
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplications of aryl-sulfinamides in the synthesis of N-heterocycles
Rose Mary Philip
a,
G. S. Susan Treesa
a,
Salim Saranya
a and
Gopinathan Anilkumar
 *abc
*abc
aSchool of Chemical Sciences, Mahatma Gandhi University, Priyadarsini Hills P O, Kottayam, Kerala 686560, India. E-mail: anilgi1@yahoo.com; anil@mgu.ac.in; Fax: (+91) 481-2731036
bAdvanced Molecular Materials Research Centre (AMMRC), Mahatma Gandhi University, Priyadarsini Hills P O, Kottayam, Kerala, 686560 India
cInstitute for Integrated Programmes and Research in Basic Sciences (IIRBS), Mahatma Gandhi University, Priyadarsini Hills P O, Kottayam, Kerala, 686560 India
First published on 8th June 2021
Abstract
Enantiopure aryl-sulfinamides are important chiral auxiliaries in the asymmetric synthesis of amines and their derivatives. Here, we provide an overview of arylsulfinamide mediated asymmetric methods towards N-heterocycle synthesis. This methodology through sulfinylimines offers general access to structurally diverse piperidines, pyrrolidines, aziridines and their derivatives which represent the structural motif of many natural products and therapeutically important compounds. The review covers articles from 2006–2020 and we have categorized the review based on the ring size as 3-, 5-, and 6- membered heterocycles and their derivatives.
 Rose Mary Philip | Rose Mary Philip was born in Kottayam, Kerala, India. She obtained her dual degree of BSMS (Bachelor of Science and Master of Science) from Indian Institute of Science Education and Research, Thiruvananthapuram in 2019. She has qualified the CSIR-UGC National Eligibility Test 2019 with a research fellowship. Currently she is pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 G. S. Susan Treesa | G. S. Susan Treesa was born in 1994 in Kerala, India. She received her B. Sc. and M. Sc. degrees from St. George College, Aruvithura in 2015 and 2017 respectively. She has completed her M. Phil degree under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University. |
 Salim Saranya | Salim Saranya was born in 1990 in Kerala, India. She received her B. Sc. and M. Sc. degrees from Christian College, Chengannur in 2012 and 2014 respectively. She qualified the CSIR-UGC National Eligibility Test in 2015 with a research fellowship and currently pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and took his Ph. D in 1996 from Regional Research Laboratory (renamed as National Institute for Interdisciplinary Science and Technology NIIST-CSIR), Trivandrum with Dr Vijay Nair. He did postdoctoral studies at University of Nijmegen, The Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin A. Davis), Leibniz-Institüt für Organische Katalyse (IfOK), Rostock, Germany (with Professor Matthias Beller) and Leibniz-Institüt für Katalyse (LIKAT), Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently he is Professor in Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry, heterocycles and catalysis. He has published more than 130 papers in peer-reviewed journals, 7 patents, 7 book chapters and edited 2 books entitled “Copper Catalysis in Organic Synthesis” (Wiley-VCH, 2020) and “Green Organic Reactions” (Springer, in press). He has received Dr S. Vasudev Award from Govt. of Kerala, India for best research (2016) and Evonik research proposal competition award (second prize 2016). |
1. Introduction
The advent of chiral sulfoxide N-protecting groups as chiral inductors opened up efficient methodologies towards the preparation of chiral amines,1 which forms part of various bioactive compounds. A series of enantiopure sulfinyl motifs were designed over time by different research groups to regulate the reactivity of the sulfinimines towards the desired direction. The first report on the synthesis of sulfinimines came from the research group of Davis through the synthesis of p-toluene-sulfinimines over 45 years ago, followed by the generation of its enantiopure form by Cinquini et al. in 1982. Later, the introduction of enantiopure tert-butanesulfinamide by Ellman and co-workers2 offered facile access to tert-butyl-sulfinimines.The highly stereodirecting nature of the sulfinyl group is applied in numerous methodologies and its easy deprotection enabled further modification of substrates. Among the different chiral sulfoxides, p-toluene- and tert butyl-sulfinimines stay well explored in asymmetric synthesis.1 One of the notable advantages of p-toluenesulfinimines is that being UV active, the reactions can be easily monitored.3 Nucleophilic addition onto enantiopure sulfinimines remains the finest method for the asymmetric construction of chiral building blocks that can be transformed to a series of nitrogen heterocycles including aziridines, pyrrolidines and piperidines.4,5 In view of the obvious presence of N-heterocycles in nucleic acids, hormones, vitamins, drugs and agrochemicals, novel methodologies towards their synthesis are always in great demand.6,7 The reports utilizing enantiopure sulfinimines are increasing day by day proving their importance in constructing bioactive molecules, natural products and pharmaceuticals.
The pioneers of the field, Davis et al. carefully summarized their major contributions till 2006.8 In the same year, Stockman et al. compiled a review that presented the significance of chiral non-racemic sulfinimines in asymmetric synthesis.1 Several informative reviews have also appeared on the preparation and applications of sulfinamides over the times.9–11 Recently, our group contributed a review outlining the synthetic methods towards N-heterocycles mediated by tert-butanesulfinamide.12 The current review aims to discuss the application of aryl sulfinimines, majorly p-toluene sulfinimine in the synthesis of N-heterocycles. The review covers the relevant articles from 2006 to 2020 and is organized based on the ring size as 3-, 5-, and 6-membered heterocycles and their derivatives.
2. Three-membered ring
Davis' group devised a method for the synthesis of 2-substituted 2H-azirine 3-carboxylates 6 in an optically pure form via the dehydrochlorination of methyl 2-chloroaziridine 2-carboxylates 5.13 Initially, lithium enolate of methyl dichloroacetate 2 was added to sulfinimines 1 to yield single enantiomers of β-amino esters 3 followed by cyclization with the use of KH to obtain 2-chloroaziridines 4 in good yields (Scheme 1). Then the aza Diels–Alder reaction between azirine 6 obtained after photodesulfinylation, and diene 7 underwent smoothly and provided with the tricyclic aziridine carboxylates 8 in enantiopure form.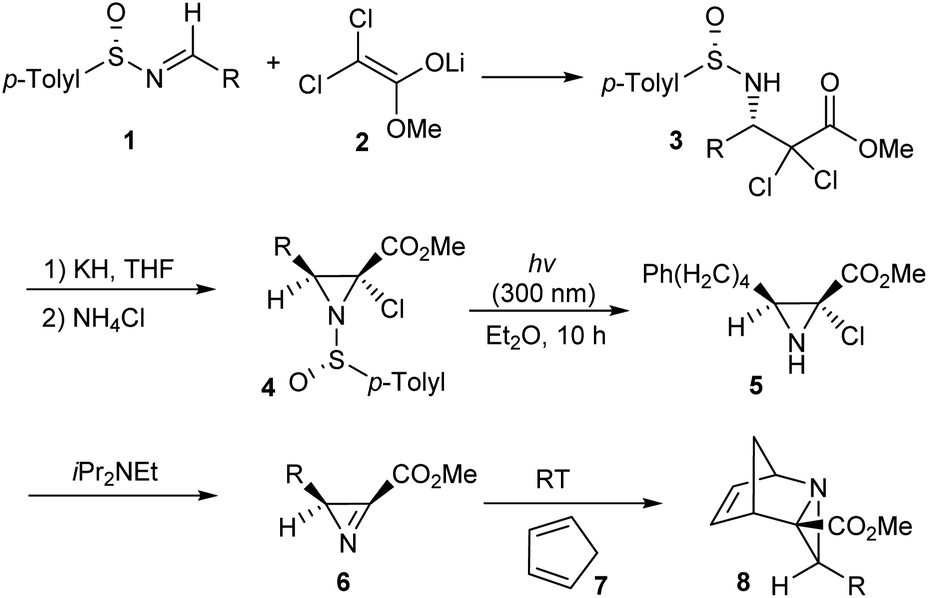 | ||
| Scheme 1 Synthesis of bicyclic and tricyclic aziridine carboxylates. | ||
In 2009, a simple protocol towards the synthesis of chiral aziridines from cyclic alkenes was developed.14 Herein, the lithium salt of p-toluenesulfinamide, 10 was added to cyclic α-haloenones 9 to afford the anticipated aziridines 11a and 11b in 30–65% of diastereomeric excess (Scheme 2). Analysis with different cyclic olefins concluded that yield and selectivity were higher with 6-membered α-bromoenones. The remarkably low diastereoselectivity observed with alkenes that are not part of the ring suggested the importance of the conformational restriction offered by cyclic alkenes in chiral induction.
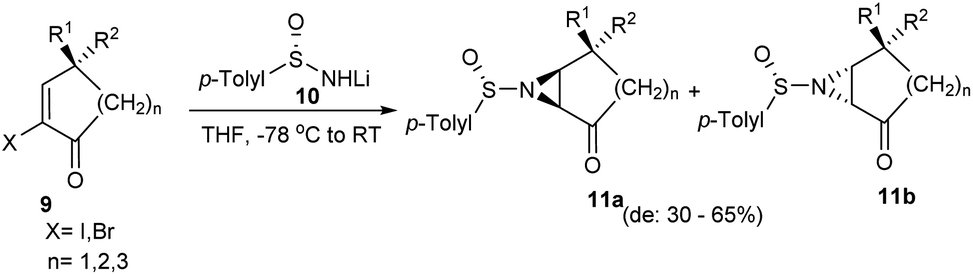 | ||
| Scheme 2 Synthesis of chiral aziridines from cyclic α-haloenones. | ||
Stockman and coworkers conducted an aza-Darzens reaction of optically active sulfinimines with substituted 2-bromoesters 12 which resulted in a wide variety of trisubstituted aziridines 15 in good stereoselectivity and high yields (Scheme 3).15 Both mesityl- and t-butanesulfinimines smoothly underwent aziridination where better cis/trans ratios were obtained when mesityl sulfinimines 13 possessing a C3-aliphatic chain were employed rather than an aromatic imine. On the other hand, Ellman's auxiliary was found suitable for aromatic imines. They also demonstrated the successive removal of the auxiliaries and predicted the applicability of the present three step protocol in preparing optically active N–H aziridines.
 | ||
| Scheme 3 Aza-Darzens reaction of chiral sulfinimines with substituted 2-bromoesters forming trisubstituted aziridines. | ||
Further to their previous studies, they examined the synthetic utility of the aforementioned vinyl aziridine 2-carboxylates in the generation of cyclic sulfoximines 18.16 They devised a one-pot strategy that employed sulfinimines as the starting material that gave cyclic sulfoximines 18 in high yields and excellent stereoselectivity (Scheme 4). They made a mechanistic hypothesis that the ester species activated the alkene to conduct the thermal sigmatropic rearrangement. To exemplify the reactivity of the products, the formal synthesis of biologically active trachelanthamidine 21 was carried out via the conversion of the cyclic sulfoximine 20 into a pyrroline.
 | ||
| Scheme 4 One-pot synthesis of chiral cyclic sulfoximines from optically active sulfinimines. | ||
3. Five-membered ring
A novel method for the synthesis of chiral N-tosyl α-amino aldehydes from N-sulfinyl α-amino 1,3-dithioacetals was developed by the research group of Davis in 2006.17 They made use of DBDMH (1,3-dibromo-5,5-dimethylhydantoin) as the hydrolyzing agent for this transformation. Besides, they presented the formal synthesis of (−)-2,3-trans-3,4-cis-dihydroxyproline 28 to demonstrate the application of the obtained α-amino aldehydes (Scheme 5). Here, the aldehyde 25 obtained after hydrolysis underwent a sequence of reactions to construct the pyrrolidine 27 after ring-closing metathesis. Further functionalization could access the dihydroxyproline derivative 28.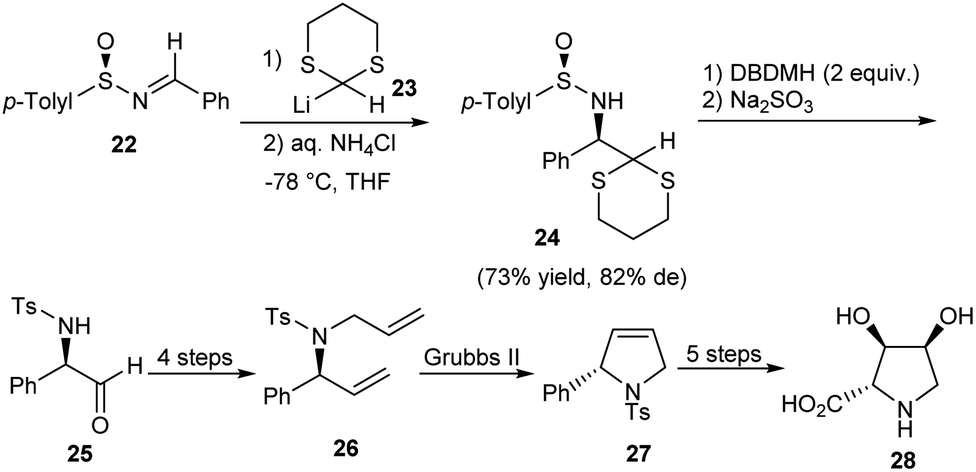 | ||
| Scheme 5 Synthesis of dihydroxyproline derivative through chiral N-tosyl α-amino aldehydes. | ||
Synthesis of trans-2,5-disubstituted pyrrolidines in enantiopure form was accomplished by the same research group through an iodocyclization strategy.18 To illustrate the utility of the method, the stereoselective synthesis of (−)-pyrrolidine 197B 33 was conducted (Scheme 6). The strategy began with the addition reaction on sulfinimine 29 leaving the ester 30 as a single diastereomer. The precursor for iodocyclization, the homoallylic sulfonamide 31 was prepared from the sulfinimine derived ester in 4 steps. The iodocyclization step using I2/K2CO3/H2O/MeCN afforded the corresponding 3-iodo trans-2,5-disubstituted pyrrolidine 32 which was then transformed to the final cyclized product 33. Hence, the present method to access the significant N-containing ring is applicable in constructing similar motifs of bioactive compounds.
 | ||
| Scheme 6 Synthetic protocol towards trans-2,5-disubstituted pyrrolidines. | ||
In 2008, Lautens' group introduced an iodide-mediated Mannich/cyclization sequence to afford trans-2,3-disubstituted pyrrolidines 36 and 38 in a single step from methylenecyclopropyl amides 35.19 They utilized magnesium iodide to conduct the reaction between diverse methylenecyclopropyl amides with aromatic, heteroaromatic imines 34 and α,β-unsaturated imines 37 which gave good to excellent yields and selectivities (Scheme 7). Later, cleavage of the auxiliary was conducted under mild reaction conditions to furnish pyrrolidines in highly enantioenriched form (dr up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
 | ||
| Scheme 7 Synthesis of trans-2,3-disubstituted pyrrolidines from methylenecyclopropyl amides. | ||
Viso and Pradilla with coworkers designed a method for the stereoselective synthesis of 3-sulfinyl and 3-sulfonyl 2,5-cis-dihydropyrroles via chiral sulfinimines.20 They performed a highly diastereoselective addition of chiral α-metalated vinyl and dienyl sulfoxides onto enantiopure N-sulfinimines 39 that offered respective allylic amines 41 in good yields (93–98% yield) and selectivity (dr up to 99:1) (Scheme 8). Herein, the highly diastereoselective construction of the new C–C bond was attributed to the stereoinduction by chiral sulfinyl groups present in both the starting compounds. Starting from 41a, cis-2,5-disubstituted dihydropyrroles 42 were prepared via an electrophilic cyclization reaction.
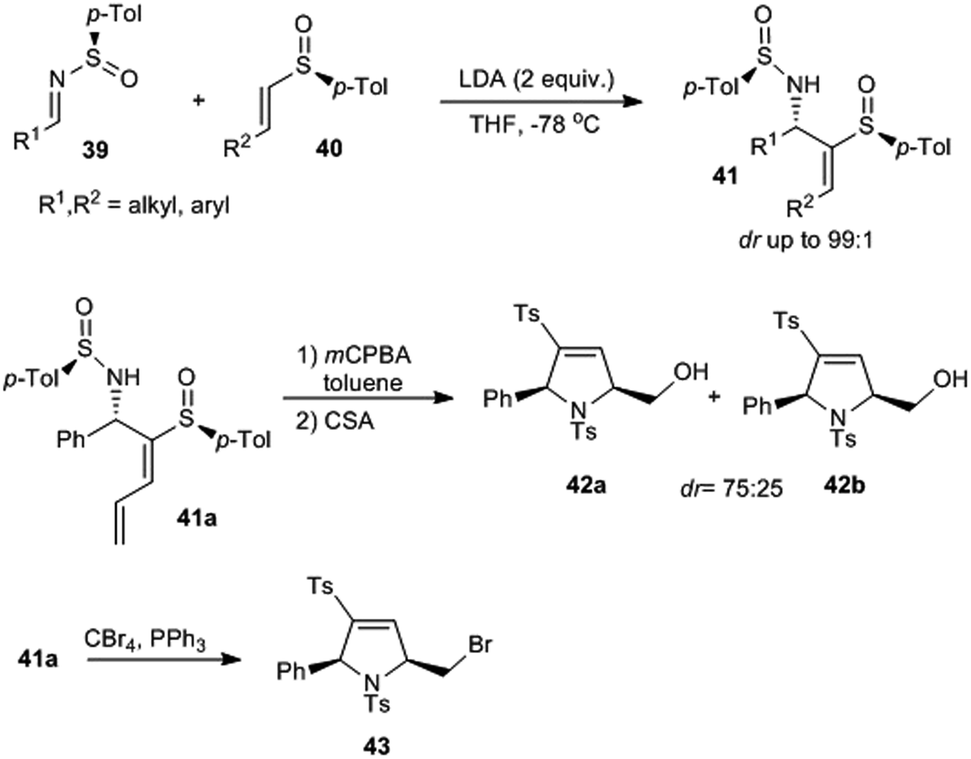 | ||
| Scheme 8 Stereoselective synthesis of 3-sulfinyl and 3-sulfonyl 2,5-cis-dihydropyrroles through chiral sulfinimines. | ||
The research group of Kamimura established an elegant methodology towards the synthesis of chiral 2-alkyl-substituted 2,5-dihydropyrroles 45.21 This method made use of their earlier protocol of Michael/iminoaldol domino reaction with an acrylate and a p-tolylsulfinimine to produce aza-Baylis–Hillman adduct 44 in high optical purity (Scheme 9).22 With 44 in hand, a short and simple three step conversion gave optically active 2,5-dihydropyrroles 45 through N-allyl-β-amino-R-methylene ester intermediate. Moreover, they demonstrated the synthetic utility of the method in preparing (−)-trachelanthamidine 52, a pyrrolizidine alkaloid with anticipated biological activity. From the chiral sulfinimine 46, a highly stereoselective formal synthesis of (−)-trachelanthamidine was accomplished in 11 steps. Since chiral 2,5-dihydropyrroles are considered a potent starting point in the synthesis of heterocyclic compounds, they predicted the synthetic applicability of the method in future.
 | ||
| Scheme 9 Synthesis of chiral 2-alkyl-substituted 2,5-dihydropyrroles and formal synthesis of (−) trachelanthamidine. | ||
Davis' group devised an efficient protocol for the preparation of indolizidine alkaloids (+)-monomorine I 58 and (−)-indolizidine 195B 57 from sulfinimine derived common intermediates.23 In the initial steps, the stable α-diazophosphonate 55 prepared from β-amino ester 53 was heated with Rh2(OAc)4 that conducted a diastereoselective intramolecular reaction which provided the 3-oxo pyrrolidine phosphonate 56 (Scheme 10). Then, 56 acted as the common intermediate for the stereoselective synthesis of the functionalized pyrrolidines 57 and 58.
 | ||
| Scheme 10 Asymmetric synthesis of indolizidine alkaloids (+)-monomorine I and (−)-indolizidine 195B. | ||
4. Six-membered ring
Substituted piperidines have shown applications in pharmaceutical chemistry forming important building units of bioactive compounds. Davis et al. established a synthetic route to Nuphar alkaloids having 2,3,6-trisubstituted piperidines via an intramolecular Mannich reaction of sulfinimine derived amino ketone (Scheme 11).24 The amino ketone 60 was afforded majorly as a single diastereomer from (R)-(−)-N-(3-furylmethylene)-p-toluenesulfinamide 59 via the addition of potassium enolate of methyl ethyl ketone. Starting from the common precursor 64 obtained after Mannich cyclization, they discussed the synthesis of Nuphar alkaloids (−)-nupharamine 65 and (−)-(5S,8R,9S)-5-(3-furyl)-8-methyloctahydroindolizidine 66.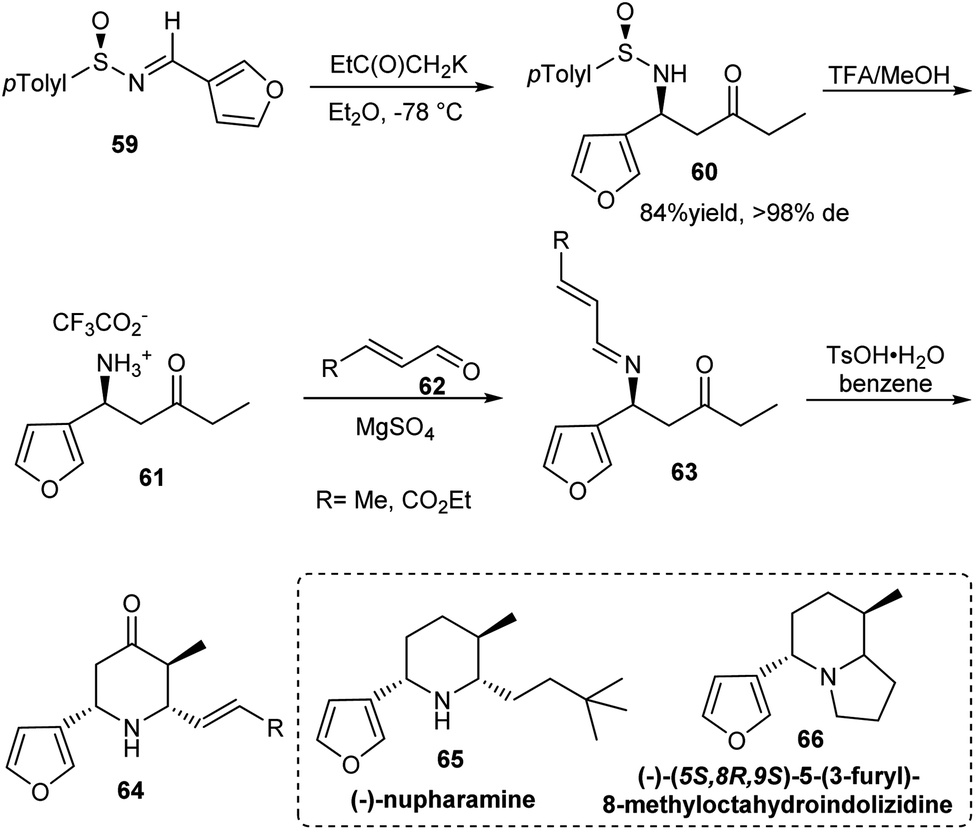 | ||
| Scheme 11 Synthetic route to Nuphar alkaloids having 2,3,6-trisubstituted piperidine core. | ||
In 2006, Kawecki presented the aza Diels–Alder reaction of chiral sulfinimines with the highly active Rawal diene 68 to furnish enantioenriched dihydropyridone.25 The reaction was performed in presence of TMSOTf and with 10-isobornylsulfinimines, 8-menthylsulfinimines, t-butyl and p-tolylsulfinimines as optically active sulfinimine partners. With p-tolylsulfinimine 67, open chain enaminone 69 was obtained in 89% ee which then underwent a cyclization step with acid and gave 2-phenyl substituted dihydropyridone 70 (Scheme 12). The method provided access to 2-aryl substituted dihydropyridones in modest stereoselectivity.
 | ||
| Scheme 12 Stereoselective synthesis of 2-phenyl dihydropyridone. | ||
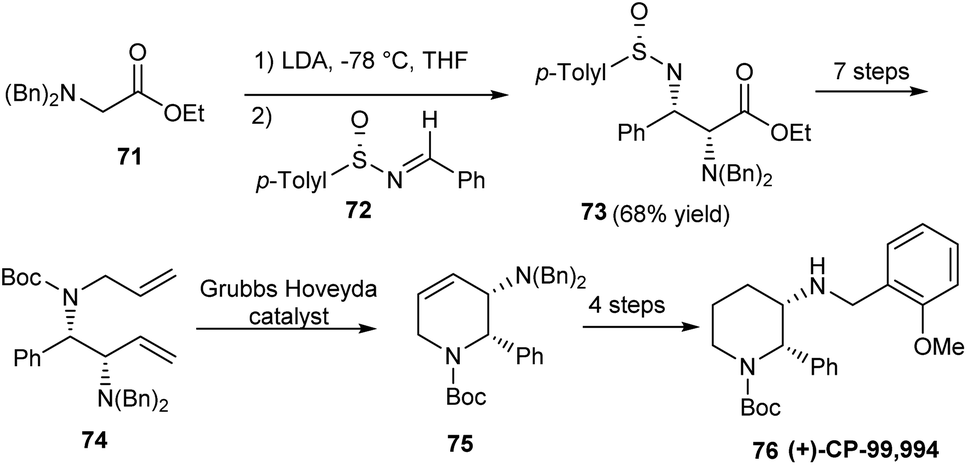
The research group of Davis established protected 2,3-diamino esters as valuable synthetic units towards the preparation of piperidine derivative (+)-CP-99,994 76, an effective neurokinin substance P receptor antagonist via a 12 step protocol.26 In the initial step sulfinimine 72 underwent the addition of a prochiral enolate moiety, constructing the compound 73 bearing different N-protecting groups and two newly generated stereogenic centers (Scheme 13). Starting from 73 various synthetic transformations followed, wherein a Kocienski-modified Julia olefination and Grubbs-Hoveyda catalyst enabled ring-closing metathesis formed the key conversions. The final stage functionalization of tetrahydropyridine 75 offered the expected amino piperidine in 4 steps.
 | ||
| Scheme 13 Asymmetric synthesis of (+)-CP-99,994. | ||
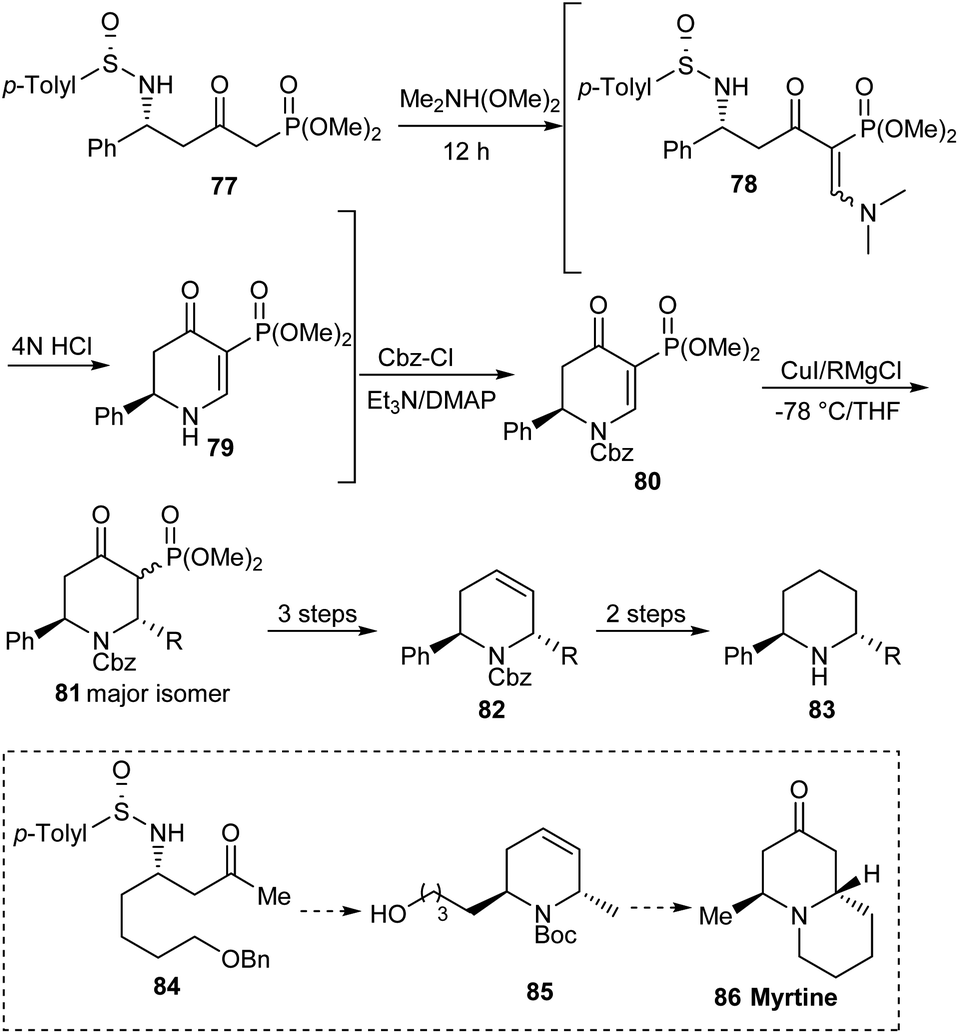
Considering the significance of 2,6-disubstituted piperidine derivatives as bioactive agents, the research group of Davis achieved a newer and general procedure for the generation of 1,2,5,6-tetrahydropyridines, important building units for the stereoselective preparation of trans-2,6-disubstituted piperidines.27 They utilized N-sulfinyl δ-amino β-ketophosphonates 77 as the precursor to initiate the reaction through a one-pot strategy involving treatment with dimethylformamide dimethyl acetal and successive addition of 4N HCl to access the dihydropyridone 79 (Scheme 14). Subjecting the dihydropyridone to a sequence of conversions comprising a stereoselective organocuprate addition provided with trans-2,6-disubstituted piperidines 83. In addition, they presented the utility of the protocol in synthesizing the quinolizidine alkaloid (−)-myrtine 86.
 | ||
| Scheme 14 Asymmetric synthesis of trans-2,6-disubstituted piperidines and the quinolizidine alkaloid (−)-myrtine. | ||
An elegant synthesis of the hydroxyl piperidines, (+)-epipinidinol 96 and (−)-pinidinol 94 was established by the same group in 2008.28 The synthesis commenced with the stereoselective addition of Weinreb amide enolate 88 on the masked oxo sulfinimine 87 to provide 89, the N-sulfinyl β-amino amide in a diastereomeric ratio of 22:1 (Scheme 15). The key step in the synthetic strategy involved a selective reduction of the common N-sulfinyl-β-amino ketone 90 with Li(t-BuO)3AlH and LiEt3BH to give the syn- and anti- 1,3-amino alcohols in high diastereoselectivity. In the final stage, they devised a newer acid-catalyzed reaction of an N-sulfinylamino silyl protected alcohol ketal which rendered 94 and 96 starting from 91 and 95 respectively.
 | ||
| Scheme 15 Synthetic route towards hydroxyl piperidines, (+)-epipinidinol and (−)-pinidinol. | ||
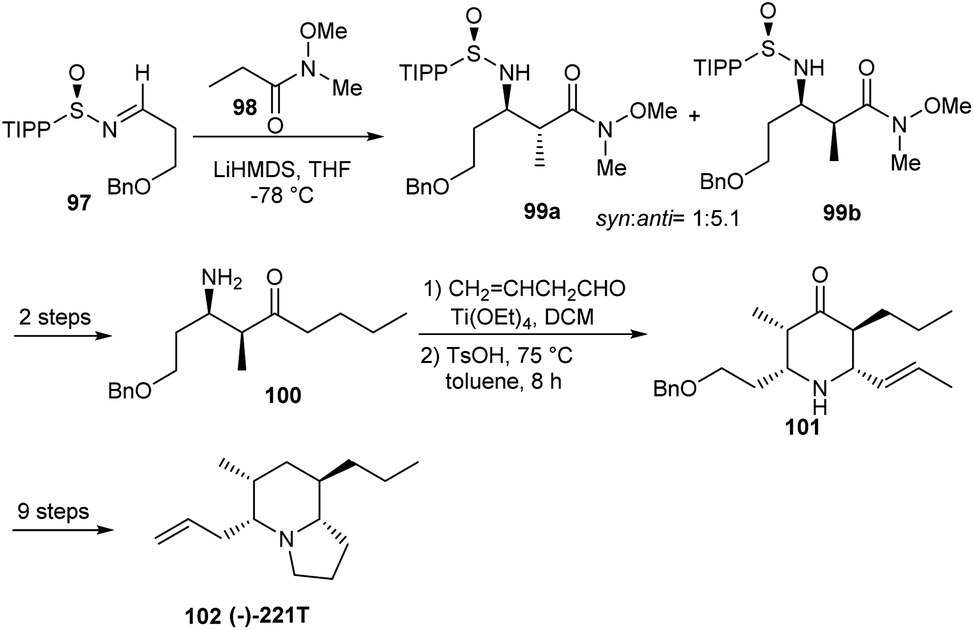
In the following year, they reported an unprecedented total synthesis of (5R,6R,8R,9S)-(−)-5,9Z-indolizidine 221T.29 The synthetic route started from sulfinimine 97, derived from (R)-(+)-2,4,6-triisopropylphenylsulfinamide which formed 1:5.1 ratio of 99 a and 99 b, anti and syn-β-amino Weinreb amides (Scheme 16). Importantly, 2,4,6-triisopropylphenyl (TIPP) sulfinamide was particularly chosen as the chiral auxiliary based on the previous studies that suggested best syn:anti selectivities with the same. Thereafter, the sulfinimine derived aminoketone 100 was reacted with crotonaldehyde and Ti(OEt)4 to afford the imine which in turn was subjected to an intramolecular Mannich cyclization in presence of pTSA in toluene to construct the piperidone 101. Finally, the targeted compound 102 was achieved after a few functional group conversions.
 | ||
| Scheme 16 Total synthesis of (5R,6R,8R,9S)-(−)-5,9Z-indolizidine 221T. | ||
In 2009, Davis employed anti-2,3-diamino esters derived from sulfinimines in the synthesis of (2S,3R)-(−)-epi-CP-99,994 109.30 Here, the syn-analog of 109 is a known neurokinin substance P receptor antagonist. The synthesis commenced with their previously disclosed protocol31 for the addition of Z-lithium enolate of N,N-(diphenylmethylene)glycine ethyl ester 103 to (S)-(+)-104 in the presence of water to yield anti-2,3-diaminoester 105 in excellent yield and high diastereoselectivity (dr > 33:1). With anti-2,3-diaminoester 105 in hand, the C-2 N,N-(diphenylmethylene) group was chemoselectively hydrolysed and then reprotected by a dibenzylamino group (Scheme 17). A diamino diene 107 obtained upon subsequent transformations underwent a ring closing metathesis to construct the piperidine core in 108. Final stage modifications of 108 presented (2S,3R)-(−)-epi-CP-99,994 109 in enantiopure form.
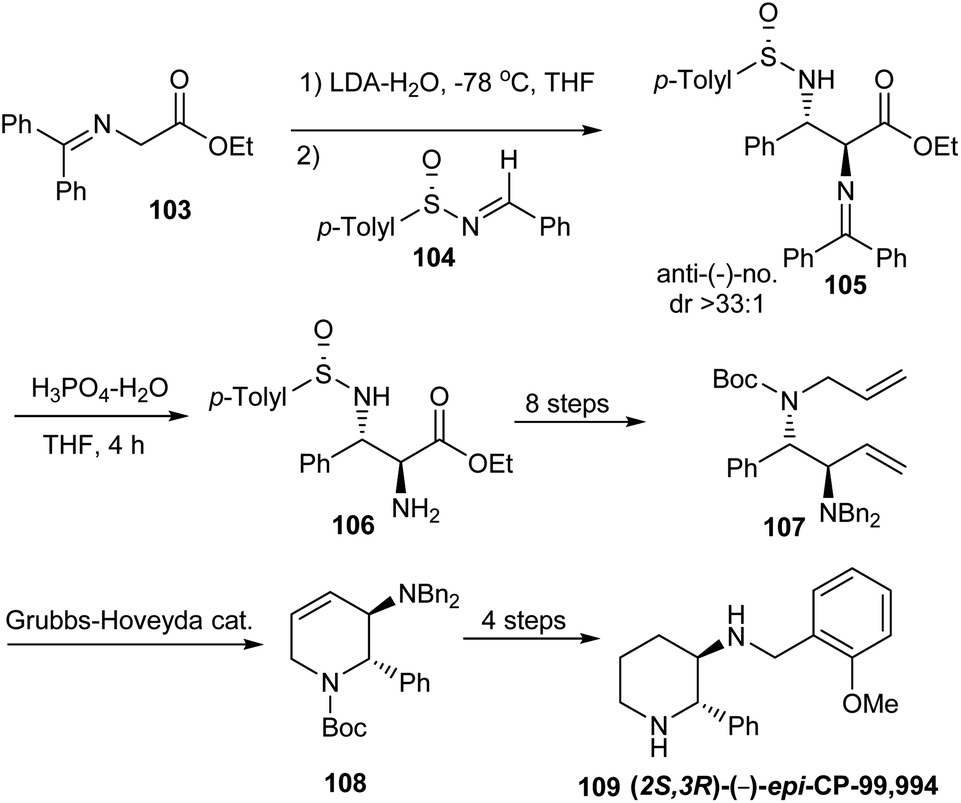 | ||
| Scheme 17 Synthesis of (2S,3R)-(−)-epi-CP-99,994. | ||
5. Miscellaneous
An effective method for the synthesis of (−)-normalindine in enantioenriched form has been introduced in 2006.32 The initial reactions provided the sulfinimine 110 which underwent diastereoselective addition of base treated 4-methyl-3-cyanopyridine 111 in the key step to furnish the sulfinamide 112 in greater than 80% diastereomeric excess (Scheme 18). Exposure of the sulfinamide 112 with MeLi and successive treatment with aqueous HCl yielded the cyclic imine 113 which undertook sequential reduction, deprotection and a final cyclization to accomplish the desired alkaloid 114. The usefulness of the method is expected in the synthesis of analogous tetrahydronaphthyridines and tetrahydroisoquinolines.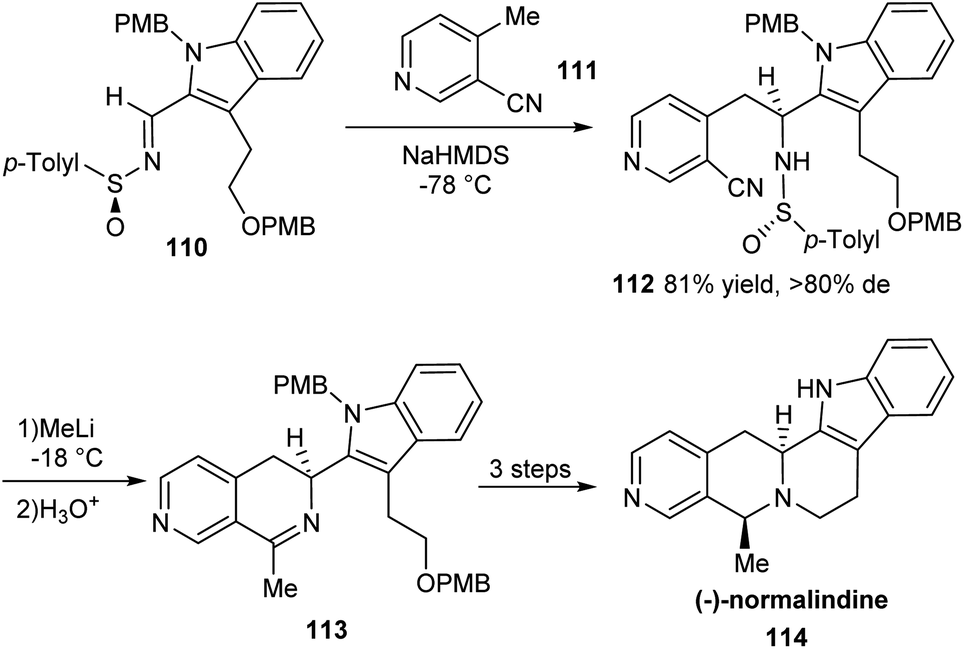 | ||
| Scheme 18 Synthesis of enantiomerically enriched (−)-normalindine. | ||
Yuste and Ruano with coworkers successfully established a novel strategy towards the asymmetric synthesis of (S)-(−)-xylopinine.33 Initially, o-sulfinyl benzyl carbanion obtained from 115 was condensed with (S)-(E)-sulfinylimine 116 to yield tetrahydroisoquinolines 117a and 117b as a 2:1 mixture. 117a and 117b differed merely in their configuration at sulfur and the mixture upon N-desulfinylation provided with the diastereomeric sulfoxides (Scheme 19). Then, these sulfoxides were subjected to Pictet–Spengler cyclization conditions to afford (S)-(−)-xylopinine 118. Importantly, this key step represented the first-time ipso electrophilic substitution of a sulfinyl group and the authors foresee more synthetic implications in this direction. The method stands superior among the known synthetic routes towards 118 due to the high stereocontrol and short sequence of transformations.
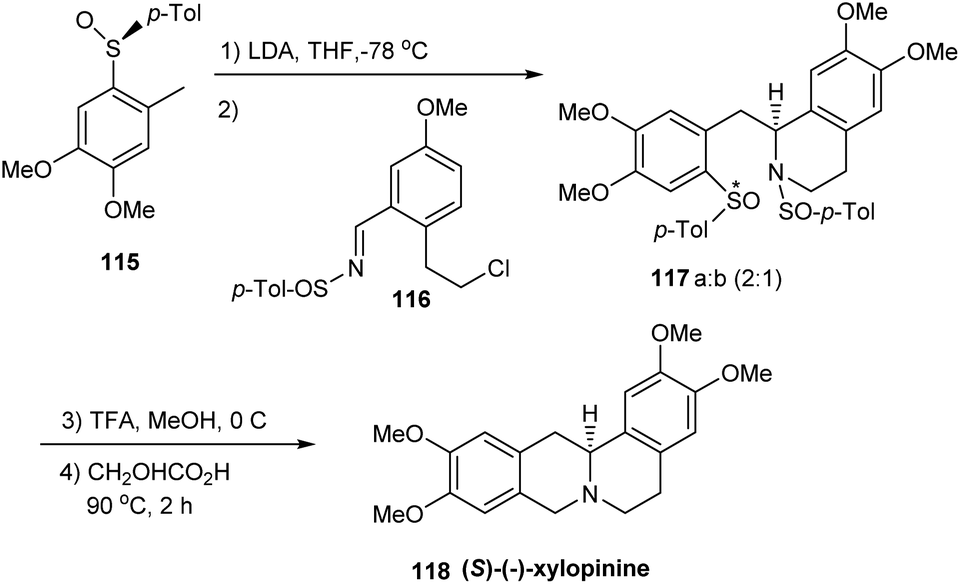 | ||
| Scheme 19 Asymmetric synthesis of (S)-(−)-xylopinine. | ||
Mastranzo and coworkers established an efficient asymmetric synthetic route to (S)-(−)-tetrahydropalmatine and (S)-(−)-canadine via a three step methodology.34 In the initial step, the carbanionic nucleophile generated from 119 was added to p-tolylsulfinylimines 120 to yield the tetrahydroisoquinolines 121 in excellent diastereoselectivity (>96% de) after an intramolecular cyclization (Scheme 20). The p-tolyl group ensured better control of selectivity during this key step. Then, N-desulfinylation followed by the Pictet–Spengler cyclization with the use of TFA and paraformaldehyde under microwave radiation at 140 °C performed cyclization and successive C-desulfinylation to achieve enantiopure (S)-(−)-tetrahydropalmatine 123 and (S)-(−)-canadine 124.
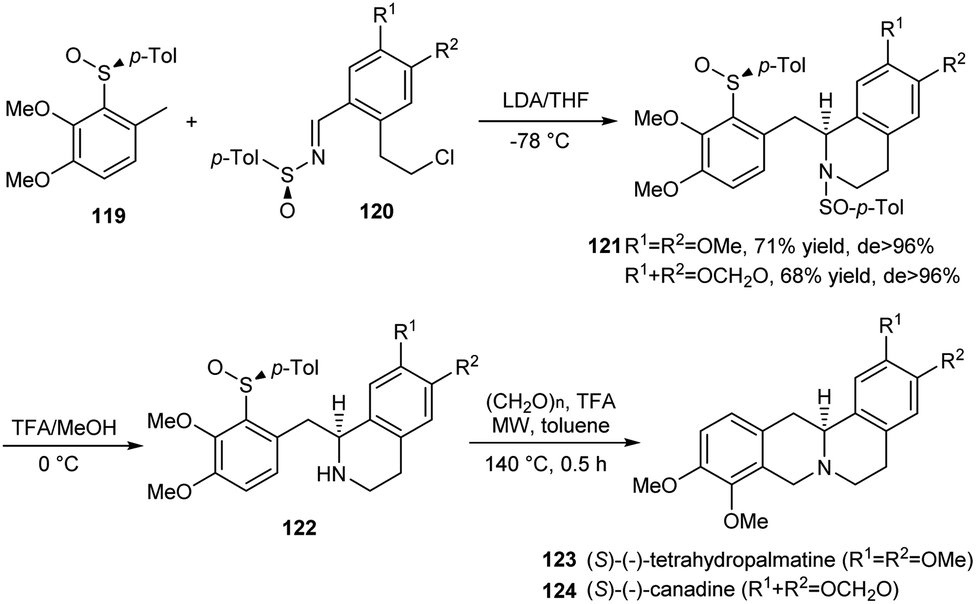 | ||
| Scheme 20 Asymmetric synthetic route to (S)-(−)-tetrahydropalmatine and (S)-(−)-canadine. | ||
An elegant protocol to access homotropinones like (−)-euphococcinine and (−)-adaline was proposed by Davis et al. in 2009.35 In the initial step, N-sulfinyl β-amino ketals 127 were synthesized as inseparable diastereomers through the addition of metal enolates of N-methoxy-N-methyl acetamide 126 onto masked oxo-sulfinimine 125 (Scheme 21). Then, a subsequent Grignard addition offered respective N-sulfinyl β-amino ketone ketals 128 in high yields and diastereoselectivity. These methyl ketones 128a and 128b upon heating with buffer solution NH4OAc:HOAc carried out a four-step intramolecular Mannich cyclization to furnish the anticipated homotropinones 129, 130 and substituted homotropinone 131.
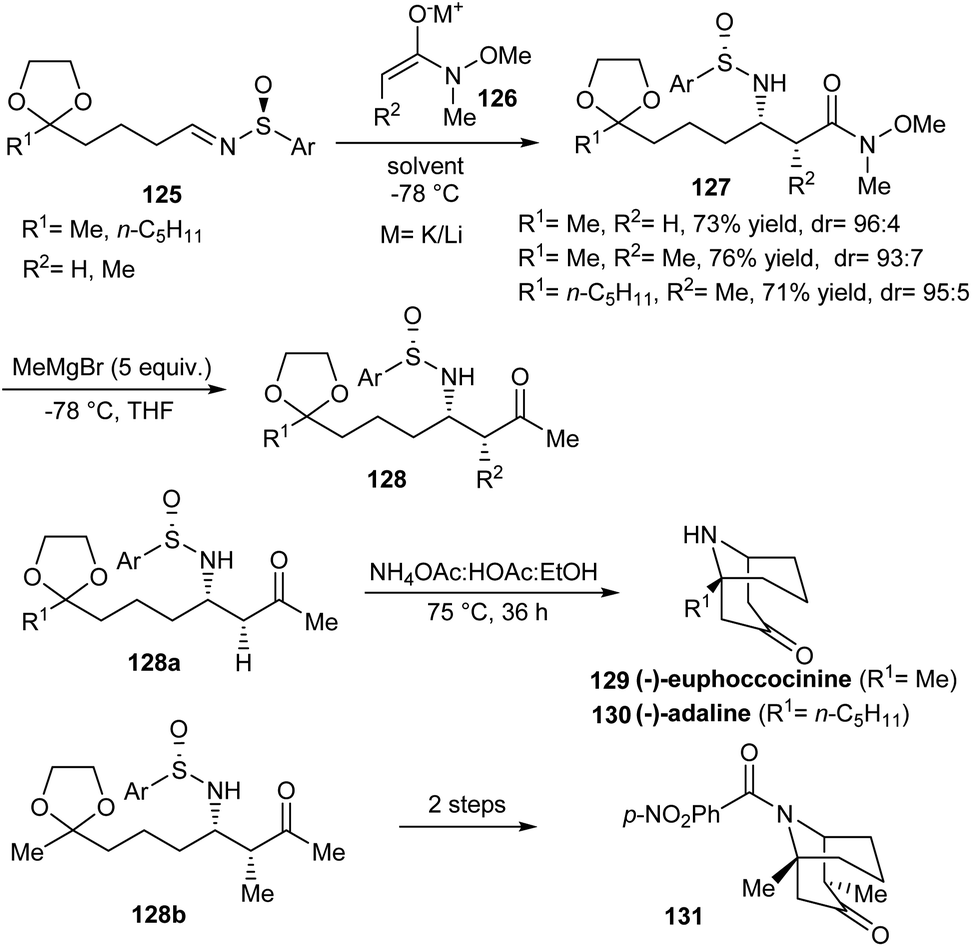 | ||
| Scheme 21 Synthetic strategy towards homotropinones like (−)-euphococcinine and (−)-adaline. | ||
Davis' group devised protocols for the synthesis of tropane alkaloids owing to their important biological properties.36 In the first step, masked oxo sulfinimines 132 after twofold treatment with an excess of the enolate of methyl acetate offered N-sulfinyl δ-amino-ketoester ketal 133 as the major diastereoisomer (Scheme 22). When 133 was subjected to hydrolysis, dehydropyrrolidine species 134 was produced. Then, 134 underwent cyclization via intramolecular Mannich reaction upon treatment with (Boc)2O/DMAP which furnished the tropinone 135 in good yields. Similarly, substituted tropanes 137 and 139 were accessed from dehydropyrrolidine ketones 136 and 138 respectively.
 | ||
| Scheme 22 Synthetic route to substituted tropanes. | ||
In 2010, Davis' group described the first synthetic route to C-1 analogs of cocaine, 146 and (S)-(+)-cocaine 148 in high optical purity.37 The synthesis comprised of nine steps commencing from masked oxo sulfinimine 140. The sulfinimines 140 on reaction with sodium enolate of methyl acetate 141 yielded single diastereoisomers of the respective N-sulfinyl β-amino ester ketals 142 at −78 °C in Et2O (Scheme 23). In the key step, α,β-unsaturated pyrrolidine nitrones 144 derived from sulfinimines underwent a highly stereoselective intramolecular [3 + 2] cycloaddition upon heating with Al(O-t-Bu)3 to furnish tricyclic isoxazolidines 145 in good yields. These tricyclic compounds after a three step conversion provided the desired C-1 analogs 146 in enantiopure form. In a similar manner, (S)-(+)-cocaine 148 was synthesized from the sulfinimine 147 in nine steps.
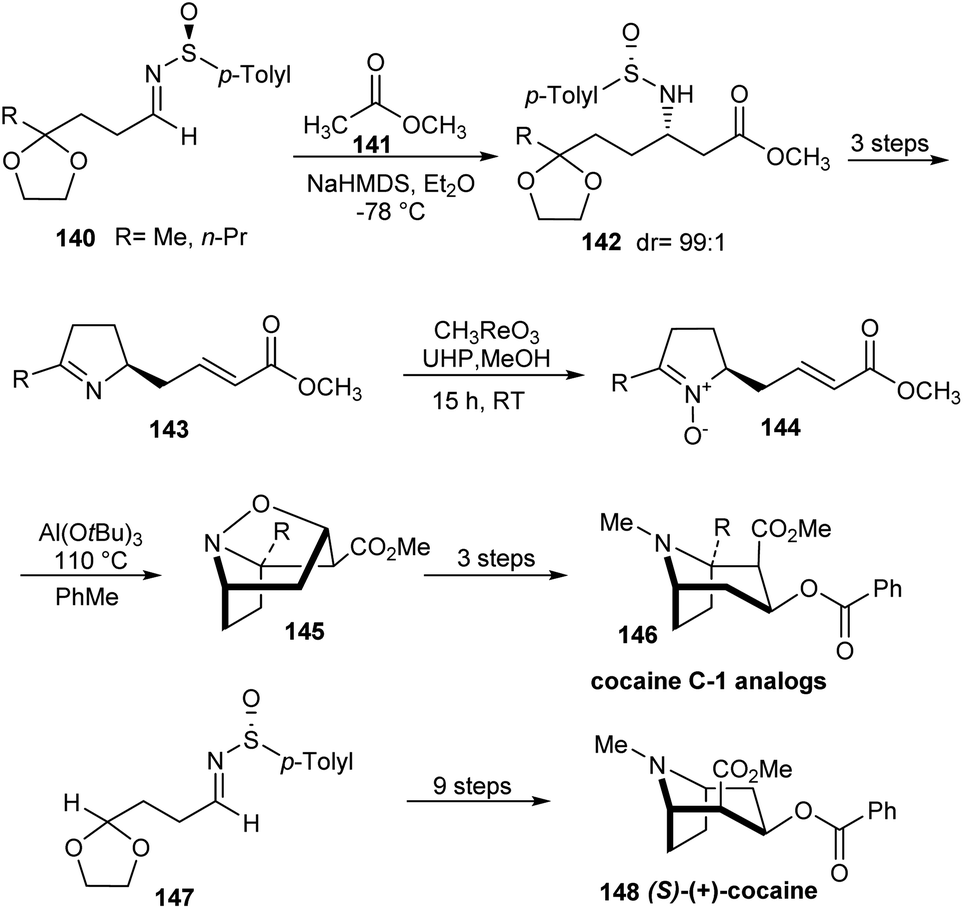 | ||
| Scheme 23 Synthetic route to C-1 analogs of cocaine and (S)-(+)-cocaine. | ||
Expanding their efforts in preparing derivatives of cocaine, they synthesized different cocaine analogues having methyl, ethyl, n-propyl, n-pentyl, and phenyl substituents at the C-1 or bridgehead position of its tropane skeleton.38 Both chiral t-butyl and p-tolyl sulfinimines were explored to access differently substituted cocaine derivatives. As described before, synthesis commenced from masked oxo-sulfinimines 140, wherein the aforementioned key conversions afforded the sulfinimine-derived α,β-unsaturated pyrrolidine nitrones 144 (Scheme 24). Lewis acid assisted intramolecular [3 + 2] cycloaddition of the nitrone 144 afforded tricyclic isoxazolidines 145 which were easily converted to the anticipated cocaine analogues 146. Differently, in the absence of any Lewis acid, the nitrones 144 underwent rearrangement to the lactam 145 via an oxaziridine intermediate. In addition, they disclosed a rare Pd- and base-promoted rearrangement of 146, to form bridged bicyclic[4.2.1] isoxazolidines 147.
 | ||
| Scheme 24 Synthesis of cocaine C-1 analogues. | ||
6. Conclusion
Methodologies employing enantiopure sulfinamides have emerged as effective synthetic routes to access N-heterocyclic compounds owing to their high stereocontrol and easy cleavage after the reaction. In this review, we have summarized the recent reports on the application of aryl sulfinamides in the synthesis of optically pure N-heterocycles through sulfinimine intermediates. Even though most of the reports are with p-toluene sulfinamide, mesityl sulfinamide has also proven useful in N-heterocycle synthesis notably in aziridine synthesis. Other aryl derivatives of sulfinamides are utilized in other asymmetric reactions to meet specific requirements.When some articles presented novel protocols towards N-heterocycles with substrate scope studies, others described the total synthesis of natural products containing N-heterocycles. Importantly, some examples achieved simple and short asymmetric total syntheses of targeted natural products including alkaloids like C-1 analogues of cocaine and other tropane alkaloids. A close look at the literature suggests that a major contribution in the field comes from Davis' group and their insightful research to unveil the chemistry of sulfinamides inspired scientists around the globe. Although being the first introduced sulfinamide, p-toluene sulfinamide is less explored in recent decades. Exploring the applicability of available aryl sulfinamides and designing differently substituted analogues to access currently unexplored heterocyclic motifs will be appreciated in future. We expect that the varied strategies discussed may benefit people across the fields of medicinal chemistry, synthetic chemistry, and agrochemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
RMP and SS thank the Council of Scientific and Industrial Research (CSIR), New Delhi for a research fellowship. GA thanks the Kerala State Council for Science, Technology and Environment (KSCSTE), Trivandrum, for the award of a research fellowship and research grant (order no. 341/2013/KSCSTE dated 15.03.2013) respectively. We greatly acknowledge the support from Department of Science and Technology (DST-New Delhi) under the DST-PURSE programme.References
- D. Morton and R. A. Stockman, Tetrahedron, 2006, 62, 8869–8905 CrossRef CAS.
- G. Liu, D. A. Cogan and J. A. Ellman, J. Am. Chem. Soc., 1997, 119, 9913–9914 CrossRef CAS.
- T. M. Solá, I. Churcher, W. Lewis and R. A. Stockman, Org. Biomol. Chem., 2011, 9, 5034–5035 RSC.
- F. A Davis, J. Org. Chem., 2006, 71, 8993–9003 CrossRef PubMed.
- M. Medjahdi, J. C. Gonzalez-Gomez, F. Foubelo and M. Yus, J. Org. Chem., 2009, 74, 7859–7865 CrossRef CAS PubMed.
- (a) N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed; (b) B. Eftekhari-Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CrossRef CAS PubMed; (c) N. Kerru, S. Maddila and S. B. Jonnalagadda, Curr. Org. Chem., 2019, 23, 3156–3192 Search PubMed; (d) Y. Ju and R. S. Varma, J. Org. Chem., 2006, 71, 135–141 CrossRef CAS PubMed; (e) D. Z. Zarate, R. Aguilar, R. I. Hernandez-Benitez, E. M. Labarrios, F. Delgado and J. Tamariz, Tetrahedron, 2015, 71, 6961–6978 CrossRef.
- B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 RSC.
- F. A. Davis, B. Yang, J. Deng, Y. Wu, Y. Zhang, A. Rao, T. Fang, R. Goswami, K. R. Prasad, M. B. Nolt and G. Anilkumar, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 1109–1117 CrossRef CAS.
- C. Achuenu, S. Carret, J.-F. Poisson and F. Berthiol, Eur. J. Org. Chem., 2020, 5901–5916 CrossRef CAS.
- E. Wojaczynska and J. Wojaczynski, Chem. Rev., 2020, 120, 4578–4611 CrossRef CAS PubMed.
- P. Dinér, A. Sadhukhan and B. Blomkvist, ChemCatChem, 2014, 6, 3063–3066 CrossRef.
- R. M. Philip, S. Radhika, P. V. Saranya and G. Anilkumar, RSC Adv., 2020, 10, 42441–42456 RSC.
- F. A. Davis and J. Deng, Org. Lett., 2007, 9, 1707–1710 CrossRef CAS PubMed.
- V. D. Bonifacio, C. Gonzalez-Bello, H. S. Rzepa, S. Prabhakar and A. M. Lobo, Synlett, 2010, 145–149 CAS.
- T. Moragas, I. Churcher, W. Lewis and R. A. Stockman, Org. Lett., 2014, 16, 6290–6293 CrossRef CAS PubMed.
- T. Moragas, R. M. Liffey, D. Regentova, J.-P. S. Ward, J. Dutton, W. Lewis, I. Churcher, L. Walton, J. A. Souto and R. A. Stockman, Angew. Chem., Int. Ed., 2016, 55, 10047–10051 CrossRef PubMed.
- F. A. Davis, T. Ramachandar, J. Chai and E. Skucas, Tetrahedron Lett., 2006, 47, 2743–2746 CrossRef CAS.
- F. A. Davis, M. Song and A. Augustine, J. Org. Chem., 2006, 71, 2779–2786 CrossRef CAS PubMed.
- M. E. Scott and M. Lautens, J. Org. Chem., 2008, 73, 8154–8162 CrossRef CAS PubMed.
- A. Viso, R. F. Fernandez de la Pradilla, M. Ureña and I. Colomer, Org. Lett., 2008, 10, 4775–4778 CrossRef CAS PubMed.
- S. Ishikawa, F. Noguchi and A. Kamimura, J. Org. Chem., 2010, 75, 3578–3586 CrossRef CAS PubMed.
- A. Kamimura, H. Okawa, Y. Morisaki, S. Ishikawa and H. Uno, J. Org. Chem., 2007, 72, 3569–3572 CrossRef CAS PubMed.
- F. A. Davis, J. Zhang and Y. Wu, Tetrahedron Lett., 2011, 52, 2054–2057 CrossRef CAS.
- F. A. Davis and M. Santhanaraman, J. Org. Chem., 2006, 71, 4222–4226 CrossRef CAS PubMed.
- R. Kawecki, Tetrahedron: Asymmetry, 2006, 17, 1420–1423 CrossRef CAS.
- F. A. Davis, Y. Zhang and D. Li, Tetrahedron Lett., 2007, 48, 7838–7840 CrossRef CAS PubMed.
- F. A. Davis, H. Xu and J. Zhang, J. Org. Chem., 2007, 72, 2046–2052 CrossRef CAS PubMed.
- F. A. Davis, P. M. Gaspari, B. M. Nolt and P. Xu, J. Org. Chem., 2008, 73, 9619–9626 CrossRef CAS PubMed.
- F. A. Davis, M. Song, H. Qiu and J. Chai, Org. Biomol. Chem., 2009, 7, 5067–5073 RSC.
- F. A. Davis and Y. Zhang, Tetrahedron Lett., 2009, 50, 5205–5207 CrossRef CAS PubMed.
- F. A. Davis, Y. Zhang and H. Qiu, Org. Lett., 2007, 9, 833–836 CrossRef CAS PubMed.
- F. A. Davis, J. Y. Melamed and S. S. Sharik, J. Org. Chem., 2006, 71, 8761–8766 CrossRef CAS PubMed.
- V. M. Mastranzo, F. Yuste, B. Ortiz, R. Sánchez-Obregón, R. A. Toscano and J. L. Garcia Ruano, J. Org. Chem., 2011, 76, 5036–5041 CrossRef CAS PubMed.
- V. M. Mastranzo, J. L. O. Romero, F. Yuste, B. Ortiz, R. Sánchez-Obregón and J. L. G. Ruano, Tetrahedron, 2012, 68, 1266–1271 CrossRef CAS.
- F. A. Davis and R. Edupuganti, Org. Lett., 2010, 12, 848–851 CrossRef CAS PubMed.
- F. A. Davis, N. Theddu and P. M. Gaspari, Org. Lett., 2009, 11, 1647–1650 CrossRef CAS PubMed.
- F. A. Davis, N. Theddu and R. Edupuganti, Org. Lett., 2010, 12, 4118–4121 CrossRef CAS PubMed.
- F. A. Davis, N. V. Gaddiraju, N. Theddu, J. R. Hummel, S. K. Kondaveeti and M. J. Zdilla, J. Org. Chem., 2012, 77, 2345–2359 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |