
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of core/shell organic–inorganic hybrid polymer nanoparticles and their application to toughening poly(methyl methacrylate)
Jia Xia,
Xia Luo,
Jin Huang,
Jiajun Ma * and
Junxiao Yang*
* and
Junxiao Yang*
State Key Laboratory of Environmental-friendly Energy Materials, School of Material Science and Engineering, Southwest University of Science and Technology, Mianyang 621010, China
First published on 20th October 2021
Abstract
On account of the utility of poly(methyl methacrylate) (PMMA) as a glass substitute, toughening of PMMA has attracted significant attention. Brittle failure can often be avoided by incorporating a small fraction of filler particles. Core–shell composite particles composed of a rubbery core and a glassy shell have recently attracted interest as a toughening agent for brittle polymers. Here, core/shell organic–inorganic hybrid polymer nanoparticles (Si-ASA HPNs) with a silicone-modified butyl acrylate copolymer (PBA) core and a styrene-acrylonitrile copolymer (SAN) shell were used to toughen PMMA. Silicone plays dual roles as a compatibilizer and a chain extender, and it not only improves interfacial adhesion between the PBA particles and SAN copolymer, but it also increases chain entanglement of poly(acrylonitrile-styrene-acrylate) (ASA). The mechanical properties of the PMMA/ASA alloys strongly depend on the Si content, and the impact strength and elongation at break greatly improve when silicone-modified ASA is added. However, this is accompanied by loss of rigidity. Specifically, the PMMA/ASA-2 composite exhibits a good balance between toughness and rigidity, indicating that ASA-2 with 5 wt% KH570 is the most suitable impact modifier. This research provides a facile and practical method to overcome the shortcomings of ASA and promote its application in a wider range of fields.
Introduction
Poly(methyl methacrylate) (PMMA) is a thermoplastic polymer with good optical properties, transparency, and chemical resistance, and it can be used as an alternative to inorganic glass. By virtue of its hardness, low weight and color versatility, PMMA can be used in a wide range of applications, such as in optical materials, sensors, the automobile field, solar cells, electronics, display units, biomedical devices, pneumatic actuation, and other fields.1 Nevertheless, PMMA is a hard and brittle plastic material with low mechanical strength, and its inherent brittleness means that it fails to meet the high requirements of certain applications as an engineering plastic.2 Therefore, the research on the improving of its toughness is of great significance.To improve the toughness of PMMA, many researchers have modified PMMA by copolymerization with suitable monomers and blending with second-phase particles. Brittle failure can often be avoided by incorporation of a small fraction of filler particles. Inorganic materials, such as alumina and silica, have been shown to significantly increase the toughness of PMMA.3,4 However, inorganic nanoparticles easily agglomerate, which makes it difficult to disperse them in polymers, and serious phase separation often occurs between the inorganic nanoparticles and the polymer. Therefore, the surface of the inorganic nanoparticles must be modified, which increases the number of experimental steps and difficulty. Anisotropic fillers, such as carbon nanotubes and nanofibers, have also been exploited to significantly augment the mechanical properties of PMMA.5 However, care must be taken to ensure that incorporation of a filler material does not lead to non-ideal interface bonding between the toughened filler and PMMA matrix, the existence of pores, refraction rate mismatch, and excessive light scattering in the material, which would reduce optical transparency and significantly affect the toughness, causing the filled material to be ill-suited as a glass substitute. A promising class of fillers are rubber particles, which can form an interfacial bond with the bulk plastic and absorb energy during fracture through multiple mechanisms.6 The toughness of PMMA can also be improved by using directional stretching technology, although directional stretching requires expensive equipment.
Nelliappan et al. used two- and three-layer composite latex particles to prepare rubber-toughened PMMA. The three-layer particles were found to be more effective in toughening PMMA than the two-layer particles.7 Guo et al. synthesized a series of methyl methacrylate-butadiene-St (MBS) copolymers by grafting polybutadiene (PB) particles with St and methyl methacrylate (MMA) through emulsion polymerization.8 The obtained MBS was then blended with PMMA to form blends. It was found that the impact strength of the blend increased and then decreased with increasing PB content in MBS. Yu et al. blended different mass fractions of acrylate copolymer (ACR) with PMMA by melt blending to prepare PMMA/ACR.9 The results showed that the Izod notched impact strength of the modified PMMA increased with increasing ACR content, while the tensile strength and bending strength decreased. The Izod notched impact strength of PMMA modified by ACR increased by 42.42% when the ACR mass fraction was 5%. Huang et al. investigated toughening modification of casted PMMA by poly(butyl acrylate) (PBA), which was prepared by polymerization.10 The results showed that PBA is a good toughener when dispersed in the PMMA matrix as nanoparticle clusters. The impact strength of PMMA modified by PBA increased by 38% when the PBA mass fraction was 0.5 wt%.
Recently, core–shell composite particles composed of a rubbery core and a glassy shell have received remarkable interest as a toughening agent for brittle polymers.11 One of remarkable characteristics of their morphological structures is that the rubbery core phase covered by the shell polymer has good compatibility with the matrix polymer. Poly(acrylonitrile (AN)-styrene (St)-acrylate) (ASA) terpolymer is a typical core/shell structure polymer, which is obtained by grafting copolymerization of St and AN monomers onto crosslinked PBA rubber particles.12 The structures of ASA and AN-butadiene-St (ABS) terpolymer are similar.13 They have the same St-AN graft shells, but the polybutadiene (PB) rubber core within ABS is substituted by the PBA rubber core in ASA.14,15 Compared with the butadiene rubber core of ABS, ASA has much better weather resistance because there are no double bonds in the main chains of the acrylic elastomer core.
One of the potential applications of ASA is toughening of polymers, because the soft core as a toughening agent allows plastic deformation, while the hard shell effectively weakens the softening caused by the core.16 To enhance the impact toughness of polymers, ASA is often introduced into a polymer system to prepare the ASA/polymer composite, which has excellent rigidity, heat resistance, and advanced weather resistance.17–24 However, ASA is very limited in providing toughness. Using ASA does not significantly increase the toughness of brittle polymers, even when the ASA content is above 30%. Moreover, although ASA can improve the toughness of PMMA, a substantial decrease in the tensile strength of the composite usually occurs. Therefore, to take advantage of the merits of ASA and meet the requirements of the high toughness required in certain applications, toughening modification of ASA is necessary.
Organic–inorganic nanocomposites have been extensively studied because its the combined characteristics of organic polymers and inorganic materials. The PBA rubber core plays a toughening role in ASA, however, PBA has the disadvantages of low-temperature brittleness and high-temperature viscosity, which seriously affect the performance of ASA. Silicone is well-known to have an extremely low glass-transition temperature, an extremely low elastic modulus, and good thermochemical stability, which make up for the disadvantages of PBA.25 In addition, the coupling effect of the silicone monomer can effectively improve adhesion of the polymer to the matrix. Hydrolysis of the silicon monomers can also establish a cross-linked silica network, which can improve the stability and mechanical strength of the polymer.26 Although ASA has excellent comprehensive performance, there is still space for further improvement of the toughness and thermal stability. According to the above reports, the focus of modification is the PBA rubber core, and it is essential to fabricate core/shell organic–inorganic hybrid polymer nanoparticles by combining PBA with silicone.
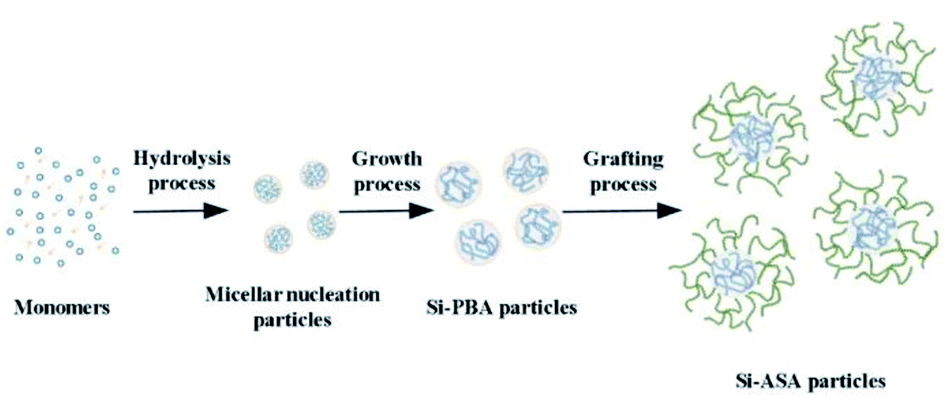
Herein, we report an effective and environment friendly protocol to prepare core/shell organic–inorganic silicone-ASA hybrid nanoparticles (Si-ASA HNPs) with a Si-PBA core and a poly(St-AN) (SAN) shell, which combine the advantages of the components and provide the nanoparticles with high toughness, temperature resistance, and other characteristics. Using tert-butyl hydroperoxide (t-BHP) as an initiator and sodium dodecyl sulfate (SDS) as an emulsifier, silicone was introduced into PBA to prepare the Si-hybrid PBA core. Using t-BHP as an initiator and sodium dodecylbenzenesulphonate (SDBS) as an emulsifier, the Si-ASA HNPs were then obtained by grafting copolymerization of St and AN monomers onto the Si-hybrid PBA core. By adjusting the amount of Si-ASA HNPs added, PMMA/ASA composites with different compositions were prepared by melt blending. The toughening effect of the Si-ASA HNPs on PMMA was studied by testing the mechanical properties of the PMMA/ASA composites. According to the test results, the relationship between the Si-ASA HNPs and the mechanical performance of the PMMA/ASA composite was revealed, and the toughening mechanism was explored. This work provides a convenient and efficient method to prepare core/shell organic–inorganic hybrid ASA nanoparticles with high performance.
Experimental
Materials measurements
3-Methacryloxypropyltrimethoxysilane (KH570), SDBS, butyl acrylate (BA), and magnesium sulfate heptahydrate (MgSO4) were provided by Kelong Chemical Reagent Co., Ltd. (Chengdu, China). 2-Butanone and ferrous sulfate (FeSO4) were purchased from J&K Scientific Co., Ltd. (China). Styrene (St) was purchased from Jinshan Chemical Test Co., Ltd. (Chengdu, China). 1,3-Butanediol dimethacrylate, tert-butyl hydroperoxide (t-BHP), sodium edetate (EDTA), and sodium formaldehyde sulfoxylate dihydrate (SFS) were provided by Aladdin Reagent Co., Ltd. (Shanghai, China). Acrylonitrile (AN) was purchased from Heyi Chemical Technology Co., Ltd. (Dalian, China). Dodecylbenzenesulphonic acid (DBSA) was provided by Sigma-Aldrich Company. Allyl methacrylate (ALMA) was purchased from Adamas Reagent, Ltd. (China). Deionized water (DDI) was produced by an ultrapure water device (ULUP-IV-100L, Youpu Ultrapure Technology Co., Ltd., Sichuan, China). All of the chemicals were used as received without further purification. PMMA (CM-205) was produced by Zhenjiang GPPC Chemical Co., Ltd. (Zhenjiang, China).Characterization
Dynamic light scattering (DLS) was performed with a ZetaPALS 90Plus zeta potential and nanoparticle size analyzer with DDI as a dispersant. The Fourier transform infrared (FTIR) spectroscopy measurements were performed with a Nicolet iS5 spectrophotometer at room temperature in the scan range of 400–4000 cm−1. Scanning electron microscopy (SEM) was performed with a Tescan MAIA3 scanning electron microscope at an accelerating voltage of 15 kV. The transmission electron microscopy (TEM) images were acquired with a Zeiss Libra 200FE transmission electron microscope (Carl Zeiss, Germany). The differential scanning calorimetric (DSC) measurements were performed with an Instruments DSCQ2000 from room temperature to 200 °C with heating and cooling speeds of 10 °C min−1 under a N2 atmosphere. Thermogravimetric analysis (TGA) was performed with a simultaneous SDT Q600 thermal analyzer (TA Instruments, USA). The temperature range was room temperature to 600 °C with a heating rate of 10 °C min−1 under a N2 atmosphere.Mechanical properties of the PMMA/ASA composites
The tensile tests were performed with an MTS C45.504E universal tensile testing machine according to GB/T1040.3-2006. The tensile rate was 2 mm min−1 and the sample size was 60.0 mm × 10.0 mm × 1.0 mm. A PIT501J LCD-type plastic impact test machine (Shenzhen Wance Test Equipment Co., Ltd., China) was used for the impact tests according to GB/T1043.1-2008. The samples were unnotched strips and their size was 80.0 mm × 10.0 mm × 4.0 mm. The thickness of the sample was measured with a micrometer. All of the samples were tested three times.Synthesis of ASA HNPs
A series of Si-ASA HNPs were designed and synthesized by emulsion polymerization. To investigate the effect of the KH570 content on the properties of the Si-ASA HNPs, by adjusting the weight ratio of KH570 to BA (5/45, 10/40, 15/35, 20/30, and 25/25), different Si-ASA latex composites were obtained. For comparison, the ASA latex without KH570 was prepared by emulsion polymerization using the same reaction recipe. The ASA latex and Si-ASA HNPs with 5, 10, 15, 20, and 25 wt% KH570 are designated as ASA-1, ASA-2, ASA-3, ASA-4, ASA-5, and ASA-6, respectively. The proportion of each component is given in Table 1.| Sample | Core | Shell | |
|---|---|---|---|
| KH570 content (wt%) | BA content (wt%) | (St/AN) contenta (wt%) | |
a (St/AN) was composed of St and AN monomer with a mass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. :1. |
|||
| ASA-1 | 0 | 50 | 50 |
| ASA-2 (KH570-5 wt%) | 5 | 45 | 50 |
| ASA-3 (KH570-10 wt%) | 10 | 40 | 50 |
| ASA-4 (KH570-15 wt%) | 15 | 35 | 50 |
| ASA-5 (KH570-20 wt%) | 20 | 30 | 50 |
| ASA-6 (KH570-25 wt%) | 25 | 25 | 50 |
All of the reactions were performed in the 5 L three-neck glass-jacketed reactor equipped with a mechanical stirrer. The reactor was connected to a water bath with thermostatic control. The stirring speed was controlled at 150 rpm. A mixture of KH570 (66.00 g), SDBS (5.28 g), and DDI (198.00 g) was pre-emulsified as K-1. The polysiloxane copolymer (K-1) was synthesized by hydrolysis of siloxane monomers followed by hydrolytic condensation. In the first stage, polymerization was performed at 75 °C. K-1 (the added amounts are given in Table 2), DBSA (1.32 g), and DDI (64.68 g) were added to the system. The solution was stirred for 2 h. In the second stage, polymerization was performed at 60 °C. SDBS (12.41 g), DDI (2679.60 g), BA (the added amounts are given in Table 2), ALMA (2.77 g), t-BHP (1.72 g), and 1,3-butanediol dimethacrylate (1.45 g) were added to the system. The solution was stirred for 1 h. M1 (135.17 g) was then added, and the solution was stirred at 78 °C for 2 h. In the third stage, polymerization was performed at 60 °C. M2 (137.28 g), AN (99.00 g), St (297.00 g), and t-BHP (2.38 g) were added to the system. The solution was stirred for 2 h. M3 (135.30 g), AN (66.00 g), St (198.00 g), and t-BHP (1.32 g) were then added, and the solution was stirred for 1.5 h. The compositions of the M1, M2, and M3 mixtures are given in Table 3.
| Sample | Addition (g) | |
|---|---|---|
| K-1 | BA | |
| ASA-1 | — | 660.00 |
| ASA-2 (KH570-5 wt%) | 335.28 | 594.00 |
| ASA-3 (KH570-10 wt%) | 670.56 | 528.00 |
| ASA-4 (KH570-15 wt%) | 1005.84 | 462.00 |
| ASA-5 (KH570-20 wt%) | 1341.12 | 396.00 |
| ASA-6 (KH570-25 wt%) | 1676.40 | 330.00 |
| Code | Addition (g) | |||
|---|---|---|---|---|
| FeSO4 | EDTA | SFS | DDI | |
| M1 | 0.0001 | 0.0003 | 0.24 | 10.00 |
| M2 | — | — | 0.40 | 10.00 |
| M3 | 0.0002 | 0.0006 | 0.25 | 10.00 |
Preparation of the ASA powders
The ASA powder was prepared by demulsification. The reaction was performed in a 5 L three-neck glass-jacketed reactor equipped with a mechanical stirrer. The reactor was connected to a water bath with thermostatic control. The stirring speed was controlled at 150 rpm. First, MgSO4 was dissolved in DDI and heated to 85 °C. The core/shell organic–inorganic hybrid ASA nanoparticle latex was then dropped within 1 h. Finally, the obtained product was washed with DDI and the sample was oven-dried at 75 °C for at least 24 h to give a white powder.Preparation of the PMMA/ASA alloys
The ASA powder and PMMA were dried at 70 °C for 5 h in a vacuum oven. The PMMA/ASA composites with weight ratios of 95/5, 90/10, 85/15, 80/20, 75/25, 70/30, 65/35, and 60/40 were alloys with a twin-screw extruder at 240 °C for 5 min with a rotation speed of 200 rpm. Neat PMMA was also subjected to the same mixing processing to create the same thermal history. The alloys were injection molded into standard bars for the mechanical tests. Rectangular specimens with size of 80.0 mm × 10.0 mm × 4.0 mm were used for the impact tests. Rectangular specimens with size of 60.0 mm × 10.0 mm × 1.0 mm were used for the tensile tests.Results and discussion
Synthesis and structure of the ASA HNPs
Emulsion polymerization is a method to prepare core–shell rubber particles that exhibit alternating rubbery and glassy layers.27,28 Two-stage emulsion synthesis is frequently performed in which the core is synthesized in the presence of a crosslinking agent followed by swelling with the shell monomer and then a second polymerization.29 These particles have been successfully used to toughen polymers. The components of these particles are a rubbery core and an outer glassy polymer shell. The rubbery core is generally based on polybutadiene and PBA.30 The outer glassy shell is required to prevent coalescence of the rubbery particles during synthesis, and it enhances formation of a good interface with the matrix. The shell is usually based on St/AN copolymers.According to the above reports, as the inner layer concept the silicon-containing polymers may function as the ductile inner layer. They are much preferred along with PBA especially for its extremely low glass transition temperatures, extremely low elastic modulus, and good thermochemical stability. However, owing to the large surface and strong aggregation of nanosilica, synthesis of stable nanosilica/polymer composites through emulsion polymerization is difficult. Surface modification is necessary to prevent formation of agglomerated nanoparticles in emulsion polymerization. Introduction of silicone oligomers does not require special treatment, and the steps are easy and non-complicated. In addition, they do not require unique environmental conditions for curing.
Owing to the monomer grafting reaction site in the ASA system is the double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), the silicone monomer for the system needs to contain CC bonds. Among the silicone monomers, KH570 contains unsaturated CC bonds, which can react with BA, St, and AN. It is an ideal silicone monomer. On the one hand, KH570 is an abundant readily available product and cheaper than other silane coupling agents with similar properties. On the other hand, KH570 also provides notable reinforcement for improving the mechanical properties of polymer composites. KH570 plays dual roles as a compatibilizer and a chain extender, and it not only improves interfacial adhesion between the PBA particles and SAN copolymer, but it also increases chain entanglement of ASA.
C), the silicone monomer for the system needs to contain CC bonds. Among the silicone monomers, KH570 contains unsaturated CC bonds, which can react with BA, St, and AN. It is an ideal silicone monomer. On the one hand, KH570 is an abundant readily available product and cheaper than other silane coupling agents with similar properties. On the other hand, KH570 also provides notable reinforcement for improving the mechanical properties of polymer composites. KH570 plays dual roles as a compatibilizer and a chain extender, and it not only improves interfacial adhesion between the PBA particles and SAN copolymer, but it also increases chain entanglement of ASA.
In this study, KH570-modified PBA (Si-PBA) oligomer, which contains double bonds, was synthesized. Using t-BHP as an initiator and SDS as an emulsifier, silicone groups were introduced into the acrylate polymer, which can combine the advantages the two components and endow the polymer have high temperature resistance and other characteristics.31 Using t-BHP as an initiator and SDBS as an emulsifier, the AN-St-acrylate terpolymer was then obtained by graft copolymerization of St and AN monomers onto PBA particles. The synthetic pathways for preparing Si-PBA and Si-ASA are shown in Fig. 1 and 2, respectively.
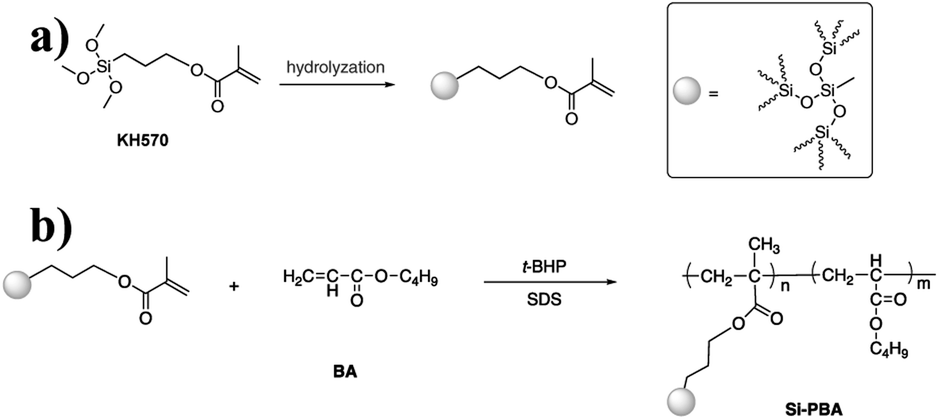 | ||
| Fig. 1 Synthetic pathway for preparing Si-PBA. | ||
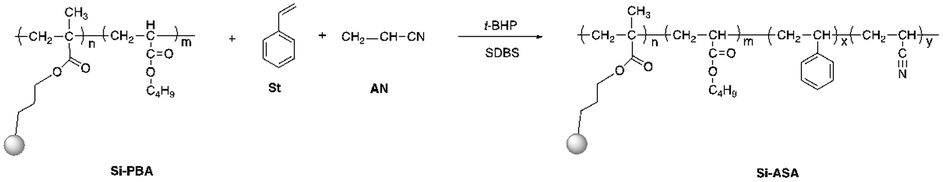 | ||
| Fig. 2 Synthetic pathway for preparing Si-ASA. | ||
The group contained in the ASA HNPs
FTIR spectroscopy was performed to analyze the various groups in the ASA HNP polymers in the scanning frequency range from 400 to 4000 cm−1 (Fig. 3). The absorption bands at 3060 and 3024 cm−1 are ascribed to the C–H stretching vibration in the aromatic phenyl ring, and the bands at 1602 and 1493 cm−1 are ascribed to the conjugated CC bonds in the ring. The C–H asymmetric stretching vibration of –CH3 appears at 2961 cm−1. The absorption bands at 2930 and 2875 cm−1 correspond to C–H symmetric and asymmetric stretching vibration and bending vibration of –CH2– in the chain backbone, respectively. In addition, the presence of the peaks at 1457, 842, 760, and 700 cm−1 is evidence of C–H bending or ring puckering.32 The absorption band at 2237 cm−1 is assigned to the CN absorption band, indicating that AS was successfully grafted onto the core phase.33 Furthermore, the relatively obvious absorption band at 1734 cm−1 is the CO stretching vibration, and the bands with peak values at around 1261, 1165, and 1064 cm−1 are assigned to the O–C vibrations of PBA.34 From the infrared spectra, the characteristic peaks of the ASA HNPs with different PBA cores did not show formation of new groups or group shifts.35 It is noteworthy that the absorption peaks all become sharper when addition of KH570 was increased from 5 to 25 wt%. This may be because addition of KH570 led to an increase of the grafting rate, so the absorption peaks became sharper.
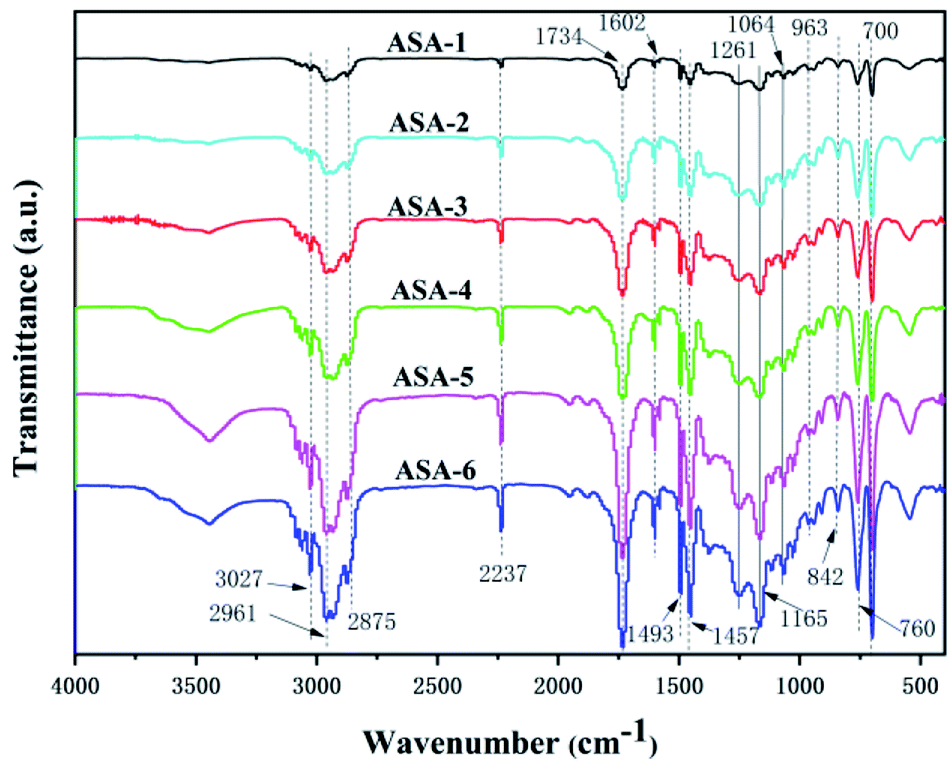 | ||
| Fig. 3 FTIR spectra of the ASA HNPs. | ||
DLS of the PBA latex particles
The aim of this work is to prepare latex particles with a narrow size distribution and good stability of the colloidal particles. A polydispersity index (PDI) value of greater than 0.7 indicates that the sample has a very broad size distribution, which means that the emulsion is not stable. In general, structure and size control of the particles is difficult in silicone-based latex materials prepared by emulsion polymerization. In addition, silicone may cause the local concentration to be too high to dissipate heat and cause agglomeration. Through the synthesis process in this study, the PDI values of the PBA latex materials were less than 0.5 with a narrow distribution and good stability. In addition, the synthesis process is simple and environmentally friendly, it is easy to control the structure and size of the particles, and the particles do not easily agglomerate.For ASA, the PBA rubber core plays the main role in toughening. One of the important factors for the toughness of the core–shell structure is the particle size of the PBA rubber core.36 The particle-size distributions of the PBA latex particles synthesized by emulsion polymerization are shown in Fig. 4 and Table 4. All of the core particles were small. The sizes of the PBA latex particles of ASA-1 were around 30 nm (Fig. 4(A)), while the Si-PBA latex particles modified by KH570 were within the range 20–40 nm. As the proportion of KH570 increased, the core particle size first steadily increased and then slightly decreased. For ASA-2 and ASA-3, the sizes of Si-PBA latex particles increased to 35 and 37 nm compared with those of ASA-1 (Fig. 4(B) and (C)). For ASA-5 and ASA-6, the sizes of Si-PBA latex particles decreased to 28 and 26 nm compared with those of ASA-1 (Fig. 4(E) and (F)). Smaller particles are more effective at initiating shear deformation and crazing, while larger particles sustain propagation of crazing by absorbing more energy. Therefore, ASA-2 and ASA-3 could show a better toughening effect, which was investigated by mechanical property tests.
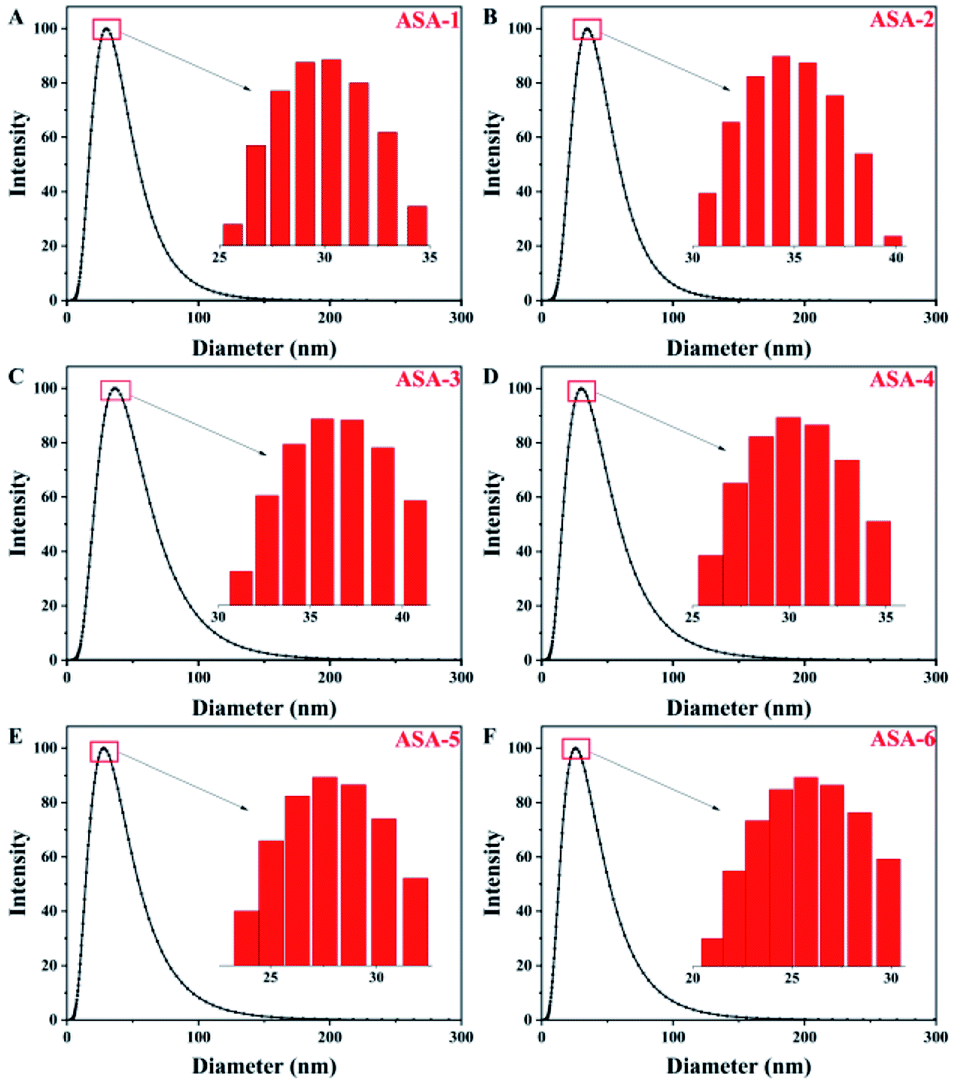 | ||
| Fig. 4 Particle-size distributions of the PBA latex particles: (A) ASA-1, (B) ASA-2, (C) ASA-3, (D) ASA-4, (E) ASA-5, and (F) ASA-6. | ||
Grafting rate of the ASA HNPs
It is generally believed that when the core particles are dispersed in the shell matrix, the grafting rate has the optimal value. If the grafting rate is lower than this optimal value, the rubber particles cannot be well covered by the shell layer. The van der Waals force between the rubber particles will cause the rubber particles to aggregate. The interface bonding force and compatibility will be affected if the grafting rate is too low, and it is easy to separate the phases, difficult to exert the toughening effect, and the impact performance of the material decreases. As the grafting rate increases, the compatibility of the material and impact performance also increase. The grafting rate of ASA with different core phases are given in Table 5. The yields of ASA for the different core phases were around 80%, which was conducive to industrial production. Most of the Si-ASA grafting rate were greater than 94%, except for ASA-1. The grafting rate of ASA-1 was around 89%, which was consistent with previous studies. The grafting rate of ASA-2 and ASA-3 were about 94%, and those of ASA-4, ASA-5, and ASA-6 were around 96%. This showed that the degree of grafting of Si-PBA increased with increasing KH570 content, which is probably because addition of KH570 increases cross-linking of the PBA core. The SAN hard-shell grafting reaction site is the double bond on the branch of the PBA core particles. The surface of the PBA core particles needs to be covered by grafting reaction sites (CC bonds). For ASA-1, the surface had relatively few grafting active sites, so the grafting rate was relatively low. With increasing KH570 content, the degree of cross-linking of the PBA core increased and the number of CC double bonds increased, which was conducive to the grafting reaction, and the grafting rate were relatively high. For the synthetic method used in this study, the grafting rate of ASA was as high as 96.76%, thereby achieving the toughening effect.
| Sample | Grafting rate (%) | Yield (%) |
|---|---|---|
| ASA-1 | 89.72 | 80.23 |
| ASA-2 (KH570-5 wt%) | 94.89 | 81.06 |
| ASA-3 (KH570-10 wt%) | 94.32 | 80.45 |
| ASA-4 (KH570-15 wt%) | 96.76 | 79.86 |
| ASA-5 (KH570-20 wt%) | 95.96 | 80.66 |
| ASA-6 (KH570-25 wt%) | 96.31 | 79.59 |
Morphologies of the ASA HNPs
The morphologies of the ASA latex particles with a core–shell structure were examined by TEM (Fig. 5), which observes the particle size directly corresponding to the above results by DLS (Fig. 4). The ASA powder was observed by SEM (Fig. 6). The Si-ASA HNPs consisted of a dark core, which was Si-PBA, and a brighter shell, which was SAN (Fig. 5), indicating that the ASA latex had a clear core–shell structure. The Si-ASA particles were not a three-layer structure, indicating that copolymerization between KH570 and butyl acrylate was sufficient. There was no obvious phase separation, and a uniform continuous rubber phase formed. The particle sizes of ASA and PBA could also be observed in the TEM images, and the ASA particle size exceeded the size of the PBA core phase. The PBA core in the TEM images was larger than the size measured by DLS. The reason is that DLS measured the average particle size and particle-size distribution, and there were other larger particles in the latex, which were the particles observed by TEM. | ||
| Fig. 5 TEM image of ASA. | ||
 | ||
| Fig. 6 SEM image of the ASA powders. | ||
SEM images of the Si-ASA powder obtained by demulsification of the Si-ASA emulsion are shown in Fig. 6. The morphology observed by TEM (Fig. 5) confirmed the spherical shape observed in the SEM image (Fig. 6). The SEM images showed that the particle sizes of the Si-ASA powder were not uniform. The large particles were surrounded by small particles in the SEM images, and the increase of the possibility of latex-particle polymerization effectively increased the particle size. The reason for this differences is that when in the demulsification process, the molecular chain of the SAN shell structure is unstable, and grafting of the SAN chain on the rubber particles is not uniform. The larger particles formed because the low value of the polarity index of the SAN medium caused growth of the particles and assembly with the highly extended SAN polymer chains in the low-polarity-index water mixture, thus forming rubber powder particles with different sizes through agglomeration. From the SEM and TEM measurements, the final ASA latex was composed of spherical particles with a narrow size distribution.
Glass-transition behavior of the ASA HNPs
The glass-transition behavior of the ASA HNPs was further investigated by DSC. The DSC curves are shown in Fig. 7 and the detailed Tg results are given in Table 6. As a core/shell structured terpolymer, poly(acrylonitrile–styrene–acrylate) shows two typical glass-transition temperatures: one at about −40 °C corresponding to the glass-transition temperature of the PBA core (Tg,1) and another at around 110 °C corresponding to the glass-transition temperature of the SAN shell (Tg,2). The two glass-transition temperatures of ASA-1 were at Tg,1 = −39.62 °C and Tg,2 = 111.30 °C. This indicates that core/shell ASA nanoparticles were successfully synthesized. It is worth noting that both the Tg,1 and Tg,2 values of the ASA HNPs improved compared with ASA-1, which is attributed to addition of KH570. Tg,1 of the PBA core increased from −39.62 to −34.46 °C when 25 wt% KH570 was added to ASA. One reason for the Tg,1 improvement of the PBA phase is related to the higher Tg,1 of silicone and their good miscibility. The other reason lies in the branching reaction between PBA and KH570, which increases the molecular weight of PBA and limits its molecular chain movement. The higher Tg,1 of PBA indicates that the heat resistance of ASA is improved and it can endure much higher deformation temperature under load. Tg,2 of the SAN shell increased from 111.30 to 115.68 °C when 5 wt% KH570 was added to ASA. However, when addition of KH570 was increased from 5 to 25 wt%, Tg,2 remained at around 115 °C. One possible explanation for this difference is that the Si content was less, so the increase was not obvious. In summary, it is possible to achieve polymer-toughening modification by using core/shell ASA as an impact modifier.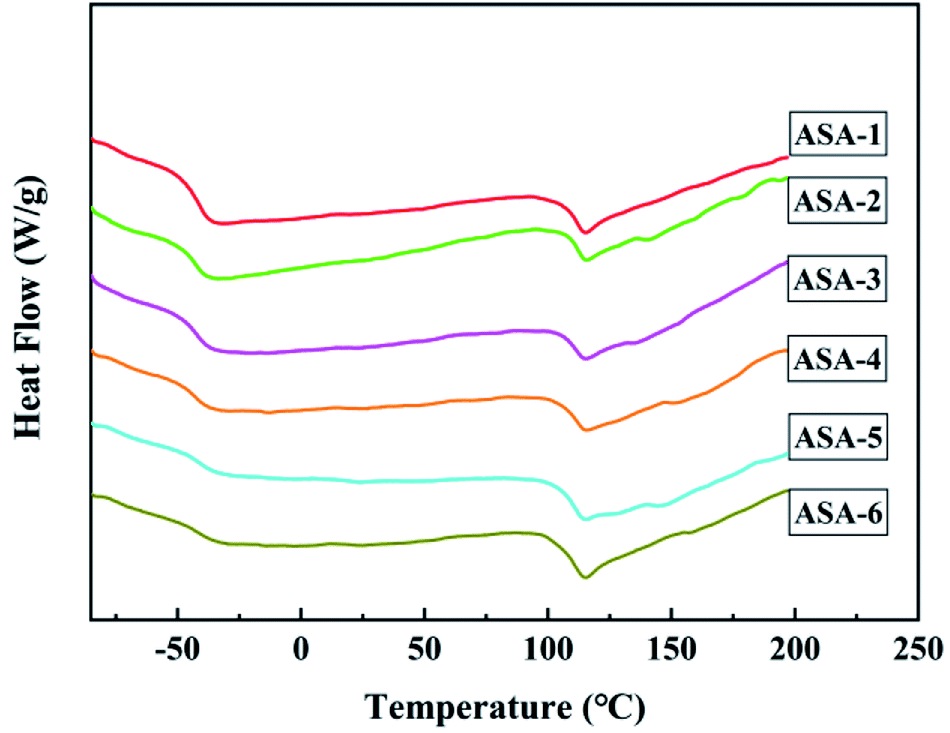 | ||
| Fig. 7 DSC curves of ASA powders. | ||
| Sample | Tg,1 | Tg,2 |
|---|---|---|
| ASA-1 | −39.62 | 111.30 |
| ASA-2 (KH570-5 wt%) | −38.39 | 115.68 |
| ASA-3 (KH570-10 wt%) | −37.44 | 115.33 |
| ASA-4 (KH570-15 wt%) | −37.44 | 115.43 |
| ASA-5 (KH570-20 wt%) | 34.47 | 115.08 |
| ASA-6 (KH570-25 wt%) | −34.46 | 115.39 |
TGA of the ASA HNPs
The TGA and derivative thermogravimetry (DTG) curves of the ASA terpolymer nanoparticles are shown in Fig. 8, and the corresponding data are given in Table 7. It was found that the difference between the decomposition temperatures of ASA and Si-ASA modified by KH570 was not very large, but Tmax and the char residue yield increased. ASA only showed one thermal degradation stage between 350 and 450 °C (Fig. 8). For ASA-1, the initial decomposition temperature (T5%) was 351 °C, the maximum thermal weight loss rate temperature (Tmax) was 409 °C, and the char residue yield at 800 °C was only 2.27%, which is consistent with previous studies. With increasing KH570 content, T5% of Si-ASA decreased. When 5 wt% KH570 was added, T5% of ASA-2 decreased to 343 °C, Tmax increased to 413 °C, and the char residue yield at 800 °C increased to 3.58%. It is worth noting that when 15 wt% KH570 was added, T5% of ASA-4 decreased to 349 °C, Tmax increased to 418 °C, and the char residue yield at 800 °C increased to 4.29%. When the amount of KH570 was 15%, Tmax and the char residue yield reached the optimal values, and with further increasing amount of KH570, the values of these performance indicators began to decrease. Polymers containing Si–O–Si segments in the main chain usually exhibit high degradation temperatures.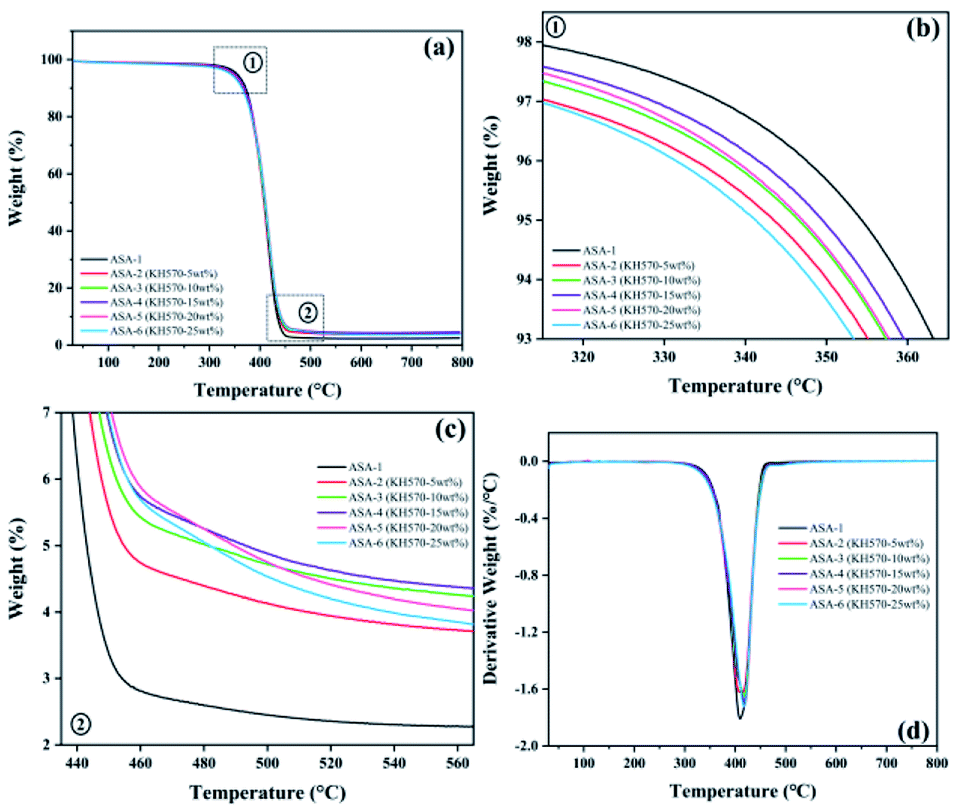 | ||
| Fig. 8 (a) TGA curves of ASA powders. (b) Enlarged view of the TGA curves around the initial decomposition temperature. (c) Enlarged view of the TGA curves in the range 440–560 °C. (d) DTG curves of ASA powders. | ||
| Sample | T5% (°C) | Tmax (°C) | Rate of Tmax (%/°C) | Char yield (%) | ||
|---|---|---|---|---|---|---|
| 600 °C | 700 °C | 800 °C | ||||
| ASA-1 | 351 | 409 | −1.81 | 2.45 | 2.31 | 2.27 |
| ASA-2 (KH570-5 wt%) | 343 | 413 | −1.63 | 3.75 | 3.62 | 3.58 |
| ASA-3 (KH570-10 wt%) | 346 | 417 | −1.65 | 4.17 | 4.13 | 4.05 |
| ASA-4 (KH570-15 wt%) | 349 | 418 | −1.68 | 4.54 | 4.32 | 4.29 |
| ASA-5 (KH570-20 wt%) | 347 | 418 | −1.72 | 4.01 | 3.88 | 3.80 |
| ASA-6 (KH570-25 wt%) | 341 | 417 | −1.72 | 3.74 | 3.68 | 3.58 |
In the initial decomposition stage of ASA, the reduction of the initial decomposition temperature is more conducive to promoting the dehydration and carbonization of the ASA surface, forming a dense carbon film, which is conducive to isolating the outside air and heat, while reducing the amount of combustible substances produced by decomposition and delaying the further decomposition of ASA.
In addition, the SAN shell structure also has a certain protective effect on the ASA, which increases the energy required for breaking the Si–O and CC. On the one hand, there are bulky phenyl groups and strong polar cyano groups in the molecular structure of ASA. The bulky phenyl groups increase the rotational resistance in the ASA molecular chain, and the strong polar cyano groups increase the interaction force of the ASA molecular chain, resulting in ASA. The decomposition activation energy of the molecular chain is improved. On the other hand, the bond energy of the phenyl group and the cyano group in the ASA molecular structure is larger, and the energy required for breaking is higher.
With the increase of KH570 content, the double bonds on the surface of Si-PBA core also increase, which increases the possibility of grafting shell layer monomers, the shell layer grafting is more complete, the performance is better, and the carbon residue rate also increases. When the ASA core–shell ratio is constant, the number of shell monomers is determined. When the content of KH570 is too high, there are too many CC bonds remaining on the surface of the Si-PBA core, the grafted shell layer cannot completely cover the Si-PBA core, and the Si-PBA core is partially exposed, which reduces the performance and the carbon residue rate decline.
With increasing KH570 content, the thermal stability of the polymer increased. The results provided additional evidence for silicone grafting onto the ASA polymer. These results indicate that addition of KH570 improves the thermal stability of ASA.
Impact properties of the PMMA/ASA alloy
The energy absorbed by the impact force is closely correlated to the PBA core phase in the material. The impact strengths of PMMA/ASA alloys with different core phases are shown in Fig. 9 and the impact strength values are given in Table 8. PMMA is a brittle material with low impact resistance (about 21 kJ m−2). As the ASA content increased, the impact strength of the PMMA/ASA alloys first steadily increased and then slightly decreased. For the PMMA/ASA-1 alloys, the impact strength increased to 44 kJ m−2 with addition of 30 wt% ASA. For the PMMA/ASA-2 alloys, the impact strength increased to 54 kJ m−2 with addition of 25 wt% ASA-2, while for the PMMA/ASA-3 alloys, the impact strength increased to 53 kJ m−2 with addition of 25 wt% ASA-3. The impact strengths of both ASA-2 and ASA-3 increased by about 100% compared with pure PMMA. With less Si-ASA addition, the impact strength can be greatly improved to achieve the purposes of cost saving and higher toughening efficiency.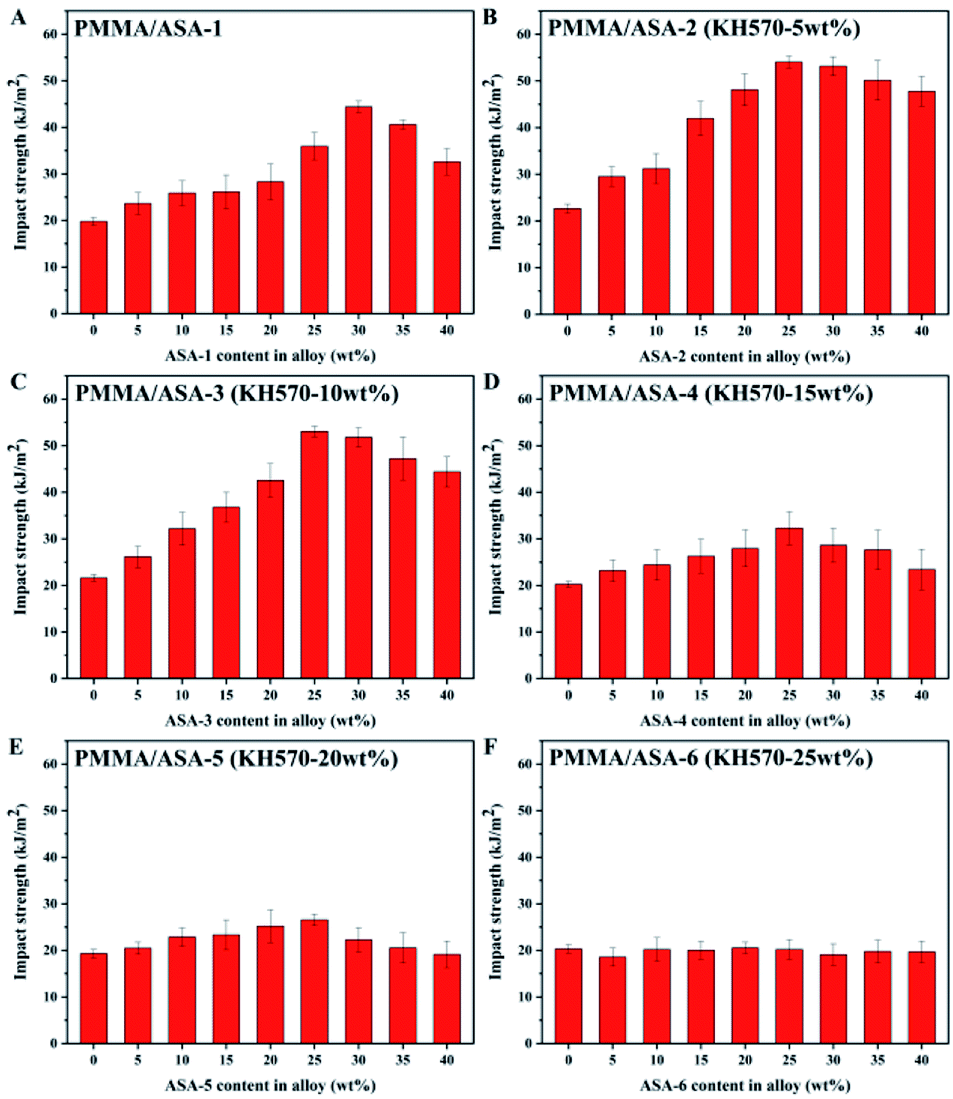 | ||
| Fig. 9 Impact strengths of the PMMA/ASA alloys. | ||
| ASA content in alloy (wt%) | Impact strength (kJ m−2) | |||||
|---|---|---|---|---|---|---|
| ASA-1 | ASA-2 | ASA-3 | ASA-4 | ASA-5 | ASA-6 | |
| 0 | 19.78 ± 0.88 | 22.66 ± 0.95 | 21.61 ± 0.72 | 20.27 ± 0.66 | 19.28 ± 0.89 | 20.30 ± 0.92 |
| 5 | 23.64 ± 2.33 | 29.52 ± 2.13 | 26.14 ± 2.31 | 23.19 ± 2.32 | 20.46 ± 1.29 | 18.56 ± 1.95 |
| 10 | 25.88 ± 2.65 | 31.21 ± 3.21 | 32.20 ± 3.54 | 24.42 ± 3.25 | 22.88 ± 1.99 | 20.20 ± 2.53 |
| 15 | 26.15 ± 3.63 | 41.98 ± 3.65 | 36.79 ± 3.22 | 26.29 ± 3.64 | 23.34 ± 3.21 | 19.99 ± 1.95 |
| 20 | 28.31 ± 3.85 | 48.13 ± 3.42 | 42.55 ± 3.63 | 27.97 ± 3.95 | 25.21 ± 3.54 | 20.52 ± 1.23 |
| 25 | 35.96 ± 2.96 | 54.08 ± 1.32 | 53.06 ± 1.13 | 32.29 ± 3.52 | 26.56 ± 1.23 | 20.19 ± 2.14 |
| 30 | 44.41 ± 1.21 | 53.16 ± 2.01 | 51.82 ± 2.03 | 28.62 ± 3.64 | 22.26 ± 2.65 | 19.07 ± 2.33 |
| 35 | 40.63 ± 0.99 | 50.15 ± 4.23 | 47.22 ± 4.63 | 27.65 ± 4.21 | 20.53 ± 3.26 | 19.76 ± 2.45 |
| 40 | 32.60 ± 2.85 | 47.79 ± 3.21 | 44.40 ± 3.24 | 23.44 ± 4.36 | 19.12 ± 2.89 | 19.66 ± 2.31 |
This result are logical because the AS phase in ASA is a brittle matrix, and acrylic rubber particles mainly rely on inducing crazing and expansion of crazing to dissipate energy, so as to achieve toughening. With increasing ASA content, the density of rubber particles in the system increases. Therefore, when subjected to an external force, interaction of the stress field occurs, which increases the density of crazing between the rubber particles, thereby absorbing more energy. In the PMMA/ASA alloys, the silicone-based impact modifiers ASA-2 and ASA-3 improved the impact properties and efficiency more than the other ASA modifiers. This is logical because KH570 improved the toughening efficiency of the Si-ASA composite and Si-ASA showed much higher impact strength than ASA when the KH570 content was below 10 wt%.
Tensile properties of the PMMA/ASA alloy
The tensile properties of the PMMA/ASA alloys are shown in Fig. 10 and the tensile strength values are given in Table 9. With increasing ASA content, the tensile strength of the alloy clearly decreased. This is logical because ASA contains a PBA rubber phase, and its strength and rigidity are smaller than those of PMMA. As a consequence, with increasing ASA content, the proportion of the PBA rubber phase in the whole system also increases, and the tensile strength of the alloys decreases.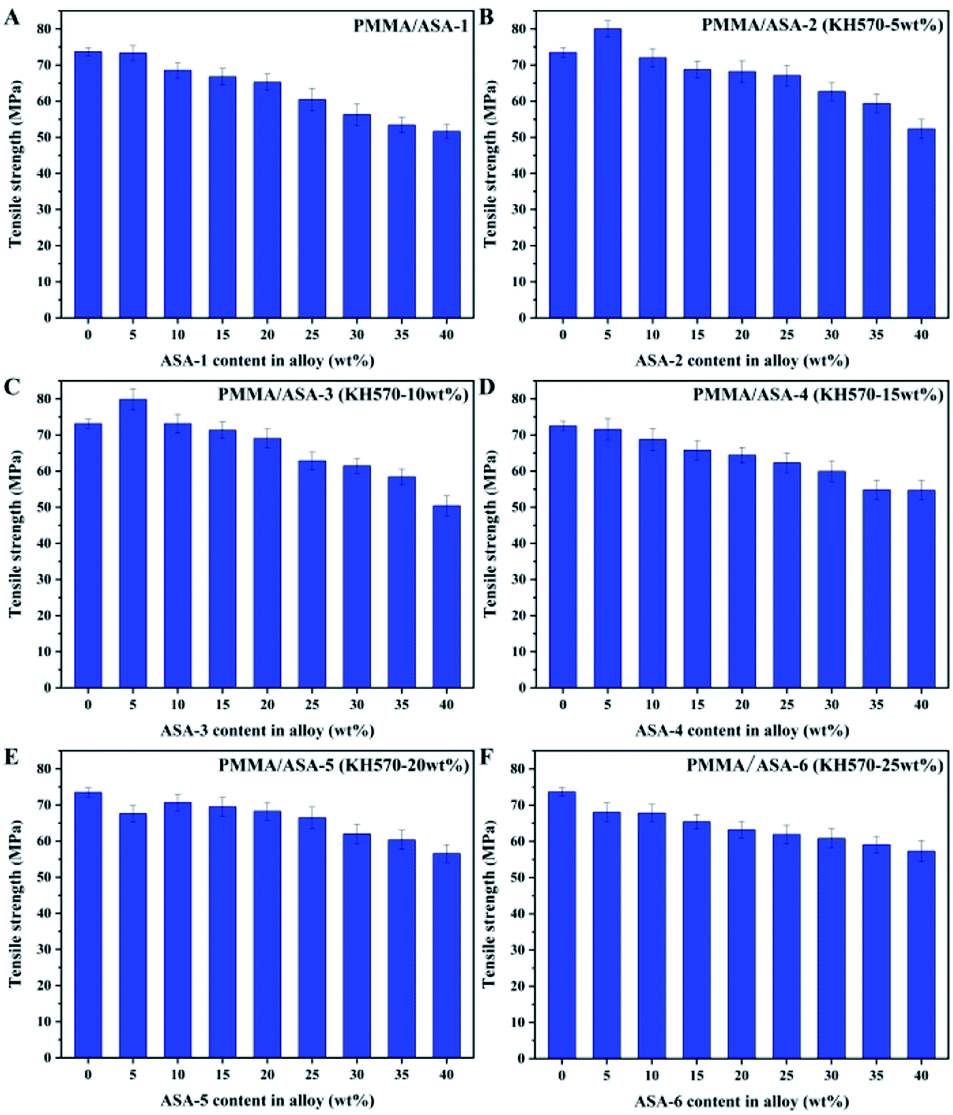 | ||
| Fig. 10 Tensile strengths of the PMMA/ASA alloys. | ||
| ASA content in alloy (wt%) | Tensile strength (Mpa) | |||||
|---|---|---|---|---|---|---|
| ASA-1 | ASA-2 | ASA-3 | ASA-4 | ASA-5 | ASA-6 | |
| 0 | 73.71 ± 1.14 | 73.48 ± 1.42 | 73.14 ± 1.23 | 72.53 ± 1.33 | 73.48 ± 1.22 | 73.66 ± 1.15 |
| 5 | 73.30 ± 2.10 | 80.05 ± 2.23 | 79.86 ± 2.95 | 71.58 ± 3.02 | 67.59 ± 2.23 | 68.06 ± 2.59 |
| 10 | 68.56 ± 2.23 | 72.04 ± 2.54 | 73.14 ± 2.61 | 68.75 ± 3.09 | 70.70 ± 2.21 | 67.85 ± 2.36 |
| 15 | 66.78 ± 2.25 | 68.78 ± 2.36 | 71.34 ± 2.31 | 65.82 ± 2.59 | 69.49 ± 2.65 | 65.43 ± 1.99 |
| 20 | 65.25 ± 2.28 | 68.19 ± 3.02 | 69.04 ± 2.65 | 64.45 ± 2.16 | 68.25 ± 2.45 | 63.25 ± 2.31 |
| 25 | 60.44 ± 3.11 | 67.12 ± 2.89 | 62.85 ± 2.44 | 62.30 ± 2.81 | 66.49 ± 2.88 | 61.91 ± 2.58 |
| 30 | 56.30 ± 3.02 | 62.68 ± 2.49 | 61.48 ± 2.13 | 59.96 ± 2.91 | 61.97 ± 2.75 | 60.84 ± 2.64 |
| 35 | 53.45 ± 2.06 | 59.42 ± 2.61 | 58.42 ± 2.19 | 54.86 ± 2.64 | 60.33 ± 2.65 | 59.12 ± 2.23 |
| 40 | 51.65 ± 1.98 | 52.33 ± 2.66 | 50.43 ± 2.73 | 54.76 ± 2.65 | 56.59 ± 2.46 | 57.31 ± 2.87 |
With addition of 5 wt% ASA, the tensile strength of the PMMA/ASA-2 alloys increased from 73.48 to 80.05 MPa, while the tensile strength increased to 79.86 MPa for the PMMA/ASA-3 alloys. However, when more than 5 wt% ASA was added, the yield strengths of the alloys decreased. This suggests that ASA-2 and ASA-3 exhibited a good balance between toughness and rigidity. More importantly, KH570 improved the toughening efficiency of the core/shell organic–inorganic hybrid polymer nanoparticles when the KH570 content was below 10 wt%.
Elongation at break of the PMMA/ASA alloy
The elongation at break is another factor that can be used to characterize the toughness of materials. The stress–strain curves of the PMMA/ASA alloys specimens are shown in Fig. 11 and the elongation at break values are given in Table 10. With increasing addition of ASA, the elongation at break increased, indicating that the toughness of the blend improved with increasing ASA content. For the PMMA/ASA-2 alloys, with the addition of 35 wt% ASA-2, the elongation at break increased from 11.92% to 65.44%, which is 5.5 and 2.1 times higher than the elongation at break for pure PMMA and the PMMA/ASA-1 (65/35) alloys. For the PMMA/ASA-3 alloys, with the addition of 35 wt% ASA-3, the elongation at break increased from 6.87% to 41.06%, which is 6.0 and 1.3 times higher than the elongation at break of pure PMMA and the PMMA/ASA-1 (65/35) alloys, indicating the high toughening efficiency of ASA-2 and ASA-3 in the PMMA/ASA alloys. The elongation at break results are consistent with the impact strength results. The alloys showed elastic–plastic deformation behavior. In general, the degree of plasticity increased with increasing ASA content. The increase in the degree of plasticity with increasing ASA content is manifested as a gradual decrease in the loading drop after yield (strain softening), which results in a gradual increase in post-yield deformation, and hence the elongation at break. The gradual increase for 40 wt% ASA content was lower than for 35 wt% ASA content.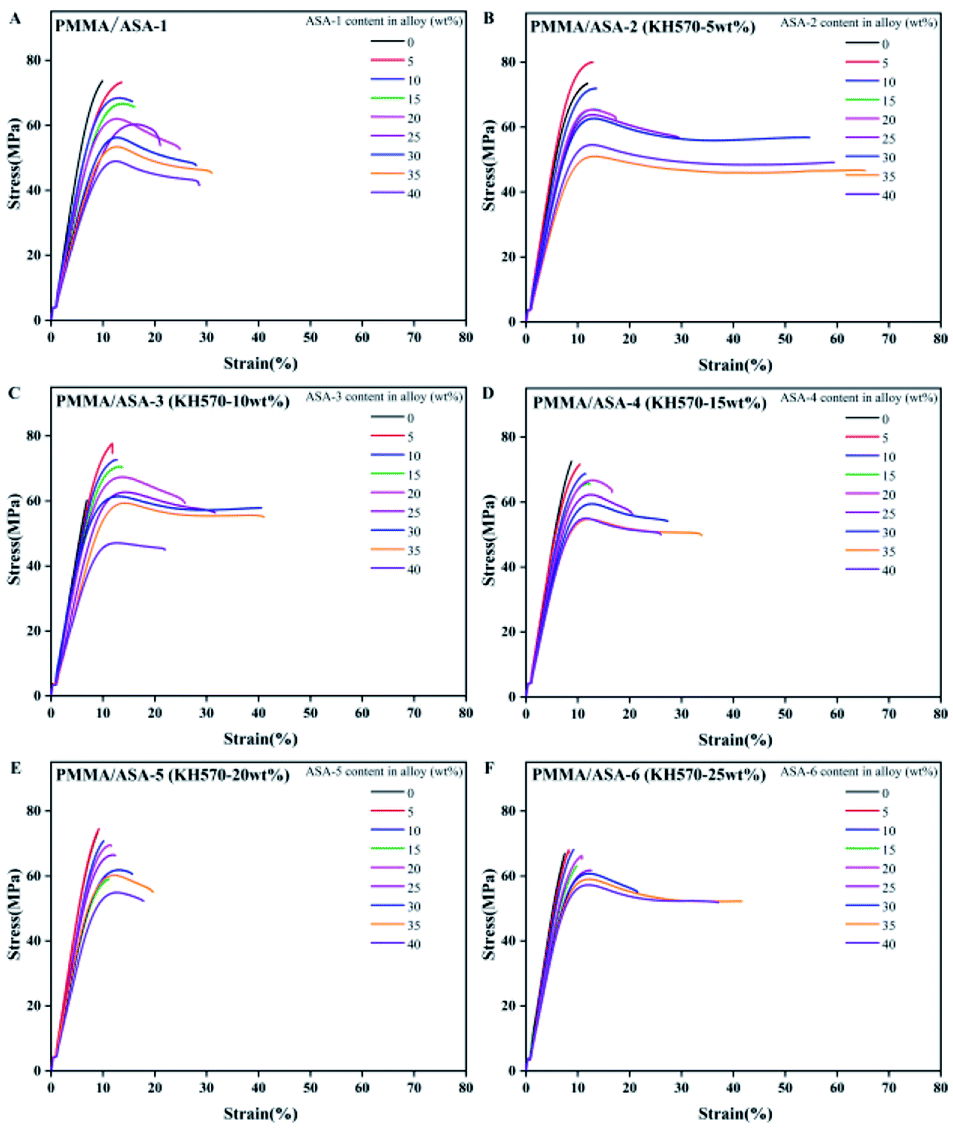 | ||
| Fig. 11 Stress–strain curves of the PMMA/ASA alloys. | ||
| ASA content in alloy (wt%) | Elongation at break (%) | |||||
|---|---|---|---|---|---|---|
| ASA-1 | ASA-2 | ASA-3 | ASA-4 | ASA-5 | ASA-6 | |
| 0 | 9.92 | 11.92 | 6.87 | 8.82 | 9.02 | 7.47 |
| 5 | 13.61 | 12.90 | 11.82 | 10.40 | 9.21 | 8.26 |
| 10 | 15.70 | 13.59 | 12.68 | 11.47 | 10.14 | 9.18 |
| 15 | 16.09 | 14.24 | 13.72 | 12.29 | 11.60 | 9.78 |
| 20 | 24.83 | 17.32 | 25.79 | 16.59 | 12.36 | 10.81 |
| 25 | 21.05 | 29.50 | 31.65 | 20.51 | 11.12 | 12.63 |
| 30 | 27.97 | 54.76 | 40.56 | 27.34 | 15.70 | 21.56 |
| 35 | 30.99 | 65.44 | 41.06 | 33.90 | 19.59 | 41.62 |
| 40 | 28.57 | 59.43 | 21.93 | 26.02 | 17.84 | 37.10 |
Toughening mechanism
The proposed structural diagram of ASA is shown in Fig. 12. For ASA-1 (without KH570 modification), the core is pure PBA rubber without the silicon polymer structure. When the PBA rubber particles are subjected to an external force, it is easy to cause crazing, generate holes, and disperse in the SAN matrix phase with the effect of stress concentration, so as to absorb the impact energy. For the Si-ASA HNPs synthesized in this study, the Si-PBA core is a copolymer of butyl acrylate and KH570. As reported in the literature, the toughening mechanism of silicone (KH570) is that some Si–O–Si network structures form in the polymer matrix. Silicone plays dual roles as a compatibilizer and a chain extender, which not only improves interfacial adhesion between the PBA particles and SAN copolymer, but also increases chain entanglement of PBA. Because the monomer grafting reaction site in the ASA system is the double bond (CC), KH570 contains unsaturated CC bonds, which can react with BA, St, and AN.
 | ||
| Fig. 12 Proposed structural diagram of ASA. | ||
As the KH570 content increases, copolymerization of KH570 and BA increases the degree of crosslinking of the polymer. In addition, owing to hydrolysis and condensation of KH570, the Si–O–Si network in the Si-PBA rubber core increases, which results in more residual double bonds on the surface of the Si-PBA rubber core, increasing the probability of grafting shell monomers and improving the mechanical properties.
When the ASA core–shell ratio is constant, the number of shell monomers is certain. When the KH570 content is too high, there are too many residual double bonds on the surface of the Si-PBA rubber core, and the grafted shell cannot completely cover the rubber core. When the rubber core is impacted by an external force, part of the force will directly act on the rubber core and it cannot be buffered, resulting in performance degradation. In addition, after the degree of polymer cross-linking reaches a certain level, the degree of hydrolysis and condensation of KH570 increases, and the compactness of the formed three-dimensional network structure decreases. When it is impacted by an external force, the molecular chain can easily slip, which results in a decrease in the performance. The difference in the toughening behavior is speculated to arise from the morphological effects caused by a thicker interphase, which in turn results in better coverage by the PMMA shell and a more uniform distribution of the toughening particles in the PMMA matrix.37
Conclusions
We have investigated the effect of KH570 modification on ASA and its composites. Core/shell organic–inorganic hybrid polymer nanoparticles were successfully prepared by emulsion polymerization and their structure was verified by FTIR spectroscopy. Various properties of ASA and its composites were investigated, and the toughening mechanism of ASA was explored. The main results are as follows.(1) With addition of KH570, the size of the PBA rubber particles increased and the grafting rate of ASA increased from 91.72% to 96.76%. Silicone played dual roles of a compatibilizer and a chain extender, which not only improved the interfacial adhesion between the PBA particles and SAN copolymer, but also increased chain entanglement of ASA.
(2) DSC and TGA analysis results suggested that KH570 not only increased the glass-transition temperature of the PBA core, but it also increased the glass-transition temperature of the SAN shell, which was the main reason for the considerable improvement in the heat resistance and mechanical performance. It is also conducive to processing and forming.
(3) Pure PMMA is very hard and brittle owing its molecular structure. The impact strength and elongation at break greatly improved by adding ASA. The impact strength and elongation at break of the PMMA/ASA-2 and PMMA/ASA-3 alloys were higher than those of the PMMA/ASA-1 alloys, indicating that the core/shell organic–inorganic hybrid polymer nanoparticles (Si-ASA) can effectively toughen brittle PMMA when the KH570 content is below 10 wt%.
(4) In particular, the PMMA/ASA-2 alloys exhibited a good balance between toughness and rigidity, indicating that ASA-2 is the most suitable impact modifier. Silicone-containing polymers acted as a compensation medium and balanced the lost strength by modifying the molecular structure and improving the thermal behavior. Thus, in the process of making high-toughness ASA, it is necessary to add a silicone monomer, such as KH570, to balance the mechanical and thermal properties.
(5) The core–shell structure of the ASA HNPs could be adjusted to tune and optimize the properties. This work provides a facile and efficient method for manufacturing high-performance ASA and promoting its application in a wider range of fields.
Conflicts of interest
There are no conflicts of interest to declare. We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled “Preparation of Core/shell Organic–inorganic Hybrid Polymer Nanoparticles and Their Application to Toughening Poly(methyl methacrylate)”.Acknowledgements
This work was financially supported by the Project of the State Key Laboratory of Environment-Friendly Energy Materials, Southwest University of Science and Technology (Nos. 17FKSY0102, 18FKSY0206, and 20fksy03). We thank the Sichuan Civil-Military Integration Institute for support with the DSC measurements.References
- U. Alia, K. J. B. A. Karima and N. A. Buanga, Polym. Rev., 2015, 55, 678–705 CrossRef.
- M. Ishikawa, H. Ogawa and I. Narisawa, J. Macromol. Sci., Part B: Phys., 1981, 19, 421–443 CrossRef.
- B. J. Ash, D. F. Rogers, C. J. Wiegand, L. S. Schadler, R. W. Siegel, B. C. Benicewicz and T. Apple, Polym. Compos., 2002, 23, 1014–1025 CrossRef CAS.
- L. Yu and M. Zhang, China Synth. Resin Plast., 2009, 26, 34–37 CAS.
- J. Zeng, B. Saltysiak, W. S. Johnson, D. A. Schiraldi and S. Kumar, Composites, Part B, 2004, 35, 245–249 CrossRef.
- A. Hassan and B. Haworth, J. Mater. Process. Technol., 2006, 172, 341–345 CrossRef CAS.
- V. Nelliappan, M. S. El-Aasser, A. Klein, E. S. Daniels, J. E. Roberts and R. A. Pearson, J. Appl. Polym. Sci., 1997, 65, 581–593 CrossRef CAS.
- L. Guo, L. Ren, M. Li, Y. Liu, M. Zhang and H. Zhang, China Plast., 2012, 1, 18–22 Search PubMed.
- L. Yu and M. Zhang, China Plast. Ind., 2008, 36, 55–56 CAS.
- C. Huang, Y. Li, K. Gong, L. Yan, S. Wang and H. Li, China Plast. Ind., 2003, 31, 22–24 CAS.
- Q. B. Si, C. Zhou, H. D. Yang and H. X. Zhang, Eur. Polym. J., 2007, 43, 3060–3067 CrossRef CAS.
- G. E. McKee, A. Kistenmacher, H. Goerrissen and M. Breulmann, Modern Styrenic Polymers: Polystyrenes and Styrenic Copolymers, 2003, pp. 341–362 Search PubMed.
- S. C. Tjong and Y. Z. Meng, Eur. Polym. J., 2000, 36, 123–129 CrossRef CAS.
- G. Iannuzzi, B. Mattsson and M. Rigdahl, Polym. Eng. Sci., 2013, 53, 1687–1695 CrossRef CAS.
- R. M. Santos, G. L. Botelho and A. V. Machado, J. Appl. Polym. Sci., 2010, 116, 2005–2014 CAS.
- M. R. Moghbeli and S. Tolue, Iran. Polym. J., 2011, 20, 137–146 CAS.
- M. J. Bijhanmanesh and N. Etesami, J. Appl. Polym. Sci., 2016, 133, 44079 CrossRef.
- J. Yu, P. Feng and H. Zhang, Polym. Eng. Sci., 2010, 50, 295–301 CrossRef CAS.
- Z. Zhang, S. Wang, J. Zhang, W. Zhu and T. Tian, Polym. Test., 2016, 51, 1–5 CrossRef CAS.
- X. Zhang and J. Zhang, J. Appl. Polym. Sci., 2018, 135, 46839 CrossRef.
- Y. G. Du, J. G. Gao, J. B. Yang and X. Q. Liu, J. Polym. Res., 2012, 19, 9993 CrossRef.
- S. Rimdusit, S. Damrongsakkul, P. Wongmanit, D. Saramas and S. Tiptipakorn, J. Reinf. Plast. Compos., 2011, 30, 1691–1702 CrossRef CAS.
- Z. Zhang, B. Li, S. Chen, J. Zhang and X. Jin, Polym. Adv. Technol., 2012, 23, 336–342 CrossRef CAS.
- L. P. Jiang, C. X. He and J. J. Fu, Constr. Build. Mater., 2019, 223, 133–141 CrossRef CAS.
- J. Qian, X. Chen and P. He, Appl. Chem., 2003, 20, 1200–1203 CAS.
- G. Mathew, J. M. Rhee and C. Nah, Polym. Eng. Sci., 2010, 46, 1455–1460 CrossRef.
- S. Tolue, M. R. Moghbeli and S. M. Ghafelebashi, Eur. Polym. J., 2009, 45, 714–720 CrossRef CAS.
- Y. L. Liang, H. J. Sue and R. Minkwitz, J. Appl. Polym. Sci., 2012, 126, 1088–1096 CrossRef CAS.
- B. Xiang and J. Zhang, Int. J. Polym. Anal. Charact., 2017, 22, 534–544 CrossRef CAS.
- H. Y. Jiang, B. J. Liu, Y. J. Deng, J. Y. Ma and H. X. Zhang, Polym. Adv. Technol., 2015, 26, 182–189 CrossRef CAS.
- X. Shi, W. Cheng, Z. Lu, Q. Du and Y. Yang, Polym. Bull., 2002, 48, 389–396 CrossRef CAS.
- A. S. Orlov, S. A. Kiselev, E. A. Kiseleva, A. V. Budeeva and V. I. Mashukov, J. Appl. Spectrosc., 2013, 80, 47–53 CrossRef CAS.
- X. Yan, W. Zhou, X. Zhao, J. Xu and P. Liu, J. Therm. Anal. Calorim., 2016, 124, 719–728 CrossRef CAS.
- R. Shanti, A. N. Hadi and Y. S. Salim, RSC Adv., 2016, 7, 112–120 RSC.
- W. Huang, Z. Mao, Z. Xu, B. Xiang and J. Zhang, Eur. Polym. J., 2019, 120, 109247 CrossRef.
- C. Zhou, H. Liu, M. Chen, G. Wu and H. Zhang, Polym. Eng. Sci., 2012, 52, 2523–2529 CrossRef CAS.
- M. Fasihi and H. Mansouri, J. Appl. Polym. Sci., 2016, 133–141 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |