
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMarkovnikov-addition of H-phosphonates to terminal alkynes under metal- and solvent-free conditions†
Nana Xin *a,
Yongjian Liana,
Yongzheng Lva,
Yongjie Wangb,
Xian-Qiang Huanga and
Chang-Qiu Zhao*a
*a,
Yongjian Liana,
Yongzheng Lva,
Yongjie Wangb,
Xian-Qiang Huanga and
Chang-Qiu Zhao*a
aInstitution of Functional Organic Molecules and Materials, Shandong Provincial Key Laboratory of Chemical Energy Storage and Novel Cell Technology, Liaocheng University, College of Chemistry and Chemical Engineering, Liaocheng, Shandong 252059, China. E-mail: xinnana2008@163.com; literabc@hotmail.com
bLiaocheng Power Supply Company, Liaocheng, Shandong 252059, China
First published on 19th July 2021
Abstract
An addition of H-phosphonates to aryl alkynes was realized under solvent- and metal-free conditions, affording Markovnikov-selective α-vinylphosphonates in moderate to good yields. A wide range of aryl alkynes could be applied for the reaction. A tentative mechanism of addition–substitution was proposed based on in situ 31P {1H} NMR studies.
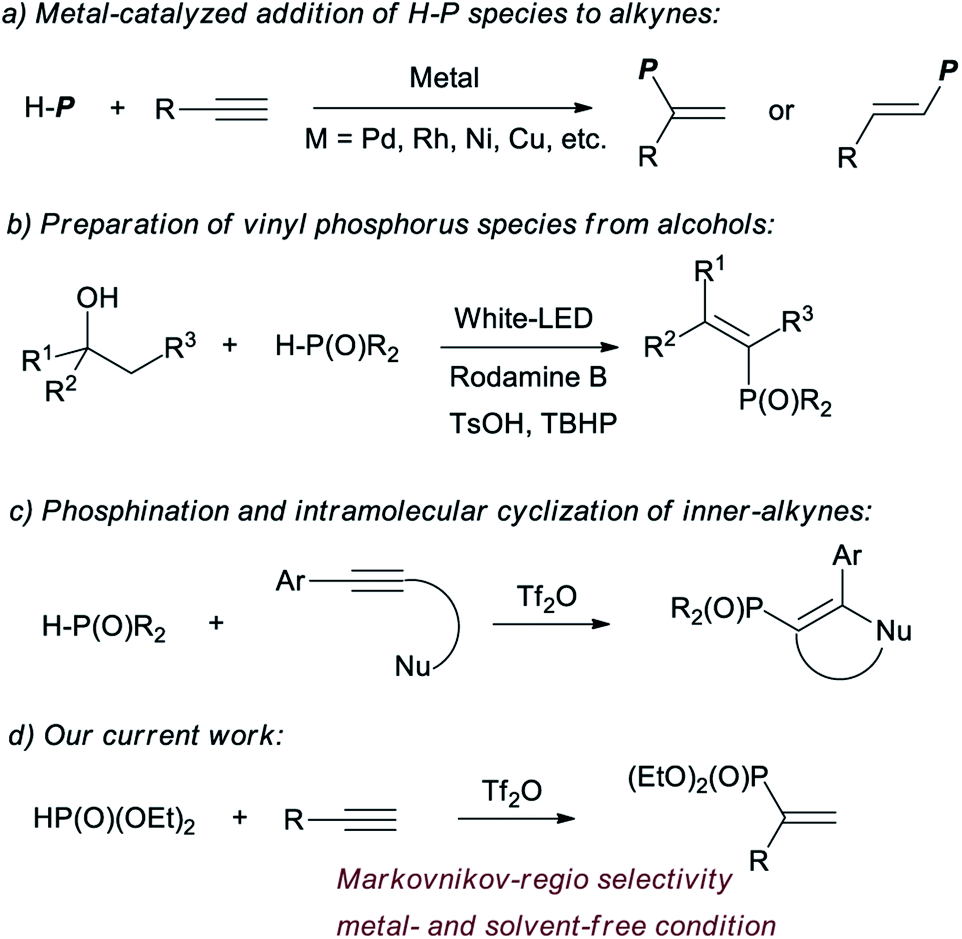
Vinyl phosphorus compounds are of high importance because of their wide application in the fields of biological metabolism,1 functional materials,2 and as synthetic blocks of organophosphorus compounds.3 Some reviews have documented the preparations and applications of the vinyl phosphorus.4 Normally, the compounds can be acquired from alkynes, via the addition with H–P species. Aromatic ketones and aldehydes are also used as the sources of vinyl phosphorus, via multi-step conversions.1b,5 The recently developed methods to obtain vinyl phosphorus involving H–P species include decarboxylation of cinnamic acid,6 dehydrogenation of active alkene,7 and dehydration of alcohols.8
The addition of H–P species to alkynes is undoubtedly the most effective and atom economical method. The addition was usually catalysed by metals.4e,9 One of the authors has reported palladium and rhodium catalysed additions of H–P species to alkynes.9a–d Similar additions could be realized by catalysis with copper, nickel, molybdenum and ytterbium (Chart 1).10 The metal-catalysed procedures also include Suzuki reaction and others.11 Although much progress has been made in this area, problems associated with narrow substrate scope, the need to use expensive transition metals and a stoichiometric amount of additives, and poor regio- and stereo-control have encouraged researchers to search for new and more efficient methods.
 | ||
| Chart 1 Comparison of the construction of C(sp2)–P bonds to our current work. | ||
Recently, triflic anhydride (Tf2O) has been applied for electrophilic activation of amides and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species, and the activated intermediates could be used for the substitutions with various nucleophilic reagents.7b,12 The conversions of PO species to C(sp2)–P bonds are quite rare. Miura and coauthors have reported a Tf2O promoted activation of secondary phosphine oxides, which is applied for phosphination and intramolecular cyclization with alkynes.13 However, the scopes are limited with inner-alkynes. The application of H-phosphonates was not reported for the reaction (Chart 1).
O species, and the activated intermediates could be used for the substitutions with various nucleophilic reagents.7b,12 The conversions of PO species to C(sp2)–P bonds are quite rare. Miura and coauthors have reported a Tf2O promoted activation of secondary phosphine oxides, which is applied for phosphination and intramolecular cyclization with alkynes.13 However, the scopes are limited with inner-alkynes. The application of H-phosphonates was not reported for the reaction (Chart 1).
Herein we enclosed a novel Tf2O promoted reaction of terminal alkyne with H-phosphonates. Vinyl phosphonates were afforded under metal- and solvent-free conditions, in excellent Markovnikov-regio selectivity.
Phenylacetylene (1a) was initially treated with diethyl phosphite (2a) in the presence of Tf2O in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. To our delight, diethyl 1-phenyl ethenylphosphonate (3aa), a Markovnikov addition product, was obtained in high regio-selectivity but in poor yield (10%). The reaction conditions were subsequently optimized with 1a and 2a. Considering Tf2O was moisture sensitive, all experiments were performed under nitrogen. Two equivalents 2a was used to ensure 1a was consumed as much as possible. The conversion and isolated yield were calculated based on 1a (Table 1).
:1:1. To our delight, diethyl 1-phenyl ethenylphosphonate (3aa), a Markovnikov addition product, was obtained in high regio-selectivity but in poor yield (10%). The reaction conditions were subsequently optimized with 1a and 2a. Considering Tf2O was moisture sensitive, all experiments were performed under nitrogen. Two equivalents 2a was used to ensure 1a was consumed as much as possible. The conversion and isolated yield were calculated based on 1a (Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Molar ratio of 1a/2a/Cat | Base | Temp. | Solvent (1 mL) | Conversion% (isolated yield%)b |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.40 mmol) and Tf2O (0.2 mmol) stirring at different temperatures for 24 h.b The conversions were estimated based on 1H NMR spectrum, and isolated yields were calculated based on 1a.c TfOH was used.d 1.0 equiv. of base was used. | |||||
| 1 | 1:2:1 |
— | Rt | — | 12 |
| 2 | 1:2:1 |
— | 40 °C | — | 46 |
| 3 | 1:2:1 |
— | 60 °C | — | 92 |
| 4 | 1:2:1 |
— | 65 °C | — | 98 (53) |
| 5 | 1:2:1 |
— | 80 °C | — | 98 (56) |
| 6 | 1:2:0.5 |
— | 65 °C | — | 79 |
| 7 | 1:2:0.8 |
— | 65 °C | — | 92 |
| 8 | 1:2:1.2 |
— | 65 °C | — | 86 |
| 9 | 1:2:1 |
— | 65 °C | — | 0c |
| 10 | 1:2:1 |
— | 65 °C | CH2Cl2 | 3 |
| 11 | 1:2:1 |
— | 65 °C | Toluene | 9 |
| 12 | 1:2:1 |
— | 65 °C | THF | 0 |
| 13 | 1:2:1 |
— | 65 °C | EtOAc | 22 |
| 14 | 1:2:1 |
Na2CO3 | 65 °C | — | 0d |
| 15 | 1:2:1 |
Pyridine | 65 °C | — | 99 (64)d |
| 16 | 1:2:1 |
2,6-Lutidine | 65 °C | — | 99 (54)d |
| 17 | 1:2:1 |
DBU | 65 °C | — | 99 (64)d |
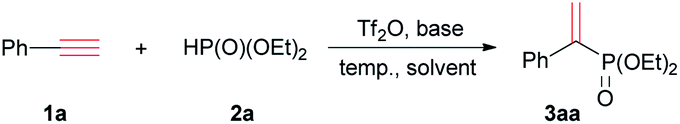
When the mixture of 1a, 2a and Tf2O was stirred at room temperature, a new signal at 17.1 ppm was observed on 31P NMR spectrum. The two doublet peaks at 6.35 and 6.17 ppm on proton NMR spectrum, assigned as 3aa, was detected in 12% (entry 1 of Table 1). The conversion of 1a was improved to 46% when the reaction was carried out at 40 °C, and to 92% at 60 °C (entries 2 and 3). At 65 °C, the conversion was detected in 98%, and 3aa was isolated, whose structure was confirmed by NMR spectrum.14 The conversion cannot be further improved at higher temperature such as 80 °C.
When 0.5 equiv. Tf2O was used, the conversion of 1a was dropped to 79%. The usage of 0.8 equivalent of Tf2O led 92% conversion (entries 6 and 7). However, excessive Tf2O, such as 1.2 equivalents, resulted in the reduced conversion (86%, entry 8).
Once triflic acid was used to replace Tf2O, 3aa was not detected (entry 9).
To carry the reaction in some solvent such as dichloromethane, toluene, THF or ethyl acetate, gave worse result than neat condition (entries 10–13). In the presence of sodium carbonate, the reaction cannot take place (entry 14). The presence of pyridine, 2,6-lutidine or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) slightly increased the conversion (entries 15–17).
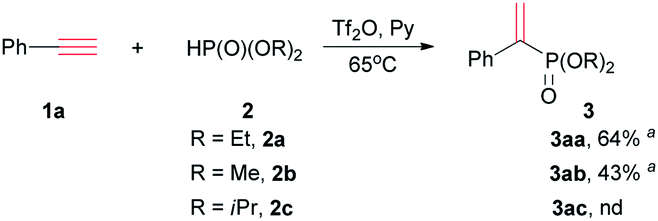
Adopting the above optimized condition, the reaction of alkyne with H-phosphonate was performed (Table 2). Dimethyl phosphite 2b reacted with phenylacetylene to afford dimethyl 1-phenylethenyl phosphonate 3ab in 43% yield, which was less than the reaction of 2a. Perhaps due to the bigger spatial hindrance, diisopropyl phosphite 2c did not reacted with 1a to afford 3ac. The reactions of diphenylphosphine oxide or ethyl phenylphosphinate cannot occur.
| a Isolated yields based on 1a. |
|---|
 |
Various terminal alkynes was then examined (Table 3). Besides of 1a, aromatic alkynes possessing electron-donating groups smoothly reacted with 2a to afford corresponding 3ba–3ha in 17–62% yields. The low yield of 3ha may be ascribed to meta-methyl that had variable electronic effect. For chloro and bromo-substituted alkynes, 3ia and 3ja were afforded in 56% and 46% yields, respectively. Perhaps due to steric hindrance, the ortho-chlorophenyl ethyne gave lower yield of 3 than para-substituted alkyne.
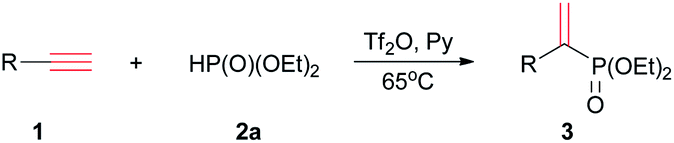
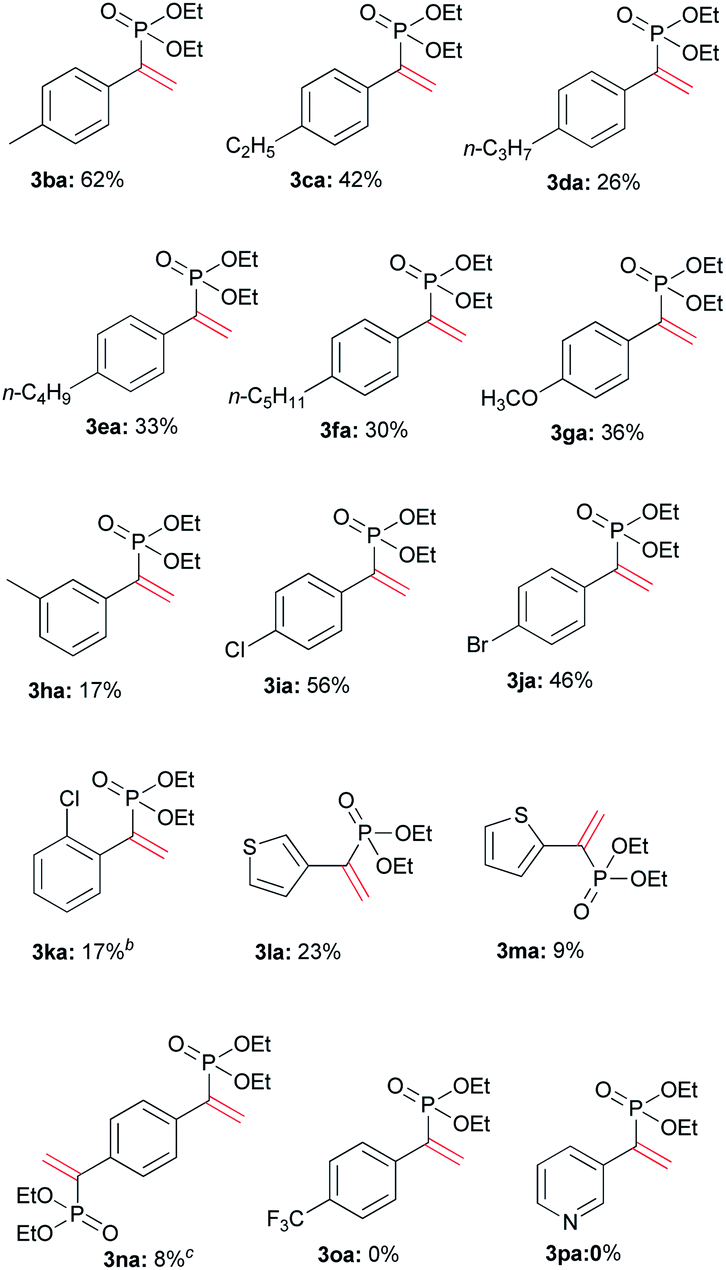
Some ethynyl heterocycles, such as 2- or 3-ethynylthiophene could be employed for the reaction, affording 3la or 3ma, respectively. For 1,4-diethynylbenzene (1n), the addition was realized when 2a and Tf2O were used in double equivalents. However, the alkynes having strong EWG, such as trifluoromethyl or 3-pyridinyl, cannot react with 2a. Aliphatic terminal alkynes, such as 1-octyne, also did not afford the product.
The mechanism of the reaction was studied via in situ NMR experiments, and was proposed in Scheme 1. 2a and Tf2O formed triflate 4 of phosphite via an equilibrium.12b,15 Meanwhile, triflic acid was generated (step 1). The addition of triflic acid to 1a formed 1-phenylvinyl triflate 5 as an intermediate (step 2). The subsequent nucleophilic substitution of 5 with 4 formed vinyl phosphonate 3aa (step 3).
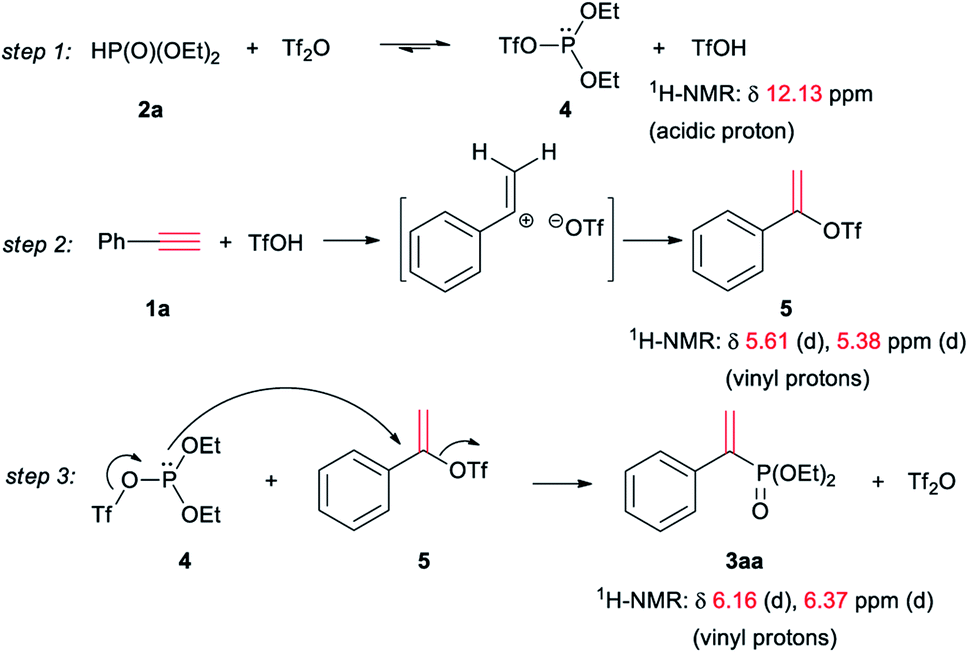 | ||
| Scheme 1 Proposed mechanism. | ||
The step 1 was confirmed by the observation of the signal of acidic proton in TfOH during the mixing of 2a with Tf2O (Fig. 1A). When 1a was added, 5 was detected, as seen the two doublet peaks at 5.61 and 5.38 ppm on 1H NMR spectrum (step 2 and Fig. 1B). When the mixture was heated at 60 °C, the signals of 3aa at 6.16–6.37 ppm emerged. The peaks were increased with prolonged heating, accompanied with the decreasing of the signals of 5 (step 3, Fig. 1C and D). Similar results were observed when Tf2O was initially mixed with 1a (Fig. 1E–H). After heating at 60 °C for 8 h, the signals of 1a, respected by the peak of terminal alkyne at 3.07 ppm, were disappeared (Fig. 1H).
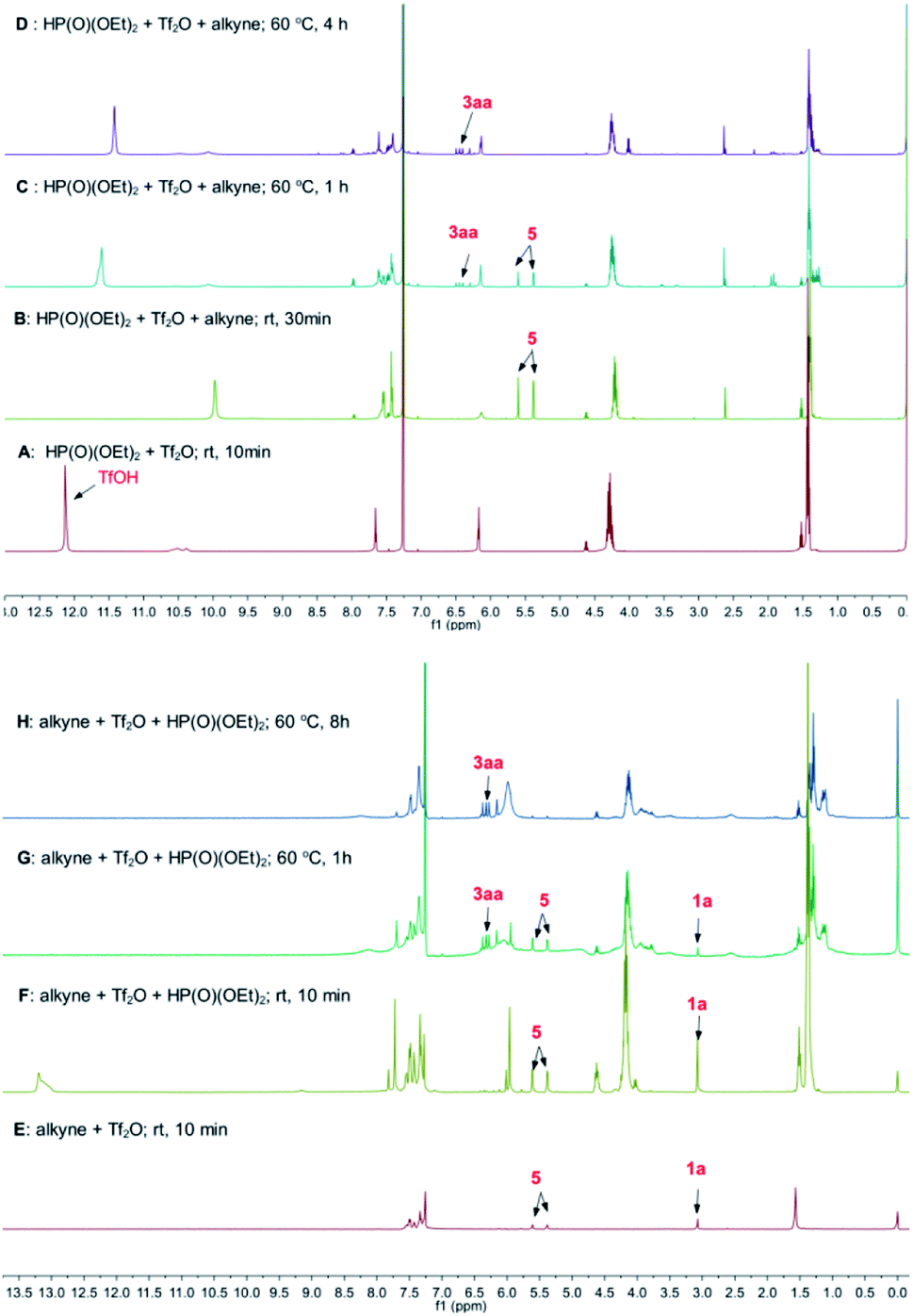 | ||
| Fig. 1 The results of in situ NMR experiments of the reaction of 1a with 2a in the presence of triflic anhydride. | ||
The addition of triflic acid to 1a probably formed α-carbon ion as intermediate, which could be stabilized by phenyl and be converted to Markovnikov-product 5. Although the nucleophilic substitution on vinyl carbon was difficulty, the strong leaving activity of trifluoromethanesulfonyloxy on 5 enabled step 3 occurred. Miura proposed a cyclic phosphirenium intermediate for the addition and cyclization,13 which could be probably stabilized by the alkyl groups of inner-alkynes and secondary phosphine oxides. In our case, the electron-withdrawing alkoxyl on phosphorus of 2a was not favorable for the formation of the phosphirenium, thus the reaction proceeded via α-carbon ion and addition–substitution occurred.
TfOH was necessary to the formation of 5 from 2a (step 2). However, triflic acid cannot sole promote the reaction, as seen in entry 9 of Table 1. The results indicated the poor nucleophilic ability of 2a. After 2a was converted to P(III) species 4 by triflic anhydride, the lone electron pair on phosphorus of 4 could attack 5 to form the product. It was not sure the small quartet peak at 4.62 ppm belonged to 4 (Fig. 1F). On 31P NMR spectrum, the peak of 2a at 17.0 ppm obviously became broad in poor resolution when mixed with triflic anhydride (as seen in ESI†). The results also supported the equilibrium between 2a and 4 (step 1).
Conclusions
In summary, a mild and convenient method to generate vinylphosphonates was developed. The method employed aryl alkynes and H-phosphonates, was performed under metal- and solvent-free conditions, and afforded Markovnikov-adducts as single regio-isomer. This methodology was compatible with a wide range of aryl alkynes. On the basis of in situ 1H NMR studies, an addition–substitution mechanism was proposed, which was different to the reported phosphirenium mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by NSFC (21502084, 21401094) and Liaocheng University (318051437).Notes and references
- For example, see: (a) R. Parmar, J. L. S. Willoughby, J. Liu, D. J. Foster, B. Brigham, C. S. Theile, K. Charisse, A. Akinc, E. Guidry, Y. Pei, W. Strapps, M. Cancilla, M. G. Stanton, K. G. Rajeev, L. S. Lorenzino, M. Manoharan, R. Meyers, M. A. Maier and V. Jadhav, ChemBioChem, 2016, 17, 985–989 CrossRef CAS PubMed; (b) X. Q. Lu, C. D. Sun, W. J. Valentine, S. E, J. X. Liu, G. Tigyi and R. Bittman, J. Org. Chem., 2009, 74, 3192–3195 CrossRef CAS; (c) R. D. Bertram, C. J. Hayes and P. Soultanas, Biochemistry, 2002, 41, 7725–7731 CrossRef CAS PubMed.
- For example, see: (a) B. S. Soller, S. Salzinger and B. Rieger, Chem. Rev., 2016, 116, 1993–2002 CrossRef CAS PubMed; (b) C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed; (c) G. David and C. Negrell-Guirao, in Phosphorus-Based Polymers: from Synthesis to Applications, RSC Polymer Chemistry Series, ed. S. Monge and G. David, Royal Society of Chemistry, United Kingdom, 2014, vol. 11, pp 35–50 Search PubMed.
- For example, see: (a) L. J. Ren, M. G. Ran, J. X. He, D. Xiang, F. Chen, P. J. Liu, C. Y. He and Q. L. Yao, Eur. J. Org. Chem., 2019, 33, 5656–5661 CrossRef; (b) A. K. Chaturvedi and N. Rastogi, J. Org. Chem., 2016, 81, 3303–3312 CrossRef CAS PubMed; (c) K. W. Dong, Z. Wang and K. L. Ding, J. Am. Chem. Soc., 2012, 134, 12474–12477 CrossRef CAS PubMed; (d) H. Ishikawa, M. Honma and Y. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 2824–2827 CrossRef CAS; (e) S. S. Mossé, M. Tissot and A. Alexakis, Org. Lett., 2007, 9, 3749–3752 CrossRef PubMed.
- For example, see: (a) D. S. Glueck, J. Org. Chem., 2020, 85, 14276–14285 CrossRef CAS PubMed; (b) Q. Xu, C.-Q. Zhao, Y. Zhou, S. Yin and L.-B. Han, Chin. J. Org. Chem., 2012, 32, 1761–1775 CrossRef CAS; (c) S. Van der Jeught and C. V. Stevens, Chem. Rev., 2009, 109, 2672–2702 CrossRef CAS; (d) A. L. Schwan, Chem. Soc. Rev., 2004, 33, 218–224 RSC; (e) I. P. Beletskaya, C. Nájera and M. Yus, Russ. Chem. Rev., 2021, 90, 70–93 CrossRef CAS.
- N. S. Goulioukina, T. M. Dolgina, I. P. Beletskaya, J.-C. Henry, D. Lavergne, V. Ratovelomanana-Vidal and J.-P. Genet, Tetrahedron: Asymmetry, 2001, 12, 319–327 CrossRef CAS.
- L. X. Liu, D. Zhou, J. Y. Dong, Y. B. Zhou, S.-F. Yin and L.-B. Han, J. Org. Chem., 2018, 83, 4190–4196 CrossRef CAS PubMed.
- (a) A.-X. Zhou, L.-L. Mao, G.-W. Wang and S.-D. Yang, Chem. Commun., 2014, 50, 8529–8532 RSC; (b) T. Yuan, S. L. Huang, C. Cai and G.-P. Lu, Org. Biomol. Chem., 2018, 16, 30–33 RSC; (c) L.-L. Liao, Y.-Y. Gui, X.-B. Zhang, G. Shen, H.-D. Liu, W.-J. Zhou, J. Li and D.-G. Yu, Org. Lett., 2017, 19, 3735–3738 CrossRef CAS PubMed.
- P. Z. Xie, J. Fan, Y. N. Liu, X. Y. Wo, W. S. Fu and T.-P. Loh, Org. Lett., 2018, 20, 3341–3344 CrossRef CAS PubMed.
- (a) T. Q. Chen, C.-Q. Zhao and L.-B. Han, J. Am. Chem. Soc., 2018, 140, 3139–3155 CrossRef CAS PubMed; (b) L.-B. Han, C.-Q. Zhao, S.-Y. Onozawa, M. Goto and M. Tanaka, J. Am. Chem. Soc., 2002, 124, 3842–3843 CrossRef CAS PubMed; (c) L.-B. Han, C.-Q. Zhao and M. Tanaka, J. Org. Chem., 2001, 66, 5929–5932 CrossRef CAS; (d) C.-Q. Zhao, L.-B. Han, M. Goto and M. Tanaka, Angew. Chem., Int. Ed., 2001, 40, 1929–1932 CrossRef CAS; (e) S. K. Nune and M. Tanaka, Chem. Commun., 2007, 2858–2860 RSC; (f) L.-B. Han and M. Tanaka, J. Am. Chem. Soc., 1996, 118, 1571–1572 CrossRef CAS.
- (a) I. G. Trostyanskaya and I. P. Beletskaya, Tetrahedron, 2014, 70, 2556–2562 CrossRef CAS; (b) M. Y. Niu, H. Fu, Y. Y. Jiang and Y. F. Zhao, Chem. Commun., 2007, 272–274 RSC; (c) V. P. Ananikov, L. L. Khemchyan, I. P. Beletskaya and Z. A. Starikova, Adv. Synth. Catal., 2010, 352, 2979–2992 CrossRef CAS; (d) L.-B. Han, C. Zhang, H. Yazawa and S. Shimada, J. Am. Chem. Soc., 2004, 126, 5080–5081 CrossRef CAS; (e) A. I. Kuramshin, A. A. Nikolaev and R. A. Cherkasov, Mendeleev Commun., 2005, 15, 155–156 CrossRef; (f) K. Takaki, M. Takeda, G. Koshoji, T. Shishido and K. Takehira, Tetrahedron Lett., 2001, 42, 6357–6360 CrossRef CAS.
- (a) L. Zhang, Y. W. Fang, X. P. Jin, H. S. Xu, R. F. Li, H. Wu, B. Chen, Y. M. Zhu, Y. Yang and Z. M. Tian, Org. Biomol. Chem., 2017, 15, 8985–8989 RSC; (b) A. N. Barrett, H. J. Sanderson, M. F. Mahon and R. L. Webster, Chem. Commun., 2020, 56, 13623–13626 RSC.
- (a) D. Kaiser and N. Maulide, J. Org. Chem., 2016, 81, 4421–4428 CrossRef CAS PubMed; (b) H. Huang and J. Y. Kang, Synlett, 2019, 30, 635–641 CrossRef CAS; (c) H. Huang, J. Denne, C.-H. Yang, H. B. Wang and J. Y. Kang, Angew. Chem., Int. Ed., 2018, 57, 6624–6628 CrossRef CAS PubMed.
- Y. Unoh, K. Hirano and M. Miura, J. Am. Chem. Soc., 2017, 139, 6106–6109 CrossRef CAS.
- D.-Y. Wang, X.-P. Hu, J. Deng, S.-B. Yu, Z.-C. Duan and Z. Zheng, J. Org. Chem., 2009, 74, 4408–4410 CrossRef CAS PubMed.
- (a) B. G. Janesko, H. C. Fisher, M. J. Bridle and J.-L. Montchamp, J. Org. Chem., 2015, 80, 10025–10032 CrossRef CAS PubMed; (b) N. N. Xin, X. Y. Xie, C.-Q. Zhao, X. Q. Huang, J. H. Zhang, J.-Y. Du, J. Yang and C. L. Ma, Synlett, 2018, 29, 1219–1222 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04306d |
| This journal is © The Royal Society of Chemistry 2021 |