
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge-scale synthesis of a monophosphonated tetrathiatriarylmethyl spin probe for concurrent in vivo measurement of pO2, pH and inorganic phosphate by EPR†
Teresa D. Gluth‡
 ab,
Martin Poncelet‡ab,
Stephen DeViencebc,
Marieta Genchevabc,
Emily. H. Hoblitzellbd,
Valery V. Khramtsovbc,
Timothy D. Eubankbd and
Benoit Driesschaert*ab
ab,
Martin Poncelet‡ab,
Stephen DeViencebc,
Marieta Genchevabc,
Emily. H. Hoblitzellbd,
Valery V. Khramtsovbc,
Timothy D. Eubankbd and
Benoit Driesschaert*ab
aDepartment of Pharmaceutical Sciences, West Virginia University, School of Pharmacy, Morgantown, WV 26506, USA. E-mail: Benoit.driesschaert@hsc.wvu.edu
bIn Vivo Multifunctional Magnetic Resonance Center, Robert C. Byrd Health Sciences Center, West Virginia University, Morgantown, WV 26506, USA
cDepartment of Biochemistry, West Virginia University, School of Medicine, Morgantown, WV 26506, USA
dDepartment of Microbiology, Immunology, and Cell Biology, West Virginia University, School of Medicine, Morgantown, WV 26506, USA
First published on 27th July 2021
Abstract
Low-field electron paramagnetic resonance spectroscopy paired with pTAM, a mono-phosphonated triarylmethyl radical, is an unmatched technique for concurrent and non-invasive measurement of oxygen concentration, pH, and inorganic phosphate concentration for in vivo investigations. However, the prior reported synthesis is limited by its low yield and poor scalability, making wide-spread application of pTAM unfeasible. Here, we report a new strategy for the synthesis of pTAM with significantly greater yields demonstrated on a large scale. We also present a standalone application with user-friendly interface for automatic spectrum fitting and extraction of pO2, pH, and [Pi] values. Finally, we confirm that pTAM remains in the extracellular space and has low cytotoxicity appropriate for local injection.
Low-field Electron Paramagnetic Resonance (EPR) with the use of a molecular spin probe is a powerful technique to non-invasively measure important physiological parameters in a living animal.1–4 EPR combines high sensitivity and good penetration depth. Stable tetrathiatriarylmethyl radicals (TAMs or trityls) are ideal spin probes for in vivo EPR applications. They exhibit unprecedented stability in vivo and ultra-narrow linewidths, which result in a high signal-to-noise ratio.5 TAM structures with spectral sensitivity to oxygen,6 pH,7,8 thiol concentration,9,10 microviscosity,11 ROS,12–14 or redox15,16 have been developed. We recently reported on a mono-phosphonated tetrathiatriarylmethyl radical pTAM (Fig. 1) whose EPR spectrum is sensitive to multiple parameters, namely oxygen concentration, pH, and inorganic phosphate concentration, [Pi].17–20 This multifunctional probe was utilized to profile the tumor microenvironment (TME) in various mouse models of cancer.19 The unmatched capability to measure [Pi] has resulted in the identification of this biomarker as a new TME marker for tumor progression.19 Moreover, the ability to measure pO2, pH, and [Pi] concurrently using the same probe allows for the direct correlation of these important parameters independent of the probe distribution, providing insight into the biological processes occurring in the TME.
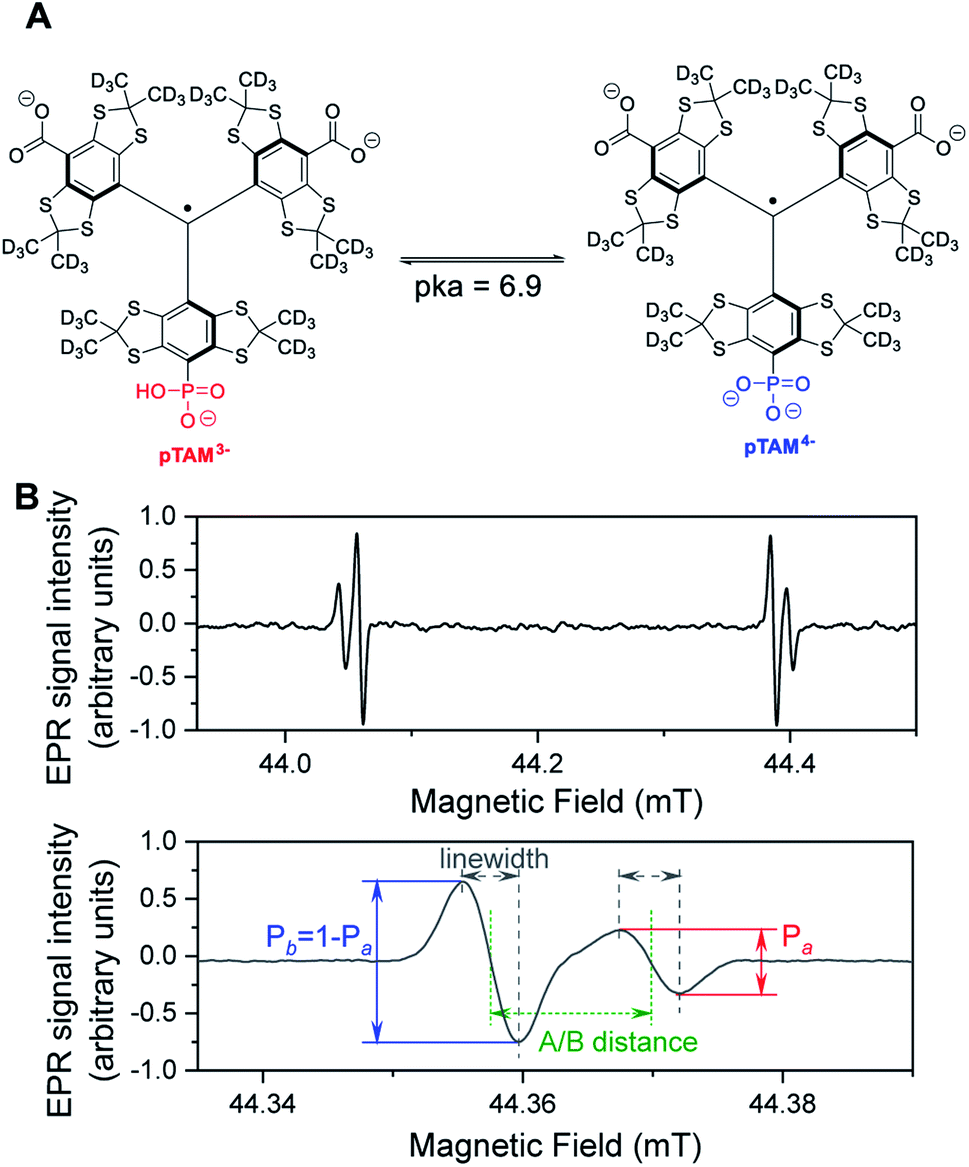 | ||
| Fig. 1 (A) Structure of pTAM spin probe and ionic forms at physiological pH. (B) L-band full spectrum (top) of pTAM at pH = 7.13 showing both ionic forms present in the spectrum and zoom on the high field component (bottom). The molar fraction of the acidic form Pa versus basic form Pb is a function of the pH of the solution while the linewidths are functions of the oxygen concentration. Inorganic phosphate modulates the exchange rate between the two ionic forms and the A/B distance. Spectral simulation allows the three parameters to be extracted from the spectrum. | ||
While this spin probe has proven to be of great importance for the study of tissue microenvironment in vivo, its current synthesis suffers from a very low yield. Indeed, the published synthesis17 (Scheme 1) uses a lithiation of tetrathiatriarylmethanol 1 and subsequent reaction with a (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture of diethyl carbonate and diethyl chlorophosphate. This reaction leads to a statistical mixture of mono-, di- and tri-phosphonated tetrathiatriarylmethanol 2n that requires tedious purification and drastically decreases the yield of the desired 2b. After hydrolysis of the ethyl esters using sodium hydroxide and deprotection of the phosphonic acid by TMSBr, the final pTAM probe was isolated with a yield of less than 5% from 1. The published procedure allowed for isolating milligram quantity of the probe for limited in vivo studies.19 However, more extensive utilization of this probe would require a synthetic method that enables gram-scale synthesis of pTAM.
:1) mixture of diethyl carbonate and diethyl chlorophosphate. This reaction leads to a statistical mixture of mono-, di- and tri-phosphonated tetrathiatriarylmethanol 2n that requires tedious purification and drastically decreases the yield of the desired 2b. After hydrolysis of the ethyl esters using sodium hydroxide and deprotection of the phosphonic acid by TMSBr, the final pTAM probe was isolated with a yield of less than 5% from 1. The published procedure allowed for isolating milligram quantity of the probe for limited in vivo studies.19 However, more extensive utilization of this probe would require a synthetic method that enables gram-scale synthesis of pTAM.
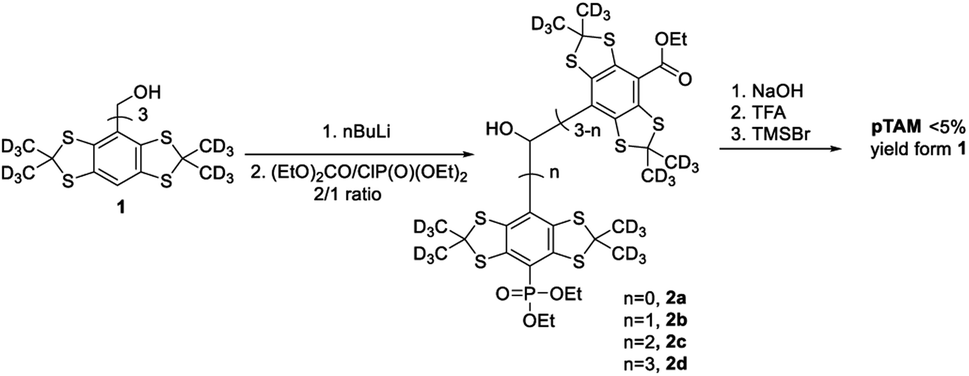 | ||
| Scheme 1 The first reported synthesis of pTAM from 1.17 | ||
Hereby we report an efficient protocol for the large-scale production of the pTAM probe as well as a MATLAB application for the automatic fitting of the EPR spectra and determination of the physiological parameters, namely pH, pO2, and [Pi]. Our new strategy takes advantage of a reaction of ipso nucleophilic substitution of an aromatic hydrogen or a carboxyl group on tetrathiatriarylmethyl derivatives reported previously.21,22 Our synthesis starts with the deuterated Finland trityl (dFT) which can be synthesized at large scale without chromatography (Fig. 2A).23–25 The one-electron oxidation of dFT with one equivalent of potassium hexachloroiridate(IV), K2IrCl6, in water leads to the trityl carbocation dFT+, which is immediately treated with ten equivalents of trimethyl phosphite. The nucleophilic addition of the phosphite in the para-position of the aryl ring triggers an oxidative decarboxylation, leading to the mono-phosphonic ester pTAM-(OMe)2 in 35% conversion, as determined by HPLC/MS (Fig. 2B and S6†). Importantly, the HPLC/MS chromatogram shows that dFT radical was also generated back from the trityl carbocation dFT+ in 65% yield, consistent with preferential oxidation of intermediate 3 by dFT+ in line with previous reports.21,22 dFT can therefore be recycled for future reactions. In addition, <5% of quinone methide (QM) was also generated from the nucleophilic addition of water on the trityl cation (see ESI† for mechanism). The use of additional equivalents of K2IrCl6 did not increase the yield of pTAM-(OMe)2 but did lead to higher conversion to the QM, TAMs with multiple phosphonates, and unidentified products. The preferential oxidation of 3 by dFT+ explains 50% of the back conversion of the trityl radical from the cation. The slightly higher formation of dFT observed (65%) could be the result of the direct reduction of the trityl cation by the trimethyl phosphite.
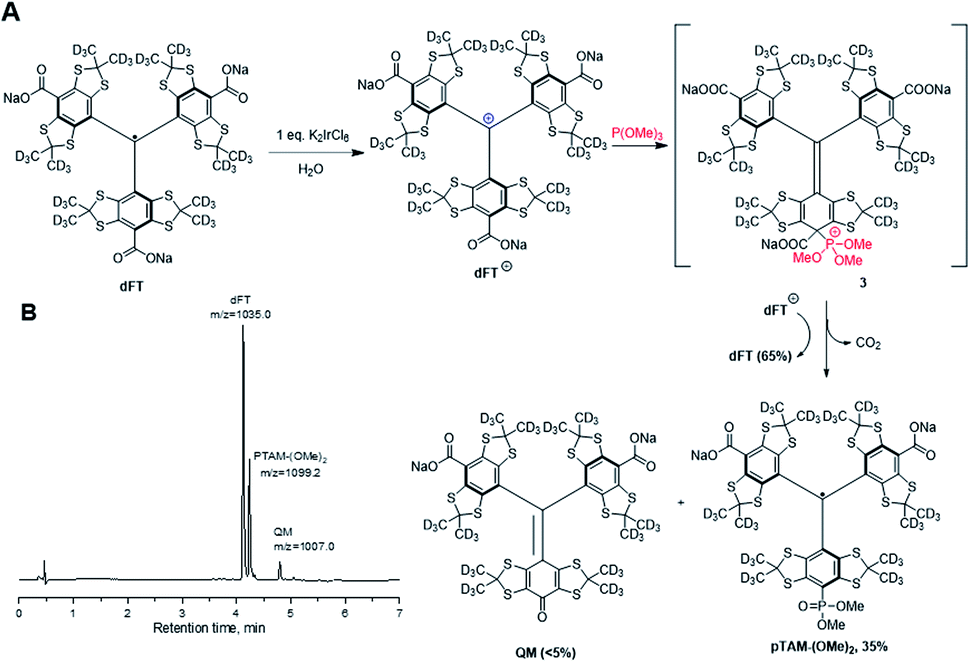 | ||
| Fig. 2 (A) Synthesis of pTAM-(OMe)2 from dFT and (B) HPLC/MS chromatogram and m/z ratio of the products after addition of P(OMe)3. | ||
The mono-phosphonated derivatives pTAM-(OMe)2, and dFT can be separated using a C18 column in 30% and 60% yield, respectively. Finally, the phosphonic acid was deprotected by treatment of pTAM-(OMe)2 with TMSBr in 95% yield (Scheme 2).
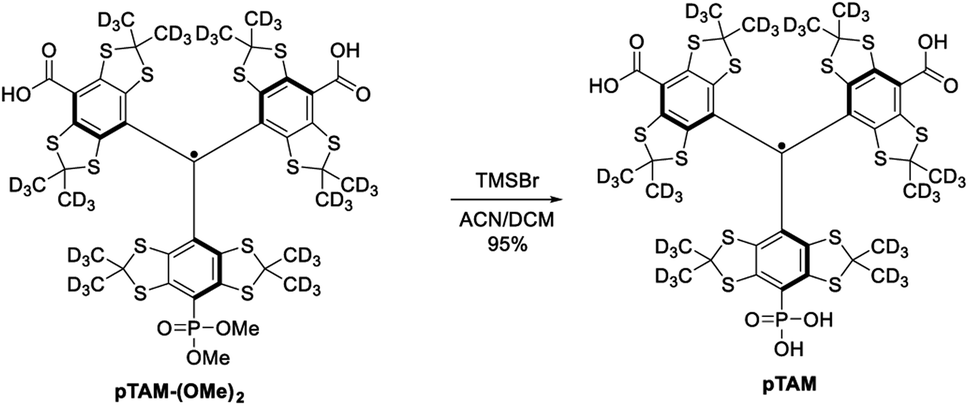 | ||
| Scheme 2 Deprotection of the phosphonic acid leading to pTAM. | ||
However, for a multigram scale, we found the separation of dFT and pTAM-(OMe)2 to be more challenging. The use of other phosphites with longer alkyl chains (triethyl-, triallyl- or tributyl phosphite), allowed for easier purification but led to smaller conversion (15–25%). On a large scale (tens of grams), the quantitative esterification of the carboxylic acids using methyl iodide and sodium carbonate in DMF directly on the dFT/pTAM-(OMe)2 mixture (Scheme 3) allowed for easy purification by flash chromatography on silica gel. The esterified pTAM-(OMe)4 was isolated in 35% yield from dFT starting material alongside with dFT-(OMe)3 (63%). Then, the phosphonic acid was deprotected by TMSBr in DCM, and the methyl esters hydrolyzed using lithium hydroxide in 1,4-dioxane/water leading to pTAM in 95% yield after purification on a C18 column. dFT-(OMe)3 was also hydrolyzed, leading to dFT in 99% yield with no purification needed. The relatively low conversion of dFT to the monophosponated ester is compensated by the recovery of the starting material. The calculated yield based on the recovery of the starting material reaches 92%. Our large scale synthesis allowed for the selective mono-phosphorylation of dFT in 4 steps and two purifications. The key step is the nucleophilic quenching of the trityl cation by trimethyl phosphite leading to the mono-phosphonated derivative.
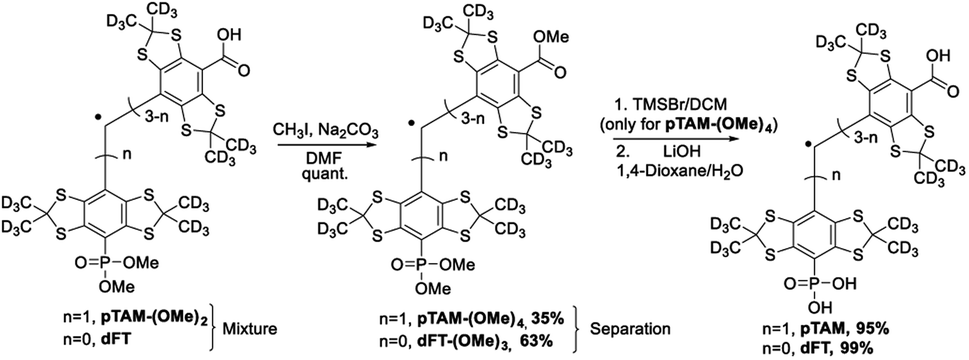 | ||
| Scheme 3 Esterification of the carboxyl groups to allow for large-scale separation of pTAM-(OMe)4 and dFT-(OMe)3. Then the carboxyl and phosphonic acids are deprotected, leading to pTAM and dFT. | ||
The extraction of pO2, pH, and [Pi] from the spectrum can be achieved using spectral fitting of the whole spectrum (see Fig. 1B, top) or only the high or low field EPR lines (Fig. 1B, bottom) using a homemade MATLAB algorithm as reported previously.18,19 However, to provide a user-friendly interface for those unfamiliar with MATLAB, we developed a graphical user interface for fitting the spectra and deriving the values for pO2, pH, and [Pi]. Fig. 3 demonstrates the use of the standalone application to fit a spectrum of pTAM administered into the mammary gland of a MMTV-PyMT mouse (see ESI† for calibration and use of the app).
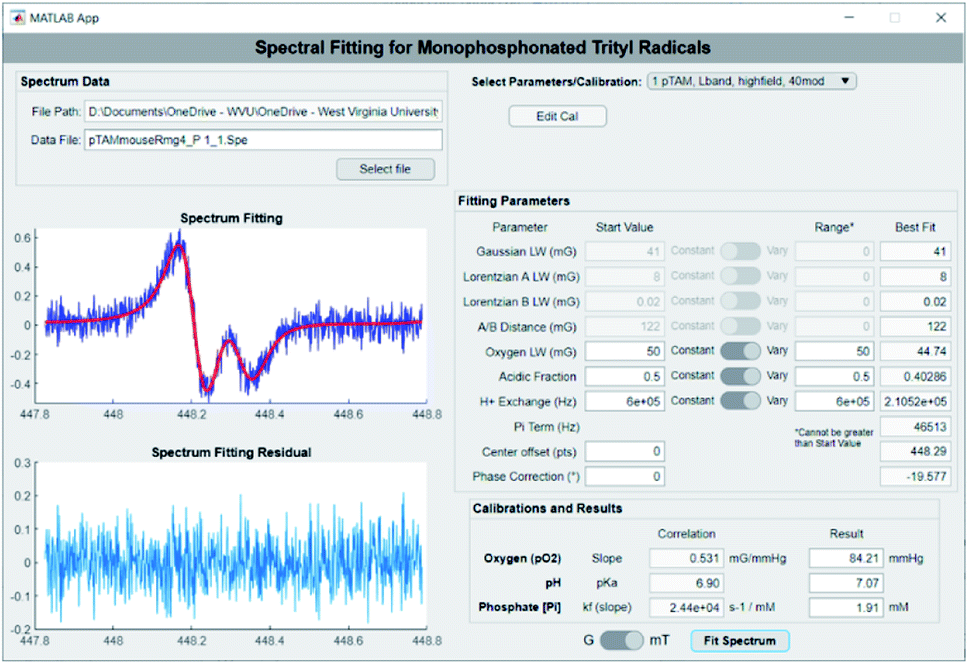 | ||
| Fig. 3 Screenshot of the pTAM spectrum fitting app developed in-house with a spectrum measured of pTAM injected directly in the mammary gland of a MMTV-PyMT mouse. Values of pO2 = 84.21 mmHg, pH = 7.07 and [Pi] = 1.91 mM are automatically calculated from the experimental spectrum. | ||
When applied in vivo, the charged nature of the probe and its large size (MW = 1073 g mol−1) is expected to prevent its diffusion through the cell membrane. In order to verify that pTAM cannot enter the cytosol, pTAM (200 μM) was incubated with 8.5 × 106 MDA-MB-231 cells (human triple negative breast cancer cells) with and without 10 mM Gd-DTPA, a paramagnetic extracellular broadening agent.7 Fig. 4 shows a large broadening of the EPR lines of pTAM upon addition of Gd-DTPA and no residual narrow component confirming the absence of pTAM spin probe in the intracellular compartment. In vivo, the physiological parameters reported by pTAM are therefore the extracellular ones.
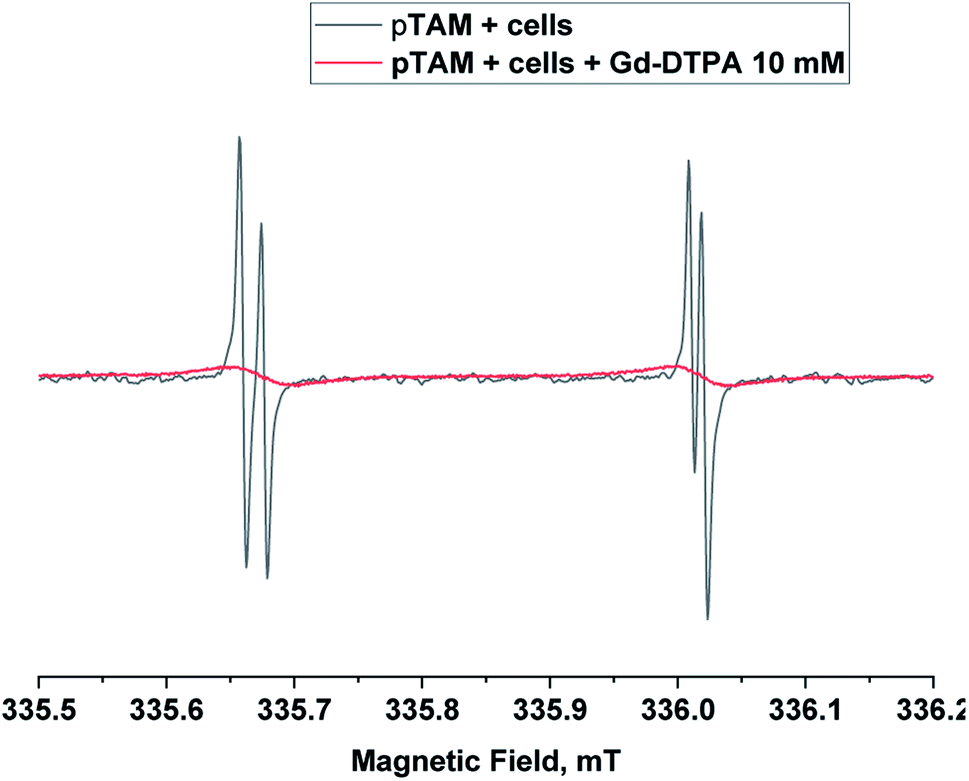 | ||
| Fig. 4 X-band EPR spectra of pTAM (200 μM, 100 μL) incubated with 8.5 × 106 MDA-MB-231 cells without (black) and with 10 mM of Gd-DTPA (red) as extracellular broadening agent. | ||
Next we assessed pTAM cell toxicity using the MTT assay for cell viability and proliferation. MDA-MB-231 cells at 60–70% confluency were incubated with various concentration of pTAM for 24 h. The result (Fig. 5) shows that up to 1 mM, the probe is well tolerated with ∼80% cell viability after 24 h. It is worth noting that only a few hundred micromolar range is required for in vivo L-band spectroscopy and the MTT results show no significant difference between 100 μM of probe and the control. Moreover, pTAM was incubated 24 h with cells while in vivo the probe is cleared from the tissue in less than 1 h.26 Therefore, the probe can be considered as non-toxic upon local injection, which is the mode of delivery for pTAM.19
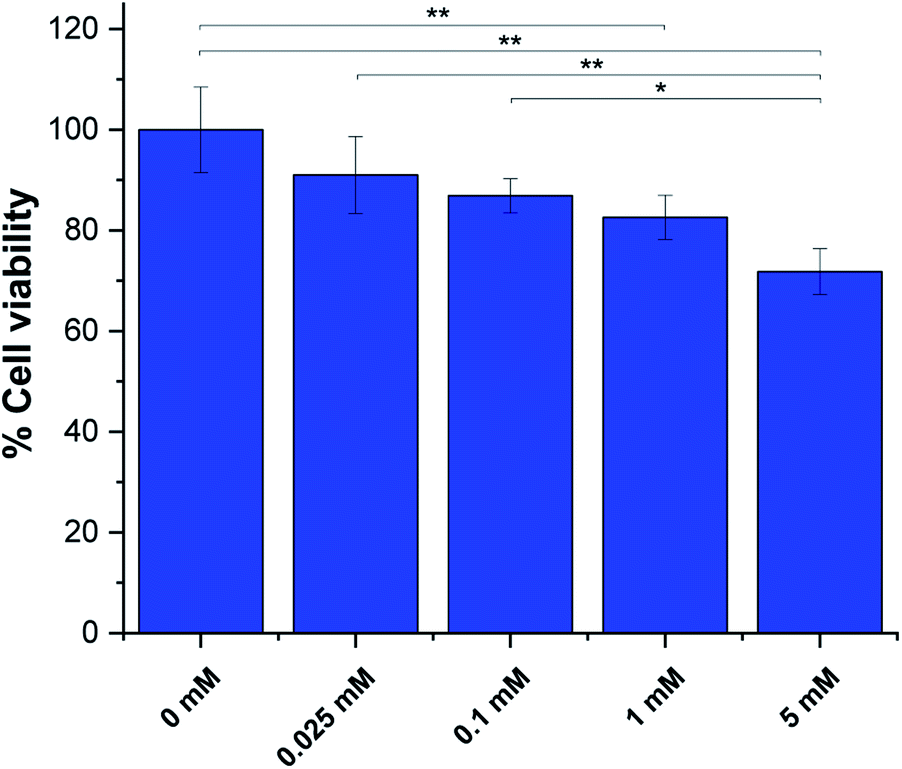 | ||
| Fig. 5 MTT assays for pTAM at various concentration incubated with MDA-MB-231 cells for 24 h. (n = 3, *p < 0.05, **p < 0.01). | ||
Conclusions
In conclusion, we have reported a procedure to synthesize gram quantities of pTAM and a MATLAB application for automatic extraction of pO2, pH, and [Pi] from an experimental spectrum. Furthermore, we showed that pTAM does not cross the cell membrane and has a low cell toxicity for local delivery.Author contributions
B. D. conceived, supervised the project, performed the experiment and wrote the manuscript. T. D. G. developed the app, performed the MTT assay and wrote the manuscript, M. P. performed the synthesis and wrote the manuscript. S. D. developed the fitting algorithm. M. G. performed the cell and animal work, E. H. H. performed the animal work. T. D. E. supervised and discussed the cell and animal work. V. V. K. provided guidance on the fitting of pTAM radical and provided the initial fitting function. All authors have read and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the NIH grants (USA): EB023990, EB028553, CA194013, CA192064, GM104942. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. West Virginia University Health Sciences Center is acknowledged for start-up fund to B. D. Andrey A. Bobko is acknowledged for sharing the initial MATLAB fitting function of pTAM.Notes and references
- V. V. Khramtsov, A. A. Bobko, M. Tseytlin and B. Driesschaert, Anal. Chem., 2017, 89, 4758–4771 CrossRef CAS PubMed.
- J. R. Biller and J. E. McPeak, Appl. Magn. Reson., 2021, 1–27, DOI:10.1007/s00723-020-01304-z.
- I. Gertsenshteyn, M. Giurcanu, P. Vaupel and H. Halpern, J. Physiol., 2021, 599, 1759–1767 CrossRef CAS PubMed.
- B. Epel, S. V. Sundramoorthy, M. Krzykawska-Serda, M. C. Maggio, M. Tseytlin, G. R. Eaton, S. S. Eaton, G. M. Rosen, J. P. Y. Kao and H. J. Halpern, J. Magn. Reson., 2017, 276, 31–36 CrossRef CAS PubMed.
- J. H. Ardenkjær-Larsen, I. Laursen, I. Leunbach, G. Ehnholm, L. G. Wistrand, J. S. Petersson and K. Golman, J. Magn. Reson., 1998, 133, 1–12 CrossRef PubMed.
- U. Sanzhaeva, M. Poncelet, O. Tseytlin, M. Tseytlin, M. Gencheva, T. D. Eubank, V. V. Khramtsov and B. Driesschaert, J. Org. Chem., 2020, 85, 10388–10398 CrossRef CAS PubMed.
- V. Marchand, P. Levêque, B. Driesschaert, J. Marchand-Brynaert and B. Gallez, Magn. Reson. Med., 2017, 77, 2438–2443 CrossRef CAS PubMed.
- B. Driesschaert, V. Marchand, P. Levêque, B. Gallez and J. Marchand-Brynaert, Chem. Commun., 2012, 48, 4049–4051 RSC.
- Y. Liu, Y. Song, A. Rockenbauer, J. Sun, C. Hemann, F. A. Villamena and J. L. Zweier, J. Org. Chem., 2011, 76, 3853–3860 CrossRef CAS PubMed.
- X. Tan, K. Ji, X. Wang, R. Yao, G. Han, F. A. Villamena, J. L. Zweier, Y. Song, A. Rockenbauer and Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 928–934 CrossRef CAS PubMed.
- M. Poncelet and B. Driesschaert, Angew. Chem., Int. Ed., 2020, 59, 16451–16454 CrossRef CAS PubMed.
- B. Driesschaert, A. A. Bobko, V. V. Khramtsov and J. L. Zweier, Cell Biochem. Biophys., 2017, 75, 241–246 CrossRef CAS PubMed.
- C. Rizzi, A. Samouilov, V. Kumar Kutala, N. L. Parinandi, J. L. Zweier and P. Kuppusamy, Free Radicals Biol. Med., 2003, 35, 1608–1618 CrossRef CAS PubMed.
- V. K. Kutala, N. L. Parinandi, J. L. Zweier and P. Kuppusamy, Arch. Biochem. Biophys., 2004, 424, 81–88 CrossRef CAS PubMed.
- Y. Liu, F. A. Villamena, Y. Song, J. Sun, A. Rockenbauer and J. L. Zweier, J. Org. Chem., 2010, 75, 7796–7802 CrossRef CAS PubMed.
- Y. Liu, F. A. Villamena, A. Rockenbauer and J. L. Zweier, Chem. Commun., 2010, 46, 628–630 RSC.
- I. Dhimitruka, A. A. Bobko, T. D. Eubank, D. A. Komarov and V. V. Khramtsov, J. Am. Chem. Soc., 2013, 135, 5904–5910 CrossRef CAS PubMed.
- A. A. Bobko, I. Dhimitruka, J. L. Zweier and V. V. Khramtsov, Angew. Chem., Int. Ed., 2014, 53, 2735–2738 CrossRef CAS PubMed.
- A. A. Bobko, T. D. Eubank, B. Driesschaert, I. Dhimitruka, J. Evans, R. Mohammad, E. E. Tchekneva, M. M. Dikov and V. V. Khramtsov, Sci. Rep., 2017, 7, 41233 CrossRef CAS PubMed.
- A. Taguchi, S. DeVience, B. Driesschaert, V. V. Khramtsov and H. Hirata, Analyst, 2020, 145, 3236–3244 RSC.
- C. Decroos, T. Prangé, D. Mansuy, J.-L. Boucher and Y. Li, Chem. Commun., 2011, 47, 4805–4807 RSC.
- O. Y. Rogozhnikova, V. G. Vasiliev, T. I. Troitskaya, D. V. Trukhin, T. V. Mikhalina, H. J. Halpern and V. M. Tormyshev, Eur. J. Org. Chem., 2013, 2013, 3347–3355 CrossRef CAS PubMed.
- I. Dhimitruka, M. Velayutham, A. A. Bobko, V. V. Khramtsov, F. A. Villamena, C. M. Hadad and J. L. Zweier, Bioorg. Med. Chem. Lett., 2007, 17, 6801–6805 CrossRef CAS PubMed.
- I. Dhimitruka, O. Grigorieva, J. L. Zweier and V. V. Khramtsov, Bioorg. Med. Chem. Lett., 2010, 20, 3946–3949 CrossRef CAS PubMed.
- J. J. Jassoy, A. Berndhäuser, F. Duthie, S. P. Kühn, G. Hagelueken and O. Schiemann, Angew. Chem., Int. Ed., 2017, 56, 177–181 CrossRef CAS PubMed.
- A. A. Gorodetskii, T. D. Eubank, B. Driesschaert, M. Poncelet, E. Ellis, V. V. Khramtsov and A. A. Bobko, Sci. Rep., 2019, 9, 12093 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analysis, calibration, in vivo and cells experiments. See DOI: 10.1039/d1ra04551b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |