
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of catalysts with fine platinum particles supported by high-surface-area activated carbons and optimization of their catalytic activities for polymer electrolyte fuel cells†
Md. Mijanur Rahman *ab,
Kenta Inabab,
Garavdorj Batnyagtb,
Masato Saikawab,
Yoshiki Katob,
Rina Awatab,
Byambasuren Delgertsetsegaab,
Yasuo Kanetac,
Kotaro Higashide,
Tomoya Urugade,
Yasuhiro Iwasawade,
Koichi Uiab and
Tatsuya Takeguchi*ab
*ab,
Kenta Inabab,
Garavdorj Batnyagtb,
Masato Saikawab,
Yoshiki Katob,
Rina Awatab,
Byambasuren Delgertsetsegaab,
Yasuo Kanetac,
Kotaro Higashide,
Tomoya Urugade,
Yasuhiro Iwasawade,
Koichi Uiab and
Tatsuya Takeguchi*ab
aFaculty of Science and Engineering, Iwate University, 4-3-5 Ueda, Morioka, Iwate 020-8551, Japan. E-mail: mijanur@iwate-u.ac.jp; Fax: +81-019-621-6329; Tel: +81-019-621-6329
bGraduate School of Arts and Sciences, Iwate University, 4-3-5 Ueda, Morioka, Iwate 020-8551, Japan. E-mail: takeguch@iwate-u.ac.jp; Fax: +81-019-621-6335; Tel: +81-019-621-6335
cJUKES Inc., 32-18-2 Osanai-cho, Kuji, Iwate 028-0041, Japan
dInnovation Research Center for Fuel Cells, University of Electro-Communications, 1-5-1 Chofugaoka, Chofu, Tokyo 182-8585, Japan
eJASRI/SPring-8, 1-1-1, Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
First published on 8th June 2021
Abstract
Herein, we demonstrated that carbon-supported platinum (Pt/C) is a low-cost and high-performance electrocatalyst for polymer electrolyte fuel cells (PEFCs). The ethanol reduction method was used to prepare the Pt/C catalyst, which was realized by an effective matching of the carbon support and optimization of the Pt content for preparing a membrane electrode assembly (MEA). For this, the synthesis of Pt/C catalysts with different Pt loadings was performed on two different carbons (KB1600 and KB800) as new support materials. Analysis of the XRD pattern and TEM images showed that the Pt nanoparticles (NPs) with an average diameter of ca. 1.5 nm were uniformly dispersed on the carbon surface. To further confirm the size of the NPs, the coordination numbers of Pt derived from X-ray absorption fine structure (XAFS) data were used. These results suggest that the NP size is almost identical, irrespective of Pt loading. Nitrogen adsorption–desorption analysis indicated the presence of mesopores in each carbon. The BET surface area was found to increase with increasing Pt loading, and the value of the BET surface area was as high as 1286 m2 gcarbon−1. However, after 40 wt% Pt loading on both carbons, the BET surface area was decreased due to pore blockage by Pt NPs. The oxidation reduction reaction (ORR) activity for Pt/KB1600, Pt/KB800 and commercial Pt/C was evaluated by Koutecky–Levich (K–L) analysis, and the results showed first-order kinetics with ORR. The favourable surface properties of carbon produced Pt NPs with increased density, uniformity and small size, which led to a higher electrochemical surface area (ECSA). The ECSA value of the 35 wt% Pt/KB1600 catalyst was 155.0 m2 gpt−1 higher than that of the Pt/KB800 and commercial Pt/C (36.7 wt%) catalysts. A Higher ECSA indicates more available active sites for catalyst particles. The single cell test with MEA revealed that the cell voltage in the high current density regions depends on the BET surface area, and the durability of the 35 wt% Pt/KB1600 catalyst was superior to that of the 30 wt% Pt/KB800 and commercial Pt/C (46.2 wt%) catalysts. This suggests that an optimal ratio of Pt to Pt/KB1600 catalyst provides adequate reaction sites and mass transport, which is crucial to the PEFC's high performance.
1. Introduction
Polymer electrolyte fuel cells (PEFCs) have attracted a great deal of attention due to their high energy-conversion efficiency, low operating-temperature, and environment friendliness.1–3 In PEFCs, Pt is used in both the anode and cathode as the most reliable catalyst because of its high catalytic activity toward the hydrogen oxidation reaction (HOR: H2 → 2H+ + 2e−) and oxygen reduction reaction (ORR: O2 + 4H+ + 4e− → 2H2O), and durability in an acidic environment under high potentials.4 However, a large amount of Pt is needed for the cathode catalyst to accelerate the sluggish kinetics of ORR, causing a higher cost of PEFCs.5 The high cost and limited availability of this valuable metal impede the large-scale commercialization of commercial vehicles, fuel cell vehicles and domestic applications. To overcome these limitations, a very low Pt load in the anode and cathode catalyst layers while maintaining high catalytic activity, acceptable performance, stability and durability of PEFC are strongly required. At present, only a few studies have been conducted in this regard. Notable among them are focused on advanced catalyst-deposition techniques, such as sputter deposition,6 ultrasonic spraying,7 electrospraying,8 and reactive spray deposition technology,9,10 to tailor the catalyst layers. These processes usually generate a high concentration of Pt in the catalyst layers. The increased Pt concentration without support material leads to the agglomeration of Pt nanoparticles (NPs), and consequently a decrease in surface area, which greatly impedes catalytic performance.11 Therefore, determining an appropriate support material with low loading Pt is essential to improve PEFC's overall performance.As a catalyst-support material, carbon black (CB), for example, Vulcan XC 72 has been extensively used in PEFCs because of its high surface area (ca. 250 m2 g−1), suitable porosity, high corrosion resistance and excellent electrical conductivity. These unique properties of the catalyst-support material make it suitable for accelerating charge transfer at the electrode interfaces.12,13 In this regard, Pt NPs supported on the surface of carbon (Pt/C) catalysts have been recognized as one of the most promising catalysts for ORR, because carbon support provides an advantageous set of features that are extremely small particles, high surface area, uniform morphology, highly dispersed and excellent interactions with the particles, resulting in a higher Pt utilization efficiency.14,15 Dogan et al. also reported that carbon-supported Pt NPs contribute to enhance catalyst utilization and improve efficiency.16,17 Therefore, all these properties of carbon support lead to reduce Pt loading and enhance catalytic activity. However, the Pt and its carbon-supported surface are oxidized during operation, especially during the start-up and shutdown of a PEFC.18,19 Substantial oxidation of the carbon support in the cathode may result in a loss of performance due to the active surface loss of the catalyst, agglomeration or detachment from carbon supports caused by carbon corrosion, pore surface characteristics, and poor electrical connectivity of the catalyst-support structure inside the electrode.20,21 In the meantime, numerous studies have been devoted to developing alternative materials such as carbon nanotubes,22,23 carbon nanofiber,24,25 mesoporous carbon,26 and graphitic carbon27,28 instead of the commonly used Vulcan XC-72R owing to improved catalytic activity and durability. Although many promising support materials have been developed over the years, the fabrication techniques applied to these catalysts are complex, expensive and time-consuming. Thus, finding a corrosion-resistant support material to enhance the catalytic activity and durability of PEFC catalyst appears as an important task in fuel cell technology.
The difficulty lies in the fact that the support material can barely meet all these requirements at the same time. Ketjen black (KB) is found to be the most efficient ESI† owing to its unique porous structure and outstanding properties, leading to high electrochemical stability, good conductivity and large specific surface area (1300 m2 g−1).29 Kim et al.30 synthesized 60 wt% Pt/C catalyst using different types of CBs, namely KB EC-600JD and KB EC-300J with different surface areas. The average size of the Pt particles was found to decrease with increasing surface area. The catalytic activity increased with increasing Pt surface area, but their relationship was not clear enough. Speder et al.31 used a colloid synthesis method to synthesize Pt/C catalysts with different Pt loadings. The impact of Pt loading on the loss of electrochemically active surface area (ECSA) was evaluated using KB EC-300J and Vulcan XC 72R. It was found that the Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C ratio may have an influence on the rate of degradation under startup/shutdown conditions. The degradation rate of Vulcan XC 72R was substantially higher than that of KB EC-300J. Although some progress has been made in the last decades on the improvement of support materials, significant drawbacks still exist. In particular, the effect of the porous structure of the KB on the preparation and electrochemical performance of Pt/C catalysts remains unclear to the best of our knowledge. Pore structure, pore size distribution and porosity are the prerequisites for understanding mass transport losses in catalyst layers that may degrade the fuel cell performance. Therefore, further systematic investigations are necessary to identify the important factors that affect the overall performance of PEFCs.
:C ratio may have an influence on the rate of degradation under startup/shutdown conditions. The degradation rate of Vulcan XC 72R was substantially higher than that of KB EC-300J. Although some progress has been made in the last decades on the improvement of support materials, significant drawbacks still exist. In particular, the effect of the porous structure of the KB on the preparation and electrochemical performance of Pt/C catalysts remains unclear to the best of our knowledge. Pore structure, pore size distribution and porosity are the prerequisites for understanding mass transport losses in catalyst layers that may degrade the fuel cell performance. Therefore, further systematic investigations are necessary to identify the important factors that affect the overall performance of PEFCs.
In this study, the effect of carbon supports on Pt distribution, cathode efficiency of carbon-supported Pt catalysts, and their different physical properties are investigated. Two different carbon materials such as KB1600 and KB800 were used as carbon supports. We prepared Pt/KB1600 and Pt/KB800 catalysts with different Pt loadings (25, 30, 35 and 40 wt%) by ethanol reduction method. This method is capable of producing particles of nanoscale size, resulting in increases in the active surface area of the Pt/C catalyst. The prepared Pt/C catalysts were examined using X-ray diffraction (XRD), transmission electron microscopy (TEM), scanning electron microscopy (SEM), and nitrogen (N2) adsorption–desorption isotherms. In situ measurements of the time-resolved quick X-ray absorption fine structure (XAFS) were conducted at the Pt LIII-edge to examine and confirm particle size for Pt/C catalysts prepared with different Pt loading during PEFC operating conditions.32 To further confirm the NPs size, the results obtained with XAFS were compared with those obtained with XRD (Scherrer formula) and TEM analysis. The results showed no significant change in the particle size of Pt/C catalysts in MEAs. This information is pertinent for the origin of the PEFC activity and its durability, and is crucial for the development of next generation PEFC catalysts. The electrochemical properties of the prepared Pt/C catalysts were assessed by means of cyclic voltammetry (CV) and linear sweep voltammetry (LSV) on a rotating disk electrode (RDE) measuring system. These findings were compared to the commercially available Pt/C catalyst (36.7 wt%, TEC10E40E). Thereafter, the performance of Japan Automobile Research Institute (JARI) standard single cells,33 prepared with low-Pt membrane electrode assembly (MEA) was investigated for PEFC applications. The MEAs used in this study consist of one anode layer, one Nafion membrane and one cathodic layer. Pt/C catalysts prepared with various Pt loadings were used as cathode catalysts, and a commercial Pt/C catalyst (36.7 wt%, TEC10E40E) was used as anode catalyst for current–voltage (I–V) measurements. The power densities and I–V polarization curves were derived using single cells to assess the performance of cathode catalyst. Subsequently, accelerated durability tests (ADTs) were performed in hydrogen (H2) and oxygen (O2) operating atmospheres at room temperature (RT) to study the durability of PEFC using low Pt loaded MEAs. The ADT results were compared to those of the commercial Pt/C catalyst (46.2 wt%, TEC10E50E). The results revealed that the Pt NPs was successfully prepared with average diameters of ca. 1.5 nm and had a uniform dispersion of NPs on the carbon supports. The ECSA for each Pt/C catalyst was calculated from carbon monoxide (CO) stripping voltammetry measurements without rotation. The results confirmed that the ECSA decreased as the loading (wt%) of Pt NPs increased. Furthermore, the results of the JARI standard single cell test showed that the voltage of the cell in the high current density region depends on the surface area determined by the Brunauer–Emmett–Teller (BET) theory. The performance and durability of the PT/KB1600 (35 wt%) cathode catalyst is significantly greater than that of the Pt/KB800 and commercial Pt/C catalyst. Therefore, the above results suggest that an appropriate ratio of Pt to Pt/KB1600 catalyst may provide adequate reaction sites and mass transports that lead to higher ORR activity, which is crucial for the high-performance of PEFC. Furthermore, this study will contribute to the development of low-cost and highly efficient catalysts for their practical applications in the areas of clean energy, energy conversion technologies and beyond.
2. Experimental
2.1 Preparation of Pt/C catalysts
Pt/C catalysts with various Pt loadings (25, 30, 35 and 40 wt%) were synthesized by ethanol reduction method. KB1600 (Ketjen Black, LION) and KB800 (Ketjen Black, LION) as the carbon supports were dissolved in 200 ml of deionized water (18.0 MΩ cm, Milli-Q Co. Ltd.) and ultrasonicated for 2 min. Subsequently, Pt (NO2)2(NH3)2 (4.555 wt% Pt, Tanaka Kikinzoku Kogyo Co. Ltd.) was included in the solution, and magnetically stirred at RT for 1 h. The weight ratios of Pt to the Pt/C catalyst were kept at 25:100, 30:100, 35:100 and 40:100. 30 ml of ethanol was added to the mixture, followed by stirring for 30 min at RT. The mixture was then stirred and heated to 95 °C for 16 h. After vacuum filtration, the residue was washed with 1000 ml deionized water. The precipitate was dried at RT during the overnight. After drying naturally, the extract was crushed and dehydrated in a tube furnace in an atmosphere of N2 at 80 °C for 10 h, to obtain Pt/C catalysts.
2.2 Physical characterization of Pt/C catalysts
Crystalline structure analysis of the Pt/C catalysts (KB1600 and KB800) with various Pt loadings (25–40 wt%) was conducted by XRD. The diffraction patterns of the powder XRD were saved on a RINT-2200 (Rigaku Corp.) with Cu Kα (λ = 1.5418 Å) radiation operating at 30 mA and 40 kV. The scan range of 2θ ranged from 10° to 80°, and had a scan rate of 2° min−1. The size of Pt NPs was estimated using Scherrer's theory as follows (1):|
D = kλ/βcosθ
| (1) |
In situ measurements of the time-resolved quick XAFS were conducted at the BL36XU beam line at SPring-8 (8 GeV, 100 mA, Japan). The detailed experimental procedures for XAFS measurements can be found elsewhere.34 The emitted X-rays were monochromatized by a standard Si (111) channel-cut crystal monochromator. Two vertical mirrors between the monochromator and incident ion chamber were used to deny the higher harmonics. The spectra derived from the XAFS measurements were saved as the mode of transmission. The incident (I0) and the transmitted (I1 and I2) X-rays were detected by fully loaded ionization chambers filled with N2 and N2/Ar (50:50), respectively. After aging, the XAFS cell with an MEA was positioned between the I0 and I1 ionization chambers at 45°. A Pt loading of 0.2 mgPt cm−2 was used as a catalyst for the anode whereas a cathode catalyst was Pd/C. The Pt/C catalyst prepared with different Pt/C ratios was used as an anode catalyst to prevent interference with XAFS measurements. Double serpentine flow channels were used for the flow of H2 and N2 or dry air to the anode and cathode, respectively. H2 and N2 flow rates were controlled by mass flow regulators, and bubbled in 351 K humidifiers with a commercial gas supply kit. In the course of the measurements, the cell voltage was controlled by a potentiostat-galvanostat and a current amplifier, and the current density was changed from 0 to 50 mA cm−2. The overall measurement time was about 6 h. A series of XAFS spectra for Pt/C anode catalysts in practical MEAs under PEFC operating conditions were measured. The coordination numbers (CNs) of the Pt derived from fitting XAFS data were used to estimate average particle size to further confirm NP size.
Furthermore, N2 adsorption–desorption isotherms were measured at −196 °C using a surface area and pore size distribution analyser (3FLEX, Micromeritics). Pore size distributions were determined using Barrett–Joyner–Halenda (BJH) method. The specific surface area (SSA) of the Pt NPs catalysts was determined in accordance with the standard BET method (BET surface area).
2.3 Electrochemical measurements
Electrochemical measurements were performed at RT using CV and LSV on a RDE measuring system with a triple electrode cell. The working electrode was a glassy carbon (GC) rotating disk electrode (surface area: 0.196 cm2, Pine Instruments). The GC electrode was smoothed for a mirror finish with alumina slurry (0.05 μm, Buehler), followed by ultrasonic cleaning with ethanol and deionized water for 10 min each. The electrode was dried well before application. The catalyst inks were then prepared using a mixture of Pt/C catalyst, 2 ml deionized water, 3 ml ethanol, and 0.05 ml Nafion solution (5 wt%, Sigma-Aldrich). The mixed solution was well sonicated for 15 min in order to generate the homogeneous ink. Similar procedures were conducted for the commercial Pt/C catalyst (36.7 wt% Pt, TEC10E40E, Tanaka Kikinzoku Kogyo Co. Ltd.) to prepare catalyst ink. Then, approximately 0.01 ml of each type of catalyst ink was uniformly spread on the polished GC, and the solvent was slowly evaporated in air followed by oven drying at 60 °C for 15 min. The Pt loading on each working electrode was kept at 17.6 μgPt cm−2.The obtained working electrodes were placed in an AFMSRCE Pine Instruments electrode rotator with standard triple-electrode cell measurement. A platinum wire was used as the counter electrode. A standard Ag/AgCl double junction electrode submerged in a saturated solution of 0.1 M HClO4 was used as a reference electrode. The electrode potential measured by the reference electrode was converted to the scale of the reverse hydrogen electrode (RHE), according to the Nernst equation as follows (2):
| ERHE = EAg/AgCl + 0.059pH + E0Ag/AgCl | (2) |
| 1/i = 1/ik + 1/(0.62 × nFAD2/3v−1/6Cω1/2) | (3) |
485 C mol−1), and A is an electrode surface (cm2). D corresponds to the diffusion coefficient of O2 into the electrolyte (1.9 × 10−5 cm2 s−1), and C is the O2 concentration for the electrolyte (1.1 × 10−6 mol cm−3). ω is the angular frequency of rotation (rad s−1), and v is the kinematic viscosity of the electrolyte (1.0 × 10−2 cm2 s−1).
For the CO-stripping voltammetry measurements, working electrodes were made by dripping catalyst ink onto the GC electrodes so that the Pt loading was 0.1 mgPt cm−2. The Pt loading was increased from 0.0176 mgPt cm−2 to 0.1 mgPt cm−2 in order to minimize the measurement error. For comparative purposes, the catalyst inks were prepared using KB1600 and KB800 catalyst with different Pt loadings (25, 30, 35 and 40 wt%), and commercial Pt/C catalyst (36.7 wt% Pt, TEC10E40E) according to the same method as described earlier, but 2-propanol was used instead of ethanol as a dispersion solvent. Afterwards, the prepared working electrodes were immersed into the CO-saturated 0.1 M HClO4 solution, and in order to make sure that all the exposed Pt atoms were covered by CO, the CO gas flow was bubbled to the solution for 20 min at a fixed current of 0.05 A. Subsequently, Ar gas was supplied in the solution for 15 min to eliminate the dissolved CO from the electrolyte solution. The CO-stripping curves were recorded with the same apparatus without rotation as described earlier. Next, CV measurements were performed in a 0.1 M HClO4 solution saturated with Ar at RT. The scan rate was 20 mV s−1 with a potential range from 0.05 V to 1.20 V vs. RHE. Initial experiments were carried out to ensure that longer CO deposition times were not necessary and that carbon neither adsorbed nor oxidised CO. The CO-stripping reaction is: Pt–CO + H2O → Pt + CO2 + 2H+ + 2e−, which gives rise to a peak at 925 mVRHE as the adsorbed CO oxidized to CO2. The ECSA for Pt/C catalysts was estimated from the charges associated with the electrochemical oxidation of CO, using the following eqn (4):
| ECSA = QCO/LPt × 0.420 mC cm−2 | (4) |
2.4 MEA preparation and characterization
The MEA is the central element in a fuel cell that helps generate the electrochemical reactions necessary to segregate the electrons. It is a stack assembled from polymer electrolyte membrane with anode and cathode catalyst layers applied to both sides, two gas diffusion layers (GDLs), and two sets of sealing gaskets. For preparation of MEA, AvCarb P50T carbon paper (0.17 mm thickness, Fuel Cell Earth) was used as the backing layer for both the anode and cathode.36 Vulcan XC-72 carbon powder (0.29 g) was added to 5.8 g ethanol, and sonication was performed for 2 min at RT. Then, 1.7 g of polytetrafluoroethylene (PTFE) solution (10.9 wt%, Sigma-Aldrich) was added to the mixture and stirred for 20 min using a glass rod. Finally, the carbon black ink was obtained. The prepared ink was coated on AvCarb P50T carbon paper followed by annealing at 350 °C for 1 h to obtain GDLs. For preparing the electrode catalyst layers, the Pt/C catalyst and Nafion solution were suspended in deionized water. A similar process was carried out for each type of catalyst, such as KB1600 and KB800 catalyst with different Pt loadings (25–40 wt%), as well as commercial Pt/C catalyst for comparison. Note that commercial Pt/C (36.7 wt% Pt, TEC10E40E) was used as the catalyst for both the cathode and anode. The wet ratio of the Nafion solution to the Pt/C catalysts was controlled at 9:1. However, the wet ratio between the Nafion solution and the Pt/KB1600 catalyst (Nafion content: Pt/KB1600) was adjusted to 8:2. The percentage for Nafion was estimated using eqn (5), as described in ref. 37| Nafion loading = Nafion (mg cm−2)/{Nafion (mg cm−2) + catalyst (mg cm−2)} × 100 | (5) |
After the ethanol was added, the mixture was sonicated for 15 min to obtain the catalyst ink. The prepared ink was painted on carbon paper, and then air-dried overnight. The electrodes were then cut into 2.2 × 2.2 cm2 for the cathode and anode. The Pt loadings for anode and cathode were 0.5 mgPt cm−2 and 0.1 mgPt cm−2, respectively. To assemble the MEA, the anode and cathodes coated with catalytic ink were positioned on both sides of a Nafion membrane (area: 4.0 × 4.0 cm2, thickness: 0.05 mm, Sigma-Aldrich), and sealed with two Teflon sheets (area: 4.0 × 4.0 cm2, thickness: 0.10 mm, AS ONE). The electrodes were then pressed hot at 125 °C for 20 min at a pressure of 2.0 MPa to obtain the MEA. Hot pressing is carried out during the MEA fabrication process to enhance the contact properties between the membrane and electrodes, leading to reduction of ohmic resistance and improvement of MEA durability.38 The as-prepared MEA was used to assemble a single cell between the bipolar plates and gaskets. The bipolar plates are prepared by graphite or metal with straight flow fields, and gaskets made of silicone rubber. A JARI standard single cell with a 5.0 cm2 geometric area was prepared where the active area of the electrode in the MEA was adjusted to 2.2 × 2.2 cm2 for both anode and cathode.
The JARI standard single cell was connected with the FC5105-NN fuel cell testing platform (Chino Corp.) to measure the I–V polarization curves at 70 °C cell temperature under ambient pressure using humidified H2 gas (100% relative humidity (RH) and 80 ml min−1 at the inlet) fed to the anode, and humidified O2 gas (100% RH and 80 ml min−1 at the inlet) fed to the cathode. Anode and cathode humidification was controlled at 70 °C. A PARSTAT MC Multichannel potentiostat-galvanostat from Princeton Applied Research (AMETEK) was used for controlling the current at a potential applied in potentiostat mode, or vice versa in galvanostat mode, to understand how electrical changes impact a chemical reaction. Prior to the recording of the polarization curves, MEAs were conditioned to a current density of 4.84 A cm−2 for at least 14 h in H2 and O2 operating atmospheres for activation. After activation of the MEA, I–V curves were recorded to evaluate the performance of the cathode catalysts by measuring the stable cell voltage at each giving current density. To determine the durability of the catalysts, ADT was performed using a standard potential cycling between 0.6 and 1.0 V vs. RHE at a scanning speed of 100 mV s−1 for 10000 cycles in H2 and O2 operating atmospheres, according to the recommendation of the Fuel Cell Commercialization Conference of Japan (FCCJ).39 Both the H2 and O2 had a flow rate of 85 ml min−1 were delivered to the anode and cathode, respectively. The cell temperature was kept constant at 80 °C, and the humidifier temperatures for the anode and cathode were controlled at 80 °C and 66 °C, respectively. For characterization of the commercial Pt/C (36.7 wt%) anode catalyst, I–V characteristic curves were recorded from open-circuit voltage (OCV) to 0.20 V every 1000 cycles. The ADT results of the prepared catalysts were compared to the commercial catalyst Pt/C (46.2 wt%, TEC10E50E, Tanaka Kikinzoku Kogyo Co. Ltd.).
3. Results and discussion
3.1 Physical characterization of Pt/C catalysts
The XRD patterns for the different Pt loaded (25, 30, 35 and 40 wt%) in the Pt/KB1600 and Pt/KB800 catalysts are presented in Fig. 1(a) and (b), respectively. The diffraction peaks of Pt NPs were obtained at 39.2°, 45.7° and 66.6°, corresponding to the planes (111), (200) and (220) of the face-centered cubic (fcc) crystalline Pt, respectively (JCPDS card no. 88-2343).40 The relative degree of crystallization of the catalysts was examined using the height-to-peak ratio of the face of the Pt crystal (111) and carbon reflection.41 Assuming spherical particles and using the Debye–Scherrer equation, the mean diameter of the Pt NPs calculated using the full width at half maximum (FWHM) of the plane (111) is approximately 1.5 ± 0.1 nm. | ||
| Fig. 1 XRD patterns of (a) Pt/KB1600 and (b) Pt/KB800 catalyst with different Pt loadings (25, 30, 35 and 40 wt%). | ||
In order to evaluate the size and distribution of the prepared Pt/C catalysts with different Pt loadings, TEM observations were conducted. Fig. 2 shows a series of TEM images of the Pt/KB1600 and Pt/KB800 catalysts with different Pt loadings (25 wt%, 30 wt%, 35 wt%, 40 wt%), respectively. In these images, the Pt NPs are shown in black, the carbon as catalyst support is grey, and the pores in the carbon are light grey. As can be seen in Fig. 2, all the catalysts are well dispersed on the surface of the carbon support with a narrow particle size distribution. The Pt/KB1600 and Pt/KB800 catalysts have Pt NPs of apparently similar size, but smaller than the commercial Pt/C catalyst (2.8 nm). The Pt NPs with an average diameter of approximately 1.5 nm were distributed almost uniformly on the carbon support. As the Pt NPs settle on the carbon support, the Pt NPs grow slightly from 1.5 nm to 1.8 nm for the 25 wt% Pt/C and 40 wt% Pt/C, respectively. Such a small particle size is essential to reach high mass activities in catalysis. In fact, there were no notable differences in the mean sizes measured from the TEM images and estimated from the XRD patterns. The morphology of the obtained Pt/C catalysts was investigated by SEM. The ESI Fig. S1† shows the formation of a probably spherical and dense morphology. No remarkable differences were observed among these samples, as indicated in the Fig. S1.† These results suggest that a well-controlled size of Pt NPs was successfully synthesized by the ethanol reduction method.
 | ||
| Fig. 2 TEM images of the Pt/KB1600 and Pt/KB800 catalyst with different Pt loadings of (a1–a4) 25–40 wt%, and (b1–b4) 25–40 wt%, respectively. | ||
Thereafter, XAFS spectra obtained in transmission mode for the Pt/C anode catalysts in practical MEAs under PEFC operating conditions were analyzed. The CNs determined by curve-fitting XAFS analyses are almost identical, irrespective of Pt loading. Finally, the CNs of the Pt on KB are used to compute the size of the NPs, based on eqn (6). The relation between the CNobs and the average diameter of the Pt NP is as follows:
| (12–1.40 × CNobs) × dXAFS = (12–9.69) × 2.18 = 5.034 = constant | (6) |
The result indicates that the theoretically estimated CN of the Pt/C anode catalyst with a diameter of 2.18 nm is 9.69 based on regular icosahedron with an edge consisting of five Pt atoms, although the CN measured in this study was 6.90. These results are consistent with the crystal size estimates from XRD and TEM. A detailed summary of these results is presented in Table 1, which is also compared.
| Carbon support | Pt loading (wt%) | Average particle size (nm) | ||
|---|---|---|---|---|
| XRD | TEM | XAFS | ||
| KB1600 | 25 | 1.5 | 1.5 | |
| 30 | 1.6 | 1.6 | ||
| 35 | 1.6 | 1.5 | ||
| 40 | 1.5 | 1.7 | ||
| KB800 | 25 | 1.4 | 1.6 | |
| 30 | 1.5 | 1.6 | 2.0 | |
| 35 | 1.5 | 1.6 | ||
| 40 | 1.5 | 1.8 | 2.1 | |
Nitrogen adsorption–desorption isotherms were measured on carbon-supported Pt catalysts (Pt/KB1600 and Pt/KB800) with different Pt loadings to evaluate the effect of the textural properties (e.g., BET surface area, pore volume, and porosity) on the growth mechanism of the Pt NPs produced by the ethanol reduction method. All samples were isothermally typed-IV characteristic, indicating the presence of mesopores in the carbon catalysts,42 as presented in ESI Fig. S2.† As can be seen in Fig. S2,† the adsorption isotherm curves are exhibited hysteresis loops of the H3 type, when the pressure ranged relatively from 0.4 to 0.8 P/P°. However, when the step of desorption was displaced to a lesser relative pressure of approximately 0.45 P/P°, the behavior changed dramatically. It can be characterized by narrow necks and wide bodies (ink-bottle pores), which means that the carbon as the catalyst supports possess hollow structures.43
The pore size distribution of carbon supports and carbon-supported Pt catalysts (Pt/KB1600 and Pt/KB800) with different loadings of Pt (25–40 wt%) were calculated from adsorption–desorption isotherms by the BJH method, as shown in ESI Fig. S3.† A sharp and broad peak (less than 10 nm) indicates the existence of numerous mesopores in KB1600 and KB800 carbon, while a low magnitude distribution curve for KB800 was found. The intensity of the pore size distribution peaks decreased for each catalyst (Pt/KB1600, Pt/KB800) even lower than that of pristine carbon, indicating the Pt loading on the carbon supports and the resulting pore filing. The pore volumes of the carbon supports and carbon-supported Pt catalysts with various Pt loadings are presented in Table 2. The mesoporous KB1600 carbon possessed a large pore volume of ca. 2.49 cm3 gcarbon−1, which is 2.4 times larger than that of the KB800 carbon (ca. 1.03 cm3 gcarbon−1). In addition, the Pt/KB1600 catalysts have relatively large pore volumes compared to those of the Pt/KB800 catalysts. However, the volume of the pores is reduced after Pt loading on the both carbons, which implies that the Pt species disperse not only on the carbon support surface, but also on the pores. These results support the idea that the degradation of the total volume can be attributed to pore blockage by the Pt NPs, which prohibits N2 adsorption on the pores. BET surface area of the bare carbons (KB1600 and KB800), and Pt/KB1600 and Pt/KB800 catalysts calculated based on BET equation are summarized in Table 2. The KB1600 carbon shows a BET surface area (1358 m2 gcarbon−1) that is ca. 1.7 times larger than that of KB800 carbon (804 m2 gcarbon−1). Following the loading of Pt onto the carbons, the BET surface area of Pt/KB1600 and Pt/KB800 decreased with respect to the bare carbons. However, Pt/KB1600 still had a BET surface area greater than Pt/KB800. Although the BET surface area of Pt-deposited KB1600 and KB800 carbons initially decreased after loading of 25 wt% Pt in both cases, it increased to 1286 m2 gcarbon−1 for 35 wt% Pt/KB1600, and 746 m2 gcarbon−1 for 30 wt% Pt/KB800 catalyst. Furthermore, the BET surface areas decrease as the Pt loading of Pt/KB1600 and Pt/KB800 catalyst increased to 40 wt%. This decrease is attributed to pore blocking by Pt NPs. Note that the larger BET surface area of the Pt/C catalysts results in higher ORR performance in the PEFCs. Thus, considering the highest value of the BET surface area described in Table 2, the optimal platinum content in Pt/C catalysts has been determined to be 35 wt% for Pt/KB1600, and 30 wt% for Pt/KB800.
| Carbon support | Pt loading (wt%) | Pore volume (cm3 gcarbon−1) | BET surface area (m2 gcarbon−1) |
|---|---|---|---|
| KB1600 | 0 | 2.49 | 1358 |
| 25 | 1.90 | 1240 | |
| 30 | 1.90 | 1260 | |
| 35 | 1.91 | 1286 | |
| 40 | 1.87 | 1253 | |
| KB800 | 0 | 1.03 | 804 |
| 25 | 0.76 | 709 | |
| 30 | 0.88 | 746 | |
| 35 | 0.77 | 728 | |
| 40 | 0.74 | 717 |
3.2 Electrochemical performance of Pt/C catalysts
The electrochemical efficiency of the Pt/C catalysts at different loadings of Pt was evaluated for the ORR. The ORR measurements were performed by CV and LSV on the RDE. The LSV measurements were performed in the potential range of 0.20 to 1.20 V vs. RHE with a scan speed of 10 mV s−1 in 0.1 M HClO4 aqueous solution saturated with O2. ORR polarization curves of different catalysts at different rotating speeds of 400, 900, 1200 and 2500 rpm are presented in the ESI Fig. S4.† The polarization curves indicate two clearly defined potential regions which are the diffusion-limited current region less than 0.6 V vs. RHE, and the mixed kinetic-diffusion control region from 0.7 and 1.0 V vs. RHE for all Pt/C catalysts. In the diffusion-limited current region, the ORR current density value for the catalyst increased with the rotational speed. The higher onset potential for ORR can be achieved by the mixed kinetic-diffusion control region. The onset potential is a key indicator that affects the performance of the ORR.44 For a more quantitative analysis of the ORR activity of all catalysts, the kinetic current densities calculated from the ORR polarization curves were analyzed using K–L equation and then standardized as a function of Pt loading on the GC electrode. Fig. 3 shows the K–L plots of the Pt/KB1600 and Pt/KB800 catalysts with different Pt loadings compared to the commercially available Pt/C catalyst. The corresponding K–L plots show excellent linearity for 1/j and ω−1/2, indicating first-order reaction kinetics. For all samples, the Levich slope showed negligible changes to the applied potentials, indicating the first order kinetics of the ORR.45 | ||
| Fig. 3 K–L plots of (a) Pt/KB1600, and (b) Pt/KB1600 catalyst with different Pt loadings in comparison with the commercial Pt/C catalyst recorded at RT. | ||
The ORR mass activity and electron transfer numbers of different catalysts at 0.8 V vs. RHE are summarized in Table 3. In comparison with commercial Pt/C, the ORR mass activities of the prepared catalysts are significantly higher. Among all the prepared catalysts, 35 wt% Pt/KB1600 and 30 wt% Pt/KB800 exhibited higher mass activities are 0.444 and 0.440 A gPt−1, respectively, which are approximately 2.9 times higher than that of the commercial Pt/C. This suggests that the 35 wt% Pt/KB1600 catalyst is more active than the 30 wt% Pt/KB800 and commercial Pt/C catalysts. Furthermore, the increase in mass activity is due to the unique structures of the mesoporous support that favours catalyst-support interaction, facilitating the process of electron and mass transfer during the reactions. This suggests that the effect of NP size on the mass activities of 35 wt% Pt/KB1600 and 30 wt% Pt/KB800 catalyst is insignificant. The NP sizes of the prepared Pt/C catalysts were similar in size (ca. 1.5 nm), as indicated by the XRD patterns and TEM image analysis. The total number of electron transfer from the prepared catalysts is ca. 3.9, fairly close to the theoretical value of 4.0.46
| Carbon supports | Pt loading (wt%) | Mass activity at 0.8 V vs. RHE (A gPt−1) | Electron no. (−) | ECSA with COads (m2 gPt−1) |
|---|---|---|---|---|
| KB1600 | 25 | 0.181 | 3.82 | 106.0 |
| 30 | 0.307 | 4.06 | 87.7 | |
| 35 | 0.444 | 3.73 | 155.0 | |
| 40 | 0.265 | 4.34 | 99.0 | |
| KB800 | 25 | 0.206 | 3.38 | 127.0 |
| 30 | 0.440 | 3.41 | 139.0 | |
| 35 | 0.171 | 4.13 | 114.0 | |
| 40 | 0.176 | 3.83 | 98.9 | |
| Comm. Pt/C | 36.7 | 0.154 | 3.39 | 79.1 |
To determine the ECSA of the Pt/C catalysts, CO-stripping voltammetry were measured in a 0.1 M HClO4 solution saturated with Ar at RT. CO-stripping spectrum was recorded in the potential range from 0.05 V to 1.20 V vs. RHE at a sweep speed of 20 mV s−1. Fig. 4(a) and (c) show the CO-stripping spectrum for 35 wt% Pt/KB1600, 30 wt% Pt/KB800 and commercial Pt/C catalysts, respectively. In addition, the CO-stripping voltammograms for 25 wt% Pt/KB1600, 30 wt% Pt/KB1600, 40 wt% Pt/KB1600, 25 wt% Pt/KB800, 35 wt% Pt/KB800 and 40 wt% Pt/KB800 catalysts are recorded under similar conditions mentioned earlier, as shown in the ESI Fig. S5(a), (b), (c), (d), (e) and (f),† respectively. The analysis identified a signal of approximately 0.90 VRHE with a low-potential flank shoulder because of the oxidation and desorption of the CO. The relationship between ECSA and Pt ration to the Pt/C catalyst is illustrated in Fig. 4(d). A similar relationship between ECSA and Pt loadings (wt%) was observed in the Pt/C catalysts, indicating that Pt NPs are distributed uniformly on the carbon support. These findings suggest that catalytic activity is independent of the intrinsic properties of the platinum, but were controlled by the properties of the carbon supports.
 | ||
| Fig. 4 CO stripping curves of (a) 35 wt% Pt/KB1600, (b) 30 wt% Pt/KB800 and (c) commercial Pt/C recorded at RT in 0.1 M HClO4 solution with Pt loading of 0.1 mgPt cm−2 on the electrode. (d) Relations between the ECSA and the ratio of Pt to Pt/C catalyst. | ||
Using the CO-stripping measurements described above, the ECSA values of the prepared catalysts were estimated by integrating the CO-stripping peak with a conversion constant of 0.420 mC cm−2.47 The detailed ECSA values are summarized in Table 3, as discussed earlier. The ECSAs of the 25 wt% Pt/KB1600, 30 wt% Pt/KB1600, 35 wt% Pt/KB1600, 40 wt% Pt/KB1600, 25 wt% Pt/KB800, 30 wt% Pt/KB800, 35 wt% Pt/KB800, 40 wt% Pt/KB800, and commercial Pt/C catalysts were found to be 106.0, 87.7, 155.0, 99.0, 127.0, 139.0, 114.0, 98.9 and 79.1 m2 gPt−1, respectively. Higher ECSA was obtained for 35 wt% Pt/KB1600 due to the high dispersion of Pt with fine particle size compared to Pt/KB800 and the commercial catalyst. Higher ECSA was also expected to lead to high Pt utilization, which is another important factor for an effective triple phase boundary at the cathode interface.48 Thus, ECSA is one of most important characteristics of Pt/C electrocatalysts.
3.3 Performance and durability of the PEFCs
As discussed earlier, our synthesis approach is very simple but highly effective for improving the ORR performance. To assess the feasibility of the catalyst for practical application at a single cell level, we prepared MEAs using Pt/KB1600, Pt/KB800 and commercial Pt/C as the cathode catalysts with a low-Pt loading of 0.1 mgPt cmMEA−2. The I–V polarization and power density curves of the single cells are shown in Fig. 5(a) and (b), respectively. The results obtained were compared with those obtained with a commercial P/C catalyst. The voltages of single cells in the low current density (0.2 A cm−2) and high current density (1.0 A cm−2) regions were tasted to evaluate the PEFC's performance. The cell voltages were approximately the same for all electrocatalysts at 0.2 A cm−2, while their cell voltages decreased by 1.0 A cm−2. Note that the Pt/KB1600 cathode catalyst prepared with 35 wt% Pt exhibits the best performance (0.58 V) at 1.0 A cm−2 due to its high ECSA. The maximum power density of the single cell with 35 wt% Pt/KB1600 catalyst is calculated at 1.0 A cm−2 to be around 0.58 W cm−2, considerably higher than the 30 wt% Pt/KB800 catalyst (ca. 0.53 W cm−2). But for the commercial Pt/C catalyst, the calculated maximum power density was ca. 0.52 W cm−2, which was 0.06 W cm−2 lower than the 35 wt% Pt/KB1600. For high power density and cell voltage in the high current density region, significant mass transport is required. Furthermore, it is suggested that the morphology of the carbon support and the distribution of Pt NPs play a significant role in regulating the efficiency of the cell.15 Note that the BET surface areas of the prepared catalysts with high Pt loading are lower than the pristine carbon, which is probably due to the partial blocking of the pores by Pt NPs. This may be one of the reasons why the performance of a single cell has been impaired. To elucidate this disputed point, the relationship between cell performance and BET surface area toward the ratios of Pt to Pt/C catalyst was investigated, as shown in Fig. 5(c). The relatively large BET surface areas of the Pt/KB1600 and Pt/KB800 catalysts were obtained from 35 and 30 wt% Pt to Pt/C, respectively. The figure shows that a higher BET surface area caused a higher cell voltage and vice versa.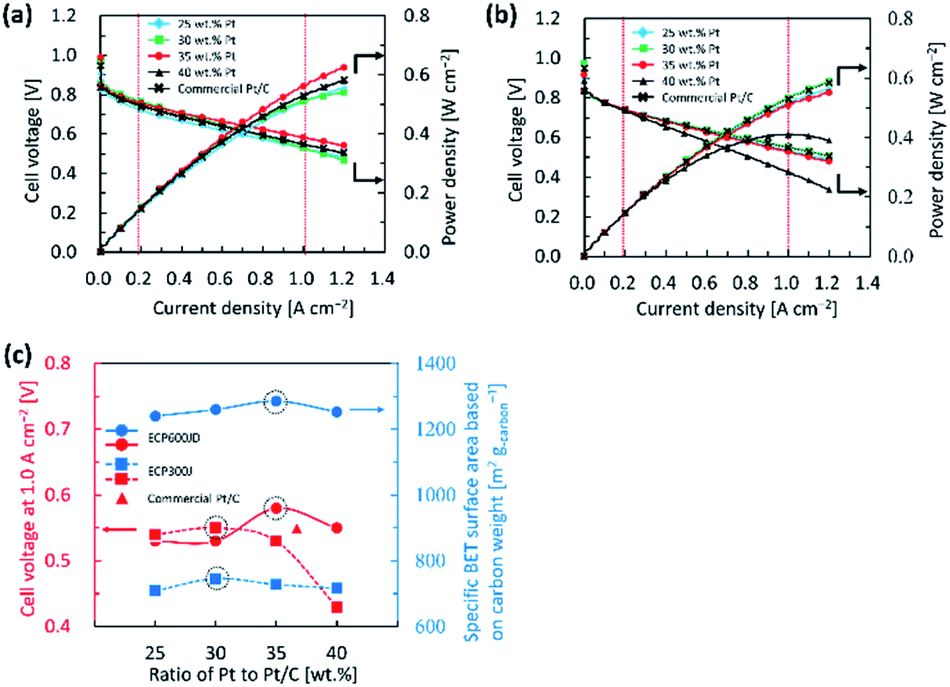 | ||
| Fig. 5 I–V characteristic curves of (a) Pt/KB1600 catalyst, and (b) Pt/KB800 catalyst in comparison with commercial Pt/C catalyst. (c) Relationship between cell voltage at 1.0 A cm−2 and BET surface area toward the ratio of Pt to Pt/C. | ||
A detailed analysis of the I–V characteristics of single cells with Pt/KB1600, Pt/KB800 and commercial Pt/C as the cathode catalysts with different loadings of Pt are summarized in Table 4. As can be seen in Table 4, the cell voltage decreased as the Pt loading in the single cells decreased. Interestingly, the MEA prepared with 35 wt% Pt/KB1600 and 30 wt% Pt/KB800 catalysts exhibited relatively higher performance in the low current density region and high current density region compared to the commercial Pt/C catalyst. The prepared catalysts also had higher power densities than the commercial Pt/C catalyst. However, the cell performance decreases as the Pt loading increases to 40 wt% for both prepared catalysts. It is therefore clear that the difference in cell performance is highly related to the Pt loading in the electrode.
| Carbon supports | Pt loading (wt%) | Cell voltage at 0.2 A cm−2 (V) | Cell voltage at 1.0 A cm−2 (V) |
|---|---|---|---|
| KB1600 | 25 | 0.72 | 0.53 |
| 30 | 0.72 | 0.53 | |
| 35 | 0.76 | 0.58 | |
| 40 | 0.74 | 0.55 | |
| KB800 | 25 | 0.73 | 0.54 |
| 30 | 0.74 | 0.55 | |
| 35 | 0.74 | 0.53 | |
| 40 | 0.73 | 0.43 | |
| Comm. Pt/C | 36.7 | 0.74 | 0.55 |
To investigate the durability of the cell prepared with 35 wt% Pt/KB1600 as cathode catalyst, the standard potential cycling tests were performed in comparison with the commercial Pt/C (46.2 wt%, TEC10E50E) catalyst. The I–V characteristic of single cells with 35 wt% Pt/KB1600 and commercial Pt/C as the cathode catalysts were measured before and after ADT, as shown in Fig. 6(a). The initial characteristics are shown by solid lines, and the performances after 10000 cycle tests are indicated as dashed lines. Changes in cell voltages at current densities of 0.2 and 1.0 A cm−2 during ADT are summarized due to assess the durability of cathode catalysts, as shown in Fig. 6(b). The estimated cell voltages at current densities of 0.2 and 1.0 A cm−2 are approximately 0.75 and 0.53 V, respectively. For the 35 wt% Pt/KB1600 catalyst, they declined to 0.72 and 0.42 V after 10000 potential cycles. However, in the case of the commercial Pt/C catalyst, the cell voltages at 0.2 and 1.0 A cm−2 were 0.75 and 0.50 V, and they decreased to 0.72 and 0.39 V after 10000 potential cycles, respectively. The performance degradation of the MEAs is much faster at the beginning, and then settle down to a slower. The lower decay rate suggests that the Pt/KB1600 catalyst is more durable. In fact, the Pt/KB1600 catalyst had a higher cell voltage, particularly in the region of high current density, compared to the commercial Pt/C catalyst during ADT. This may be primarily due to the uniform distribution of Pt NPs on the carbon support inhibiting Pt NPs agglomeration.
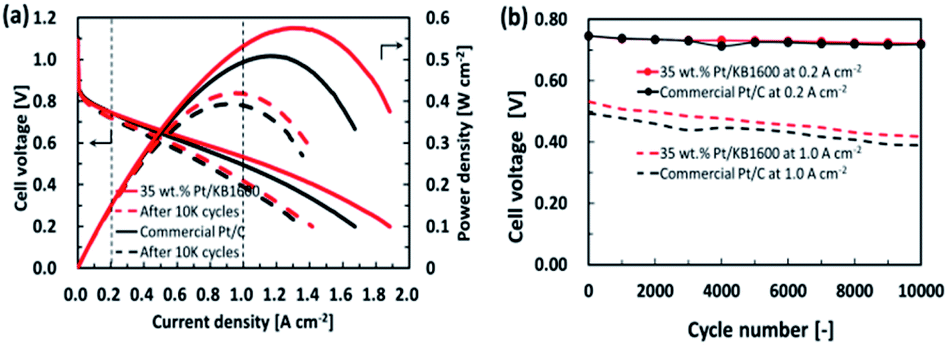 | ||
| Fig. 6 (a) I–V characteristic curves of 35 wt% Pt/KB1600 and commercial Pt/C (46.2 wt%, TEC10E50E) cathode catalyst before and after the ADT. (b) The change in the cell voltage at 0.2 and 1.0 A cm−2 during ADT for the case of 35 wt% Pt/KB1600 and commercial Pt/C catalyst. | ||
3.4 Microstructure dependence of performance degradation
The electrode microstructure directly affects the electron transport and has a strong relationship with the cell performance and cell degradation.49 Fig. 7 shows a schematic illustration of the microstructures of the Pt/KB1600 catalyst with different loadings of Pt. One can notice that the Pt is well scattered on the scaffolding surface and does not block the pores severely after loading a small amount of Pt on the carbon support. This implies that there are large amounts of carbon that would lead to a higher thickness of MEA. However, the thick electrode strongly affects the electrical resistance and causes a long gas diffusion pathway.50 Accordingly, 20–30 wt% Pt/KB1600 catalyst inhibited uniformly diffusion pathway for the oxygen gas, and therefore, the contribution of triple phase boundary (Pt, membrane electrolyte and gas phase) in cathodic reactions was decreased, resulting in the degradation of the cell performance. On the other hand, upon increasing the amount of Pt loading to 40 wt%, the pores in the carbon support were mostly blocked. A high amount of Pt loading in the catalyst implies that a small amount of carbon, which would lead to a thinner MEA. A thin electrode layer influences gas dispersion and pore blockage may cause obstruction to mass transport results in lower ORR activity. In this study, significant performance degradation due to mass transport losses was also detected in the region of high current density when a low or high amount of Pt (25 or 40 wt%) on the carbon support was used. | ||
| Fig. 7 Schematic illustration showing microstructural diagram of Pt/KB1600 catalyst with different loading of Pt. | ||
4. Conclusions
We successfully prepared Pt/KB1600 and Pt/KB800 catalysts with different loadings of Pt (25, 30, 35 and 40 wt%) by using ethanol reduction method. The BET surface area, porosity and surface defect of the carbon support greatly influenced the nucleation of Pt NPs. The structural characterization showed a greater uniformity and a high dispersion of the Pt with a fine grain size formed on KB1600 because of its high surface area with a large volume of pores and structural defects, as compared to those of the KB800 with the same weight percentage. Compared to pristine carbon, the average pore size, pore volume and BET surface area of the prepared Pt/C catalysts decreased. This phenomenon can likely be attributed to pore blocking by Pt NPs. The uniform structure of the Pt/KB1600 catalyst made it possible to obtain an ECSA superior to that of the Pt/KB800 catalyst and the commercial Pt/C catalyst (36.7 wt%, TEC10E40E). Moreover, the mass activities of the prepared catalysts (35 wt% Pt/KB1600, and 30 wt% Pt/KB800) are significantly higher than that of the commercial Pt/C catalyst. Subsequently, the effect of carbon support on the cathode performance of PEFC in terms of Pt loading was investigated in comparison with a commercial Pt/C catalyst. The maximum power density and cell voltage of the single cell prepared by 35 wt% Pt/KB1600 catalyst are calculated at 1.0 A cm−2 to be approximately 0.58 V and 0.58 W cm−2, respectively. Finally, durability tests for the 35 wt% Pt/KB1600 catalyst were conducted using a single cell test, and revealed that it was superior to that of the commercial Pt/C catalyst due to the uniform distribution of Pt NPs on a carbon support that inhibited NPs aggregation. This suggests that an appropriate Pt/C catalyst ratio provides adequate reaction sites and mass transport, resulting in higher Pt/C ORR activity. Thus, it can be concluded that control of the morphology, size distribution, composition and structure of the catalyst support is crucial to facilitate the improvement of catalytic activity, long-term durability and overall performance of the PEFC.Author contributions
Md. Mijanur Rahman – conceptualization, methodology, validation, investigation, visualization, data curation, writing original draft and manuscript revision; Kenta Inaba – investigation, data discussion, manuscript writing and revision; Garavdorj Batnyagt – investigation, data discussion and manuscript revision; Masato Saikawa – investigation and post-mortem analysis; Yoshiki Kato – investigation and data curation; Rina Awata – data analysis, discussion and suggestions; Byambasuren Delgertsetsega – data discussion, writing – review and editing; Yasuo Kaneta – data discussion and suggestions; Kotaro Higashi – XAFS measurements, data analysis, suggestions and manuscript revision; Tomoya Uruga – XAFS measurements, data discussion, suggestions and manuscript revision; Yasuhiro Iwasawa – XAFS data discussion, suggestions and manuscript revision; Koichi Ui – data discussion, suggestion and manuscript revision; Tatsuya Takeguchi – conceptualization, methodology, investigation, discussion and suggestions, project administration, funding acquisition, writing – review and editing.Conflicts of interest
The authors declare no conflicts of interest associated with this manuscript.Acknowledgements
The authors would like to thank the New Energy and Industrial Technology Development Organization (NEDO) for financial support of this work. We also gratefully acknowledge the instrumental support and dedicated efforts of the staff of the National Institute for Materials Science (NIMS) Battery Research Platform. The XAFS experiments were performed at BL09XU of SPring-8 under the approval of Japan Synchrotron Radiation Research Institute (Proposals No. 2016A7901, 2016B7901 and 2017A7901). We want to thank Mr Yuta Kato, a former student at Iwate University, for his support in some of the experiments.Notes and references
- W. S. Jung and B. N. Popov, ACS Sustainable Chem. Eng., 2017, 5, 9809 CrossRef CAS.
- H. Yano, M. Watanabe, A. Iiyama and H. Uchida, Nano Energy, 2016, 29, 323 CrossRef CAS.
- N. Jha, P. Ramesh, E. Bekyarova, X. Tian, F. Wang, M. E. Itkis and R. C. Haddon, Sci. Rep., 2017, 3, 2257 CrossRef PubMed.
- Y. Cai, P. Gao, F. Wang and H. Zhu, Electrochim. Acta, 2017, 245, 924 CrossRef CAS.
- F. Godínez-Salomon, F. Godínez-Salomon, M. J. Arellano-Jimenez, M. Jose-Yacaman and C. P. Rhodes, ACS Appl. Mater. Interfaces, 2017, 9, 18660 CrossRef PubMed.
- M. D. Gasda, R. Teki, T.-M. Lu, N. Koratkar, G. A. Eisman and D. Gall, J. Electrochem. Soc., 2009, 156, B614 CrossRef CAS.
- B. Millington, V. Whipple and B. G. Pollet, J. Power Sources, 2011, 196, 8500 CrossRef CAS.
- L. I. Şanlı, B. Yarar, V. Bayram and S. A. Gürsel, J. Mater. Sci., 2017, 52, 2091 CrossRef.
- H. Yu, J. M. Roller, W. E. Mustain and R. Maric, J. Power Sources, 2015, 283, 84 CrossRef CAS.
- T. Myles, S. Kim, R. Maric and W. E. Mustain, Catalysts, 2015, 5, 1673 CrossRef CAS.
- N. Jung, D. Y. Chung, J. Ryu, S. J. Yoo and Y.-E. Sung, Nano Today, 2014, 9, 433 CrossRef CAS.
- M. A. Molina-García and N. V. Rees, RSC Adv., 2016, 6, 94669 RSC.
- S. Gouse Peera, A. Arunchander and A. K. Sahu, Carbon, 2016, 107, 667 CrossRef.
- E. Daş and A. B. Yurtcan, Int. J. Hydrogen Energy, 2016, 41, 13171 CrossRef.
- S. Sui, X. Wang, X. Zhou, Y. Su, S. Riffat and C. J. Liu, J. Mater. Chem. A, 2017, 5, 1808 RSC.
- D. C. Dogan, S. Cho, S. M. Hwang, Y. M. Kim, H. Guim, T. H. Yang, S. H. Park, G. G. Park and S. D. Yim, ACS Appl. Mater. Interfaces, 2016, 8, 27730 CrossRef CAS PubMed.
- C. Wang, W. Tian, Y. Ding, Y. Q. Ma, Z. L. Wang, N. M. Markovic, V. R. Stamenkovic, H. Daimon and S. Su, J. Am. Chem. Soc., 2010, 132, 6524 CrossRef CAS PubMed.
- Q. He, D. C. Joy and D. J. Keffer, RSC Adv., 2013, 3, 15792 RSC.
- H. Kuroki, T. Tamaki, M. Matsumoto, M. Arao, Y. Takahashi, H. Imai, Y. Kitamoto and T. Yamaguchi, ACS Appl. Energy Mater., 2018, 1, 324 CrossRef CAS.
- N. Macauley, D. D. Papadias, J. Fairweather, D. Spernjak, D. Langlois, R. Ahluwalia, K. L. More, R. Mukundan and R. L. Borup, J. Electrochem. Soc., 2018, 165, F3148 CrossRef CAS.
- B. Avasarala, R. Moore and P. Haldar, Electrochim. Acta, 2010, 55, 4765 CrossRef CAS.
- S. Murata, M. Imanishi, S. Hasegawa and R. Namba, J. Power Sources, 2014, 253, 104 CrossRef CAS.
- Y. C. Chiang, M. K. Hsieh and H. H. Hsu, Thin Solid Films, 2014, 570, 221 CrossRef CAS.
- Y. Jeon, Y. Ji, Y. I. Cho, C. Lee, D. H. Park and Y. G. Shul, ACS Nano, 2018, 12, 6819 CrossRef CAS PubMed.
- Y. Wang, J. Jin, S. Yang, G. Li and J. Qiao, Electrochim. Acta, 2015, 177, 181 CrossRef CAS.
- C. Alegre, M. E. Gálvez, R. Moliner, V. Baglio, A. S. Aricò and M. J. Lázaro, Appl. Catal., B, 2014, 147, 947 CrossRef CAS.
- A. Ghosh, S. Basu and A. Verma, Fuel Cells, 2013, 13, 355 CrossRef CAS.
- A. Marinkas, F. Arena, J. Mitzel, G. M. Prinz, A. Heinzel, V. Peinecke and H. Natter, Carbon, 2013, 58, 139 CrossRef CAS.
- D. Wang, J. Wang, X. Luo, Z. Wu and L. Ye, ACS Sustainable Chem. Eng., 2018, 6, 983 CrossRef CAS.
- M. Kim, J. N. Park, H. Kim, S. Song and W. H. Lee, J. Power Sources, 2006, 163, 93 CrossRef CAS.
- J. Speder, A. Zana, I. Spanos, J. J. K. Kirkensgaard, K. Mortensen, M. Hanzlik and M. Arenz, J. Power Sources, 2014, 261, 14 CrossRef CAS.
- N. Ishiguro, T. Saida, T. Uruga, O. Sekizawa, K. Nagasawa, K. Nitta, T. Yamamoto, S.-i. Ohkoshi, T. Yokoyama and M. Tada, Phys. Chem. Chem. Phys., 2013, 15, 18827 RSC.
- Y. Hashimasa, T. Numata, K. Moriya and S. Watanabe, J. Power Sources, 2006, 155, 182 CrossRef CAS.
- N. Ishiguro, S. Kityakarn, O. Sekizawa, T. Uruga, H. Matsui, M. Taguchi, K. Nagasawa, T. Yokoyama and M. Tada, J. Phys. Chem. C, 2016, 120, 19642 CrossRef CAS.
- T. Vidaković, M. Christov and K. Sundmacher, Electrochim. Acta, 2007, 52, 5606 CrossRef.
- J. Zhao, A. Ozden, S. Shahgaldi, I. E. Alaefour, X. Li and F. Hamdullahpur, Energy, 2018, 150, 69 CrossRef CAS.
- İ. Fırtına, S. Güner and A. Albostan, Int. J. Energy Res., 2011, 35, 146 CrossRef.
- J. Hack, T. M. M. Heenan, F. Iacoviello, N. Mansor, N. Mansor, P. Shearing, N. Brandon and D. J. L. Brett, J. Electrochem. Soc., 2018, 165, F3045 CrossRef CAS.
- K. Shinohara, A. Ohma, A. Iiyama, T. Yoshida and A. Daimaru, Membrane and Catalyst Performance Targets for Automotive Fuel Cells by FCCJ Membrane, Catalyst, MEA WG, ECS Trans., 2011, 41, 775 Search PubMed.
- J. Zhang, C. Chen, S. Chen, Q. Hu, Z. Gao, Y. Li and Y. Qin, Catal. Sci. Technol., 2017, 7, 322 RSC.
- E. Antolini and F. Cardellini, J. Alloys Compd., 2001, 315, 118 CrossRef CAS.
- W. S. Jung and B. N. Popov, Catal. Today, 2017, 295, 65 CrossRef CAS.
- Y.-C. Park, H. Tokiwa, K. Kakinuma, M. Watanabe and M. Uchida, J. Power Sources, 2016, 315, 179 CrossRef CAS.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302 CrossRef CAS PubMed.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruof, Energy Environ. Sci., 2012, 5, 7936 RSC.
- Z. Tang, W. Wu and K. Wang, Catalysts, 2018, 8, 65 CrossRef.
- S. Rudi, C. Cui, L. Gan and P. Strass, Electrocatalysis, 2014, 5, 408 CrossRef CAS.
- Y. Jeon, D. J. Kim, J. K. Koh, Y. Ji, J. H. Kim and Y.-G. Shul, Sci. Rep., 2015, 5, 16394 CrossRef CAS PubMed.
- L. R. Jordan, A. K. Shukla, T. Behrsing, N. R. Avery, B. C. Muddle and M. Forsyth, J. Power Sources, 2000, 86, 250 CrossRef CAS.
- H.-K. Lee, J.-H. Park, D.-Y. Kim and T.-H. Lee, J. Power Sources, 2004, 131, 200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02156g |
| This journal is © The Royal Society of Chemistry 2021 |