
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of a mononuclear zinc(II) Schiff base complex: on the importance of C–H⋯π interactions†
Tanmoy Basaka,
Antonio Frontera b and
Shouvik Chattopadhyay*a
b and
Shouvik Chattopadhyay*a
aDepartment of Chemistry, Inorganic Section, Jadavpur University, Kolkata-700032, India. E-mail: shouvik.chem@gmail.com
bDepartamento de Química, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma, Baleares, Spain
First published on 9th September 2021
Abstract
A zinc(II) complex, [ZnL(H2O)]·H2O {H2L = 2,2′-[(2,2-dimethyl-1,3-propanediyl)bis(nitrilomethylidyne)]bis[6-ethoxyphenol]} has been synthesized and characterized by UV-vis and IR spectroscopy. The structure of the complex has been confirmed by X-ray crystallography and the noncovalent interactions characterized using Hirshfeld surface analysis. In addition to the conventional H-bonds involving the Zn-coordinated and non-coordinated water molecules, interesting C–H⋯π interactions between the H-atoms belonging to aliphatic part of the ligand (2,2-dimethyl-1,3-propanediyl) and the Zn-coordinated aromatic rings are established. These interactions have been studied using DFT calculations (PBE0-D3/def2-TZVP) and characterized using molecular electrostatic potential (MEP) surfaces and the noncovalent interaction (NCI) plot index analyses.
Introduction
Zinc(II) complexes have lots of important applications as useful electronic and opto-electronic materials.1–5 Many zinc(II) complexes have also been prepared recently to explore their potential use in photonic devices.6 The importance of zinc(II) in biology is well known. Several zinc(II) complexes have been shown to have the ability to mimic the active sites of many zinc metalloenzymes.7–10 Many zinc(II) complexes are shown to have the ability to degrade organic dyes in aqueous solution.11–13 To show strong fluorescence is a typical behaviour of any zinc(II) complex, and the fluorescence intensity usually decreases in presence of different nitroaromatic compounds.14–17 So, many zinc(II) complexes have been used for the detection of nitroaromatic explosives by turn off fluorescence response.18–20 The d10 electronic configuration of zinc(II) also favours a variety of coordination geometries, e.g. tetrahedral,21 square pyramidal,22 octahedral,23 trigonal prism,24 trigonal dodecahedral25 etc. Obviously, tetrahedral is by far the most common geometry of zinc(II). These complexes show varieties of molecular and supra-molecular architectures.26–30A variety of supramolecular interactions are responsible for the crystal packing of coordination compounds, leading to a broad range of dimensional frameworks (0D to 3D) through a variety of forces.31,32 Apart from coordination and hydrogen bonds,33 forces associated with aromatic π-systems like π-stacking, lone pair⋯π, ion⋯π and C–H⋯π are important guiding the formation of supramolecular polymeric architectures in solid state.34 In particular, the C–H⋯π interaction is a weak attraction between a C–H bond and a π-system (aromatic, double or triple bond), and its roles and significance in chemistry and biochemistry have been widely discussed.35,36
Salen type N2O2 donor Schiff bases have been widely used to synthesize many zinc(II) complexes.37–40 This manuscript reports the synthesis and X-ray characterization and Hirshfeld surface analysis of a new Zn(II) complex, [ZnL(H2O)]·H2O with a salen type Schiff base (H2L). The structure directing role of C–H⋯π interactions between the H-atoms belonging to aliphatic part of the ligand (2,2-dimethyl-1,3-propanediyl) and the Zn-coordinated aromatic rings has been studied energetically using DFT calculations at the PBE0-D3/def2-TZVP level of theory. These interactions have been further rationalized using molecular electrostatic potential (MEP) surfaces and characterized using the noncovalent interaction (NCI) plot index analysis.
Experimental section
3-Ethoxysalicylaldehyde, 2,2-dimethylpropane-1,3-diamine, methanol, zinc(II) acetate dihydrate and DMF were commercially available, reagent grade, and used as purchased from Sigma-Aldrich without further purification.Preparation of ligand
Preparation of complex
Yield: 0.347 g (∼70%) based on zinc(II). Colour: yellow; anal. calc. for C23H32N2O6Zn (FW 497.88): C, 55.48; H, 6.48; N, 5.63; found: C, 55.44; H, 6.34; N, 5.73%. IR (KBr, cm−1): 1625 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 2840–2960 (νC–H), 3351 (νO–H). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (DMF): 232 (6.06 × 103), 272 (2.4 × 103), 356 (7.8 × 102); 1H NMR (DMSO-d6) δ ppm: 8.12 (s, 2H, –CHN), 6.73 (d, 4H, J = 7.8 Hz, Ar-H); 6.26 (t, 2H, J = 7.7 Hz, Ar-H); 3.98 (q, 4H, J = 7.0 Hz, –CH2); 3.44 (s, 2H, –CH2); 1.23 (t, 6H, J = 7.0, –CH3); 0.87 (s, 6H, –CH3).
N), 2840–2960 (νC–H), 3351 (νO–H). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (DMF): 232 (6.06 × 103), 272 (2.4 × 103), 356 (7.8 × 102); 1H NMR (DMSO-d6) δ ppm: 8.12 (s, 2H, –CHN), 6.73 (d, 4H, J = 7.8 Hz, Ar-H); 6.26 (t, 2H, J = 7.7 Hz, Ar-H); 3.98 (q, 4H, J = 7.0 Hz, –CH2); 3.44 (s, 2H, –CH2); 1.23 (t, 6H, J = 7.0, –CH3); 0.87 (s, 6H, –CH3).
Physical measurement
Elemental analyses (carbon, hydrogen and nitrogen) were performed using a PerkinElmer 240C elemental analyzer. IR spectra in KBr (4500 to 500 cm−1) were recorded with a PerkinElmer Spectrum Two spectrophotometer. Electronic spectra in DMF were recorded on a Shimadzu UV-1700 UV-vis spectrophotometer. The 1H NMR spectra at 400 MHz were recorded in DMSO-d6 on a JEOL-JNM-ECZ400 S/L1.X-ray crystallography
Suitable crystals of the complex was used for data collection using a ‘Bruker D8 QUEST area detector’ diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The molecular structures were solved by direct method and refined by full-matrix least squares on F2 using the SHELX-18 package.41 Non-hydrogen atoms were refined with anisotropic thermal parameters. The hydrogen atoms attached to oxygen atoms of coordinated water molecules were located by difference Fourier maps and were kept at fixed positions. All other hydrogen atoms were placed in their geometrically idealized positions and constrained to ride on their parent atoms. Multi-scan empirical absorption corrections were applied to the data using the program SADABS.42 The hydrogen atoms attached with the oxygen atoms of lattice water molecules could not be fixed. Crystallographic data and refinement details of the complex are given in Table 1.| Formula | C23H30N2O6Zn |
|---|---|
| Formula weight | 495.88 |
| Temperature (K) | 273 |
| Crystal system | Orthorhombic |
| Space group | Aba2 |
| a (Å) | 26.205(11) |
| b (Å) | 25.903(11) |
| c (Å) | 14.652(6) |
| Z | 16 |
| dcal (g cm−3) | 1.325 |
| μ (mm−1) | 1.026 |
| F(000) | 4160 |
| Total reflection | 166![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 664 664 |
| Unique reflections | 10962 |
| Observe data [I > 2σ(I)] | 9714 |
| R(int) | 0.091 |
| R1, wR2 (all data) | 0.0564, 0.1327 |
| R1, wR2 [I > 2σ(I)] | 0.0450, 0.1204 |
Hirshfeld surface analysis
Hirshfeld surfaces43–45 and the associated two-dimensional (2D) fingerprint46–48 plots were calculated using Crystal Explorer,49 with bond lengths to hydrogen atoms set to standard values.50 Two distances, de (the distance from the point to the nearest nucleus external to the surface) and di (the distance to the nearest nucleus internal to the surface), are defined for each point on the Hirshfeld surface. The normalized contact distance (dnorm) based on de and di is defined as:where rvdwi and rvdwe are the van der Waals radii of the atoms. The dnorm value is negative or positive depending on intermolecular contacts being shorter or longer than the van der Waals separations. The parameter dnorm displays a surface with a red-white-blue colour design, where bright red spots highlight shorter contacts, white areas in the same surface represent contacts around the van der Waals separation and blue regions are devoid of close contacts. For a given CIF, the Hirshfeld surface is said to be unique.51
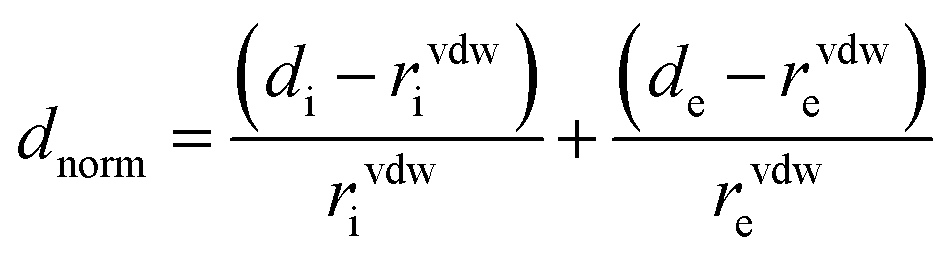
Theoretical methods
The energies of the assemblies studied herein were computed at the PBE0 (ref. 52)-D3 (ref. 53)/def2-TZVP54 level of theory using the crystallographic coordinates and the Gaussian-16 program.55 Grimme's D3 dispersion53 correction has been used since it is convenient for the correct evaluation of non-covalent interactions where dispersion effects are important like C–H⋯π interactions. The basis set superposition error for the calculation of interaction energies has been corrected using the counterpoise method.56 The MEP surfaces have been computed using the same level of theory and plotted using the 0.001 a.u. isosurface. The NCI plot57 isosurfaces have been used to characterize non-covalent interactions. They correspond to both favorable and unfavorable interactions, as differentiated by the sign of the second density Hessian eigenvalue and defined by the isosurface color. The color scheme is a red-yellow-green-blue scale with red for ρcut+ (repulsive) and blue for ρcut− (attractive).Results and discussion
Synthesis
In the present work, a Schiff base ligand, H2L, was synthesized by the condensation of 2,2-dimethyl-1,3-diaminopropane with 3-ethoxysalicylaldehyde.58 The ligand was not purified and used directly for preparation of the zinc(II) complex. The reaction of the ligand, H2L, with zinc(II) acetate in 1:1 ratio results the complex. The formation of the complex is shown in Scheme 1.
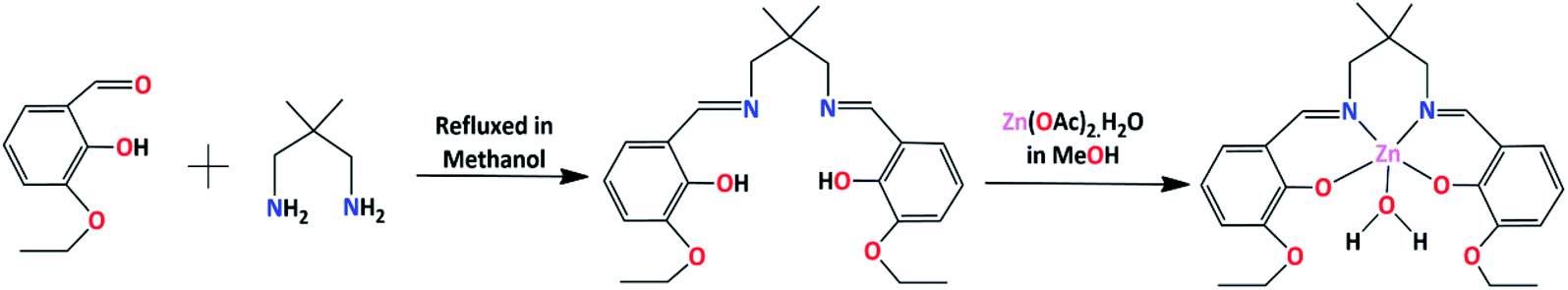 | ||
| Scheme 1 Synthetic route to the complex. Solvent water molecule has not been shown. | ||
Description of structures
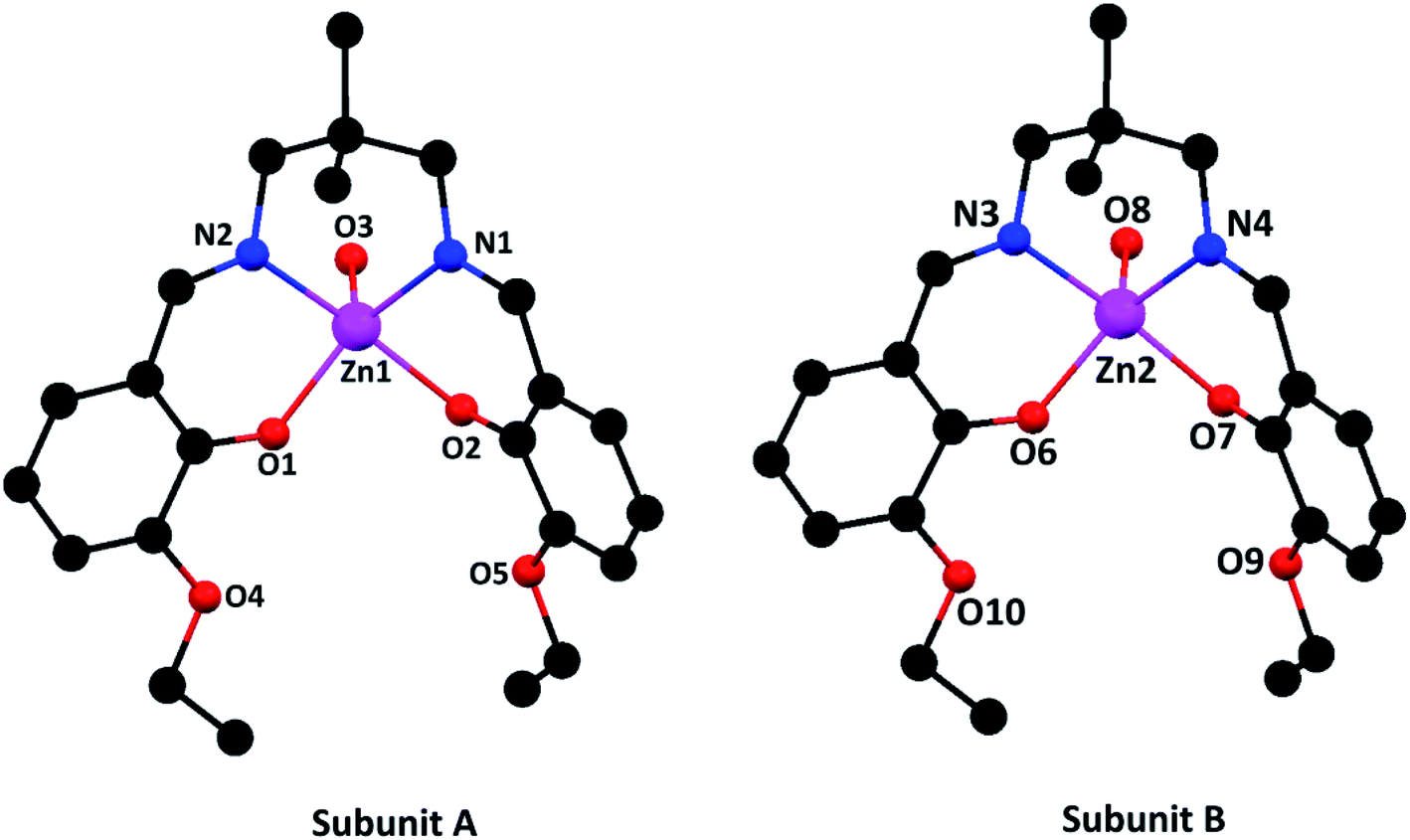 | ||
| Fig. 1 Perspective views of the complex with selective atom numbering scheme. Hydrogen atoms and solvent molecules have been omitted for clarity. | ||
In subunit A, the square pyramidal zinc(II) center, Zn(1), resides in the inner N2O2 compartment of the di-anion of the compartmental Schiff base ligand, L2−. Two imine nitrogen atoms, N(1) and N(2), and two phenoxo oxygen atoms, O(1) and O(2), of the deprotonated di-Schiff base constitute the equatorial plane. An oxygen atom, O(3), from a water molecule is coordinated in the apical position. Deviations of the coordinating atoms O(1), O(2), N(1), N(2) from the least-square mean planes through them are 0.219(4), −0.219(4), 0.211(4), 0.211(5) Å, respectively. As is usual for square pyramid structures, the zinc(II) is displaced 0.464 Å from the plane towards the apically coordinated oxygen (O3) of the water molecule.
In case of subunit B, the zinc(II) center, Zn(2), shows similar coordination mode, in which two imine nitrogen atoms, N(3) and N(4), and two phenoxo oxygen atoms, O(6) and O(7), of the deprotonated di-Schiff base constitute the equatorial plane and an oxygen atom, O(8), from a water molecule is coordinated in the apical position. Deviations of the coordinating atoms O(6), O(7), N(3) and N(4) from the least-square mean planes through them are 0.201, −0.200, −0.194, 0.193 Å, respectively. Here also the zinc(II) is displaced 0.462 Å from the plane towards the apically coordinated oxygen (O8) of the water molecule.
The selected bond lengths and bond angles of the complex are given in Tables 2 and 3 respectively. It is found that Zn(II)–O and Zn(II)–N distances vary from 1.998(4) to 2.037(4) Å and 2.105(5) to 2.121(4) Å, respectively. The bond lengths and angles are comparable with those for the related zinc(II) complexes of the salen type di-Schiff base ligands and are close to the expected values.7–9,15,16,37–39,58,60 The saturated six membered chelate rings, Zn(1)–N(1)–C(12)–C(11)–C(10)–N(2) {for subunit A} and Zn(1)–N(3)–C(33)–C(35)–C(38)–N(4) {for subunit B}, assume chair conformations with puckering parameters,61 q = 0.605(5) Å, θ = 6.3(5)°, φ = 138(4)° and q = 0.603(5) Å, θ = 6.8(5)°, φ = 277(4)°, respectively.
| Zn(1)–O(1) | 2.037(4) | Zn(2)–O(6) | 2.029(4) |
| Zn(1)–O(2) | 1.998(4) | Zn(2)–O(7) | 2.007(4) |
| Zn(1)–O(3) | 2.005(4) | Zn(2)–O(8) | 2.006(4) |
| Zn(1)–N(1) | 2.107(4) | Zn(2)–N(3) | 2.121(4) |
| Zn(1)–N(2) | 2.113(5) | Zn(2)–N(4) | 2.105(5) |
| O(1)–Zn(1)–O(2) | 88.38(17) | O(6)–Zn(2)–O(7) | 89.06(17) |
| O(1)–Zn(1)–O(3) | 96.98(16) | O(6)–Zn(2)–O(8) | 97.37(17) |
| O(1)–Zn(1)–N(1) | 166.15(16) | O(6)–Zn(2)–N(3) | 86.13(16) |
| O(1)–Zn(1)–N(2) | 86.75(17) | O(6)–Zn(2)–N(4) | 165.19(17) |
| O(2)–Zn(1)–O(3) | 112.27(18) | O(7)–Zn(2)–O(8) | 110.56(17) |
| O(2)–Zn(1)–N(1) | 88.15(17) | O(7)–Zn(2)–N(3) | 142.75(17) |
| O(2)–Zn(1)–N(2) | 141.40(18) | O(7)–Zn(2)–N(4) | 87.73(17) |
| O(3)–Zn(1)–N(1) | 96.75(17) | O(8)–Zn(2)–N(3) | 106.68(18) |
| O(3)–Zn(1)–N(2) | 106.33(18) | O(8)–Zn(2)–N(4) | 97.30(18) |
| N(1)–Zn(1)–N(2) | 87.62(17) | N(3)–Zn(2)–N(4) | 87.69(17) |
Hirshfeld surfaces analysis
The Hirshfeld surfaces of the complex is mapped over dnorm (range −0.1 Å to 1.5 Å), shape index and curvedness (Fig. 2). Red spots on the Hirshfeld surfaces mapped with dnorm denote the dominant interactions. Shape index can be used to identify complementary hollows (red) and bumps (blue) where two molecular surfaces touch one another. Curvedness is a function of the root-mean-square curvature of the surface and maps of curvedness typically show large regions of green (relatively flat) separated by dark blue edges (large positive curvature).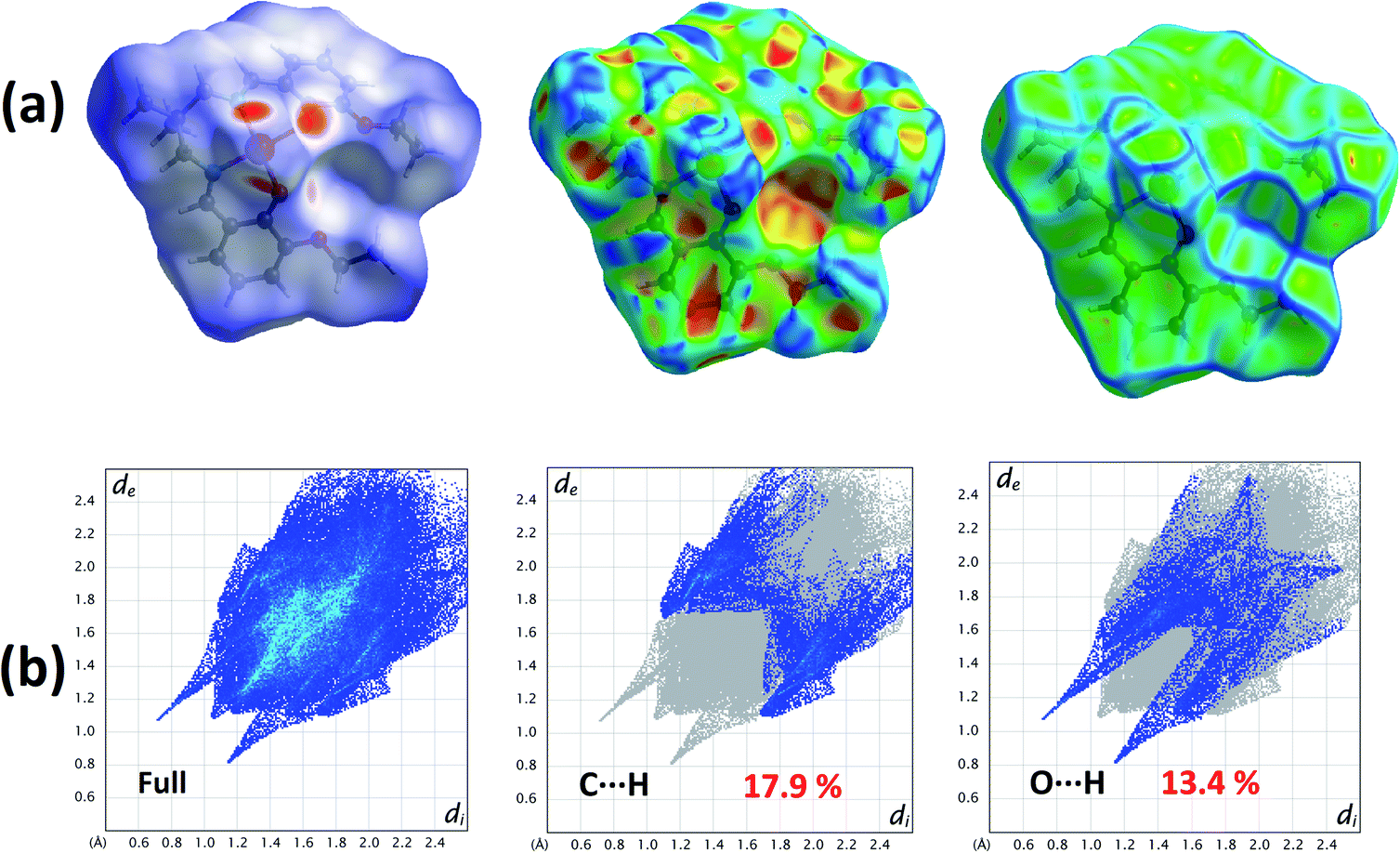 | ||
| Fig. 2 (a) Hirshfeld surfaces of the complex mapped with dnorm (left), shape index (middle) and curvedness (right); (b) the proportion of O⋯H/H⋯O interactions comprise 13.4% of the Hirshfeld surfaces and the proportion of C⋯H/H⋯C interactions comprise 17.9% of the Hirshfeld surfaces. | ||
The intermolecular interactions appear as distinct spikes in the 2D fingerprint plot (Fig. 2). The main interactions are observed between the oxygen and hydrogen atoms (O⋯H). Other visible spots in Hirshfeld surfaces correspond to C⋯H contacts.
IR and electronic spectra
In the IR spectrum of the complex (Fig. 3), a distinct band appears around 1625 cm−1 due to stretching vibrations of azomethine (CN) groups.62 The bands in the range of 2840–2960 cm−1 due to alkyl C–H bond stretching vibrations are customarily noticed in the IR spectrum of the complex.63 A distinct band is observed at 3351 cm−1 due to O–H stretching vibrations of coordinating water molecule and lattice water molecule.64
Electronic spectrum of the complex has been recorded in DMF medium. There are only three bands in the UV-vis spectrum of the complex. Bands at 232 nm and 272 nm may be attributed as π → π* transitions.65–67 The band at 356 nm may be attributed to the n → π* charge transfer transition.68,69 There is no band corresponding to d–d electronic transitions, as expected for zinc(II) complexes with d10 electronic configuration.55
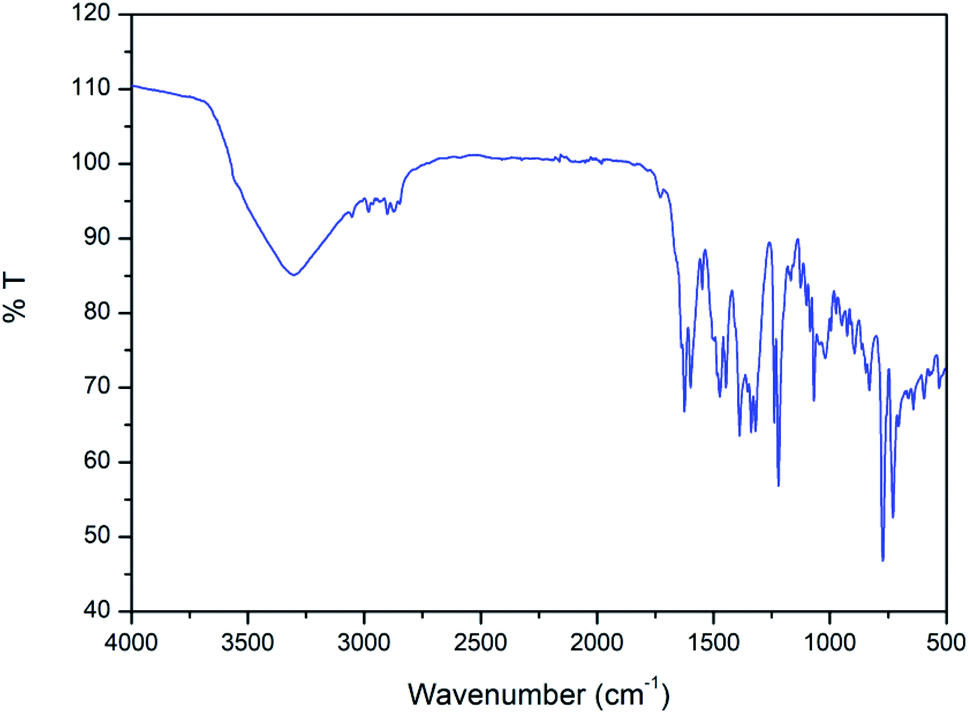 | ||
| Fig. 3 IR spectrum of the complex. | ||
1H NMR spectrum
1H NMR Spectra of complex is shown in Fig. 4. Imine protons (–CHN) of the complex appeared as a singlet at 8.12 ppm. Aromatic protons of the complex have been noticed in the range of 6.24–6.74 ppm. The methylene protons of diamine moiety of the complex appeared as singlet at 3.46 ppm another methylene protons of ethoxy group appeared as quartet in the range of 3.96–4.01 ppm. Methyl protons of ethoxy group appeared as triplet in the range of 1.21–1.24 ppm and another methyl protons of diamine appeared as singlet at 0.87 ppm.
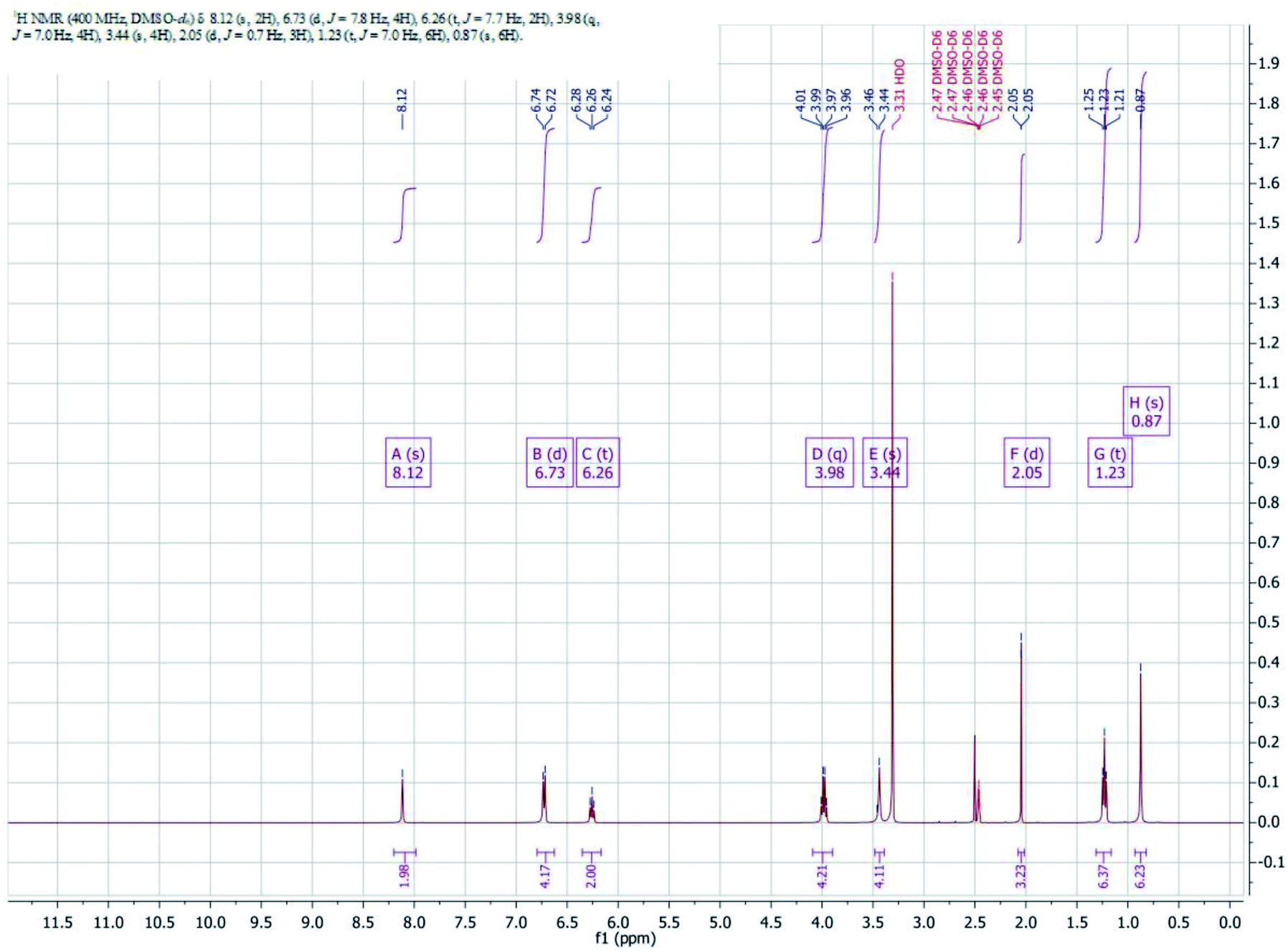 | ||
| Fig. 4 1H NMR of the complex. | ||
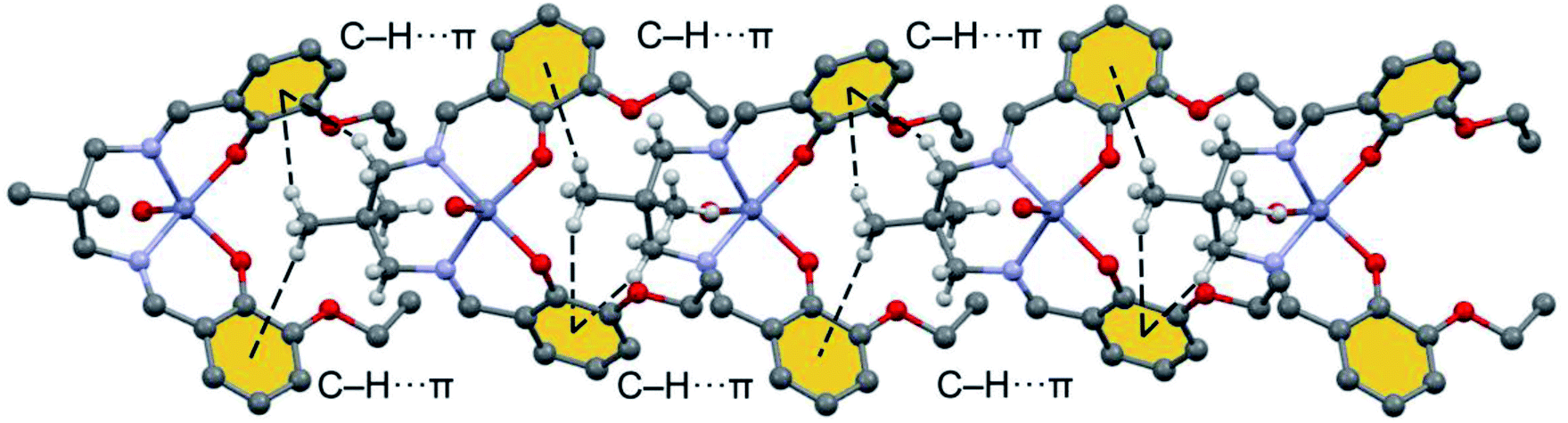 | ||
| Fig. 5 Detail 1D infinite chain observed in the solid state of [Zn(L)H2O]·H2O with indication of the C–H⋯π interactions. H-atoms omitted for clarity apart from those of the aliphatic linker. | ||
To rationalize the interaction, the MEP surface (Fig. 6) of [ZnL(H2O)] complex has first been computed since it is convenient to investigate the most nucleophilic and electrophilic groups of the complex. The MEP maximum is located at the H-atoms of the coordinated water molecule (+70 kcal mol−1). This large MEP value is due to the enhanced acidity of water H-atoms upon complexation to Zn(II). The minimum is also very large (−74 kcal mol−1) because it is located at the region where the four O-atoms of the ligand converge. Furthermore, the MEP values at the H-atoms of the methyl groups are positive (+12 and +18 kcal mol−1) and those over the center of the phenoxido ligands are negative (−31 kcal mol−1), thus anticipating a strong C–H⋯π interaction energies. Moreover, the van der Waals surface shows that the methyl group fits well into the cleft formed by both aromatic surfaces.
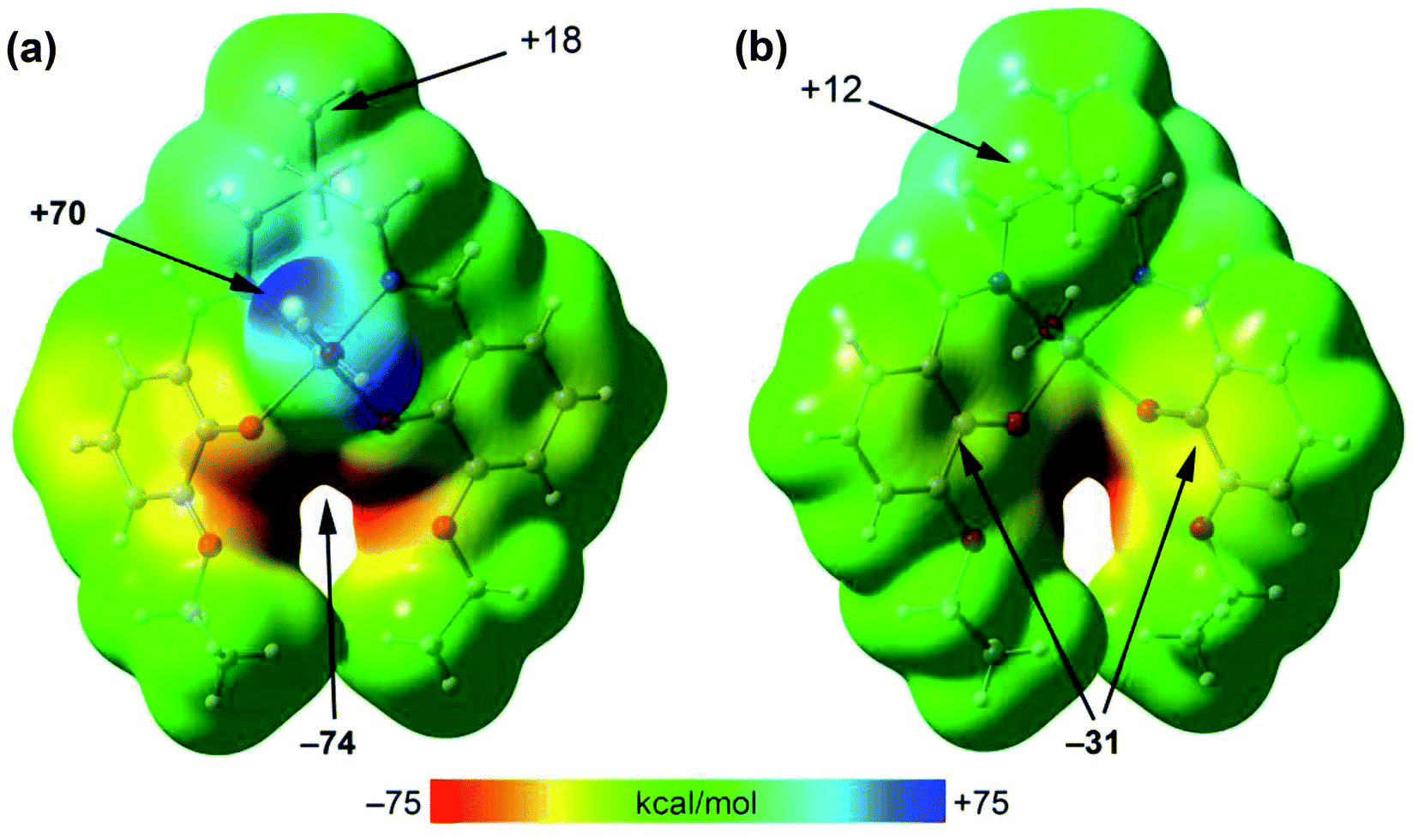 | ||
| Fig. 6 MEP surface of f [Zn(L)H2O] complex at the PBE0-D3/def2-TZVP level of theory and using two different orientations: coordinated molecule pointing up (a) or down (b). The values at selected points of the surface are indicated in kcal mol−1. | ||
Fig. 7 shows a dimer extracted from the infinite 1D chain commented above (see Fig. 5) where the network of C–H⋯π interactions is represented. Four C–H⋯π contacts are formed with H⋯C distances ranging from 3.30 to 3.56 Å, which are in the typical range described before for this type of bonding. One of the C–H bonds points to one exocyclic O-atom. The formation energy is very large ΔE = −13.9 kcal mol−1 confirming the importance of this network of interactions in the solid state of [ZnL(H2O)].
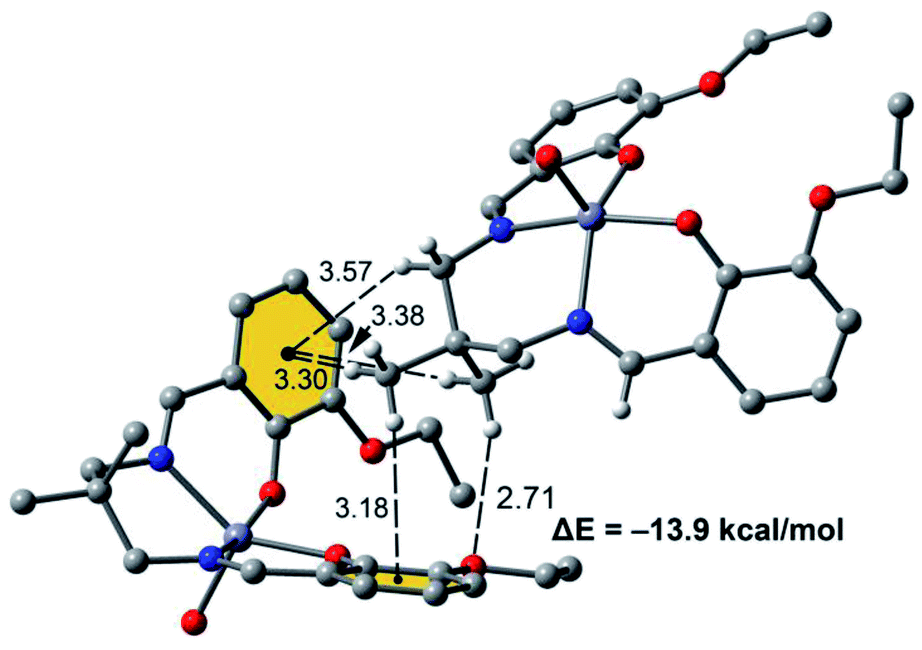 | ||
| Fig. 7 Dimer of [Zn(L)H2O] with indication of the C–H⋯π interactions. Distances are in Å. | ||
The noncovalent interaction plot index (NCI Plot) has been used further to characterize the C–H⋯π assembly in [ZnL(H2O)]. The NCI plot is an intuitive method that shows which spatial regions between molecules interact. Consequently, it is very convenient to analyze noncovalent interactions. Moreover, it gives hints regarding the strength of the interactions by using a colour code, where green is used for weak interactions and yellow for repulsive ones. Strongly attractive and repulsive interactions are represented by blue and red colors, respectively. The NCI plot of the dimer is given in Fig. 8, which confirms the existence of the C–H⋯π interactions that are characterized by several green isosurfaces located between the π-surfaces and the H-atoms. It also confirms the C–H⋯O contact also characterized by a green isosurface located between the C–H group and the O-atom of the ethoxy substituent. Despite all individual contacts are weak (green isosurface) the sum of all contributions is strong enough to dictate the formation of the 1D assembly shown in Fig. 5.
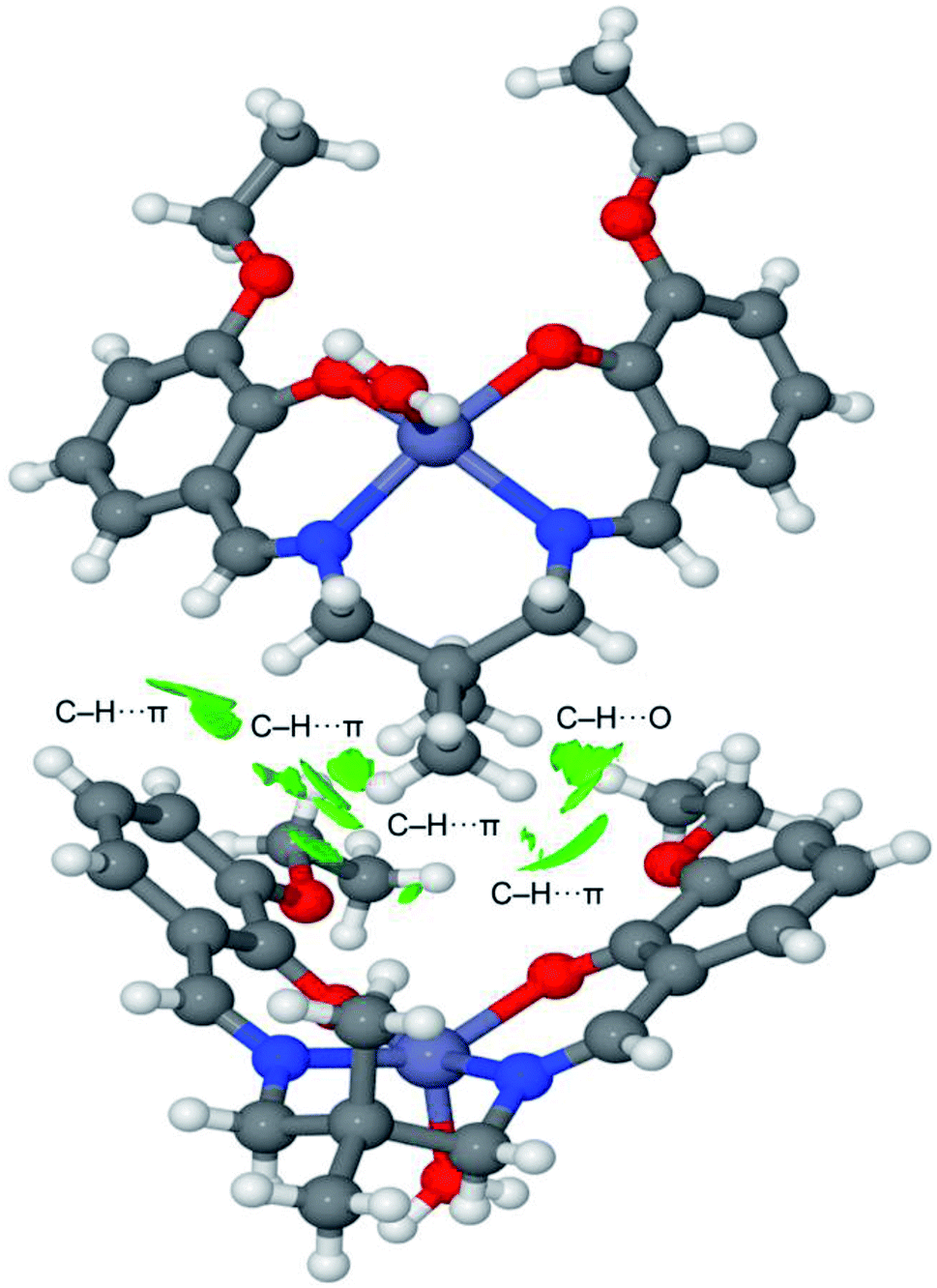 | ||
| Fig. 8 NCI Plot of the dimer of [Zn(L)H2O]. Isosurface 0.4 density cut-off 0.04 a.u. Colour scale −0.035 a.u. ≤ sign(λ2) ρ ≤ 0.035 a.u. | ||
Concluding remarks
A zinc(II) complex, [ZnL(H2O)]·H2O {H2L = 2,2′-[(2,2-dimethyl-1,3-propanediyl)bis(nitrilomethylidyne)]bis[6-ethoxyphenol]} has been synthesized and characterized by single crystal X-ray diffraction study, elemental analysis, FT-IR and electronic spectroscopic techniques. Crystal structure analysis reveals that C–H⋯π interactions lead to the formation of 1D supramolecular assembly in the solid state. The strength of the C–H⋯π interaction has been evaluated using DFT calculations and also analysed using the MEP surface and NCI plot index computational tool. The large dimerization energy confirms the importance of C–H⋯π interactions in the solid state of [ZnL(H2O)]·H2O.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. B. thanks the CSIR, India, for awarding a Senior Research Fellowship [Sanction No. 09/096(0861)/2016-EMR-I]. We also acknowledge the MCIU/AEI of Spain (project CTQ2017-85821-R, FEDER, UE funds) for financial support.References
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC.
- R.-G. Xiong, J.-L. Zuo, X.-Z. You, B. F. Abrahams, Z.-P. Bai, C.-M. Chec and H.-K. Fun, Chem. Commun., 2000, 2061–2062 RSC.
- M. D. Heinemann, R. Mainz, F. Österle, H. Rodriguez-Alvarez, D. Greiner, C. A. Kaufmann and T. Unold, Sci. Rep., 2017, 7, 45463 CrossRef CAS PubMed.
- V. Nishal, D. Singh, R. K. Saini, S. Bhagwan, V. Tanwar, Sonika, R. Srivastava and P. S. Kadya, J. Mater. Sci.: Mater. Electron., 2015, 26, 6762–6768 CrossRef CAS.
- H.-J. Son, W.-S. Han, J.-Y. Chun, B.-K. Kang, S.-N. Kwon, J. Ko, S. J. Han, C. Lee, S. J. Kim and S. O. Kang, Inorg. Chem., 2008, 47, 5666–5676 CrossRef CAS PubMed.
- R. T. Kuznetsova, I. V. Aksenova, A. A. Prokopenkoa, V. A. Pomogaev, E. V. Antina, M. B. Berezin, L. A. Antina and N. A. Bumagina, J. Mol. Liq., 2019, 278, 5–11 CrossRef CAS.
- T. Joshi, B. Graham and L. Spiccia, Acc. Chem. Res., 2015, 48, 2366–2379 CrossRef CAS PubMed.
- T. Basak, A. Bhattacharyya, M. Das, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, ChemistrySelect, 2017, 2, 6286–6295 CrossRef CAS.
- M. Karmakar and S. Chattopadhyay, Polyhedron, 2020, 184, 114527 CrossRef CAS.
- E. Y. Tirel, Z. Bellamy, H. Adams, V. Lebrun, F. Duarte and N. H. Williams, Angew. Chem., Int. Ed., 2014, 53, 8246–8250 CrossRef CAS PubMed.
- J. Huo, D. Yu, H. Li, B. Luoa and N. Arulsamy, RSC Adv., 2019, 9, 39323–39331 RSC.
- T. Basak, M. G. B. Drew and S. Chattopadhyay, Inorg. Chem. Commun., 2018, 98, 92–98 CrossRef CAS.
- Y. Zhao, L. Wang, N.-N. Fan, M.-L. Han, G.-P. Yang and L.-F. Ma, Cryst. Growth Des., 2018, 18, 7114–7121 CrossRef CAS.
- S. Li Yao, S. Jun Liu, C. Cao, X.-M. Tian, M.-N. Bao and T.-F. Zheng, J. Solid State Chem., 2019, 269, 195–202 CrossRef.
- M. Karmakar, S. Roy and S. Chattopadhyay, New J. Chem., 2019, 43, 10093–10102 RSC.
- A. Das, S. Jana and A. Ghosh, Cryst. Growth Des., 2018, 18, 2335–2348 CrossRef CAS.
- S. Shanmugaraju, C. Dabadie, K. Byrne, A. J. Savyasachi, D. Umadevi, W. Schmitt, J. A. Kitchenc and T. Gunnlaugsson, Chem. Sci., 2017, 8, 1535–1546 RSC.
- S. Roy, I. Mondal, K. Harms and S. Chattopadhyay, Polyhedron, 2019, 159, 265–274 CrossRef CAS.
- M. Karmakar, T. Basak and S. Chattopadhyay, New J. Chem., 2019, 43, 4432–4443 RSC.
- S. Dasgupta, E. Zangrando and I. Majumder, ChemistrySelect, 2017, 2, 7073–7081 CrossRef CAS.
- A.-C. Chamayou, S. Ludeke, V. Brecht, T. B. Freedman, L. A. Nafie and C. Janiak, Inorg. Chem., 2011, 50, 11363–11374 CrossRef CAS PubMed.
- G. Parkin, Chem. Rev., 2004, 104, 699–767 CrossRef CAS PubMed.
- H. Sakiyama, R. Mochizuki, A. Sugawara, M. Sakamoto, Y. Nishida and M. Yamasaki, J. Chem. Soc., Dalton Trans., 1999, 997–1000 RSC.
- S. Aoki, M. Zulkefeli, M. Shiro and E. Kimura, Proc. Natl. Acad. Sci. U. S. A., 2002, 9, 4894–4899 CrossRef PubMed.
- S. Roy, I. Mondal, K. Harms and S. Chattopadhyay, Polyhedron, 2019, 159, 265–274 CrossRef CAS.
- T. Basak, R. M. Gomila, A. Frontera and S. Chattopadhyay, CrystEngComm, 2021, 23, 2703–2710 RSC.
- M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed., 1997, 36, 1725–1727 CrossRef CAS.
- M. Schmittel and S. Saha, Adv. Inorg. Chem., 2018, 71, 135–175 CrossRef CAS.
- M. Karmakar, A. Frontera and S. Chattopadhyay, CrystEngComm, 2021, 23, 1918–1928 RSC.
- M. Nisa, M. Sirajuddin, S. Ali, M. N. Tahir and M. Iqbal, Polyhedron, 2020, 177, 114273 CrossRef.
- T. Basak, A. Bhattacharyya, K. Harms and S. Chattopadhyay, Polyhedron, 2019, 157, 449–457 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- S. Saha, M. K. Mishra, C. M. Reddy, C. Malla and G. R. Desiraju, Acc. Chem. Res., 2008, 51, 2957–2967 CrossRef PubMed.
- M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction, Wiley-VCH, New York, 1998 Search PubMed.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- C. R. Bhattacharjee, G. Das, P. Mondal and N. V. S. Rao, Polyhedron, 2010, 29, 3089–3096 CrossRef CAS.
- J. Gradinaru, A. Forni, V. Druta, F. Tessore, S. Zecchin, S. Quici and N. Garbalau, Inorg. Chem., 2007, 46, 884–895 CrossRef CAS PubMed.
- I. Mondal, T. Basak, S. Banerjee and S. Chattopadhyay, CrystEngComm, 2020, 22, 3005–3019 RSC.
- X.-X. Zhou, H.-C. Fang, Y.-Y. Ge, Z. Yuan Zhou, Z.-G. Gu, X. Gong, G. Zhao, Q.-G. Zhan, R.-H. Zeng and Y.-P. Cai, Cryst. Growth Des., 2010, 10, 4014–4022 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, SADABS, V2014/5, Software for Empirical Absorption Correction, University of Göttingen, Institute fur Anorganische Chemieder Universitat, Gottingen, Germany, 1999–2003 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- H. F. Clausen, M. S. Chevallier, M. A. Spackman and B. B. Iversen, New J. Chem., 2010, 34, 193 RSC.
- A. L. Rohl, M. Moret, W. Kaminsky, K. Claborn, J. J. McKinnon and B. Kahr, Cryst. Growth Des., 2008, 8, 4517 CrossRef CAS.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Crystal Explorer 2.0, University of Western Australia, Perth, Australia, 2007, http://hirshfeldsurfacenet.blogspot.com Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. J. Taylor, J. Chem. Soc., Perkin Trans. 1, 1987, 2, S1 Search PubMed.
- J. J. Kinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef CAS PubMed.
- T. Basak, K. Ghosh and S. Chattopadhyay, Polyhedron, 2018, 146, 81–92 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J Chem. Soc., Dalton Trans, 1984, 1349–1356 RSC.
- S. Roy, S. Bhattacharya and S. Chattopadhyay, J. Coord. Chem., 2016, 69, 112–122 CrossRef CAS.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- T. Basak, D. Das, P. P. Ray, S. Banerjee and S. Chattopadhyay, CrystEngComm, 2020, 22, 5170–5181 RSC.
- S. Roy, T. Basak, S. Khan, M. G. B. Drew, A. Bauzá, A. Frontera and S. Chattopadhyay, ChemistrySelect, 2017, 2, 9336–9343 CrossRef CAS.
- T. K. Ghosh, S. Jana and A. Ghosh, Inorg. Chem., 2018, 57, 15216–15228 CrossRef CAS PubMed.
- T. Basak, C. J. Gòmez-Garcia, R. M. Gomila, A. Frontera and S. Chattopadhyay, RSC Adv., 2021, 11, 3315–3323 RSC.
- S. Roy, M. G. B. Drew, A. Bauzá, A. Frontera and S. Chattopadhyay, New J. Chem., 2018, 42, 6062–6076 RSC.
- D. N. Kumar and B. S. Garg, Spectrochim. Acta, Part A, 2006, 64, 141–147 CrossRef PubMed.
- T. Basak, A. Frontera and S. Chattopadhy, Inorg. Chim. Acta, 2021, 516, 120081 CrossRef CAS.
- V. Nishal, D. Singh, A. Kumar, V. Tanwar, I. Singh, R. Srivastava and P. Singh Kadyan, J. Org. Semicond., 2014, 2, 15–20 CrossRef.
Footnote |
| † CCDC 2082611. For crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03943a |
| This journal is © The Royal Society of Chemistry 2021 |