
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization and application of natural and recombinant butelase-1 to improve industrial enzymes by end-to-end circularization†
Xinya Hemu a,
Xiaohong Zhanga,
Giang K. T. Nguyenb,
Janet Toa,
Aida Serracd,
Shining Looa,
Siu Kwan Szea,
Chuan-Fa Liua and
James P. Tam*a
a,
Xiaohong Zhanga,
Giang K. T. Nguyenb,
Janet Toa,
Aida Serracd,
Shining Looa,
Siu Kwan Szea,
Chuan-Fa Liua and
James P. Tam*a
aSchool of Biological Sciences, Nanyang Technological University, 637551, Singapore. E-mail: jptam@ntu.edu.sg
bWIL@NUS Corporate Lab, MD6 Centre for Translational Medicine, Wilmar International Limited, National University of Singapore, 117599, Singapore
cIMDEA Food Research Institute, +Pec Proteomics, Campus of International Excellence UAM+CSIC, Old Cantoblanco Hospital, 8 Crta. Cantoblanco, Madrid 28049, Spain
dIproteored – Instituto de Salud Carlos III (ISCIII), Campus UAM, Cantoblanco, Madrid 28049, Spain
First published on 30th June 2021
Abstract
Butelase-1, an asparaginyl endopeptidase or legumain, is the prototypical and fastest known Asn/Asp-specific peptide ligase. It is highly useful for engineering and macrocyclization of peptides and proteins. However, certain biochemical properties and applications of naturally occurring and recombinant butelase-1 remain unexplored. Here we report methods to increase the yield of natural and bacterial expressed recombinant butelase-1 and how they can be used to improve the stability and activity of two important industrial enzymes, lipase and phytase, by end-to-end circularization. First, the yield of natural butelase-1 was increased 3-fold to 15 mg kg−1 by determining its highest distribution which is found in young tissues, such as shoots. The yield of recombinantly-produced soluble butelase-1 was improved by promoting cytoplasmic disulfide folding, codon changes, and truncation of the N-terminal pro-domain. Natural and recombinant butelase-1 displayed similar ligase activity, physical stability, and salt tolerance. Furthermore, the processing and glycosylation sites of natural and recombinant butelase-1 were determined by proteomic analysis. Storage conditions for both forms of butelase-1, frozen or lyophilized, were also optimized. Cyclization of lipase and phytase mediated by either soluble or immobilized butelase-1 was highly efficient and simple, and resulted in increased thermal stability and enhanced enzymatic activity. Overall, improved production of butelase-1 can be exploited to improve the biocatalytic efficacy of lipase and phytase by end-to-end cyclization. In turn, ligase-improved enzymes could be a general and environmentally friendly strategy for producing more stable and efficient industrial enzymes.
Introduction
Enzymes, particularly proteinaceous enzymes, have attracted increasing interest as environmentally friendly, aqueous-compatible, and highly efficient biocatalysts for a wide range of industrial applications.1 Two popular industrial enzymes, lipase and phytase, are produced in metric tons each year to meet the needs of billion-dollar global markets.2,3 Lipases are enzymes which digest lipids into fatty acids and glycerol. Thermostable lipases have found wide applications in food, detergent, pharmaceutical, biofuel, leather industries, and especially, waste management.4 Phytases are enzymes which release inorganic phosphorous from phytate, a dominant, insoluble and indigestible form of phosphate in plants. They are extensively used as additives for plant-based animal feed as well as human nutrition to digest phytate, a major phosphorous source.5,6 Sustained efforts to boost the production, efficiency, stability, and specificity of industrial enzymes include approaches such as site-directed mutagenesis, directed evolution, post-production modifications, and immobilization.7–13 Another emerging environmentally friendly approach to improve industrial enzymes is through other enzymes.Our laboratory has a long-standing interest in chemoenzymatic ligation to form peptide bonds through a proximity-driven O/S–N acyl migration mechanism, which is shared by both chemical and enzymatic peptide ligation.14–21 We recently discovered butelase-1, a prototypic peptide asparaginyl ligase (PAL).22 PALs belong to the C13 subfamily of cysteine proteases (EC 3.4.22.34), which are well-represented by legumains, also known as asparaginyl endopeptidases (AEPs).23 AEPs cleave the peptide bond after an Asn/Asp (Asx) residue. In contrast, PALs catalyse peptide bond formation after an Asx. PALs recognize the simple tripeptide motif Asx–Xaa–Yaa and form a new Asx–Zaa peptide bond either intramolecularly to give a cyclized product or intermolecularly to give a linear ligated product.
End-to-end or N-to-C cyclization of the peptidyl backbone is an accepted approach for improving protein stability which minimizes proteolytic degradation by exopeptidases and reduces the flexibility of less-structured N- and C-terminal ends.24 However, the molecular size and complexity of enzymes make cyclization by chemical methods challenging. On the other hand, cyclizing a proteinaceous enzyme could be efficiently accomplished by a peptide bond forming-ligase, such as butelase-1 because of its exquisitely high site-specificity.22,25,26 Currently, only a few examples of enzyme-mediated cyclization of industrial enzymes have been reported. They include N-to-C cyclization of phytase by a self-splicing intein27 and by a SpyLigase forming an isopeptide bond.28 In both examples, intein and SpyLigase are co-expressed with the target enzyme as a fusion protein, rendering them non-reusable, in contrast to free-standing and reusable butelase-1.
Here we report the preparation, characterization and use of the natural and recombinant butelase-1 to improve the thermostability and activity of two industrial enzymes by N-to-C cyclization. This work aims to provide a simple, efficient and environmentally friendly ligase-mediated approach to meet the growing needs for improving industrial enzymes. In addition, we show the high level of natural butelase-1 in young plant tissues and the optimized storage condition of butelase-1.
Results and discussion
High level of butelase-1 in young shoots of Clitoria ternatea leads to a 3-fold increase in yield
Previously, we reported an isolation yield of 5 mg kg−1 butelase-1 from the pods of C. ternatea.22 To improve yield, tissues of C. ternatea were profiled to determine their expression level of butelase-1. Nine different parts were collected, including shoots, young leaves, old leaves, flowers, as well as 1-, 2-, 3-week-old pods (shells and seeds). A synthetic 31-residue cyclotide precursor of kalata-B1 (kB1-HV, 3044 Da) containing the C-terminal tripeptide butelase-1 recognition motif NHV was used as a substrate for the butelase-mediated ligation yielding the 29-residue cyclic kB1 (2891 Da). The level of butelase-1 activity in each tissue extract was determined by the yield of cyclic kB1 using matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) (Fig. S1†).The amount of butelase-1 in various crude tissue extracts was estimated to range from 20 to >60 mg kg−1 by comparing the ligase activity of tissue extracts with purified natural butelase-1 (nBu1) in the cyclization of kB1-HV. Fig. 1A shows that young tissues, such as shoots, displayed the highest level of butelase-1 activity, followed by flowers and 1-week-old pods; aged tissues, such as the 3-week-old pods, produced the lowest ligase activity. It is worth noting that most tissues produced a low level of Asn-specific protease activity, which yielded <5% linear kB1 at pH 6.0. The only exception was old leaves, which produced an equal amount of cyclic and linear kB1. An increasing amount of hydrolysed product (linear kB1) in old leaves suggested high expression level of AEPs, which may be associated with protein degradation and cell death in old tissues.29,30
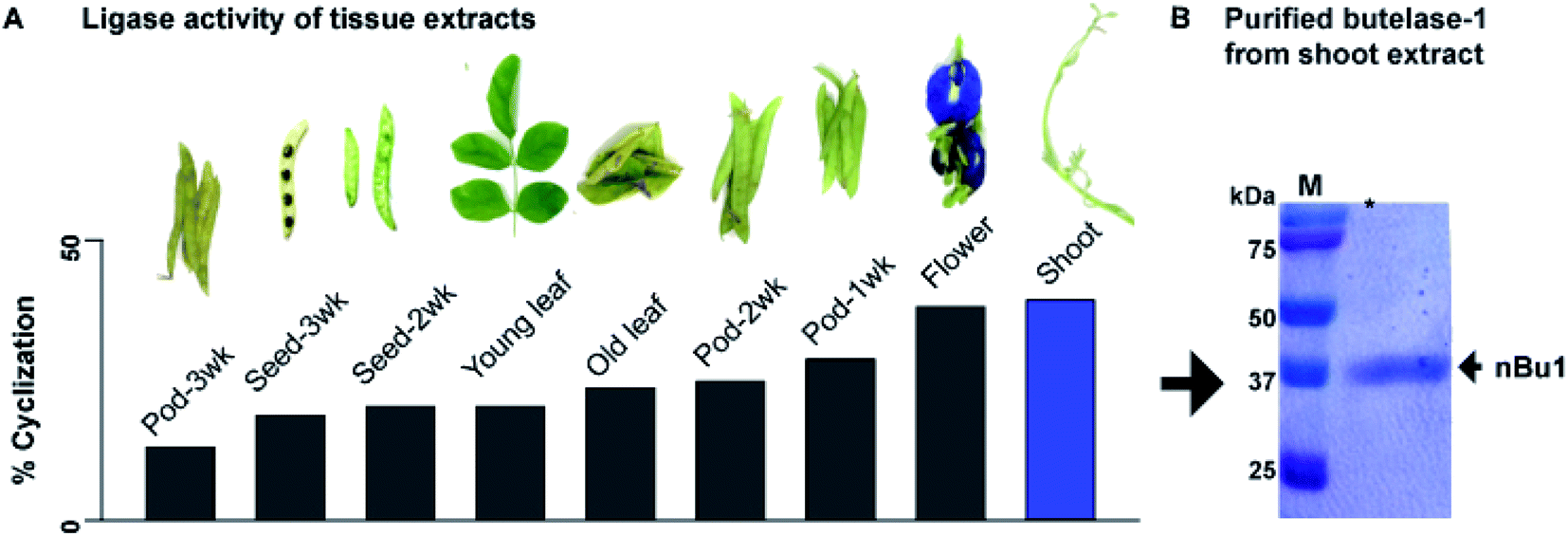 | ||
| Fig. 1 Isolation of butelase-1 from C. ternatea shoot extracts. (A) Ligase activity screening suggested young shoots express the highest level of butelase-1. (B) Purity of isolated nBu1 determined by SDS-PAGE. | ||
Since shoot extracts contained the highest ligase activity, these tissues were used to isolate nBu1 instead of pods. The previous isolation protocol31 was simplified to a 3-step chromatography process that includes anion exchange flash chromatography, anion-exchange fast protein liquid chromatography (FPLC), and size exclusion FPLC (Fig. S2†). In addition, the ammonium sulfate precipitation step that leads to butelase-1 degradation was omitted and polyvinylpolypyrrolidone was added to prevent enzyme oxidation by phenolic compounds in plant tissues.32 Fig. 1B shows the purity of the isolated butelase-1 by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). These accumulative improvements resulted in an isolation yield of 15 mg kg−1 of nBu1 from young shoots, a 3-fold increase compared to the previous protocol.31
Optimizing recombinant expression of butelase-1
Previous reports of recombinant platforms for the production of PALs and AEPs include Escherichia coli, Pichia pastoris, parasites (LEXSY system), and insect cells (Baculovirus system).33–37 Our early attempts to produce rBu1 in E. coli Rosetta (DE3) with the signal peptide M1-G20 replaced by a His6 affinity tag were unsatisfactory. Although rBu1 was expressed in high yield (200 mg L−1), it was largely produced as an insoluble form.22 In a continued effort to produce rBu1, various avenues for improvement were explored, four of which gave useful results. First, the expression strain was changed to disulfide-promoting Shuffle-T7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 to facilitate formation of three conserved disulfide bonds, which significantly reduced the amount of insoluble form produced. Second, the natural cDNA sequence of butelase-1 was used to construct rBu1(ΔSP) with its signal peptide substituted by a His6 tag instead of the previous codon optimization strategy which gave mainly misfolded insoluble products. It is likely that codon-optimized RNA fold into unexpected structures that affect translation efficiency39 although another group has reported bacterial expression of butelase-1 using a codon-optimized sequence.35 In the present case, rBu1(ΔSP) afforded >10 times higher expression level than the codon-optimized sequence Opt-rBu1(ΔSP-Ubi) which contains a His6-ubiquitin tag to replace its signal peptide (Fig. 2B and S3†). Third, the first five residues (IRDDF) were deleted from the pro-domain to derive the truncated construct rBu1(Δ1–25) which further improved expression levels (Fig. 2B). Fourth, acid-induced auto-activation was required to activate rBu1 proenzyme by removing both the cap domain which served as an inhibitor of the active core domain and parts of the N-terminal pro-domain. Using the optimized auto-activation condition of pH 4.0, at 37 °C for 2 h, a single band corresponding to the active form was obtained and the cap domain was completely degraded (Fig. 3A and S4†).
38 to facilitate formation of three conserved disulfide bonds, which significantly reduced the amount of insoluble form produced. Second, the natural cDNA sequence of butelase-1 was used to construct rBu1(ΔSP) with its signal peptide substituted by a His6 tag instead of the previous codon optimization strategy which gave mainly misfolded insoluble products. It is likely that codon-optimized RNA fold into unexpected structures that affect translation efficiency39 although another group has reported bacterial expression of butelase-1 using a codon-optimized sequence.35 In the present case, rBu1(ΔSP) afforded >10 times higher expression level than the codon-optimized sequence Opt-rBu1(ΔSP-Ubi) which contains a His6-ubiquitin tag to replace its signal peptide (Fig. 2B and S3†). Third, the first five residues (IRDDF) were deleted from the pro-domain to derive the truncated construct rBu1(Δ1–25) which further improved expression levels (Fig. 2B). Fourth, acid-induced auto-activation was required to activate rBu1 proenzyme by removing both the cap domain which served as an inhibitor of the active core domain and parts of the N-terminal pro-domain. Using the optimized auto-activation condition of pH 4.0, at 37 °C for 2 h, a single band corresponding to the active form was obtained and the cap domain was completely degraded (Fig. 3A and S4†).
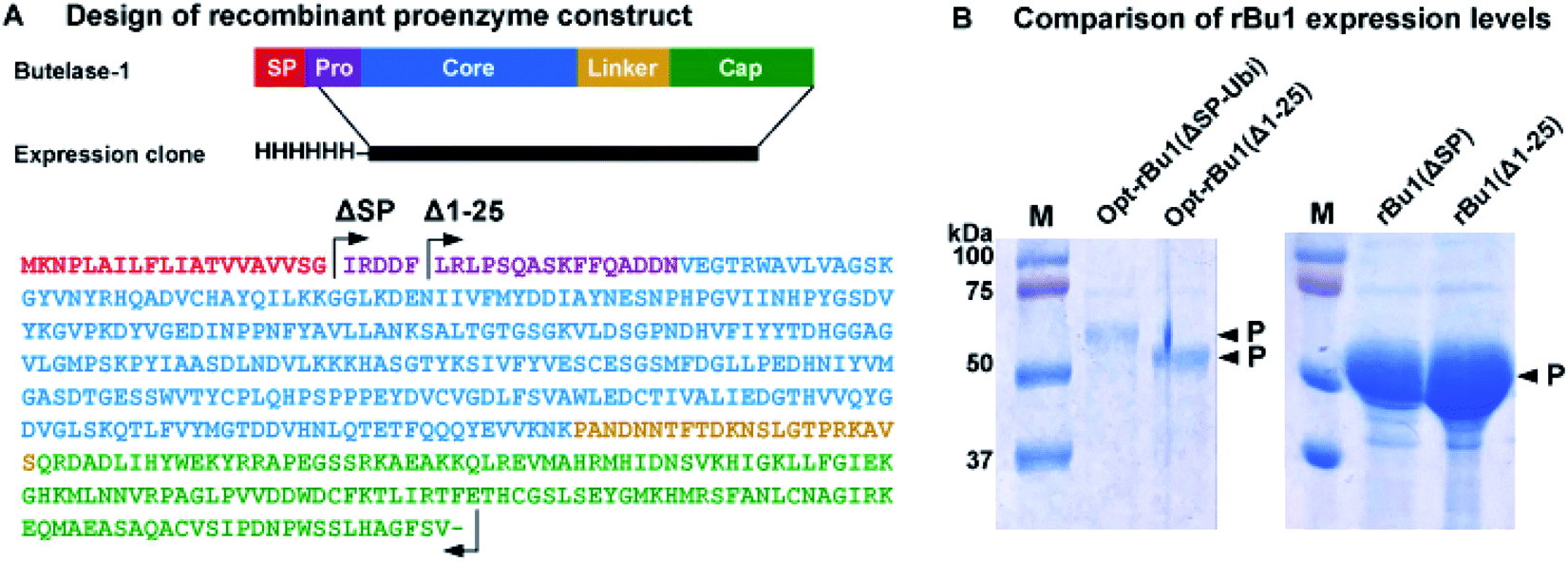 | ||
| Fig. 2 Recombinant expression and sequence analysis of butelase-1. (A) Constructs of rBu1 contains an N-terminal His6-tag followed by the proenzyme sequence from I21 or Leu26 to Val482. Protein domains were predicted based on the model structure of the butelase-1 proenzyme. SP, signal peptide. Pro, pro-domain. Core, core or active domain. Cap, cap or inhibitory domain. (B) Comparison of expression levels of four rBu1 constructs under the same expression conditions. | ||
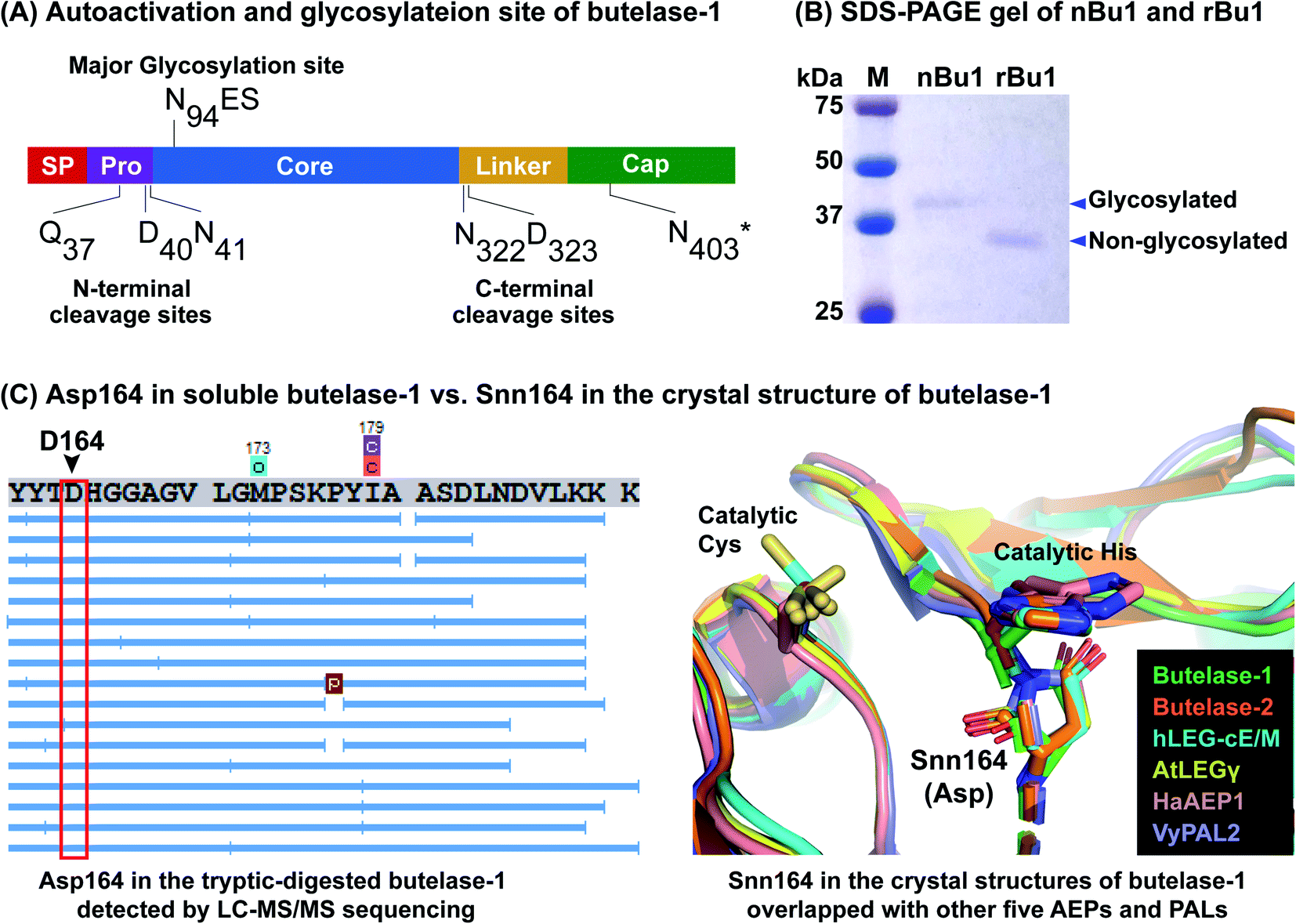 | ||
| Fig. 3 Proteomic characterization of nBu1 and activated rBu1. (A) Determination of auto-activation sites and N-glycosylation site. N403*, estimated cleavage site based on intermediate band size and LC-MS/MS sequencing data. (B) Coomassie blue-stained SDS-PAGE gel of nBu1 and the activated rBu1. nBu1, calculated molecular weight (MW) = 32 kDa, observed MW ≈ 38 kDa. rBu1, calculated MW = 31 kDa, observed MW ≈ 37 kDa. (C) Detection of Asp164 in the soluble butelase-1 by LC-MS/MS sequencing in contrast to the Snn164 in the crystal structure of rBu1 superimposed with other AEPs and PALs. Left panel shows one example of MS/MS sequencing of tryptic digested butelase-1 fragments (image obtained by PEAK Studio after DB search and PTM analysis). Right panel includes structures with PDB codes: butelase-1 (6DHI), butelase-2 (6L4V), human legumain–cystatin E/M complex (4N6O), AtLEGγ (5OBT), HaAEP1 (6AZT), and VyPAL2 (6IDV). | ||
Comparative proteomic analysis of activated rBu1 and nBu1
To determine the processing sites of activated rBu1 and nBu1, amino acid sequencing was performed using a previously developed proteomic approach.37,40 To this end, the SDS-PAGE bands corresponding to nBu1 (38 kDa), rBu1-proenzyme (51 kDa), rBu1-intermediate (43 kDa) and activated rBu1 (37 kDa) were analysed. These bands were cut out, subjected to in-gel trypsin digestion, and then analysed by LC-MS/MS. The proteomic data were analysed using the butelase-1 proenzyme sequence as a template to obtain the sequence and post-translational modifications of each peptide fragment.Fig. 3A summarizes the processing sites of both nBu1 and rBu1. The major processing sites of both are at Gln37 in the pro-domain and Asn331 in the linker region (Fig. S5†). In 38 kDa-nBu1, additional cleavage also occurred at Asp40 or Asn41 in the N-terminus and Asn322 or Asp323 in the C-terminus. Sequencing of the 43 kDa-rBu1 intermediate suggested that the processing of the rBu1-proenzyme involve an additional step in the cap domain, most likely occurring at Asn403 located after the α8 helix. These data agreed with previous reports that the processing of butelase-1 is similar to other PALs and AEPs, which involve multiple steps and sites on both ends.33,37,41,42
LC-MS/MS analysis revealed the major difference between nBu1 and rBu1, which is the presence of post-translational N-linked glycosylation at Asn94 of nBu1 (Fig. 3A). The N-linked glycans on Asn94 of nBu1 included a complex glycan core (1216 Da) and several truncated forms including Hex3HexNac2 (892 Da) and a single hex-N-acylation (203 Da) (Fig. S6†). Consequently, a 1–2 kDa difference was observed by SDS-PAGE between ∼38 kDa-nBu1 and ∼37 kDa-rBu1, although they share the same amino acid sequence. As such, the active forms of butelase-1, either natural or recombinant, migrated slower in SDS-PAGE gel due to the negative net charge (isoelectric points <5.0),43 which produces a 6–7 kDa mass increase in the gel (Fig. 3B). These results agree with our previous study showing that nBu1 are glycosylated and can be non-covalently immobilized on concanavalin A beads and the immobilized nBu1 can be reused for >100 times without diminishing its catalytic activity.22,44
Thus far, 31 crystal structures belonging to 12 different AEPs and PALs have been determined (Protein Data Bank, Table S1†), 21 of which show a succinimide (Snn) at the conserved Asp164 (butelase-1 numbering) preceding the catalytic His165. Snn, also known as aspartimide, is a dehydrated Asp derivative which forms a 5-membered ring with its neighbouring amino acid, and is particularly facile in Asp–Gly and Asp–His sequences.45 The presence of Snn164–His165 in the catalytic site of AEPs and PALS found in crystal structures of different mammalian and plant proenzymes and active forms has been proposed to play a catalytic role in the religation of cap and core domains at basic pH to re-form an AEP zymogen.46,47 However, our proteomic analysis show that no Asp164-to-Snn conversion (−18 Da by dehydration) was detected in either nBu1 or rBu1 (Fig. 3C and S7†). No mass of iso-Asp (c ion +57 Da or z ion −57 Da) was determined in the MS/MS spectra of any Asp164-containing fragments as demonstrated by DeGraan-Weber et al.48 Since the Snn164 is not found in both nBu1 and rBu1 soluble enzymes but found in crystal structure of rBu1, we speculated that the dehydration of Asp164 commonly observed in the crystal structures of AEPs and PALs may occur under specific conditions, such as exposing to a high-energy source in the crystallization–determination procedure, rather than a post-translational modification.
rBu1 is functionally as active and stable as the natural form
The catalytic efficiency of nBu1 and rBu1 was compared in the intramolecular ligation of the 16-residue peptide substrate, GN14-HV (GISTKSIPPISYRNHV, 1768 Da) with a C-terminal NHV tripeptide motif to form the 14-residue cyclic peptide GN14. A fixed enzyme-to-substrate molar ratio of 1:1000 in a series of conditions was used, including eight different pH (4.5 to 8.0), and three temperatures (25, 37, and 42 °C). Reactions were quenched after 5 min, and the product profiles analysed using MALDI-TOF MS (Fig. S8†) showed that rBu1 is equally as potent as nBu1. Fig. 4A shows that both forms display optimal catalytic activity at pH 6–6.5 and 37–42 °C to give 75–80% cyclized cGN14 product within 5 min. At 25 °C, pH 7 was found to be optimal for yielding 40% cGN14. No hydrolytic product GN14 (1532 Da) was observed at pH 5.5–8.0. However, at a pH lower than 5.5, trace amounts (<3%) of the hydrolytic product GN14 were observed after 10 min, consistent with the auto-proteolytic ability of butelase-1 at an acidic pH of 4 to 4.5.
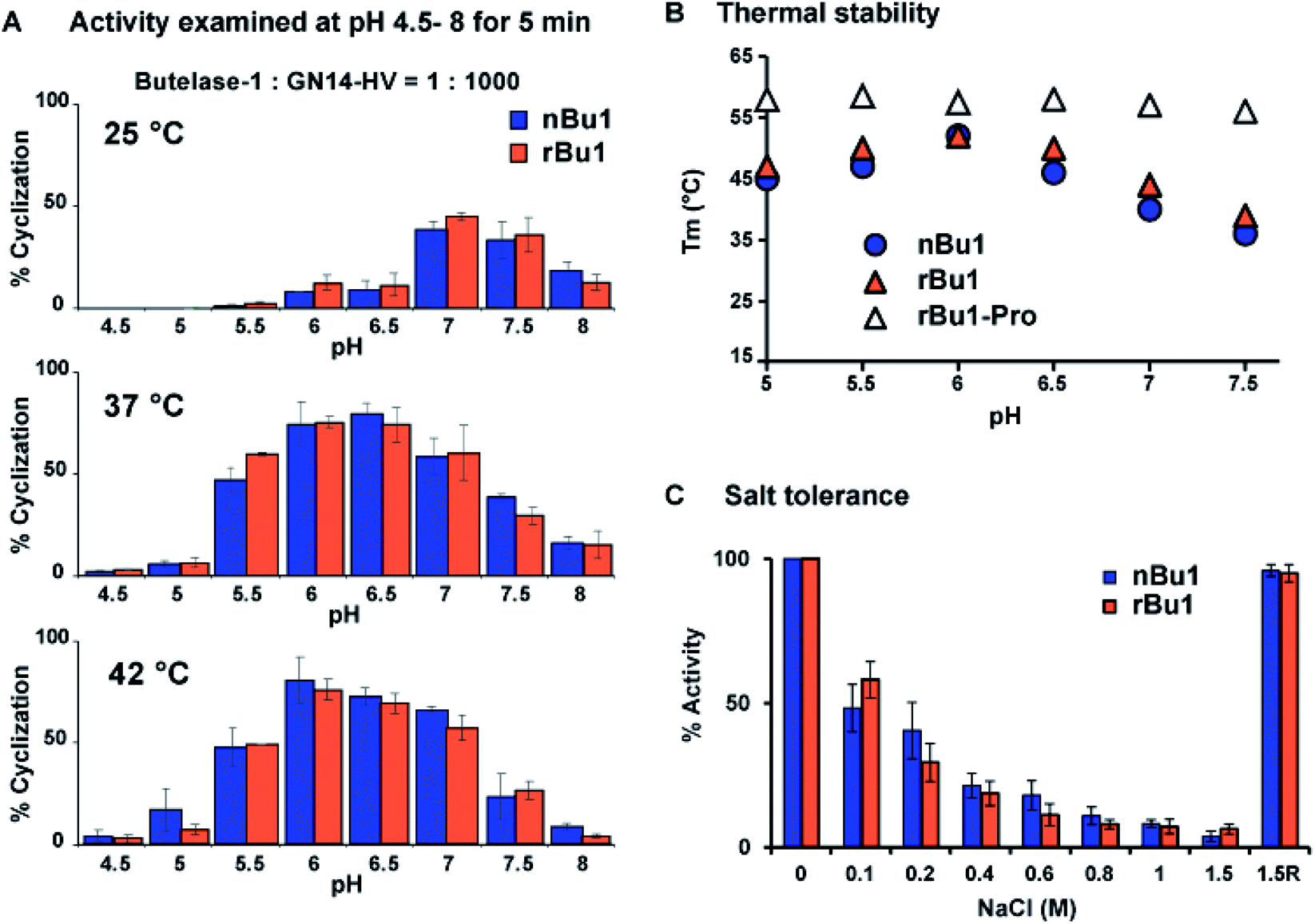 | ||
| Fig. 4 Activity and stability comparison of nBu1, rBu1 and rBu1-pro (rBu1 proenzyme). (A) Summary of yield of cGN14 mediated by nBu1 and rBu1. (B) Thermal shift assays were conducted using SYPRO Orange dye with salt-free buffers at a pH ranging from 5 to 7.5. (C) Ligase activity of butelase-1 in the reaction buffer with 0 to 1.5 M NaCl. 1.5R, enzyme was regenerated by dialysis to remove 1.5 M NaCl. | ||
To examine the thermal stability of nBu1, rBu1, and the rBu1-proenzyme, thermal shift assays were performed at pH 5–7.5 using enzyme solutions prepared with six different reaction buffers. Fig. 4B shows that the thermal stability of rBu1 and nBu1 is similar and pH-dependent. The highest melting temperature (Tm) was at 52 °C and pH 6.0, while the lowest Tm was at 38–40 °C and pH 7.5. The pH-dependent thermal stability of butelase-1 correlates with its pH-dependent catalytic activity, and provides an explanation for the shift of optimal condition from pH 7.0 at 25 °C to pH 6.0 at 42 °C (Fig. 3). Similar to other PALs,44 the rBu1-proenzyme with Tm at 55–57 °C is more stable and less pH-sensitive than the active forms. The Tm of nBu1 in salt-free reaction buffers was about 1–5 °C higher than the previously reported Tm obtained using buffers containing 0.1 M NaCl,44 suggesting that butelase-1 may be destabilized by salt. The effect of ionic strength was then evaluated by comparing the enzymatic activity in 20 mM sodium phosphate buffers containing varying amounts of NaCl from 0 to 1.5 M. The addition of salt exerted an inhibitory effect: butelase-1 activity decreased with an increasing concentration of NaCl. Only 20% butelase-1 catalytic activity remained in the presence of 1.5 M NaCl, but enzymatic activity was restored by removing NaCl (Fig. 4C). Overall, the absence of surface glycan in E. coli-produced rBu1 does not affect its stability or function.
Optimal conditions for storing butelase-1
For storage of butelase-1-like PALs, 20% (w/v) sucrose was used to prevent formation of ice crystals in frozen stocks.37 The same enzyme storage solution was used to test the stability of nBu1 in multiple rounds of freeze–thaw cycles. The activities before and after each freeze (−20 or −80 °C) and thaw (0 °C) cycle were compared by the cyclization of GN14-HV assay as described above. There was no significant activity loss (<3%) after five cycles. In contrast, without the protection by 20% sucrose, ∼5% activity loss per freeze–thaw cycle was observed (Fig. S9†).We also explored conditions for butelase-1 lyophilization. Freeze-drying of proteins involved two main denaturing stresses, freezing stress due to the formation of dendritic ice crystals, increased ionic strength, changed pH, or phase separation and drying stress due to the removal of the protein hydration shell.49 Since the freeze–thaw cycle of butelase-1 caused only about 5% reduction in activity without protectants, the drying phase during lyophilization was expected to be the main cause of activity loss. 16 different conditions include common lyoprotectants disaccharides (sucrose or trehalose), detergents (Tween-20), and bulking reagents (glycine) were examined (Table 1).50 In the absence of additives, <5% activity was recovered upon reconstitution. In contrast, the presence of additives generally improved recovered ligase activities, with a combination of 12% sucrose and 0.01% Tween-20 being the best condition, recovered 93% activity.
| No. | Sucrose (%) | Trehalose (%) | Tween-20 (%) | Glycine (M) | Activity (%) |
|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 0 | <5 |
| 2 | 0 | 0 | 0.01 | 0 | 8 |
| 3 | 6 | 0 | 0 | 0 | 72 |
| 4 | 12 | 0 | 0 | 0 | 81 |
| 5 | 12 | 0 | 0.1 | 0 | 45 |
| 6 | 12 | 0 | 0.05 | 0 | 75 |
| 7 | 12 | 0 | 0.01 | 0 | 93 |
| 8 | 24 | 0 | 0.1 | 0 | 60 |
| 9 | 24 | 0 | 0.05 | 0 | 75 |
| 10 | 24 | 0 | 0.01 | 0 | 49 |
| 11 | 0 | 10 | 0.1 | 0 | 78 |
| 12 | 0 | 10 | 0.05 | 0 | 82 |
| 13 | 0 | 20 | 0.1 | 0 | 65 |
| 14 | 0 | 20 | 0.05 | 0 | 79 |
| 15 | 12 | 0 | 0.01 | 0.1 | 54 |
| 16 | 12 | 0 | 0.05 | 0.2 | 33 |
Efficient cyclization by butelase-1 improved thermal stability and activity of industrial enzymes
The presence of bulky surface glycans on nBu1 can be exploited for immobilization on lectin beads.44 Thus, glycosylated nBu1 was immobilized on concanavalin-A beads to prepare conA–nBu1 beads for the N-to-C cyclization of lipase. A thermoalkalophilic lipase T1.2 isolated from Geobacillus species was selected and shared 93% sequence identity with lipase T1.51,52 The recombinant lipase T1.2 was expressed with butelase-1 recognition signals as a 414-residue linear precursor His6-TEV-GV-T1.2-NHV (Fig. S10†). The purified linear substrate GV-T1.2-NHV was obtained after immobilized metal affinity chromatography (IMAC) purification combined with tobacco etch virus (TEV) protease cleavage. Fig. 5A shows that cyclization of GV-T1.2-NHV by conA–Bu1 beads was completed in 30 min with a 1:500 Bu1-to-lipase ratio at pH 6.5 at room temperature.
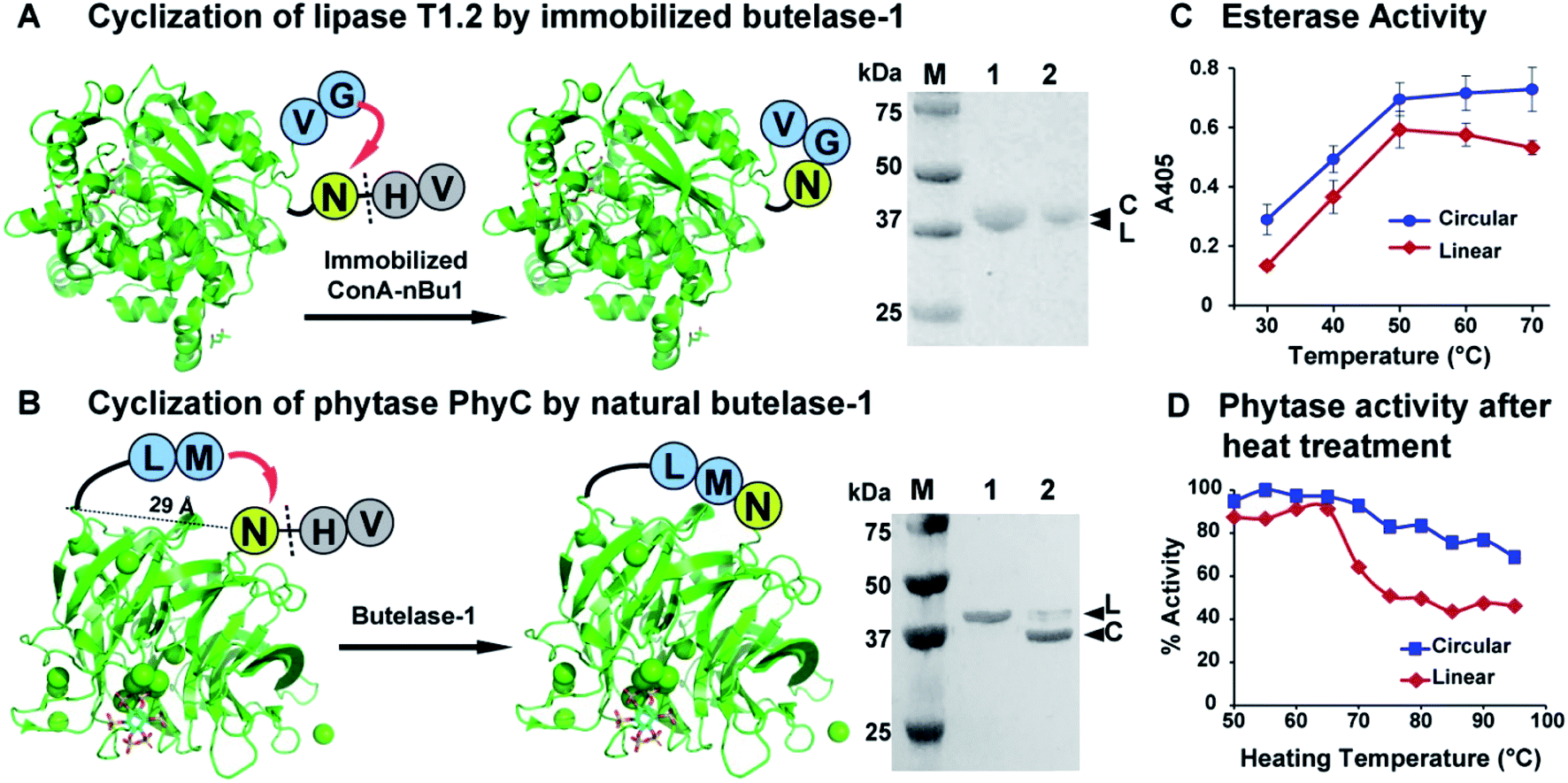 | ||
| Fig. 5 Butelase-mediated cyclization of industrial enzymes and their improvement in thermal stability. (A) Reaction scheme and SDS-PAGE of the lipase T1.2 before and after cyclization. (B) Reaction scheme and SDS-PAGE of the PhyC phytase before and after cyclization. Lane 1 was the purified linear enzyme precursors (L) and lane 2 was the cyclization products. (C) Comparison of esterase activity of linear and circular T1.2. (D) Comparison of phytase activity recovery after heat treatment at 50–95 °C. The activity of linear phytase under optimum conditions at pH 7 and 50 °C was set at 100%. | ||
To circularize phytase, Bacillus phytase PhyC, a well-characterized phytase was selected.53,54 The crystal structure of PhyC reveals a 29 Å gap between the N- and C-termini.55 To produce a cyclic phytase, nine residues were added to the N-terminus, starting with a ligation-promoting dipeptide (Met–Leu), containing a hydrophobic Leu at the P2′′ position to facilitate butelase-1 recognition,22 followed by a His6-tag to facilitate purification (Fig. S11†). The butelase-1 tripeptide recognition signal NHV was added to the C-terminus. Fig. 5B shows that the bacteria-expressed 370-residue linear precursor ML-His6-phytase-NHV was cyclized by nBu1 in 90% yield within 30 min with a Bu1-to-phytase molar ratio of 1:100 at pH 6.0 at 37 °C in the presence of 2 mM CaCl2. The success in cyclization between Asn and Met supported the earlier observation that butelase-1 has broad substrate tolerance to the P1′′ residue which could be any amino acids except Pro.22
The thermal stability of linear and circular enzymes were examined by a thermal shift assay using SYPRO Orange. Lipase T1.2 is thermophilic with a high melting temperature of 65 °C. Since the N- and C-termini are close to each other and flexible, the effect of cyclization on thermal stability is mild that resulted in 1 °C increase in melting temperature (Fig. S12A†). Nevertheless, the esterase activity of lipase could be significantly improved by cyclization. In a colorimetric assay measuring the lipase-mediated hydrolysis of p-nitrophenyl palmitate (pNPP),56 circular T1.2 produced 35% more hydrolytic product p-nitrophenol (λmax = 405, yellow colour) than the linear T1.2 at the optimal reaction condition (70 °C, pH 9) (Fig. 5C).
The melting temperature of circular PhyC increased 6 °C in the salt-free buffer and 10 °C in the presence of 0.2 M NaCl (Fig. S12B†). This improved thermostability of circular PhyC resulted in an improved tolerance against heat treatment, which is a common disinfection practice in feed and food industries. We treated both linear and circular PhyC at 50–95 °C for 5 min followed by renaturation at room temperature for 1 h. A colorimetric phytase activity assay was performed by determining the amount of inorganic phosphate released from phytate via measuring the formation of phosphomolybdate at 700 nm.57 In the post-treatment activity test performed at 50 °C, circular PhyC retained at least 75% activity. In contrast, linear PhyC lost more than half of its activity after heating at 75–90 °C (Fig. 5D).
Significance and conclusions
The present study reports various avenues for improving the production and yield of a free-standing and reusable enzyme, the asparaginyl-specific ligase butelase-1. In turn, this enzyme ligase was used to improve the efficiency and stability of two globally important industrial enzymes via N-to-C circularization. Such an enzyme-to-improve-enzymes approach has practical and fundamental significances.The major fundamental significance is the discovery that butelase-1 is present in every plant tissue parts tested, and particularly high in young tissues. Since butelase-like ligases are responsible for the maturation of host defence cyclotides, their abundant occurrence in young tissues is in agreement with their defence role in young and vulnerable tissues.58–61 Moreover, using shoots as the source to increase the isolation yield to 15 mg kg−1 is a viable approach to obtaining already activated peptide ligases. Glycosylation of nBu1 gives an additional advantage for immobilization on lectin columns.
The major practical significance is that this study provides the first example of a reusable ligase-mediated cyclization of industrial enzymes, lipase and phytase. Furthermore, the results show that either free-standing or immobilized butelase-1 can be used. The N-to-C circularized enzymes gain increased stability, heat tolerance, and activity, which are needed for both production and pollution management. This butelase-mediated cyclization represents a simple, efficient, and environmentally friendly enzyme-improving-enzymes approach that is applicable to other important industrial proteins.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Academic Research Grant Tier 3 (MOE2016-T3-1-003) from the Singapore Ministry of Education (MOE) and a Nanyang Technological University internal funding-Synzymes and Natural Products Center (SYNC). We thank Wilmar International Limited for providing the sequence of lipase T1.2 from their in-house collection.Notes and references
- J. M. Choi, S. S. Han and H. S. Kim, Biotechnol. Adv., 2015, 33, 1443–1454 CrossRef CAS PubMed.
- U. Mrudula Vasudevan, A. K. Jaiswal, S. Krishna and A. Pandey, Bioresour. Technol., 2019, 278, 400–407 CrossRef CAS PubMed.
- P. Chandra, Enespa, R. Singh and P. K. Arora, Microb. Cell Fact., 2020, 19, 169 CrossRef CAS PubMed.
- K.-E. Jaeger and M. T. Reetz, Trends Biotechnol., 1998, 16, 396–403 CrossRef CAS PubMed.
- X. G. Lei, J. M. Porres, E. J. Mullaney and H. Brinch-Pedersen, in Industrial Enzymes: Structure, Function and Applications, ed. J. Polaina and A. P. MacCabe, Springer Netherlands, Dordrecht, 2007, pp. 505–529, DOI: DOI:10.1007/1-4020-5377-0_29.
- X. G. Lei, J. D. Weaver, E. Mullaney, A. H. Ullah and M. J. Azain, Annu. Rev. Anim. Biosci., 2013, 1, 283–309 CrossRef PubMed.
- C. Schmidt-Dannert and F. H. Arnold, Trends Biotechnol., 1999, 17, 135–136 CrossRef CAS PubMed.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- H. Yang, J. Li, H. D. Shin, G. Du, L. Liu and J. Chen, Appl. Microbiol. Biotechnol., 2014, 98, 23–29 CrossRef CAS PubMed.
- I. J. Minten, N. Abello, M. E. Schooneveld-Bergmans and M. A. van den Berg, Appl. Microbiol. Biotechnol., 2014, 98, 6215–6231 CrossRef CAS PubMed.
- J. Chapman, A. Ismail and C. Dinu, Catalysts, 2018, 8, 238 CrossRef.
- Z. Xu, Y. K. Cen, S. P. Zou, Y. P. Xue and Y. G. Zheng, Crit. Rev. Biotechnol., 2020, 40, 83–98 CrossRef CAS PubMed.
- C.-F. Liu and J. P. Tam, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6584–6588 CrossRef CAS PubMed.
- J. P. Tam, Y. A. Lu, C.-F Liu and J. Shao, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 12485–12489 CrossRef CAS PubMed.
- C.-F. Liu, C. Rao and J. P. Tam, J. Am. Chem. Soc., 1996, 118, 307–312 CrossRef CAS.
- C.-F. Liu, C. Rao and J. P. Tam, Tetrahedron Lett., 1996, 37, 933–936 CrossRef CAS.
- L. Zhang and J. P. Tam, Tetrahedron Lett., 1997, 38, 3–6 CrossRef CAS.
- L. Zhang and J. P. Tam, J. Am. Chem. Soc., 1997, 119, 2363–2370 CrossRef CAS.
- J. P. Tam and Y. A. Lu, Protein Sci., 1998, 7, 1583–1592 CrossRef CAS PubMed.
- J. P. Tam, Y. A. Lu and Q. Yu, J. Am. Chem. Soc., 1999, 121, 4316–4324 CrossRef CAS.
- G. K. T. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Nat. Chem. Biol., 2014, 10, 732–738 CrossRef CAS PubMed.
- J. A. Barrett and D. N. Rawlings, Biol. Chem., 2001, 382, 727–733 Search PubMed.
- A. Purkayastha and T. J. Kang, Biotechnol. Bioprocess Eng., 2019, 24, 702–712 CrossRef CAS.
- G. K. Nguyen, X. Hemu, J. P. Quek and J. P. Tam, Angew. Chem., Int. Ed., 2016, 55, 12802–12806 CrossRef CAS PubMed.
- J. P. Tam, N.-Y. Chan, H. T. Liew, S. J. Tan and Y. Chen, Sci. China: Chem., 2020, 63, 296–307 CrossRef CAS.
- S.-Z. PingZhang, W. Lin, M. Yan, Y.-L. Zhao, Z.-L. Lu, W.M. Chen, People′s Republic of China Pat., CN101638642A, 2010.
- C. Schoene, S. P. Bennett and M. Howarth, Sci. Rep., 2016, 6, 21151 CrossRef CAS PubMed.
- I. Hara-Nishimura, N. Hatsugai, S. Nakaune, M. Kuroyanagi and M. Nishimura, Curr. Opin. Plant Biol., 2005, 8, 404–408 CrossRef CAS PubMed.
- N. Hatsugai, M. Kuroyanagi, M. Nishimura and I. Hara-Nishimura, Apoptosis, 2006, 11, 905–911 CrossRef CAS PubMed.
- G. K. Nguyen, Y. Qiu, Y. Cao, X. Hemu, C.-F. Liu and J. P. Tam, Nat. Protoc., 2016, 11, 1977–1988 CrossRef CAS PubMed.
- J. D. Jones, A. C. Hulme and L. S. C. Wooltorton, Phytochemistry, 1965, 4, 659–676 CrossRef CAS.
- E. Dall and H. Brandstetter, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2012, 68, 24–31 CrossRef CAS PubMed.
- L. Zhao, T. Hua, C. Crowley, H. Ru, X. Ni, N. Shaw, L. Jiao, W. Ding, L. Qu, L. W. Hung, W. Huang, L. Liu, K. Ye, S. Ouyang, G. Cheng and Z. J. Liu, Cell Res., 2014, 24, 344–358 CrossRef CAS PubMed.
- A. M. James, J. Haywood, J. Leroux, K. Ignasiak, A. G. Elliott, J. W. Schmidberger, M. F. Fisher, S. G. Nonis, R. Fenske, C. S. Bond and J. S. Mylne, Plant J., 2019, 98, 988–999 CAS.
- N. Pi, M. Gao, X. Cheng, H. Liu, Z. Kuang, Z. Yang, J. Yang, B. Zhang, Y. Chen, S. Liu, Y. Huang and Z. Su, Biochemistry, 2019, 58, 3005–3015 CrossRef CAS PubMed.
- X. Hemu, A. El Sahili, S. Hu, K. Wong, Y. Chen, Y. H. Wong, X. Zhang, A. Serra, B. C. Goh, D. A. Darwis, M. W. Chen, S. K. Sze, C.-F. Liu, J. Lescar and J. P. Tam, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11737–11746 CAS.
- J. Chen, J. L. Song, S. Zhang, Y. Wang, D. F. Cui and C. C. Wang, J. Biol. Chem., 1999, 274, 19601–19605 CrossRef CAS PubMed.
- Y. Wan, M. Kertesz, R. C. Spitale, E. Segal and H. Y. Chang, Nat. Rev. Genet., 2011, 12, 641–655 CrossRef CAS PubMed.
- A. Serra, X. Hemu, G. K. Nguyen, N. T. Nguyen, S. K. Sze and J. P. Tam, Sci. Rep., 2016, 6, 23005 CrossRef CAS PubMed.
- D. N. Li, S. P. Matthews, A. N. Antoniou, D. Mazzeo and C. Watts, J. Biol. Chem., 2003, 278, 38980–38990 CrossRef CAS PubMed.
- K. S. Harris, T. Durek, Q. Kaas, A. G. Poth, E. K. Gilding, B. F. Conlan, I. Saska, N. L. Daly, N. L. van der Weerden, D. J. Craik and M. A. Anderson, Nat. Commun., 2015, 6, 10199 CrossRef CAS PubMed.
- Y. Shi, R. A. Mowery, J. Ashley, M. Hentz, A. J. Ramirez, B. Bilgicer, H. Slunt-Brown, D. R. Borchelt and B. F. Shaw, Protein Sci., 2012, 21, 1197–1209 CrossRef CAS PubMed.
- X. Hemu, J. To, X. Zhang and J. P. Tam, J. Org. Chem., 2020, 85, 1504–1512 CrossRef CAS PubMed.
- T. Geiger and S. Clarke, J. Biol. Chem., 1987, 262, 785–794 CrossRef CAS.
- E. Dall, J. C. Fegg, P. Briza and H. Brandstetter, Angew. Chem., Int. Ed., 2015, 54, 2917–2921 CrossRef CAS PubMed.
- B. Elsasser, F. B. Zauner, J. Messner, W. T. Soh, E. Dall and H. Brandstetter, ACS Catal., 2017, 7, 5585–5593 CrossRef PubMed.
- N. DeGraan-Weber, J. Zhang and J. P. Reilly, J. Am. Soc. Mass Spectrom., 2016, 27, 2041–2053 CrossRef CAS PubMed.
- B. S. Bhatnagar, R. H. Bogner and M. J. Pikal, Pharm. Dev. Technol., 2007, 12, 505–523 CrossRef CAS PubMed.
- W. Wang, Int. J. Pharm., 2000, 203, 1–60 CrossRef CAS PubMed.
- T. C. Leow, R. N. Rahman, M. Basri and A. B. Salleh, Extremophiles, 2007, 11, 527–535 CrossRef CAS PubMed.
- C. Carrasco-Lopez, C. Godoy, B. de Las Rivas, G. Fernandez-Lorente, J. M. Palomo, J. M. Guisan, R. Fernandez-Lafuente, M. Martinez-Ripoll and J. A. Hermoso, J. Biol. Chem., 2009, 284, 4365–4372 CrossRef CAS PubMed.
- N.-C. Ha, B.-C. Oh, S. Shin, H.-J. Kim, T.-K. Oh, Y.-O. Kim, K. Y. Choi and B.-H. Oh, Nat. Struct. Biol., 2000, 7, 147–153 CrossRef CAS PubMed.
- S. Fu, J. Sun, L. Qian and Z. Li, Appl. Biochem. Biotechnol., 2008, 151, 1–8 CrossRef CAS PubMed.
- Y. F. Zeng, T. P. Ko, H. L. Lai, Y. S. Cheng, T. H. Wu, Y. Ma, C. C. Chen, C. S. Yang, K. J. Cheng, C. H. Huang, R. T. Guo and J. R. Liu, J. Mol. Biol., 2011, 409, 214–224 CrossRef CAS PubMed.
- J. Guo, C. P. Chen, S. G. Wang and X. J. Huang, Enzyme Microb. Technol., 2015, 71, 8–12 CrossRef CAS PubMed.
- M. Nassiri and H. Ariannejad, Rep. Biochem. Mol. Biol., 2015, 4, 10–18 Search PubMed.
- S. M. Simonsen, L. Sando, D. C. Ireland, M. L. Colgrave, R. Bharathi, U. Goransson and D. J. Craik, Plant Cell, 2005, 17, 3176–3189 CrossRef CAS PubMed.
- G. K. T. Nguyen, S. Zhang, N. T. K. Nguyen, P. Q. T. Nguyen, M. S. Chiu, A. Hardjojo and J. P. Tam, J. Biol. Chem., 2011, 286, 24275–24287 CrossRef CAS PubMed.
- J. S. Mylne, L. Y. Chan, A. H. Chanson, N. L. Daly, H. Schaefer, T. L. Bailey, P. Nguyencong, L. Cascales and D. J. Craik, Plant Cell, 2012, 24, 2765–2778 CrossRef CAS PubMed.
- E. K. Gilding, M. A. Jackson, A. G. Poth, S. T. Henriques, P. J. Prentis, T. Mahatmanto and D. J. Craik, New Phytol., 2016, 210, 717–730 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, supplementary tables and figures. See DOI: 10.1039/d1ra03763c |
| This journal is © The Royal Society of Chemistry 2021 |