
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe development and validation of a sensitive HPLC-MS/MS method for the quantitative and pharmacokinetic study of the seven components of Buddleja lindleyana Fort.†
Xia Zhang a,
Zhi-qing Zhanga,
Li-cang Zhanga,
Ke-xin Wanga,
Lan-tong Zhangb and
De-qiang Li*a
a,
Zhi-qing Zhanga,
Li-cang Zhanga,
Ke-xin Wanga,
Lan-tong Zhangb and
De-qiang Li*a
aDepartment of Pharmacy, The Second Hospital of Hebei Medical University, Shijiazhuang 050000, P. R. China. E-mail: deqli@163.com; Tel: +86 0311-66636302 Tel: +86 18132685779
bDepartment of Pharmaceutical Analysis, School of Pharmacy, Hebei Medical University, P. R. China
First published on 28th July 2021
Abstract
Buddleja lindleyana Fort., a traditional Chinese medicine, has demonstrated anti-inflammatory, immunomodulatory, antidementia, neuroprotective, antibacterial, and antioxidant effects. Its flowers, leaves, and roots have been used as traditional Chinese medicines. A simple and rapid high-performance liquid chromatography method coupled with mass spectrometry (HPLC-MS/MS) was applied in the multicomponent determination of Buddleja lindleyana Fort., and the discrepancies in the contents from ten different habitats were analyzed. The present study simultaneously determined the concentrations of seven chemical compounds of Buddleja lindleyana Fort. extract in rat plasma via HPLC-MS/MS, which was applied in the pharmacokinetic (PK) study of Buddleja lindleyana Fort. A C18 column was used for chromatographic separation, and ion acquisition was achieved by multiple-reaction monitoring (MRM) in negative ionization mode. The optimized mass transition ion-pairs (m/z) for quantization were 591.5/282.8 for linarin, 609.4/300.2 for rutin, 284.9/133.0 for luteolin, 300.6/151.0 for quercetin, 268.8/116.9 for apigenin, 283.0/267.9 for acacetin, 623.3/160.7 for acteoside, and 252.2/155.8 for sulfamethoxazole (IS). A double peak appeared in the drug–time curve of apigenin, which was associated with entero-hepatic recirculation. There were discrepancies in the contents of seven chemical compounds from 10 batches of Buddleja lindleyana Fort., which were associated with the growth environments. Herein, the pharmacokinetic parameters of seven analytes in Buddleja lindleyana Fort. extract are summarized. The maximum plasma concentration (Cmax) of linarin, rutin, luteolin, quercetin, apigenin, acacetin and acteoside were 894.12 ± 9.34 ng mL−1, 130.76 ± 18.33 ng mL−1, 77.37 ± 25.72 ng mL−1, 20.15 ± 24.85 ng mL−1, 146.42 ± 14.88 ng mL−1, 31.92 ± 17.58 ng mL−1, and 649.78 ± 16.42 ng mL−1, respectively. The time to reach Cmax for linarin, rutin, luteolin, quercetin, apigenin, acacetin, and acteoside were 10, 5, 5, 5, 180, 10 and 10 min, respectively. This is the first report on the simultaneous determination of seven active components for 10 different growing environments and the pharmacokinetic studies of seven active components in rat plasma after the oral administration of Buddleja lindleyana Fort. extract. This study lays the foundation for a better understanding of the absorption mechanism of Buddleja lindleyana Fort., and the evaluation of its clinical application.
1. Introduction
Buddleja lindleyana Fort. is the general name of the Buddle-ja plants in the Loganiaceae family, which are distributed in tropical and subtropical areas such as South America, Asia and southern Africa.1 There are more than 100 species, with 20 species and hybrids found in China.2 Buddleja lindleyana Fort. is famous for its toxicity to fish as well as their exotic flowers.3 Its flowers, leaves and roots have been used as traditional Chinese medicine, and have been applied in the treatment of rheumatism, cough, and blood stasis among other.4–8 The main chemical components of Buddleja lindleyana Fort. include triterpenoids,9,10 flavonoids,11 iridoid glycosides12,13 and phenylethaoids.14In recent years, high-performance liquid chromatography coupled with mass spectrometry (HPLC-MS/MS) has been routinely used to determine the contents of chemical compounds in traditional chemical materials and also applied in pharmacokinetic studies in rats and humans.15,16 Moreover, it has been used to study drug metabolism,17–22 toxicokinetics,23,24 lipidomics,25,26 proteomics27,28 and metabolomics.29,30 The main advantage of mass spectrometry (MS/MS) is the ability to detect a broad range of drugs with high sensitivity and specificity in a single analytical run.31–34 Chemical compounds are recognized by comparing their retention times and parent-daughter ions with those of reference substances.
Pharmacokinetics (PK) is a discipline that quantitatively studies the absorption, distribution, metabolism and excretion in vivo, which uses mathematical theory and methods to make an exposition of the law for changing drug concentrations in blood over time. With the development of medicinal chemistry and the continuous improvement of human health, the requirements for the PK of drugs are getting higher and higher, which will determine the development trends of drugs, especially in the foreground of the market. The therapeutic effects of a drug must be strong, with few side effects and good PK parameters.
The contents of chemical compounds (linarin, luteolin, acacia-7-O-β-D-glucopyranoside, acacetin, rutin, quercetin, apigenin, clinoposaponin III, desrhamnoverbascosaponin and mimengoside I) were determined by HPLC and UPLC according to previous research.5,35,36 In this study, a sensitive and rapid HPLC-MS/MS method was developed for the simultaneous determination of seven compounds in different batches of medicinal materials and in rat plasma after oral treatment with Buddleja lindleyana Fort. extract. The HPLC-MS/MS technology demonstrated the advantages of being rapid and efficient. This is the first systematic multicomponent quantification and PK study of seven chemical constituents of Buddleja lindleyana Fort. Multiple-reaction monitoring (MRM) was employed and the ESI source was operated in negative mode. The contents of seven components of Buddleja lindleyana Fort. from ten different sources were compared and the PK parameters were summarized. These results laid the foundation for clinical application.
2. Materials and methods
2.1. Chemicals and materials
Linarin (137-200702), luteolin (137-200708), apigenin (137-200928), acacetin (137-200619) and acteoside (137-200901) were purchased from Nanjing Guangrun Biotechnology Co. Ltd (Nanjing, China). Rutin (100080-200707), quercetin (100081-200907) and sulfamethoxazole (100025-200904) were obtained from the National Institutes for Food and Drug Control. The purities of these standards were higher than 98%.HPLC-grade methanol was purchased from J. T. Baker Chemical Company (Phillipsburg, NJ, USA). Formic acid (HPLC grade) was provided by Diamond Technology (Dikma Technologies Inc., Lake Forest, CA, USA). Ethanol (analytical grade) was provided by Tianjin Guangfu Technology Development Co. Ltd (Tianjin, China). Purified water was obtained from Guangzhou Watson's Food & Beverage Co. Ltd (Guangzhou, China).
The entire Buddleja lindleyana Fort. plant was collected from Anguo Chinese Medicinal Materials Wholesale Market and identified by Zengke Kong, Professor of Pharmacognosy, Hebei Province Institute for Drug Control. The sources of Buddleja lindleyana Fort. are listed in Table 1.
| Sample number | Source |
|---|---|
| 1 | Guangxi |
| 2 | Xizang |
| 3 | Sichuan |
| 4 | Anhui-Bozhou |
| 5 | Zhejiang-Huzhou |
| 6 | Yunnan |
| 7 | Anhui-Bengbu |
| 8 | Zhejiang-Hangzhou |
| 9 | Jiangxi |
| 10 | Henan |
2.2. Instrumentation and conditions
HPLC-MS/MS analysis was performed on a Shimadzu HPLC system (Shimadzu Corp., Kyoto, Japan) that was coupled with an API 4000+ MS/MS system (AB Sciex, CA, USA). Nitrogen was obtained from Shijiazhuang Fulite Gas Co. Ltd The chromatographic separation was conducted on a Symmetry® C18(4.6 × 150 mm, 3.5 μm) column with a SecurityGuard® HPLC C18 pre-column (Agilent Corp, Santa Clara, CA, USA). The column temperature was maintained at 25 °C. The mobile phase consisted of water containing 0.1% formic acid (A) and acetonitrile (B). The gradient elution program was optimized for the separation, and the program was as follows: 0–2 min, 25–40% B; 2–5 min, 40–90% B; 5–8 min, 90–95% B. The column was returned to its starting conditions in 1 min and gradient elution was carried out after pre-equilibration for 6 min. The flow rate of the mobile phase was set to 0.8 mL min−1, and the injection volume was 10 μL. Data acquisition was carried out using Analyst software (version 1.6.2) from Applied Biosystems/MDS Sciex. The typical extracted-ion chromatograms (XIC) of MRM chromatograms of standards and samples obtained are shown in Fig. 1.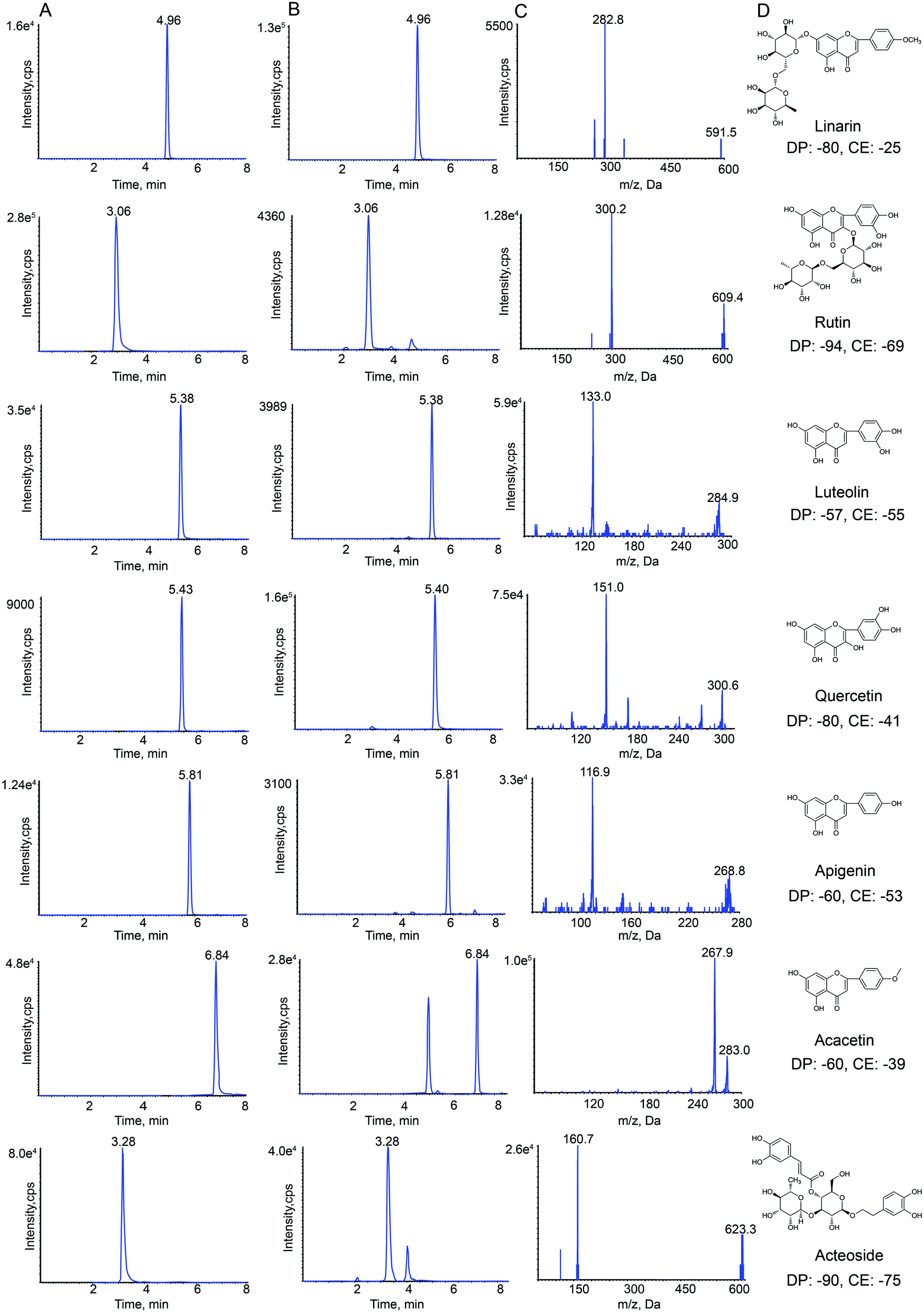 | ||
| Fig. 1 Representative extraction chromatograms (XIC) of multiple-reaction monitoring (MRM) chromatograms of linarin, rutin, luteolin, quercetin, apigenin, acacetin and acteoside (A) standard and (B) Buddleja lindleyana Fort. extract; (C) monitored MRM transitions of the seven standards; (D) chemical structure, declustering potential (DP) and collision energy (CE). | ||
Negative electrospray ionization mode was used for detecting analytes by API 4000+ MS. The following MS/MS conditions were used: ion spray voltage, −4.5 kV; the turbo spray temperature, 550 °C. Nitrogen was used as the nebulizer and auxiliary gas. Furthermore, the flows of the nebulizer gas (gas 1), heater gas (gas 2) and curtain gas were set to 55, 50, and 25 psi, respectively. The declustering potential (DP) and collision energy (CE) of all analytes are summarized in Fig. 1.
2.3. Preparation of Buddleja lindleyana Fort. extract
For quantitative analysis in different batches of medical materials, the powdered Buddleja lindleyana Fort. (1.0 g, 60 mesh sieve) was approximately weighed and placed in a 50 mL conical flask with a cover, then, 25 mL of 70% ethanol was precisely added and both were accurately weighed. After ultrasound treatment for 40 min, the lost weight was made up using 70% ethanol. The resultant was filtered through a 0.22 μm millipore filter and 10 μL was used for HPLC-ESI-MS/MS analysis. Buddleja lindleyana Fort. materials from ten different sources were treated in the same way, which were used for quantitative analysis.For pharmacokinetics study, Buddleja lindleyana Fort. (Xizang) was cut up and soaked for 12 h with 70% ethyl alcohol.37 The ratios of medicinal materials and 70% ethyl alcohol were 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15, 1:10 and 1:10, respectively. The medicinal materials were extracted three times by heating under reflux. The extraction solutions were filtered and merged, and were concentrated by reducing the pressure. Finally, the residuary solution was concentrated to get the Buddleja lindleyana Fort. extract with a concentration equivalent to 2.5 g mL−1 of the raw Buddleja lindleyana Fort. material. The content of linarin, rutin, luteolin, quercetin, apigenin, acacetin and acteoside were 14828.24, 422.1, 510.99, 336.45, 665.01, 1840.24 and 1555.87 ng mL−1, respectively. For the PK study, Buddleja lindleyana Fort. (Xizang) was treated with this method.
:15, 1:10 and 1:10, respectively. The medicinal materials were extracted three times by heating under reflux. The extraction solutions were filtered and merged, and were concentrated by reducing the pressure. Finally, the residuary solution was concentrated to get the Buddleja lindleyana Fort. extract with a concentration equivalent to 2.5 g mL−1 of the raw Buddleja lindleyana Fort. material. The content of linarin, rutin, luteolin, quercetin, apigenin, acacetin and acteoside were 14828.24, 422.1, 510.99, 336.45, 665.01, 1840.24 and 1555.87 ng mL−1, respectively. For the PK study, Buddleja lindleyana Fort. (Xizang) was treated with this method.
2.4. Preparation of standard solutions and quality control (QC) samples
The appropriate amounts of linarin, rutin, luteolin, quercetin, apigenin, acacetin, acteoside and sulfamethoxazole were accurately weighed and dissolved in methanol to make stock solutions. The concentrations of each standard in the stock solutions were 0.91, 1.05, 0.92, 1.08, 0.88, 0.65, 0.94 and 0.88 mg mL−1, which were diluted with methanol to plot the standard curve for quality control of Buddleja lindleyana Fort.For the PK study, the mixed standard solution was made by mixing the seven stock solutions, which contained 2894.93 ng mL−1 of linarin, 410.28 ng mL−1 of rutin, 248.68 ng mL−1 of luteolin, 65.52 ng mL−1 of quercetin, 467.72 ng mL−1 of apigenin, 104.68 ng mL−1 of acacetin and 2077.28 ng mL−1 of acteoside. A series of standard mixture working solutions with concentrations in the range of 6.85–14474.65 ng mL−1 for linarin, 6.05–2051.40 ng mL−1 for rutin, 7.90–1243.40 ng mL−1 for luteolin, 6.60–327.60 ng mL−1 for quercetin, 8.85–2338.60 ng mL−1 for apigenin, 11.60–523.40 ng mL−1 for acacetin, and 10.15–10386.40 ng mL−1 for acteoside, were obtained by the attenuation of stock standard solutions with methanol. The concentration of the IS (sulfamethoxazole) standard solution was 1.18 μg mL−1, which was dissolved in methanol.
Working solutions of the corresponding concentrations (20 μL) and IS solution (20 μL) were added to 100 μL of blank rat plasma. The ranges of the final plasma concentrations were 1.37–2894.93 ng mL−1 for linarin, 1.21–410.28 ng mL−1 for rutin, 1.58–248.68 ng mL−1 for luteolin, 1.32–65.52 ng mL−1 for quercetin, 1.77–467.72 ng mL−1 for apigenin, 2.32–104.68 ng mL−1 for acacetin, and 2.03–2077.28 ng mL−1 for acteoside.
The quality control (QC) samples containing low, medium and high concentrations were prepared at the concentrations of 6.85, 723.73 and 2861.18 ng mL−1 for linarin, 6.05, 102.57 and 418.53 ng mL−1 for rutin, 6.32, 62.17 and 247.58 ng mL−1 for luteolin, 2.64, 16.38 and 64.48 ng mL−1 for quercetin, 7.08, 116.93 and 468.54 ng mL−1 for apigenin, 4.62, 26.17 and 102.14 ng mL−1 for acacetin, 2.03, 519.32 and 2027.29 ng mL−1 for acteoside.
2.5. Preparation of plasma samples
All rat plasma samples were prepared by protein precipitation with methanol. IS (20 μL) and methanol (20 μL, volume of the corresponding working solution for the calibration curve and QC sample) were spiked into the plasma samples (100 μL). After that, 500 μL of methanol was added to the mixture, followed by vortexing for 5 min and centrifugation at 12726.2×g for 10 min. The supernatant was then transferred into a 1.5 mL tube and dried under nitrogen at 37 °C, then reconstituted with 100 μL of methanol. The mixture was then vortexed for 5 min and centrifuged at 12726.2×g for 10 min. Finally, the supernate (10 μL) was injected for HPLC-MS/MS analysis.
2.6. Method validation for the quantitative study of Buddleja lindleyana Fort
The validation guidelines for the quantitative study of Buddleja lindleyana Fort. were taken from the Pharmacopoeia of the People's Republic of China, and other literature.38–40443.86 μg; rutin, 24.29, 30.36 and 36.44 μg; luteolin, 208.35, 260.44 and 312.52 μg; quercetin, 5.79, 7.24 and 8.69 μg; apigenin, 174.86, 218.57 and 262.29 μg; acacetin, 3290.89, 4113.61 and 4936.34 μg; acteoside, 943.75, 1179.69 and 1415.62 μg. The average recoveries were calculated by the formula: recovery (%) = (detected values − original amount)/amount spiked × 100%, and RSD (%) = (SD/mean) × 100%. The range of sample recovery was 95–105%, and the RSD value was not more than 5%.In order to investigate the stability of the samples, they were analyzed at 0, 2, 4, 8, 12, and 24 h at room temperature, respectively.
The RSD values of precision and stability were not more than 5%.
2.7. Method validation of the pharmacokinetics study of Buddleja lindleyana Fort. extract
The validation guidelines adopted in this study were from the Guiding Principle of ICH M10 “Validation of Bioanalytical Methodology” 2019 edition.The lower limit of quantification (LLOQ, S/N = 10) of the assay served as the lowest concentration of the standard curve.42 The LLOQ should be less than 10–5% of Cmax. The accuracy of six samples continuously determined should be 80–120%, and the RSD values should be less than 20%.
| SE = (measured value − actual value)/actual value × 100% |
| RSD = SD/X × 100%; SD = Sqr (∑Xn − X)2/(n − 1) (SD – standard deviation; X – mean; n – number of samples) |
The intra- and inter-day RSD of high and medium concentrations were less than 15%, and the intra- and inter-day RSD of the low concentration was less than 20%. The RE of high and medium concentrations were less than ±15%, and the low concentration was not more than ±20%.
A non-compartmental model was used to calculate the pharmacokinetic parameters of analytes by using the DAS 3.0 software.44,45 The PK parameters calculated in this study included the maximum concentration (Cmax), time of achieving maximum concentration (Tmax), half-time (T1/2), area of the concentration–time curve at 0–24 h (AUC0–t), area of the concentration–time curve at 0–∞(AUC0–∞) and clearance rate (CL), which were expressed as mean ± standard deviation (mean ± SD).
3. Results and discussion
3.1. Optimization of the extraction procedure
Methanol (50%, 60%, 70% 80%, 90% and 100%) and alcohol (50%, 60%, 70% 80%, 90% and 100%) of different proportions were compared in this study, which displayed that the extraction efficiency of 70% alcohol was highest. This study also compared ultrasonication with refluxing, which revealed that ultrasonic extraction was simpler and more effective for seven analytes. Four sample-solvent ratios (1:15, 1:25, 1:35 and 1:60, w/v) were compared, and the sample-solvent ratio of 1:25 was chosen. Different ultrasonication times (15, 30, 40, 45 and 60 min) were also compared. The results displayed that the extraction efficiency would no longer increase when the time of extraction was higher than 40 min.
3.2. Optimization of bio-sample preparation
Sample preparation is a key step for the precise analysis of bio-samples by HPLC-MS/MS. In this study, solid-phase extraction (SPE), liquid–liquid extraction (LLE) and protein precipitation (PPT) methods for sample preparation were compared. The SPE method is complicated and expensive; PPT was used to prepare bio-samples for greater extraction recovery than LLE. This study also compared different precipitants (methanol and acetonitrile) and volumes of precipitant. The results showed that 500 μL of methanol serving as the protein precipitant for 5 min could obtain the highest extraction recovery.3.3. LC-MS/MS optimization
The precursor and product ions of seven analytes and IS were detected by infusing 200 ng mL−1 standard solution under MRM mode, respectively. The results showed that the seven chemical components and IS had higher intensities under the negative ion mode, which could be associated with the fact that these analytes were flavonoid and phenylpropanoid glycosides. Because of the stability and high abundance, the deprotonated molecular ions [M–H]− were chosen as precursor ions for the MS/MS fragmentation analysis of analytes. The optimized mass transition ion-pairs (m/z) for quantitation were 591.5/282.8 for linarin, 609.4/300.2 for rutin, 284.9/133.0 for luteolin, 300.6/151.0 for quercetin, 268.8/116.9 for apigenin, 283.0/267.9 for acacetin, 623.3/160.7 for acteoside, and 252.2/155.8 for IS, which are shown in Fig. 1.Different mobile phases (methanol–water and acetonitrile–water) were compared in order to obtain a better peak shape and shorter elution time, which showed that the effect of using acetonitrile–water as the mobile phase was better. Moreover, different concentrations of formic acid (0.1%, 0.2%, 0.5%, 0.01%, 0.02% and 0.05%) were compared. Finally, acetonitrile–water (0.1% formic acid) was chosen as the mobile phase in this study.
3.4. Method validation for the quantitative study of seven analytes
| Component | Regression equation | r | Linear range/(μg mL−1) | LOD (ng mL−1) | LOQ (ng mL−1) |
|---|---|---|---|---|---|
| Linarin | Y = 1999X + 5742 | 0.999 8 | 10.24–655.38 | 0.40 | 1.30 |
| Rutin | Y = 14952X − 2106 |
0.999 4 | 0.01875–2.360 | 0.37 | 1.02 |
| Luteolin | Y = 14102X + 15678 |
0.999 7 | 0.02625–26.75 | 0.27 | 0.96 |
| Quercetin | Y = 29099X + 295.1 |
0.999 8 | 0.003875–2.47 | 0.38 | 1.00 |
| Apigenin | Y = 30536X + 12621 |
0.999 5 | 0.02625–13.62 | 0.34 | 1.02 |
| Acacetin | Y = 11197X + 79447 |
0.999 4 | 1.400–212.2 | 0.32 | 0.74 |
| Acteoside | Y = 32671X + 19740 |
0.999 6 | 2.750–352.5 | 0.27 | 0.66 |
| Analyte | Precision (n = 6) | Accuracy (n = 3) | ||||||
|---|---|---|---|---|---|---|---|---|
| Intra-day RSD (%) | Inter-day RSD (%) | Stability (%) | Original quantity (μg) | Spiked quantity (μg) | Detected (μg) | Recovery (%) | RSD (%) | |
| Linarin | 1.82 | 2.39 | 2.19 | 9536.98 | 7629.24 | 17141.04 |
99.67 | 1.09 |
| 9536.55 | 18934.30 |
98.54 | 0.98 | |||||
| 11443.86 |
20909.89 |
99.38 | 0.82 | |||||
| Rutin | 1.03 | 3.39 | 3.02 | 30.38 | 24.29 | 54.53 | 99.43 | 0.79 |
| 30.36 | 60.31 | 98.57 | 0.48 | |||||
| 36.44 | 66.85 | 100.1 | 1.17 | |||||
| Luteolin | 1.79 | 2.94 | 1.55 | 260.52 | 208.35 | 466.23 | 98.73 | 0.56 |
| 260.44 | 520.41 | 99.79 | 0.64 | |||||
| 312.52 | 569.67 | 98.92 | 0.39 | |||||
| Quercetin | 0.56 | 1.75 | 3.68 | 7.24 | 5.79 | 13.12 | 101.5 | 0.84 |
| 7.24 | 14.34 | 98.02 | 1.03 | |||||
| 8.69 | 15.86 | 99.26 | 0.98 | |||||
| Apigenin | 2.17 | 3.88 | 3.79 | 218.59 | 174.86 | 391.79 | 99.05 | 0.67 |
| 218.57 | 438.92 | 100.8 | 1.13 | |||||
| 262.29 | 476.82 | 98.45 | 0.78 | |||||
| Acacetin | 1.98 | 4.01 | 4.02 | 4113.61 | 3290.89 | 7380.15 | 99.26 | 1.27 |
| 4113.61 | 8187.73 | 99.04 | 1.31 | |||||
| 4936.34 | 9049.95 | 100.0 | 0.59 | |||||
| Acteoside | 1.64 | 3.22 | 2.37 | 1179.68 | 943.75 | 2105.59 | 98.11 | 0.46 |
| 1179.69 | 2356.54 | 99.76 | 0.63 | |||||
| 1415.62 | 2580.44 | 98.95 | 0.86 | |||||
| Content (μg g−1,n = 3) | Linarin | Rutin | Luteolin | Quercetin | Apigenin | Acacetin | Acteoside |
|---|---|---|---|---|---|---|---|
| 1 | 6581.903 | 15.080 | 79.359 | 2.563 | 12.660 | 2091.259 | 837.118 |
| 2 | 9536.981 | 30.377 | 260.522 | 7.238 | 218.592 | 4113.609 | 1179.681 |
| 3 | 7967.634 | 47.280 | 134.832 | 11.376 | 104.280 | 2705.533 | 3268.070 |
| 4 | 6962.231 | 16.364 | 167.611 | 36.405 | 67.599 | 2907.661 | 673.124 |
| 5 | 7700.850 | 19.088 | 462.755 | 2.710 | 120.592 | 2625.502 | 999.367 |
| 6 | 5341.558 | 6.743 | 1.071 | 0.825 | 2.441 | 1530.265 | 445.744 |
| 7 | 5363.830 | 6.263 | 1.723 | 0.525 | 2.690 | 102.032 | 981.599 |
| 8 | 5713.082 | 23.612 | 14.470 | 1.842 | 21.240 | 645.446 | 6949.254 |
| 9 | 5908.544 | 13.550 | 28.033 | 0.898 | 63.274 | 879.504 | 1571.813 |
| 10 | 6529.122 | 8.779 | 54.185 | 0.440 | 35.725 | 413.231 | 606.707 |
Cluster analysis was performed for Buddleja lindleyana Fort. from ten habitats, as displayed in Fig. 2. The results showed that Buddleja lindleyana Fort. from ten habitats were divided into five classes (Class I: 1, 4 and 5; Class II: 6, 7, 9 and 10, Class III: 2, Class IV: 3 and Class V: 8). Buddleja lindleyana Fort. from Tibet (Xizang) was used as an extract for intragastric administration to perform the PK study. Buddleja lindleyana Fort. material from Xizang was selected as the representative plant for pharmacokinetic study because of its highest content of the main compounds. The results revealed that the contents of compounds in Buddleja lindleyana Fort. were associated with their places of origin and planting environments, which would guide the safety and rationalization of clinical use of traditional Chinese medicine.
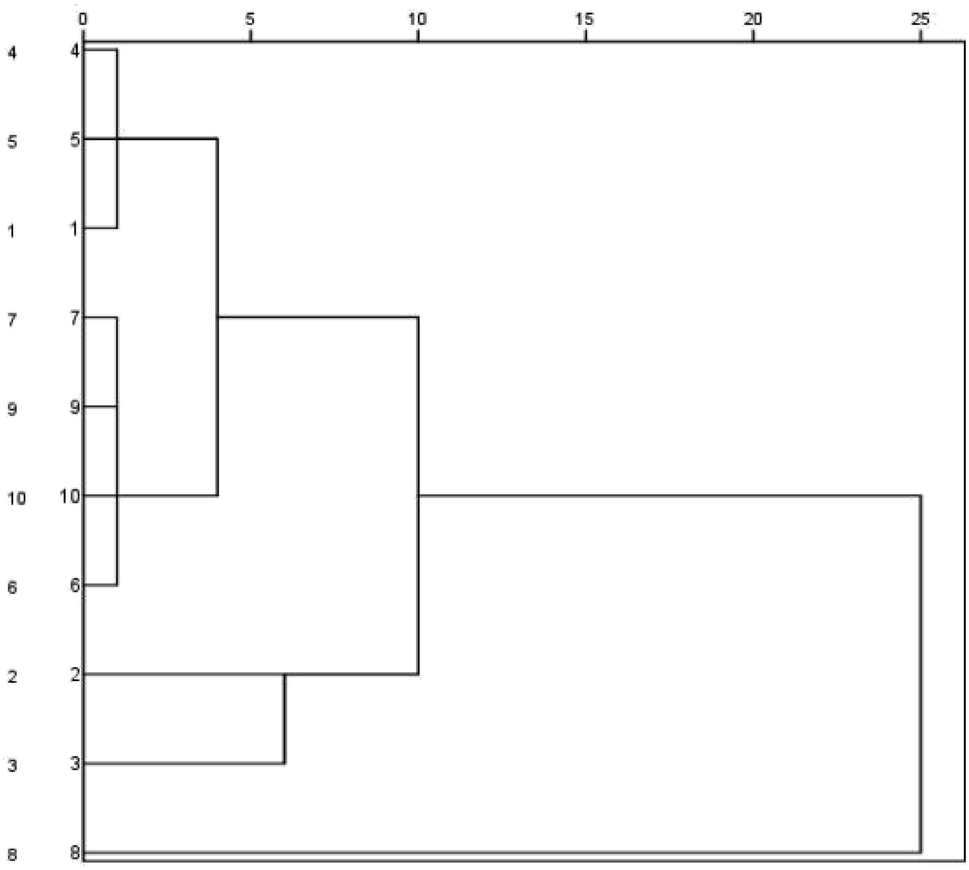 | ||
| Fig. 2 Dendrograms of hierarchical clustering for the ten batches of samples of Buddleja lindleyana Fort. from different sources using the furthest neighbour method. | ||
3.5. Method validation of PK study of Buddleja lindleyana Fort.
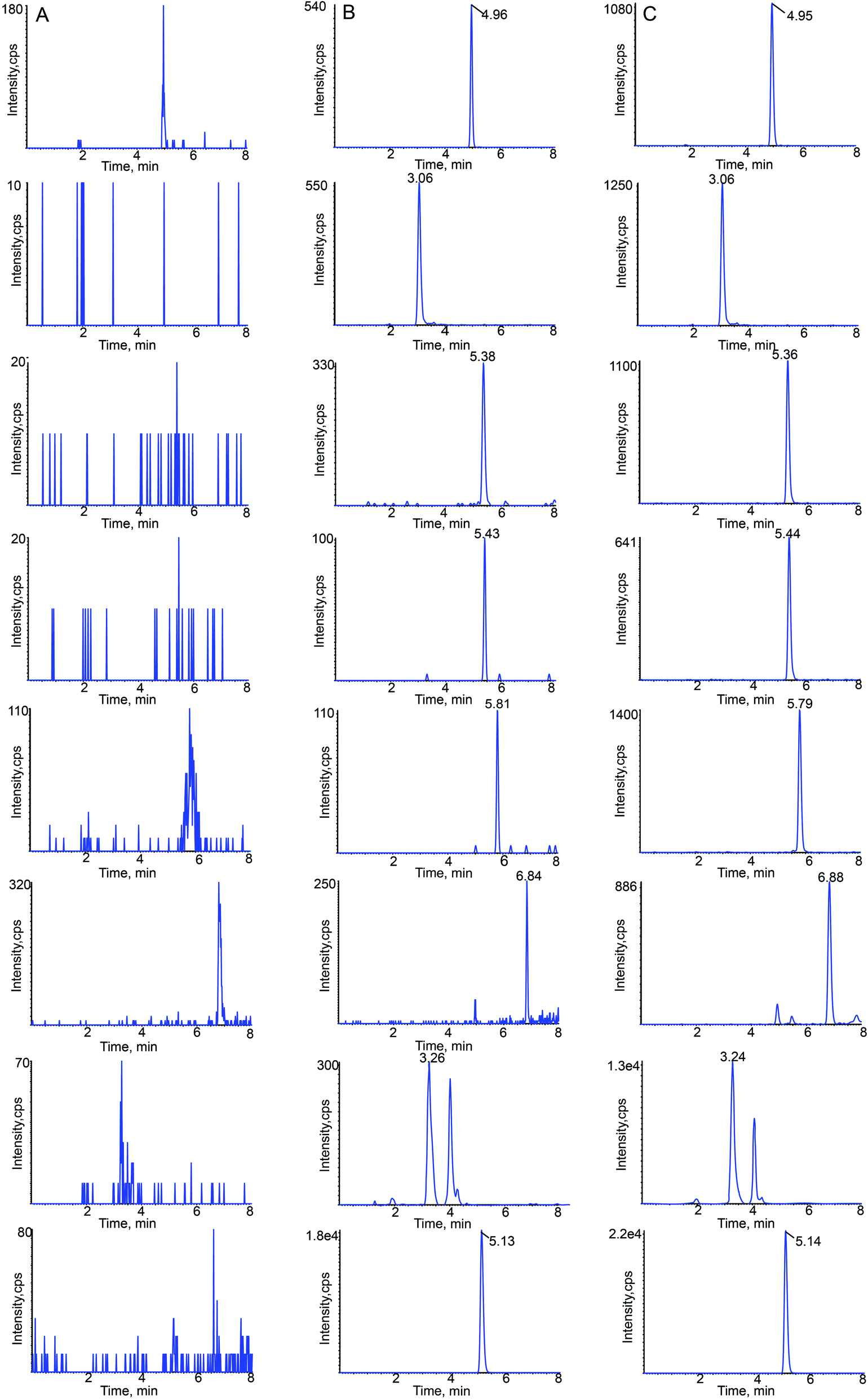 | ||
| Fig. 3 Typical chromatograms of linarin, rutin, luteolin, quercetin, apigenin, acacetin, acteoside and sulfamethoxazole (IS) in rat plasma: (A) blank plasma; (B) blank plasma spiked with the seven analytes at LLOQ and IS; (C) 15 min sample plasma after a single oral administration of Buddleja lindleyana Fort. extract. | ||
| Analyte | Regression equation | R2 | Linear range (ng mL−1) | LLOQ (ng mL−1) |
|---|---|---|---|---|
| Linarin | Y = 0.0008X + 0.0034 | 0.9998 | 1.37–2894.93 | 1.37 |
| Rutin | Y = 0.0024X + 0.0001 | 0.9989 | 1.21–410.28 | 1.21 |
| Luteolin | Y = 0.2120X + 0.1311 | 0.9979 | 1.58–248.68 | 1.58 |
| Quercetin | Y = 0.0259X + 0.0007 | 0.9974 | 1.32–65.52 | 1.32 |
| Apigenin | Y = 0.0085X + 0.0002 | 0.9981 | 1.77–467.72 | 1.77 |
| Acacetin | Y = 0.0545X + 0.0232 | 0.9978 | 2.32–104.68 | 2.32 |
| Acteoside | Y = 0.0166X − 0.0056 | 0.9989 | 2.03–2077.28 | 2.03 |
| Compounds spiked concentration (ng mL−1) | Intra-day (n = 6) | Inter-day (n = 6) | ||||
|---|---|---|---|---|---|---|
| Measured concentration (ng mL−1) | Accuracy (%) | Precision (%) | Measured concentration (ng mL−1) | Accuracy (%) | Precision (%) | |
| Linarin | ||||||
| 6.85 | 6.97 ± 1.78 | 1.75 | 8.19 | 7.05 ± 3.36 | 2.92 | 4.57 |
| 723.73 | 735.74 ± 3.45 | 1.66 | 5.04 | 736.68 ± 4.34 | 1.79 | 3.19 |
| 2861.18 | 2969.33 ± 5.86 | 3.78 | 7.93 | 2958.17 ± 2.01 | 3.39 | 4.86 |
|
||||||
| Rutin | ||||||
| 6.05 | 6.20 ± 4.24 | 2.48 | 3.47 | 6.15 ± 6.19 | 1.65 | 3.67 |
| 102.57 | 106.55 ± 5.83 | 3.88 | 6.23 | 105.08 ± 7.42 | 2.45 | 8.25 |
| 418.53 | 424.81 ± 9.09 | 1.50 | 5.01 | 438.12 ± 4.89 | 4.68 | 5.62 |
|
||||||
| Luteolin | ||||||
| 6.32 | 6.60 ± 1.07 | 4.43 | 9.16 | 6.69 ± 6.09 | 5.85 | 7.47 |
| 62.17 | 64.11 ± 3.33 | 3.12 | 7.31 | 64.27 ± 5.12 | 3.38 | 4.85 |
| 247.58 | 252.68 ± 4.47 | 2.06 | 6.04 | 253.40 ± 8.48 | 2.35 | 5.79 |
|
||||||
| Quercetin | ||||||
| 2.64 | 2.79 ± 3.14 | 5.68 | 6.85 | 2.78 ± 7.04 | 5.30 | 8.74 |
| 16.38 | 16.84 ± 4.57 | 2.81 | 4.16 | 17.14 ± 6.46 | 4.64 | 7.88 |
| 64.48 | 65.38 ± 6.81 | 1.40 | 3.58 | 68.70 ± 8.43 | 6.54 | 6.89 |
|
||||||
| Apigenin | ||||||
| 7.08 | 7.27 ± 4.13 | 2.68 | 5.75 | 7.41 ± 6.44 | 4.66 | 4.69 |
| 116.93 | 121.70 ± 3.49 | 4.08 | 5.89 | 123.76 ± 8.66 | 5.84 | 6.59 |
| 468.54 | 485.55 ± 5.23 | 3.63 | 7.51 | 480.49 ± 5.18 | 2.55 | 5.86 |
|
||||||
| Acacetin | ||||||
| 4.62 | 4.71 ± 3.15 | 1.95 | 7.48 | 4.93 ± 7.14 | 6.71 | 6.46 |
| 26.17 | 27.24 ± 4.65 | 4.09 | 4.46 | 27.14 ± 4.68 | 3.71 | 4.61 |
| 102.14 | 105.46 ± 7.65 | 3.25 | 5.17 | 107.08 ± 6.23 | 4.84 | 3.83 |
|
||||||
| Acteoside | ||||||
| 2.03 | 2.14 ± 8.18 | 5.42 | 5.58 | 2.13 ± 8.02 | 4.93 | 5.79 |
| 519.32 | 536.20 ± 7.54 | 3.25 | 4.52 | 564.14 ± 9.98 | 8.63 | 7.98 |
| 2027.29 | 2167.78 ± 6.25 | 1.39 | 6.93 | 2100.48 ± 5.24 | 3.61 | 8.75 |
| Compounds spiked concentration (ng mL−1) | Short-tern stability (room temperature for 8 h) | Long-tern stability (−20 °C for 21 d) | Free-thaw stability (3 free thaw cycles) | Post-preparation stability (room temperature for 4 h) | ||||
|---|---|---|---|---|---|---|---|---|
| Measured concentration (ng mL−1) | Accuracy (%) | Measured concentration (ng mL−1) | Accuracy (%) | Measured concentration (ng mL−1) | Accuracy (%) | Measured concentration (ng mL−1) | Accuracy (%) | |
| Linarin | ||||||||
| 6.85 | 7.07 ± 1.76 | 3.21 | 7.10 ± 17.48 | 3.65 | 7.02 ± 0.73 | 2.48 | 7.22 ± 1.36 | 5.40 |
| 723.73 | 755.28 ± 10.77 | 4.36 | 766.72 ± 17.29 | 5.94 | 754.99 ± 16.54 | 4.32 | 740.38 ± 7.66 | 2.30 |
| 2861.18 | 2930.71 ± 17.78 | 2.43 | 3056.88 ± 21.55 | 6.84 | 2962.18 ± 18.46 | 3.53 | 2986.21 ± 12.59 | 4.37 |
|
||||||||
| Rutin | ||||||||
| 6.05 | 6.36 ± 6.81 | 5.12 | 6.31 ± 5.88 | 4.30 | 6.2 ± 1.88 | 2.48 | 6.26 ± 0.48 | 3.47 |
| 102.57 | 108.48 ± 7.41 | 5.76 | 107.37 ± 9.98 | 4.68 | 110.30 ± 8.24 | 7.54 | 106.61 ± 13.32 | 3.94 |
| 418.53 | 426.82 ± 18.15 | 1.98 | 429.20 ± 19.79 | 2.55 | 437.7 ± 10.02 | 4.58 | 440.50 ± 16.46 | 5.25 |
|
||||||||
| Luteolin | ||||||||
| 6.32 | 6.76 ± 0.18 | 6.96 | 6.7 ± 0.76 | 6.01 | 6.60 ± 0.18 | 4.43 | 6.50 ± 0.16 | 2.85 |
| 62.17 | 64.88 ± 0.92 | 4.36 | 67.34 ± 3.63 | 8.32 | 66.03 ± 2.64 | 6.21 | 64.29 ± 0.37 | 3.41 |
| 247.58 | 260.55 ± 0.49 | 5.24 | 261.02 ± 4.65 | 5.43 | 258.35 ± 4.86 | 4.35 | 253.42 ± 2.77 | 2.36 |
|
||||||||
| Quercetin | ||||||||
| 2.64 | 2.80 ± 1.62 | 6.06 | 2.72 ± 0.32 | 3.03 | 2.78 ± 0.89 | 5.30 | 2.74 ± 0.32 | 3.79 |
| 16.38 | 17.12 ± 1.22 | 4.52 | 17.27 ± 2.69 | 5.43 | 17.48 ± 1.19 | 6.72 | 16.78 ± 1.77 | 2.44 |
| 64.48 | 66.98 ± 7.47 | 3.88 | 68.36 ± 3.68 | 6.02 | 67.46 ± 2.42 | 4.62 | 66.99 ± 4.58 | 3.89 |
|
||||||||
| Apigenin | ||||||||
| 7.08 | 7.17 ± 1.91 | 1.27 | 7.58 ± 0.51 | 7.06 | 7.34 ± 0.32 | 3.67 | 7.33 ± 0.16 | 3.53 |
| 116.93 | 123.78 ± 2.47 | 5.86 | 124.51 ± 4.02 | 6.48 | 124.68 ± 6.33 | 6.63 | 122.61 ± 7.12 | 4.86 |
| 468.54 | 490.89 ± 7.31 | 4.77 | 484.98 ± 5.09 | 3.51 | 494.36 ± 4.99 | 5.51 | 485.92 ± 3.94 | 3.71 |
|
||||||||
| Acacetin | ||||||||
| 4.62 | 4.69 ± 0.36 | 1.52 | 4.81 ± 0.27 | 4.11 | 4.81 ± 0.22 | 4.11 | 4.78 ± 0.18 | 3.46 |
| 26.17 | 27.31 ± 5.34 | 4.36 | 26.53 ± 3.21 | 1.38 | 26.99 ± 4.36 | 3.13 | 27.25 ± 3.96 | 4.13 |
| 102.14 | 107.02 ± 3.38 | 4.78 | 105.68 ± 9.65 | 3.46 | 108.48 ± 6.51 | 6.21 | 107.29 ± 6.25 | 5.04 |
|
||||||||
| Acteoside | ||||||||
| 2.03 | 2.11 ± 0.13 | 3.94 | 2.07 ± 0.12 | 1.97 | 2.1 ± 0.14 | 3.45 | 2.16 ± 0.35 | 6.40 |
| 519.32 | 548.82 ± 2.92 | 5.68 | 540.92 ± 9.44 | 4.16 | 544.87 ± 4.54 | 4.92 | 542.64 ± 4.56 | 4.49 |
| 2027.29 | 2150.14 ± 11.92 | 6.06 | 2167.98 ± 18.52 | 6.94 | 2132.91 ± 11.91 | 5.21 | 2140.01 ± 9.32 | 5.56 |
| Compounds spiked concentration (ng mL−1) | Extraction recovery | Matrix effect | ||
|---|---|---|---|---|
| Mean (%) | RSD (%) | Mean (%) | RSD (%) | |
| Linarin | ||||
| 6.85 | 89.02 | 3.16 | 80.67 | 3.67 |
| 723.73 | 85.42 | 5.94 | 85.34 | 7.97 |
| 2861.18 | 87.57 | 2.95 | 91.27 | 4.45 |
|
||||
| Rutin | ||||
| 6.05 | 87.68 | 2.86 | 101.5 | 3.65 |
| 102.57 | 88.75 | 5.64 | 95.10 | 4.18 |
| 418.53 | 86.76 | 3.53 | 90.13 | 5.29 |
|
||||
| Luteolin | ||||
| 6.32 | 83.43 | 4.42 | 83.56 | 3.13 |
| 62.17 | 96.21 | 6.57 | 85.86 | 6.71 |
| 247.58 | 88.84 | 7.32 | 87.47 | 7.16 |
|
||||
| Quercetin | ||||
| 2.64 | 87.39 | 2.64 | 96.49 | 5.48 |
| 16.38 | 88.64 | 4.89 | 88.77 | 4.23 |
| 64.48 | 90.05 | 7.13 | 97.64 | 6.26 |
|
||||
| Apigenin | ||||
| 7.08 | 89.77 | 2.39 | 101.2 | 5.87 |
| 116.93 | 87.16 | 3.42 | 93.28 | 2.16 |
| 468.54 | 96.89 | 4.08 | 90.09 | 1.93 |
|
||||
| Acacetin | ||||
| 4.62 | 85.48 | 4.42 | 84.07 | 2,67 |
| 26.17 | 89.66 | 7.21 | 87.59 | 5.26 |
| 102.14 | 84.06 | 2.95 | 91.28 | 4.98 |
|
||||
| Acteoside | ||||
| 2.03 | 89.10 | 3.95 | 87.31 | 5.18 |
| 519.32 | 90.69 | 4.78 | 84.23 | 7.34 |
| 2027.29 | 85.25 | 5.23 | 90.79 | 5.87 |
|
||||
| Sulfamethoxazole (IS) | ||||
| 476 | 98.41 | 3.78 | 95.67 | 3.28 |
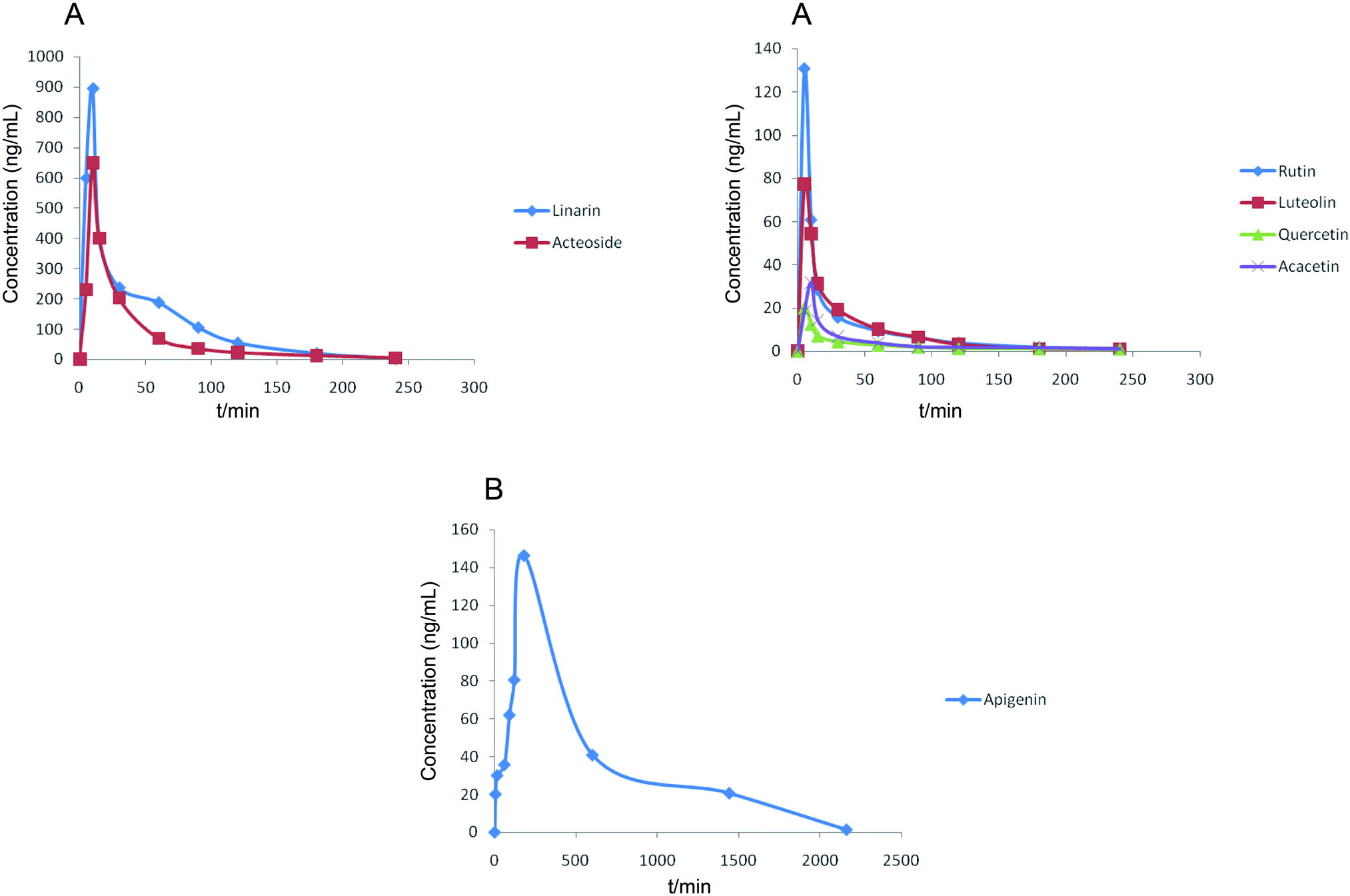 | ||
| Fig. 4 Mean plasma concentration–time curves of seven analytes after the oral administration of Buddleja lindleyana Fort. extract. (A) Linarin, rutin, luteolin, quercetin, acacetin and acteoside, (B) apigenin. | ||
| Compounds | Cmax (ng mL−1) (mean ± SD) | Tmax (min) | T1/2 (min) | AUC0–t (ng L−1 h−1) (mean ± SD) | AUC0–∞ (ng L−1 h−1) (mean ± SD) | CL (L h−1 kg−1) |
|---|---|---|---|---|---|---|
| Linarin | 894.12 ± 9.34 | 10 ± 0.45 | 36.49 ± 0.07 | 28447.88 ± 11.16 |
28770.18 ± 27.33 |
695.164 |
| Rutin | 130.76 ± 18.33 | 5 ± 0.35 | 49.57 ± 0.18 | 3039.40 ± 26.08 | 3039.62 ± 54.07 | 6464.93 |
| Luteolin | 77.37 ± 25.72 | 5 ± 0.41 | 36.08 ± 0.24 | 2106.15 ± 28.81 | 2122.72 ± 34.23 | 9421.89 |
| Quercetin | 20.15 ± 24.85 | 5 ± 0.33 | 46.03 ± 0.09 | 558.58 ± 33.33 | 1093.45 ± 55.91 | 18290.81 |
| Apigenin | 146.42 ± 14.88 | 180 ± 0.38 | 298.50 ± 0.16 | 85202.18 ± 58.54 |
85759.09 ± 32.94 |
233.21 |
| Acacetin | 31.92 ± 17.58 | 10 ± 0.35 | 96.87 ± 0.12 | 916.00 ± 51.59 | 1062.19 ± 50.61 | 18829.04 |
| Acteoside | 649.78 ± 16.42 | 10 ± 0.22 | 46.80 ± 0.35 | 18381.44 ± 65.07 |
18704.54 ± 59.21 |
1069.26 |
The results showed that the mean plasma concentration–time curves of the seven compounds were made up of two parts, which included (A) linarin, rutin, luteolin, quercetin, acacetin and acteoside and (B) apigenin. Linarin, rutin, luteolin, quercetin, acacetin and acteoside were rapidly absorbed, and the Tmax values were 10, 5, 5, 5, 10 and 10 min, respectively. However, the Tmax of apigenin was 180 min. The T1/2 of the seven chemical compounds were 36.49, 49.57, 36.08, 46.03, 298.50, 96.87 and 46.80 min.
The concentrations of linarin and acteoside were highest. Acteoside could be obtained by the hydrolysis of linarin after oral administration of Buddleja lindleyana Fort. extract, which indicated that the amount of acteoside absorbed in blood included the chemical compound acteoside and part of the hydrolysed linarin. A further study should be performed, such as on the metabolism and excretion of the linarin monomer. A double-peak phenomenon of apigenin appeared in Fig. 4, which was associated with entero-hepatic recirculation. The first peak of apigenin appeared at 5 min, and the second at 180 min, which is higher than the first. In general, the results of this study might be helpful for the study of metabolism, excretion and activity screening after oral administration of Buddleja lindleyana Fort. extract and monomer linarin, which would be beneficial for the application of Buddleja lindleyana Fort. in clinical therapy.
4. Conclusion
Herein, a powerful analytical and sensitive HPLC-MS/MS method was developed for the simultaneous determination of seven chemical compounds of Buddleja lindleyana Fort., and PK studies were conducted after oral administration of Buddleja lindleyana Fort. extract. This is the first quantitative and pharmacokinetic study of Buddleja lindleyana Fort via HPLC-MS/MS. The excellent selectivity, sensitivity, precision, accuracy and extraction recovery proved that the method is fast and highly efficient. This study revealed that the contents of seven chemical compounds were different because of different growth environments. In general, the highest contents of the seven chemical compounds were found in Buddleja lindleyana Fort. obtained from Xizang, so this was chosen for conducting the PK study. Linarin, rutin, luteolin, quercetin, acacetin and acteoside were rapidly absorbed in the blood, while apigenin was absorbed very slowly. The main absorption substances after the administration of Buddleja lindleyana Fort. extract were linarin and acteoside. The maximum plasma concentrations (Cmax) of linarin, rutin, luteolin, quercetin, apigenin, acacetin and acteoside were 894.12 ± 9.34 ng mL−1, 130.76 ± 18.33 ng mL−1, 77.37 ± 25.72 ng mL−1, 20.15 ± 24.85 ng mL−1, 146.42 ± 14.88 ng mL−1, 31.92 ± 17.58 ng mL−1 and 649.78 ± 16.42 ng mL−1, respectively. The times to reach Cmax of linarin, rutin, luteolin, quercetin, apigenin, acacetin and acteoside were 10, 5, 5, 5, 180, 10, and 10 min, respectively. A double-peak phenomenon appeared in the drug–time curve of apigenin. These results might be helpful for investigating the bioactive mechanism and clinical application of Buddleja lindleyana Fort.Conflicts of interest
All the authors have declared no conflict of interest.Acknowledgements
The work received financial support from Hebei Administration of Traditional Chinese Medicine (No. 2021147), and the Natural Science Foundation of Hebei Province of China (H2020206091).References
- Flora of China Editorial Committee, Flora Reipublicae Popularis Sinicae. Science Press, Beijing, 1992, vol. 61, p. 308 Search PubMed.
- G. Chen, W. B. Sun and H. Sun, Bot. J. Linn. Soc., 2007, 154, 305–312 CrossRef.
- W. Zhang, Z. Li, F. Q. Xu, Y. S. Ren, S. W. Xu, T. S. Wang, J. S. Liu and D. L. Wu, J. Asian Nat. Prod. Res., 2019, 21(5), 426–434 CrossRef CAS PubMed.
- Jiangsu new medical college, Traditional Chinese medicine dictionary. Vol II: Shanghai Scientific and Technical Publisher. Shanghai, 1986, p. 3575 Search PubMed.
- P. Y. Wu, Y. S. Ren, D. L. Wu and J. W. Qiao, China J. Chin. Mater. Med., 2016, 41, 1218–1221 Search PubMed.
- A. Y. Mensan, J. Nat. Prod., 2000, 2000(63), 1210–1213 CrossRef PubMed.
- J. Lu, X. Pu, Y. Li, Y. Zhao and G. Tu, Z. Naturforsch., B: J. Chem. Sci., 2005, 60, 211–214 CrossRef CAS.
- T. Yoshida, J. Nobuhara, N. Fuji and T. Okuda, Chem. Pharm. Bull., 1978, 26, 2543 CrossRef CAS.
- H. Z. Guo, K. Kazuo, W. Li, S. Tadaaki, D. A. Guo and N. Tamotsu, J. Nat. Prod., 2004, 67, 10–13 CrossRef CAS PubMed.
- D. L. Wu, Y. K. Wang, J. S. Liu, X. C. Wang and W. Zhang, J. Asian Nat. Prod. Res., 2012, 14, 342–347 CrossRef CAS PubMed.
- A. M. Emam, R. Elias, A. M. Moussa, R. Faure, L. Debrauwer and G. Balansard, Phytochemistry, 1998, 48, 739–742 CrossRef CAS PubMed.
- J. H. Lu, X. P. Pu, G. Z. Tu and Y. P. Zhao, J. Chin. Pharm. Sci., 2004, 13, 151–154 CAS.
- B. H. Tai, N. X. Nhiem, T. H. Quang, N. T. T. Ngan, N. H. Tung, Y. Kim, J. J. Lee, C. S. Myung, N. M. Cuong and Y. H. Kim, Bioorg. Med. Chem. Lett., 2011, 21, 3462–3466 CrossRef CAS PubMed.
- J. G. A. Acevedo, C. M. C. Castaneda, F. J. C. Benitez, D. A. Duran, V. R. Barroso, C. G. Martinez, L. J. L. Munoz, C. A. Martinez and A. R. De Vivar, Fitoterapia, 2005, 76, 301–309 CrossRef PubMed.
- L. N. Moreira, C. Feltrin, J. E. Gonçalves, W. V. de Castro, C. M. O. Simoes, R. M. de Pádua, S. F. Cortes and F. C. Braga, Biomed. Pharmacother., 2020, 132, 110900, DOI:10.1016/j.biopha.2020.110900.
- A. L. Nijstad, M. M. Tibben, A. Gebretensae, H. Rosing, E. de Vos-Kerkhof, C. M. Zwaan, A. D. R. Huitema and J. H. Beijnen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2021, 1171, 122639–122645 CrossRef CAS PubMed.
- X. Zhang, J. T. Yin, C. J. Liang, Y. P. Sun and L. T. Zhang, J. Agric. Food Chem., 2017, 65, 10959–10972 CrossRef CAS PubMed.
- R. Feng, X. W. Zhang, J. T. Yin, Y. Q. Zhang, Y. L. Ma, X. Zhang, L. T. Zhang and D. Q. Li, J. Pharm. Biomed. Sci., 2021, 197, 113905, DOI:10.1016/j.jpba.2021.113905.
- X. Zhang, M. Liao, X. Y. Cheng, C. J. Liang, X. P. Diao and L. T. Zhang, Rapid Commun. Mass Spectrom., 2018, 32, 1451–1461 CrossRef CAS PubMed.
- M. Y. Liu, S. H. Zhao, Z. Q. Wang, Y. F. Wang, T. Liu, S. Li, C. C. Wang, H. T. Wang and P. F. Tu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 949–950, 115–126 CrossRef CAS PubMed.
- S. Wang, P. C. Qi, N. Zhou, M. M. Zhao, W. J. Ding, S. Li, M. Y. Liu, Q. Wang and S. M. Jin, Anal. Bioanal. Chem., 2016, 408, 7423–7436 CrossRef CAS PubMed.
- X. W. Shi, M. Liu, M. Zhang, K. R. Zhang, S. C. Liu, S. Qian, R. Shi, X. J. Jiang and Q. Wang, Food Chem., 2013, 141, 357–365 CrossRef CAS PubMed.
- J. Nie, P. Yaro, K. F. He and S. Zeng, J. Pharm. Biomed. Sci., 2019, 72, 78–85 CrossRef PubMed.
- C. P. Yang, M. H. Liu, W. Zou, X. L. Guan, L. Lai and W. W. Su, J. Asian Nat. Prod. Res., 2012, 14, 68–75 CrossRef CAS PubMed.
- N. Ge, L. Kong, A. H. Zhang, Y. Sun, M. Q. Zhao, B. Zhang, L. Xu, X. Ke, H. Sun and X. J. Wang, RSC Adv., 2021, 11, 5491–5505 RSC.
- C. He, J. Cao, Y. Y. Bao, Z. X. Sun, Z. Y. Liu and C. Li, Food Chem., 2021, 347, 129057, DOI:10.1016/j.foodchem.2021.129057.
- M. Afzal, M. Sielaff, V. Curella, M. Neerukonda, K. E. Hassouni, D. Schuppan, S. Tenzer and C. F. H. Longin, Plants, 2021, 10, 424–442 CrossRef CAS PubMed.
- D. Wei, W. L. Fan and Y. Xu, Food Chem., 2021, 353, 129521, DOI:10.1016/j.foodchem.2021.129521.
- J. Gao, N. Shi, H. J. Guo, J. F. Gao, X. Tang, S. Y. Yuan, J. H. Qian and B. Y. Wen, ACS Omega, 2021, 6, 5348–5358 CrossRef CAS PubMed.
- K. Liu, B. J. Jia, L. P. Zhou, L. Xing, L. Y. Wu, Y. Y. Li, J. G. Lu, L. Zhang and S. Guan, J. Proteome Res., 2021, 20, 1371–1381 CrossRef CAS PubMed.
- X. Wang, S. S. Johansen, M. K. K. Nielsen and K. Linnet, Drug Test. Anal., 2017, 9, 1137–1151 CrossRef CAS PubMed.
- M. Cortese, C. Delporte, D. Dufour, C. Noyon, M. Chaumont, B. De Becker, F. Reye, A. Rousseau, E. Omer, J. Neve, M. Piagnerelli, K. Z. Boudjeltia, B. Robaye and P. Van Antwerpen, Talanta, 2019, 193, 206–214 CrossRef CAS PubMed.
- S. L. Parker, S. Pandey, F. B. Sime, J. Lipman, J. A. Roberts and S. C. Wallis, Anal. Bioanal. Chem., 2019, 411, 7831–7840 CrossRef CAS PubMed.
- H. Jungen, H. Andresen- Streichert, A. Muller and S. Iwersen-Bergman, J. Anal. Toxicol., 2017, 41, 22–31 CrossRef CAS PubMed.
- Z. Li, D. L Wu, H. S. Zhao, F. Q. Xu and W. Zhang, Chin. J. Exp. Tradit. Med. Formulae, 2017, 23(6), 74–77 Search PubMed.
- X. C. Wang and J. H. Lu, Chin. J. Hosp. Pharm., 2018, 38(10), 1056–1058 Search PubMed.
- Z. Y. Zhang, P. P. Jia, X. X. Zhang, Q. Y. Zhang, H. T. Yang, H. Shi and L. T. Zhang, J. Ethnopharmacol., 2014, 158, 66–75 CrossRef CAS PubMed.
- H. J. Xu, X. W. Shi, X. Ji, Y. F. Du, H. Zhu and L. T. Zhang, J. Sep. Sci., 2011, 34, 308–316 CrossRef CAS PubMed.
- X. W. Shi, Y. B. Wu, T. Lv, Y. F. Wang, Y. Fu, M. M. Sun, Q. W. Shi, C. H. Huo, Q. Wang and Y. C. Gu, Anal. Bioanal. Chem., 2017, 409, 4669–4679 CrossRef CAS PubMed.
- X. Zhang, C. J. Liang, C. L. Li, M. D. Bu, L. Y. Bu, Y. D. Xiao, H. G. Sun and L. T. Zhang, J. Chromatogr. Sci., 2018, 56(7), 582–594 CAS.
- Y. Sun, Y. F. Du, K. Yang, L. Chang, L. Cao, Y. P. Ren, Q. Sun, Q. Wang, L. T. Zhang and P. T. Lv, J. Pharm. Biomed. Sci., 2013, 81, 34–43 CrossRef PubMed.
- C. J. Liang, X. Zhang, X. P. Diao, M. Liao, Y. P. Sun and L. T. Zhang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1084, 69–79 CrossRef CAS PubMed.
- X. Zhang, C. J. Liang, J. T. Yin, Y. P. Sun and L. T. Zhang, RSC Adv., 2018, 8, 11813–11827 RSC.
- L. J. Zhu, L. Chen, C. F. Bai, A. G. Wu, S. C. Liang, F. H. Huang, S. S. Cao, L. Yang, W. J. Zou and J. M. Wu, Biomed. Pharmacother., 2020, 123, 109756, DOI:10.1016/j.biopha.2019.109756.
- C. H. Du, Y. Yan, C. X. Shen, X. F. Cui, X. P. Pei and X. M. Qin, J. Pharm. Anal., 2020, 10, 385–395 CrossRef PubMed.
- F. Cai, W. Xu, H. Wei, L. N. Sun, S. H. Gao, Q. Yang, J. Feng, F. Zhang and W. S. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 1845–1854 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The validation information on the content analysis. Table repeatability and matrix effect of seven analytes. See DOI: 10.1039/d1ra04154a |
| This journal is © The Royal Society of Chemistry 2021 |