
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Aryl iminochromenes inhibit cyclooxygenase enzymes via π–π stacking interactions and present a novel class of anti-inflammatory drugs†
Ha Thi Nguyenab,
Thien-Y. Vuc,
A. Vijay Kumard,
Vo Nguyen Huy Hoangc,
Pham Thi Ngoc Myc,
Prashant S. Mandal d and
Vinay Bharadwaj Tatipamula*ab
d and
Vinay Bharadwaj Tatipamula*ab
aInstitute of Research and Development, Duy Tan University, Da Nang 550000, Vietnam. E-mail: vinaybharadwajtatipamula@duytan.edu.vn
bFaculty of Medicine, Duy Tan University, Da Nang 550000, Vietnam
cFaculty of Pharmacy, Ton Duc Thang University, Ho Chi Minh City 700000, Vietnam
dDepartment of Chemistry, Institute of Chemical Technology, Mumbai 400019, India
First published on 2nd September 2021
Abstract
Cyclooxygenase enzymes (COX1/2) have been widely studied and noted for their role in the biosynthesis of inflammation-induced proteins, prostaglandins and thromboxane. Multiple anti-inflammatory drugs have been developed to target these two enzymes, but most of them appeared to have notable adverse effects, especially on the cardiovascular system and lower gastrointestinal tract, suggesting an urgent need for new potent anti-inflammatory drugs. In this study, we screened twenty-two previously synthesized N-aryl iminochromenes (NAIs) for their anti-inflammatory activity by performing COX-1/2 inhibitory assays. Five compounds (1, 10, 14, 15, and 20) that gave the best in vitro anti-inflammatory results were subjected to an in vivo anti-inflammatory assay using the formalin-induced hind rat paw oedema method, followed by in silico studies using indomethacin and celecoxib as standard drugs. Among them, compound 10 stood out as the best candidate, and the percentage reduction in paw oedema at the dose of 20 mg kg−1 body weight was found to be substantially higher with compound 10 than that with indomethacin. This is mostly due to the excellent suitability of the chromene-phenyl scaffold with a highly concentrated area of aromatic residues, which produced good π–π stacking interactions. Taken together, this study strongly suggests compound 10 as a potential candidate for anti-inflammatory drug research.
Introduction
Inflammation is a normal response of the immune system against injury to neutralize the invading pathogens and repair the injured tissues, thereby promoting wound healing.1 Although inflammation is a self-limiting process, it can become chronic and cause several other serious complications,2,3 including cancer,4 atherosclerosis,5 Alzheimer's disease,6 and rheumatoid arthritis.7 Historically, non-steroidal anti-inflammatory drugs (NSAIDs), including organic acids and non-acidic compounds, have been largely used in the treatment of inflammation.8 Indomethacin (a potent NSAID) is a COX-1/2 inhibitor that has been widely used as an anti-inflammatory, antipyretic, and analgesic agent for decades with several reported severe adverse effects on the GI tract.9,10In 1990s, several first in vivo studies involving anti-inflammatory agents were investigated, resulting in the successful discovery of anti-inflammatory drugs with fewer side effects on the gastrointestinal (GI) tract commonly caused by NSAIDs.11 Thereafter, numerous molecular and cellular pharmacological studies have been carried out, leading to the identification of the cyclooxygenase-1/2 (COX-1/2) enzymes. These enzymes are responsible for the physiological production of prostaglandins (PGs) and thromboxane (TXA2), which primarily cause inflammation.11 COX-1 plays a crucial role in the regulation of GI, renal, and vascular functions, while COX-2 regulates cytokines, endotoxins, and mitogens in inflammation, pain, and fever.12,13 These findings served as the basis for the development of selective COX-2 inhibitor drugs, such as celecoxib and rofecoxib. However, these drugs appeared to have serious adverse effects on the cardiovascular14 and lower GI15 systems. To address these issues, many attempts have been made in the last decade to develop new natural product-based/like-anti-inflammatory drugs with fewer and milder adverse effects.16–20
Noticeably, “chromene”-based compounds have drawn much attention from researchers and are considered as an important source for developing new anti-inflammation drugs.19,21 Different series of chromene analogues have been reported to have pharmacological activities, including anti-inflammatory, anticancer, and antimicrobial actions.21,22 For decades, various 2H-chromene-containing scaffolds like coumarins and 2-imino/oxo-2H-1-benzopyran-3-carboxamides have been well-acknowledged for their anti-inflammatory properties.23–25 Further, the structure–activity relationship (SAR) of 2H-chromene at various positions, including C-3, C-6, and C-7, have been studied.24 Markedly, via a cross-coupling strategy, our group has successfully synthesized twenty-two N-aryl iminochromenes (NAIs),26 which belong to a class of novel 2H-chromene ring-containing molecules with different N-arylimino substituents vicinal to the oxygen (Fig. 1).
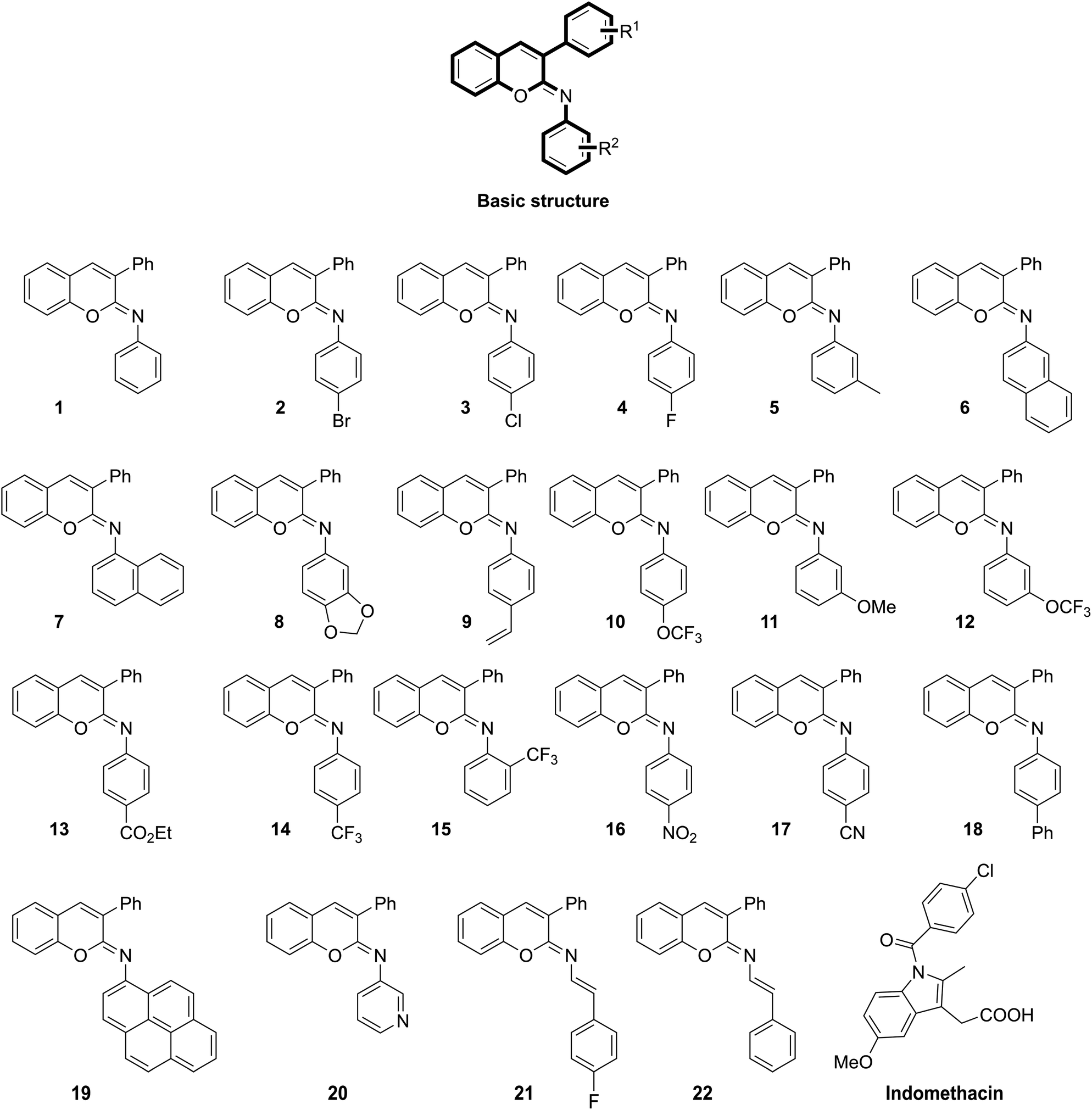 | ||
| Fig. 1 Chemical structures of the previously reported N-aryl iminochromene derivatives26 and the standard drug indomethacin. | ||
In this study, we aimed to investigate the anti-inflammatory activities of these NAIs by performing an in vitro anti-inflammatory assay against COX-1 and COX-2 enzymes, followed by an in vivo anti-inflammatory assay using the formalin-induced hind rat paw oedema method and in silico studies using indomethacin as the reference drug.
Results and discussion
COX-1 inhibitory assay
The COX-1 inhibitory assay results revealed that 20 out of 22 tested compounds had better inhibition capacity against the COX-1 enzyme (with lower IC50 values) compared with that of indomethacin (15.4 ± 0.6 nM). Among these, compounds 10, 15, 5, 14, 16, 1, 20 and 2 exhibited the highest inhibition capacity with IC50 values in the range of 7.3 ± 0.5 to 10.1 ± 0.7 nM, which were significantly lower than that of indomethacin (p < 0.001) (Table 1).| Compound | Half-maximal inhibitory concentration (IC50) valuesa (nM) | |
|---|---|---|
| Cyclooxygenase-1 | Cyclooxygenase-2 | |
| a Values are presented as the mean ± standard deviation of three individual experiments (n = 3). Statistical analyses were done by one-way analysis of variance followed by Tukey's test; *p < 0.05, **p < 0.001 and ***p < 0.0001 indicate a significant reduction compared with that of the standard drug, indomethacin. | ||
| 1 | 9.0 ± 0.5*** | 9.7 ± 1.2* |
| 2 | 10.1 ± 0.7*** | 12.4 ± 0.4 |
| 3 | 10.9 ± 0.5** | 8.5 ± 0.2** |
| 4 | 11.8 ± 0.4 | 11.1 ± 0.5 |
| 5 | 8.4 ± 0.3*** | 10.2 ± 1.1 |
| 6 | 20.7 ± 0.4 | 16.2 ± 0.3 |
| 7 | 12.6 ± 1.3 | 8.3 ± 0.7*** |
| 8 | 12.3 ± 0.6 | 14.0 ± 0.1 |
| 9 | 13.7 ± 0.5 | 12.7 ± 0.5 |
| 10 | 7.3 ± 0.5*** | 9.5 ± 0.5* |
| 11 | 16.0 ± 0.4 | 8.7 ± 0.6** |
| 12 | 18.4 ± 0.6 | 11.3 ± 0.9 |
| 13 | 14.4 ± 1.0 | 10.0 ± 1.2* |
| 14 | 8.7 ± 0.8*** | 8.7 ± 0.7** |
| 15 | 7.4 ± 0.5*** | 7.8 ± 0.6*** |
| 16 | 9.7 ± 0.7*** | 13.1 ± 0.9 |
| 17 | 19.5 ± 0.9 | 19.2 ± 0.6 |
| 18 | 20.6 ± 1.1 | 11.3 ± 0.5 |
| 19 | 9.9 ± 0.9*** | 10.6 ± 1.7 |
| 20 | 9.7 ± 1.2*** | 9.6 ± 1.0* |
| 21 | 11.5 ± 0.8* | 14.0 ± 0.6 |
| 22 | 15.1 ± 0.2 | 18.3 ± 0.9 |
| Indomethacin | 15.4 ± 0.6 | 14.7 ± 1.7 |
| Celecoxib | — | 7.1 ± 0.5 |
COX-2 inhibitory assay
Similarly, the COX-2 inhibitory assay outcomes pointed out that 18 out of 22 compounds in this study displayed higher inhibition ability toward COX-2 than indomethacin (IC50: 14.7 ± 1.7 nM). Of these, compounds 15, 7, 3, 14, 11, 10, 20 and 1 possessed the best inhibition capacity against COX-2 (with the IC50 values varying from 7.8 ± 0.6 to 9.7 ± 1.2 nM), which were significantly lower than that of indomethacin and marginally higher than celecoxib (7.1 ± 0.5) (Table 1).Acute toxicity studies
Compound 1 was used to evaluate the median lethal dose (LD50) in male albino rats using the oral acute toxic class method.27 Accordingly, no clinical signs were observed in the male rats, and the LD50 of compound 1 was found to be above 200 mg kg−1 body weight (b.w), and the low and high dosages were respectively fixed at 10 and 20 mg kg−1 b.w.In vivo studies
The best five compounds, namely 1, 10, 14, 15 and 20, which had the highest protection capacity against both COX-1 and COX-2 enzymes in vitro, were selected for further in vivo studies using the standard protocol of formalin-induced rat paw oedema by plethysmography, as described previously.28 The effects of these compounds and the standard drugs (indomethacin and celecoxib) on the rat paw oedema were measured relative to the control (Fig. 2). The results are presented as the percentage reduction of oedema relative to the basal paw volume at two doses: 10 and 20 mg kg−1 b.w (Fig. 3). Noticeably, the tested NAIs showed good potency in reducing paw oedema at both tested doses compared with indomethacin and celecoxib (Fig. 3). The paw-inflamed rats dosed with compounds 1, 10, 14, 15 and 20 exhibited a remarkable reduction in paw oedema. Of these, compound 14 presented with the lowest percentage reduction at both low and high doses for all the measured time-points (1, 2 and 4 h) (Fig. 3).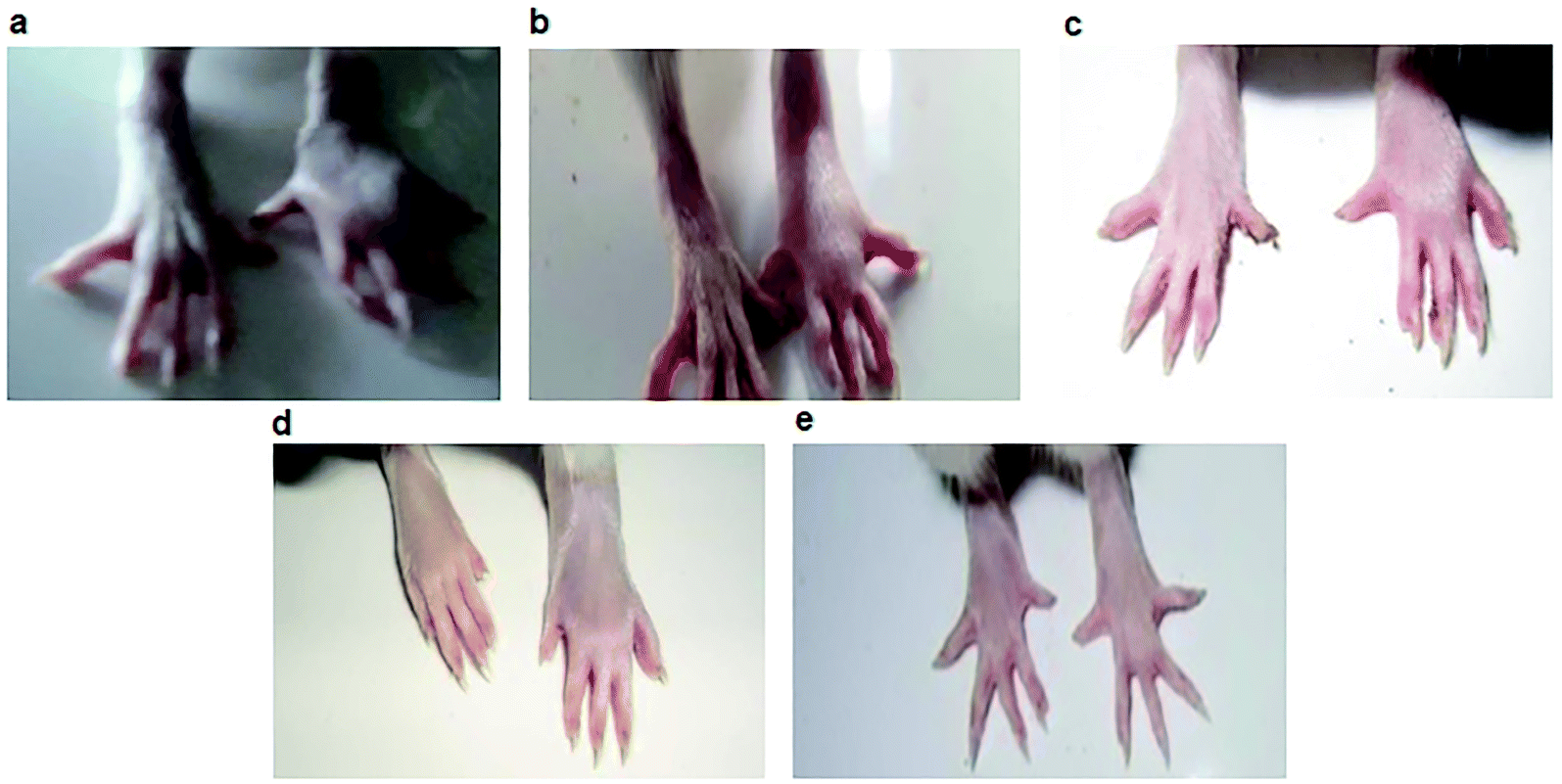 | ||
| Fig. 2 Paw oedema images of albino rats. (a) Control group; (b) indomethacin-treated group; (c) celecoxib-treated group; (d) compound 10-treated group; (e) compound 15-treated group at 20 mg kg−1 body weight. | ||
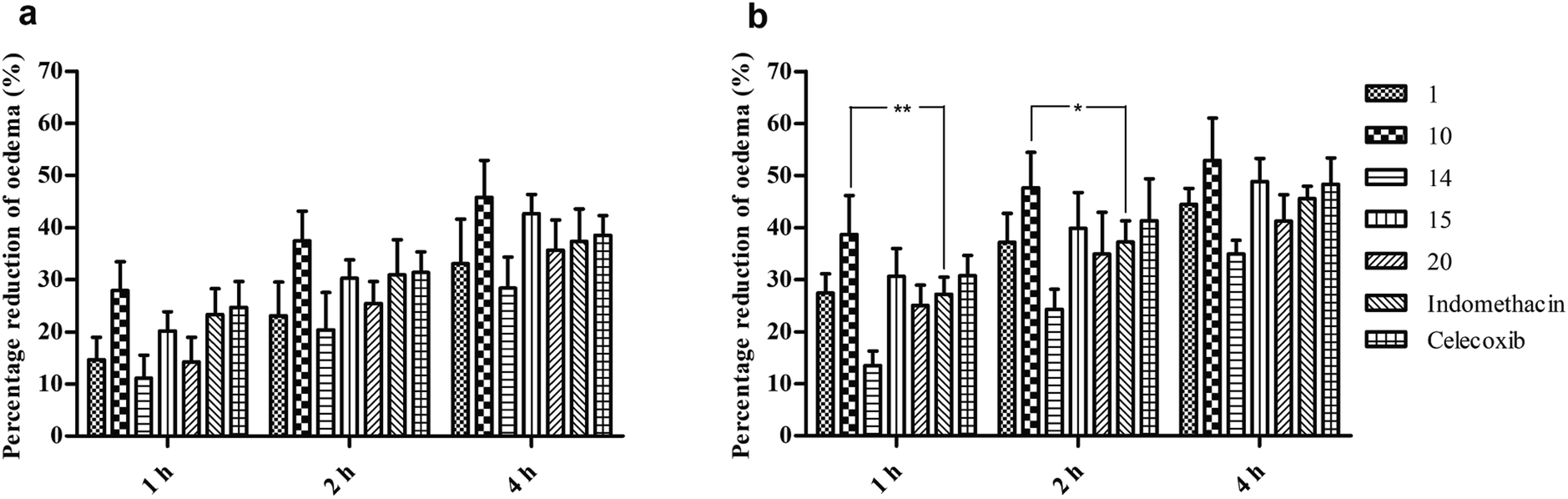 | ||
| Fig. 3 In vivo anti-inflammatory activity of N-aryl iminochromenes. (a) The percentage reduction of rat paw oedema at a low dose (10 mg kg−1 body weight (b.w)), and (b) the percentage of reduction of rat paw oedema at a high dose (20 mg kg−1 b.w) at 1, 2 and 4 h. The data are presented as the mean ± standard error of mean of five biological replicates. The statistical analysis was performed using two-way analysis of variance followed by Bonferroni post-tests. The star (*) mark indicates a significant difference between the two groups with *p < 0.05 or **p < 0.01. | ||
On the other hand, compounds 10 and 15 were the most effective compounds in reducing paw oedema in the rats. Specifically, treatment with a low dose of compound 10 showed a higher reduction in rat paw oedema at all the three time-points (27.9 ± 2.5, 37.5 ± 2.5, and 45.8 ± 3.2%) compared with those of indomethacin (23.3 ± 2.2, 31.0 ± 3.0, and 37.4 ± 2.8%) and celecoxib (24.7 ± 2.2, 31.4 ± 1.8, and 38.5 ± 1.7) though the differences were not significant (Fig. 3a). At a high dose, the compound 10-treated group showed potent activity with 38.7 ± 3.4, 47.6 ± 3.1, and 52.9 ± 3.6% reduction in rat paw oedema at 1, 2 and 4 h, respectively. Noticeably, the percentage of paw oedema inhibition in the compound 10-treated group was significantly higher than that of the indomethacin-treated group at 1 h (p < 0.01) and 2 h (p < 0.05) after treatment (Fig. 3b). Similarly, compound 15 exhibited a good reduction in paw oedema, and the reduction rates were found to be 20.2 ± 1.7, 30.3 ± 1.6, and 42.7 ± 1.6% for the low dose and 30.7 ± 2.4, 39.9 ± 3.1, and 48.9 ± 2.0% for the high dose at 1, 2 and 4 h, respectively, equivalent to that of the indomethacin and celecoxib (Fig. 3).
Structure–activity relationship (SAR)
According to the results, most of the tested NAIs were potent against inflammation. From the COX-1/2 inhibitory studies, we explored the SAR of the new class of NAIs that have various substituents at different R2 positions for their anti-inflammatory activities.• The synthesis of fluoro-substituted derivatives (like –CF3) at the ortho or para position of R2 improved the biological activity.
• Substitution with electron-donating groups (like –CH3) at the meta position of R2 improved the pharmacological activity than did electron-withdrawing groups, such as –OCF3 and –OCH3.
• The placement of electron-withdrawing groups, such as phenyl, nitro, esters, and cyano groups, at the para position of R2 produces less activity.
• The presence of heterocyclic aromatic moieties (like 1,3-benzodioxole and pyridine) in place of R2 decreased the potency.
• The substitution of halogens at the para position of R2 produced activity in the order Br > Cl > F.
• The replacement of the phenyl moiety with styrene at R2 produced less pharmacological activity than its para-fluoro derivative.
• The replacement of the phenyl moiety with polycyclic/bicyclic rings like pyrene/naphthalene at R2 was found to decrease the biological activity.
• The para substitution of a phenyl moiety at R2 shortened the activity.
Docking studies
To understand the possible mechanisms of action through which the NAIs inhibit COX-1/2 functions, the five ligands were subjected to molecular modelling. Three numerical values for the Glide rigid docking, induced fit docking (IFD) top 1 and IFD top 2 poses with COX-1 and COX-2 are presented in Table 2.| Ligands | Cyclooxygenase-1 | Cyclooxygenase-2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MM-GBSA estimation (kcal mol−1) | ΔGexp (kcal mol−1) | RMSD of rigid and IFD top 1 | MM-GBSA estimation (kcal mol−1) | ΔGexp (kcal mol−1) | RMSD of rigid and IFD top 1 | |||||
| Rigid | IFD (top 1) | IFD (top 2) | Rigid | IFD (top 1) | IFD (top 2) | |||||
| a MM-GBSA: molecular mechanics/generalized born surface area; IFD: induced fit docking; RMSD: root-mean-square deviation; ΔGexp: experimental binding free energy. | ||||||||||
| 1 | −80.6 | −84.3 | −84.3 | −11.0 | 0.1 | −73.7 | −95.0 | −84.7 | −11.0 | 0.2 |
| 10 | −73.8 | −86.2 | −96.6 | −11.2 | 1.0 | −58.2 | −89.0 | −89.0 | −11.0 | 0.4 |
| 14 | −78.5 | −88.0 | −88.0 | −11.1 | 1.2 | −53.6 | −88.7 | −88.7 | −11.1 | 0.9 |
| 15 | −66.2 | −95.6 | −95.6 | −11.2 | 0.6 | −61.5 | −106.9 | −106.9 | −11.1 | 0.6 |
| 20 | −78.4 | −87.9 | −83.1 | −11.0 | 0.1 | −73.6 | −83.1 | −83.1 | −11.0 | 0.5 |
| Indomethacin | −32.3 | −76.2 | −55.2 | −11.0 | 0.4 | −26.4 | −53.9 | −44.6 | −10.8 | 0.9 |
| Celecoxib | — | — | — | — | — | −37.6 | −49.8 | −60.9 | −11.2 | 0.9 |
| Average | −68.3 | −88.1 | −87.4 | −11.0 | 0.8 | −54.9 | −84.2 | −84.3 | −11.0 | 0.7 |
Firstly, the five studied compounds had good mean experimental binding energies at −11.0 kcal mol−1 similar to those of indomethacin and celecoxib. This was a typical case where all five compounds were with nearly flat SAR. In other words, even with different substituents on the central scaffold, these compounds still matched the binding sites of the COX-1/2 enzyme, but no key interactions could be generated. Therefore, a correlation between the theoretical and experimental energies has not been performed. This finding, however, highlighted the decisive role of the central scaffolds and suggested the need for further theoretical analyses.
For the COX-1 protein, the theoretically estimated values showed a good correlation (R2) with the experimental results, and the R2 values of rigid docking, IFD top 1 and IFD top 2 were 0.61, 0.14, and 0.84, respectively (Table 2). Generally, due to a higher degree of accuracy, IFD top 1 or the flexible selection-based IFD top 2 will give a better correlation than that of rigid docking.29 In this study, the correlations were typically high for rigid docking and IFD top 2 and moderate for IDF top 1. Moreover, all these five ligands exhibited stronger interactions with the COX-1 enzyme with the average free binding energies of −68.3, −88.1, and −87.4 kcal mol−1 using the rigid, IFD top 1, and IFD top 2 methods, respectively, much higher compared to those of indomethacin (−32.3, −76.2, and −55.2 kcal mol−1). Obviously, the binding energy calculated by IFD was much stronger than that from rigid docking mostly due to the ability to alter the binding site in IFD to conform with the shape and binding mode of the ligands.
According to the binding poses of the five ligands predicted by IDF top 2, hydrophobic interactions were the main interactions of this congeneric series (Fig. 4). The three aromatic rings chromene, phenyl, and benzenaminium were fused together in these ligands (Fig. 1). They were strongly positioned by π–π stacking with Tyr385, Trp387, Phe518, and/or Tyr348. A marked improvement was recorded when the aromatic electrophile of –OCF3 was substituted at the para position of benzenaminium in ligand 10 since anchoring by the –OCF3 group can create many interesting interactions, which were further observed by molecular dynamics (MD) simulation. The Arg120 residue, which is the usual target for H-bond formation in anti-inflammatory drugs, such as indomethacin, was attenuated by π–cation interactions in this congeneric series. The decline inevitably raised questions about the superior inhibitory capacity of these compounds relative to the reference drug and the factors that actually govern its activity.
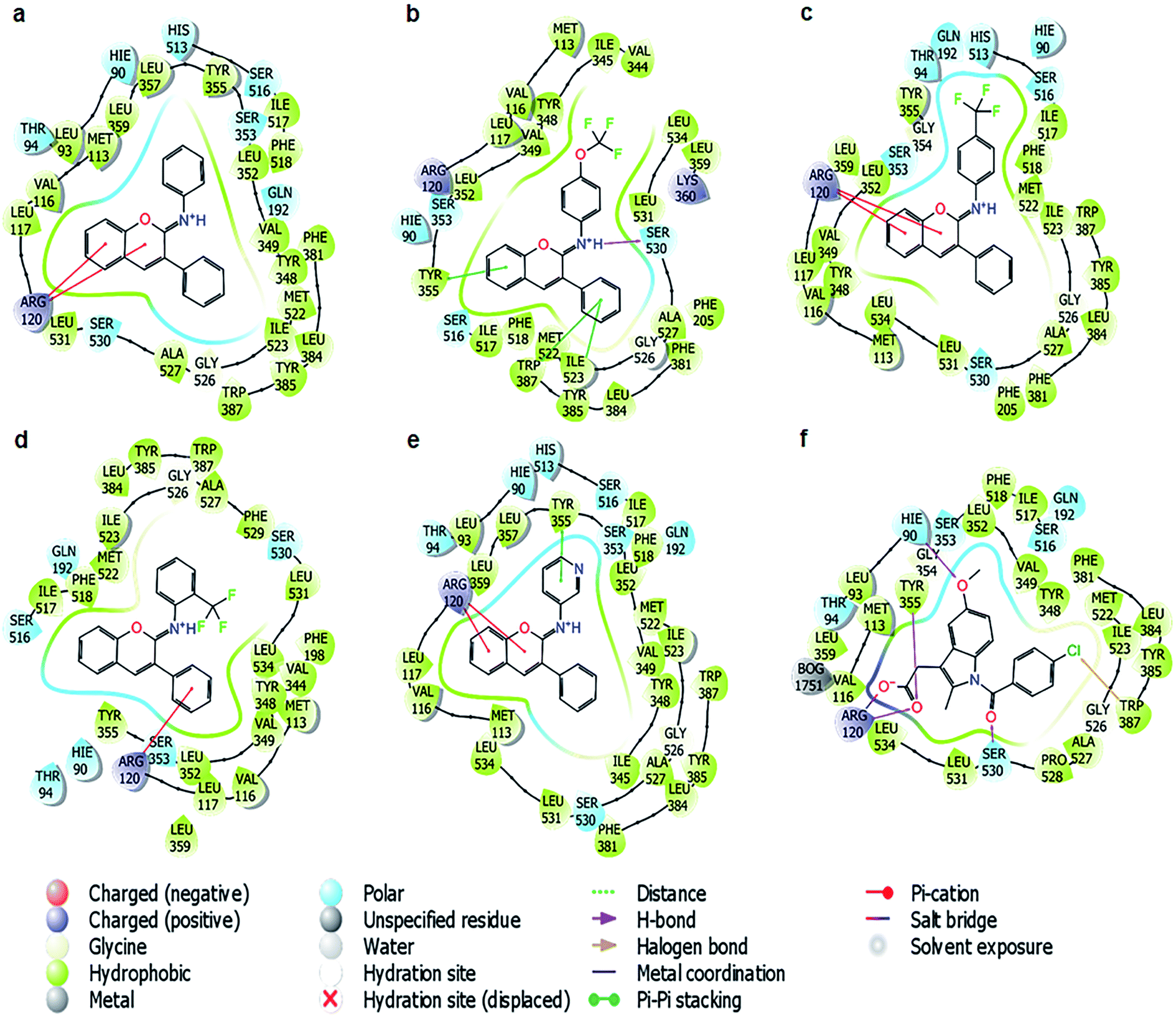 | ||
| Fig. 4 The binding poses of the five ligands and indomethacin with the cyclooxygenase-1 protein predicted in IFD top 2. (a) Ligand 1; (b) ligand 10; (c) ligand 14; (d) ligand 15; (e) ligand 20; (f) standard drug, indomethacin. | ||
Similarly, for the COX-2 protein, a good correlation was observed, with the R2 of both IFD top 1 and IFD top 2 at 0.64, while that of rigid docking was stable around 0.52. The mean value of the free binding energy increased by more than 30 kcal mol−1 while using the IFD methods. Following a similar trend to COX-1, when these ligands could not form a direct H-bond with the Arg120 residue of the protein, they induced π–π stacking with Tyr385, Trp387 or Tyr355 on one side and gained access to Arg120 on the other side. This binding mode was completely different from that of celecoxib, in which the hydrophobic interactions are overshadowed by three hydrogen bonds with Arg120, Tyr355, and Val116 (Fig. S1†), suggesting a different role of the hydrophobic interactions and hydrogen bonds in the COX-1/2 active sites and the need for dynamic simulation analysis to understand this congeneric series better.
Since the theoretical calculations showed good coherence with the in vitro and in vivo assays, the ligand-COX-1 complexes were further analyzed using MD simulations to shed light on the inhibitory mechanisms of these compounds on the target proteins.
The first question was whether the π–cation interactions of these ligands with Arg120 were sufficient to inhibit the COX-1 protein? Statistics showed that the π–cation interaction was not a dominant interaction in all these cases. Specifically, only ligand 1 showed a 100% interaction fraction for the π–cation interaction with Arg120, while the rest of the ligands showed rates below 50%, declining in the order of 20 (50%), 14 (40%), 10 (10%), and 15 (0%). In the opposite direction, the hydrophobic interactions, particularly π–π stacking with several aromatic residues, emerged as the substitute for the π–cation interaction of Arg120. From 30% in the case of ligand 1, these interactions rapidly rose to 50% for ligand 20 and almost 100% for the remaining ligands 14, 15, and 10 (Fig. 5 and S2–S5†). Meanwhile, the inhibition capacity was also proportional to these π–π stacking interactions. Hence, the next question was whether these interactions really played an essential role even in pairing with Arg120.
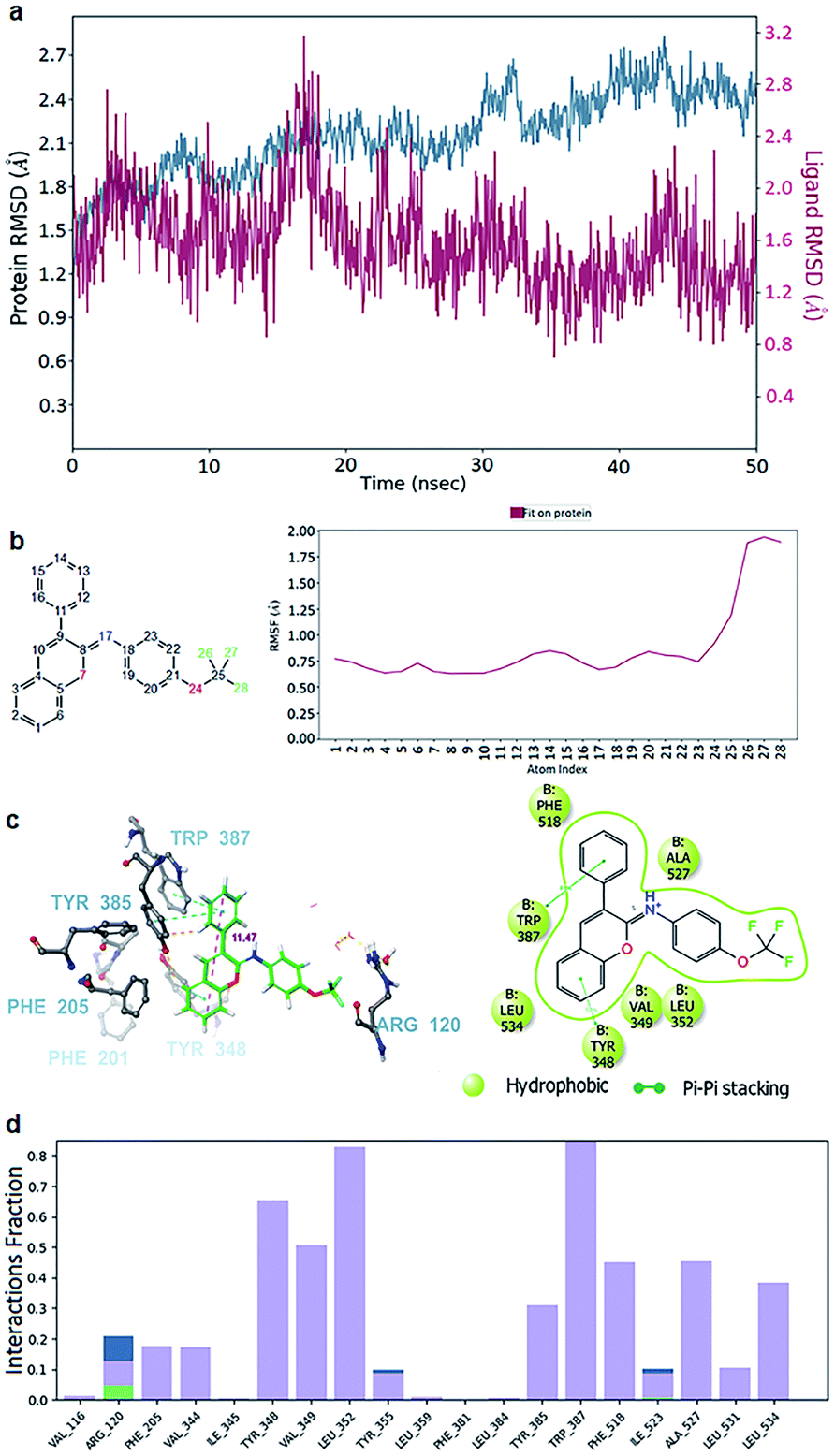 | ||
| Fig. 5 Molecular dynamics simulation (MDS) of the ligand 10–cyclooxygenase-1 complex. (a) Root-mean-square deviation of the protein (azure) and ligand 10 (red signal). (b) The root mean square fluctuation (RMSF) of molecule 10 when fitted on the protein (red line). The atom numbers of ligand 10 (left) corresponding to the X-axis of the RMSF plot (right). (c) Snapshots 3D (left) from the stable segments of MDS show that ligand 10 formed two π–π stacking interactions with Tyr 385, Trp 387 and Tyr 348 (right). The red dashed line is the measured length between the farthest atoms of the phenyl and chromene groups. The interactions that occur for more than 30.0% of the simulation time in the selected trajectory (0.00 through 50.05 nanoseconds) are shown. (d) Interaction diagram demonstrating the percentage interaction of ligand 10 with the surrounding residues. The red dashed line was the measured length between the farthest atoms of the phenyl and chromene groups. | ||
The answers could be found at the binding sites of COX-1 and COX-2, which were quite similar to a triangle plane ABC (Fig. 6), in which the AB edge (17 Å in length approximately between –CH3–hydrogen of Met 522 and –CH–hydrogen of ile 345) was the most concentrated place of aromatic residues, such as Phe518, Trp387, and Tyr385., and the position of Arg120 was at vertex C of the triangle.
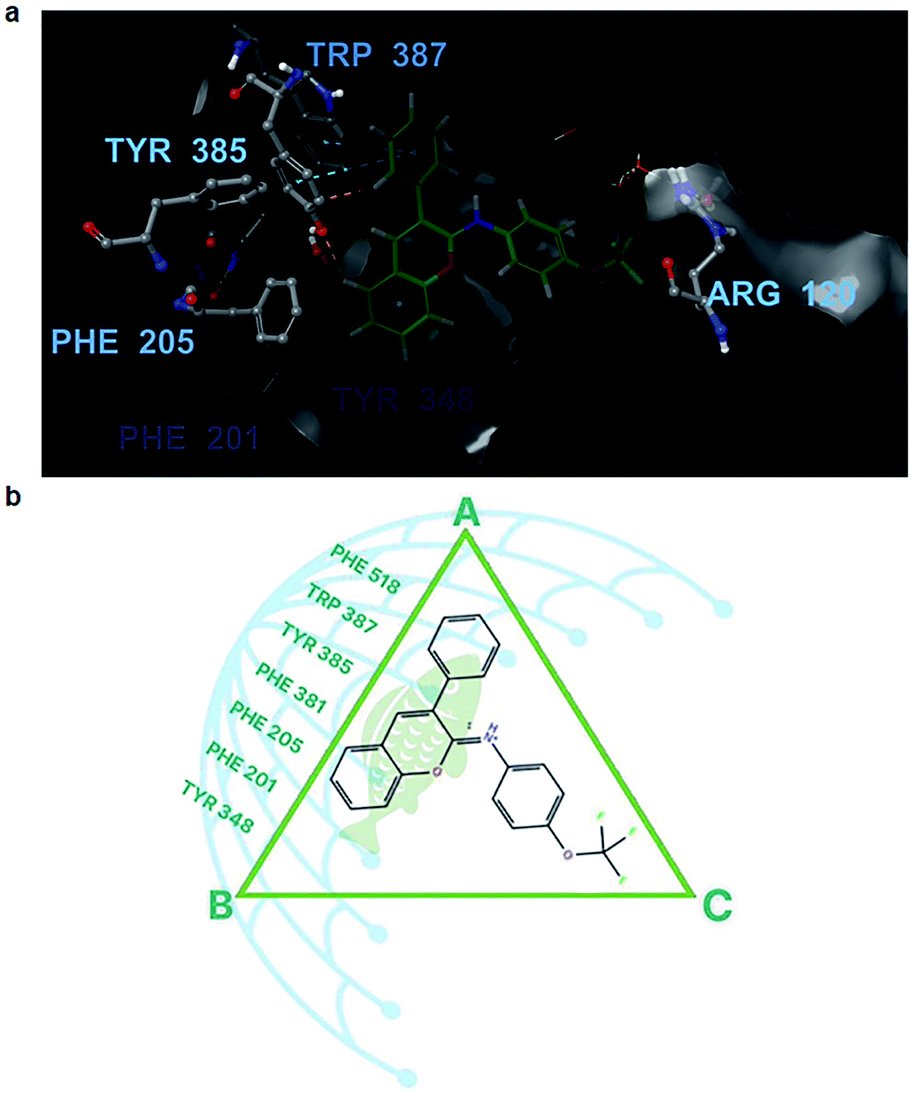 | ||
| Fig. 6 Ligand 10–cyclooxygenase-1 (COX-1) interactions. (a) The binding site of COX-1 (grey shape) is inhibited by ligand 10 (green ball and sticks). The surrounding residues are presented as grey balls and sticks. Oxygen is presented in red, nitrogen in blue, carbon in grey, and hydrogen in white. (b) The active site of COX-1 with the distribution of aromatic fused residues on the left side. The fish scales represent the aromatic scaffold of ligand 10, and each mesh of the fishnet is an aromatic ring that forms a sturdy π–π stacking system on the fish scales. | ||
With ligand 10 as the example, the 50 ns molecular dynamic simulation demonstrated many aspects. Firstly, the root-mean-square deviation (RMSD) of ligand 10 was stable under 2.0 Å (Fig. 5a). Next, the chromene or phenyl atoms were the least volatile with the root mean square fluctuation (RMSF) under 1 Å (Fig. 5b). Finally, the hydrophobic interactions due to the π–π stacking of ligand 10 with the aromatic residues Tyr348, Tyr385, Trp387, and Phe518 were all observed over 40% of the interactive time (Fig. 5d). Meanwhile, the length of the chromene frame with the phenyl group was about 12 Å, which perfectly suited their position along the edge AB (Fig. 5c). Therefore, the chromene scaffolds and phenyl groups were squeezed by the π–π stacking interactions with Phe518, Trp387, Tyr385, Phe381, Phe205, Phe201, and/or Tyr348 (Video S1†). Molecules with lengths greater than 12 Å, however, will not be able to produce a drastic reduction in activity because they form bad interactions with the surrounding residues even if they have many polycyclic/bicyclic rings (i.e., compounds 6, 18, and 19). Additionally, a small but worthy contribution was from the interactions of ligand 10 with Arg120. This ligand formed a direct H-bond with Arg120 through the –OCF3 oxygen several ns after the beginning of the simulation, and then it was quickly replaced by hydrophobic interactions or H-bonds via water brigdes (Fig. 5c and 6a). Although they only contributed for 20% of the interaction time, ligand 10 was slightly better than the other compounds in terms of free binding energy and presented the best in vitro and in vivo results of all the tested compounds.
The remaining compounds differed in the distribution of the three principal groups, but they had a common feature in the arrangement such that the number of π–π stacking interactions with the AB edge of the triangle were as high as possible. The MD simulations of the remaining ligands also presented significant hydrophobic interactions at edge AB (Fig. S2–S5 and Videos S2–S5†).
In short, this study has identified a group of NAIs that have potent anti-inflammatory activity and explored the mechanism underlying their inhibitory activity, which was mostly by forming π–cation or π–π stacking interactions with the Arg120 residue. It also demonstrates, for the first time, the role of aromatic moieties in the COX-1 binding site which are as important as the Arg120 residue.
Experimental methods
Materials
Twenty-two NAIs (1–22) previously synthesized via a cross-coupling strategy26 were used in this study (Fig. 1). Indomethacin and celecoxib were purchased from Sigma Aldrich (USA). All the chemicals used in this study were of analytical grade.COX-1/2 inhibitory assays
By using the COX inhibitor screening assay kit (no. 560131, Cayman Chemical Company, MI, USA), the COX-1/2 inhibitory activities of the NAIs were estimated.30 Indomethacin was used as the reference drug. The test was carried out according to the manufacturer's instructions with some changes. In brief, to 10 μL of the enzyme (COX-1 or COX-2), 10 μL of each test compound solution at four different concentrations (2.5, 5.0, 7.5, 10.0 mg mL−1) and 960 μL of a reaction buffer (0.1 M Tris–HCl buffer, pH 8.0, containing 5 mM EDTA and 2 mM phenol) were added. The mixture was incubated at 37 °C for 10 min. Next, 10 μL of 100 μM arachidonic acid (substrate) was added, and the mixture was incubated for 2 min at 37 °C. Afterward, 50 μL of 1 M HCl was added to stop the reaction. The absorbance was spectrophotometrically measured at 410 nm against the blank. This inhibitory assay was performed thrice, and the IC50 values were attained via logistic regression analysis.Animals
Healthy albino rats of both sexes matured between 2-3 months and weighing 180–200 g were procured from the Animal House of Duy Tan University, Vietnam, and fed with the standard pellet diet and water (ad libitum) for at least seven days before the study. The experimental protocols were maintained as per the guidelines of the Control and Supervision of Experiments on Animals for experimental clearance by Duy Tan University, Vietnam (code: DTU.2020.934).Acute toxicity study
By using the oral acute toxic class method,27 compound 1 was subjected to an acute toxicity study in healthy male adult albino rats as per the guidelines of the Organization for Economic Co-operation and Development. Compound 1 at 200 mg kg−1 b.w was administered orally to five male rats (n = 5), and they were examined for physiological and biological changes for 24 h.In vivo anti-inflammatory activity
By using the formalin-induced hind rat paw oedema assay,28 the in vivo anti-inflammatory activity of the NAIs was estimated. The healthy adult albino rats of both sexes were grouped into thirteen batches containing five adult albino rats (n = 5) each. The first batch served as the normal control (dosed only with 0.5% w/v carboxymethyl cellulose (CMC)), while the next two batches were administered with the standard drug indomethacin at 10 and 20 mg kg−1 b.w, respectively, and the remaining batches were treated with the five selected compounds (1, 10, 14, 15, and 20) at 10 and 20 mg kg−1 b.w. All the test samples were deliquesced in 0.5% w/v CMC and administered intraperitoneally. After 30 min of test sample administration, 0.1 mL of formalin (1% w/v) was administered in the sub-plantar region of the left paw of the rats, while the right paw (non-inflamed) was used as a reference for inflammation. The rat paw oedema volume of all the tested adult albino rats was measured at 1, 2, and 4 h after the test sample dosage using plethysmography. Finally, the measured percentage variation in rat paw oedema (using the below equation) was compared with that caused by indomethacin.| Percentage reduction (%) = (C − T)/C × 100 |
Statistics
The in vitro and in vivo results are presented as the mean ± SD and mean ± standard error of three and five independent experiments, respectively. The statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey's test for in vitro studies and a two-way ANOVA followed by Bonferroni post-tests for in vivo studies. The differences were considered statistically significant if *p < 0.05 or **p < 0.01 or ***p < 0.0001 when compared to positive control.Compound docking and molecular dynamics simulations
In silico docking, the MM-GBSA free binding energy evaluations, and rendering the model outputs were executed using the Schrödinger software 2020-3. The correlation scatter plots were generated by Tableau 2020.2. The published crystal structure of the COX-1 (PDB: 3KK6) and COX-2 (PDB: 1CX2) receptors were imported and prepared using the Protein Preparation Wizard31 (Maestro software, Schrödinger Release 2020-3). Next, the structures of molecules 1–22 and the reference drug indomethacin were created and prepared by Ligprep.32 The general process of standard precision (SP), extra precision (XP) docking and molecular mechanics/generalized born surface area (MM-GBSA) free binding energy estimation have been previously described.31 Notably, the Ligand Filtering operation was performed to refine all ligands using the SP method with Glidescore ≤−8.5 kcal mol−1 before proceeding with XP docking. An induced fit docking (IFD) protocol33–35 was used in tandem with the Glide SP, XP dockings to predict the accurate complex structures of the ligand and proteins. From there, the system set-up for molecular dynamics simulations for docked ligands was built with COX-1 and COX-2 by using Desmond[i]. The general conditions of Desmond were established according to the previous study.36 The solvent model was set with a flexible simple point-charge water model with the OPLS3e force field. 50 nanoseconds (ns) and 50 picoseconds (ps) were set as the total simulation time and the trajectory recording interval, respectively. The temperature was 300.0 K, and the pressure was 1.01325 bar, while the Relax model system was the default option. The experimental binding energies (ΔGexp) were calculated using the equation ΔGexp = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnIC50, where the gas constant R = 1.987 cal mol−1 K−1 and the temperature T = 300 K.
lnIC50, where the gas constant R = 1.987 cal mol−1 K−1 and the temperature T = 300 K.
Conclusions
Twenty-two previously synthesized NAIs were screened for their anti-inflammatory activity by performing an in vitro anti-inflammatory assay against COX-1/2 enzymes. Five compounds (1, 10, 14, 15, and 20) that gave the best in vitro anti-inflammatory results were subjected to an in vivo anti-inflammatory assay using the formalin-induced hind rat paw oedema method, followed by in silico studies using indomethacin as the standard drug. Among these, compound 10 exhibited an outstanding anti-inflammatory property in all the in vitro, in vivo and in silico studies. This achievement was due to the excellent suitability of the chromene-phenyl scaffold with a highly concentrated nucleated area of aromatic residues, which produced good π–π stacking interactions. The results of this study show that compound 10 is a potent anti-inflammatory agent that can be considered as a candidate for anti-inflammatory drug research.Funding
This study was supported by the Department of Science and Technology (India) for the research grant, BRICS (Grant no. DST/ICP-BIO/1798) for the research funding. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.Ethical statement
The animal experiments were performed in accordance with the guidelines of Control and Supervision of Experiments on Animals and approved by the ethics committee at the Duy Tan University, Vietnam (code: DTU.2020.934).Author contributions
HTN and VBT designed and conducted the in vitro and in vivo experiments and data analysis and co-wrote the manuscript. T-YV, VNHH, and PTNM designed and conducted the in silico experiments and data analysis. VKA and PSM synthesized the chemical compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Mr Toai Tuyn Phan (a technical staff, the High Potential Computer group, Ton Duc Thang University) and Dr Duc Duy Vo (Uppsala University, Sweden) for their supports.References
- M. Y. Huang, J. Lin, Z. J. Huang, H. G. Xu, J. Hong, P. H. Sun, J. L. Guo and W. M. Chen, Medchemcomm, 2016, 7, 658–666 RSC.
- L. Ferrero-Miliani, O. H. Nielsen, P. S. Andersen and S. E. Girardin, Clin. Exp. Immunol., 2007, 147, 227–235 CAS.
- L. Z. Chen, W. W. Sun, L. Bo, J. Q. Wang, C. Xiu, W. J. Tang, J. B. Shi, H. P. Zhou and X. H. Liu, Eur. J. Med. Chem., 2017, 138, 170–181 CrossRef CAS PubMed.
- X. Y. Lu, Z. C. Wang, S. Z. Ren, F. Q. Shen, R. J. Man and H. L. Zhu, Bioorg. Med. Chem. Lett., 2016, 26, 3491–3498 CrossRef CAS PubMed.
- P. Libby, Nature, 2002, 420, 868–874 CrossRef CAS PubMed.
- H. Y. Kim, H. V. Kim, S. Jo, C. J. Lee, S. Y. Choi, D. J. Kim and Y. Kim, Nat. Commun., 2015, 6, 1–14 Search PubMed.
- C. Rommel, M. Camps and H. Ji, Nat. Rev. Immunol., 2007, 7, 191–201 CrossRef CAS PubMed.
- C. A. Velázquez, Q. H. Chen, M. L. Citro, L. K. Keefer and E. E. Knaus, J. Med. Chem., 2008, 51, 1954–1961 CrossRef PubMed.
- S. Lucas, Headache, 2016, 56, 436–446 CrossRef PubMed.
- M. P. Draper, R. L. Martell and S. B. Levy, Br. J. Cancer, 1997, 75, 810–815 CrossRef CAS PubMed.
- G. A. FitzGerald and C. Patrono, N. Engl. J. Med., 2001, 345, 433–442 CrossRef CAS PubMed.
- K. D. Rainsford, Am. J. Med., 1999, 107, 27–35 CrossRef.
- K. D. Rainsford, Subcell. Biochem., 2007, 42, 3–27 CAS.
- G. A. FitzGerald, N. Engl. J. Med., 2004, 351, 1709–1711 CrossRef CAS PubMed.
- J. Jacob P, S. L. Manju, K. R. Ethiraj and G. Elias, Eur. J. Pharm. Sci., 2018, 121, 356–381 CrossRef PubMed.
- J. Bost, A. Maroon and J. Maroon, Surg. Neurol. Int., 2010, 1, 80 CrossRef PubMed.
- N. Sultana and Z. Saeed Saify, Anti-Inflammatory Anti-Allergy Agents Med. Chem., 2012, 11, 3–19 CrossRef CAS.
- M. B. Palkar, A. S. Singhai, P. M. Ronad, A. H. M. Vishwanathswamy, T. S. Boreddy, V. P. Veerapur, M. S. Shaikh, R. A. Rane and R. Karpoormath, Bioorg. Med. Chem., 2014, 22, 2855–2866 CrossRef CAS PubMed.
- S. A. Patil, S. A. Patil and R. Patil, Future Med. Chem., 2015, 7, 893–909 CrossRef CAS PubMed.
- D. Kashyap, A. Sharma, H. S. Tuli, S. Punia and A. K. Sharma, Recent Pat. Inflammation Allergy Drug Discovery, 2016, 10, 21–33 CrossRef CAS PubMed.
- M. Costa, T. A. Dias, A. Brito and F. Proença, Eur. J. Med. Chem., 2016, 123, 487–507 CrossRef CAS PubMed.
- P. Proksch and E. Rodriguez, Phytochemistry, 1983, 22, 2335–2348 CrossRef CAS.
- I. E. Bylov, M. V. Vasylyev and Y. V. Bilokin, Eur. J. Med. Chem., 1999, 34, 997–1001 CrossRef CAS PubMed.
- J. Grover and S. M. Jachak, RSC Adv., 2015, 5, 38892–38905 RSC.
- H. M. Revankar, S. N. A. Bukhari, G. B. Kumar and H. L. Qin, Bioorg. Chem., 2017, 71, 146–159 CrossRef CAS PubMed.
- P. S. Mandal and A. V. Kumar, Synlett, 2016, 27, 1408–1412 CrossRef CAS.
- V. B. Tatipamula, M. K. Kolli, S. B. Lagu, K. R. Paidi, P. R. Reddy and R. P. Yejella, Pharmacol. Rep., 2019, 71, 233–242 CrossRef CAS PubMed.
- V. B. Tatipamula and G. S. Vedula, Nat. Prod. J., 2020, 10, 87–93 CAS.
- E. B. Miller, R. B. Murphy, D. Sindhikara, K. W. Borrelli, M. J. Grisewood, F. Ranalli, S. L. Dixon, S. Jerome, N. A. Boyles, T. Day, P. Ghanakota, S. Mondal, S. B. Rafi, D. M. Troast, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2021, 17, 2630–2639 CrossRef CAS PubMed.
- H. T. Nguyen, T.-Y. Vu, V. Chandi, H. Polimati and V. B. Tatipamula, Sci. Rep., 2020, 10, 15965 CrossRef CAS PubMed.
- J. Li, R. Abel, K. Zhu, Y. Cao, S. Zhao and R. A. Friesner, Proteins: Struct., Funct., Bioinf., 2011, 79, 2794–2812 CrossRef CAS PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- R. Farid, T. Day, R. A. Friesner and R. A. Pearlstein, Bioorg. Med. Chem., 2006, 14, 3160–3173 CrossRef CAS PubMed.
- W. Sherman, H. S. Beard and R. Farid, Chem. Biol. Drug Des., 2006, 67, 83–84 CrossRef CAS PubMed.
- W. Sherman, T. Day, M. P. Jacobson, R. A. Friesner and R. Farid, J. Med. Chem., 2006, 49, 534–553 CrossRef CAS PubMed.
- T. V. Pham, H. N. T. Hoang, H. T. Nguyen, H. M. Nguyen, C. T. Huynh, T. Y. Vu, A. T. Do, N. H. Nguyen and B. H. Do, BioMed Res. Int., 2021, 2021, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04407a |
| This journal is © The Royal Society of Chemistry 2021 |