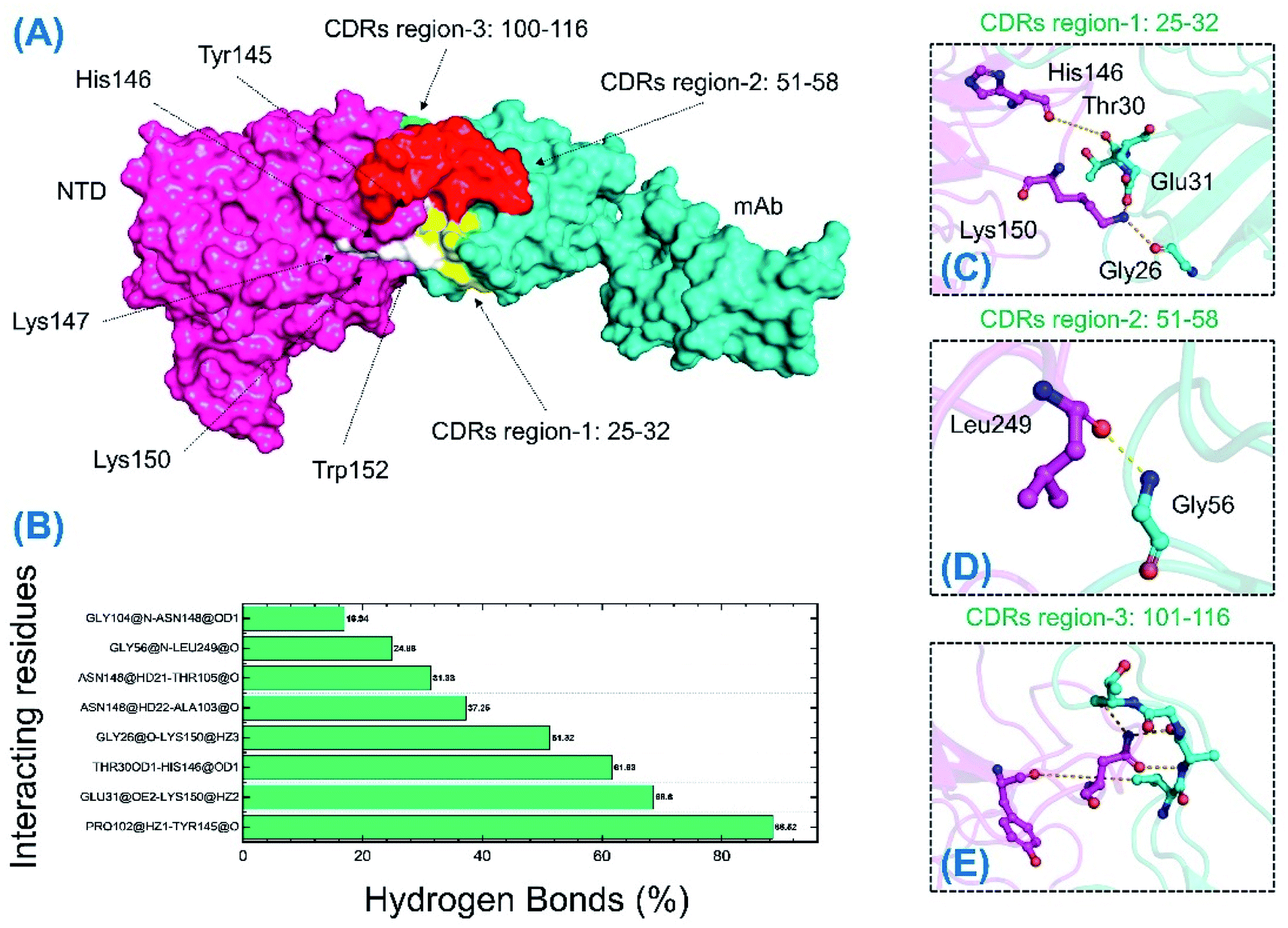
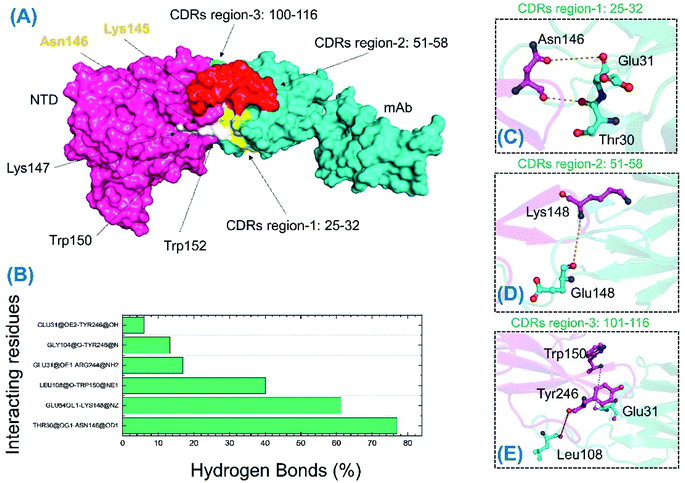
The binding differences during the MD simulation and the half-life of each bond between mAb and the wild-type and B.1.618 NTDs were also estimated. Measurement of such interactions improves understanding of the key residues that could be targeted for future therapeutics. As shown in Table 3 and Fig. 9, the interaction pattern of the wild type was mostly conserved as the docking conformation. The availability of three CDRs on the surface of mAb and the two important loops, N3 (141–156) and N5 (246–260), mediated the interaction. The residues of the CDR1 region interacted primarily with the N3 loop residues Glu31–Lys150, Thr30–His146, and Gly26–Lys150; these interactions were sustained for 68.60%, 61.63%, and 51.32% of the simulations. The only interaction between CDR3 and the N5 loop (Gly56–Leu249) was sustained for only 24% of the simulations. Among the other residues (CDR3), Gly26, Thr30, and Glu31 interacted with His146 and Lys150 of loop 3. The only interaction formed by CDR3 between Pro102 and Tyr145 was sustained in 88.52% of the trajectories. Similar findings (i.e., similar interacting residues) were reported in a previous study.24
Table 3 Hydrogen bond occupancy of the wild-type-NTD and B.1.618-NTD complexes during the 500 ns simulation time
| mAb |
Wild-type-NTD |
Frames |
Percentage |
| Pro102@HZ1 |
Tyr145@O |
22130 |
88.52 |
| Glu31@OE2 |
Lys150@HZ2 |
17150 |
68.60 |
| Thr30@OD1 |
His146@OD1 |
15409 |
61.63 |
| Gly26@O |
Lys150@HZ3 |
12830 |
51.32 |
| Ala103@O |
Asn148@HD22 |
9314 |
37.25 |
| Thr105@O |
Asn148@HD21 |
7846 |
31.38 |
| Gly56@N |
Leu249@O |
6221 |
24.88 |
| Gly104@N |
Asn148@OD1 |
4235 |
16.94 |
| mAb |
B.1.618-NTD |
Frames |
Percentage |
| Thr30@OG1 |
Asn146@OD1 |
19241 |
76.96 |
| Glu54OE1 |
Lys148@NZ |
15312 |
61.24 |
| Leu108@O |
Trp150@NE1 |
10008 |
40.03 |
| Glu31@OE1 |
Arg244@NH2 |
4213 |
16.85 |
| Gly104@O |
Tyr246@N |
3324 |
13.29 |
| Glu31@OE2 |
Tyr246@OH |
1508 |
6.03 |
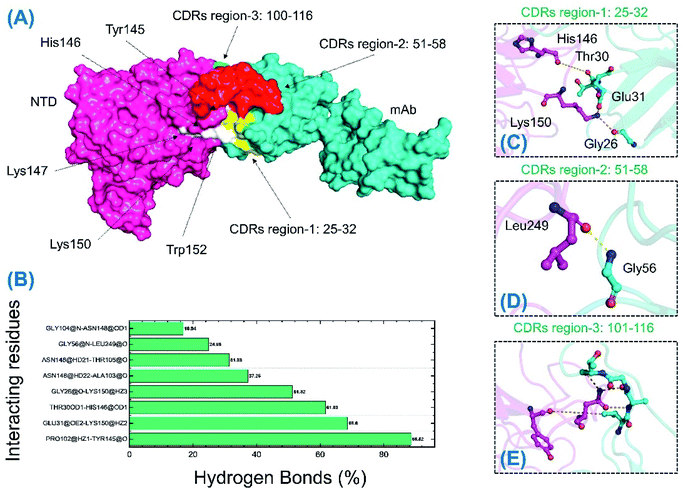 |
| | Fig. 9 H-bonding analysis of the 500 ns trajectory of the wild-type-NTD. (A) The interacting hotspots. (B) The hydrogen bond percentages. (C), (D) and (E) The key interactions observed to be sustained during the simulation. | |
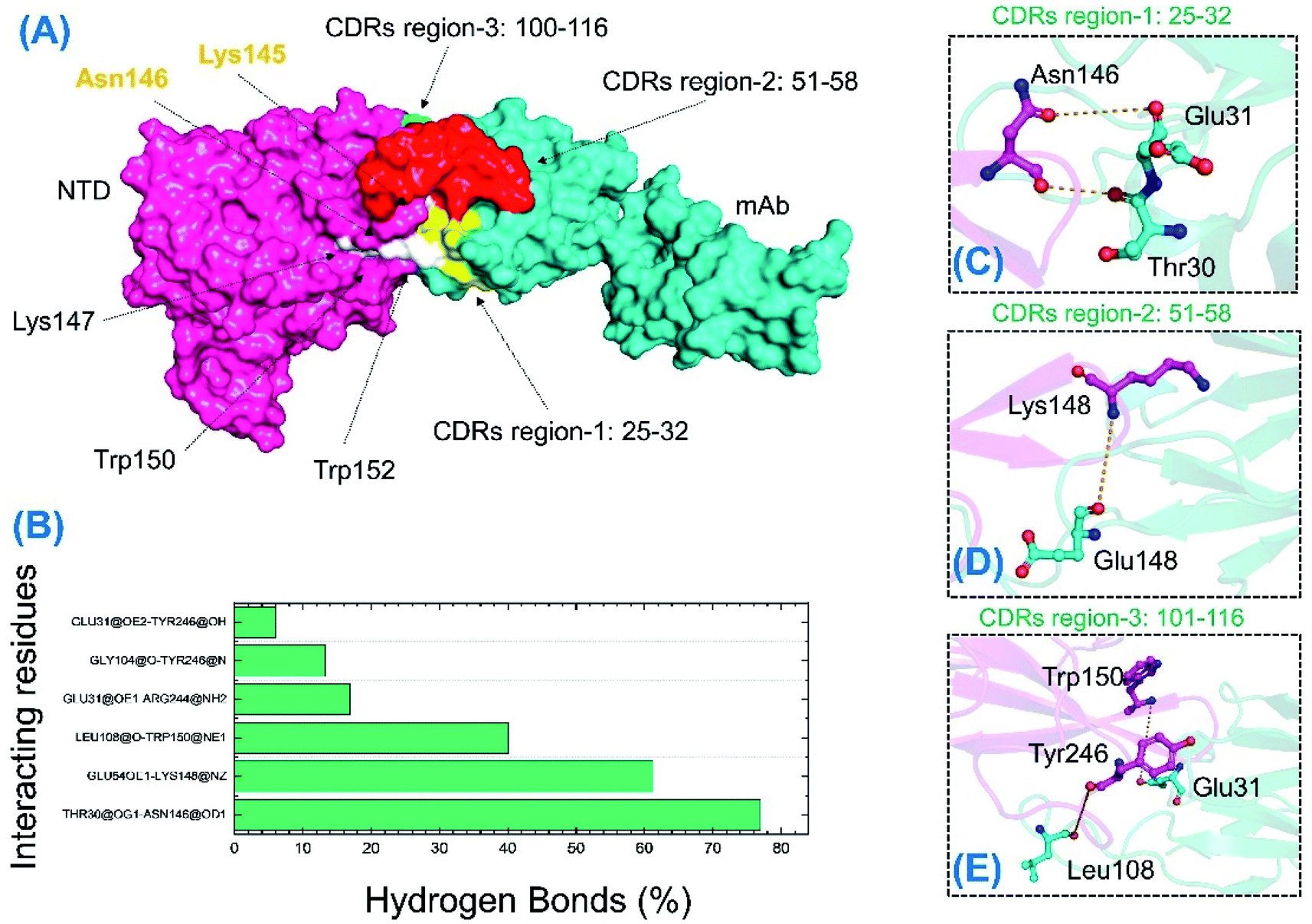
Unlike the wild-type and B.1.618 NTDs, only six intermolecular hydrogen bonds were sustained for a longer time. Among the interactions given in Table 3 and Fig. 10, the bond between Thr30 and Asn146 was sustained in 76.96% of the total simulation trajectories. Herein, the key interactions with Tyr145 and His146 are lost during the simulation. Most of the interacting residues were conserved between the wild-type-NTD and B.1.618-NTD; however, interaction with Tyr246 was observed to be present only in the B.1.618-NTD complex. This shows that the loss of key residues impacts the interaction pattern, alters the hydrogen bonding network and eventually the interaction of mAb to the NTD, which decreases the binding affinity and thus potentially escapes neutralizing antibodies.
 |
| | Fig. 10 H-bonding analysis of the 500 ns trajectory of the B.1.618-NTD. (A) The interacting hotspots. (B) The hydrogen bond percentages. (C–E) The key interactions observed to be sustained during the simulation. | |
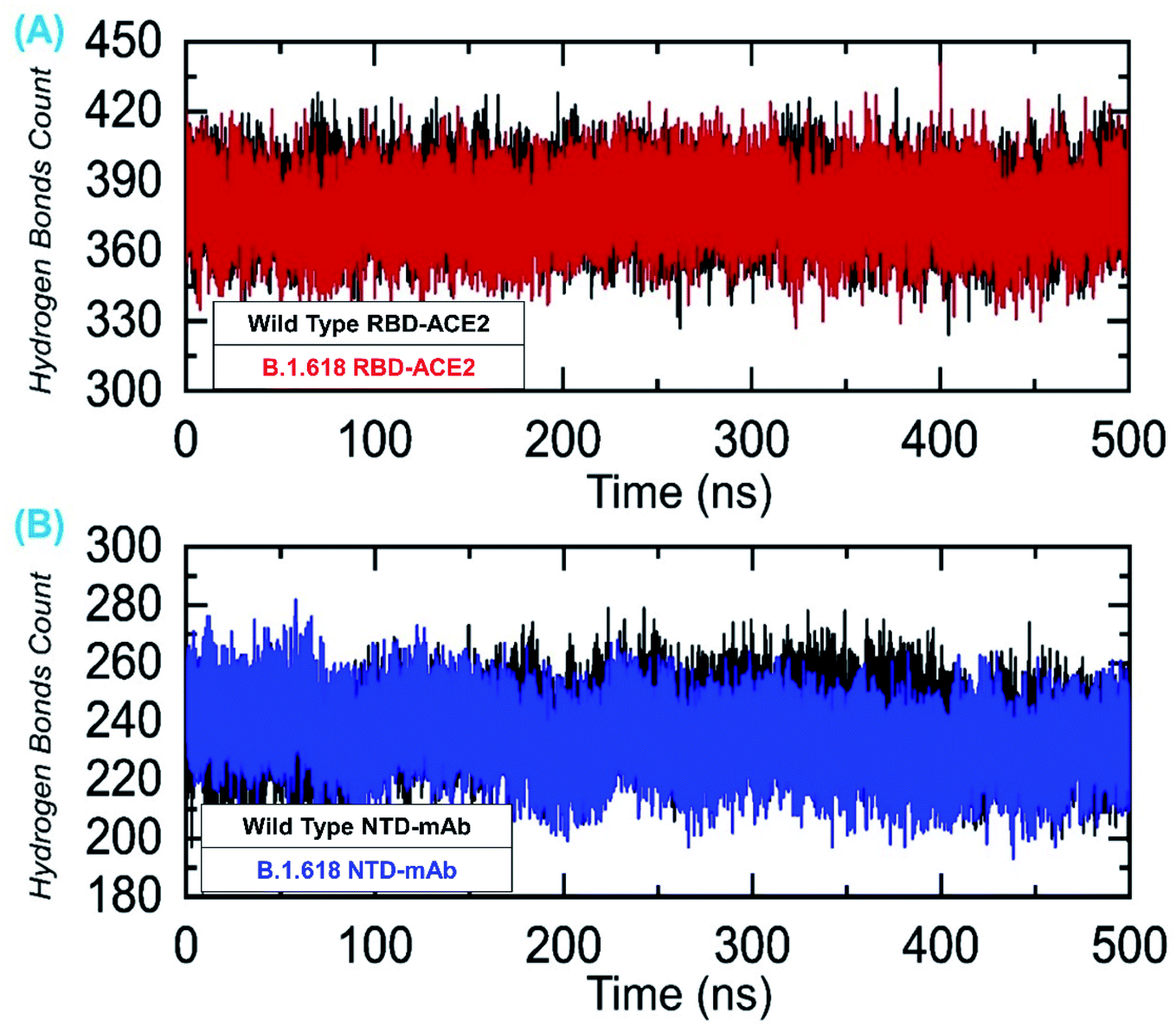
We also evaluated the total number of hydrogen bonds, both intermolecular and intramolecular, for the 500 ns simulation trajectories. The results shown in Fig. 11A represent the total number of hydrogen bonds in the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2 complexes. The average numbers of hydrogen bonds in the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2 were 388 and 391, respectively. Contrastingly, the total numbers of hydrogen bonds in the wild-type-NTD and B.1.618-NTD were 252 and 246 on average (Fig. 11B). Thus, the mutations in these variants seem to have altered their hydrogen bonding networks and they may use a different strategy for infection. All H-bond results are presented in Fig. 11.
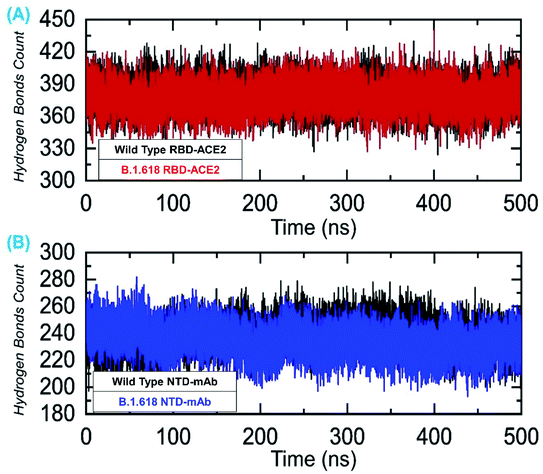 |
| | Fig. 11 Hydrogen bonding analysis of all the complexes. (A) The H-bonds for the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2 complexes and (B) the H-bonds for the wild-type-NTD and B.1.618-NTD complexes. | |
3.10 MM-GBSA binding energies
Estimating the binding affinity of a protein-small molecule or two macromolecules can help determine the strength of the biomolecular association. Recent computational estimations of binding free energy via the MM-GBSA approach are arguably the most frequently used methods by which to reevaluate docking conformations through predictions of structural-dynamic stability, the strength of interacting key hotspots, and total binding affinities. The abovementioned approach is computationally cheaper than any alternative approach, i.e., alchemical free energy estimation methods. The MM-GBSA method is regarded as more precise and accurate than the conformist scoring functions. Considering the higher applicability of this method, we estimated the effects of the reported substitution in the RBD (E484K) and the two deletions (Tyr145 and His146) in the NTD on binding to ACE2 and mAb. The different energy components estimated by the MM-GBSA method for wild and variant complexes are provided in Tables 4 and 5, respectively. In total, 25000 structural frames were used while the binding free energy was calculated at different time intervals, i.e., 0–100, 101–200, 201–300, 301–400, and 401–500 ns. Finally, the averages of each energy term were calculated and are presented in the aforementioned tables. Comparative binding analysis of the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2 revealed that the mutation E484K increases the binding affinity toward the ACE2 receptor. For the complexes of wild-type-RBD–ACE2 and B.1.618-RBD–ACE2, the vdW energies were −92.36 and −75.92 kcal mol−1, respectively, whereas the electrostatic energies were −623.28 and −885.42 kcal mol−1, respectively. These findings are consistent with those from previously reported studies, which also claimed that electrostatic interactions are the key drivers of stronger binding and increased infectivity.16,38 Furthermore, the total binding energy was also associated with the docking scores, which revealed the average binding energies as −62.43 kcal mol−1 for the wild type and −67.14 kcal mol−1 for the B.1.618-RBD–ACE2 complex. Overall, these results show that the binding affinity toward the host cellular receptor is increased, although not significantly, and that infectivity rate is changed.
Table 4 Free energy calculation (MM-GBSA) results obtained from the MD simulation trajectories of the wild type-RBD–ACE2 and B.1.618-RBD–ACE2 complexes. All the energies given here are calculated in kcal mol−1
| Complex |
MM-GBSA (wild-type-RBD–ACE2) |
| 0–100 ns |
101–200 ns |
201–300 ns |
301–400 ns |
401–500 ns |
Averages |
| vdW |
−104.28 |
−86.62 |
−90.78 |
−92.65 |
−87.51 |
−92.368 |
| Electrostatic |
−542.96 |
−593.63 |
−593.14 |
−687.7 |
−698.97 |
−623.28 |
| GB |
599.63 |
624.7 |
639.33 |
729.74 |
733.15 |
665.31 |
| SA |
−12.88 |
−11.7 |
−11.87 |
−12.21 |
−11.78 |
−12.088 |
| Total binding energy |
−60.5 |
−67.26 |
−56.45 |
−62.82 |
−65.12 |
−62.43 |
| Complex |
MM-GBSA (B.1.618-RBD–ACE2) |
| 0–100 ns |
101–200 ns |
201–300 ns |
301–400 ns |
401–500 ns |
Averages |
| vdW |
−71.19 |
−51.07 |
−84.06 |
−88.54 |
−84.78 |
−75.92 |
| Electrostatic |
−829.34 |
−839.54 |
−971.6 |
−908.54 |
−878.09 |
−885.42 |
| GB |
855.89 |
820.86 |
997.19 |
945.28 |
900.34 |
903.91 |
| SA |
−8.91 |
−6.43 |
−10.99 |
−11.69 |
−10.55 |
−9.714 |
| Total binding energy |
−53.55 |
−76.18 |
−69.45 |
−63.48 |
−73.08 |
−67.14 |
Table 5 Free energy calculation (MM-GBSA) results obtained from MD simulation trajectories of the wild-type and mutant complexes. All the energies given here are calculated in kcal mol−1
| Complex |
MM-GBSA (wild-type-NTD) |
| 0–100 ns |
101–200 ns |
201–300 ns |
301–400 ns |
401–500 ns |
Averages |
| vdW |
−69.37 |
−48.77 |
−72.76 |
−50.93 |
−79.84 |
−64.33 |
| Electrostatic |
−697.24 |
−627.88 |
−756.52 |
−993.07 |
−768.24 |
−768.59 |
| GB |
733.02 |
651.83 |
759.45 |
995.92 |
795.21 |
787.08 |
| SA |
−9.66 |
−6.52 |
−8.98 |
−8.34 |
−7.99 |
−8.29 |
| Total binding energy |
−43.25 |
−31.34 |
−78.81 |
−56.42 |
−60.86 |
−54.13 |
| Complex |
MM-GBSA (B.1.618-NTD) |
| 0–100 ns |
101–200 ns |
201–300 ns |
301–400 ns |
401–500 ns |
Averages |
| vdW |
−58.12 |
−64.10 |
−58.93 |
−56.03 |
−44.47 |
−56.33 |
| Electrostatic |
−754.45 |
−536.94 |
−843.79 |
−995.79 |
−952.23 |
−816.64 |
| GB |
772.45 |
583.94 |
845.03 |
1002.30 |
980.77 |
836.89 |
| SA |
−7.92 |
−7.74 |
−9.00 |
−8.82 |
−5.27 |
−7.75 |
| Total binding energy |
−48.04 |
−24.84 |
−66.69 |
−58.33 |
−21.20 |
−43.82 |
We also explored the binding differences between the wild-type-NTD and B.1.618-NTD using MM-GBSA. The aforementioned time intervals were used to compute the binding energy at different times and then the averages were estimated for each complex. As shown in Table 5, the data suggest that the binding affinity of the B.1.618-NTD was substantially decreased. For the wild-type-NTD, the average vdW and electrostatic energies were −64.33 and −768.59 kcal mol−1, respectively; for the B.1.618-NTD, these respective values were −56.33 and −816.44 kcal mol−1. Interestingly, differences in the total binding energy were higher than the RBD domain interaction energy. The average total binding energy for the wild-type-NTD was −54.13 kcal mol−1, whereas for the B.1.618-NTD it was −43.82 kcal mol−1. Consequently, the binding of mAb to the new variant was reduced; this explains how the B.1.618-NTD variant is an antibody-escaping variant.
4. Conclusions
In conclusion, using macromolecular docking and biomolecular approaches, we discovered that the new SARS-CoV-2 variant B.1.618 is an antibody-escaping variant. Via deep-bonding network analysis, we revealed that the hydrogen bonding network between the wild type and mutant (both RBD and NTD) is reprogrammed and that structural-dynamics features exhibit significant variations. We also found that the flexibility of three important loops and the increment in electrostatic energy in the RBD are the primary determinants of the observed variations between the wild-type-RBD–ACE2 and B.1.618-RBD–ACE2. In an analysis of the NTD, the binding of mAb was found to be reduced drastically by two deletions, and the hydrogen bonding network was also altered. As indicated by our data, the B.1.618 variant slightly alters the binding affinity to the host while potentially emerging as an antibody-escaping variant.
Data availability
All the data is available on RCSB and UniProt, and any simulation data will be provided on demand.
Funding
Dong-Qing Wei is supported by grants from the Key Research Area Grant 2016YFA0501703 of the Ministry of Science and Technology of China, the National Science Foundation of China (Grant No. 32070662, 61832019, 32030063), the Science and Technology Commission of Shanghai Municipality (Grant No.: 19430750600), the Natural Science Foundation of Henan Province (162300410060), as well as SJTU JiRLMDS Joint Research Fund and Joint Research Funds for Medical and Engineering and Scientific Research at Shanghai Jiao Tong University (YG2017ZD14). The computations were partially performed at the Pengcheng Lab and the Center for High-Performance Computing, Shanghai Jiao Tong University.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The computations were partially performed at the Center for High-Performance Computing, Shanghai Jiao Tong University. We acknowledge their help.
References
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian and Y.-Y. Pei, Nature, 2020, 579, 265–269 CrossRef CAS PubMed.
- N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang, J. Song, X. Zhao, B. Huang, W. Shi and R. Lu, N. Engl. J. Med., 2020, 727–733 CrossRef CAS PubMed.
- W.-j. Guan, Z.-y. Ni, Y. Hu, W.-h. Liang, C.-q. Ou, J.-x. He, L. Liu, H. Shan, C.-l. Lei and D. S. Hui, medRxiv, 2020, 1–30 Search PubMed.
- C. Huang, Y. Wang, X. Li, L. Ren, J. Zhao, Y. Hu, L. Zhang, G. Fan, J. Xu and X. Gu, Lancet, 2020, 395, 497–506 CrossRef CAS.
- S. R. Weiss and J. L. Leibowitz, in Advances in virus research, Elsevier, 2011, vol. 81, pp. 85–164 Search PubMed.
- Z. Zhu, X. Lian, X. Su, W. Wu, G. A. Marraro and Y. Zeng, Respir. Res., 2020, 21, 1–14 CrossRef PubMed.
- Z. Song, Y. Xu, L. Bao, L. Zhang, P. Yu, Y. Qu, H. Zhu, W. Zhao, Y. Han and C. Qin, Viruses, 2019, 11, 59 CrossRef CAS PubMed.
- T. Kirby, Lancet Respir. Med., 2021, 9, e20–e21 CrossRef CAS PubMed.
- S. M. Haque, O. Ashwaq, A. Sarief and A. K. Azad John Mohamed, Future Virol., 2020, 15, 625–648 CrossRef CAS PubMed.
- W. Li, M. J. Moore, N. Vasilieva, J. Sui, S. K. Wong, M. A. Berne, M. Somasundaran, J. L. Sullivan, K. Luzuriaga, T. C. Greenough, H. Choe and M. Farzan, Nature, 2003, 426, 450–454 CrossRef CAS PubMed.
- S. F. Ahmed, A. A. Quadeer and M. R. McKay, Viruses, 2020, 12, 254 CrossRef CAS PubMed.
- F. Li, J. Virol., 2015, 89, 1954–1964 CrossRef PubMed.
- A. Kawana, Jpn. J. Clin. Med., 2016, 74, 1967–1972 Search PubMed.
- S. Günther, P. Y. A. Reinke, Y. Fernández-García, J. Lieske, T. J. Lane, H. M. Ginn, F. H. M. Koua, C. Ehrt, W. Ewert, D. Oberthuer, O. Yefanov, S. Meier, K. Lorenzen, B. Krichel, J.-D. Kopicki, L. Gelisio, W. Brehm, I. Dunkel, B. Seychell, H. Gieseler, B. Norton-Baker, B. Escudero-Pérez, M. Domaracky, S. Saouane, A. Tolstikova, T. A. White, A. Hänle, M. Groessler, H. Fleckenstein, F. Trost, M. Galchenkova, Y. Gevorkov, C. Li, S. Awel, A. Peck, M. Barthelmess, F. Schluenzen, P. Lourdu Xavier, N. Werner, H. Andaleeb, N. Ullah, S. Falke, V. Srinivasan, B. A. França, M. Schwinzer, H. Brognaro, C. Rogers, D. Melo, J. J. Zaitseva-Doyle, J. Knoska, G. E. Peña-Murillo, A. R. Mashhour, V. Hennicke, P. Fischer, J. Hakanpää, J. Meyer, P. Gribbon, B. Ellinger, M. Kuzikov, M. Wolf, A. R. Beccari, G. Bourenkov, D. von Stetten, G. Pompidor, I. Bento, S. Panneerselvam, I. Karpics, T. R. Schneider, M. M. Garcia-Alai, S. Niebling, C. Günther, C. Schmidt, R. Schubert, H. Han, J. Boger, D. C. F. Monteiro, L. Zhang, X. Sun, J. Pletzer-Zelgert, J. Wollenhaupt, C. G. Feiler, M. S. Weiss, E.-C. Schulz, P. Mehrabi, K. Karničar, A. Usenik, J. Loboda, H. Tidow, A. Chari, R. Hilgenfeld, C. Uetrecht, R. Cox, A. Zaliani, T. Beck, M. Rarey, S. Günther, D. Turk, W. Hinrichs, H. N. Chapman, A. R. Pearson, C. Betzel and A. Meents, Science, 2021, eabf7945, DOI:10.1126/science.abf7945.
- A. Khan, S. S. Ali, M. T. Khan, S. Saleem, A. Ali, M. Suleman, Z. Babar, A. Shafiq, M. Khan and D.-Q. Wei, J. Biomol. Struct. Dyn., 2020, 1–12 Search PubMed.
- I. Hussain, N. Pervaiz, A. Khan, S. Saleem, H. Shireen, D.-Q. Wei, V. Labrie, Y. Bao and A. A. Abbasi, Genes Immun., 2020, 1–11 CAS.
- A. Khan, M. Khan, S. Saleem, Z. Babar, A. Ali, A. A. Khan, Z. Sardar, F. Hamayun, S. S. Ali and D.-Q. Wei, Interdiscip. Sci.: Comput. Life Sci., 2020, 12, 335–348 CrossRef CAS PubMed.
- S. Kang, M. Yang, Z. Hong, L. Zhang, Z. Huang, X. Chen, S. He, Z. Zhou, Z. Zhou and Q. Chen, Acta Pharm. Sin. B, 2020, 10, 1228–1238 CrossRef CAS PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181, 281–292.e286 CrossRef CAS PubMed.
- L. Yurkovetskiy, X. Wang, K. E. Pascal, C. Tomkins-Tinch, T. P. Nyalile, Y. Wang, A. Baum, W. E. Diehl, A. Dauphin and C. Carbone, Cell, 2020, 183, 739–751.e738 CrossRef CAS PubMed.
- Y. Cai, J. Zhang, T. Xiao, H. Peng, S. M. Sterling, R. M. Walsh, S. Rawson, S. Rits-Volloch and B. Chen, Science, 2020, 369, 1586–1592 CrossRef CAS PubMed.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang and L. Zhang, Nature, 2020, 581, 215–220 CrossRef CAS PubMed.
- Y. Huang, C. Yang, X.-f. Xu, W. Xu and S.-w. Liu, Acta Pharmacol. Sin., 2020, 41, 1141–1149 CrossRef PubMed.
- X. Chi, R. Yan, J. Zhang, G. Zhang, Y. Zhang, M. Hao, Z. Zhang, P. Fan, Y. Dong and Y. Yang, Science, 2020, 369, 650–655 CrossRef CAS PubMed.
- A. Khan, S. Khan, S. Saleem, N. Nizam-Uddin, A. Mohammad, T. Khan, S. Ahmad, M. Arshad, S. S. Ali and M. Suleman, Comput. Biol. Med., 2021, 104420 CrossRef CAS PubMed.
- S. S. Abdool Karim and T. de Oliveira, N. Engl. J. Med., 2021, 1866–1868 CrossRef PubMed.
- Y. Dehouck, J. M. Kwasigroch, M. Rooman and D. Gilis, Nucleic Acids Res., 2013, 41, W333–W339 CrossRef PubMed.
- C. H. Rodrigues, Y. Myung, D. E. Pires and D. B. Ascher, Nucleic Acids Res., 2019, 47, W338–W344 CrossRef CAS PubMed.
- C. H. Rodrigues, D. E. Pires and D. B. Ascher, Protein Sci., 2021, 30, 60–69 CrossRef CAS PubMed.
- C. Dominguez, R. Boelens and A. M. Bonvin, J. Am. Chem. Soc., 2003, 125, 1731–1737 CrossRef CAS PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 CAS.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- R. Zwanzig, J. Stat. Phys., 1973, 9, 215–220 CrossRef.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69–82 CrossRef CAS PubMed.
- M. Ylilauri and O. T. Pentikäinen, J. Chem. Inf. Model., 2013, 53, 2626–2633 CrossRef CAS PubMed.
- A. Khan, T. Zia, M. Suleman, T. Khan, S. S. Ali, A. A. Abbasi, A. Mohammad and D.-Q. Wei, J. Cell. Physiol., 2021, 7045–7057 CrossRef CAS PubMed.
- A. Khan, W. Heng, Y. Wang, J. Qiu, X. Wei, S. Peng, S. Saleem, M. Khan, S. S. Ali and D.-Q. Wei, Phytother. Res., 2021, 1–5 CAS.
- A. Khan, M. Khan, S. Saleem, Z. Babar, A. Ali, A. A. Khan, Z. Sardar, F. Hamayun, S. S. Ali and D.-Q. Wei, Interdiscip. Sci.: Comput. Life Sci., 2020, 1–14 Search PubMed.
- A. Khan, M. T. Khan, S. Saleem, M. Junaid, A. Ali, S. S. Ali, M. Khan and D.-Q. Wei, Comput. Struct. Biotechnol. J., 2020, 2174–2184 CrossRef CAS PubMed.
- A. Prerna, A. Kempf, I. Nehlmeier, A. Sidarovich, N. Krüger, L. Graichen and A.-S. Moldenhauer, et al., bioRxiv, 2021 Search PubMed.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2016, 54, 5.6.1–5.6.37 Search PubMed.
- T. N. Starr, A. J. Greaney, S. K. Hilton, D. Ellis, K. H. Crawford, A. S. Dingens, M. J. Navarro, J. E. Bowen, M. A. Tortorici and A. C. Walls, Cell, 2020, 182, 1295–1310.e1220 CrossRef CAS PubMed.
- A. Ali and R. Vijayan, Sci. Rep., 2020, 10, 1–12 CrossRef PubMed.
- A. Khan, D.-Q. Wei, K. Kousar, J. Abubaker, S. Ahmad, J. Ali, F. Al-Mulla, S. S. Ali, N. Nizam-Uddin and A. M. Sayaf, ChemBioChem, 2021, 2641–2649 CrossRef CAS PubMed.
- Q. Wang, Y. Zhang, L. Wu, S. Niu, C. Song, Z. Zhang, G. Lu, C. Qiao, Y. Hu and K.-Y. Yuen, Cell, 2020 Search PubMed.
- J. P. Landry, Y. Fei and X. Zhu, Assay Drug Dev. Technol., 2012, 10, 250–259 CrossRef CAS PubMed.
- J. P. Landry, Y. Fei and X. Zhu, Int. Drug Discovery, 2011, 8 Search PubMed.
- J. Landry, Y. Sun, X. Guo and X. Zhu, Appl. Opt., 2008, 47, 3275–3288 CrossRef CAS PubMed.
- X. Tian, C. Li, A. Huang, S. Xia, S. Lu, Z. Shi, L. Lu, S. Jiang, Z. Yang and Y. Wu, Emerging Microbes Infect., 2020, 9, 382–385 CrossRef CAS PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 183(6), 1735 CrossRef CAS PubMed.
- J. L. Bernal, N. Andrews, C. Gower, E. Gallagher, R. Simmons, S. Thelwall, J. Stowe, E. Tessier, N. Groves, G. Dabrera, R. Myers, C. Campbell, G. Amirthalingam, M. Edmunds, M. Zambon, K. Brown, S. Hopkins, M. Chand and M. Ramsay, N. Engl. J. Med., 2021, 385(no. 7), 585–594 CrossRef CAS PubMed.
- T. M. Davenport, J. Gorman, M. G. Joyce, T. Zhou, C. Soto, M. Guttman, S. Moquin, Y. Yang, B. Zhang, N. A. Doria-Rose, S.-L. Hu, J. R. Mascola, P. D. Kwong and K. K. Lee, Structure, 2016, 24, 1346–1357 CrossRef CAS PubMed.
- V. Ovchinnikov, J. E. Louveau, J. P. Barton, M. Karplus and A. K. Chakraborty, eLife, 2018, 7, e33038 CrossRef PubMed.
- D. Chen, N. Oezguen, P. Urvil, C. Ferguson, S. M. Dann and T. C. Savidge, Sci. Adv., 2016, 2(3), e1501240 CrossRef PubMed.
- J. D. Chodera and D. L. Mobley, Annu. Rev. Biophys., 2013, 42, 121–142 CrossRef CAS PubMed.
- R. Patil, S. Das, A. Stanley, L. Yadav, A. Sudhakar and A. K. Varma, PLoS One, 2010, 5, e12029 CrossRef PubMed.
- T. S. Olsson, J. E. Ladbury, W. R. Pitt and M. A. Williams, Protein Sci., 2011, 20, 1607–1618 CrossRef CAS PubMed.
- J. de Andrade, P. F. B. Gonçalves and P. A. Netz, ChemBioChem, 2021, 22, 865–875 CrossRef CAS PubMed.
- A. Malik, D. Prahlad, N. Kulkarni and A. Kayal, bioRxiv, 2020 DOI:10.1101/2020.06.15.152892.
- A. Ali and R. Vijayan, Dynamics of the ACE2-SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms, Nature Publishing Group, 2020 Search PubMed.
Footnote |
| † Abbas Khan and Jianjun Gui contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jianjun Gui†b,
Waqar Ahmadc,
Inamul Haq
a,
Jianjun Gui†b,
Waqar Ahmadc,
Inamul Haq