
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRed to orange thermally activated delayed fluorescence polymers based on 2-(4-(diphenylamino)-phenyl)-9H-thioxanthen-9-one-10,10-dioxide for efficient solution-processed OLEDs†
Praetip Khammultri a,
Pongsakorn Chasinga,
Chirawat Chitpakdeeb,
Supawadee Namuangrukb,
Taweesak Sudyoadsuka and
Vinich Promarak*ac
a,
Pongsakorn Chasinga,
Chirawat Chitpakdeeb,
Supawadee Namuangrukb,
Taweesak Sudyoadsuka and
Vinich Promarak*ac
aDepartment of Material Science and Engineering, School of Molecular Science & Engineering, Vidyasirimedhi Institute of Science and Technology, Wangchan, Rayong 21210, Thailand. E-mail: vinich.p@vistec.ac.th
bNational Nanotechnology Center (NANOTEC), National Science and Technology Development Agency, Pathum Thani, 12120, Thailand
cResearch Network of NANOTEC-VISTEC on Nanotechnology for Energy, Vidyasirimedhi Institute of Science and Technology, Wangchan, Rayong 21210, Thailand
First published on 16th July 2021
Abstract
Most highly efficient thermally activated delayed fluorescence (TADF)-based organic light-emitting diodes (OLEDs) are multi-layer devices fabricated by thermal vacuum evaporation techniques, which are unfavorable for real applications. However, there are only a few reported examples of efficient solution-processed TADF OLEDs, in particular TADF polymer OLEDs. Herein, a series of solution-processable TADF conjugated polymers (PCTXO/PCTXO-Fx (x = 25, 50 and 75)) were designed and synthesized by copolymerization of 2-(4-(diphenylamino)-phenyl)-9H-thioxanthen-9-one-10,10-dioxide (TXO-TPA) as a red/orange emissive TADF unit, 9,9′-((fluorene-9,9-diyl)-bis(octane-8,1-diyl))-bis(3,6-di-tert-butylcarbazole) as host/hole-transporting unit and 2,7-N-(heptadecan-9-yl)carbazole as a conjugated linker and solubilizing group. They possessed a conjugated backbone with donor TPA-carbazole/fluorene moieties and a pendent acceptor 9H-thioxanthen-9-one-10,10-dioxide (TXO) forming a twisted donor–acceptor structure. These polymers in neat films displayed red/orange color emissions (601–655 nm) with TADF properties, proved by theory calculations and transient PL decay measurements. Their hole-transporting capability was improved when the content of 9,9′-((fluorene-9,9-diyl)-bis(octane-8,1-diyl))-bis(3,6-di-tert-butylcarbazole) within the polymers increased. All polymers were successfully employed as emitters in solution-processed OLEDs. In particular, the doped OLED fabricated with PCTXO exhibited an intense deep orange emission at 603 nm with the best electroluminescence performance (a maximum external quantum efficiency 10.44%, a maximum current efficiency of 14.97 cd A−1 and a turn-on voltage of 4.2 V).
Introduction
Thermally activated delayed fluorescence (TADF), recognized as the third-generation light-emitting mechanism for organic light-emitting diodes (OLEDs), has gained a lot of attention over the past few years.1 Because TADF emissive molecules can reach the theoretical 100% internal quantum efficiency (IQE) by utilizing up-conversion of non-radiative triplet excitons to radiative singlet excitons via a reverse intersystem crossing (RISC) process. Considering that they are pure organic molecules, the TADF emitters are capable of effectively tempering the electroluminescent (EL) performance of the phosphorescent OLEDs without the utilization of noble metals. Owing to the synthetic versatility of the organic compounds, moreover, the photophysical and electronic properties of TADF molecules can be fine-tuned by delicately modifying their molecular structures. So far, many TADF materials have been developed using various strategies, such as intramolecular donor–acceptor (D–A) TADF,2 through-space D–A TADF,3 excited-state intramolecular proton transfer (ESIPT) induced D–A TADF,4 organometallic complex TADF,5 and TADF exciplex.6 To date, the OLEDs based on TADF emitters can produce the emissive colors covering the whole visible spectrum,2b,7 especially in the high-energy spectral region (deep-blue emission).8 For blue and green TADF emitters, the EQE of OLEDs has now exceeded 30%,9 while that for red TADF emitters has surpassed 25%.10 Despite the tremendous progress in EL properties of the TADF OLEDs, new TADF materials are still essential to be developed for commercial applications, in particular, the solution-processable TADF materials, since most of the highly efficient devices are based on small-molecule TADF materials. The inherent physicochemical properties of low solubility, easy crystallization, and poor film-forming ability and morphology of a small-molecule material render most of the efficient small-molecule TADF emitters to be only fabricated by thermal vacuum deposition. Since this thin film deposition technique involves heating a solid material inside a high vacuum chamber at high temperatures and in a small area, it is not suitable for fabricating flexible and large area-size devices. In responding to such requirements, polymers might be a promising alternative. Unlike small molecules, polymeric materials commonly made up of a mixture of different sized molecules can be cast into a high-quality thin film by a solution-based process, such as spin-coating, slot-die coating, and roll-to-roll printing, which is simple, cost-effective, and easy to operate and scale-up for commercial applications. Besides, polymeric emitters have become one of the key findings or developments contributing to the advancement of modern OLEDs since the first report of poly(p-phenylene-vinylene) (PPV)-based OLEDs by Friend et al. in 1990.11 Once the TADF concept is introduced into a polymer platform, several types of solution-processable TADF polymers and macromolecules have been designed and synthesized using a variety of tactics, such as conjugated main-chain TADF polymers,12 non-conjugated main-chain TADF polymers,13 conjugated side-chain TADF polymers,1f non-conjugated side-chain TADF polymers,14 through-space charge transfer (TSCT) TADF polymers,15 and dendrimers.16 However, up till now, only limited examples of TADF polymers have been reported, while their EL efficiencies are still inferior to small-molecule TADF.2a,17 This could be due to the fact that TADF polymers are more susceptible to suffer triplet–triplet annihilation (TTA) via intra- and inter-chain interactions than a small molecule,18 while it is challenging to concurrently realize a minimal spatial overlap of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), and a sufficiently small energy gap (ΔEST) between the lowest singlet (S1) and triplet (T1) excited states particularly in the conjugated polymers. These are a couple of recently reported high-performance TADF polymers. Ren et al. synthesized a series of conjugated side-chain TADF polymer (Copo1), in which a small-molecule TADF of 2-(10H-phenothiazin-10-yl)dibenzothiophene-S,S-dioxide as a pendant group was attached to the insulating styrene backbone.14a Green emitting OLED device based on one of these polymers exhibits a high EQE of over to 20%. Most recently, Wang et al. reported the conjugated TADF polymers comprising of small molecular TADF of (4-(9,9-dioctylacridin-10(9H)-yl))(4-(9H-carbazol-9-yl))benzophenone (AcBPCz) and methyl-substituted phenylene linker.12e It was found that the conjugated linkage decreased the local excited triplet state caused by the delocalized polymer backbone. This polymer shows the highest EQE of over 23% with green emission. Despite these progressive improvements in device EL efficiencies, it is still essential and inspiring to realize suitable TADF polymeric emitters for high-performing solution-processed OLEDs.In this study, therefore, a series of conjugated main-chain polymers were designed and synthesized based on a classical small-molecule TADF of 2-(4-(diphenylamino)-phenyl)-9H-thioxanthen-9-one-10,10-dioxide (TXO-TPA).19 In these polymers, the TXO-TPA unit was implanted into the backbone by copolymerization with 2,7-N-(heptadecan-9-yl)carbazole and 9,9′-((fluorene-9,9-diyl)-bis(octane-8,1-diyl))-bis(3,6-di-tert-butylcarbazole) through the diphenylamino group of its triphenylamine (TPA) donor moiety, forming a backbone-donor/pendant-acceptor type π-conjugated TADF polymer (Scheme 1). In this design, both carbazole and fluorene as comonomers will provide a rigid π-conjugated polymer backbone which could suppress a non-radiative decay giving rise to the enhancement of the photoluminescence efficiency while linking via their 2,7 positions will not strongly affect the donor property of the TXO-TPA unit. Furthermore, the 2,7-N-(heptadecan-9-yl)carbazole comonomer is also intended to act as a linker capable of the hole-transporting channel and improving the solubility of the polymers. It has been reported that branched N-alkyl-substituted-carbazole-2,7-diyl-based copolymers with high molecular weight demonstrate high hole-transporting ability and good processability.20 Besides, the grafted 3,6-di-tert-butylcarbazole units on the side chain of the comonomer would also enable the hole-transporting ability and the host without altering the electronic properties of the polymer backbone as it has been noticed in some of its derivatives.1f,21 By managing the content of the 9,9′-((fluorene-9,9-diyl)-bis(octane-8,1-diyl))-bis(3,6-di-tert-butylcarbazole) unit in the polymers, series of TADF polymers with tuning optical and electronic properties, namely PCTXO and PCTXO-Fx (x represents the molar content of this fluorene monomer) were attained (Scheme 1). Indeed, all polymers exhibit TADF property with an improved hole-transporting capability as the content of x increased. The red/orange TADF polymer OLED peaked at 603 nm reaches an EQEmax of over 10% and an current efficiency (CEmax) of 14.97 cd A−1.
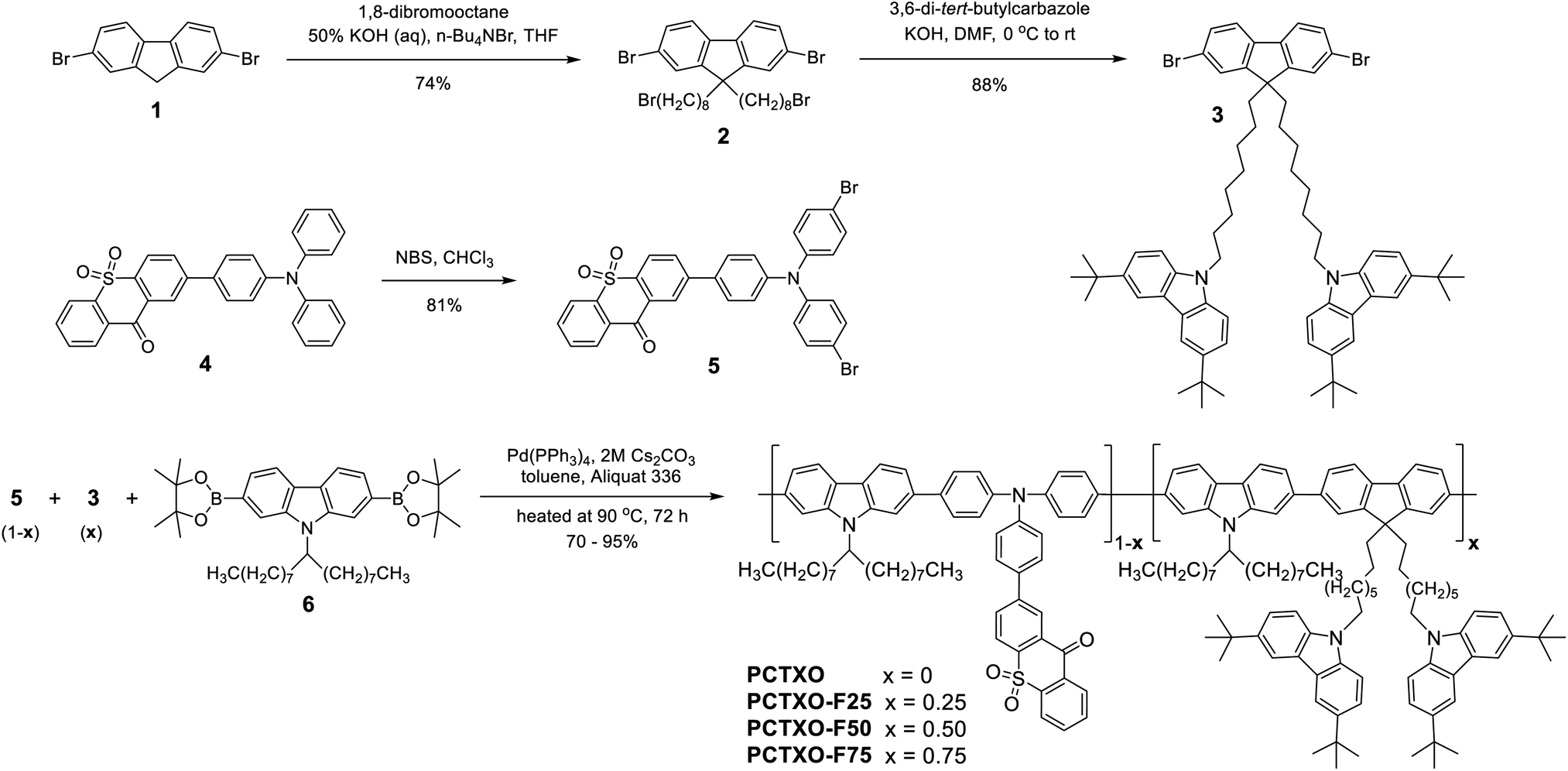 | ||
| Scheme 1 The synthesis and structures of polymers PCTXO and PCTXO-Fx (x = 25, 50, and 75). | ||
Results and discussion
Synthesis and characterizations
The designed TADF polymers were synthesized via Suzuki cross-coupling polycondensation between relevant 2-(4-(bis(4-bromophenyl)amino)phenyl)-9H-thioxanthen-9-one-10,10-dioxide (5), 9,9′-((2,7-dibromofluorene)-bis(octane-8,1-diyl))-bis(3,6-di-tert-butylcarbazole) (3) and 9-(9-heptadecanyl)-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)carbazole (6) monomers, as described in Scheme 1. The monomer 5 was prepared in good yield from an NBS bromination of its precursor 4. The monomer 3 was synthesized by alkylation of dibromofluorene 1 with an excess of 1,8-dibromooctane in the presence of KOH followed by a nucleophilic substitution reaction of the resultant 2 and 3,6-di-tert-butylcarbazole catalyzed by KOH. In the polymerization, three monomers 5, 3 and 6 were fed in different molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0:1, 0.75:0.25:1, 0.50:0.50:1, and 0.25:0.75:1 in the presence of Pd(PPh3)4/Cs2CO3(aq.) as catalysts in toluene/Aliquat at 90 °C. The resulting polymers denoted as PCTXO and PCTXO-Fx (x = 25, 50 and 75) were subjected to sequential Soxhlet extraction with acetone and chloroform and purified to remove unreacted monomer and low molecular weight species as well as the reaction catalyst to afford red to orange solids in 70–95% yields, respectively. The chemical structures of these polymers were verified by proton nuclear magnetic resonance (1H NMR) and Fourier-transform infrared spectroscopy (FTIR) analysis, as depicted in Fig. 1 (Fig. S1, ESI†). In terms of structural characterization, the contents of monomers 5 and 3 in each polymer could be quantified using both 1H NMR and FTIR spectra. As shown in Fig. 1, by comparing the 1H NMR spectra of PCTXO-Fx to that of PCTXO, it can be noticed that the signal peaks at chemical shifts of 8.06 (d), 4.11 (a), 2.12 (b) and 1.73 ppm (c) originated from the characteristic protons of monomer 3 are visible, confirming the existence of this monomer in the polymers. Meanwhile, the signal peaks derived from the protons of the monomers 5 and 6 are observed at chemical shifts of 8.57 (g) and 8.38 ppm (i), and 4.69 (e) and 0.79 ppm (f) in all cases, respectively. The intensities of protons (g) and (i) of monomer 5 decrease gradually with the decrease in the percentage of monomer 5 in the polymers. The actual molar ratios of monomer 3 in the polymers were estimated from analysis of their 1H NMR spectra (Fig. S2, ESI†) to be 23.5%, 50.3%, 76.1% for PCTXO-F25, PCTXO-F50, PCTXO-F75, respectively, which is in good agreement with the feed amount of monomer 3. Analysis of the FTIR spectra of these polymers also revealed that the relative intensities of absorption peaks of C
:0:1, 0.75:0.25:1, 0.50:0.50:1, and 0.25:0.75:1 in the presence of Pd(PPh3)4/Cs2CO3(aq.) as catalysts in toluene/Aliquat at 90 °C. The resulting polymers denoted as PCTXO and PCTXO-Fx (x = 25, 50 and 75) were subjected to sequential Soxhlet extraction with acetone and chloroform and purified to remove unreacted monomer and low molecular weight species as well as the reaction catalyst to afford red to orange solids in 70–95% yields, respectively. The chemical structures of these polymers were verified by proton nuclear magnetic resonance (1H NMR) and Fourier-transform infrared spectroscopy (FTIR) analysis, as depicted in Fig. 1 (Fig. S1, ESI†). In terms of structural characterization, the contents of monomers 5 and 3 in each polymer could be quantified using both 1H NMR and FTIR spectra. As shown in Fig. 1, by comparing the 1H NMR spectra of PCTXO-Fx to that of PCTXO, it can be noticed that the signal peaks at chemical shifts of 8.06 (d), 4.11 (a), 2.12 (b) and 1.73 ppm (c) originated from the characteristic protons of monomer 3 are visible, confirming the existence of this monomer in the polymers. Meanwhile, the signal peaks derived from the protons of the monomers 5 and 6 are observed at chemical shifts of 8.57 (g) and 8.38 ppm (i), and 4.69 (e) and 0.79 ppm (f) in all cases, respectively. The intensities of protons (g) and (i) of monomer 5 decrease gradually with the decrease in the percentage of monomer 5 in the polymers. The actual molar ratios of monomer 3 in the polymers were estimated from analysis of their 1H NMR spectra (Fig. S2, ESI†) to be 23.5%, 50.3%, 76.1% for PCTXO-F25, PCTXO-F50, PCTXO-F75, respectively, which is in good agreement with the feed amount of monomer 3. Analysis of the FTIR spectra of these polymers also revealed that the relative intensities of absorption peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1590 cm−1) and OSO groups (1303 and 1153 cm−1)22 came from the monomer 5 slowly decreased, while the relative intensities of absorption peaks of alkyl groups (2920 and 2851 cm−1) progressively increased (Fig. S1, ESI†), proving that the synthesized polymers contained various monomer proportions. The number-average molecular weight (Mn) and polydispersity indices (PDI) of PCTXO and PCTXO-Fx were determined by gel permeation chromatography (GPC) eluting with THF to be 11.71–21.65 kDa and 2.21–2.39, respectively (Table 1 and Fig. S3, ESI†). It is noted that the Mn values of these polymers are increased with the increased percentage of monomer 3 units. Owing to the existence of alkyl chains, PCTXO and PCTXO-Fx can be dissolved nicely in common polar organic solvents, such as toluene, dichloromethane, chlorobenzene, and THF, which is helpful to form good-quality thin film using solution processing.
O (1590 cm−1) and OSO groups (1303 and 1153 cm−1)22 came from the monomer 5 slowly decreased, while the relative intensities of absorption peaks of alkyl groups (2920 and 2851 cm−1) progressively increased (Fig. S1, ESI†), proving that the synthesized polymers contained various monomer proportions. The number-average molecular weight (Mn) and polydispersity indices (PDI) of PCTXO and PCTXO-Fx were determined by gel permeation chromatography (GPC) eluting with THF to be 11.71–21.65 kDa and 2.21–2.39, respectively (Table 1 and Fig. S3, ESI†). It is noted that the Mn values of these polymers are increased with the increased percentage of monomer 3 units. Owing to the existence of alkyl chains, PCTXO and PCTXO-Fx can be dissolved nicely in common polar organic solvents, such as toluene, dichloromethane, chlorobenzene, and THF, which is helpful to form good-quality thin film using solution processing.
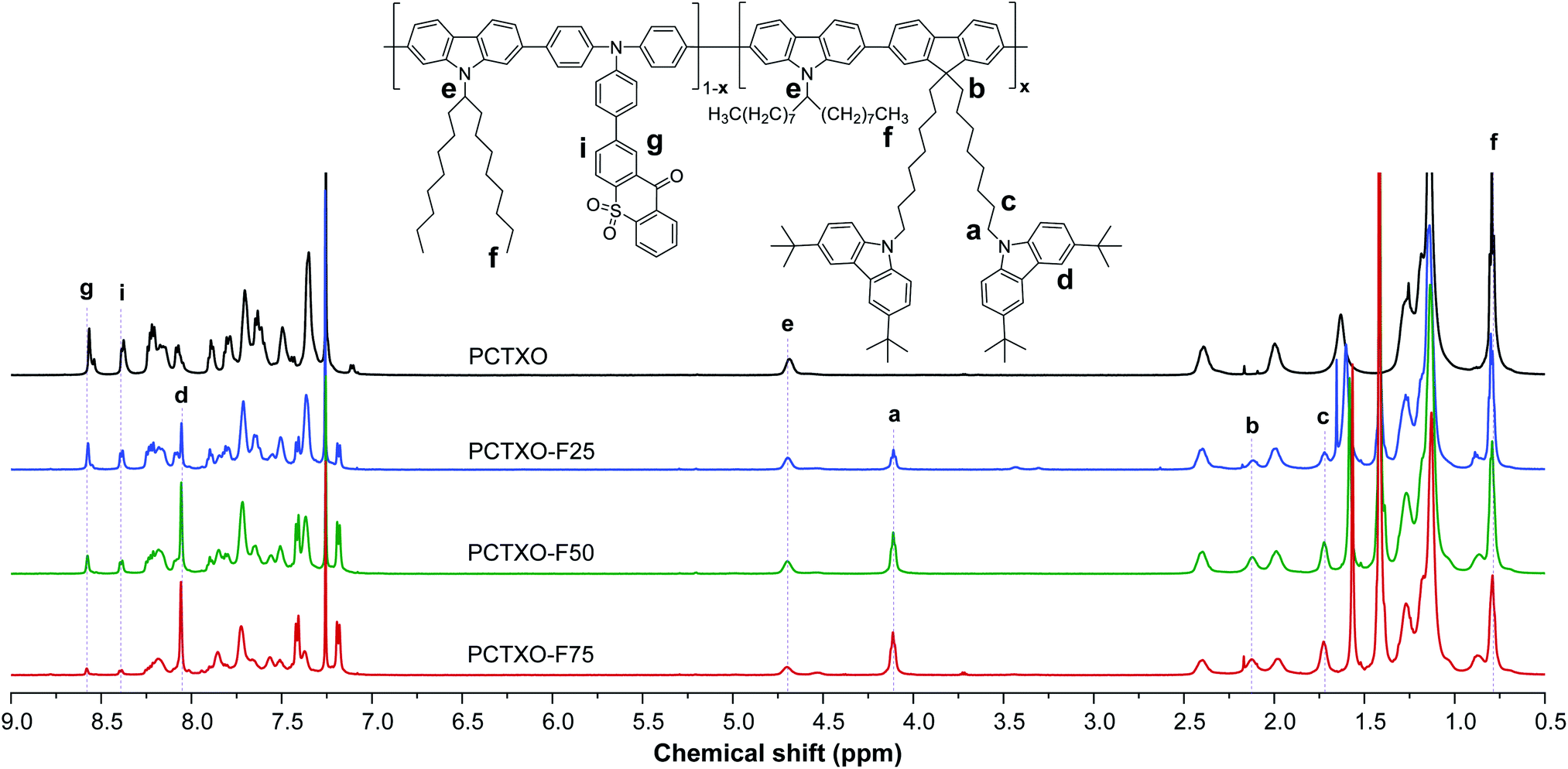 | ||
| Fig. 1 600 MHz 1H-NMR spectra (CDCl3) of polymers PCTXO and PCTXO-Fx (x = 25, 50 and 75). | ||
| Polymer | Mn/PDIa | λabs (nm) solb/nfc | λem (nm) solb/nfc/dfd | ΦPLe (%) solb/nfc/dfd | τf (ns) solb,g(solb,h)/nfc | τi (μs) | Eoptgj (eV) | HOMO/LUMOk (eV) | ΔESTl (eV) | μhm (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by GPC, eluting with THF, by comparison with polystyrene standards.b Recorded in toluene solution (10−6 M).c Measured as neat film coated on fused silica substrates.d Measured as polymer 5 wt% doped mCP films coated on fused silica substrates.e Absolute photoluminescence quantum yield measured using an integrating sphere in air.f Prompt fluorescence lifetime measured in ambient conditions.g Obtained in air-saturated conditions.h Obtained in degassed conditions.i Delayed fluorescence lifetime measured as polymer 5 wt% doped mCP films at 300 K.j Deduced from the absorption onset of a neat film: Eoptg = 1240/λonset.k HOMO measured by AC-2 of a neat film and LUMO = HOMO + Eoptg.l Excited singlet–triplet energy gap (ΔEST) obtained from the fluorescent and phosphorescent spectra of 1 wt% polymer blended Zeonex films measured at 300 K and 77 K, respectively.m Hole mobility (ITO/PEDOT:PSS (35 nm)/polymer (130 nm)/MoO3 (10 nm)/Al (80 nm)). | ||||||||||
| PCTXO | 11713/2.21 |
378/384 | 605/655/590 | 22/25/78 | 3.1(17.4)/8.2 | 12.5 (90.3%), 47.9 (9.7%) | 2.32 | −5.50/−3.18 | 0.15 | 1.35 × 10−7 |
| PCTXO-F25 | 15628/2.39 |
380/386 | 603/641/586 | 19/21/70 | 4.0(13.4)/16.7 | 11.0 (87.8%), 42.9 (12.2%) | 2.37 | −5.53/−3.16 | 0.13 | 3.86 × 10−7 |
| PCTXO-F50 | 17324/2.37 |
381/387 | 602/622/583 | 32/27/81 | 3.3(22.0)/24.1 | 12.3 (85.7%), 48.1 (14.3%) | 2.43 | −5.56/−3.13 | 0.12 | 8.22 × 10−6 |
| PCTXO-F75 | 21648/2.34 |
382/389 | 602/601/572 | 26/28/71 | 3.7(30.6)/32.3 | 11.1 (77.8%), 47.4 (22.2%) | 2.45 | −5.69/−3.24 | 0.12 | 1.36 × 10−5 |
Theoretical calculation
In order to evaluate the effect of the comonomer on the TXO-TPA TADF unit in the polymer backbone, the HOMO and LUMO distributions and S1 and T1 energies of the four polymer fragments (A, B, C, and D) consisting of a TXO-TPA unit and its adjacent carbazole/TPA moieties were calculated by the time-dependent density functional theory (TD)-B3LYP/6-31g(d) and the Tamm–Dancoff approximation (TDA)-M062X/6-31G(d) methods, respectively.23 As shown in Fig. 2, in all cases, the LUMO orbitals are localized entirely on the electron acceptor 9H-thioxanthen-9-one-10,10-dioxide (TXO) moiety, while a clear difference in the distribution of the HOMO orbital is seen in the polymer fragments. For fragment A, the HOMO orbitals are distributed on the donor TPA moiety and spread over the adjacent carbazole units and slightly on the adjoining phenyl ring of the TXO unit. However, by further copolymerization of the repeating fragment A with monomer 3 forming fragment B, the two connected fluorene rings to A barely makes any effect in the distribution of the HOMO, while the distribution of the HOMO is spread over the central TPA (TPA-TXO) and the two carbazole-TPA moieties in the fragment C when A is further polymerized with monomer 5. For fragment D, where A is copolymerized with both monomers 3 and 5, the HOMO orbitals holding a feature of both B and C are delocalized on the central TPA and the carbazole-TPA moiety with no contribution from the fluorene unit from another side. Hence, the TADF emissive features of the polymers largely depend on the fragment comprising of A and its adjacent TPA unit. According to the calculated HOMO and LUMO energy levels of A–D (Fig. 2), there are roughly two possible TADF fragments of A/B and C/D that could be responsible for the TADF property in the polymers. Besides, there is a slight overlap between both orbitals of the fragments, suggesting that the HOMO → LUMO transition possesses a strong intramolecular charge transfer (ICT) between the donor and acceptor parts, which is beneficial for enhancing the photoluminescence efficiency due to the escalating radiative rate from the overlap.1c Such effective HOMO–LUMO separation can also induce a small electron exchange energy. The calculated ΔEST values of B and C were 0.15 and 0.26 eV, respectively, which are somewhat higher than that of its original small-molecule, TXO-TPA (ΔEST = 0.04–0.09 eV).19,24 This could be ascribed to a noticeable π-electron conjugation between the TXO-TPA and the comonomers in the ground state.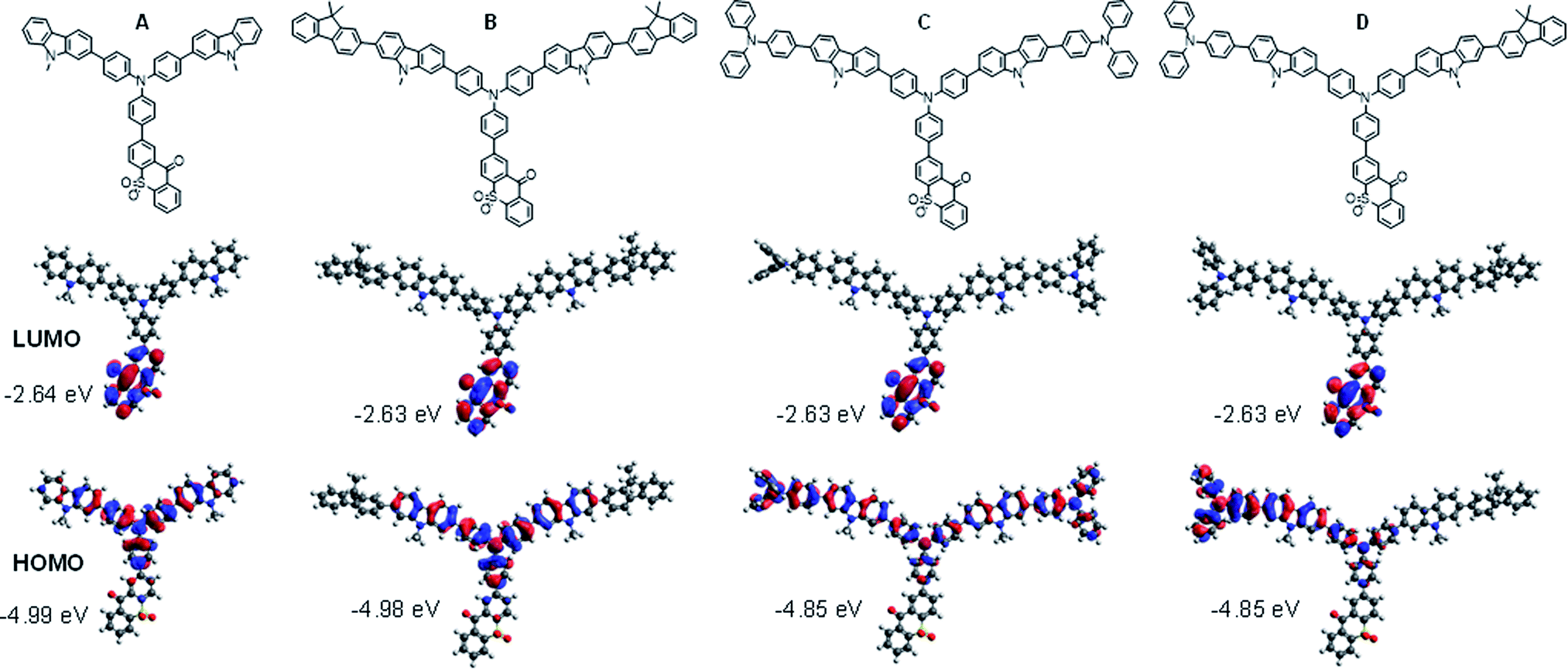 | ||
| Fig. 2 Molecular structures, and frontier orbital distributions of HOMO and LUMO of the polymer fragments (A–D) calculated by DFT-B3LYP/6-31g(d) method. | ||
Photophysical properties
The UV-vis absorption and photoluminescence (PL) spectra of PCTXO and PCTXO-Fx in both toluene and neat film are given in Fig. 3, and their photophysical data are listed in Table 1. In solution, all four polymers showed an intense absorption band peaked at ∼378–382 nm, correlating to the π–π* transition of the conjugated polymer backbone,25 whereas a peak at ∼299 nm is only clearly seen in the spectra of PCTXO-Fx, which corresponds to π–π* transition of the carbazole unit in the monomer 3 (Fig. 3a). Besides, a much less intense broad absorption band at >420 nm could be assigned to the CT absorption, which is similarly observed in the absorption spectrum of the TXO-TPA TADF molecule.19,24 The intensity of this band gradually decreased with a decrease in the content of the monomer 5 unit in the polymers. Their PL spectra in toluene displayed single CT emission bands from the TADF emissive unit at ∼603 nm when excited at ∼400 nm (corresponding to the absorption peak of polymer backbone), indicating an efficient intra- and intermolecular charge transfer from the conjugated backbone to the TADF fragment.26 A similar phenomenon was also noticed for the polymers in neat films, as described in Fig. 3b. This also means that the excited energy levels (LUMO or S1/T1) of the incorporated copolymer fragment match well with those of the emissive TADF one allowing an energy transfer to take place with very high efficiency. Thus, in the designed polymers, the comonomer of 9,9′-((fluorene-9,9-diyl)-bis(octane-8,1-diyl))-bis(3,6-di-tert-butylcarbazole) (monomer 3) can act as successful support for the organization of the host matrix for the TADF unit along with the conjugated copolymer backbone. In film state, both UV-vis absorption and PL spectra of polymers were red-shifted towards the longer wavelength compared to their corresponding spectra in solution (Table 1). Besides, the emission peaks were steadily blue-shifted with a decrease in the TADF unit (monomer 5) content, owing to aggregation in the condensed solid state. These photophysical phenomena are also identical to previous reports on the conjugated copolymers comprising a long-wavelength emissive unit.12a,c,27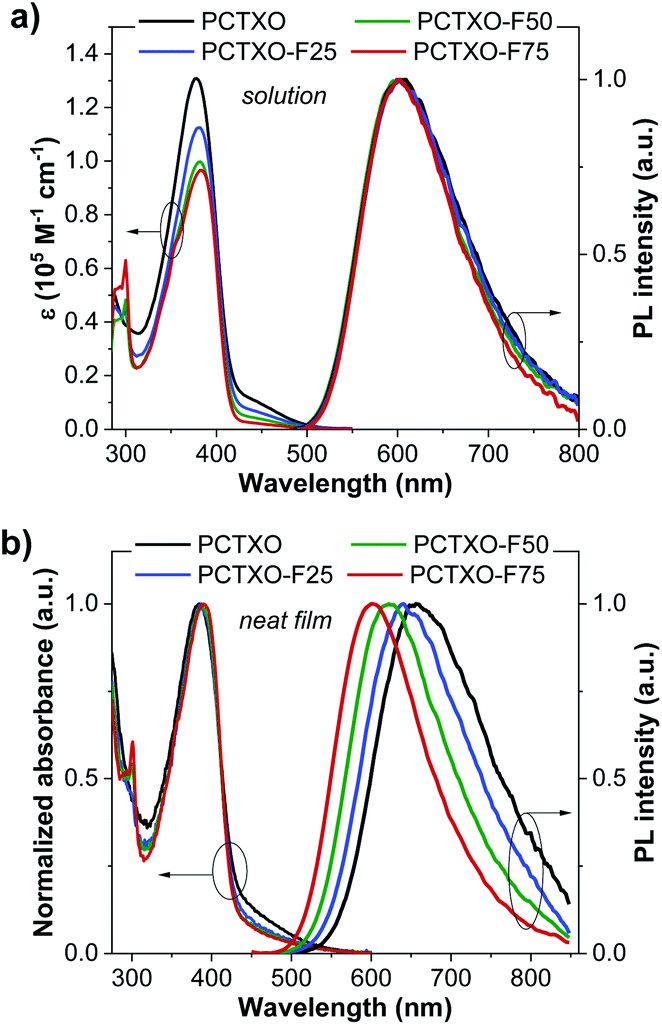 | ||
| Fig. 3 UV-Vis absorption and PL spectra of the polymers in (a) toluene and (b) as neat film. | ||
In toluene solution under ambient conditions, all polymers were moderate emitters with absolute photoluminescence quantum yield (ΦPL) of 19–32% (Table 1). The transient PL decay plots indicated a short mean PL lifetime (τ = 3.1–4.0 ns) without noticeably delayed fluorescence. However, both PL emission intensities and PL lifetimes of the polymer solutions under degassed conditions were significantly higher and longer (τ = 13.4–30.6 ns) than in air-saturated environments, respectively (Fig. 4a, b, S4, ESI† and Table 1). This is because of the suppression of the oxygen quenching effect on the triplet state, signifying the delayed fluorescence character. As shown in Fig. 4c, the transient PL decay spectra of the polymer in the neat film display a longer PL lifetime relative to those in the solution (Table 1). Notably, these τ values incredibly increased with the increasing content of monomer 3 in the polymers from τ = 8.2 ns for PCTXO, τ = 16.7 ns for PCTXO-F25, τ = 24.1 ns for PCTXO-F50 to τ = 32.2 ns for PCTXO-F75. This suggests that the presence of 9,9′-((fluorene-9,9-diyl)-bis(octane-8,1-diyl))-bis(3,6-di-tert-butylcarbazole) (monomer 3) as copolymer has the capability of suppressing aggregation quenching between the TADF emissive fragments presumably due to its crowded molecular structure and molecular rigidity in the conjugated backbone. To further study the TADF characteristics of PCTXO and PCTXO-Fx, the transient PL decay measurements were performed on 5 wt% doped mCP films at room temperature (Fig. 4d and S5, ESI†). All spectra displayed double exponential decay of the nanosecond-scale prompt fluorescence (PF) and the microsecond-scale delayed fluorescence (DF). As a result, their ΦPL values (70–81%) in doped mCP films were considerably higher than those in solution and neat film (Table 1). The microsecond-scale or TADF decay is a multi-exponential that can be roughly split into two components (Fig. 4d and S5, ESI†). The fast component (77.8–90.3%) of TADF has a single-exponential decay time in the range of 11.0–12.5 μs, while the slow one (9.7–22.2%) has a single-exponential decay time in the range of 42.9–48.1 μs (Table 1). The two TADF components appear to arise from two different TADF fragments in the doped film because of the different spectra observed for these two components (Fig. 4d inset). This result is well consistent with a previous observation in the theoretical calculations. Two sets of TADF fragments (A/B and C/D) can contribute to the TADF emissions in the polymers. Notably, these DF lifetime values (42.9–48.1 μs) are somewhat lower than that of the TXO-TPA TADF small molecule (78.0 μs),19 probably due to the extended conjugation in the HOMO of the TADF part.28 Furthermore, the polymers were also characterized by the temperature-dependent transient PL decay measurements of the polymers doped mCP:Zeonex films from 100 to 300 K (Fig. 4e and S6, ESI†). All of them obviously display the PF and DF components. The DF intensity increases with the increase in temperature from 100 to 300 K, suggesting that the up-conversion from T1 to S1 states is activated by supplying thermal energy. This phenomenon corroborates that PCTXO and PCTXO-Fx are TADF emitters.
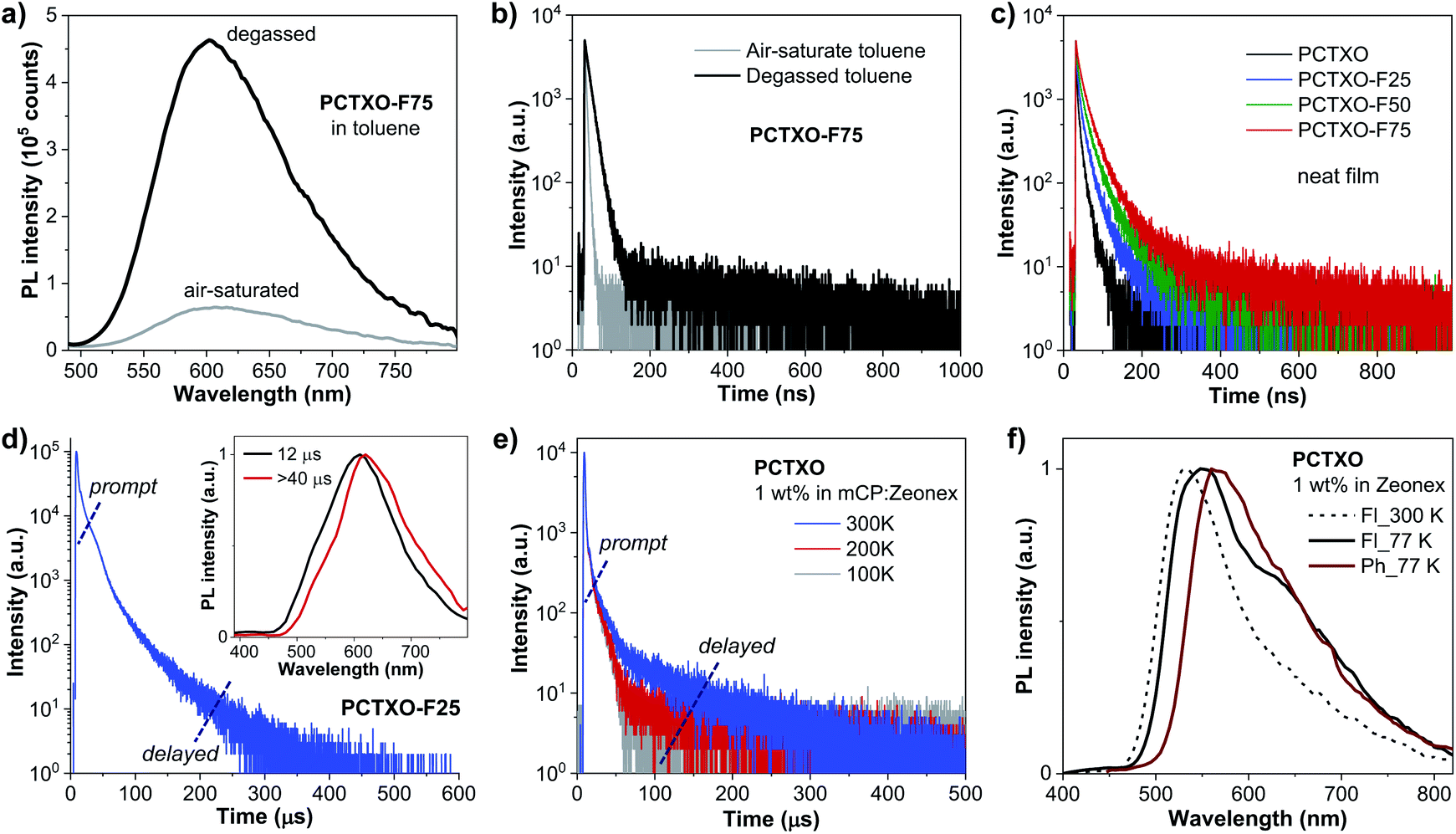 | ||
| Fig. 4 (a) PL spectra and (b) transient PL decay spectra of air-saturated and degassed PCTXO-F75 toluene solutions. (c) Transient PL decay spectra of the polymer as neat films. (d) Transient PL decay spectra of PCTXO-F25 5 wt% doped mCP films at room temperature (inset: PL spectra of TADF components at 12 μs and >40 μs). (e) Transient PL decay spectra of PCTXO 1 wt% doped in mCP:Zeonex film recorded at various temperatures in N2. (f) Fluorescence (Fl) and phosphorescence (Ph) spectra of PCTXO 1 wt% doped in Zeonex film recorded at 300 K and 77 K. The wide impulse response function (micro flash lamp) causes the distortion of the prompt component in the decay spectra of (d) and (e). | ||
To ensure the TADF properties of PCTXO and PCTXO-Fx, the phosphorescent (Ph) and fluorescent (Fl) spectra of the 1 wt% doped in Zeonex films were recorded at 77 K, as shown in Fig. 4f and S7 (ESI†). By taking the maximum peak wavelengths of the Ph spectra to determine the triplet energy (ET) and the maximum peak wavelengths of the Fl spectra to calculate the singlet energy (ES), the ΔEST values were estimated to be in the range of 0.12–0.15 eV, which are beneficial for efficient TADF (Table 1). Such a relatively small ΔEST value (<0.2 eV) is one of the crucial obligations to accomplish an efficient reverse intersystem crossing (RISC) process from the T1 to S1.
Thermal, electrochemical, charge transport and morphological properties
The thermal properties of PCTXO and PCTXO-Fx were investigated by TGA and DSC under a nitrogen atmosphere at a heating rate of 10 °C min−1. All polymers showed good thermal stability with high decomposition temperatures (Td, with 5% weight loss) of 358–432 °C and glass transition temperatures (Tg) of 112–158 °C (Fig. S8, ESI†). Moreover, no apparent exothermic crystallization and endothermic melting peaks over the heating range were detected in the DSC traces, suggesting that the crystallization of TADF units is suppressed by the comonomer units in the polymer backbone.The electrochemical behavior of these polymers was studied by cyclic voltammetry (CV) in an argon-saturated anhydrous dichloromethane solution containing 0.1 M n-Bu4NPF6 as a supporting electrolyte. As depicted in Fig. 5a, all polymers exhibit two quasi-reversible and one reversible reduction process. The reduction wave of all four polymers occurred at the same reduction half-wave potential (E1/2) of ∼−1.04 V, which could be attributed to a radical anion formation of the carbonyl oxygen atoms of the TXO moiety.29 The first oxidation wave at E1/2 of ∼0.91 V could be ascribed to the generation of radical cations on a donor moiety of the TADF fragment, while the second oxidation wave at E1/2 of ∼1.11 V observed in the polymers PCTXO-Fx could be assigned to the oxidation of the 3,6-di-tert-butylcarbazole pendant in the comonomer 3.21 Apparently, as the content of monomer 5 in the polymers is reduced from PCTXO to PCTXO-F75, the currents of both reduction and first oxidation waves are steadily decreased, whereas the current of the oxidation wave at 1.11 V is gradually enhanced. The HOMO energy levels of PCTXO and PCTXO-Fx in film state were then measured by UV photoelectron spectroscopy in air (Model AC-2) to be −5.50, −5.53, −5.56, and −5.69 eV, respectively (Table 1 and Fig. S9, ESI†). From the HOMO values and the optical band gap energies (Eoptg) estimated from an absorption onset value of the neat film UV-vis absorption spectra, the LUMO levels of these polymers were deduced to be in the range of −3.13 to −3.24 eV (Table 1).
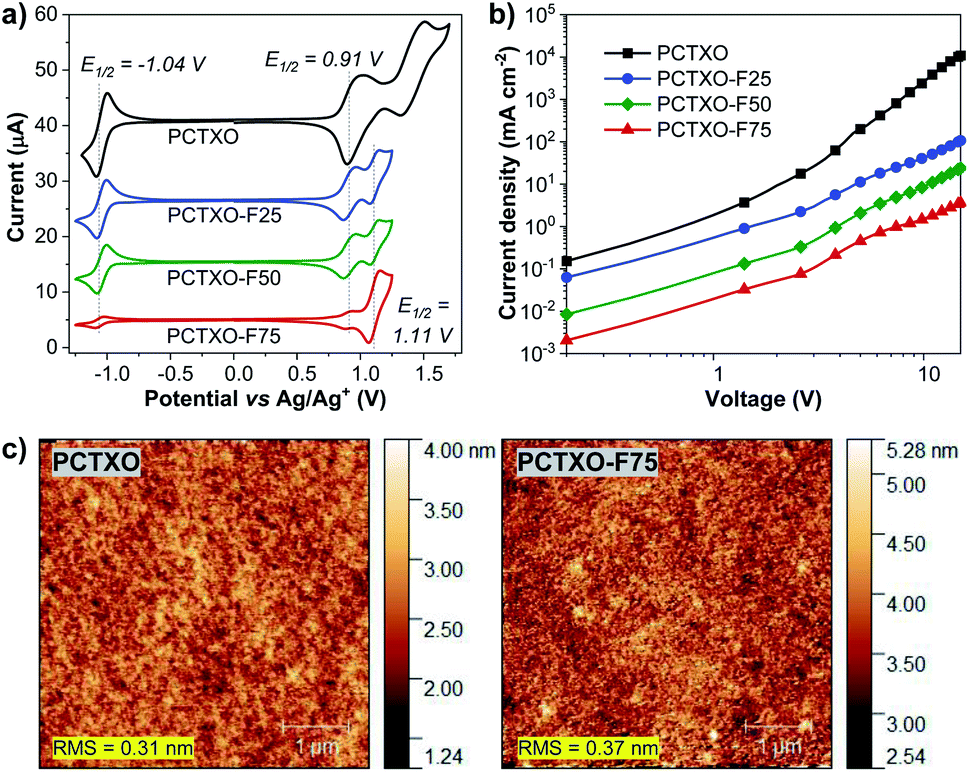 | ||
| Fig. 5 (a) Cyclic voltammograms of the polymers in dichloromethane solutions measured at a scan rate of 50 mV s−1. (b) Current density–voltage (J–V) plots of the hole only device (ITO/PEDOT:PSS (35 nm)/polymer (130 nm)/MoO3 (10 nm)/Al (80 nm)). (c) AFM images of PCTXO and PCTXO-F75 5 wt% doped in mCP films. | ||
To evaluate the hole-transporting property of these polymers, the hole-only device (HOD) was fabricated with the structure of ITO/PEDOT:PSS (35 nm)/polymer (130 nm)/MoO3 (10 nm)/Al (80 nm), where MoO3 was employed to ensure that only hole carrier is injected and transported in the device due to its work function of 6.90 eV being high enough to block electron injection from the Al cathode.30 The hole motility (μh) of each polymer was estimated by means of the space-charge-limited current (SCLC) method and extracted by fitting the J–V curves in the SCLC regime where the applied voltage range follows the Mott–Gurney model (Fig. 5b).31 The μh values were calculated to be in the range of 1.35 × 10−7 to 1.36 × 10−5 cm2 V−1 s−1 (Table 1). The hole-transporting ability of the polymers is improved as the content of comonomer 3 in their conjugated backbone increased. It is well understood that superior hole mobility can be profitable for a high-performance OLED device since higher charge mobility through the emitter could provide a wider recombination zone in an emissive layer (EML) and lead to a longer device lifetime as well as lower driving voltages.32
Electroluminescence properties
To explore the potential application of these TADF polymers as the emitters for solution-processed OLEDs, the non-doped devices (I–IV) were firstly fabricated with the optimized architecture of ITO/PEDOT:PSS (35 nm)/polymers (60 nm)/TmPyPB (40 nm)/LiF (0.5 nm)/Al (100 nm), where poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) and 1,3,5-tri(m-pyrid-3-yl-phenyl) benzene (TmPyPB) serves as the hole injection layer and the hole- and electron-transporting layer, respectively. The neat films of PCTXO and PCTXO-Fx as the emissive layer (EML) are spin-coated from chlorobenzene solution. As shown in Fig. 6a, the OLEDs (device I–IV) exhibit their inherent emissions with similar emissive wavelengths to PL spectra of the neat films. The device (I) using PCTXO as EML gave saturated red electroluminescence centered at 662 nm with the satisfied color coordinates of CIE (0.63, 0.36). Markedly, the emissive wavelengths of this PCTXO-based OLED showed a red-shifted EL with respect to those of the TXO-TPA19 and its TADF polymeric emitters.12a,c The emission colors of the devices gradually blue-shifted as the ratio of monomer 5 in the polymers decreased, as illustrated in Fig. 6b. The PCTXO-F75-based OLED (device IV) emitted orange emission peaked at 611 nm with CIE coordinates (0.55, 0.44). Clearly, among these four polymers, the PCTXO-F75 (device IV) achieved the best device EL performance showing both the highest maximum external quantum efficiency (EQEmax) of 1.54% and the lowest turn-on voltage (Von) of 5.1 V. The EL performance of these non-doped OLEDs (device I–IV) (CE, and EQEmax values) increases in the order of I < II < III < IV (Fig. 6c and Table 2), correlated well with the hole mobility of their corresponding emissive polymers, which is observed to be PCTXO-F75 > PCTXO-F50 > PCTXO-F25 > PCTXO (Table 1). Unfortunately, the EL efficiencies of these non-doped devices were still inferior, with EQEmax values in the range of 0.13–1.54%, considering that they were fabricated with TADF emitters. This is presumably due to the imbalanced charge injection and transport and aggregation-caused quenching (ACQ) owing to high proportions of TADF units in the neat films, consistent with their low thin-film ΦPL values.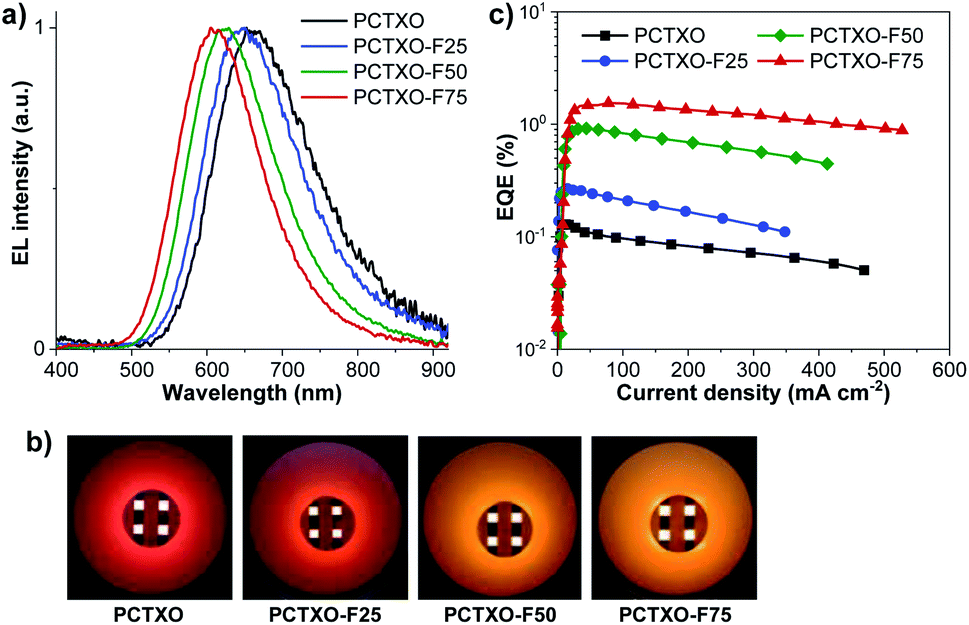 | ||
| Fig. 6 (a) Normalized EL spectra, (b) EQE–current density plots, and (c) photographs of the non-doped OLEDs (devices I–IV) based on PCTXO and PCTXO-Fx. | ||
| Device | Polymer | Von (V) | λEL(nm) | Lmax (cd m−2) | Jmax (mA cm−2) | FWHM (nm) | CEmaxc (cd A−1) | EQEmax (%) | CIE (x, y) |
|---|---|---|---|---|---|---|---|---|---|
| a ITO/PEDOT:PSS (35 nm)/polymer (60 nm)/TmPyPB (40 nm)/LiF (0.5 nm)/Al (100 nm).b ITO/PEDOT:PSS (35 nm)/polymer 5 wt% doped mCP (60 nm)/TmPyPB (40 nm)/LiF (0.5 nm)/Al (100 nm).c Maximum current efficiency. | |||||||||
| Ia | PCTXO | 8.6 | 662 | 319 | 348 | 158 | 0.19 | 0.13 | 0.63, 0.36 |
| IIa | PCTXO-F25 | 6.2 | 648 | 302 | 447 | 148 | 0.20 | 0.27 | 0.62, 0.38 |
| IIIa | PCTXO-F50 | 5.7 | 624 | 1617 | 389 | 136 | 0.51 | 0.92 | 0.58, 0.41 |
| IVa | PCTXO-F75 | 5.1 | 611 | 1447 | 501 | 130 | 0.88 | 1.54 | 0.55, 0.44 |
| Vb | PCTXO | 4.2 | 603 | 6836 | 273 | 117 | 14.97 | 10.44 | 0.56, 0.43 |
| VIb | PCTXO-F25 | 5.6 | 600 | 6987 | 274 | 121 | 11.98 | 7.86 | 0.54, 0.45 |
| VIIb | PCTXO-F50 | 5.8 | 596 | 4696 | 249 | 121 | 10.06 | 6.31 | 0.52, 0.46 |
| VIIIb | PCTXO-F75 | 6.2 | 587 | 7008 | 209 | 121 | 9.72 | 5.10 | 0.50, 0.48 |
To attain the optimal TADF properties of the synthesized polymers in the device, OLEDs were fabricated using the polymer doped mCP films as the EML (Fig. 7a). The device structure was ITO/PEDOT:PSS (35 nm)/EML (60 nm)/TmPyPB (40 nm)/LiF (0.5 nm)/Al (100 nm), where the EML was spin-coated from PCTXO/PCTXO-Fx:mCP in chlorobenzene solution. The doping concentration ratio between the polymers and mCP host material was initially optimized by varying from 1 wt% to 20 wt%. Fig. S11 (ESI†) presents PL and transient PL decay spectra of all polymers doped in mCP. The films containing polymer 5 wt% doped in mCP exhibited the highest ΦPL values of 70–81% (Table 1) with the PL spectra purely represent the emission of the polymer, indicating an effective energy transfer from the host to dopant. In comparison to their PL in the neat film, these PL spectra are somewhat blue shifts (29–65 nm), suggesting that diluting of the polymers in mCP host well repress intra-/inter-polymer chain aggregation and interactions in the film state. Furthermore, in all cases, the transient PL decay spectra obviously displayed both PF and DF components, signifying the existence of the TADF feature in the doped film. To assess the morphological stability and the compatibility of the polymers in the mCP matrix, the surface of these doped films was examined by atomic force microscopy (AFM). As shown in Fig. 5c (Fig. S12, ESI†), all thin films display smooth and homogeneous film morphologies with no pinholes and crystalline islands. The root-mean-square (RMS) roughness values were in the range of 0.31–0.42 nm, indicating high-quality nanoscale thin films.
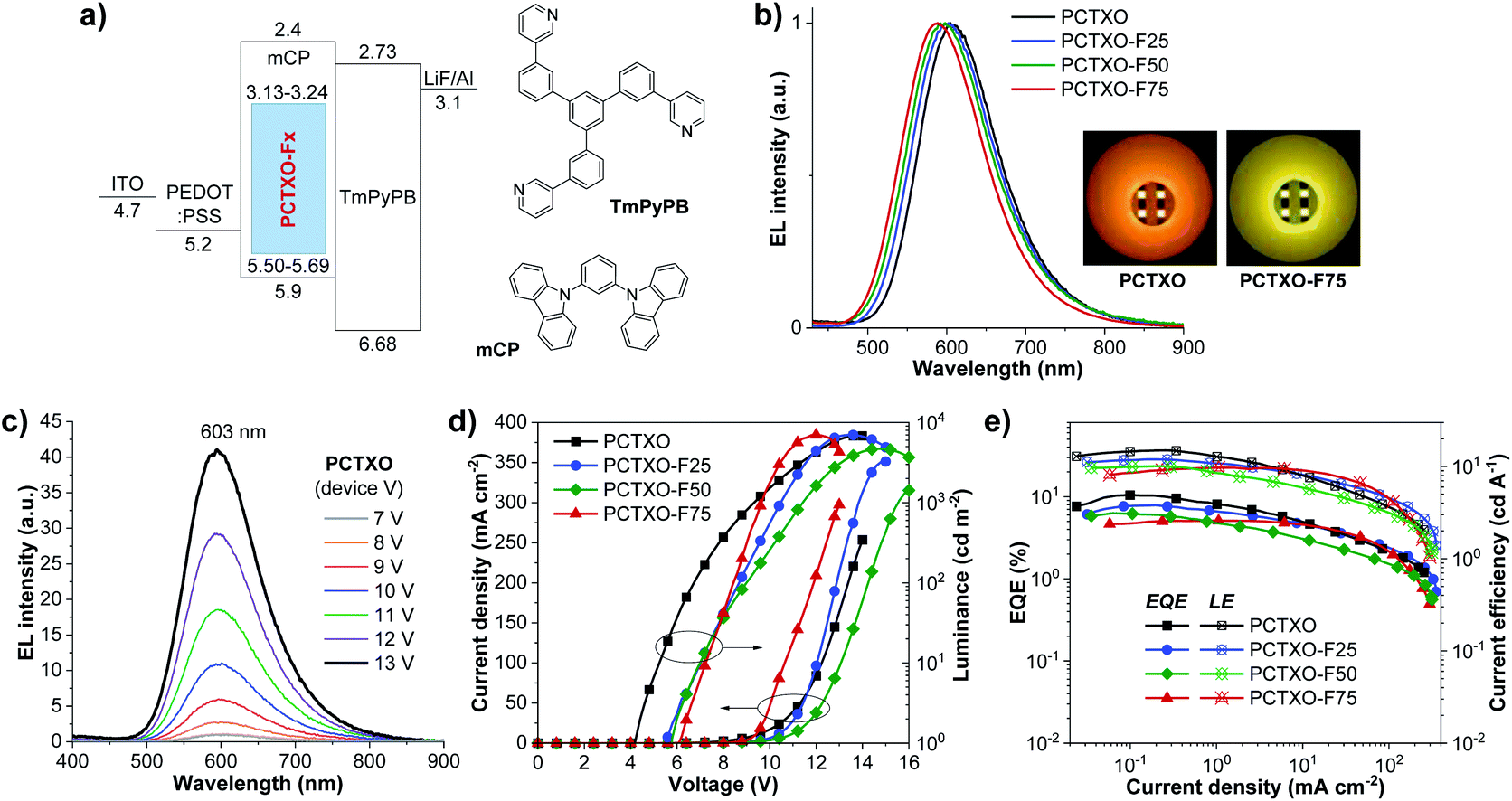 | ||
| Fig. 7 (a) Energy level diagram of the OLEDs and organic materials used in this study. (b) Normalized EL spectra (Inset: photographs of PCTXO and PCTXO-F75-based OLEDs (devices V–VIII)), (c) EL spectra of PCTXO-based OLED (device V) at different applied voltages. (d) Current density–voltage–luminance (J–V–L) characteristics and (e) EQE–current density–current efficiency plots of the doped OLEDs (devices V–VIII) based on PCTXO and PCTXO-Fx. | ||
The EL characteristics and parameters of the doped devices (V–VIII) are shown in Fig. 7 and are summarized in Table 2, respectively. Remarkably, all fabricated OLEDs (device V–VIII) exhibited a significant enhancement in the EL performances compared with those of their corresponding non-doped devices (I–IV). Such superior EL performances in the doped devices imply that the TADF facet of the polymers and their triplet excitons could be effectively utilized in the dispersed dopant/host systems. As illustrated in Table 2, the EL properties (EQEmax, CEmax, and Von) of the devices (V–VIII) tend to drop steadily as the ratio of monomer 5 in the polymer decreases, probably due to a reduction in the proportions of TADF fragments in the EML. Nevertheless, the EQEmax values of these devices were over a theoretical limit of 5% for the efficiency of the conventional fluorescent OLEDs, certainly demonstrating the efficient RISC contribution from the TADF fragments in the polymers. Among them, the device V using PCTXO 5 wt% doped mCP as an EML showed the best EL performance with an EQEmax of 10.44%, a maximum current efficiency (CEmax) of 14.97 cd A−1, a Von of 4.2 V, and a luminescence over 6836 cd m−2. This OLED gave a deep orange EL emission with the peak wavelength at 603 nm and color coordinates of CIE (0.56, 0.43) (Fig. 7b). The PCTXO-F25-based device (VI) exhibited a bit lower EL property with EQEmax/CEmax values of 7.86%/11.98 cd A−1, a Von of 5.6 V, and a luminescence over 6987 cd m−2 at the peak wavelength of 600 nm. Device VIII based on 5 wt% PCTXO-F75 doped mCP having the minor content of the TADF unit in the film delivered the lowest EL performance with EQEmax/CEmax values of 5.10%/9.72 cd A−1 and deep yellow EL emission peaking at 587 nm. In addition, the EL spectra of all doped devices (V–VIII) showed single peak emissions, which coincided nicely with the PL profiles of the EML. This result clearly demonstrates the complete transfer of energy from the mCP host/polymer conjugated backbone to the TADF emissive units. Their EL spectra were not also influenced by the changes in the applied voltages over the 7–13 V range, as shown in Fig. 7c (Fig. S13, ESI†), meaning that these fabricated devices are not suffered by the excimer or exciplex emissions from EML/TmPyPB interface and possess excellent operating stability. To the best of our knowledge, the EL performance of PCTXO-based device (V) is one of the best achieved based on TXO-TPA derivatives by solution-process technique so far.12a,c,24 Although its EQE of 10.44% is moderate compared with the reported state-of-the-art multi-layer thermally evaporated small-molecule TADF-based OLEDs,1a,10b,19,33 this OLED offers a straightforward device structure (only two organic layers) with a low-cost fabrication process, which is suitable for a practical application.
Conclusions
In summary, a series of red/orange emissive TADF conjugated polymers PCTXO/PCTXO-Fx (x = 25, 50 and 75) have been designed and synthesized by Suzuki coupling polycondensation, in which 2-[4-(diphenylamino)phenyl]-9H-thioxanthen-9-one-10,10-dioxide (TXO-TPA) as a red/orange emissive TADF unit was polymerized with 2,7-carbazole as host and solubilizing component via its TPA donor moiety forming a backbone-donor/pendant-acceptor type TADF polymer. The TADF behaviours were tuned by copolymerization of TXO-TPA with 9,9′-((fluorene-9,9-diyl)-bis(octane-8,1-diyl))-bis(3,6-di-tert-butylcarbazole) as host/hole-transporting unit in various ratios. Photophysical and theoretical data verified that all polymers possess efficient TADF characteristics with small ΔEST values (0.12–0.15 eV). All polymers exhibited a single emission band from the TADF component with emission colors ranging from saturated red to orange. Their hole-transporting abilities steadily improved as the content of the 9,9′-((fluorene-9,9-diyl)-bis(octane-8,1-diyl))-bis(3,6-di-tert-butylcarbazole) in the backbone increased. As a result, the solution-processed OLED fabricated with PCTXO-F75 as a non-doped emissive layer showed the best device electroluminescence (EL) property with a maximum external quantum efficiency (EQEmax) of 1.54%. Further doping these polymers (5 wt%) in mCP host was significantly enhanced the EL performances of their OLED devices. All solution-processed OLEDs showed high EQEmax values of over 5%. Particularly, the doped device employing PCTXO as the emitter attained the finest EL results (an EQEmax of 10.44%, a maximum current efficiency of 14.97 cd A−1, a Von of 4.2 V) with an intense orange color emission peaked at 603 nm. These findings demonstrate a helpful design strategy to enhance the TADF performance of TXO-TPA-based red/orange emissive materials for highly efficient solution-processed OLEDs and will be a valuable guide for the further development of new polymeric TADF materials.Experimental
Materials and methods
All chemicals were purchased from Tokyo Chemical Industry and Sigma-Aldrich and used without further purification. Solvents were dried using standard techniques.The polymers were confirmed molecular weight by gel permeation chromatography (GPC) on Malvern's Viscotek TDAmax using THF as a solvent system and polystyrene (PS) as a standard material. 1H-NMR and 13C-NMR spectra were recorded on Bruker AVANCE III HD 600 MHz. IR spectra were carried out on PerkinElmer model Frontier FT-IR, Universal-ATR. High-resolution mass spectrometry (HRMS) was performed on Bruker Autoflex speed MALDI-TOF mass spectrometer (MALDI-TOF-MS). Thermogravimetric analysis (TGA) was performed on Rigaku model Thermoplus TG8120 under N2 flow at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) was recorded on PerkinElmer model Lab System-DSC-8500 under N2 flow at a heating rate of 10 °C min−1. UV-vis absorption spectra of solution and neat film were performed on PerkinElmer model Lambda 1050. Fluorescence and phosphorescence spectra were recorded on the Edinburgh fluorescence spectrometer (FLS980). The absolute photoluminescence quantum yields (ΦPL) of solution and film were measured by integrating spheres under ambient air. To expel the dissolved oxygen, nitrogen gas was perfused through the solution. The delay fluorescence spectra were recorded as a neat film using EPL picosecond pulsed diode lasers as the excitation sources at room temperature. Phosphorescence spectra were measured under nitrogen atmosphere in cryoprobe with microsecond flashlamp as the excitation sources at 77 K. Atomic force microscopy (AFM) images of the thin film were taken by non-contact mode with Park Systems model NX-10. Cyclic voltammetry (CV) was performed on Autolab PGSTAT101 using dichloromethane as solvent under argon atmosphere and tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) as a supporting electrolyte. The HOMO energy level of the neat film was measured by photoelectron spectroscopy (PES) on RIKEN model AC-2 under ambient air. The theoretical calculations were carried out with Gaussian 09 program package.23 The ground state molecular geometries, HOMO and LUMO distributions, and HOMO, LUMO energy levels of the polymer fragment were computed by the B3LYP/6-31g(d) method. For the excited state calculations, the lowest-lying singlet (S1) and triplet (T1) energy levels were optimized by the TDA-M062X/6-31G(d) method.
The solution-processed OLED devices were fabricated with both non-doped and doped configurations of ITO/PEDOT:PSS (35 nm)/polymer (60 nm)/TmPyPB (40 nm)/LiF (0.5 nm)/Al (100 nm) and ITO/PEDOT:PSS (35 nm)/polymer 5 wt% doped mCP (60 nm)/TmPyPB (40 nm)/LiF (0.5 nm)/Al (100 nm), respectively. The thoroughly cleaned ITO-coated glass substrates were cured with UV ozone for 20 min. The PEDOT:PSS was spin-coated on the substrates in ambient air, followed by annealing at 160 °C for 30 min. In a nitrogen glove box, the synthesized polymers as a non-doped emissive layer and polymers 5 wt% doped in mCP as a doped emissive layer were dissolved in chlorobenzene. The polymer solutions (2% w/v) were spin-coated on top of the PEDOT:PSS coated ITO substrates at a spin speed of 3000 rpm for 30 seconds to obtain a 60 nm-thick film. Film thickness was measured by Dektak XTL stylus profiler. Then TmPyPB, LiF, and Al were successively coated on top by evaporation technique using Kurt J. Lasker mini SPECTROS 100. The devices were encapsulated with epoxy resin and curing with a UV curing lamp. The EL spectra were recorded by an Ocean Optic USB4000 multi-channel spectrometer in ambient air at room temperature. The current density–voltage–luminance (J–V–L) characteristics were measured by Keithley 2400 source meter and a Hamamatsu Photonics PMA-12 multi-channel analyzer. The absolute external quantum efficiency (EQE) was measured by Hamamatsu Photonics External Quantum Efficiency Measurement C9920-12, utilizing an integrating sphere. All measurements were performed under an ambient atmosphere at room temperature.
Synthesis and characterization
2-(4-(Bis(4-bromophenyl)amino)phenyl)-9H-thioxanthen-9-one-10,10-dioxide (5) was synthesized according to literature method.12g: 0, 0.75:0.25, 0.50:0.50 and 0.25:0.75, to of 9-(9-heptadecanyl)-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)carbazole (6) (145 mg, 0.221 mmol), trioctylmethyl ammonium chloride (Aliquat 336) (4.45 mg, 0.011 mmol), 2 M Cs2CO3 (aq.) (0.90 mL) and toluene (4 mL) in Schlenk tube was degassed with N2 for 10 min. Then, Pd(PPh3) (8.1 mg, 0.007 mmol) was added, and the mixture was degassed with N2 for another 5 min. The reaction mixture was stirred vigorously under nitrogen at 90 °C for 72 h. It was then poured into methanol (50 mL) and stirred. The precipitate was collected by filtration and washed with 2 M HCl and MeOH, respectively. It was subsequently purified by Soxhlet extraction with acetone followed by chloroform. The chloroform extract was treated with a metal scavenger (SiliaMetS Thiourea) to remove the remaining palladium catalyst and stirred for 2 h at room temperature. The mixture was filtered through a plug of silica eluting with chloroform, and the solvent was removed by a rotary evaporator. The crude product was reprecipitated in methanol, filtrated, and air-dried.PCTXO (187 mg, 95%) as red solids. Mn = 11713 Da, PDI = 2.21; 1H NMR (600 MHz, CDCl3) δ 8.57 (d, Ar H), 8.38 (d, Ar H), 8.27–8.11 (m, Ar H), 8.07 (d, Ar H), 7.89 (t, Ar H), 7.83–7.75 (m, Ar H), 7.70 (s, Ar H), 7.67–7.56 (m, Ar H), 7.50 (s, Ar H), 7.43 (d, Ar H), 7.35 (s, Ar H), 7.10 (d, Ar H), 4.69 (s, CH), 2.39 (s, CH2), 2.00 (s, CH2), 1.63 (s, CH2), 1.32–0.99 (m, CH2), 0.79 (t, CH3) ppm.
PCTXO-F25 (283 mg, 70%) as red solids. Mn = 15628 Da, PDI = 2.39; 1H NMR (600 MHz, CDCl3) δ 8.57 (s, Ar H), 8.38 (d, Ar H), 8.25–8.16 (m, Ar H), 8.08 (d, Ar H), 8.06 (s, Ar H), 7.90 (t, Ar H), 7.84–7.79 (m, Ar H), 7.71 (s, Ar H), 7.64 (d, Ar H), 7.55 (s, Ar H), 7.50 (s, Ar H), 7.41 (d, Ar H), 7.36 (s, Ar H), 7.18 (d, Ar H), 4.69 (s, CH), 4.11 (t, CH2), 2.40 (s, CH2), 2.11 (s, CH2), 1.99 (s, CH2), 1.74 (s, CH2), 1.60 (s, CH2), 1.40 (s, CH3), 1.27–1.14 (m, CH2), 1.22–1.14 (m, CH2), 0.89–0.87 (m, CH2), 0.80 (t, CH3) ppm.
PCTXO-F50 (290 mg, 72%) as red solids. Mn = 17324 Da, PDI = 2.37; 1H NMR (600 MHz, CDCl3) δ 8.58 (s, Ar H), 8.38 (d, Ar H), 8.25–8.18 (m, Ar H), 8.06 (s, Ar H), 7.89 (t, Ar H), 7.85–7.80 (m, Ar H), 7.71 (s, Ar H), 7.64 (s, Ar H), 7.56 (s, Ar H), 7.50 (s, Ar H), 7.41 (d, Ar H), 7.36 (s, Ar H), 7.18 (d, Ar H), 4.70 (s, CH), 4.11 (s, CH2), 2.40 (s, CH2), 2.12 (s, CH2), 1.97 (s, CH2), 1.72 (s, CH2), 1.57 (s, CH2), 1.42 (s, CH3), 1.26 (s, CH2), 1.14 (s, CH2), 0.86 (br, CH2), 0.80 (s, CH3) ppm.
PCTXO-F75 (315 mg, 78%) as orange solids. Mn = 21648 Da, PDI = 2.34; 1H NMR (600 MHz, CDCl3) δ 8.58 (s, Ar H), 8.39 (d, Ar H), 8.25–8.18 (m, Ar H), 8.06 (s, Ar H), 7.90–7.80 (m, Ar H), 7.72 (s, Ar H), 7.66–7.65 (m, Ar H), 7.57 (s, Ar H), 7.50 (s, Ar H), 7.42 (d, Ar H), 7.37 (s, Ar H), 7.19 (d, Ar H), 4.70 (s, CH), 4.11 (s, CH2), 2.40 (s, CH2), 2.12 (s, CH2), 1.98 (s, CH2), 1.73 (s, CH2), 1.57 (s, CH2), 1.42 (s, CH2), 1.26–1.24 (m, CH2), 1.18–1.13 (m, CH2), 0.87 (bs, CH2), 0.79 (s, CH3) ppm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was financially supported by the Thailand Research Fund (RTA6080005) and the National Nanotechnology Center, National Science and Technology Development Agency, Ministry of Higher Education, Science, Research and Innovation, Thailand, through its program of Research Network National Nanotechnology Center. Thanks also go to Vidyasirimedhi Institute of Science and Technology for the support of both Postdoctoral Fellowship (to T. S.) and equipment.Notes and references
- (a) M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed; (b) Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed; (c) K. Shizu, M. Uejima, H. Nomura, T. Sato, K. Tanaka, H. Kaji and C. Adachi, Phys. Rev. Appl., 2015, 3, 014001 CrossRef CAS; (d) Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS; (e) M. Godumala, S. Choi, M. J. Cho and D. H. Choi, J. Mater. Chem. C, 2019, 7, 2172–2198 RSC; (f) J. Luo, G. Xie, S. Gong, T. Chen and C. Yang, Chem. Commun., 2016, 52, 2292–2295 RSC.
- (a) X. Yin, Y. He, X. Wang, Z. Wu, E. Pang, J. Xu and J. Wang, Front. Chem., 2020, 8, 725 CrossRef CAS PubMed; (b) J. Kumsampao, C. Chaiwai, P. Chasing, T. Chawanpunyawat, S. Namuangruk, T. Sudyoadsuk and V. Promarak, Chem.–Asian J., 2020, 15, 3029–3036 CrossRef CAS; (c) M. Godumala, S. Choi, M. J. Cho and D. H. Choi, J. Mater. Chem. C, 2016, 4, 11355–11381 RSC.
- (a) E. Spuling, N. Sharma, I. D. W. Samuel, E. Zysman-Colman and S. Bräse, Chem. Commun., 2018, 54, 9278–9281 RSC; (b) F. Ma, H. Ji, D. Zhang, K. Xue, P. Zhang, Z. Qi and H. Zhu, Dyes Pigm., 2021, 188, 109208 CrossRef CAS; (c) Z. M. Hudson, J. Poisson, C. M. Tonge, N. R. Paisley, E. R. Sauvé, H. McMillan and S. V. Halldorson, Macromolecules, 2021, 54, 2466–2476 CrossRef; (d) H. Tsujimoto, D. G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CrossRef CAS PubMed.
- (a) K. Wu, T. Zhang, Z. Wang, L. Wang, L. Zhan, S. Gong, C. Zhong, Z. H. Lu, S. Zhang and C. Yang, J. Am. Chem. Soc., 2018, 140, 8877–8886 CrossRef CAS; (b) M. Mamada, K. Inada, T. Komino, W. J. Potscavage, H. Nakanotani and C. Adachi, ACS Cent. Sci., 2017, 3, 769–777 CrossRef CAS.
- (a) J. M. López-De-Luzuriaga, M. Monge, M. E. Olmos, M. Rodríguez-Castillo, I. Soldevilla, D. Sundholm and R. R. Valiev, Inorg. Chem., 2020, 59, 14236–14244 CrossRef PubMed; (b) D. Zhou, W. P. To, G. S. M. Tong, G. Cheng, L. Du, D. L. Phillips and C. M. Che, Angew. Chem., Int. Ed., 2020, 59, 6375–6382 CrossRef CAS PubMed; (c) J. C. Lima and L. Rodríguez, Inorganics, 2019, 7, 124 CrossRef CAS.
- (a) D. Dou, P. Wu, Z. Liao, J. Hao, J. Zhang and Z. Wang, RSC Adv., 2019, 9, 23810–23817 RSC; (b) Z. Wang, H. Wang, J. Zhu, P. Wu, B. Shen, D. Dou and B. Wei, ACS Appl. Mater. Interfaces, 2017, 9, 21346–21354 CrossRef CAS PubMed.
- (a) S. J. Woo, Y. Kim, S. K. Kwon, Y. H. Kim and J. J. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 7199–7207 CrossRef CAS PubMed; (b) H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC; (c) H. Wang, C. Zang, G. Shan, Z. Yu, S. Liu, L. Zhang, W. Xie and H. Zhao, Adv. Opt. Mater., 2019, 7, 1801718 CrossRef; (d) S. Kothavale, W. J. Chung and J. Y. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 18730–18738 CrossRef CAS PubMed; (e) C. Li, R. Duan, B. Liang, G. Han, S. Wang, K. Ye, Y. Liu, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 11525–11529 CrossRef CAS PubMed; (f) T. Huang, D. Liu, J. Jiang and W. Jiang, Chem.–Eur. J., 2019, 25, 10926–10937 CrossRef CAS PubMed.
- (a) J. Rao, C. Zhao, Y. Wang, K. Bai, S. Wang, J. Ding and L. Wang, ACS Omega, 2019, 4, 1861–1867 CrossRef CAS PubMed; (b) H. Lim, H. J. Cheon, S. J. Woo, S. K. Kwon, Y. H. Kim and J. J. Kim, Adv. Mater., 2020, 32, 2004083 CrossRef CAS PubMed; (c) P. Rajamalli, D. Chen, W. Li, I. D. W. Samuel, D. B. Cordes, A. M. Z. Slawin and E. Zysman-Colman, J. Mater. Chem. C, 2019, 7, 6664–6671 RSC.
- (a) Y. Xiang, P. Li, S. Gong, Y. H. Huang, C. Y. Wang, C. Zhong, W. Zeng, Z. Chen, W. K. Lee, X. Yin, C. C. Wu and C. Yang, Sci. Adv., 2020, 6, eaba7855 CrossRef CAS PubMed; (b) T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed; (c) D. R. Lee, B. S. Kim, C. W. Lee, Y. Im, K. S. Yook, S. H. Hwang and J. Y. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 9625–9629 CrossRef CAS PubMed; (d) D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540–546 CrossRef CAS.
- (a) W. Zeng, H. Y. Lai, W. K. Lee, M. Jiao, Y. J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K. T. Wong, C. C. Wu and C. Yang, Adv. Mater., 2018, 30, 1704961 CrossRef PubMed; (b) J. Liang, C. Li, Y. Cui, Z. Li, J. Wang and Y. Wang, J. Mater. Chem. C, 2020, 8, 1614–1622 RSC.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- (a) Y. Wang, Y. Zhu, G. Xie, H. Zhan, C. Yang and Y. Cheng, J. Mater. Chem. C, 2017, 5, 10715–10720 RSC; (b) Y. Liu, G. Xie, Z. Ren and S. Yan, ACS Appl. Polym. Mater., 2019, 1, 2204–2212 CrossRef CAS; (c) Y. Wang, Y. Zhu, G. Xie, Q. Xue, C. Tao, Y. Le, H. Zhan and Y. Cheng, Org. Electron., 2018, 59, 406–413 CrossRef CAS; (d) Y. Hu, W. Cai, L. Ying, D. Chen, X. Yang, X. F. Jiang, S. Su, F. Huang and Y. Cao, J. Mater. Chem. C, 2018, 6, 2690–2695 RSC; (e) J. Rao, X. Liu, X. Li, L. Yang, L. Zhao, S. Wang, J. Ding and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 1320–1326 CrossRef CAS PubMed; (f) Y. Zhu, Y. Zhang, B. Yao, Y. Wang, Z. Zhang, H. Zhan, B. Zhang, Z. Xie, Y. Wang and Y. Cheng, Macromolecules, 2016, 49, 4373–4377 CrossRef CAS; (g) P. Khammultri, W. Kitisriworaphan, P. Chasing, S. Namuangruk, T. Sudyoadsuk and V. Promarak, Polym. Chem., 2021, 12, 1030–1039 RSC; (h) Y. Liu, S. Yan and Z. Ren, Chem. Eng. J., 2020, 417, 128089 CrossRef.
- (a) X. Liu, J. Rao, X. Li, S. Wang, J. Ding and L. Wang, iScience, 2019, 15, 147–155 CrossRef CAS PubMed; (b) H. J. Kim, C. Lee, M. Godumala, S. Choi, S. Y. Park, M. J. Cho, S. Park and D. H. Choi, Polym. Chem., 2018, 9, 1318–1326 RSC; (c) A. E. Nikolaenko, M. Cass, F. Bourcet, D. Mohamad and M. Roberts, Adv. Mater., 2015, 27, 7236–7240 CrossRef CAS PubMed; (d) S. Y. Lee, T. Yasuda, H. Komiyama, J. Lee and C. Adachi, Adv. Mater., 2016, 28, 4019–4024 CrossRef CAS PubMed.
- (a) Z. Ren, R. S. Nobuyasu, F. B. Dias, A. P. Monkman, S. Yan and M. R. Bryce, Macromolecules, 2016, 49, 5452–5460 CrossRef CAS; (b) C. Li, Y. Xu, Y. Liu, Z. Ren, Y. Ma and S. Yan, Nano Energy, 2019, 65, 104057 CrossRef CAS; (c) C. Li, R. S. Nobuyasu, Y. Wang, F. B. Dias, Z. Ren, M. R. Bryce and S. Yan, Adv. Opt. Mater., 2017, 5, 1700435 CrossRef; (d) Y. Yang, L. Zhao, S. Wang, J. Ding and L. Wang, Macromolecules, 2018, 51, 9933–9942 CrossRef CAS; (e) X. Zhou, M. Huang, X. Zeng, T. Chen, G. Xie, X. Yin and C. Yang, Polym. Chem., 2019, 10, 4201–4208 RSC.
- (a) S. Shao, J. Hu, X. Wang, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2017, 139, 17739–17742 CrossRef CAS PubMed; (b) J. Hu, Q. Li, X. Wang, S. Shao, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2019, 58, 8405–8409 CrossRef CAS PubMed.
- (a) Y. Li, T. Chen, M. Huang, Y. Gu, S. Gong, G. Xie and C. Yang, J. Mater. Chem. C, 2017, 5, 3480–3487 RSC; (b) X. Ban, W. Jiang, T. Lu, X. Jing, Q. Tang, S. Huang, K. Sun, B. Huang, B. Lin and Y. Sun, J. Mater. Chem. C, 2016, 4, 8810–8816 RSC; (c) K. Sun, D. Chu, Y. Cui, W. Tian, Y. Sun and W. Jiang, Org. Electron., 2017, 48, 389–396 CrossRef CAS; (d) K. Sun, Y. Sun, D. Liu, Y. Feng, X. Zhang, Y. Sun and W. Jiang, Dyes Pigm., 2017, 147, 436–443 CrossRef CAS.
- (a) T. Jiang, Y. Liu, Z. Ren and S. Yan, Polym. Chem., 2020, 11, 1555–1571 RSC; (b) Q. Wei, Z. Ge and B. Voit, Macromol. Rapid Commun., 2019, 40, 1800570 CrossRef PubMed.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- H. Wang, L. Xie, Q. Peng, L. Meng, Y. Wang, Y. Yi and P. Wang, Adv. Mater., 2014, 26, 5198–5204 CrossRef CAS PubMed.
- (a) J. F. Morin and M. Leclerc, Macromolecules, 2002, 35, 8413–8417 CrossRef CAS; (b) R. He, S. Hu, J. Liu, L. Yu, B. Zhang, N. Li, W. Yang, H. Wu and J. Peng, J. Mater. Chem., 2012, 22, 3440–3446 RSC; (c) F. Dumur, Org. Electron., 2015, 25, 345–361 CrossRef CAS.
- (a) W. Kitisriworaphan, T. Chawanpunyawat, T. Manyum, P. Chasing, S. Namuangruk, T. Sudyoadsuk and V. Promarak, RSC Adv., 2021, 11, 12710–12719 RSC; (b) Y. Thathong, P. Chasing, T. Manyum, S. Namuangruk, S. Saengsuwan, T. Sudyoasuk and V. Promarak, New J. Chem., 2021, 45, 7694–7704 RSC.
- L. Hao, M. Wang, W. Shan, C. Deng, W. Ren, Z. Shi and H. Lü, J. Hazard. Mater., 2017, 339, 216–222 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., 2016 Search PubMed.
- M. Kumar and L. Pereira, ACS Omega, 2020, 5, 2196–2204 CrossRef CAS PubMed.
- V. Sannasi and D. Jeyakumar, Des. Monomers Polym., 2016, 19, 719–729 CrossRef CAS.
- G. Tu, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Appl. Phys. Lett., 2004, 85, 2172–2174 CrossRef CAS.
- (a) Y. Wang, Y. Zhu, X. Lin, Y. Yang, B. Zhang, H. Zhan, Z. Xie and Y. Cheng, J. Mater. Chem. C, 2018, 6, 568–574 RSC; (b) J. Huang, Y. Niu, W. Yang, Y. Mo, M. Yuan and Y. Cao, Macromolecules, 2002, 35, 6080–6082 CrossRef CAS.
- Q. Wei, P. Kleine, Y. Karpov, X. Qiu, H. Komber, K. Sahre, A. Kiriy, R. Lygaitis, S. Lenk, S. Reineke and B. Voit, Adv. Funct. Mater., 2017, 27, 1605051 CrossRef.
- (a) P. Lundberg, Y. Tsuchiya, E. M. Lindh, S. Tang, C. Adachi and L. Edman, Nat. Commun., 2019, 10, 5307 CrossRef CAS PubMed; (b) R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Adv. Opt. Mater., 2016, 4, 597–607 CrossRef CAS.
- (a) P. C. Kao, J. H. Lin, J. Y. Wang, C. H. Yang and S. H. Chen, J. Appl. Phys., 2011, 109, 094505 CrossRef; (b) C. J. Zheng, J. Ye, M. F. Lo, M. K. Fung, X. M. Ou, X. H. Zhang and C. S. Lee, Chem. Mater., 2012, 24, 643–650 CrossRef CAS; (c) N. B. Kotadiya, H. Lu, A. Mondal, Y. Ie, D. Andrienko, P. W. M. Blom and G. J. A. H. Wetzelaer, Nat. Mater., 2018, 17, 329–334 CrossRef CAS PubMed.
- (a) Y. Li, T. Dutta, N. Gerasimchuk, S. Wu, K. Shetye, L. Jin, R. Wang, D. M. Zhu and Z. Peng, ACS Appl. Mater. Interfaces, 2015, 7, 9372–9384 CrossRef CAS PubMed; (b) M. Nikolka, K. Broch, J. Armitage, D. Hanifi, P. J. Nowack, D. Venkateshvaran, A. Sadhanala, J. Saska, M. Mascal, S. H. Jung, J. K. Lee, I. McCulloch, A. Salleo and H. Sirringhaus, Nat. Commun., 2019, 10, 2122 CrossRef PubMed.
- (a) S. W. Culligan, A. C. A. Chen, J. U. Wallace, K. P. Klubek, C. W. Tang and S. H. Chen, Adv. Funct. Mater., 2006, 16, 1481–1487 CrossRef CAS; (b) R. A. K. Yadav, D. K. Dubey, S. Z. Chen, T. W. Liang and J. H. Jou, Sci. Rep., 2020, 10, 9915 CrossRef CAS PubMed; (c) P. Therdkatanyuphong, P. Chasing, C. Kaiyasuan, S. Boonnab, T. Sudyoadsuk and V. Promarak, Adv. Funct. Mater., 2020, 30, 2002481 CrossRef CAS.
- (a) A. Kumar, W. Lee, T. Lee, J. Jung, S. Yoo and M. H. Lee, J. Mater. Chem. C, 2020, 8, 4253–4263 RSC; (b) F. M. Xie, P. Wu, S. J. Zou, Y. Q. Li, T. Cheng, M. Xie, J. X. Tang and X. Zhao, Adv. Electron. Mater., 2020, 6, 1900843 CrossRef CAS; (c) B. Li, Z. Li, T. Hu, Y. Zhang, Y. Wang, Y. Yi, F. Guo and L. Zhao, J. Mater. Chem. C, 2018, 6, 2351–2359 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04599g |
| This journal is © The Royal Society of Chemistry 2021 |