
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclic oxygen exchange capacity of Ce-doped V2O5 materials for syngas production via high-temperature thermochemical-looping reforming of methane†
Asim Riaz ,
Wojciech Lipiński* and
Adrian Lowe*
,
Wojciech Lipiński* and
Adrian Lowe*
Research School of Electrical, Energy and Materials Engineering, The Australian National University, Canberra, ACT 2601, Australia. E-mail: wojciech.lipinski@anu.edu.au; adrian.lowe@anu.edu.au; Tel: +61 2 612 57896 Tel: +61 2 612 54881
First published on 1st July 2021
Abstract
Synthesis gas production via solar thermochemical reduction-oxidation reactions is a promising pathway towards sustainable carbon-neutral fuels. The redox capability of oxygen carriers with considerable thermal and chemical stability is highly desirable. In this study, we report Ce-doped V2O5 structures for high-temperature thermochemical-looping reforming of methane coupled to H2O and CO2 splitting reactions. Incorporation of fractional amounts of large cerium cations induces a V5+ to V3+ transition and partially forms a segregated CeVO4 phase. More importantly, the effective combination of efficient ion mobility of cerium and high oxygen exchange capacity of vanadia achieves synergic and cyclable redox performance during the thermochemical reactions, whereas the pure vanadia powders undergo melting and show non-cyclic redox behaviour. These materials achieve noteworthy syngas production rates of up to 500 mmol molV−1 min−1 during the long-term stability test of 100 CO2 splitting cycles. Interestingly, the cerium ions are mobile between the lattice and the surface of the Ce-doped vanadia powders during the repeated reduction and oxidation reactions and contribute towards the cyclic syngas production. However, this also causes the formation of the CeVO4 phase in Ce-rich powders, which increases the H2/CO ratios and lowers fuel selectivity, which can be controlled by optimizing the cerium concentration. These findings are noteworthy towards the experimental approach of evaluating the oxygen carriers with the help of advanced characterization techniques.
1. Introduction
Cerium oxide (CeO2) is a state-of-the-art oxygen carrier for thermochemical synthesis gas (syngas) production mainly due to its high ion mobility, fast redox kinetics and structure stability at temperatures up to 1600 °C.1 Various morphologies and structural features of ceria such as foams,2 reticulated porous ceramics3 and nanostructures,4 and metal doping in ceria1,5,6 have been briefly explored to achieve considerable oxygen exchange capacity at T < 1600 °C. These research efforts have accomplished high cyclic redox capacity at relatively low temperatures. However, issues like low chemical and structural stability, low redox kinetics and inferior fuel selectivity arise after the structural alteration of ceria. Hence, further advances in redox material performance are required for scalable thermochemical syngas production.Recently, a few perovskites oxides7–10 have been demonstrated to exhibit promising thermochemical syngas production performance. For instance, structural engineering of the LSM perovskites with optimally incorporated lanthanum resulted in faster reaction kinetics compared to state of the art pure ceria, during isothermal CO2 and H2O splitting cycles coupled to the methane partial oxidation reaction at 780 °C.7 Earth abundant redox materials are now the focus of the research for “sustainable” syngas production via thermochemical redox reactions. Recently, a study demonstrated a way to achieve considerable syngas production performance via NiFe2O4 during H2O and CO2 splitting cycles coupled to methane partial oxidation reaction.11
Vanadium-based oxides have been studied for catalytic conversion of hydrocarbons into H2 and hydrocarbon commodities at temperatures lower than those of typical thermochemical processes.12–16 For instance, partial oxidation of methane to formaldehyde has been reported at temperatures 500–640 °C with insights into the mechanism of sequential oxidation of methane.17–19 Recently, Fu et al. reported thermodynamic analysis of vanadia redox materials for thermochemical ammonia production with high fuel selectivity and a high solar-to-fuel efficiency of up to 38.1% is achieved.20
In our previous work, we reported CeO2/CeVO4 mixed-oxides systems21,22 and V-doped ceria23 for syngas production via the solar thermochemical looping reforming of methane, where the high ionic conductivity of cerium and catalytic nature of vanadium resulted in remarkable redox characteristics than the pure ceria structures. A CeVO4/CeVO3 redox pair is formed during the reduction and oxidation reactions (eqn (1)), which contributes towards the overall oxygen exchange capacity of the CeO2–CeVO4 system, where the achieved syngas production yields are 68% higher than that of pure ceria structures for up to 200 cycles. While the activation temperature for methane partial oxidation reaction is significantly reduced when vanadium is added to the ceria lattice and cycle capacity of ceria is improved due to the reducing sites provided by V5+ ions in the ceria lattice.
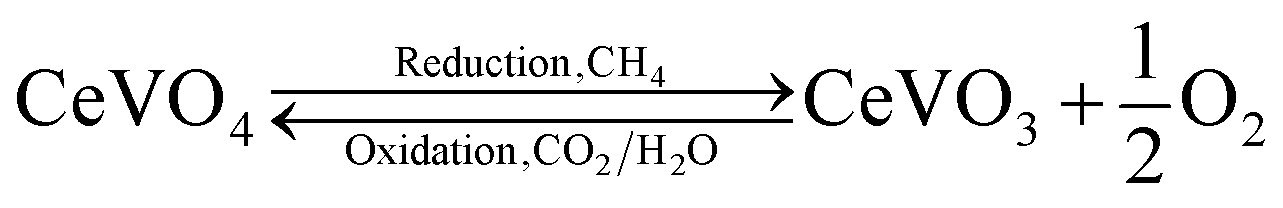 | (1) |
Giving the vast potential of vanadium for thermochemical syngas production, we report here a mechanism to utilize vanadia as an active oxygen carrier for cyclic syngas production via thermochemical chemical looping reforming of methane. Fractional amounts of cerium are incorporated into the V2O5 lattice via a facile liquid phase precursor decomposition method. The structural analysis revealed critical information about the phase changes occurred in the vanadia structures with cerium doping, where V2O5 can be transformed into V2O3 phase along with a segregated “CeVO4” phase. Cyclic reduction and oxidation reactions during the thermochemical looping reforming of methane result in formation of a reversible V2O3/VO redox pair, as given by eqn (2)–(5). This is possible when large cerium cations facilitate the charge mobility and weaken the V–O bonds, which enhances the redox activity in the Ce-doped V2O5 materials during the methane partial oxidation and H2O/CO2 splitting reactions.21,22
| V2O5 + 2CH4 → V2O3 + 2CO + 4H2 | (2) |
| V2O3 + CH4 → 2VO + CO + 2H2 | (3) |
| 2VO + CO2 → V2O3 + CO | (4) |
| 2VO + H2O → V2O3 + H2 | (5) |
Chemical nature of the powder surfaces is analysed via the XPS technique before and after the cycling. A mechanism for the redox activity of Ce-doped vanadia structures is proposed and correlated with the syngas production performance. Structural analysis of the after-cycling materials revealed a reversible V2O3/VO cycle capable of achieving enhanced syngas production performance for up to 100 consecutive cycles. Here, we propose to dope metal cations into vanadia to explore the thermochemical syngas production via redox cycling and chemical looping reforming of methane.
2. Experimental
2.1 Materials preparation
Pure and xCe-doped V2O5 powders with a range of Ce![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V molar ratios (x = 1%:1CeV, 3%:3CeV, 5%:5CeV, 7%:7CeV and 9%:9CeV) were synthesized via a liquid phase precursor decomposition method.24 Cerium(III) nitrate hexahydrate (CeN3O9–6H2O, Aldrich) and vanadium(V) oxytripropoxide (C9H21O4V, Aldrich) were used as precursors. Different stoichiometric amounts of the Ce and V precursors were separately dissolved in ethanol, mixed and stirred until a homogeneous sol was acquired. Finally, the mixture was dried under an air atmosphere at 80 °C and then fired at 600 °C for 2 h.
:V molar ratios (x = 1%:1CeV, 3%:3CeV, 5%:5CeV, 7%:7CeV and 9%:9CeV) were synthesized via a liquid phase precursor decomposition method.24 Cerium(III) nitrate hexahydrate (CeN3O9–6H2O, Aldrich) and vanadium(V) oxytripropoxide (C9H21O4V, Aldrich) were used as precursors. Different stoichiometric amounts of the Ce and V precursors were separately dissolved in ethanol, mixed and stirred until a homogeneous sol was acquired. Finally, the mixture was dried under an air atmosphere at 80 °C and then fired at 600 °C for 2 h.
2.2 Thermochemical testing
Thermochemical performance of the pure and Ce-doped V2O5 powders was evaluated in a vertical-tube reactor placed inside an infrared gold image furnace (P4C-VHT, Advance Riko). Two layers (2 mm thick and 1 cm apart) of highly porous and high temperature resistant alumina-based fibrous wool (97% Al2O3 and 3% SiO2), were placed in the middle of tube, with powder samples placed between them. The gas composition was analysed by a quadrupole mass spectrometer (OmniStar™ GSD 320, Pfeiffer Vacuum). The schematic diagram of the thermochemical testing setup is shown in Fig. SI1.†The reactor was purged with Ar (500 mL min−1: grade 5.0) before and after each reduction and oxidation reaction step to remove any residual reactant/product gases. Isothermal thermochemical reactions were carried out at an optimized temperature of 900 °C, with the ramp rate set at 100 °C min−1. Fast heating rates were used to achieve the 900 °C target temperature without melting of the V2O5-containing powders, while methane was introduced at 900 °C resulting in the onset of the reduciton reaction. The flow rates of 20 mL min−1 of CH4 (grade 4.5) and 10 mL min−1 of CO2 (grade 4.5) were set for the methane partial oxidation (MPO) and CO2 splitting (CDS) reactions, respectively. The flow rate of the gas phase (including Ar as the carrier gas) was kept constant at 250 mL min−1. For the H2O splitting (WS) reactions, steam vapor was generated at 95 °C in a bubbler filled with deionized water. The vapor was carried by flowing a 30 mL min−1 of Ar through the bubbler and further diluted with 220 mL min−1 flow of Ar.
2.3 Materials characterization
Samples were characterized as prepared and after the thermochemical cycling processes. The morphology of the powder samples was investigated by a field emission scanning electron microscopy (FESEM, Zeiss Ultraplus). Lattice spacing and particle size distribution in powders were estimated by a high-resolution transmission electron microscope (HR-TEM, JEOL 2100F). Energy dispersive spectroscopy (EDS) analysis was carried out by the scanning transmission electron microscopy (STEM-EDS) technique. For the TEM sample preparation, dispersion of the powders was drop casted onto the lacy carbon-coated 200 mesh Cu grids. ImageJ software was used to perform initial image processing as well as particle size and lattice planar spacing measurements.A Hitachi HF500 Cs-STEM/TEM was utilized to perform electron energy loss spectroscopy (EELS) analysis for the atomically resolved estimation of lattice spacing and positions of cerium and vanadium atoms. The instrument was equipped with a W (310) cold-cathode FEG electric field filament and a Cs probe STEM corrector. An accelerating voltage of up to 200 kV was set for the high-resolution imaging and EELS analysis.
Phase and structural parameters of the powder samples were analysed by a D2 phaser diffractometer (Bruker), equipped with a Cu Kα (1.54 Å) radiation source and operated with a powder of 300 W (30 kV × 10 mA). Powders were scanned at a rate of 0.75° min−1 in a Bragg's angle range of 10–80°, with a 0.02° increment per data point. A Match3 V.3.8.1.143 (Crystal Impact) software was used to perform the phase and crystal structure identification, while phase quantification was approximated by applying a Rietveld refinement technique to the XRD patterns.
Chemistry of the powder surfaces was investigated by utilizing an X-ray photoelectron spectrometer (XPS: ESCALAB 250 Xi, Thermo Fisher). A microprobe and an aluminium Kα radiation source with a spot size of 200–900 μm was attached to the instrument and operated at 15 kV voltage and 12 mA current, while the energy range for scanning was 160–40 eV. Advantage software (ver. 5.978, Thermo Fisher) was utilized to deconvolute the spectra. Advantageous carbon C 1s peak situated at 284.8 eV was used as a reference for the binding energies acquired in the sample spectra.
Synchrotron light X-ray absorption spectroscopy (XAS) technique was utilized to investigate the Ce L3 and V K edges in the X-ray absorption near edge structure (XANES) region. The synchrotron electron energy beam energy was 1.2 GeV and the range of current was 80–150 mA, while the photon flux was maximum at 1.7 × 1011 photons cm−2 s−1 A monochromator with two germanium (220) single crystal was used with an X-ray beam size of 14 mm width and 1 mm height, while the spatial resolution was 0.2 eV. Samples were analysed in transmission and fluorescence modes and the obtained spectra were processed using Athena software.
A Raman imaging microscope (Renishaw plc, model 2000) was utilized to investigate the structural features of the oxygen carriers. The instrument was attached to an optical microscope (Olympus BH2) with a motorized XYZ stage, an air-cooled CCD detector and a CCD camera. Samples were excited with a 785 nm NIR laser source and Raman shift spectra were recorded in the 100–1200 cm−1 range, while the laser power was adjusted between 0.01–0.5% (<6 MW) and exposure time was set at 20 s.
3. Results
3.1 Pre-cycling materials characterization
Structural, chemical, and morphological studies were carried out to investigate the effects of cerium doping. The FESEM analysis showed a decline in the particle size of V2O5 powders after cerium incorporation. Furthermore, the highly faceted and crystalline features approach to a near-spherical morphology, as shown in Fig. SI2a–c,† which can be further confirmed from the HRTEM micrographs shown in Fig. 1a and c. The insets in the HRTEM images revealed an increased lattice spacing from 0.237 nm in pure V2O5 to 0.307 nm in 1CeV powders caused by the distortions in the V2O5 lattice after the incorporation of large cerium cations. Further increase in the cerium concentration reduced the lattice spacing to 273 nm in 5CeV and 250 nm in 9CeV powders, possibly due to the formation of CeVO4 phase. This phenomenon will be briefly discussed in the coming paragraphs.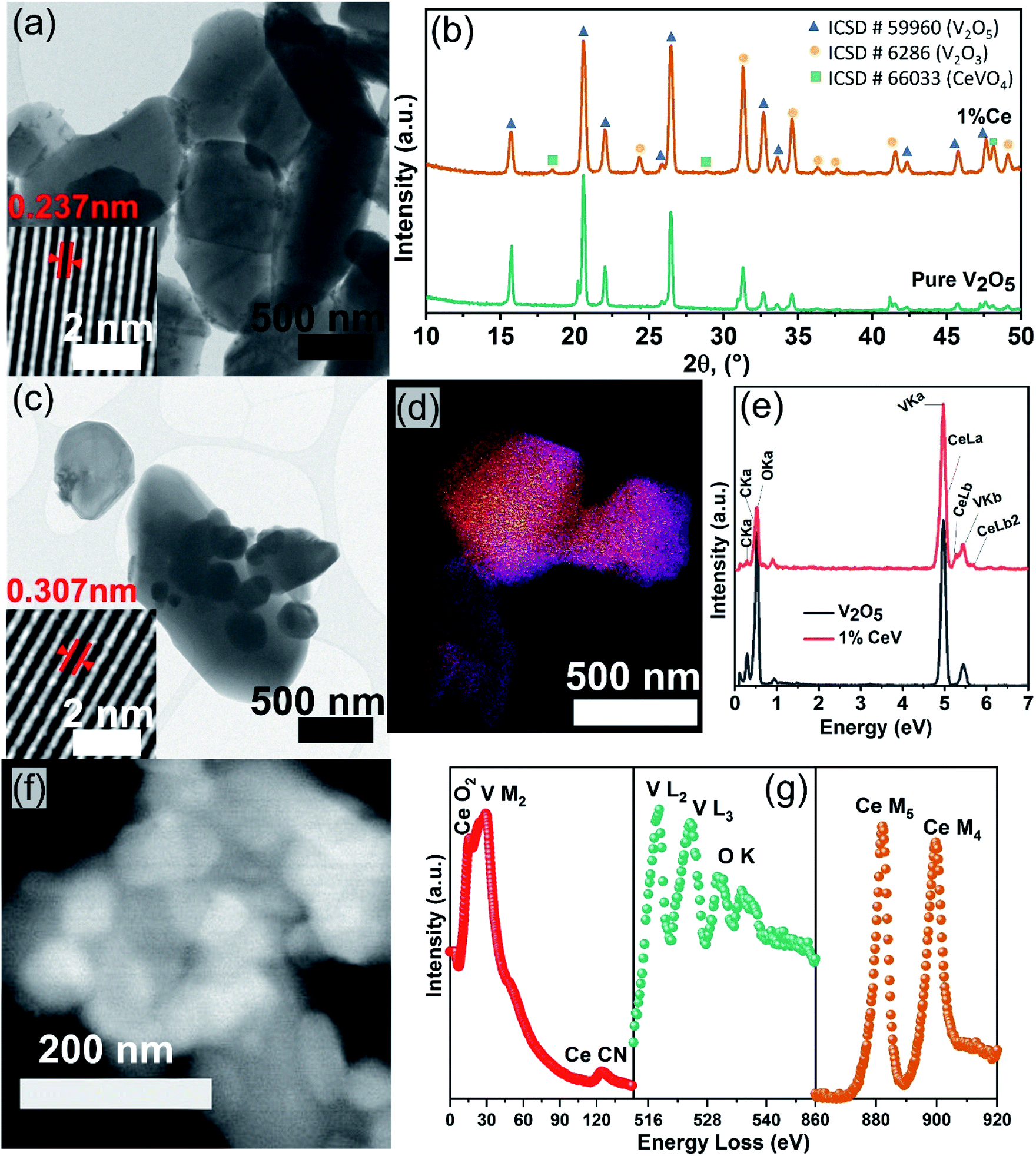 | ||
| Fig. 1 Structural and chemical analysis of as-prepared pure and Ce-doped V2O5 powders: HRTEM micrographs of (a) pure V2O5 and (c) 1CeV powders with insets showing the inner lattice spacing. (b) XRD patterns of pure V2O5 and 1CeV powders, showing the evolving V2O3 and CeVO4 phases after cerium doping. (d) Overlay image of STEM-EDS maps of 1CeV powders and (e) EDS spectra of pure V2O5 and 1CeV powders, showing presence of cerium in the V2O5 powders. (f) HAADF image and (g) EELS spectra of 1CeV powders. | ||
X-ray diffraction analysis showed the structural changes induced in the V2O5 lattice after cerium doping (Fig. 1b). Rietveld refinement method was applied on the XRD patterns to quantify phases in the pure and Ce-doped V2O5 powders. Lattice expansion caused by the large cerium cations reduced the V5+ species to V3+ and up to 77.1% of V2O3 phase was observed for samples with 9% of cerium doping. In addition, cerium cations migrated to the surface of Ce-rich V2O5 powders and form up to 10.3% of CeVO4 phase in 9CeV sample, with remaining 12.5% of V2O5 phase. The evolving V2O3 and CeVO4 phases with the increasing Ce concentration can be observed in the XRD patterns of pure and Ce-doped V2O5 powders, Fig. SI3.†
STEM-EDS technique was used to carry out chemical analysis on pure and Ce-doped V2O5 samples. Fig. 1d shows the overlay image of STEM-EDS elemental mapping of 1CeV sample, where yellow colour represents cerium. The EDS spectra of pure V2O5 and 1CeV samples are shown in Fig. 1e, where shoulder peak at 5.23 eV is associated with Ce Lb.25 The presence of cerium in doped V2O5 was further confirmed by the electron energy loss spectroscopy (EELS) technique. The Ce O2, Ce CN, Ce M5 and Ce M4 peaks at 15 eV, 120 eV, 883 eV and 801 eV, respectively, showed the fingerprint EELS signals of cerium, which aligns with the reported EELS features of Ce, (Fig. 1f and g).26,27 The V M2, V L2 and V L3 peaks situated at 38 eV, 519 eV and 525 eV, respectively, corroborate the EELS features of vanadium.28
Formation of V2O3 is further confirmed with the Raman spectroscopy technique. Fig. 2a shows the growing peak of V2O3 at Raman shift of 538 cm−1, while distinct peaks of CeVO4 at 860 cm−1 and 798 cm−1 are also observed in the Ce-doped V2O5 powders.13 The presence of cerium species onto the surface of as-prepared V2O5 powders is analysed using the XPS technique. Evidently, the concentration of cerium ions on the V2O5 surfaces increased with increasing the cerium contents, as shown in Fig. 2b.
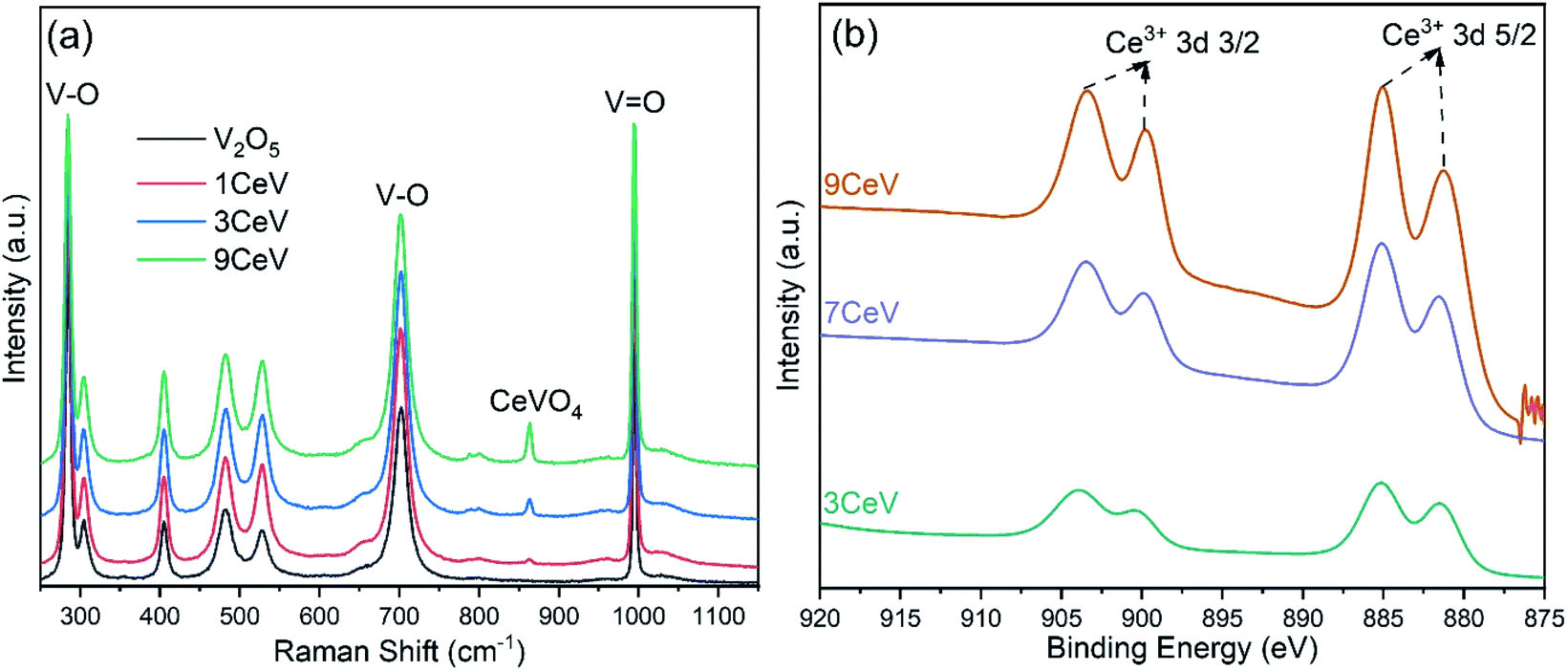 | ||
| Fig. 2 (a) Raman spectra of pure and Ce-doped V2O5 powders depicting the evolution of V2O3 and CeVO4 phases with cerium doping. (b) XPS spectra of pure and Ce-doped V2O5 powders showing Ce3+ content onto the powder surfaces with the various doping concentrations. | ||
The activation temperature for chemical looping of methane was investigated by fast heating (100 °C min−1) the powder samples from ambient temperature to 400 °C in Ar flow, followed by heating from 400 °C to 1000 °C at a rate of 3 °C in CH4 flow (20 mL min−1). Fig. 3a and b show the instantaneous rates of CO and H2 produced during the activation test, respectively. It is evident that the CO and H2 production rates significantly increased for Ce-doped materials. Furthermore, temperature for the peak rates decreased by up to 75 °C for materials with 5% of Ce content. Further increase in Ce contents elevated the temperature for decent syngas production rates, which may be due to a phase shift induced by higher concentrations of CeVO4 in Ce-rich V2O5 powders.
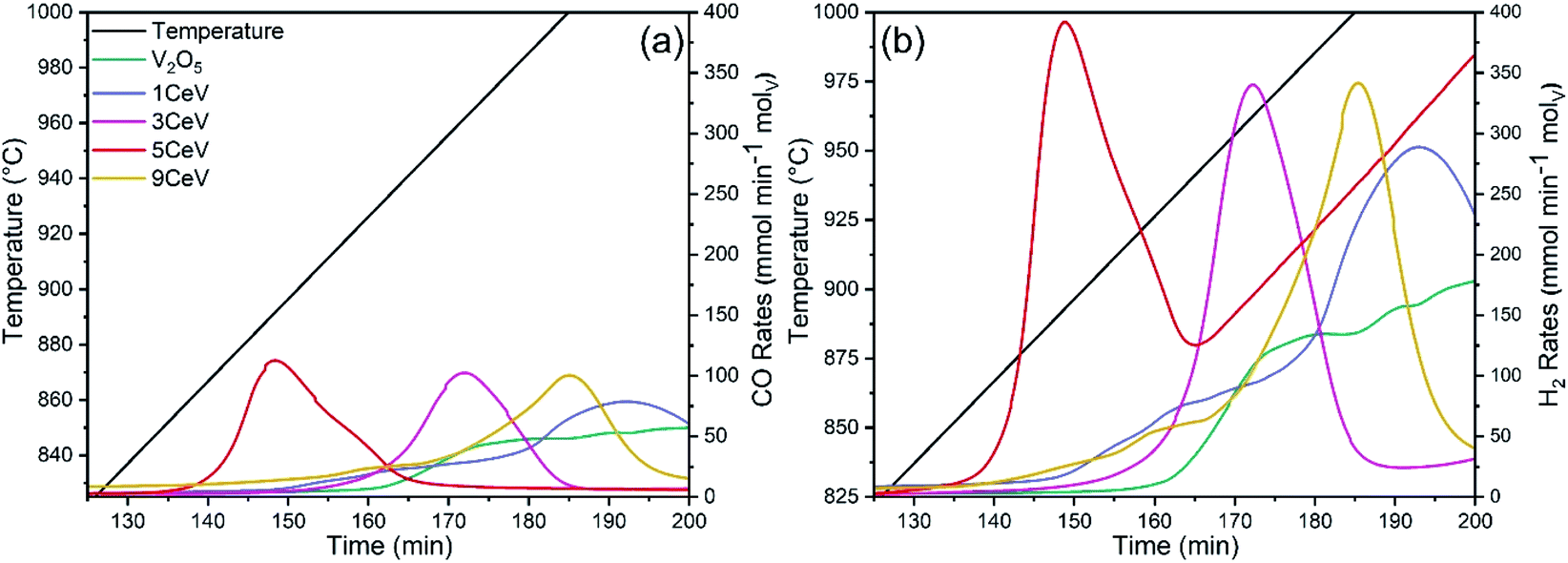 | ||
| Fig. 3 Instantaneous (a) CO and (b) H2 production rates obtained for pure and Ce-doped V2O5 powders during a continuous heating from ambient temperature to 1000 °C under a methane atmosphere. | ||
Ten preliminary WS and CDS cycles were performed to investigate the syngas production performance of the pure and Ce-doped V2O5 powders. A blank test was carried out to investigate the amounts of CO and H2 produced due to possible thermal decomposition of reactant gases. Fig. SI4† compares the syngas produced during MPO–CDS cycles with a blank reactor and with 1CeV sample. Negligible amount of syngas is observed during the blank run, which clearly highlights the role of the redox materials toward achieving efficient and stable syngas production rates. Fig. 4a and b compare the syngas production rates of the pure V2O5 with 1CeV powders during MPO–CDS redox cycles. Clearly, the pure V2O5 powders showed inferior and unstable syngas production rates when compared to that of the Ce-doped vanadia samples, where the rates of H2 and CO produced by 1CeV are up to 500% higher than those for the pure V2O5 sample. Interestingly, the syngas production rates obtained for the pure V2O3 sample were as low as those for the pure V2O5 sample (Fig. SI5†), which indicates the irreversible oxygen exchange in the V2O3 structure.
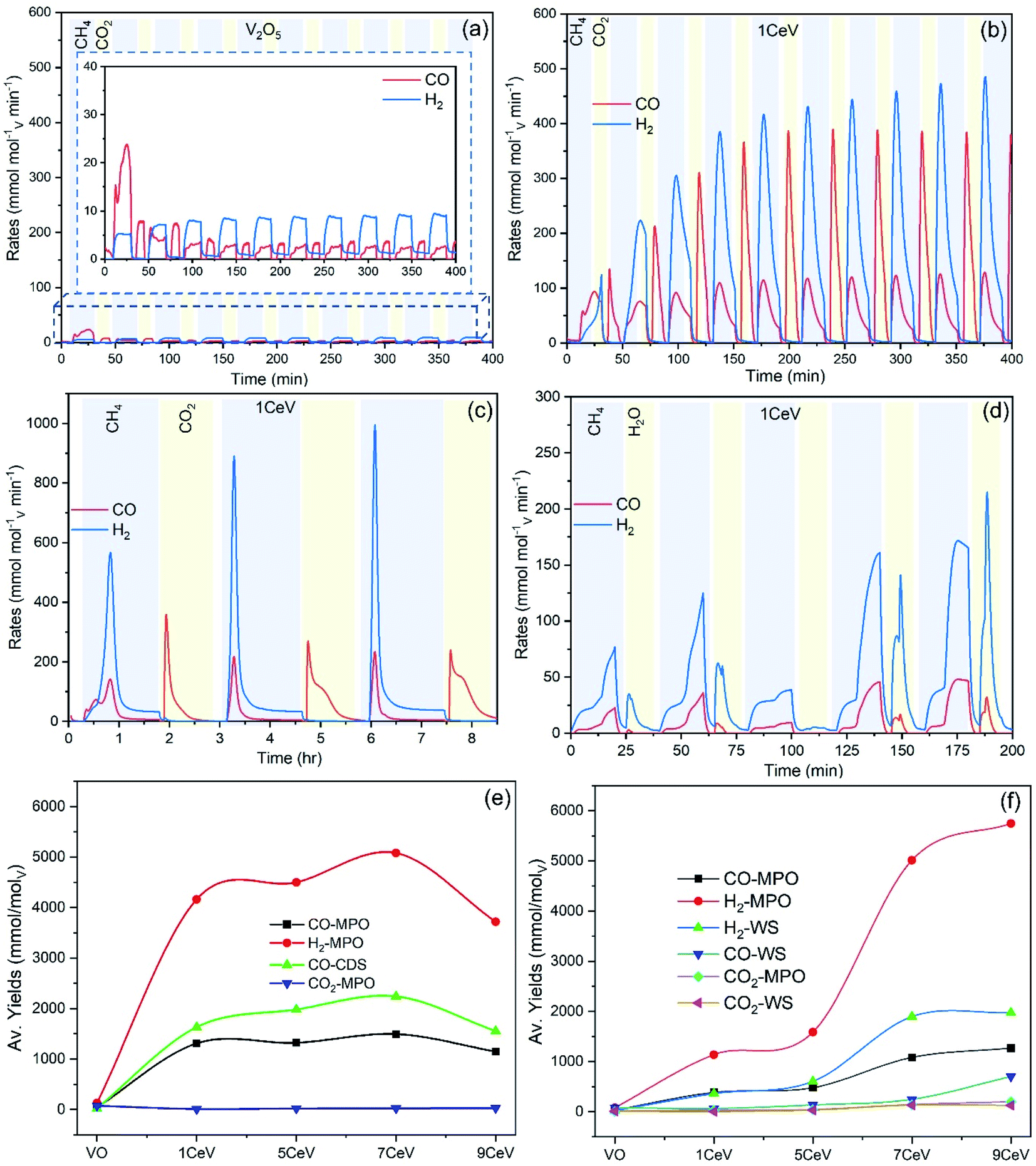 | ||
| Fig. 4 Thermochemical fuel production performance: syngas production rates obtained for (a) pure V2O5 and (b) 1CeV powders during 10 MPO–CDS cycles. (c) Syngas production rates obtained for 1CeV powders during (c) MPO–CDS and (d) MPO–WS cycles. Syngas production yields obtained for pure and Ce-doped V2O5 samples during (e) MPO–CDS and (f) MPO–WS cycling. | ||
Here, the initial slow rates were due to the slow activation of Ce-doped vanadia powders. However, the rates = increased and stabilized at 450 mmolH2 molV min−1 and 120 mmolCO molV min−1 during the MPO reaction, while the average rates of 400 mmolCO molV min−1 are achieved during the CO2 splitting step of MPO–CDS cycles. Notably, the syngas production rates achieved during the long-duration reduction–oxidation reactions are up to 80% higher than those for the shorter MPO–CDS cycles, as shown in Fig. 4c. This is attributed to the fact that a longer cycle duration results in better re-oxidation of materials, for which more oxygen molecules are available for the subsequent methane partial oxidation reaction.
The activation of the pure and Ce-doped V2O5 powders is slower during the H2O splitting reactions as compared to that of CO2 splitting, as shown in Fig. 4d. This may be due to a low affinity of these reduced oxygen carriers towards steam compared to CO2. The highest rates of 180 mmolH2 molV min−1 and 45 mmolCO molV min−1 during MPO and 210 mmolH2 molV min−1 upon H2O splitting are achieved by the 1CeV powder samples, while the rates were considerably higher for the 9CeV samples, as shown in Fig. SI6.† However, considerable amounts of CO were observed in the H2O splitting step, which is attributed to the oxidation of carbon deposited on the powder surface.
The average syngas yields produced during the MPO–CDS and MPO–WS cycling are presented in Fig. 4c and d, respectively. High CO yields up to 1.5 mol molV−1 were achieved during the methane partial oxidation reaction of MPO–CDS cycling as compared to that of MPO–WS cycling with maximum CO yields up to 1.02 mol molV−1. Similarly, a higher (ca. 0.6) methane conversion was achieved for the MPO–CDS cycling as compared to those of 0.41 during the MPO–WS cycling. Note that the H2:CO ratios can be controlled by adjusting the CH4:O2 ratios (R) relative to the amount of CH4 purged in the reactor and available oxygen per gram of powder. R < 2 results in complete combustion of methane while R > 2 results in carbon formation.6 In this study, the oxygen concentration varied for each sample due to different sample phase composition, which in turn resulted in CH4:O2 ratios in a range of 2–3.58 range.
The recorded CO2 to CO conversion was 74% for 1CeV sample, 76% for 5CeV sample and 83% for the 7CeV sample. The CO2 conversion decreased in the 9CeV sample to 73% due the increasing CeVO4 phase concentration. The materials with high Ce concentration showed low purity of H2 during the H2O splitting reaction due to considerable amounts of CO produced upon the oxidation of solid carbon. The high amount of solid carbon deposited on the powder surface for a high Ce concentration of about 0.31 mmol g−1 of powder supports these findings. Notably, negligible amounts of CO2 are produced during the methane partial oxidation reaction, which leads to high fuel selectivity achieved by the Ce-doped vanadia materials. The maximum CO selectivity achieved for 1CeV powders is 99.3% and 92.7% during the MPO–CDS and MPO–WS cycling, respectively, which was decreased with an increasing Ce content. In addition, improved purity of H2 of up to 94%, was obtained in presence of small amounts of CO2 and CO.
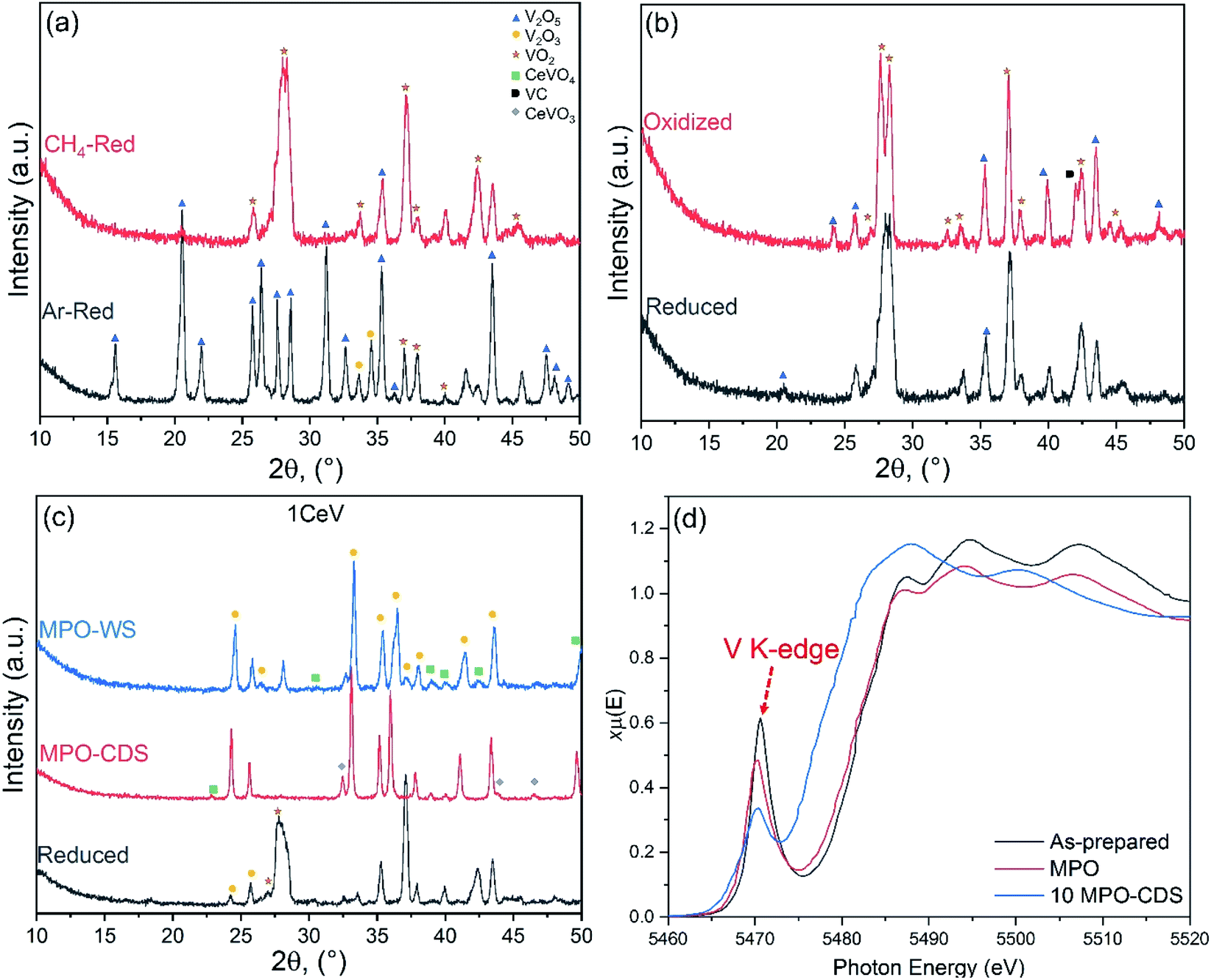 | ||
| Fig. 5 Post cycling structural characterization of 1CeV powders: XRD patterns acquired after (a) reduction under CH4 and Ar atmospheres, and redox reactions during (b) MPO–CDS and (c) MPO–WS cycles. (d) Comparison of XANES V5+ K-edge spectrum of as prepared, reduced and after MPO–CDS cycling. | ||
The inferior syngas production performance of the pure V2O3 powders identified in this study is inconsistent with the former efficient redox behaviour of Ce-doped vanadia powders, where V2O3 was the major constituent. Ce plays a crucial role in facilitating the ion mobility and ensuring cyclic and efficient oxygen exchange due to a lattice distortions caused by its large cationic radii. In addition, the after-cycled pure V2O3 powder revealed the presence of V2O5 and melting of powder (V2O5) was observed on contact with the alumina wool, which largely contributed to the observed low syngas production rates of pure vanadia samples. Thus, addition of Ce enhanced the structural stability of vanadia powders as well.
The XANES analysis revealed a growing V2O3 phase in the Ce-doped V2O5 powders after cycling, where intensity of the V5+ K-edge peak decreased by up to 50% after 10 MPO–CDS cycles, as shown in Fig. 5d. The surface interactions during the repeated redox reactions, promoted the formation of CeVO4 phase on the powder surfaces, corresponding to the residual V5+ K-edge peak in the XANES spectra, as seen in Fig. 5d. This can be further confirmed from the backscattered and FESEM analysis of the Ce-doped V2O5 powders after cycling, where the CeVO4 particles can be observed on the powders, as shown in Fig. SI7.†
The distribution of phases in the Ce-doped V2O5 powders was significantly affected by the duration of redox reactions, oxidation atmosphere and concentration of cerium ions. For instance, high Ce content promoted the formation of V2O3 phase after reduction and oxidation reactions. The formation of the VC phase was prominent in the Ce-rich powders after CDS cycling as compared to the WS cycling (see, Fig. 5c). Furthermore, the concentration of CeVO3 phase was higher after CDS cycling when compared to the WS cycling, which indicates to formation of a cyclic CeVO4/CeVO3 redox pair during the WS cycling, while the carbonaceous atmosphere (CO2) suppressed the oxidation of reduced powders.
XPS analysis revealed the nature of oxygen species present onto the powder surfaces before and after cycling. The XPS spectra of 5CeV powders after reduciton under methane and Ar atmospheres are shown in Fig. 6a. Large area of OII peak depicts high extents of oxygen exchange in samples during the MPO reaction as compared to the reduction under an Ar atmosphere. However, the formation of CO increased surface species adsorption during the MPO reaction, as shown in Fig. 6a. The XPS spectra of reduced and reoxidized 5CeV powders are shown in Fig. 6b, where changes in OI and OII species are associated with the exchange of oxygen during the reduction and oxidation reactions. A split in the V 2p3/2 is observed after reduction, which may correspond to the multiple valence states of vanadium formed during the MPO reaction.23
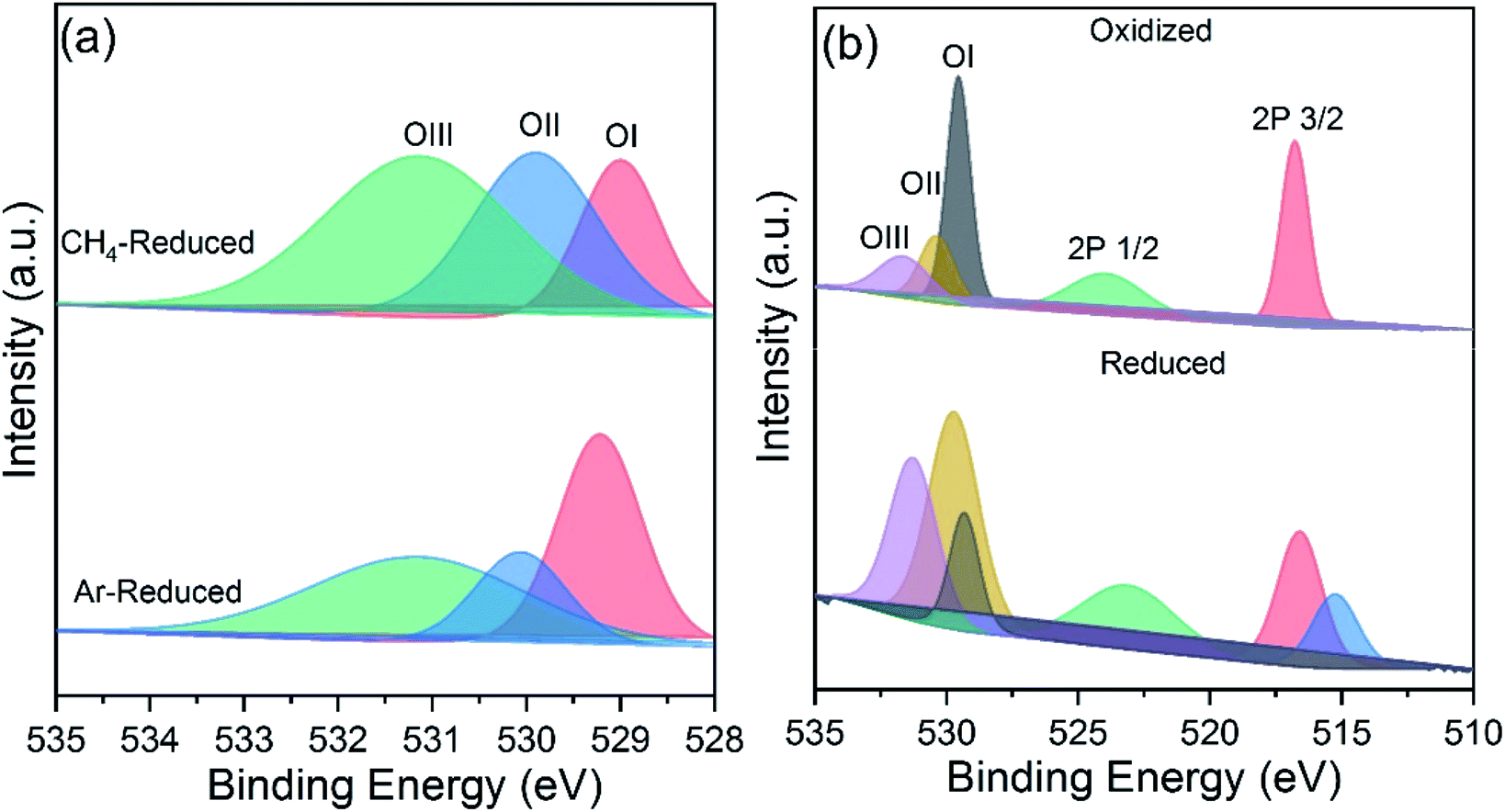 | ||
| Fig. 6 Surface analysis of oxygen carriers after cycling: (a) XPS spectra of reduced 3CeV powders under CH4 and Ar atmospheres. (b) XPS spectra of 3CeV powders, acquired after the reduction and oxidation reactions during MPO–CDS cycles. | ||
A long-term stability test of up to 100 MPO–CDS cycles was performed on the Ce-doped V2O5 powders, as shown in Fig. 7a. The average rates of up to 360 mmolH2 g−1 min−1 and 150 mmolCO g−1 min−1 are achieved during the methane partial oxidation reaction, while the splitting of CO2 resulted in 180 mmolCO g−1 min−1, as seen in Fig. 7b. The repeated reduction and oxidation reactions promoted the cerium ions to interact with the reactant gases on the powder surfaces. The presence of cerium on the powder surface could be confirmed in the XPS analysis, as shown in Fig. 7c. The origin of cerium may be associated with the formation of CeVO4 phase on surface, as shown in Fig. SI7.†
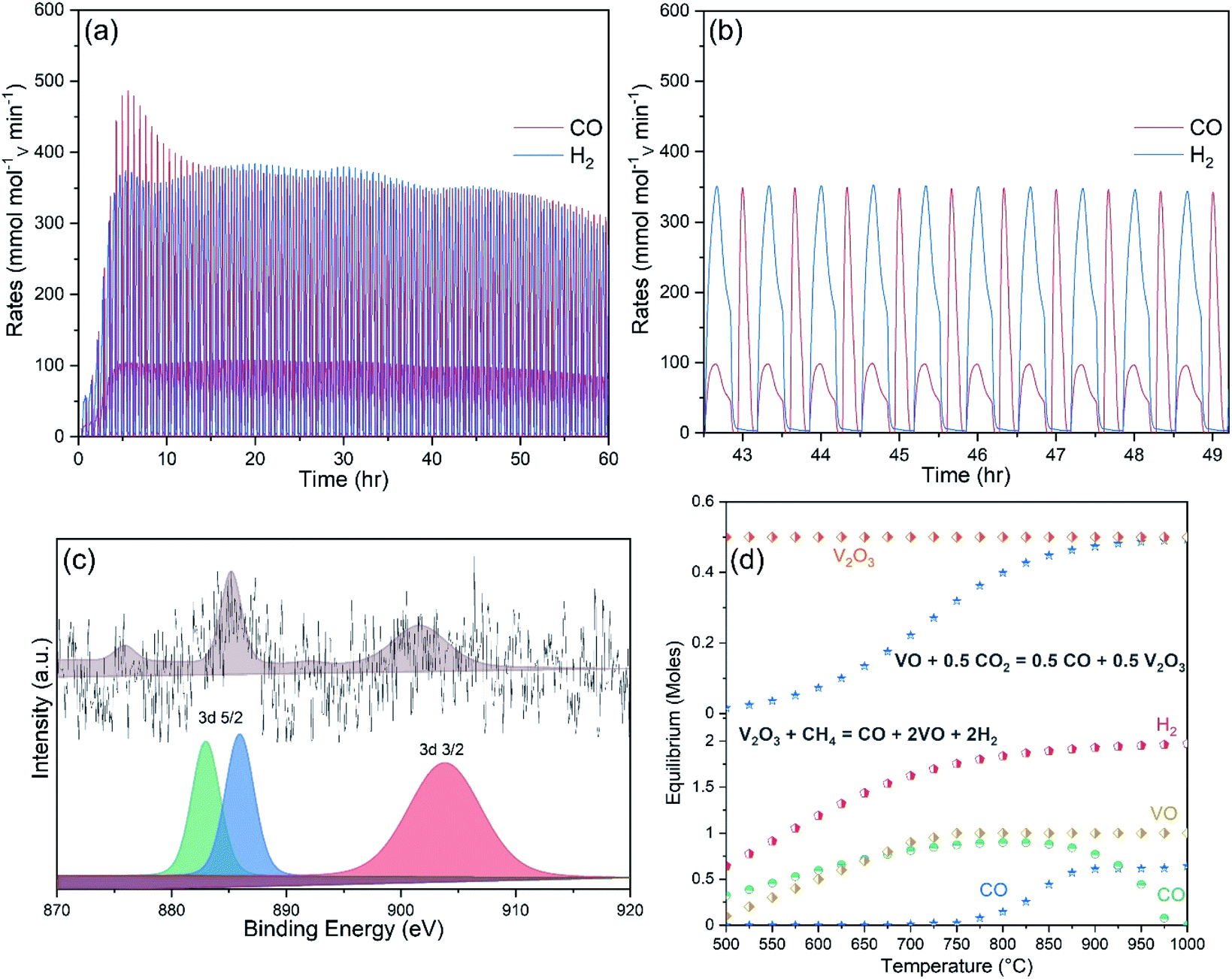 | ||
| Fig. 7 (a) Syngas production rates achieved by 1CeV powders during 200 continuous MPO–CDS cycles. (b) The expanded view of part (a) from 63rd to 72th cycle. (c) XPS spectra of 1CeV sample, acquired after the 100 MPO–CDS cycles. (d) Equilibrium distribution of products formed during the MPO and CDS reactions, produced by the V2O3/VO redox pair. The reactions were balanced and the equilibrium composition was obtained using Factsage. | ||
4. Discussion
The structural study revealed that fractional incorporation of cerium into the V2O5 lattice shifts the oxidation state from +5 to +3. The surface analysis showed cerium on the surface of powders with cerium doping concentrations greater than 5%, which is further confirmed by the presence of CeVO4 phase observed in the XRD analysis. Repeated reduction and oxidation reactions resulted in a V2O3/VO redox pair. The equilibrium phases calculated by Factsage equilibrium module in the V2O3/VO cycle during the reduction and oxidation reactions are shown in Fig. 7d. The V2O3 phase was reduced to VO during methane reforming and reoxidised by CO2 into V2O3. The stability of V2O3/VO cycle was greatly influenced by the concentration of cerium ions that were responsible for a complete conversion of V2O5 to V2O3 phase, and resulted in a reversible redox pair (V2O3/VO) without the issue of powder melting, and consequently considerable rates of syngas production during the thermochemical redox cycling. However, Ce ions present inside the lattice and on the surface of the vanadia structure may have entirely different characteristics for the redox reactions, depending on the cerium concentration. For instance, lattice cerium ions enhance the structural stability and ion conductivity of the vanadia powders, while surface cerium ions lower the methane cracking. A segregated phase “CeVO4” is formed on the surface of Ce-rich vanadia powders, which has adverse effects on the syngas production performance. For instance, formation of a CeVO4/CeVO3 redox pair is cyclic when surrounded by cerium cations, as reported in our previous study.29 whereas the reoxidation of the CeVO3 is slow on vanadia powders and impedes the oxygen flow into the vanadia structure, which in turn results in inferior syngas production performance. In addition, carbonaceous atmospheres further slowdown the reoxidation of CeVO3 phase, which result in lower overall syngas yields and fuel selectivity. Hence, Ce-doped vanadia powders are independent of the CeVO4. In fact formation of the CeVO4 phase leaves few cerium cations, required to achieve high oxygen exchange capacity in vanadia structure. Non-cyclic and unstable redox behaviour of pure V2O5 and V2O3 materials also highlights the role of cerium incorporation for repeated and efficient oxygen exchange capacity during the methane partial oxidation and CO2/H2O splitting reactions.5. Conclusions
In this study, a way to utilize vanadium-based oxygen carriers for syngas production via high-temperature chemical-looping reforming of methane has been demonstrated. Fractional amounts of large cerium cations were incorporated into the V2O5 lattice, which resulted in a V2O5 to V2O3 phase transition and formed a segregated CeVO4 phase on the surface. Compared to the pure V2O3 phase, combination of cerium and V2O3 showed as excellent redox capability and chemical stability at high temperatures. The structural and phase analysis revealed a VO/V2O3 redox pair formed during the repeated reduction and oxidation reactions for up to 200 cycles. Formation of this redox pair is resulted after the initial slow syngas production. Cerium cations tend to move between the surface and lattice of the vanadia powders, which significantly affected the syngas production performance, depending on the doping concentration. For instance, low concentrations of cerium improve the rates and yields of syngas produced by the vanadia powders. Large quantity of the CeVO4 phase was formed in the cerium rich vanadia powders, which slowed down the reoxidation rates and resulted in high carbon contents and low syngas yields.Funding
This project was supported by the Australian Research Council (ARC Future Fellowship FT140101213 by W. Lipiński).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study used the facilities and the scientific and technical assistance at the Centre of Advanced Microscopy at the Australian National University.References
- J. R. Scheffe and A. Steinfeld, Oxygen Exchange Materials for Solar Thermochemical Splitting of H2O and CO2: A Review, Mater. Today, 2014, 17(7), 341–348, DOI:10.1016/j.mattod.2014.04.025.
- A. Haeussler, S. Abanades, A. Julbe, J. Jouannaux, M. Drobek, A. Ayral and B. Cartoixa, Remarkable Performance of Microstructured Ceria Foams for Thermochemical Splitting of H2O and CO2 in a Novel High–Temperature Solar Reactor, Chem. Eng. Res. Des., 2020, 156, 311–323, DOI:10.1016/j.cherd.2020.02.008.
- S. Ackermann, M. Takacs, J. Scheffe and A. Steinfeld, Reticulated Porous Ceria Undergoing Thermochemical Reduction with High-Flux Irradiation, Int. J. Heat Mass Transfer, 2017, 107, 439–449, DOI:10.1016/j.ijheatmasstransfer.2016.11.032.
- X. Gao, A. Vidal, A. Bayon, R. Bader, J. Hinkley, W. Lipiński and A. Tricoli, Efficient Ceria Nanostructures for Enhanced Solar Fuel Production via High-Temperature Thermochemical Redox Cycles, J. Mater. Chem. A, 2016, 4, 9614–9624, 10.1039/C6TA02187E.
- J. E. Miller, A. H. McDaniel and M. D. Allendorf, Considerations in the Design of Materials for Solar-Driven Fuel Production Using Metal-Oxide Thermochemical Cycles, Adv. Energy Mater., 2014, 4(2), 1300469, DOI:10.1002/aenm.201300469.
- P. T. Krenzke, J. R. Fosheim and J. H. Davidson, Solar Fuels via Chemical-Looping Reforming, Sol. Energy, 2017, 156, 48–72, DOI:10.1016/j.solener.2017.05.095.
- A. Riaz, T. Tsuzuki, F. Kremer, S. Sattayaporn, M. U. Ali, W. Lipiński and A. Lowe, Structural Rearrangement in LSM Perovskites for Enhanced Syngas Production via Solar Thermochemical Redox Cycles, ACS Catal., 2020, 8263–8276, DOI:10.1021/acscatal.0c02439.
- L. Neal, A. Shafiefarhood and F. Li, Effect of Core and Shell Compositions on MeOxLaySr1−yFeO3 Core-Shell Redox Catalysts for Chemical Looping Reforming of Methane, Appl. Energy, 2015, 157, 391–398, DOI:10.1016/j.apenergy.2015.06.028.
- E. h. Yang, Y. S. Noh, G. H. Hong and D. J. Moon, Combined Steam and CO2 Reforming of Methane over La1−xSrxNiO3 Perovskite Oxides, Catal. Today, 2018, 299, 242–250, DOI:10.1016/j.cattod.2017.03.050.
- A. Riaz, P. Kreider, F. Kremer, H. Tabassum, J. S. Yeoh, W. Lipiński and A. Lowe, Electrospun Manganese-Based Perovskites as Efficient Oxygen Exchange Redox Materials for Improved Solar Thermochemical CO2 Splitting, ACS Appl. Energy Mater., 2019, 2, 2494–2505, DOI:10.1021/acsaem.8b01994.
- B. Guene Lougou, Y. Shuai, Z. Guohua, G. Chaffa, C. Ahouannou and H. Tan, Analysis of H2 and CO Production via Solar Thermochemical Reacting System of NiFe2O4 Redox Cycles Combined with CH4 Partial Oxidation, Int. J. Hydrogen Energy, 2018, 43(12), 5996–6010, DOI:10.1016/j.ijhydene.2018.01.197.
- C. A. Chagas, L. C. Dieguez and M. Schmal, Investigation of the Stability of CeO2, V2O5 and CeV Mixed Oxide on the Partial Oxidation of Propane, Catal. Lett., 2012, 142(6), 753–762, DOI:10.1007/s10562-012-0800-x.
- M. V. Martínez-Huerta, J. M. Coronado, M. Fernández-García, A. Iglesias-Juez, G. Deo, J. L. G. Fierro and M. A. Bañares, Nature of the Vanadia-Ceria Interface in V5+/CeO2 Catalysts and Its Relevance for the Solid-State Reaction toward CeVO4 and Catalytic Properties, J. Catal., 2004, 225(1), 240–248, DOI:10.1016/j.jcat.2004.04.005.
- M. Lamers, S. Fiechter, D. Friedrich, F. F. Abdi and R. Van De Krol, Formation and Suppression of Defects during Heat Treatment of BiVO4 Photoanodes for Solar Water Splitting, J. Mater. Chem. A, 2018, 6(38), 18694–18700, 10.1039/c8ta06269b.
- V. V. Guliants, Structure-Reactivity Relationships in Oxidation of C4 Hydrocarbons on Supported Vanadia Catalysts, Catal. Today, 1999, 51(2), 255–268, DOI:10.1016/S0920-5861(99)00049-8.
- C. A. Carrero, C. J. Keturakis, A. Orrego, R. Schomäcker and I. E. Wachs, Anomalous Reactivity of Supported V2O5 nanoparticles for Propane Oxidative Dehydrogenation: Influence of the Vanadium Oxide Precursor, Dalton Trans., 2013, 42(35), 12644–12653, 10.1039/c3dt50611h.
- H. Launay, S. Loridant, D. Nguyen, A. Volodin, J. Dubois and J. Millet, Vanadium Species in New Catalysts for the Selective Oxidation of Methane to Formaldehyde: Activation of the Catalytic Sites, Catal. Today, 2007, 128(3–4), 176–182, DOI:10.1016/j.cattod.2007.07.014.
- Q. Sun, J. M. Jehng, H. Hu, R. G. Herman, I. E. Wachs and K. Klier, In Situ Raman Spectroscopy during the Partial Oxidation of Methane to Formaldehyde over Supported Vanadium Oxide Catalysts, J. Catal., 1997, 165(1), 91–101, DOI:10.1006/jcat.1997.1446.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. L. Haller, Catalytic Performance of Vanadium Incorporated MCM-41 Catalysts for the Partial Oxidation of Methane to Formaldehyde, Appl. Catal., A, 2006, 302(1), 48–61, DOI:10.1016/j.apcata.2005.12.013.
- M. Fu, J. Jin, H. Ma, L. Wang, T. Ma, L. Dou, Z. Chang, X. Li and Y. Lu, Thermodynamic Analysis of Novel Vanadium Redox Materials for Solar Thermochemical Ammonia Synthesis from N2 and CH4, Int. J. Hydrogen Energy, 2020, 45(4), 2569–2577, DOI:10.1016/j.ijhydene.2019.11.122.
- A. Riaz, M. U. M. U. Ali, W. Lipiński and A. Lowe, Enhanced Oxygen Exchange Capacity in Nano-Structured Vanadia–Ceria Multi-Phase Oxygen Carriers for Solar Thermal Fuel Production, J. Mater. Chem. A, 2019, 7(48), 27347–27360, 10.1039/C9TA06471K.
- A. Riaz, M. U. Ali, T. G. Enge, T. Tsuzuki, A. Lowe and W. Lipiński, Concentration-Dependent Solar Thermochemical CO2/H2O Splitting Performance by Vanadia–Ceria Multiphase Metal Oxide Systems, Research, 2020, 2020, 1–12, DOI:10.34133/2020/3049534.
- A. Riaz, F. Kremer, T. Kim, S. Sattayaporn, T. Tsuzuki, W. Lipiński and A. Lowe, Experimental Demonstration of Vanadium-Doped Nanostructured Ceria for Enhanced Solar Thermochemical Syngas Production, Nano Energy, 2020, 105639, DOI:10.1016/j.nanoen.2020.105639.
- Y. Y. Luo, D. S. Jo, K. Senthil, S. Tezuka, M. Kakihana, K. Toda, T. Masaki and D. H. Yoon, Synthesis of High Efficient Ca2SiO4:Eu2+ Green Emitting Phosphor by a Liquid Phase Precursor Method, J. Solid State Chem., 2012, 189, 68–74, DOI:10.1016/j.jssc.2011.11.046.
- M. Abbasi, A. A. Mirzaei and H. Atashi, Hydrothermal Synthesis of Fe-Ni-Ce Nano-Structure Catalyst for Fischer-Tropsch Synthesis: Characterization and Catalytic Performance, J. Alloys Compd., 2019, 799, 546–555, DOI:10.1016/j.jallcom.2019.05.314.
- D. R. R. Mullins, S. H. H. Overbury and D. R. R. Huntley, Electron Spectroscopy of Single Crystal and Polycrystalline Cerium Oxide Surfaces, Surf. Sci., 1998, 409(2), 307–319, DOI:10.1016/S0039-6028(98)00257-X.
- R. Sinclair, S. C. Lee, Y. Shi and W. C. Chueh, Structure and Chemistry of Epitaxial Ceria Thin Films on Yttria-Stabilized Zirconia Substrates, Studied by High Resolution Electron Microscopy, Ultramicroscopy, 2017, 176, 200–211, DOI:10.1016/j.ultramic.2017.03.015.
- D. S. Su, M. Wieske, E. Beckmann, A. Blume, G. Mestl and R. Schlögl, Electron Beam Induced Reduction of V2O5 Studied by Analytical Electron Microscopy, Catal. Lett., 2001, 75(1–2), 81–86, DOI:10.1023/A:1016754922933.
- A. Riaz, M. U. Ali, W. Lipiński and A. Lowe, Enhanced Oxygen Exchange Capacity in Nano-Structured Vanadia-Ceria Multi-Phase Oxygen Carriers for Solar Thermal Fuel Production, J. Mater. Chem. A, 2019, 7(48), 27347–27360, 10.1039/c9ta06471k.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02234b |
| This journal is © The Royal Society of Chemistry 2021 |