DOI:
10.1039/D1RA02358F
(Paper)
RSC Adv., 2021,
11, 20580-20590
UV-enhanced nano-nickel ferrite-activated peroxymonosulfate for the degradation of chlortetracycline hydrochloride in aqueous solution
Received
25th March 2021
, Accepted 3rd June 2021
First published on 8th June 2021
Abstract
In this study, nano-nickel ferrite (NiFe2O4) was successfully prepared by hydrothermal synthesis and applied to the oxidative removal of chlortetracycline hydrochloride (CTH) in the presence of ultraviolet radiation (UV) and peroxymonosulfate (PMS). Several characterization methods were used to reveal the morphology and surface properties of nano-NiFe2O4, including X-ray diffraction (XRD), scanning electron microscopy (SEM), and Fourier transform infrared absorption (FTIR) spectroscopy. The removal efficiency of CTH, the factors affecting the reaction process and the reaction mechanism of PMS activated by UV combined with nano-NiFe2O4 (UV + nano-NiFe2O4/PMS) in aqueous solution were systematically studied. The results showed that the UV + nano-NiFe2O4/PMS system led to a higher removal efficiency of CTH than other parallel systems. The results also showed that the CTH removal efficiency was enhanced under optimal conditions ([nano-NiFe2O4] = 1 g L−1, [PMS] = 1 g L−1, [UV wavelength] = 254 nm and [pH] = 11) and that a removal efficiency of 96.98% could be achieved after 60 min. In addition, the influence of the PMS concentration, CTH concentration, dosage of added nano-NiFe2O4 and pH on the PMS activation efficiency and CTH oxidative degradation effect was studied. Inorganic anions such as Cl−, HCO3−, CO32− and NO3− increased the removal efficiency of CTH by 21.29%, 27.17%, 25.32% and 5.96% respectively, while H2PO4− inhibited CTH removal, and the removal efficiency of CTH decreased 6.08% after 60 min. Free radical identification tests detected SO4−˙, OH˙ and 1O2 and showed that these species participated in the degradation reaction of CTH. The results of LC-MS and TOC analysis showed that CTH was degraded in the UV + nano-NiFe2O4/PMS system through hydroxylation, demethylation, deamination, and dehydration reaction and finally mineralized into CO2. These findings confirmed that nano-NiFe2O4 is a green and efficient heterogeneous catalyst for activation of PMS and demonstrates potential applicability in the treatment of antibiotic wastewater.
1. Introduction
Antibiotics are commonly used anti-infective drugs and have been widely used in human disease prevention and animal husbandry. Although they have made great contributions to the development of health care, antibiotics also threaten the environment.1–4 Chlortetracycline hydrochloride (CTH) is a tetracyclic broad-spectrum antibiotic, and due to its low price and potential for promoting the growth of livestock, it has been widely used in animal husbandry.5–7 Nevertheless, the absorption capacity of CTH by humans and animals is limited. These high concentrations of CTH have adverse effects on the ecosystem and human health. This compound is responsible for the inhibition of microbial activity and growth,8 phytoplankton toxicity,9 inhibition of protein synthesis,10 and endangering the life activities of aquatic organisms. Therefore, CTH is considered an important pharmaceutical pollutant, and its removal from water has become an urgent issue.
Various physical, chemical and biological methods such as electrodynamic technology,11 photocatalytic technology,12 microbial treatment13 and so on have been reported for the removal of CTH from water. Although these methods exhibit a certain removal effect, there are still problems such as high energy consumption and low degradation efficiency. In recent years, advanced oxidation processes (AOPs) have received widespread attention. The oxidation process has proven to be an effective method to degrade stubborn emerging pollutants,14,15 such as aromatic amine16 and aromatic alcohols.17 Almost all traditional oxidation processes involve the generation of hydroxyl radicals (OH˙), which are nonselective for most organic pollutants.18 Advanced persulfate oxidation technology produces sulfate radicals (SO4−˙), which can overcome the limitations of OH˙. As an advanced oxidation method, at room temperature, persulfate ions (S2O82−) have the advantages of strong stability, solubility, and high oxidation potential (E0 = 2.01 V).19 Recently, SO4−˙ radical-based processes have attracted significant attention among researchers. S2O82− can be further activated by external energy activators (such as ultrasound, heat and ultraviolet radiation), transition metals (such as iron, cobalt and manganese) and chemical activators (such as alkali, phenol and quinone)20 to produce SO4−˙.
Iron is a transition metal element and is less toxic than copper, manganese and other metal elements. Iron-based metal activation has been the most studied and reported method for efficient SO4−˙ production.21,22 In recent years, iron ions have been loaded into ceramics, molecular sieves and other carriers and then applied to advanced oxidation technology.23 Nickel ferrite (NiFe2O4) is a promising catalyst for advanced oxidation in practical applications because it is easily immobilized, separated, recovered and reused through application of an external magnetic field.24
In this research, we used a hydrothermal synthesis method to prepare nano-NiFe2O4 as a solid catalyst for the activation of peroxymonosulfate (PMS) and coactivated it with ultraviolet (UV) radiation to explore the oxidative removal of CTH. The nanomaterials were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM) and Fourier transform infrared (FTIR) spectroscopy. Then we first investigated the removal efficiency of CTH by different systems and the reaction mechanism of PMS activated by UV irradiation combined with nano-NiFe2O4 (UV + nano-NiFe2O4/PMS) in aqueous solution. Additionally, the effects of the initial solution pH, catalyst dosage, CTH concentration, PMS concentration and coexisting inorganic ions on the degradation efficiency were discussed. The identification and formation mechanism of oxygen species participated in the degradation reaction of CTH were analyzed in this study. Through LC-MS data analysis, the CTH degradation products were determined. The roles of the catalyst, related oxidative radicals and application potential in the UV + nano-NiFe2O4/PMS system were also discussed in detail. This study provides a feasible approach to remove CTH for catalytic oxidation in environmental remediation.
2. Experimental
2.1 Chemicals and materials
Standard samples of CTH (98%), potassium monopersulfate (KHSO5·0.5KHSO4·0.5K2SO4, ≥42% KHSO5 basis), nickel chloride hexahydrate (NiCl2·6H2O, 99%), ferric chloride hexahydrate (FeCl3·6H2O, 97%), sodium hydrosulfide hydrate (Na2HS, 99%), sodium hydroxide (NaOH, 96%), potassium chloride (KCl, 96%), sodium bicarbonate (NaHCO3, 99.5%), sodium carbonate (Na2CO3, 99.8%), tert-butanol (TBA, GR, ≥99.5%), methanol (MeOH, AR, ≥99.5%), ethanol (EtOH, AR, ≥99.5%), and furfuryl alcohol (FFA, CP, ≥97%) were purchased from Aladdin Industrial Corporation (Shanghai, China). Ultrapure water was used in the experiments, and all chemical reagents in the experiments were analytically pure.
2.2 Synthesis of reduced nano-NiFe2O4
Nano-NiFe2O4 was synthesized by a hydrothermal synthesis method. First, 3.5652 g of NiCl2·6H2O, 8.1002 g of FeCl3·6H2O and 5.12 g of NaOH were dissolved in 250 mL of deionized water. Then, the dissolved NiCl2 solution and dissolved FeCl3 solution were mixed thoroughly, 8.65 g of Na2HS was added, and the solution was stirred until the mixture became a creamy yellow (JB-3 timing thermostatic magnetic mixer, Leicixinjing Shanghai, China). The pH of the mixed solution was adjusted to 11 with NaOH while the solution was stirred continuously and refluxed at 120 °C (DHG-9023AD, Qixin Shanghai, China) for 2 h. After the product temperature decreased to room temperature, it was cleaned with deionized water three times and then cleaned with absolute ethanol three times. Next, the product was dried at 100 °C. Finally, the black powder was calcined at 600 °C for 4 h and grinded the sample until could pass through a 2 nm sieve, the sample was labelled “nano-NiFe2O4”. The entire reaction was completed in about 2 hours time.
2.3 Experimental procedures
Degradation experiments of CTH were carried out to evaluate the catalytic performance of the doped nano-NiFe2O4 catalysts. All of the experiments were carried out at room temperature and performed under a UV lamp (254 nm, ZF-8, YuMing Shanghai, China). The UV lamp was turned on 20 min before the beginning of the experiments to obtain a stable level of UV irradiation. The initial concentration of CTH was 120 mg L−1, and 50 mL of this solution was added to the reaction flask. Then, the initial concentrations of PMS (1 g L−1) and the initial of nano-NiFe2O4 (1 g L−1) were added to the experimental system at the initial pH = 2.7. The pH of the experimental system was adjusted by using 1 M NaOH, 0.1 M NaOH and 0.1 M HCl. The samples were collected at set time points (10, 20, 30, 40, 50, and 60 min) with a plastic syringe and filtered through a 0.45 μm filter. Then, the absorbance of CTH in the solution was measured at 364 nm using a UV-vis spectrophotometer. Finally, the concentration of CTH was calculated from the standard curve, and then, the removal efficiency of CTH was calculated.
2.4 Analytical methods
A UV-vis spectrophotometer (UV-1800, HACH, USA) was employed to measure the concentrations of CTH using a spectrophotometric method at the characteristic wavelength of 364 nm. The pH values were determined by using an FE20 precision pH meter (Mettler Toledo, Zurich, Switzerland). TOC measurements were carried out with a TOC-LCPN analyzer (TD-3500, Dandong, China).
A UV-vis spectrophotometer (TU-1901, Beijing General Analysis, China) was used to scan from 190 to 500 nm to obtain the ultraviolet-visible (UV-vis) absorption spectrum. The surface morphology and microstructure of synthetic materials were observed by scanning electron microscopy (SEM) (su8020, Hitachi, Japan). X-ray diffraction (XRD) (TD-3500, Dandong, China) was used to determine the crystal structure of the material. The surface functional groups of the material were measured by Fourier transform infrared (FTIR) spectrometer (iS10 FT-IR spectrometer, Nicolet, USA). The free radicals produced during the process were determined using electron paramagnetic resonance (EPR) spectroscopy (A300-10/12, Bruker, Germany). A liquid chromatography-mass spectrometry (LC-MS) system (chromatography: U3000, Thermo Fisher Scientific, USA; mass spectrometry: Q Exactive, Thermo-FID, USA) was used to detect intermediate substances produced during the reaction.
3. Results and discussion
3.1 Characterization of nano-NiFe2O4
The morphology of nano-NiFe2O4 was observed by field emission SEM.25 The crystalline structure of nano-NiFe2O4 was investigated by XRD.26 FTIR27 spectroscopy was used to detect the forms of the main elements in the sample.
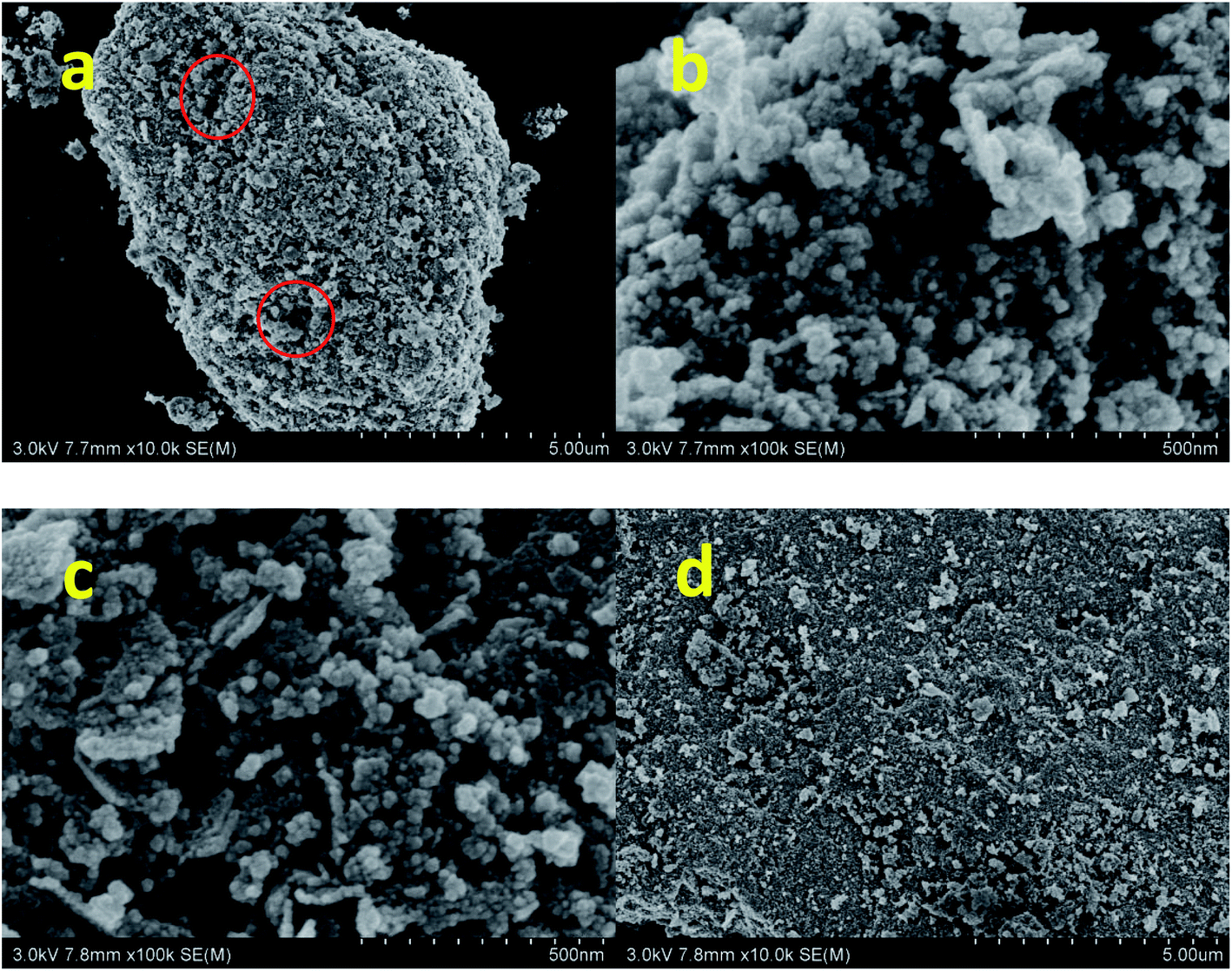
The morphology of nano-NiFe2O4 was observed by SEM. As shown in Fig. 1a and b, the material had a loose nano-microstructure with a primary particle size of 20–50 nm and a secondary particle size of 5–10 μm. The particle sizes reveal the agglomeration of particles, which can be ascribed to the heat released from combustion. Due to the heat generated during the combustion reaction, not only agglomeration but also the sintering of small particles is observed.28 After the agglomeration of nanoparticles, micron-scale particles were formed. The particle structure was relatively loose, and there were many gaps, as shown in the red circle in Fig. 1a. Loose nanostructural particles had a higher specific surface area and could therefore come into contact with more reactants; hence, the catalytic effect was better than that of other catalysts. Fig. 1c and d shows SEM images of the nano-NiFe2O4 recovered after the reaction. It can be seen from the figure that the pores of nano-NiFe2O4 decreased after the reaction and that the reactant was adsorbed and then reacted on the catalyst surface. It could be proved that the nano-NiFe2O4 has a adsorption effect.
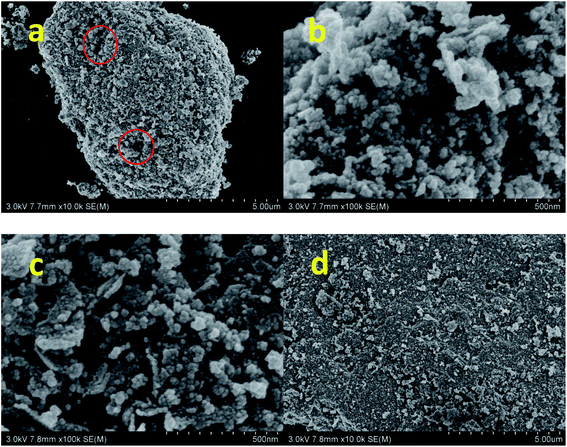 |
| | Fig. 1 SEM image of nano-NiFe2O4 before the reaction (a and b) and the nano-NiFe2O4 after the reaction (c and d). | |
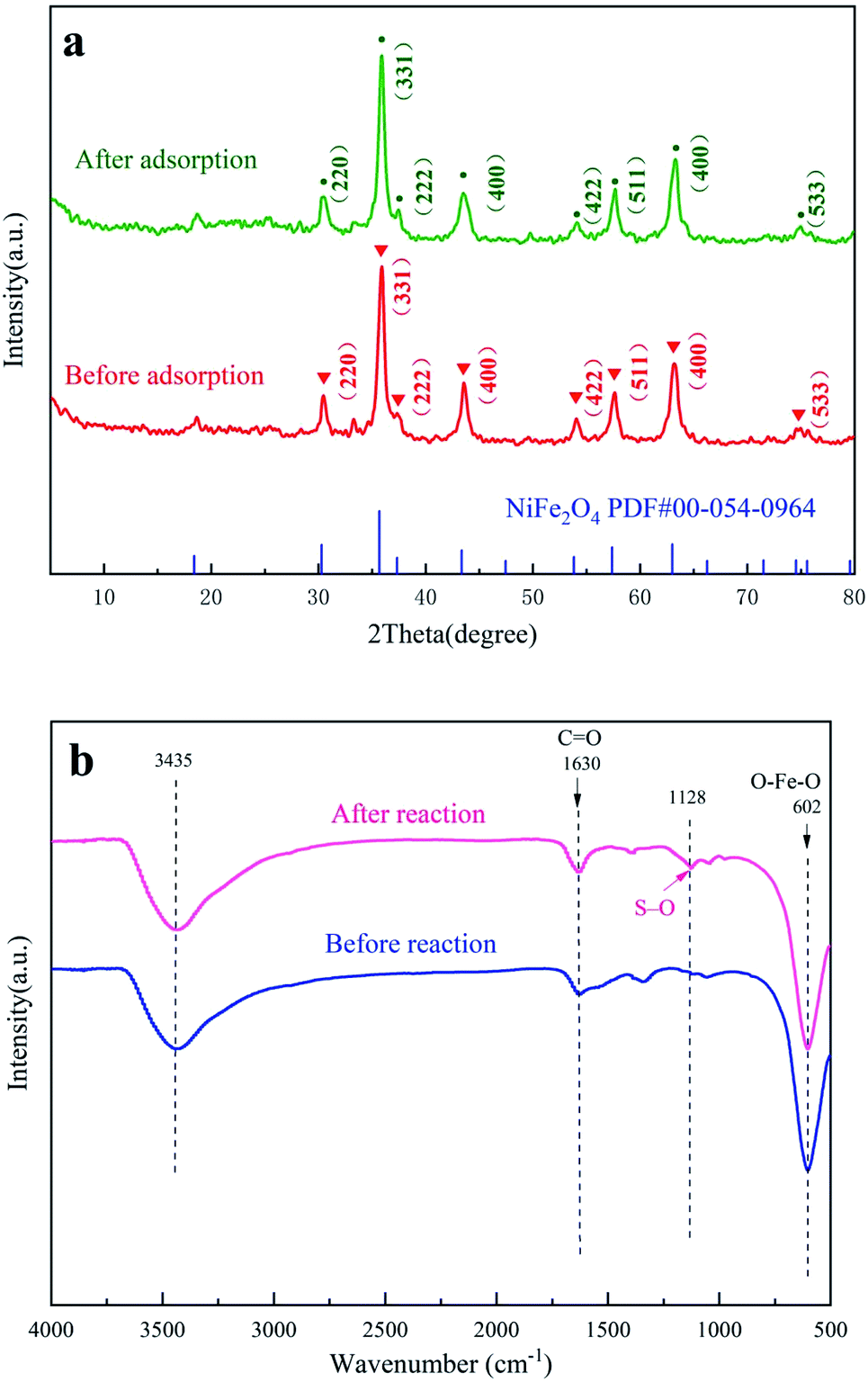
The XRD patterns of nano-NiFe2O4 before and after the reaction are shown in Fig. 2a. By comparing the XRD patterns of nano-NiFe2O4, it can be seen that the nanoparticles did not undergo a phase change after the catalysis treatment.29 The corresponding card is PDF# 00-54-0964. According to the card, the corresponding phase was a cubic phase, the space group was Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (227), and the lattice parameters of the corresponding PDF card were A = B = C = 8.3393 and α = β = γ = 90°. The diffraction peaks of nano-NiFe2O4 show that the structure of NiFe2O4 is a hexagonal crystal structure with a miller index specific at (220), (311), (222), (400), (422), (511), (440) and (533). There is almost no impurities in Fig. 2a, which proves that the sample is relatively pure. As shown in Fig. 2a, the measured XRD was offset to the right, indicating that the lattice parameters of the material were relatively small. By comparing the peak positions before and after treatment, it could be seen that the lattice constant of the material did not change after adsorption, which indicated that the stability of nano-NiFe2O4 was high. Moreover, the peak strength showed that the peak after adsorption was relatively high and sharp, which might be caused by grain agglomeration. Grain agglomeration might lead to a decrease in the adsorption capacity of the material.
m (227), and the lattice parameters of the corresponding PDF card were A = B = C = 8.3393 and α = β = γ = 90°. The diffraction peaks of nano-NiFe2O4 show that the structure of NiFe2O4 is a hexagonal crystal structure with a miller index specific at (220), (311), (222), (400), (422), (511), (440) and (533). There is almost no impurities in Fig. 2a, which proves that the sample is relatively pure. As shown in Fig. 2a, the measured XRD was offset to the right, indicating that the lattice parameters of the material were relatively small. By comparing the peak positions before and after treatment, it could be seen that the lattice constant of the material did not change after adsorption, which indicated that the stability of nano-NiFe2O4 was high. Moreover, the peak strength showed that the peak after adsorption was relatively high and sharp, which might be caused by grain agglomeration. Grain agglomeration might lead to a decrease in the adsorption capacity of the material.
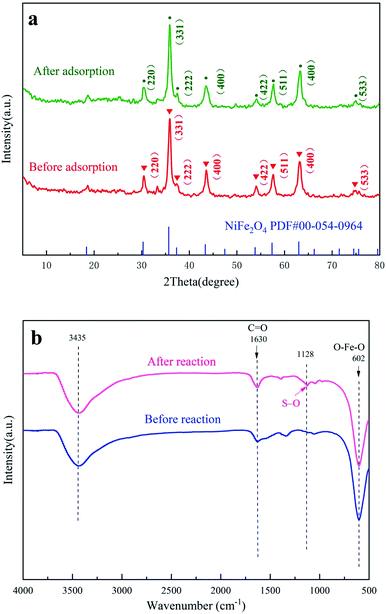 |
| | Fig. 2 XRD patterns of nano-NiFe2O4 (a), FT-IR diagram of nano-NiFe2O4 (b). | |
As shown in Fig. 2b, there were obvious infrared stretching vibration peaks at approximately 602, 1630 and 3435 cm−1, of which 602 cm−1 was the stretching vibration of O–Fe–O, 3435 cm−1 was the stretching vibration peak of hydroxyl-adsorbed water on the surface, and 1630 cm−1 was the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration peak. The stretching vibration of O–Fe–O did not change significantly after the reaction, which indicated that the structure of nano-NiFe2O4 was not destroyed after advanced oxidation. In addition, the nano-NiFe2O4 had an infrared peak at 1128 cm−1 that appeared only after the reaction, which belonged to an S–O vibration. This signal might have been caused by the adsorption of SO42− generated after the activation of persulfate on the surface of nano-NiFe2O4.
O vibration peak. The stretching vibration of O–Fe–O did not change significantly after the reaction, which indicated that the structure of nano-NiFe2O4 was not destroyed after advanced oxidation. In addition, the nano-NiFe2O4 had an infrared peak at 1128 cm−1 that appeared only after the reaction, which belonged to an S–O vibration. This signal might have been caused by the adsorption of SO42− generated after the activation of persulfate on the surface of nano-NiFe2O4.
3.2 Removal efficiency of CTH in different systems
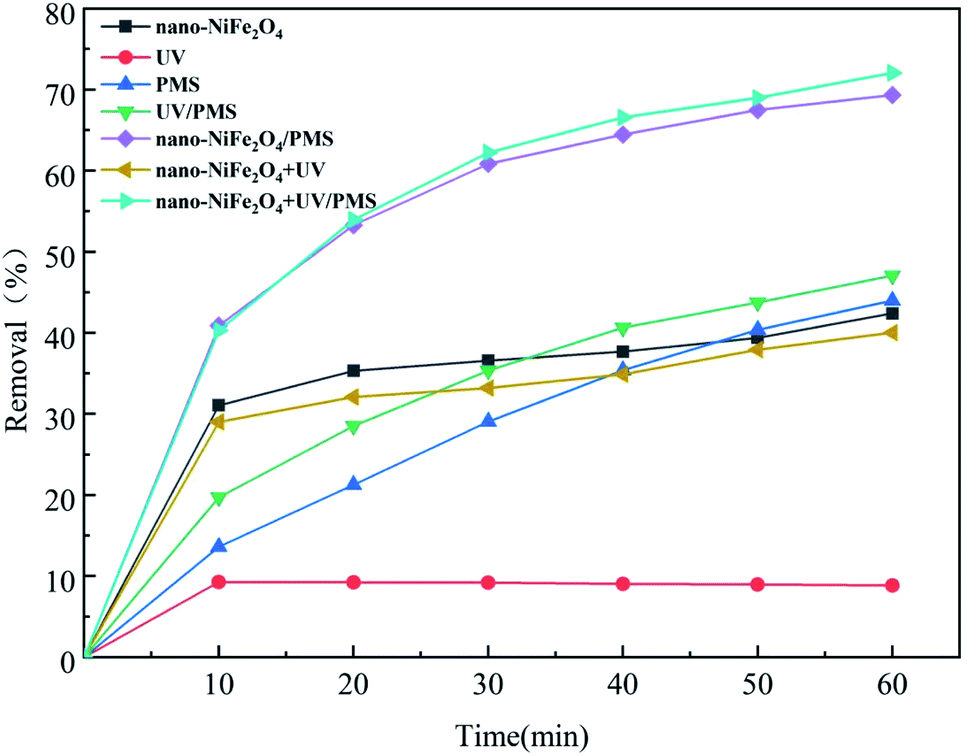
To investigate the removal efficiency of CTH by different systems, experiments were conducted under the following conditions: the dosage of nano-NiFe2O4 was 1 g L−1, the initial concentration of PMS was 1 g L−1, the initial concentration of CTH was 60 mg L−1, pH = 2.7 and the reaction time was 60 min. Fig. 3 depicts the removal efficiency of CTH under different reaction conditions.
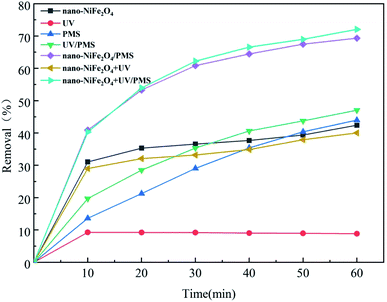 |
| | Fig. 3 Effects of different systems on the removal of CTH; the above experimental basic condition: [nano-NiFe2O4] = 1 g L−1, [CTH] = 60 mg L−1, [PMS] = 1 g L−1, [UV wavelength] = 254 nm, pH = 2.7. | |
This result shows that UV irradiation alone was the least efficient method of removing CTH. This result indicated that CTH did not easily photodegrade in the absence of catalyst, which could be explained by its UV photostability. The removal efficiency of CTH was over 30% after 10 min in both the nano-NiFe2O4 system and UV/nano-NiFe2O4 system; however, after that point, there was no significant change in the removal of CTH in the reaction, which could be explained by the removal of CTH being mainly due to the adsorption of CTH by nano-NiFe2O4.30 When only PMS was present in the system, the removal efficiency reached 43.98% after 60 min. Previous studies have found that CTH cannot be oxidized efficiently with PS used alone.31 By the addition of UV irradiation to activate PMS, the CTH concentration decreased, and the removal efficiency reached 47.07%, still at a low level, which indicated that the efficiency of PMS generated by UV activation alone was limited. In contrast, when nano-NiFe2O4 was added to the PMS system, the removal efficiency reached 69.31%. It was found that the new system could effectively activate PMS and produce SO4−˙, thus rapidly oxidizing and removing CTH. In the UV + nano-NiFe2O4/PMS system, the removal efficiency reached 72.07% after 60 min, which was higher than the removal efficiencies of PMS, nano-NiFe2O4/PMS, UV/PMS and other systems. This result indicated that nano-NiFe2O4 combined with UV irradiation has a good coordination effect; consequently, the efficiency of the combination to activate PMS to produce SO4−˙ could be improved.
3.3 Influences of several parameters on CTH removal in the UV + nano-NiFe2O4/PMS system
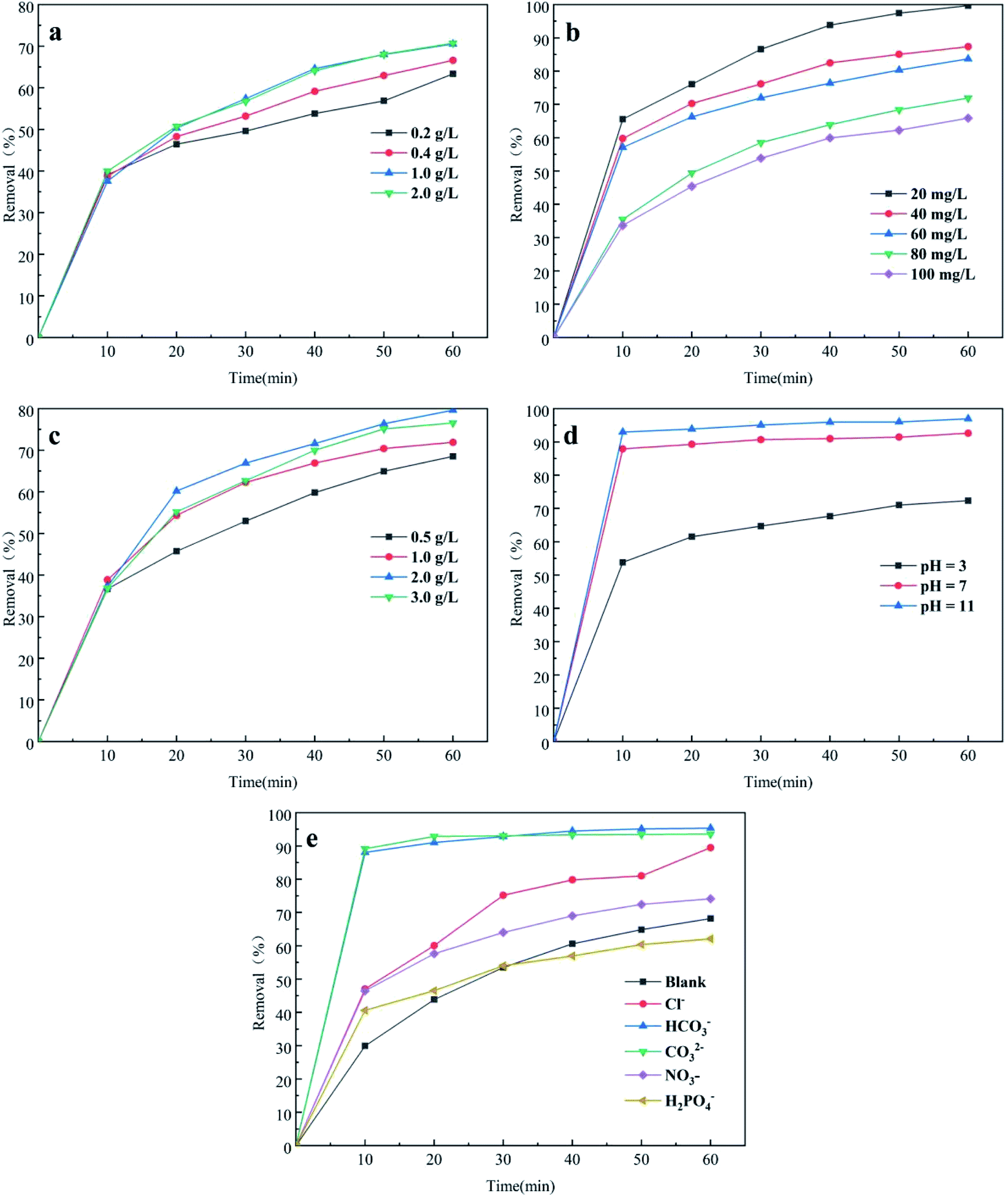
3.3.1 PMS concentration. The removal efficiency of CTH affected by different PMS concentrations (0.2, 0.4, 1.0 and 2.0 g L−1) under initial conditions (dosage of nano-NiFe2O4 was 1 g L−1, concentration of CTH was 60 mg L−1) is illustrated in Fig. 4a. With increasing PMS concentration from 0.2 g L−1 to 1 g L−1, the removal efficiency of CTH gradually increased from 63.35% to 70.73%. The degradation rate was faster at higher initial PMS concentration. It can be explained that the production of free radicals (SO4−˙, HO˙) was in proportion to PMS concentration at a certain temperature. More active radicals (SO4−˙, HO˙) were produced at high PMS concentrations to removal CTH. However, Fig. 4a shows that there was little difference in the CTH removal efficiency when the PMS concentration was 1 g L−1 and 2 g L−1. Thus, increasing the concentration of PMS did not simultaneously increase the speed of removal indefinitely. The reason was that increased concentrations of PMS generated more reactive oxygen species as well as SO4−˙, but PMS could react with SO4−˙ to form radicals with weaker oxidative capacity and lead to self-quenching,32 as shown in eqn (1) and (2). As the PMS concentration increased, the scavenging rate of SO4−˙ might have been faster than the production rate, leading to a decrease in the concentration of SO4−˙, and therefore, the removal of CTH could have become inhibited.33,34| | |
SO4−˙ + SO4−˙ → S2O82−
| (1) |
| | |
SO4−˙ + S2O82− → SO42− + S2O8−˙
| (2) |
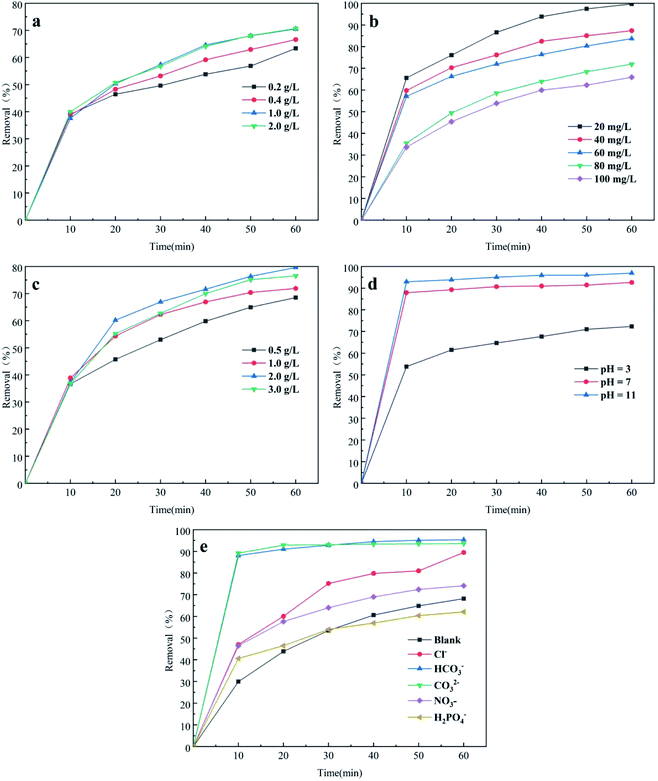 |
| | Fig. 4 Effect of PMS concentrations (a), initial CTH concentration(b), nano-NiFe2O4 dosage (c), initial solution pH (d), co-existing inorganic ions (e) on UV/nano-NiFe2O4 activated PMS system for the removal of CTH; the above experimental basic condition: pH = 2.7. | |
3.3.2 CTH initial concentration. The effect of the CTH concentration on removal was studied, and the results are presented in Fig. 4b. The experiment was conducted under the following conditions: the dosage of nano-NiFe2O4 was 1 g L−1 and the concentration of PMS was 1 g L−1.When the initial concentration was increased from 20 to 100 mg L−1, the removal efficiency of CTH decreased from 99.7% to 66.87%. With the initial conditions unchanged, the removal efficiency of CTH was negatively correlated to its initial concentration. Increasing the concentration of CTH might promote the effective collision between CTH and radicals (SO4−˙, HO˙); nevertheless, the total radical (SO4−˙, HO˙) yields were constant once the dosage of PMS became constant under the corresponding conditions. As a consequence, when the initial CTH concentration was increased, the removal efficiency of CTH decreased during the reaction time.
3.3.3 Nano-NiFe2O4 dosage. The removal efficiency of CTH in the UV + nano-NiFe2O4/PMS system with different nano-NiFe2O4 dosages was studied when the concentration of PMS was 1 g L−1 and the initial concentration of CTH was 60 mg L−1. The results are presented in Fig. 4c. The influence of the nano-NiFe2O4 dosage on CTH removal was not obvious in the first 10 min. Then, increasing the nano-NiFe2O4 dosage from 0.5 to 2 g L−1 led to a faster and more efficient removal of CTH with the efficiency increasing from 68.53% to 79.65%. These results could be explained by the high concentration of SO4−˙ formed under the condition of a higher nano-NiFe2O4 dosage, thereby leading to an increase in the removal efficiency of CTH. Secondly, the observed increase could be attributed to the presence of a large surface area of the adsorbent when the dosage was increased which provided more active binding sites.35 Nevertheless, as the nano-NiFe2O4 dosage was further increased to 2 g L−1, the removal efficiency decreased. An excessive concentration of SO4−˙ formed, enabling these radical ions to react with each other, causing a decrease in the SO4−˙ concentration that led to a decreased removal efficiency of CTH. Excessive Fe2+ release from nano-NiFe2O4 at a relatively high dosage would also consume a large amount of SO4−˙,36–38 as shown in eqn (3). In addition, high levels of nano-NiFe2O4 in solution resulted in an increase in self-aggregation, thus reducing the total number of active sites.| | |
Fe2+ + SO4−˙ → SO42− + Fe3+
| (3) |
3.3.4 Initial solution pH. The process of removing CTH in the UV + nano-NiFe2O4/PMS system was directly influenced by the pH value. In this research, the influence of initial pH was investigated under the following conditions: the initial pH was set to 3.0, 5.0 and 11.0; the dosage of nano-NiFe2O4 was 1.0 g L−1; and the initial CTH concentration was 60 mg L−1 at room temperature. Fig. 4d shows that the removal efficiency of CTH improved with increasing pH. The maximum CTH removal efficiency at pH = 11 was 98%. Previous studies have shown that SO4−˙ yields remain constant under different pH conditions39,40 and oxidation of 1,1,1-trichloroethane by thermally activation persulfate demonstrated that the reaction rate decreased with increasing pH.41 It suggest that the different performances in pH effects could be ascribed to the molecule structure of contaminants and mechanisms of degradation; however, SO4−˙ can react with water and OH− to form OH˙.42 Since the rate constant of the reaction between SO4−˙ and OH− is 5 orders of magnitude greater than that between SO4−˙ and water, alkaline conditions could promote the generation of OH˙ in the system.33 Nevertheless, inhibition experiments43 have shown that SO4−˙ and OH˙ play a major role under weakly basic conditions (pH < 9), but OH˙ and 1O2 are the main active species under strongly basic conditions (pH > 9).44,45 PMS was activated by the base to produce 1O2, which removed CTH [eqn (4)–(14)].46 In addition, the reactivity of 1O2 could be enhanced due to the smaller size of nano-NiFe2O4;47 these trends explain why the removal efficiency of CTH is increased under alkaline conditions.| | |
HSO5− + H2O → HSO4− + H2O2
| (5) |
| | |
SO52− + H2O → SO42− + H2O2
| (6) |
| | |
HSO5− + HO2− → H2O + SO4−˙ + O2−˙
| (8) |
| | |
H2O2 + OH˙ → HO2˙ + H2O
| (10) |
| | |
OH˙ + O2−˙ → 1O2 + OH−
| (12) |
| | |
2O2−˙ + 2H+ → 1O2 + H2O2
| (13) |
| | |
HSO5− + SO52− → HSO4− + SO42− + 1O2
| (14) |
3.3.5 Co-existing inorganic ions. Cl−, HCO3−, CO32−, NO3−, and H2PO4− are widely present in various natural waters and wastewaters. These five ions were added to the system at a concentration of 50 mM and their effects on CTH removal in the UV + nano-NiFe2O4/PMS system under other fixed experimental conditions are shown in Fig. 4e. Three anions, Cl−, HCO3− and CO32−, noticeably increased the removal efficiency of CTH. Compared with the system without any anion addition, the UV + nano-NiFe2O4/PMS system with Cl−, HCO3−, and CO32− added at the end of the reaction increased the removal efficiency by 21.29%, 27.17% and 25.32%, respectively. However, the removal process of CTH differed in the presence of those three different ions. The promotion of the CTH removal efficiency by HCO3− and CO32− was immediately apparent at the beginning 10 min of the reaction, while the influence of Cl− was gradually revealed as the reaction proceeded and was not initially obvious. It is well known that the OH˙ or H+ species can be consumed by CO32− or HCO3− to produce CO3−˙, which is much more selective than OH˙ for the degradation of organic species such as phenols, anilines, or amino acids [eqn (15)–(17)]. In this context, SO4−˙, CO3−˙, 1O2 work together to improved removal efficiency of CTH.48 On the other hand, HCO3− and CO32− underwent hydrolysis in water, increasing the pH of the solution; hence, this process could promote the activation of PMS and improve the removal efficiency of CTH.49 In addition, PMS could oxidize HCO3− to produce a two-electron oxide HCO4− [eqn (18) and (19)], which could promote CTH removal.50,51 Cl− reacted with HSO5− to produce strongly oxidizing HClO, which was further ionized to obtain ClO−, thereby enhancing the removal of CTH [eqn (20)–(23)].52,53 Fig. 4e shows that the removal efficiency of CTH increased slightly with the addition of NO3− Previous studies have shown that NO3− can react with OH˙ and SO4−˙ to generate NO3˙ [eqn (24) and (25)]. The propagation reaction of NO3˙ generates secondary radicals such as NO˙, which then reacts with contaminants.54,55 The addition of phosphate species (H2PO4−) inhibited CTH removal. Compared with the system without any addition, the removal efficiency of CTH decreased by 6.08% after 60 min. Iron ions can rapidly form different complexes with H2PO4− and decrease the available soluble Fe2+ for PMS activation.56 In addition, H2PO4− is ionized in water to produce H+, leading to a decrease in pH. According to previous experiments, the removal efficiency of CTH was low under acidic conditions.| | |
OH˙ + HCO3− → H2O + CO3−˙
| (15) |
| | |
OH˙ + CO32− → OH− + CO3−˙
| (16) |
| | |
OH− + HSO5− → SO52− + H2O
| (18) |
| | |
SO52− + HCO3− → HCO4− + SO42−
| (19) |
| | |
HSO5− + Cl− → SO42− + HClO
| (20) |
| | |
HSO5− + 2Cl− + H+ → SO42− + Cl2 + H2O
| (21) |
| | |
Cl2(aq) + H2O → HClO + H+ + Cl−
| (22) |
| | |
NO3− + OH˙ → NO3˙ + OH−
| (24) |
| | |
NO3− + SO4−˙ → NO3˙ + SO42−
| (25) |
3.4 Identification and generation mechanism of active species
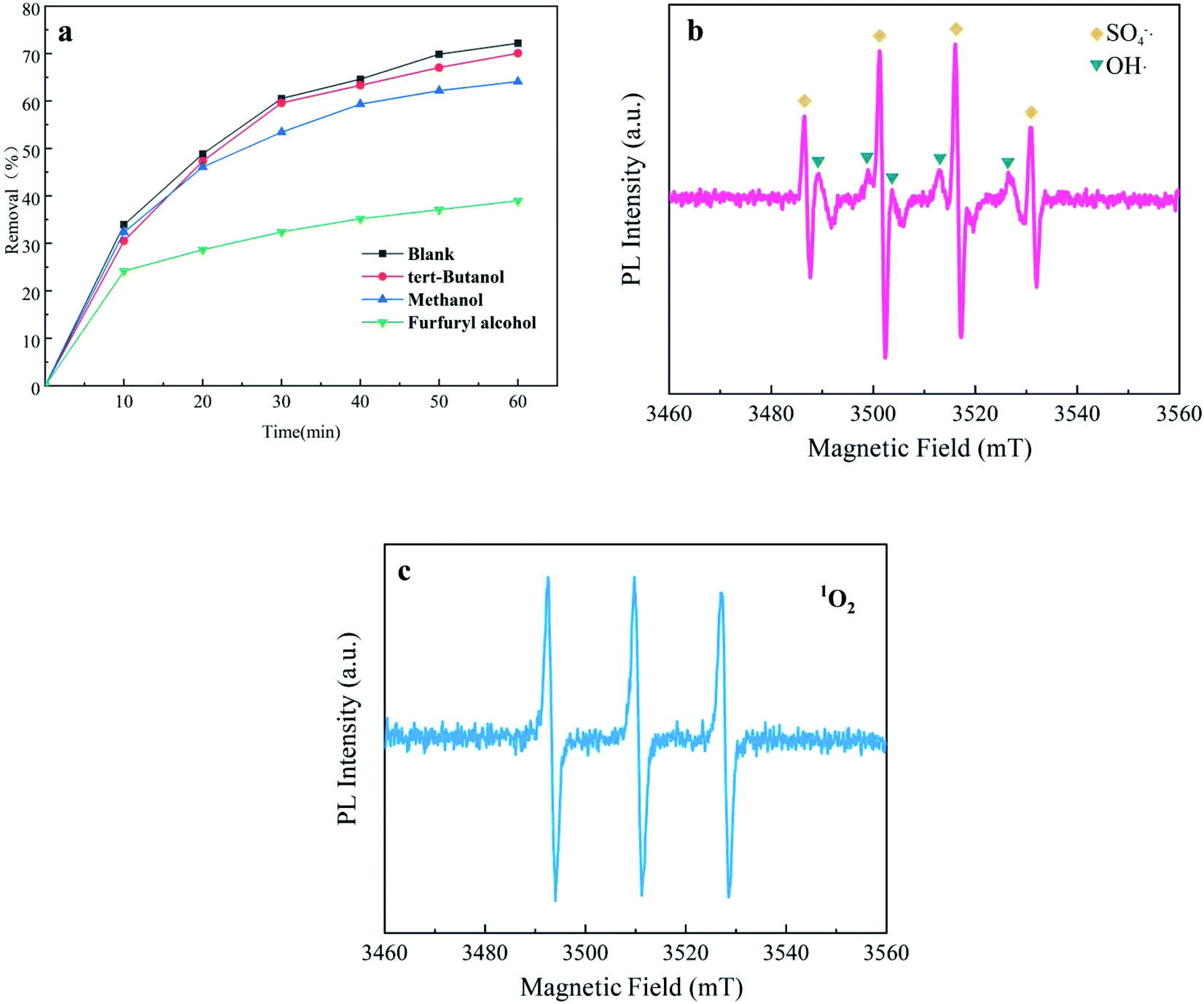
3.4.1 Identification of active species in systems. An active species quenching test was carried out to identify important free radicals and explore their reaction mechanism in the UV + nano-NiFe2O4/PMS system. First, 50 mM methanol (MeOH) was used to capture SO4−˙ and OH˙,57 50 mM tert-Butanol (TBA) was used to quench OH˙ (ref. 58) and 50 mM furfuryl alcohol was employed to capture 1O2. As presented in Fig. 5a, in the UV + nano-NiFe2O4/PMS systems with TBA and MeOH added, the removal efficiency of CTH was slightly reduced that respectively decreased from 72.0% to 70.01% and from 72.0% to 64.11%, proving that OH˙ and SO4−˙ played a smaller role. However, compared with that in the system without any addition, the removal efficiency of CTH decreased from 72.0% to 38.9% after adding furfuryl alcohol into the system, indicating that 1O2 greatly contributed to the CTH degradation process. To speculate on the mechanism of PMS activation by the UV + nano-NiFe2O4/PMS system, an EPR test was performed. EPR spectroscopy coupled with DMPO and TEMP as spin-trapping agents was applied to identify the free radicals generated in the system. The results of the EPR study are presented in Fig. 5b and c. It is obvious that characteristic signals are present in the EPR spectra of experiments conducted with DMPO and TEMP. DMPO–SO4 and DMPO–OH signals were found after PMS activation by UV + nano-NiFe2O4 (Fig. 5b). More importantly, a strong characteristic triplet signal of TEMP-1O2 was observed (Fig. 5c), which further confirmed the presence of 1O2 in the UV + nano-NiFe2O4/PMS oxidation system. The EPR results were consistent with those of the active species quenching test.
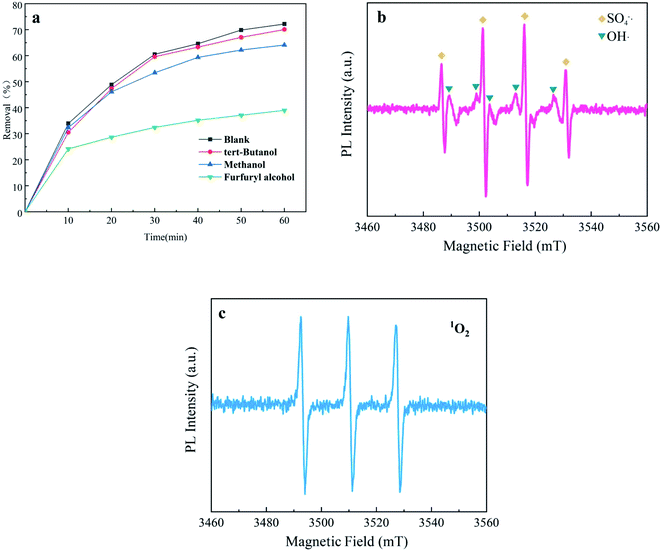 |
| | Fig. 5 Identification of radicals by different quenching agents (a); EPR spectra of OH˙, SO4−˙ (b) and 1O2 (c) formed after 60 min in the UV + nano-NiFe2O4/PMS system; the above experimental condition: [nano-NiFe2O4] = 1 g L−1, [CTH] = 60 mg L−1, [PMS] = 1 g L−1, [UV wavelength] = 254 nm, pH = 2.7. | |
3.4.2 Generation mechanism of active species. On the basis of the above analysis, a possible mechanism for free radical generation over the UV + nano-NiFe2O4/PMS system was proposed. The Ni2+ and Fe3+ ions on the surface of the catalyst could directly interact with S2O82− to produce SO4−˙ [eqn (26)–(29)]. Some of the dissolved Ni2+ could interact with water to produce the intermediate NiOH+, which could promote the conversion of PMS to SO4−˙ [eqn (30) and (31)]; SO4−˙ reacted with H2O to generate a large amount of OH˙ [eqn (32)]; OH˙ reacted with SO4−˙ to produce HSO5−; and HSO5− reacted with OH˙ and SO4−˙ to form SO5−˙. At the same time, the Ni3+ on the surface of the catalyst and in the aqueous solution could react with HSO5− to form Ni2+ and SO5−˙ [eqn (33)–(36)], and SO5−˙ in the solution could also produce the same amount of SO4−˙ [eqn (37)]. Then, a large amount of 1O2 could be produced under the action of HSO5− and SO52− and SO5−˙ and water [eqn (38) and (39)]. Finally, under the action of SO4−˙, OH˙ and 1O2, CTH was degraded and mineralized into CO2 and H2O and thereby completely degraded [eqn (40)]. The proposed mechanism of PMS activation and CTH degradation is shown in eqn (26)–(40) as follows:| |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe3+ + S2O82− → Fe2+ + S2O8−˙ Fe3+ + S2O82− → Fe2+ + S2O8−˙
| (26) |
| | |
Fe2+ + S2O82− → Fe3+ + SO4−˙ + SO42−
| (27) |
| | |
Ni2+ + S2O82− → Ni3+ + SO4−˙ + SO42−
| (28) |
| | |
Ni3+ + S2O82− → Ni2+ + S2O8−˙
| (29) |
| | |
Ni2+ + H2O → NiOH+ + H+
| (30) |
| | |
NiOH+ + HSO5− → NiOH2+ + SO4−˙ + OH−
| (31) |
| | |
SO4−˙ + H2O → OH˙ + SO42− + H+
| (32) |
| | |
HSO5− + SO4−˙ → SO5−˙ + SO42− + H+
| (34) |
| | |
HSO5− + OH˙ → SO5−˙ + H2O
| (35) |
| | |
Ni3+ + HSO5− → Ni2+ + SO5−˙ + H+
| (36) |
| | |
HSO5− + SO52− → SO42− + HSO4− + 1O2
| (38) |
| | |
2SO5−˙ + H2O → 2HSO4− + 1.51O2
| (39) |
| | |
SO4−˙ + OH˙ + 1O2 + CTH → CO2 + H2O
| (40) |
3.5 Degradation mechanism of CTH in the UV + nano-NiFe2O4/PMS system
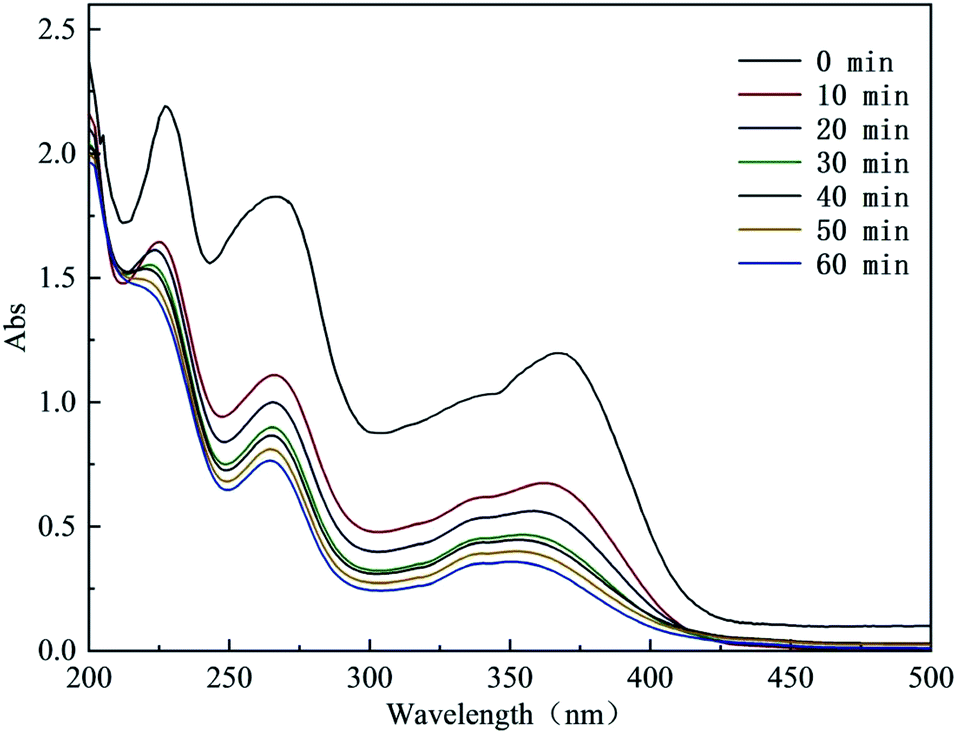
3.5.1 UV-vis spectral analysis in the degradation process of CTH. The UV-vis spectrum of the degradation process of CTH is shown in Fig. 6. CTH contains a conjugate composed of a benzene ring, ketone group and enol system, and its structure has 2 chromophores. At the beginning of the reaction, there were 3 obvious absorption bands at approximately 229 nm, 269 nm and 366 nm. These bands corresponded to 4 different ring structures in the molecular structure. After the reaction started, the intensities of these characteristic peaks continued to decrease. This decrease showed that the degradation of CTH was a gradual oxidative degradation process. In particular, the absorption peak at 366 nm showed a certain degree of blueshift after 30 min of reaction. This blueshift indicated that the benzene ring structure of CTH was progressively destroyed. This damage first occurred at the unsaturated bond position of the benzene ring, and then, the benzene ring generated a series of small-molecule intermediate products through ring opening and oxidation.31
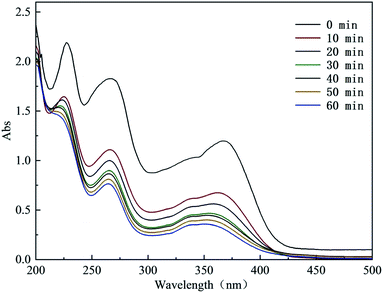 |
| | Fig. 6 UV-visible spectrum in the degradation process of CTH. | |
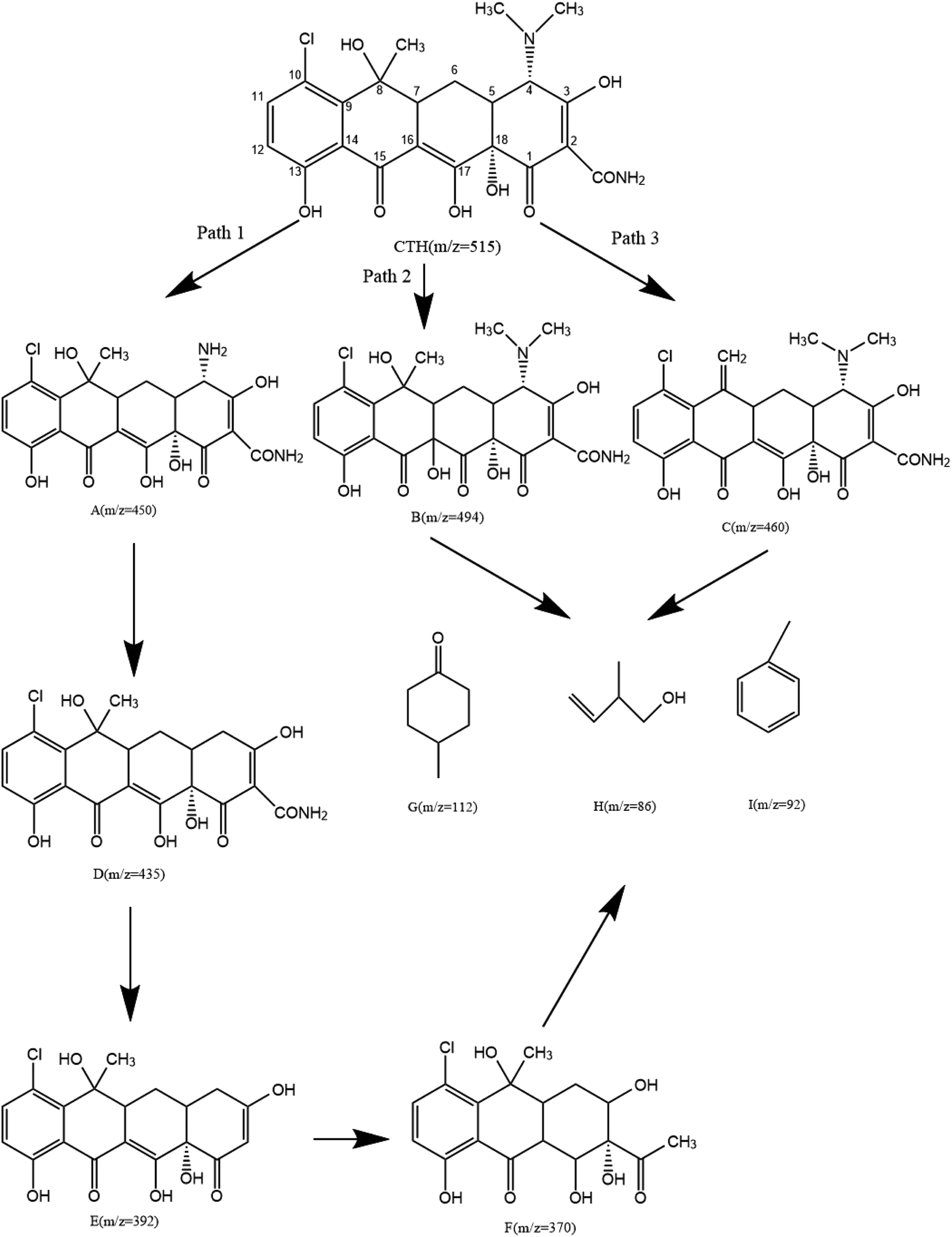
3.5.2 Degradation products and degradation pathway. The possible intermediate products formed during the CTH degradation process in the UV + nano-NiFe2O4/PMS system were determined by LC-MS. As shown in Fig. 7, nine reactive intermediates were possibly observed, and the structures are listed; the transformation pathway was proposed accordingly. In transformation pathway 1, first, the two methyl groups on the N atom were removed to form product A (m/z = 450), and then, C4 underwent deamination to produce product D (m/z = 435), after which C2 underwent deamination to produce product E (m/z = 392) and was finally oxidized to open the ring to produce product F (m/z = 370). The methyl group in the dimethylamino moiety was eliminated, which might be due to direct UV photolysis59 or OH˙ and SO4−˙ induced hydrogen atom extraction from the methyl substituent.60 The hydroxylation of transformation pathway 2 is a common reaction route in the SO4−˙ reaction mechanism that produces product B (m/z = 494). SO4−˙ has a similar reaction mechanism to HO˙, namely, (i) hydrogen abstraction; (ii) addition of hydroxyl groups to unsaturated carbons including double bonds and aromatic rings; and (iii) electron transfer.61,62 The unsaturated double bond of CTH was attacked to form a hydroxylate species. The C6 and C8 sites were prone to undergo dehydration reactions;60 hence, dehydration occurred and produced product C (m/z = 460) in transformation pathway 3. Eventually, CTH was degraded into structurally simple products [product G (m/z = 112), product H (m/z = 86), and product I (m/z = 92)] and was ultimately mineralized into CO2 and H2O.
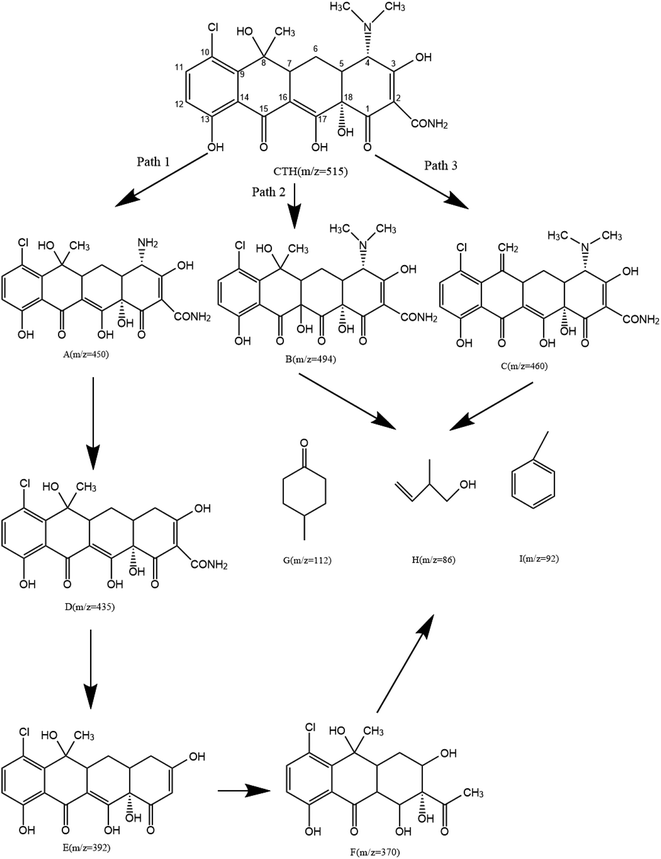 |
| | Fig. 7 The degradation pathway of CTH in the UV + nano-NiFe2O4/PMS system. | |
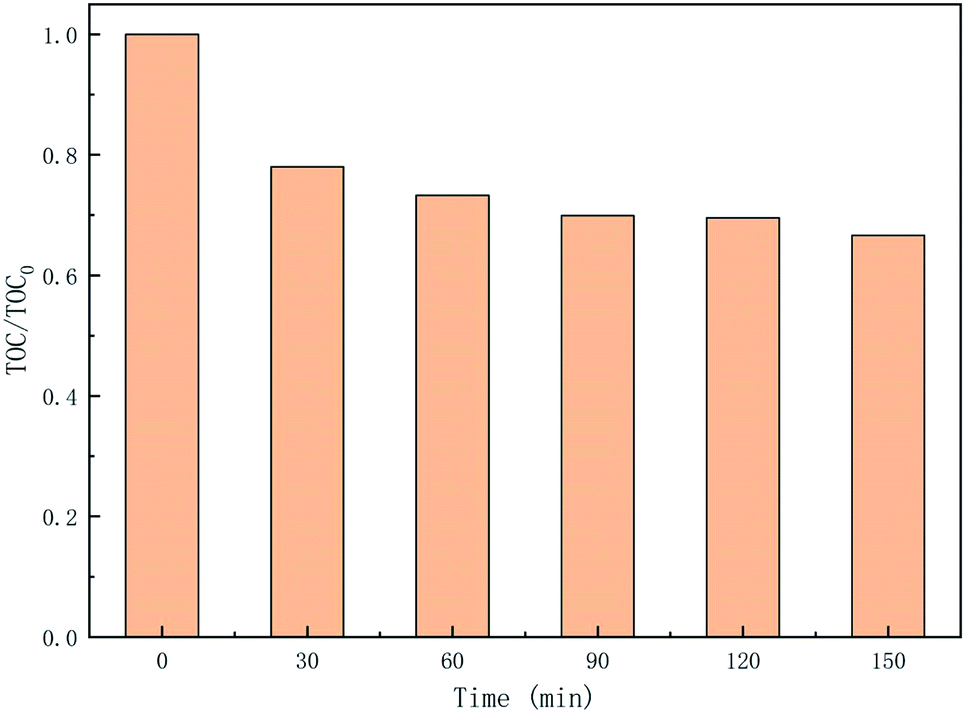
3.5.3 TOC removal in the degradation process of CTH. As an indicator of water quality, the mineralization test is an important assessment to obtain information on the degradation of the TOC.63 To explore the mineralization of CTH in the UV + nano-NiFe2O4/PMS system, the TOC was monitored from 0 to 150 min, and the results are shown in Fig. 8. As the reaction progressed, the TOC in the system showed a slow downward trend, decreasing by 22% after 30 min, and then, the TOC ultimately tended to stabilize, which may be caused by insufficient oxidant in the system. With time, the mineralization rate of TOC reached 33%, indicating that the UV + nano-NiFe2O4/PMS system could degrade CTH to generate CO2 to some extent under the conditions employed for testing the performance of this process. While the TOC decreased and reached stability, CTH experienced three possible degradation pathways, namely, (i) demethylation and deamination; (ii) hydroxylation; and (iii) dehydration. Finally, CTH was oxidized to open the ring to produce simple products, some of which were mineralized into CO2 and H2O. However, only part of the CTH was completely mineralized, and the rest have been converted into other products that were more difficult to degrade than CTH.
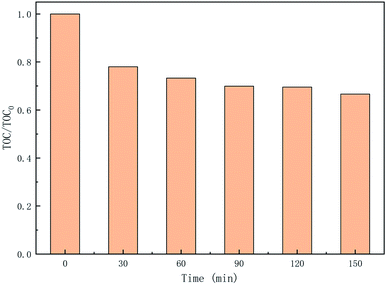 |
| | Fig. 8 TOC removal in the UV + nano-NiFe2O4/PMS system; the experimental condition: [nano-NiFe2O4] = 1 g L−1, [CTH] = 60 mg L−1, [PMS] = 1 g L−1, [UV wavelength] = 254 nm, pH = 2.7. | |
4. Conclusions
In summary, we successfully synthesized nano-NiFe2O4 by via a hydrothermal synthesis method and demonstrated that nano-NiFe2O4 as a catalyst combined with UV irradiation activation PMS can effectively degrade CTH. The following were our main conclusions:
(1) The SEM, XRD and FTIR results confirmed that nano-NiFe2O4 consisted of loose nano-microstructure particles and exhibited high stability.
(2) The heterogeneous system of UV + nano-NiFe2O4/PMS had a better effect on the removal of CTH than other parallel systems. By exploring the factors affecting the reaction process, it was found that the optimum dosages of nano-NiFe2O4 (1 g L−1), PMS (1 g L−1), CTH (60 mg L−1) and pH (11) at UV wavelength of 254 nm afforded a high removal efficiency of approximately 96%. The five anions coexisting in the solution, Cl−, HCO3−, CO32− and NO3−, could increase CTH degradation by 21.29%, 27.17%, 25.32% and 5.96% respectively, while H2PO4− could inhibit CTH degradation and the removal efficiency decreased 6.08%.
(3) The active species quenching tests and EPR analysis proved that 1O2 contributed greatly to the CTH removal process and that OH˙ and SO4−˙ had a small impact on the CTH removal efficiency.
(4) Nine different byproducts were identified via LC-MS. The degradation of CTH was mainly caused by hydroxylation, demethylation, deamination and dehydration. This study showed that only 33% of TOC was mineralized, proving that only part of the CTH was completely mineralized and that the rest might have been converted into other products that were more difficult to degrade than CTH.
This work systematically investigated the effect of different reaction parameters on the degradation of CTH and provided a comprehensive description of the degradation of CTH in the UV + nano-NiFe2O4/PMS system. Therefore, our results provide a theoretical basis for the development of large-scale degradation of CTH. Nevertheless, to apply these processes, not only should the degradation efficiency be considered but also the overall investment, operation, and maintenance costs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Undergraduate Training Program of Innovation and Entrepreneurship (202010225090), and the Heilongjiang Provincial Natural Science Foundation of China (LH2019D002).
References
- V. K. Gupta, I. Ali, T. A. Saleh, A. Nayaka and S. Agarwalc, RSC Adv., 2012, 2, 6380–6388 RSC.
- A. A. Ahmed, T. A. Saleh, A. M. Ajeebi, M. S. A. Gharsan and N. F. Aldahmash, Biotechnol. Prog., 2019, 36, e2907 Search PubMed.
- A. A. Alswat, M. B. Ahmad and T. A. Saleh, Colloid Interface Sci., 2017, 16, 19–24 CrossRef CAS.
- V. K. Gupta, D. Pathania, M. Asif and G. Sharma, J. Mol. Liq., 2014, 196, 107–112 CrossRef CAS.
- Y. J. Tian, J. R. Zou, F. Li, L. Q. Zhang and Y. Z. Liu, Water Res., 2019, 149, 35–41 CrossRef CAS PubMed.
- H. Wang, Y. Wu, M. B. Feng, W. G. Tu and T. Xiao, Water Res., 2018, 144, 215–225 CrossRef CAS PubMed.
- R. Pulicharla, R. K. Das, S. K. Brar, P. Drogui and R. Y. Surampalli, Chem. Eng. J., 2018, 347, 828–835 CrossRef CAS.
- B. Liu, Y. X. Li, X. L. Zhang, J. Wang and M. Gao, Eur. J. Soil Biol., 2015, 68, 69–76 CrossRef CAS.
- R. X. Guo and J. Q. Chen, Chemosphere, 2012, 87, 1254–1259 CrossRef CAS PubMed.
- S. C. Anderson, S. Subbiah, A. Gentles, D. Oliver, P. Stonum, T. A. Brooks and E. E. Smith, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1092, 237–243 CrossRef CAS PubMed.
- X. H. Peng, J. R. Cao, B. L. Xie, M. S. Duan and J. C. Zhao, Ecotoxicol. Environ. Saf., 2020, 188, 109869 CrossRef CAS PubMed.
- J. L. Zhang and Z. Ma, J. Chem. Eng., 2018, 26, 753–760 CAS.
- Y. Sun, X. J. Wang, S. Q. Xia and J. F. Zhao, Chem. Eng. J., 2021, 416, 129085 CrossRef CAS.
- Y. T. Lin, C. Liang and C. W. Yu, Ind. Eng. Chem. Res., 2016, 55, 2302–2308 CrossRef CAS.
- T. A. Saleha, S. Agarwal and V. K. Guptaa, Appl. Catal., B, 2011, 106, 46–53 Search PubMed.
- S. Karthikeyan, V. K. Gupta, R. Boopathy, A. Titus and G. Sekaran, J. Mol. Liq., 2012, 173, 153–163 CrossRef CAS.
- M. Qamar, M. O. Fawakhiry, A. M. Azad, M. I. Ahmed, A. Khana and T. A. Saleh, RSC Adv., 2016, 6, 71108–71116 RSC.
- Y. H. Lee and V. G. Urs, Water Res., 2010, 44, 555–566 CrossRef CAS PubMed.
- C. j. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1213–1223 CrossRef CAS PubMed.
- B. Muhammad, A. Muhammad, R. Tahir and Y. Q. Zha, Environ. Int., 2019, 124, 336–353 CrossRef PubMed.
- S. Y. Oh, H. W. Kim, J. M. Park, H. S. Park and C. Yoon, J. Hazard. Mater., 2009, 168, 346–351 CrossRef CAS PubMed.
- J. M. Monteagudo, A. Durán, R. González and A. J. Expósito, Appl. Catal., B, 2015, 176, 120–129 CrossRef.
- B. Rishikesh and S. W. Jeong, Chemosphere, 2021, 270, 128627 CrossRef PubMed.
- M. J. Xu, H. Y. Zhou, Z. L. Wu, N. W. Li, Z. K. Xiong, G. Yao and B. Lai, J. Hazard. Mater., 2020, 399, 123103 CrossRef CAS PubMed.
- A. O. Salawudeen, B. S. Tawabini, A. M. Al-Shaibani and T. A. Saleh, Environ. Nanotechnol. Monit. Manage., 2020, 13, 100288 Search PubMed.
- T. A. Saleh, Appl. Surf. Sci., 2011, 257, 7746–7751 CrossRef CAS.
- T. A. Saleh, J. Cleaner Prod., 2018, 172, 2123–2132 CrossRef CAS.
- T. Prabhakaran and J. Hemalatha, Ceram. Int., 2014, 40, 3315–3324 CrossRef CAS.
- G. Fadillah, W. P. Wicaksono and T. A. Saleh, Microchem. J., 2020, 159, 105353 CrossRef CAS.
- Y. M. Lee, G. Lee and K. Duk, J. Hazard. Mater., 2021, 403, 123591 CrossRef CAS PubMed.
- H. X. Huang, T. Guo, K. Wang, Y. Li and G. K. Zhang, Sci. Total Environ., 2021, 758, 143957 CrossRef CAS PubMed.
- J. He, J. C. Tang, Z. Zhang, L. Wang, Q. L. Liu and X. M. Liu, Chem. Eng. J., 2021, 404, 126997 CrossRef CAS.
- B. Y. Yan, D. C. Xu, Z. Q. Liu, J. Z. Tang, R. Huang, M. Zhang, F. Y. Cui, W. X. Shi and C. Hu, Chem. Eng. J., 2021, 404, 127041 CrossRef CAS.
- V. C. Mora, J. A. Rosso, G. C. L. Roux, D. O. Mártire and M. C. Gonzalez, Chemosphere, 2009, 75, 1405–1409 CrossRef CAS PubMed.
- A. T. Pereira, P. C. Henriques, P. C. Costa, M. C. L. Martins, F. D. Magalhães and I. C. Gonçalves, Compos. Sci. Technol., 2019, 184, 107819 CrossRef CAS.
- P. He, J. Y. Zhu, Y. Z. Chen, F. Chen, Z. L. Zhu, M. F. Liu, K. Zhang and M. Gan, Chem. Eng. J., 2021, 406, 126758 CrossRef CAS.
- C. K. O. d. Silva-Rackov, W. A. Lawal, P. A. Nfodzo, M. M. G. R. Vianna, C. A. O. d. Nascimento and H. Choi, Appl. Catal., B, 2016, 192, 253–259 CrossRef.
- C. Q. Gao, W. Yu, Y. C. Zhu, M. Wang, Z. Z. Tang, L. Du, M. Y. Hu, L. Fang and X. C. Xiao, Sci. Total Environ., 2021, 754, 142131 CrossRef CAS PubMed.
- A. Ghauch, A. Baalbaki, M. Amasha, R. E. Asmar and O. Tantawi, Chem. Eng. J., 2017, 317, 1012–1025 CrossRef CAS.
- C. S. D. Rodrigues and L. M. Madeira, Environ. Technol. Innovation, 2021, 21, 101265 CrossRef CAS.
- X. Gu, S. Lu, L. Li, Z. Qiu, Q. Sui, K. Lin and Q. Luo, Ind. Eng. Chem. Res., 2011, 50, 11029–11036 CrossRef CAS.
- H. P. Gao, J. B. Chen, Y. L. Zhang and X. F. Zhou, Chem. Eng. J., 2016, 306, 522–530 CrossRef CAS.
- Z. Wang, G. H. Chen, X. R. Wang, G. H. Yang, Y. Liu and C. Zhang, J. Hazard. Mater., 2021, 401, 123378 CrossRef CAS PubMed.
- C. M. Dominguez, V. Rodriguez, E. Montero, A. Romero and A. Santos, Sep. Purif. Technol., 2020, 241, 116679 CrossRef CAS.
- P. Xiao, L. An and D. Wu, New Carbon Mater., 2020, 35, 667–683 CrossRef.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- A. Ahmad, X. G. Gu, L. Li, S. G. Lv, Y. S. Xu and X. H. Guo, Environ. Sci. Pollut. Res., 2015, 22, 17876–17885 CrossRef CAS PubMed.
- J. Q. Wang, Q. R. Qian, Q. H. Chen, X. P. Liu, Y. J. Luo, H. Xue and Z. H. Li, Chin. J. Catal., 2020, 41, 1511–1521 CrossRef CAS.
- M. H. Nie, Y. Yang, Z. J. Zhang, C. X. Yan, X. N. Wang, H. J. Li and W. B. Dong, Chem. Eng. J., 2014, 246, 373–382 CrossRef CAS.
- G. D. Fang, D. D. Dionysiou, D. M. Zhou, Y. Wang, X. D. Zhu, J. X. Fan, L. Cang and Y. J. Wang, Chemosphere, 2013, 90, 1573–1580 CrossRef CAS PubMed.
- F. Aqsa, S. Karunamoorthy, K. Talukdar, Y. Kim, Y. Yoon and C. M. Park, Chem. Eng. J., 2021, 407, 127842 CrossRef.
- T. Zhang, H. B. Zhu and C. J. Philippe, Environ. Sci. Technol., 2013, 47, 2784–2791 CrossRef CAS PubMed.
- H. Chen and K. C. Carroll, Environ. Pollut., 2016, 215, 96–102 CrossRef CAS PubMed.
- P. Neta, V. Madhavan, H. Zemel and R. W. Fessenden, J. Am. Chem. Soc., 1977, 99, 163–164 CrossRef CAS.
- S. Padmaja, Z. Alfassi, P. Neta and R. Huie, Int. J. Chem. Kinet., 1993, 25, 193–198 CrossRef CAS.
- M. H. Nie, C. X. Yan, M. Li, X. N. Wang, W. L. Bi and W. B. Dong, Chem. Eng. J., 2015, 279, 507–515 CrossRef CAS.
- P. Neta, J. Grodkowski and A. B. Ross, ChemInform, 1996, 27, 1027–1284 Search PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- F. Yuan, C. Hu, X. X. Hu, D. B. Wei, Y. Chen and J. H. Qu, J. Hazard. Mater., 2011, 185, 1256–1263 CrossRef CAS PubMed.
- J. P. Wang, M. Zhang, R. J. Zhou, J. Y. Li, W. Zhao and W. Y. Chen, J. Environ. Chem. Eng., 2021, 9, 104907 CrossRef CAS.
- T. C. Liu, K. Yin, C. B. Liu, J. M. Luo, C. John, W. Q. Zhang, S. L. Luo, Q. Y. He, Y. X. Deng, H. Liu and D. Y. Zhang, Water Res., 2018, 147, 204–213 CrossRef CAS PubMed.
- Y. Q. Liu, X. X. He, Y. S. Fu and D. D. Dionysiou, J. Hazard. Mater., 2016, 305, 229–239 CrossRef CAS PubMed.
- C. Y. Shao, R. B. Zhao, S. Q. Jiang, S. S. Yao, Z. F. Wu, B. Jin, Y. L. Yang, H. H. Pan and R. K. Tang, Adv. Mater., 2018, 30, 1704867 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*