
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDestabilization potential of beta sheet breaker peptides on Abeta fibril structure: an insight from molecular dynamics simulation study†
Vinod Jani ,
Uddhavesh Sonavane and
Rajendra Joshi*
,
Uddhavesh Sonavane and
Rajendra Joshi*
Centre for Development of Advanced Computing (C-DAC), Panchavati, Pashan, Pune, India. E-mail: rajendra@cdac.in
First published on 5th July 2021
Abstract
Alzheimer's disease is characterized by amyloid-β aggregation. Currently, all the approved medications are to treat the symptoms but there is no clinically approved treatment for the cure or to prevent the progression of Alzheimer's disease (AD). Earlier reports suggest the use of small molecules and peptides to target and destabilize the amyloid fibril. The use of Beta Sheet Breaker (BSB) peptides seems to be a promising and attractive therapeutic approach as it can strongly bind and destabilize the preformed amyloid fibril. There are experimental studies describing the destabilization role of various BSB peptides, but the exact mechanism remains elusive. In the current work, an attempt is made to study the destabilization mechanism of different BSB peptides on preformed amyloid protofibril using molecular docking and simulations. Molecular docking of eight different BSB peptides of varying length (5-mer to 10-mer) on the Abeta protofibril was done. Docking was followed by multiple sets of molecular simulations for the Abeta protofibril–BSB peptide complex for each of the top ranked poses of the eight BSB peptides. As a control, multiple sets of simulations for the Abeta protofibril (APO) were also carried out. An increase in the RMSD, decrease in the number of interchain hydrogen bonds, destabilization of important salt bridge interactions (D23–K28), and destabilization of interchain hydrophobic interactions suggested the destabilization of Abeta protofibril by BSB peptides. The MM-GBSA free energy of binding for each of the BSB peptides was calculated to measure the binding affinity of BSB peptides to Abeta protofibril. Further residue wise contribution of free energy of binding was also calculated. The study showed that 7-mer peptides tend to bind strongly to Abeta protofibril as compared to other BSB peptides. The KKLVFFA peptide showed better destabilization potential as compared to the other BSB peptides. The details about the destabilization mechanism of BSB peptides will help in the design of other peptides for the therapeutic intervention for AD.
Introduction
Alzheimer's disease (AD) is a complex neurodegenerative disorder, characterized by aggregation of Abeta (Aβ) protein and thereby its deposition in the brain in the form of amyloid plaques and neurofibrillary tangles.1–3 The process starts with the proteolytic cleavage of full length amyloid precursor protein (APP) by beta and gamma secretase to release fragments of length between 39 and 42.4,5 These fragments form part of the amyloid cascade hypothesis and their fibrillary form is the basic component of the amyloid plaques found in neuronal cells.6 The two most common fragments are Aβ40 and Aβ42, out of which Aβ42 is more toxic.4 The process of formation of the Abeta fibril involves a series of events where the normal monomeric alpha helical structure is transformed into the beta structure followed by its aggregation in the pathological fibrillar insoluble state.4–7 Hayden et al., 2017 have highlighted the contribution of individual amino acid residues responsible for maintaining the native helical or random coil conformation of Abeta.8 It has been seen that the intermediate state oligomers are more toxic as compared to final state insoluble fibrils.9–12 Thus it becomes important to stop the aggregation of Abeta at a very early state.Studies to develop inhibitors for Abeta aggregates are being carried out since decades. The inhibitors designed range from organic molecules, antibodies and peptides.13–20 These inhibitors may work by preventing the formation of soluble oligomers or by destabilizing preformed oligomers.21 The major issue with the inhibitors is the toxicity which has led to their discards in the early phase of a clinical trial.22 Besides this, the exact mechanism of action of such inhibitors is also not known. Also, the destabilization strategy is gaining importance over the time as the destabilized Abeta fibrils become non-neurotoxic and also helps in the inhibition of higher order aggregates.23,24 There are multitude of studies on small molecules describing their role in the inhibition of aggregation, for example, small molecules like tramiprosate an ionic compound was able to maintain Abeta in non-fibrillar form and thereby decreasing Aβ42 induced cell death in neuronal cell cultures.25 However, in recent decades the peptide based inhibitors for the amyloid formation and its destabilization are gaining importance over the time and the same has been shown in the review by Sciaretta et al.26
Several peptide fragments have been designed to bind to important regions of Abeta involved in aggregations of the protein thereby inhibiting amyloid aggregation.27–30 The peptide based inhibitors have an advantage over the other class of inhibitors as the binding of such peptides would be through a self-recognition mechanism, mimicking the fibril growth process.31–37 Also, the peptide based inhibitors are easy to synthesis, broadly available and are less toxic.34–37 The peptide based inhibitors are mostly designed based on the sequence derived from Abeta itself. The studies have tried to identify regions of the N-terminus,38,39 central hydrophobic core,40,41 hinge or turn regions,42 and C-terminus43 of Abeta which are responsible for aggregation and fibrillization. Such studies have helped in designing the region specific peptide inhibitor. The peptide KLVFF identified by Tjernberg et al.28 binds to full-length Abeta and prevents assembly into fibrils. Soto and co-workers43 showed that the fragment LPFFD obtained by modifying LVFFA (replacement of Val at the second position by Pro and Ala at the fifth position by Asp) showed enhanced activity. The study showed that the LPFFD peptide was able to inhibit the Abeta aggregation and was also able to disassemble the preformed Abeta protofibrils in vitro.40,43,44 Minicozzi et al.45 in their work, experimentally and through molecular dynamics simulation showed the binding of Ac-LPFFD-NH2 a modified version of Ac-LPFFN-NH2 to the Aβ. The study showed that both the beta breaker peptides Ac-LPFFD-NH2 and Ac-LPFFN-NH2 were able to stabilize the native secondary structure of Abeta. Datki et al.46 obtained LPYFD-amide by the modification of LPFFD (substitution of Pro at the third position by Tyr) and its amidation at the C-terminus. In vitro study of LPYFD-amide peptide showed reduced nerve cell decay and Abeta induced cell mortality. Similarly, in vitro studies carried by Liu et al.47 on the decamer HKQLPFYEED showed better stability and inhibitory effect on Abeta aggregation inhibition as compared to HKQLPFFEED. The peptide (RGTFEGKF-NH2) designed by Liu et al.48 demonstrated that fibrillization can be disrupted by targeting specific residues of the amyloid fibril. Jagota et al.49 designed D-peptides viz. PGKLVYA, KKLVFFARRRRA and KKLVFFA based on the central hydrophobic region of Abeta (residues 16–20). In their study they evaluated inhibitory action, toxicity and pharmacological properties of these peptides on transgenic C. elegans model and showed that the two peptides PGKLVYA and KKLVFFA were able to improve survival in C. elegans.
Kanchi et al.50 in their study found that tryptophan modified LPFFD peptides were able to bind better to Abeta protofibril. Besides this, peptides with varying lengths have been designed targeting critical regions of Abeta and were found to be effective in preventing oligomerization of Abeta protein.51–53
The earlier studies by various researchers have described the destabilization role of various BSB peptides, however the mechanism of action of these peptides to disaggregate the preformed Abeta protofibril remains obscure. Secondly, the peptides of varying length and composition are reported to inhibit the fibrillization and the destabilization of preformed protofibrils. Hence in the present study, an attempt is made to study the mechanism of destabilization of various BSB peptides. Also, the study aims to find what could be the minimal length of the peptide which can effectively destabilize the preformed protofibrils. In the current study, eight BSB peptides of varying lengths were considered for docking. After docking, molecular simulations were performed for top ranked docked pose of each of the BSB peptides. Further, the interaction mechanism and destabilization effect of the peptides with Abeta protofibril was studied to identify the most potential destabilizing BSB peptide. The MM-GBSA method has been employed to estimate the free energy contribution of the residues of Abeta protofibril involved in binding with BSB peptides. These studies may provide a rationale for designing the new peptide based inhibitors against AD.
Methodology
Molecular docking
In order to study the effect of peptides on destabilization of Abeta protofibril, molecular docking of various peptides having the size in the range of five amino acids to ten amino acids was carried out. The peptides docked were HKQLPFFEED, HKQLPFYEED, RGTFEGKF, PGKLVYA, KKLVFFA, KLVFF, LPFFN and LPFFD. Prior to docking, all the peptide structures were generated in the trans configuration using Chimera software. All the peptides were energy minimized using the steepest descent and conjugate gradient method to remove any steric clashes. The molecular docking studies were carried out using NMR structure coordinates of Abeta protofibril PDB ID 2BEG.54 The structure selected was U-shaped model consisting of two beta strands. Strand 1 (residue range 17 to 26) and strand 2 (residue range 31 to 42) are connected by a bend having residues range 27 to 30. The model is a pentamer consisting of five identical chains denoted as A to E. There is growing evidence in regard to polymorphic character (U-shape and S-shape model) of Abeta protofibril, especially Abeta 1–42. A number of fibril structures with morphologically different fibril structures of Ab have been reported recently.55–58 Abeta being polymorphic in nature each of the fibril structure have its own importance and which have been highlighted in the recent experimental and computational studies.59–63 Also, studies by Cheon M et al.62 & Kahler A et al.63 suggest formation of U-shaped fibrillar structure from potent on-pathway intermediates of the Ab17-42 pentamer and hexamer oligomers (paranuclei). The U-shaped model (2BEG) has been part of most of the previously reported molecular simulation studies.64–70 Thus, 2BEG model was selected for the study.The amberff14SB force field was applied to parameterize protein (Abeta protofibril).71 As the specific binding site is not known for Abeta protofibril, hence blind docking of BSB peptides (peptide) were carried out onto the Abeta protofibril. Blind docking ensures that peptide search the entire protein surface area for possible docking sites. All the docked protofibril–peptide systems have been defined in Table 1 and indexed as S0 to S8, where S0 represents the APO system. Docking was carried out using DOCK6 software using standard parameters.72 Analyses of docking results were carried out using chimera by visual inspection along with the docking scores. Rigid docking was carried for these complexes. The relative binding affinities of the docked poses of the BSB peptides with Abeta protofibril were determined by using a grid score.
| System index | System name | Protein | Ligand (peptide) | Simulation time |
|---|---|---|---|---|
| S0 | APO | Abeta protofibril | None | 2 × 100 ns |
| S1 | HFD | Abeta protofibril | HKQLPFFEED | 2 × 100 ns |
| S2 | HYD | Abeta protofibril | HKQLPFYEED | 2 × 100 ns |
| S3 | RGT | Abeta protofibril | RGTFEGKF | 2 × 100 ns |
| S4 | PGK | Abeta protofibril | PGKLVYA | 2 × 100 ns |
| S5 | KKL | Abeta protofibril | KKLVFFA | 2 × 100 ns |
| S6 | KLV | Abeta protofibril | KLVFF | 2 × 100 ns |
| S7 | LPN | Abeta protofibril | LPFFN | 2 × 100 ns |
| S8 | LPD | Abeta protofibril | LPFFD | 2 × 100 ns |
Molecular dynamics (MD) simulations
The molecular simulation studies were performed for the protofibril–peptide complex using the AMBER 16 suite of the program.73 Based on grid score the top ranked docked pose of the protofibril–peptide complex for each of the peptide were considered for the simulations to investigate the destabilization effect. Molecular simulations were carried out for nine systems (S0 to S8) as mentioned in Table 2. The protofibril–peptide complexes were parameterized using the amberff14SB force field. The systems were put in an octahedral water box with TIP3P water model with a minimum distance between the solute and the box around 10 Å.| Peptide | Dock score (kcal mol−1) |
|---|---|
| HKQLPFFEED | −26.597866 |
| HKQLPFYEED | −29.543624 |
| RGTFEGKF | −28.651105 |
| PGKLVYA | −35.047874 |
| KKLVFFA | −33.654152 |
| KLVFF | −32.981491 |
| LPFFN | −27.007414 |
| LPFFD | −31.188615 |
Each of the systems was neutralized by adding sufficient counter ions. This was followed by minimization of systems with 5000 steps comprising of initial 2500 steps of steepest descent and remaining 2500 steps of the conjugate gradient. These minimized structures were heated till 300 K using a Langevin thermostat and then equilibrated for 1 ns at the same temperature. The hydrogen atoms were constrained using the SHAKE algorithm. The system was equilibrated for 1 ns at a constant temperature of 300 K and pressure of 1 atm. Long-range electrostatic interactions were calculated using the Particle-Mesh Ewald (PME) method. Newtonian equation of motion was solved using the Leapfrog integrator with the integration time step of 2 fs. The equilibrated structures were then subjected to production runs for 100 ns. All the systems were subjected to two runs of 100 ns each. Details of all simulations are given in Table 1. Additional set of simulations was carried out for best two peptide systems viz. PGK and KKL and APO systems upto 200 ns to observe trend of destabilization events.
Analysis of MD generated trajectories
The analysis of the trajectories were carried out using CPPTRAJ module of AMBERTOOLS.74 Various parameters were calculated to assess the destabilization of Abeta protofibril. The salt bridge distances between Asp23–Lys28 (aspartic acid at 23rd position and lysine at 28th position) between the neighboring chains were calculated. Besides these few other important distances were calculated to assess destabilization effect of peptides on Abeta protofibril. Secondary structure analysis was carried out using DSSP. The binding free energy between the Abeta protofibril and peptides were calculated using MM-GBSA method.75 In order to determine binding mode of peptides and to determine energetic contribution of individual residue, energy contribution of Abeta protofibril residues actively involved in binding with peptides were determined. Ligplot76 and PLIP77 were used to study Abeta protofibril and peptide interactions. ShiftX2 (ref. 78) was used to calculated the chemical shifts.Results
Molecular docking
In the present study, molecular docking of multiple peptides of different lengths on Abeta protofibril was carried out using DOCK6. The top ranked docked pose for each peptide was chosen based on grid score. The grid score quantifies the binding of the peptide to protein based on non-bonded interactions. Thus, a more negative grid score indicates better binding. The best docked protofibril–peptide complex for each of the docked peptides is shown in Fig. 1. The blind docking was carried out considering the docking grid around the entire protein. The binding modes for each of the peptides were investigated to gain insights into the molecular interactions that affect the potency and mode of their binding. The Fig. 1 shows that most of the peptides were found to be docked along with chain E of Abeta protofibril. The peptide HKQLPFFEED, HKQLPFFEED, RGTFEGKF, KKLVFFA, KLVFF and LPFFD were found to be docked close to chain E, while PGKLVYA and LPFFN were found to be docked close to chain A. The lowest value of grid score for PGKLVYA peptide (−35.047 kcal mol−1) suggests its strong binding with Abeta protofibrils as compared to the other peptides investigated in the present study. The grid score for all the peptides is given in Table 2. The LIGPLOT analysis of the docked complex of Abeta protofibril and peptides shown in the Fig. 2. The figure represents various interactions between Abeta protofibril and the peptides. The Fig. 2 pictorially depicts hydrogen bond (represented by the dashed line) and hydrophobic contacts (represented by arc). Details about the interactions have been given in ESI Table SI.† The importance of docking for the evaluation of the best binding pose and binding affinity of various drugs towards Abeta protofibril to test their anti-fibrilization property have been mentioned in several studies.79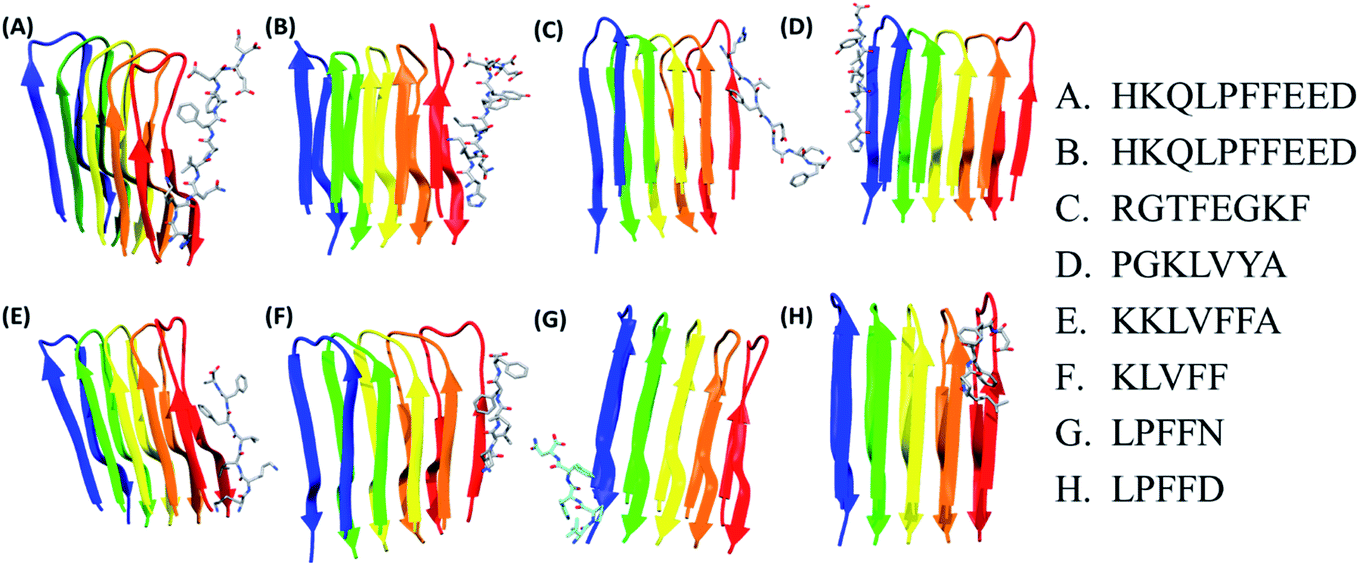 | ||
| Fig. 1 Docking representation of Abeta oligomer with best pose of different peptides. Abeta protofibril. Each Abeta protofibril consist of five chains represented as blue: chain A, green: chain B, yellow: chain C, orange: chain D and red: chain E. | ||
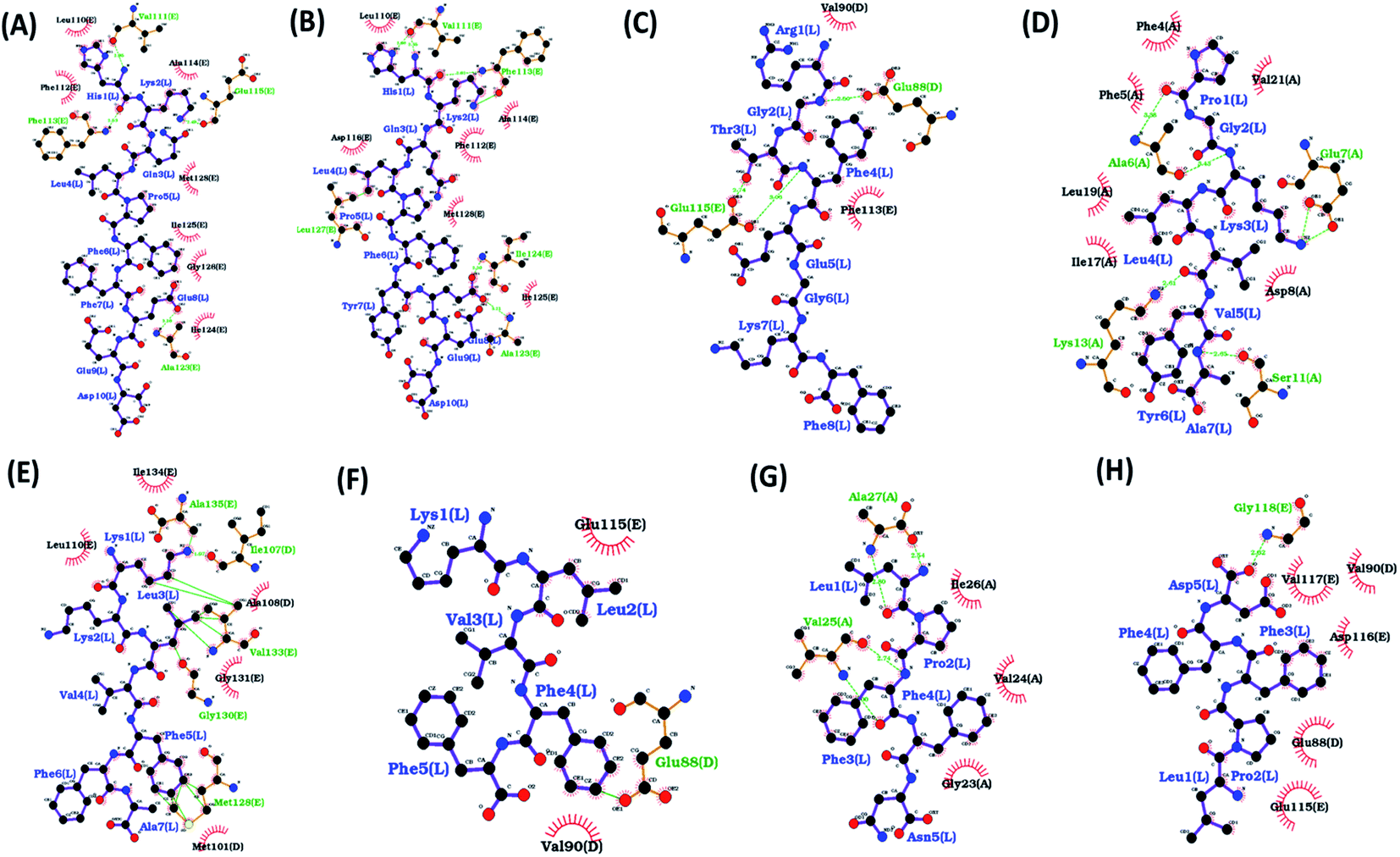 | ||
| Fig. 2 Ligplot showing various interactions between residues of Abeta protofibril and different docked peptides. Hydrogen bonds are represented by green lines and hydrophobic interactions are represented by red arc. The figure shows protofibril with docked peptide (A) HKQLPFFEED, (B) HKQLPFFEED, (C) RGTFEGKF, (D) PGKLVYA, (E) KKLVFFA, (F) KLVFF(G), LPFFN, (H) LPFFD. | ||
The results of docking for the first three top poses for each of the peptides have been given in ESI Fig. S1 and Table S2.† It was observed that binding of the peptide occur either along chain A or chain E of the protofibril. As the residues along both the chains are identical we considered the top pose for the simulations. Followed by docking two sets of molecular dynamics simulations were carried to explore the conformational space of the peptides.
Molecular dynamics (MD) simulations
The docking poses give only a static picture of the interactions between peptide and Abeta protofibril, hence MD simulations were carried out for the best docked pose of each protofibril–peptide complex. The MD simulations help to understand whether the interactions formed during docking are stable or transient. Molecular simulations of the APO system were also carried out as a control system.Comparison of the simulation data with experimental results
Prior to the analysis of simulation results, the molecular dynamics trajectories were validated by comparing with chemicals shifts of NMR structure used for simulations. The Cα and Cβ chemical shifts were computed using SHIFTX2 packages and compared. The correlation coefficient for Cα atoms was 0.98 (ESI Fig. S2a†) and for the Cβ atoms were 0.99 (ESI Fig. S2b†). The high correlation between the theoretical and experimental NMR chemical shift values indicates that MD simulations are able to reproduce the structural ensemble of Aβ42 reasonably and is supported by other studies.67Root mean square deviation
Root mean square deviation (RMSD) gives an idea about how much a structure has deviated from the original structure. The RMSD was calculated using the start structure as a reference frame. The Fig. 3 shows the histogram plot for the RMSD for the APO and all protofibril–peptide complex systems. From the figure, it can be seen that for the APO system, for the majority of the population the RMSD lies in the range of 2 Å to 3 Å. For the HFD, HYD and LPN systems it lies in the range of 4 Å to 6 Å. For the RGT and LPD systems, it lies in the range of 4 Å to 8 Å. For the PGK, KKL and KLV systems it is in the range of 4 Å to 9 Å. The increase in the RMSD values in protofibril–peptide as compared to the APO system shows that the peptides are able to destabilize the Abeta protofibril. The maximum deviation was observed in PGK, KKL and KLV systems, the PGK system showed major peaks around 5 Å and 7.7 Å, KKL system showed peaks around 3 Å and 7 Å, and KLV showed peaks around 3 Å, 5 Å and 7.5 Å.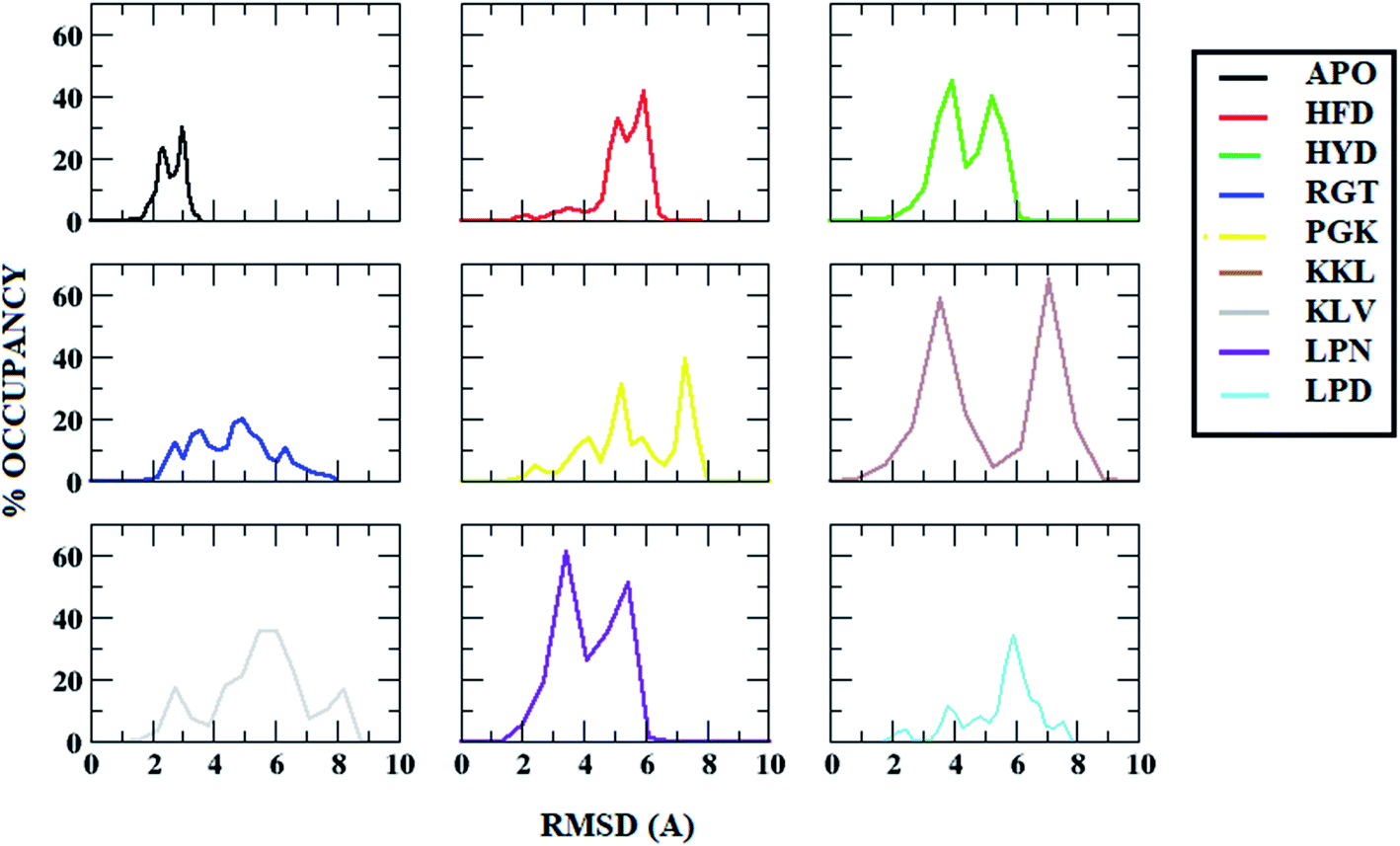 | ||
| Fig. 3 Root Mean Square Deviation (RMSD) distribution in Abeta protofibril apo structure and Abeta protofibril–peptide complexes. | ||
Root mean square fluctuation
In order to identify the regions of Abeta protofibril that showed maximum fluctuations, root mean square fluctuation (RMSF) was calculated. It gives an idea about how much a particular residue or region had fluctuated from an average structure. The Fig. 4 shows the RMSF plot for all the systems. The APO system showed minimum deviation as compared to all other systems. For the APO system, the maximum deviation was observed for the C-terminal region of chain A which was around 3 Å. All other systems showed deviation throughout the protein with an average deviation of 3 Å. The C-terminal of chain A (residue 37 to 42) and beta strand 1 of chain E (residue 24 to 33) in all protofibril–peptide systems showed maximum deviation. Among the protofibril–peptide system, maximum deviation in the C-terminal region of chain A was shown by the PGK system and deviation was around 10 Å. The KKL system showed maximum deviation in beta strand 1 of chain E and was around 8 Å. Thus maximum deviation was observed in the protofibril–peptide system consisting of 7-mer peptides viz. PGKLVYA and KKLVFFA. | ||
| Fig. 4 Root Mean Square Fluctuation (RMSF) for in Abeta protofibril apo structure and Abeta protofibril–peptide complexes for all the chains. | ||
Secondary structure analysis
The first step in the formation of Abeta protofibril is the conversion of the alpha helix to beta structure. Thus the first step in the destabilization of protofibril may be the destabilization of beta structure. Experimental and computational studies by researchers have shown the conversion of beta to coil and helix during destabilization of Abeta protofibril in presence of ligand molecules.80–82 The most important aspect for the stability of Abeta protofibrils is the higher β-sheet content and it plays an important role in maintaining the organized β-fibrillar structure. Hence secondary structure analysis was done to check the average percentage of different secondary structures during the course of simulations. Table 3 shows the percentage content of various secondary structures for APO and each of the protofibril–peptide systems. The APO system has a maximum percent of beta structure i.e. 61% and the lowest coil structure i.e. 26% among all the simulated systems. In all the protofibril–peptide systems the loss in beta structure content and gain in the coil region has been observed. The maximum loss in the beta structure content was observed for the PGK system, where the percent beta structure content was reduced to 45% and the percent coil structure was increased to 34%. Thus the interaction of the peptides with Abeta protofibril has resulted in the loss of beta structure and gain of coil structure. This is in coherence with previous studies showing decreases in beta content in presence of small ligands.67,70,82| System | B-structure (B-sheet/B-bridge) | Coil | Turn/bend | Alpha |
|---|---|---|---|---|
| APO | 61 | 26 | 10 | 0 |
| HFD | 53 | 28 | 14 | 1 |
| HYD | 55 | 30 | 12 | 0 |
| RGT | 50 | 32 | 15 | 1 |
| PGK | 45 | 34 | 13 | 1 |
| KKL | 50 | 32 | 13 | 0 |
| KLV | 52 | 30 | 15 | 1 |
| LPN | 51 | 32 | 14 | 1 |
| LPD | 53 | 30 | 14 | 0 |
Hydrogen bond analysis
The hydrogen bonds are important in maintaining the stability of the secondary structure and folding/unfolding of the protein. The biological function of a protein can be affected owing to the loss or gain of hydrogen bonds. Hence the total number of hydrogen bonds responsible for maintaining the oligomer structure were calculated. The plot for the average number of the interchain hydrogen bond between the adjacent chains and for the entire protofibril for all the protofibril–peptide systems are shown in Fig. 5. The Fig. 5A shows the average number of inter chain hydrogen bonds for the entire protofibril. As compared to the APO system where the average number of hydrogen bonds were around 55 for the entire protofibril, for all the other systems the hydrogen bond ranged from 40 to 46. Thus the presence of peptides destabilized the hydrogen bond network in the Abeta protofibril. The maximum destabilization was observed for the PGK and KKL systems, where maximum conformations had an average number of hydrogen bonds for the entire protofibril reduced to 40.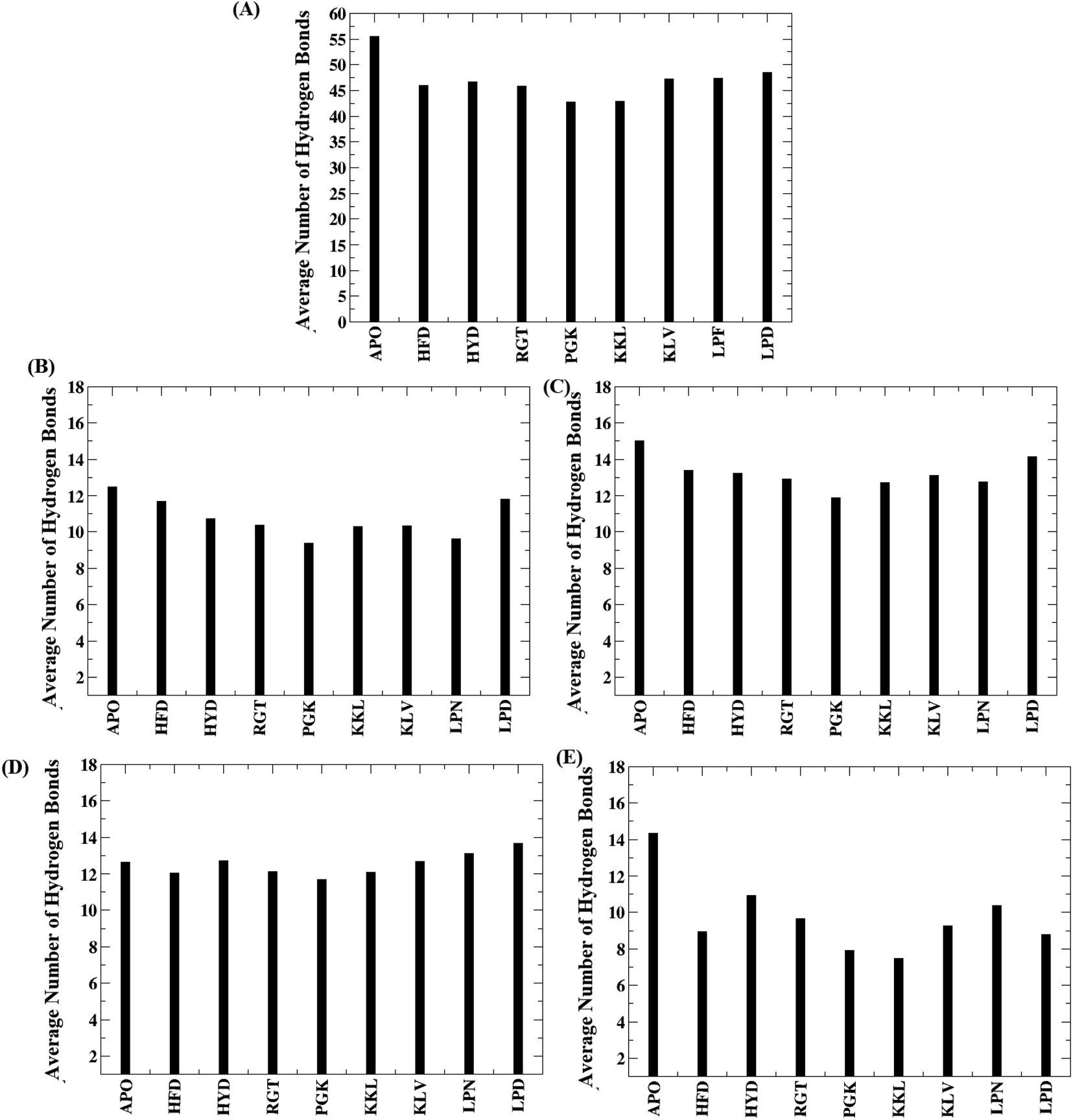 | ||
| Fig. 5 Average number of hydrogen bond for all the systems. (A) Entire system, (B) between chain A–chain B, (C) between chain B-chain C, (D) between chain C-chain D, (E) between chain D-chain E. | ||
Further, to check hydrogen bonds between which neighboring chains are affected most, the average number of hydrogen bonds between the neighboring chains are plotted in the Fig. 5B–E. From the Fig. 5B, it can be seen that the average number of hydrogen bonds between chain A and chain B varies in the range of 12 to 9 with the PGK system having the minimum number. Similarly, Fig. 5C shows the average number of hydrogen bonds between chain B and chain C with the range varying between 15 to 12. The PGK system showed the least average hydrogen bond number between chain B and chain C i.e. 12. For chain C and chain D (Fig. 5D), the average number of hydrogen bonds varies in the range of 14 to 12 with the lowest for PGK and KKL systems. For chain D and chain E (Fig. 5E), the average number of hydrogen bonds varies in the range of 14 to 7 with the lowest for the KKL system. Thus, maximum destabilization was seen between chain D and chain E followed by chain A and chain B.
Salt bridge (Asp23–Lys28)
The interchain salt bridge (Asp23–Lys28) present between the bend region for all the chains are known to stabilize protofibril structure.83,84 The salt bridge between Cγ of Asp23 and Nξ of Lys28 plays an important role in the stabilization of cross beta structure between chains of Abeta oligomer.83 As the peptides tend to bind to either chain A or chain E of the protofibril, the interchain salt bridge between chain A and chain B, and between chain D and chain E was calculated.The distance distribution of salt bridges between chain A and chain B for all the systems are given in the Fig. 6. In the case of the APO system, the salt bridge distance between chain A and B was maintained around 3 Å for most of the conformations. In the case of the HFD system, it showed a peak at around 3 Å but around 20% of conformations showed a distance of more than 4 Å. For the HYD system, most of the conformations had a distance in the range of 3 Å to 4 Å. For the RGT system, two zones were observed one with conformations around 3 Å and another around 5 Å. For the PGK system also, two peaks were observed one with conformations around 3 Å and another around 5.5 Å was observed. For other systems viz. KKL, KLV, LPN, and LPD, the conformations mostly have distances in the range of 3 Å to 4 Å. From the distance calculation, it can be seen that the RGT and the PGK systems showed comparatively more destabilization of salt-bridge between chain A and chain B.
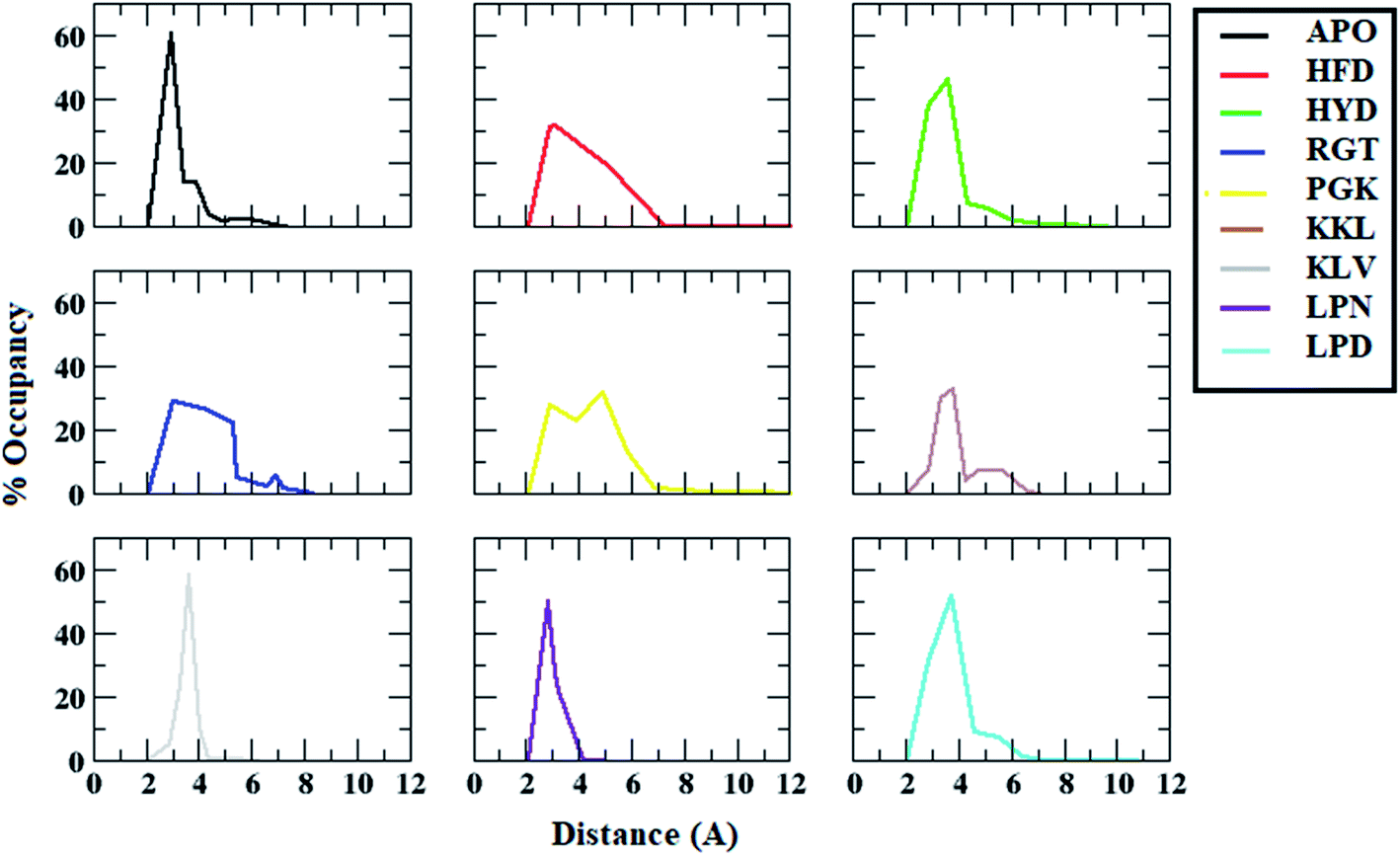 | ||
| Fig. 6 Distance distributions between ASP23 (chain A) and LYS28 (chain B) residues for salt bridge formation in Abeta protofibril apo structure and Abeta protofibril–peptide complexes. | ||
Similarly, the distance distribution of salt bridge interaction between chain D and chain E for all the systems was plotted and is shown in the Fig. 7. For the APO system, the average salt bridge distance was around 3.5 Å. However, all the protofibril–peptide systems showed two peaks, with one having a distance of more than 5 Å representing the destabilized salt bridge. The maximum destabilization was seen in the KKL system where two dominant peaks were seen one around 3.5 Å with 45% of the population, and the other around 18 Å with 30% population. From the salt bridge distance calculation it can be seen that the presence of peptides tends to affect the salt bridge stability. Based on the distance it can be seen that the salt bridge between chain A and chain B was affected most in the PGK system. Similarly, the salt bridge between chain D and chain E was affected most in the KKL system. Thus, it was observed that the 7-mer peptides (PGKLVYA and KKLVFFA) tend to affect the salt bridge the most.
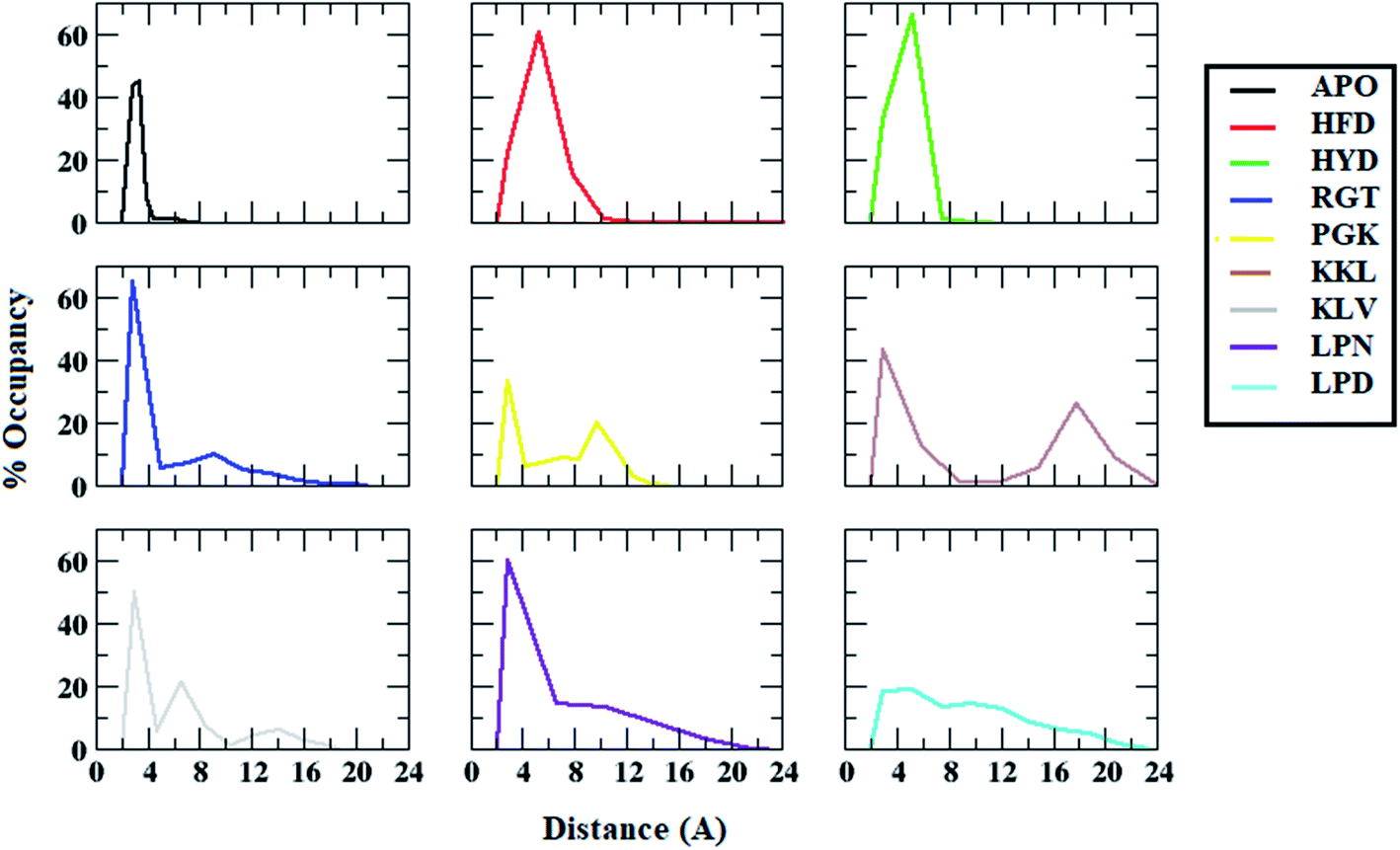 | ||
| Fig. 7 Distance distributions between ASP23 (chain D) and LYS28 (chain E) residues for salt bridge formation in Abeta protofibril apo structure and Abeta protofibril–peptide complexes. | ||
Distance between Ala21 and Val36
Besides salt-bridge interactions, hydrophobic interactions play an important role in maintaining the stability and overall folding of the protein.85,86 The hydrophobic residues tend to get buried inside the solvent inaccessible cavity and tend to provide stability to amyloid fibrils.86 Thus, the interchain distance between the hydrophobic residues Ala21 and Val36, which is one of the important parameters that determine the destabilization of Abeta protofibril, was calculated.67,87 Fig. 8 shows the average interchain distance between Ala21 (chain A)–Val36 (chain B). The average Ala21 (chain A)–Val36 (chain B) distance in the APO structure was around 8 Å. However, the average interchain distance tends to increase in all the protofibril–peptide systems with the maximum being in the PGK and the KKL systems. The average interchain distance in the PGK system tends to increase to 20 Å, while in the KKL system it tends to rise to 17 Å. Similarly, the average interchain distance between Ala21 (chain D)–Val36 (chain E) was calculated for all the systems (Fig. 9). For the APO system, the average interchain distance was around 6 Å. For all other protofibril–peptide systems it was on the higher side as compared to the APO system. The PGK and KKL systems showed maximum average interchain distance in the range of 7 Å to 15 Å. Thus, similar to salt-bridge interactions the 7-mer peptides tend to show more destabilizing effect for hydrophobic interactions as compared to all other peptides.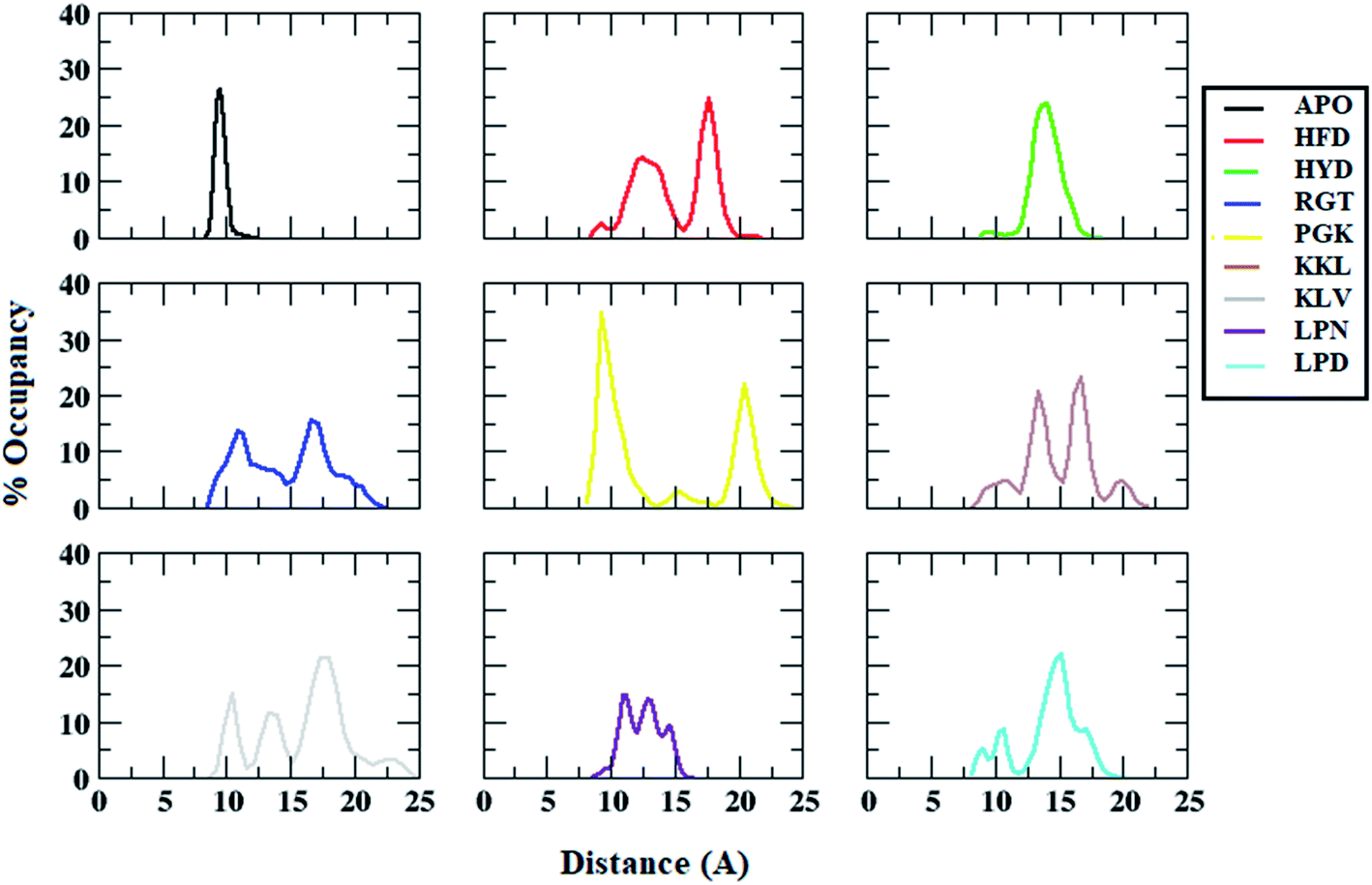 | ||
| Fig. 8 Distance distributions between ALA21 (chain A) and VAL36 (chain B) residues in Abeta protofibril apo structure and Abeta protofibril–peptide complexes. | ||
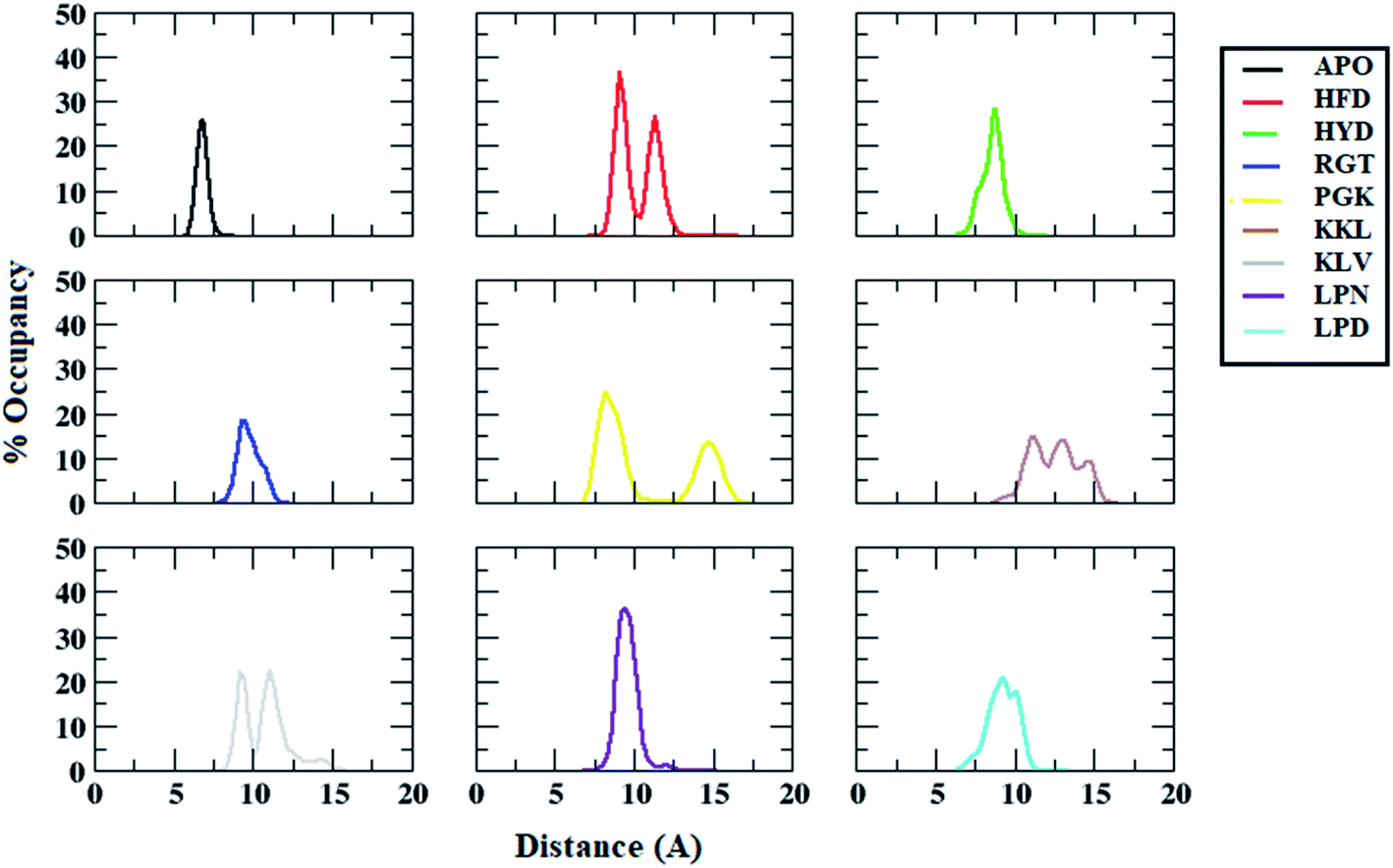 | ||
| Fig. 9 Distance distributions between ALA21 (chain A) and VAL36 (chain B) residues in Abeta protofibril apo structure and Abeta protofibril–peptide complexes. | ||
Interaction between protofibril and peptides
Interaction analysis between protofibril and peptides were carried out to identify key residues involved in the destabilization of Abeta protofibril. The interactions analysis of protofibril–peptide systems reveals the formation of hydrogen bonds and non-bonded interaction between Abeta protofibril and peptides. The key residues involved in the binding for each of the peptides and having occupancy of more than 20% have been reported in Table 4. The residues involved are represented in the form residuenumber_residuename_chain for Abeta protofibril and residuenumber_residuename for peptides.| Hydrogen bond | Hydrophobic | |
|---|---|---|
| HFD | 22GLU_C:2LYS | 19PHE_E:4LEU |
| 22GLU_E:3GLN | 39VAL_D:6PHE | |
| 23ASP_E:2LYS | ||
| HYD | 20PHE_E:3GLN | 18VAL_D:6PHE |
| 22GLU_E:3GLN | 19PHE_E:3GLN | |
| 22GLU_E:2LYS | 20PHE_D:8GLU | |
| 37GLY_E:3GLN | 20PHE_C:6PHE | |
| 20PHE_E:1HIS | 20PHE_E:2LYS | |
| RGT | 20PHE_E:3THR | 20PHE_E:3THR |
| 22GLU_C:7LYS | ||
| 22GLU_E:1ARG | ||
| 22GLU_D:1ARG | ||
| PGK | 21ALA_A:5VAL | 21ALA_A:5VAL |
| 21ALA_A:4LEU | 20PHE_C:4LEU | |
| 23ASP_A:2GLY | ||
| 20PHE_A:7ALA | ||
| KKL | 35MET_E:3LEU | 19PHE_E:6PHE |
| 35MET_E:1LYS | 36VAL_E:6PHE | |
| 37GLY_E:6PHE | 19PHE_D:6PHE | |
| 33GLY_E:1LYS | 24VAL_E:4VAL | |
| 22GLU_E:4VAL | ||
| 41ILE_E:2LYS | ||
| 28LYS_E:2LYS | ||
| 23ASP_E:1LYS | ||
| 39VAL_E:6PHE | ||
| KLV | 35MET_E:2LEU | |
| LPN | 40VAL_A:3PHE | 40VAL_A:3PHE |
| 42ALA_A:1LEU | ||
| 38GLY_A:5ASN | ||
| 25GLY_A:5ASN | ||
| LPD | 28LYS_E:4PHE | 30ALA_E:3PHE |
The HKQLPFFEED peptide tends to form three hydrogen bonds and two hydrophobic interactions. The residues involved in the formation of hydrogen bonds were 22Glu_C with 2Lys, 22Glu_E with 3Gln, and 23Asp_E with 2Lys. The residues involved in the formation of hydrophobic interactions were 19Phe_E with 4Leu and 39Val_D with 6Phe. In the case of the HKQLPFYEED peptide five hydrogen bonds and five hydrophobic interactions were observed. The residues involved in the formation of hydrogen bonds were 20Phe_E with 3Gln, 22Glu_E with 3Gln, 22Glu_E with 2Lys, 37Gly_E with 3Gln, and 20Phe_E with 1His. The residues involved in the formation of hydrophobic interactions 18Val_D with 6Phe, 19Phe_E with 3Gln, 20Phe_D with 8Glu, 20Phe_C with 6Phe and 20Phe_E with 2Lys. The RGTFEGKF peptide formed four hydrogen bonds and one hydrophobic interaction. The residues involved in the formation of hydrogen bonds are 20Phe_E with 3Thr, 22Glu_C with 7Lys, 22Glu_E with 1ARG, and 22Glu_D with 1ARG. The residues involved in the formation of hydrophobic interactions 20Phe_E with 3Thr. The PGKLVYA peptide formed four hydrogen bonds and two hydrophobic interactions. The residues involved in the formation of hydrogen bonds are 21Ala_A with 5Val, 21Ala_A with 4Leu, 23Asp_A with 2Gly and 20Phe_A with 7Ala. The residues involved in the formation of hydrophobic interactions 21Ala_A with 5Val and 20Phe_C with 4Leu. The KKLVFFA peptide formed nine hydrogen bonds and four hydrophobic interactions. The residues involved in the formation of hydrogen bonds are 35Met_E with 3Leu, 35Met_E with 1Lys, 37Gly_E with 6Phe, 33Gly_E with 1Lys, 22Glu_E with 4Val, 41Ile_E with 2Lys, 28Lys_E with 2Lys, 23Asp_E with 1Lys, and 39Val_E with 6Phe. The residues involved in the formation of hydrophobic interactions are 19Phe_E with 6Phe, 36Val_E with 6Phe, 19Phe_D with 6Phe, and 24Val_E with 4Val. The KLVFF peptide formed one stable hydrogen bond. The residues involved in the formation of hydrogen bonds are 35Met_E with 2Leu. The LPFFN peptide formed four hydrogen bonds and one hydrophobic interaction. The residues involved in the formation of hydrogen bonds are 40Val_A with 3Phe, 42Ala_A with 1Leu, 38Gly_A with 5Asn, and 25Gly_A with 5Asn. The residues involved in the formation of hydrophobic interactions are 40Val_A with 3Phe. The LPFFD peptide formed one hydrogen bond and one hydrophobic interaction. The residues involved in the formation of hydrogen bonds are 28Lys_E with 4Phe. The residues involved in the formation of hydrophobic interactions are 30Ala_E with 3Phe.
Thus, KKL a 7-mer peptide tend to form the maximum number of stable interaction with the Abeta protofibril. The peptides tend to forms hydrogen bond interaction with 23Asp or 28Lys and destabilize the salt bridge between 23Asp and 28Lys. Similarly, the studies by Barale et al.87 and Agrawal et al.88 have shown the destabilization of Abeta protofibril due to binding of the ligand to the salt bridge forming residues.
Binding free energy analysis between Abeta protofibril and peptides
The binding affinity calculation of peptides with Abeta protofibril was carried out using the MM-GBSA method. The average delta energy of binding for all the systems has been given in Fig. 10 and ESI Table S3.† From the figure, it can be seen that the KKL system has the lowest deltaG binding energy and it was −37.31 kcal mol−1. The second lowest average deltaG of binding was observed for PGK and it was around −26.43 kcal. PGK system was followed by RGT (−18.54 kcal mol−1), HFD (−16.61 kcal mol−1), KLV (−15.98 kcal mol−1), LPN (−10.41 kcal mol−1), LPD (−8.00 kcal mol−1) and HYD (−3.40 kcal mol−1).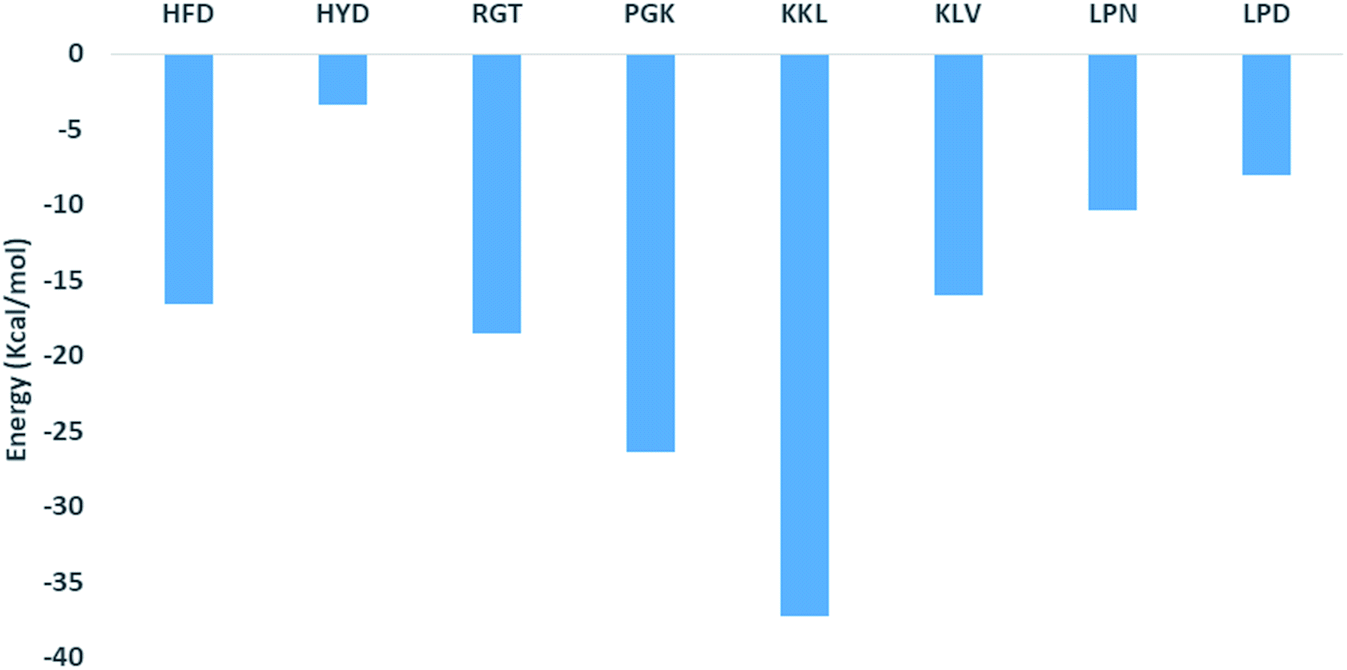 | ||
| Fig. 10 Free energy of binding of Abeta protofibril–peptide complexes. | ||
The analysis of binding free energy per-residue has been shown in Fig. 11. The residues whose contribution in binding energy is more than 0.2 kcal mol−1 have been listed here. In the HFD system, the contributing residues were Glu22 of chain B, C, D, and E and Asp23 of chain E. For the HYD system, the contributing residues were Phe20 of chain E and Glu22 of chain A and E. In the RGT system the Phe20 from chain D and E and Glu22 from chain B, C, D, and E were the contributing residues. For the PGK system, the residues were Phe19 of chain A, Phe20 of chain A and C, Ala21 of chain A, B and C, and Asp23 of chain D. For the KKL the residues mainly belonged to chain E, the residues were Phe19 of chain D and E and the residues Glu22, Asp23, Lys28, Gly29, Ile31, Gly33, Leu34, Met35, Val36, Gly37, Gly38, Val39, Ala40, Ile41 and Ala42 of the chain E. In the KLV system, the residues were Leu17 of chain D and E, Val18 chain D and E, Phe20 chain C, D, and E, Glu22 chain B, C, D, and E. For the LPN system, the residues were Met35 of chain A, B, and C, Gly38, Val39, Val40, Ile41 and Ala 42 of the chain A. In the LPD system, the residues were Leu17 of the chain D and E, Val18 of the chain C, D and E, Phe20 of the chain B, C and D, and Ala21 chain B and C. Thus the KKL system tends to form interaction with the maximum number of residues of chain E and also has lowest deltaG of binding.
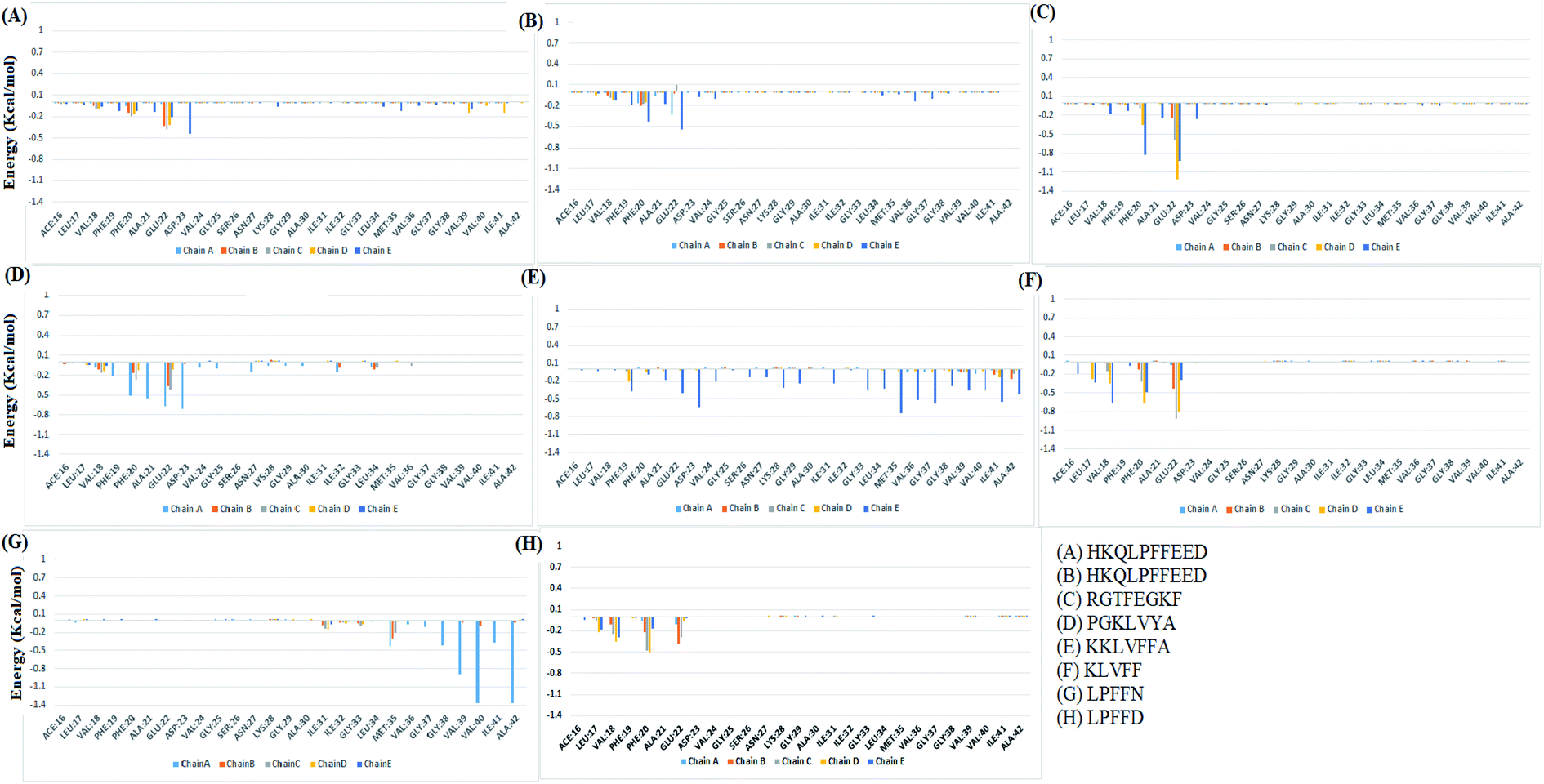 | ||
| Fig. 11 Individual residue wise energy decomposition for all the systems. Different colours represents residues from different chains. | ||
The results of the additional set of simulations for best two peptide systems viz. PGK and KKL and APO systems are presented in the ESI document Section B.† It was observed that these additional set of simulations shows similar trend as that of results of two sets of 100 ns simulations reported.
Discussion
In one of the earlier studies on design of BSB peptides Soto et al. designed the LPFFD (iaβ5) penta–peptide and in their in vitro studies observed that this peptide is able to dissolve preformed fibrils. This study led to other researchers to design various peptides of different length and study the destabilization potential of these designed peptides. The designed peptides ranged from 5-mer to 13-mer in length. Besides peptides various small molecules have also been studied experimentally and computationally for their destabilization effect.Saini et al.89 carried out simulation studies of Abeta protofibril (2BEG) in presence of fluorinated compound, D744. In the study they showed that the presence of ligand leads to decrease in β-sheet content and simultaneously increase in coil structure. They also showed that it destabilize the protofibril by disrupting the native inter-chain D23–K28 salt bridges, decreasing inter-chain backbone hydrogen bonds and rearranges the tightly compact β-strand–bend-β-strand motifs by increasing the interchain A21–V36 distance. In another study, Barale et al.87 showed that binding of arginine hybrid peptides to Abeta protofibril destabilize it by breaking of important salt bridge between D23–K28. Gupta et al.65 showed the destabilization ellagic acid (REF) using molecular dynamic simulations. They showed that REF leads to the decrease in beta-structure content, disruption of salt bridges and decline in the number of hydrogen bonds. They also showed the F19, L34, M35, V36, V40, I41, and A42 residues played major role in binding with REF. The major factors that were considered by most of the researchers for evaluating the destabilization potential of the ligands are secondary structure content especially beta and coil content, salt bridge distance between Asp23–Lys28 (especially in U-shaped model), inter-chain hydrogen and hydrophobic interaction.64,65,81–87 In the current work, it was observed that binding of peptides with Abeta protofibril has resulted in the loss of beta structure and gain of coil structure. This is in accordance with previous studies showing increase of coil and decrease of beta structures. The number of inter chain hydrogen bond also decreased in the presence of the peptides. The destabilization of protofibrils was observed owing to increase in the inter-chain distance of important salt bridge D23–K28 and hydrophobic interaction A21–V36.
In the study carried out by Shuaib et al.81 they showed that the Phe 20 has important share in binding free energy to modified iaβ5 peptide PPFFE. On similar line Jarmuła et al.61 showed that addition of Phe residues into BSB peptide (LPFFFD) and docked around Phe 20 in the CHC region of Aβ42 fibrils help to improve the destabilization potential of peptide. The importance of Phe 19 and Phe 20 was shown by Dutta et al.84 through mutation studies. In present study, residue wise free energy decomposition showed that the Phe 20 of chain C, chain D and chain E of Abeta protofibril contributes in protofibril peptide binding energy. The Phe 20 from different chains of protofibril transiently formed pi–pi interaction with Phe 4 of the LPFFD. Besides LPD system, in other peptide systems also role of Phe 20 in protofibril peptide binding energy was predominantly seen. Along with Phe 20 and Phe 19, Glu 22 was also seen to have contribution in free energy of binding. Dutta et al.84 also showed the importance of Glu 22 residue in maintaining the D23–K28 salt bridge.
Conclusion
In this study, the destabilization potential of eight BSB peptides namely, HKQLPFFEED, HKQLPFFEED, RGTFEGKF, PGKLVYA, KKLVFFA, KLVFF, LPFFN, and LPFFD have been studied using molecular docking and MD simulations. The docking study revealed the PGKLVYA peptide as the most strong binding peptide based on grid score. The BSB peptides tend to bind to the Abeta protofibril along the chain A or chain E. In order to study the dynamics of the docked complexes, top ranked docked pose of BSB peptides and protofibril complex were subjected to molecular simulations. It was observed that the presence of BSB peptides tends to destabilize the Abeta protofibril, mainly the terminal chains. The structural parameters like RMSD, RMSF, secondary structure content, and important contacts were calculated. The presence of the BSB peptides led to an increase in the overall RMSD and RMSF values of Abeta protofibril as compared to the APO system. Also the percentage beta structure content decrease for the Abeta protofibril in the presence of the BSB peptides. The change in the secondary structure was associated with a decrease in the number of interchain hydrogen bonds. The increase in the salt bridge distance between the Asp23 and Lys28 and hydrophobic distance between Ala21 and Val36 further confirms the destabilization of the Abeta protofibril by the BSB peptides. The maximum destabilization potential was observed for 7-mer peptides as compared to other BSB peptides studied in current work. The KKLVFFA peptide showed the lowest MM-GBSA binding energy and maximum perturbations in structural parameters. The hydrogen bond and hydrophobic interactions between peptides and Abeta protofibril played an important role in the destabilization of the protofibril structure. The key interacting residues of Abeta protofibril with KKLVFFA peptide were Phe19 of chain D and E and the residues Glu22, Asp23, Lys28, Gly29, Ile31, Gly33, Leu34, Met35, Val36, Gly37, Gly38, Val39, Ala40, Ile41 and Ala42 of the chain E. As peptides form safe options for therapeutics, the KKLVFFA peptide and its modifications may be considered for the destabilization of preformed higher order toxic aggregates of Abeta. The current work provides information on the structural changes in the Abeta protofibril owing to the binding of the BSB peptides. It also provides information on key interacting residues of Abeta protofibril and BSB peptides and paves the way for designing new peptide based inhibitors of 7-mer length for the destabilization of Abeta protofibril and thereby a probable treatment for Alzheimer's disease.Author contributions
Vinod Jani: formulation of the work, performing simulations, analysis, manuscript writing, Uddhavesh Sonavane and Rajendra Joshi: formulation of the work, review of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the Ministry of Electronics and Information Technology (MeitY), Government of India, New Delhi, and National Supercomputing Mission (NSM) for providing financial support. This work was performed using the “Bioinformatics Resources and Applications Facility (BRAF)”.References
- E. Masliah, Neuropathology: Alzheimer's in real time, Nature, 2008, 451(7179), 638–639, DOI:10.1038/451638a . PMID: 18256653..
- D. J. Selkoe, Folding proteins in fatal ways, Nature, 2003, 426(6968), 900–904, DOI:10.1038/nature02264 . Erratum in: Nature, 2004, 428 (6981) 445. PMID: 14685251..
- R. B. Knowles, C. Wyart, S. V. Buldyrev, L. Cruz, B. Urbanc, M. E. Hasselmo, H. E. Stanley and B. T. Hyman, Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer's disease, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(9), 5274–5279, DOI:10.1073/pnas.96.9.5274 . PMID: 10220456; PMCID: PMC21854..
- T. Qiu, Q. Liu, Y. X. Chen, Y. F. Zhao and Y. M. Li, Aβ42 and Aβ40: similarities and differences, J. Pept. Sci., 2015, 21(7), 522–529, DOI:10.1002/psc.2789 . Epub 2015 May 28. PMID: 26018760..
- F. Chiti and C. M. Dobson, Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade, Annu. Rev. Biochem., 2017, 86, 27–68, DOI:10.1146/annurev-biochem-061516-045115 . Epub 2017 May 12. PMID: 28498720..
- L. C. Serpell, Alzheimer's amyloid fibrils: structure and assembly, Biochim. Biophys. Acta, 2000, 1502(1), 16–30, DOI:10.1016/s0925-4439(00)00029-6 . PMID: 10899428..
- C. M. Dobson, Protein folding and misfolding, Nature, 2003, 426(6968), 884–890, DOI:10.1038/nature02261 . PMID: 14685248..
- E. Y. Hayden, J. L. Conovaloff, A. Mason, G. Bitan and D. B. Teplow, Preparation of pure populations of covalently stabilized amyloid β-protein oligomers of specific sizes, Anal. Biochem., 2017, 518, 78–85, DOI:10.1016/j.ab.2016.10.026 . Epub 2016 Oct. 31. PMID: 27810329; PMCID: PMC5474095..
- R. Pouplana and J. M. Campanera, Energetic contributions of residues to the formation of early amyloid-β oligomers, Phys. Chem. Chem. Phys., 2015, 17(4), 2823–2837, 10.1039/c4cp04544k . Epub 2014 Dec. 15. PMID: 25503571..
- B. Chandra, S. Halder, J. Adler, A. Korn, D. Huster and S. Maiti, Emerging structural details of transient amyloid-β oligomers suggest designs for effective small molecule modulators, Chem. Phys. Lett., 2017, 675, 51–55 CrossRef CAS.
- J. T. Jarrett and P. T. Lansbury Jr, Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie?, Cell, 1993, 73(6), 1055–1058, DOI:10.1016/0092-8674(93)90635-4 . PMID: 8513491..
- S. Chimon, M. A. Shaibat, C. R. Jones, D. C. Calero, B. Aizezi and Y. Ishii, Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's β-amyloid, Nat. Struct. Mol. Biol., 2007, 14(12), 1157–1164, DOI:10.1038/nsmb1345 . PMID: 18059284..
- F. Mangialasche, A. Solomon, B. Winblad, P. Mecocci and M. Kivipelto, Alzheimer's disease: clinical trials and drug development, Lancet Neurol., 2010, 9(7), 702–716, DOI:10.1016/S1474-4422(10)70119-8 . Erratum in: Lancet Neurol., 2011, 10 (6) 501. PMID: 20610346..
- H. W. Klafki, M. Staufenbiel, J. Kornhuber and J. Wiltfang, Therapeutic approaches to Alzheimer's disease, Brain, 2006, 129(Pt 11), 2840–2855, DOI:10.1093/brain/awl280 . Epub 2006 Oct. 3. PMID: 17018549..
- N. Guzior, A. Wieckowska, D. Panek and B. Malawska, Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer's disease, Curr. Med. Chem., 2015, 22(3), 373–404, DOI:10.2174/0929867321666141106122628 . PMID: 25386820; PMCID: PMC4435057..
- W. Xu, X. B. Wang, Z. M. Wang, J. J. Wu, F. Li, J. Wang and L. Y. Kong, Synthesis and evaluation of donepezil–ferulic acid hybrids as multi-target-directed ligands against Alzheimer's disease, MedChemComm, 2016, 7(5), 990–998 RSC.
- A. J. Doig and P. Derreumaux, Inhibition of protein aggregation and amyloid formation by small molecules, Curr. Opin. Struct. Biol., 2015, 30, 50–56, DOI:10.1016/j.sbi.2014.12.004 . Epub 2015 Jan. 2. PMID: 25559306..
- F. Belluti, A. Rampa, S. Gobbi and A. Bisi, Small-molecule inhibitors/modulators of amyloid-β peptide aggregation and toxicity for the treatment of Alzheimer's disease: a patent review (2010–2012), Expert Opin. Ther. Pat., 2013, 23(5), 581–596, DOI:10.1517/13543776.2013.772983 . Epub 2013 Feb. 21. PMID: 23425062..
- D. Goyal, S. Shuaib, S. Mann and B. Goyal, Rationally Designed Peptides and Peptidomimetics as Inhibitors of Amyloid-β (Aβ) Aggregation: Potential Therapeutics of Alzheimer's Disease, ACS Comb. Sci., 2017, 19(2), 55–80, DOI:10.1021/acscombsci.6b00116 . Epub 2017 Jan. 18. PMID: 28045249..
- W. Hoyer, C. Grönwall, A. Jonsson, S. Ståhl and T. Härd, Stabilization of a beta-hairpin in monomeric Alzheimer's amyloid-beta peptide inhibits amyloid formation, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(13), 5099–5104, DOI:10.1073/pnas.0711731105 . Epub 2008 Mar. 28. PMID: 18375754; PMCID: PMC2278213..
- J. Cao, J. Hou, J. Ping and D. Cai, Advances in developing novel therapeutic strategies for Alzheimer's disease, Mol. Neurodegener., 2018, 13(1), 64, DOI:10.1186/s13024-018-0299-8 . PMID: 30541602; PMCID: PMC6291983..
- D. Mehta, R. Jackson, G. Paul, J. Shi and M. Sabbagh, Why do trials for Alzheimer's disease drugs keep failing? A discontinued drug perspective for 2010–2015, Expert Opin. Invest. Drugs, 2017, 26(6), 735–739, DOI:10.1080/13543784.2017.1323868 . PMID: 28460541; PMCID: PMC5576861..
- C. Rivière, J. C. Delaunay, F. Immel, C. Cullin and J. P. Monti, The polyphenol piceid destabilizes preformed amyloid fibrils and oligomers in vitro: hypothesis on possible molecular mechanisms, Neurochem. Res., 2009, 34(6), 1120–1128, DOI:10.1007/s11064-008-9883-6 . Epub 2008 Nov. 23. PMID: 19030989..
- H. Shoval, D. Lichtenberg and E. Gazit, The molecular mechanisms of the anti-amyloid effects of phenols, Amyloid, 2007, 14(1), 73–87, DOI:10.1080/13506120601116674 . PMID: 17453627..
- F. Gervais, J. Paquette, C. Morissette, P. Krzywkowski, M. Yu, M. Azzi, D. Lacombe, X. Kong, A. Aman, J. Laurin, W. A. Szarek and P. Tremblay, Targeting soluble Abeta peptide with tramiprosate for the treatment of brain amyloidosis, Neurobiol. Aging, 2007, 28(4), 537–547, DOI:10.1016/j.neurobiolaging.2006.02.015 . Epub 2006 May 3. PMID: 16675063..
- K. L. Sciarretta, D. J. Gordon and S. C. Meredith, Peptide-based inhibitors of amyloid assembly, Methods Enzymol., 2006, 413, 273–312, DOI:10.1016/S0076-6879(06)13015-3 . PMID: 17046402..
- J. Ghanta, C. L. Shen, L. L. Kiessling and R. M. Murphy, A strategy for designing inhibitors of beta-amyloid toxicity, J. Biol. Chem., 1996 Nov, 271(47), 29525–29528, DOI:10.1074/jbc.271.47.29525 . PMID: 8939877..
- L. O. Tjernberg, J. Näslund, F. Lindqvist, J. Johansson, A. R. Karlström, J. Thyberg, L. Terenius and C. Nordstedt, Arrest of beta-amyloid fibril formation by a pentapeptide ligand, J. Biol. Chem., 1996, 271(15), 8545–8548, DOI:10.1074/jbc.271.15.8545 . PMID: 8621479..
- R. Liu, R. Su, M. Liang, R. Huang, M. Wang, W. Qi and Z. He, Physicochemical strategies for inhibition of amyloid fibril formation: an overview of recent advances, Curr. Med. Chem., 2012, 19(24), 4157–4174, DOI:10.2174/092986712802430018 . PMID: 22830338..
- T. L. Lowe, A. Strzelec, L. L. Kiessling and R. M. Murphy, Structure-function relationships for inhibitors of beta-amyloid toxicity containing the recognition sequence KLVFF, Biochemistry, 2001, 40(26), 7882–7889, DOI:10.1021/bi002734u . PMID: 11425316..
- S. A. Funke and D. Willbold, Peptides for therapy and diagnosis of Alzheimer's disease, Curr. Pharm. Des., 2012, 18(6), 755–767, DOI:10.2174/138161212799277752 . PMID: 22236121; PMCID: PMC3426787..
- N. Sun, S. A. Funke and D. Willbold, A survey of peptides with effective therapeutic potential in Alzheimer's disease rodent models or in human clinical studies, Mini-Rev. Med. Chem., 2012, 12(5), 388–398, DOI:10.2174/138955712800493942 . PMID: 22303971; PMCID: PMC3426789..
- T. Takahashi and H. Mihara, Peptide and protein mimetics inhibiting amyloid beta-peptide aggregation, Acc. Chem. Res., 2008, 41(10), 1309–1318, DOI:10.1021/ar8000475 . PMID: 18937396..
- J. Liu, W. Wang, Q. Zhang, S. Zhang and Z. Yuan, Study on the efficiency and interaction mechanism of a decapeptide inhibitor of β-amyloid aggregation, Biomacromolecules, 2014, 15(3), 931–939, DOI:10.1021/bm401795e . Epub 2014 Feb. 10. PMID: 24443821..
- S. Kumar, A. Paul, S. Kalita, A. K. Ghosh, B. Mandal and A. C. Mondal, Protective effects of β-sheet breaker α/β-hybrid peptide against amyloid β-induced neuronal apoptosis in vitro, Chem. Biol. Drug Des., 2017, 89(6), 888–900, DOI:10.1111/cbdd.12912 . Epub 2016 Dec. 20. PMID: 27995757..
- M. H. Viet, S. T. Ngo, N. S. Lam and M. S. Li, Inhibition of aggregation of amyloid peptides by beta-sheet breaker peptides and their binding affinity, J. Phys. Chem. B, 2011, 115(22), 7433–7446, DOI:10.1021/jp1116728 . Epub 2011 May 12. PMID: 21563780..
- A. S. Gardberg, L. T. Dice, S. Ou, R. L. Rich, E. Helmbrecht, J. Ko, R. Wetzel, D. G. Myszka, P. H. Patterson and C. Dealwis, Molecular basis for passive immunotherapy of Alzheimer's disease, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(40), 15659–15664, DOI:10.1073/pnas.0705888104 . Epub 2007 Sep. 25. PMID: 17895381; PMCID: PMC1994138..
- J. McLaurin, R. Cecal, M. E. Kierstead, X. Tian, A. L. Phinney, M. Manea, J. E. French, M. H. Lambermon, A. A. Darabie, M. E. Brown, C. Janus, M. A. Chishti, P. Horne, D. Westaway, P. E. Fraser, H. T. Mount, M. Przybylski and P. St George-Hyslop, Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis, Nat. Med., 2002, 8(11), 1263–1269, DOI:10.1038/nm790 . Epub 2002 Oct. 15. PMID: 12379850..
- C. Wasmer, A. Lange, H. Van Melckebeke, A. B. Siemer, R. Riek and B. H. Meier, Amyloid fibrils of the HET-s (218–289) prion form a beta solenoid with a triangular hydrophobic core, Science, 2008, 319(5869), 1523–1526, DOI:10.1126/science.1151839 . Erratum in: Science, 2008, 320 (5872) 50. PMID: 18339938..
- B. Permanne, C. Adessi, G. P. Saborio, S. Fraga, M. J. Frossard, J. Van Dorpe, I. Dewachter, W. A. Banks, F. Van Leuven and C. Soto, Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer's disease by treatment with a beta-sheet breaker peptide, FASEB J., 2002, 16(8), 860–862, DOI:10.1096/fj.01-0841fje . Epub 2002 Apr. 10. PMID: 11967228..
- N. L. Fawzi, A. H. Phillips, J. Z. Ruscio, M. Doucleff, D. E. Wemmer and T. Head-Gordon, Structure and dynamics of the Abeta (21–30) peptide from the interplay of NMR experiments and molecular simulations, J. Am. Chem. Soc., 2008, 130(19), 6145–6158, DOI:10.1021/ja710366c . Epub 2008 Apr. 16. Erratum in: J. Am. Chem. Soc., 2011, 133 (30) 11816. PMID: 18412346; PMCID: PMC3474854..
- J. McLaurin, M. E. Kierstead, M. E. Brown, C. A. Hawkes, M. H. Lambermon, A. L. Phinney, A. A. Darabie, J. E. Cousins, J. E. French, M. F. Lan, F. Chen, S. S. Wong, H. T. Mount, P. E. Fraser, D. Westaway and P. St George-Hyslop, Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model, Nat. Med., 2006, 12(7), 801–808, DOI:10.1038/nm1423 . Epub 2006 Jun. 11. PMID: 16767098..
- C. Soto, E. M. Sigurdsson, L. Morelli, R. A. Kumar, E. M. Castaño and B. Frangione, Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy, Nat. Med., 1998, 4(7), 822–826, DOI:10.1038/nm0798-822 . PMID: 9662374..
- M. A. Chacón, M. I. Barría, C. Soto and N. C. Inestrosa, Beta-sheet breaker peptide prevents Abeta-induced spatial memory impairments with partial reduction of amyloid deposits, Mol. Psychiatry, 2004, 9(10), 953–961, DOI:10.1038/sj.mp.4001516 . PMID: 15098004..
- V. Minicozzi, R. Chiaraluce, V. Consalvi, C. Giordano, C. Narcisi, P. Punzi, G. C. Rossi and S. Morante, Computational and experimental studies on β-sheet breakers targeting Aβ1-40 fibrils, J. Biol. Chem., 2014, 289(16), 11242–11252, DOI:10.1074/jbc.M113.537472 . Epub 2014 Feb. 28. PMID: 24584938; PMCID: PMC4036262..
- Z. Datki, R. Papp, D. Zádori, K. Soós, L. Fülöp, A. Juhász, G. Laskay, C. Hetényi, E. Mihalik, M. Zarándi and B. Penke, In vitro model of neurotoxicity of Abeta 1-42 and neuroprotection by a pentapeptide: irreversible events during the first hour, Neurobiol. Dis., 2004, 17(3), 507–515, DOI:10.1016/j.nbd.2004.08.007 . PMID: 15571986..
- W. Liu, F. Sun, M. Wan, F. Jiang, X. Bo, L. Lin, H. Tang and S. Xu, β-Sheet Breaker Peptide-HPYD for the Treatment of Alzheimer's Disease: Primary Studies on Behavioral Test and Transcriptional Profiling, Front. Pharmacol., 2018, 8, 969, DOI:10.3389/fphar.2017.00969 . PMID: 29358920; PMCID: PMC5766670..
- W. Liu, E. Crocker, W. Zhang, J. I. Elliott, B. Luy, H. Li, S. Aimoto and S. O. Smith, Structural role of glycine in amyloid fibrils formed from transmembrane alpha-helices, Biochemistry, 2005, 44(9), 3591–3597, DOI:10.1021/bi047827g . PMID: 15736968..
- S. Jagota and J. Rajadas, The Role of Pro, Gly Lys, and Arg Containing Peptides on Amyloid-Beta Aggregation, Int. J. Pept. Res. Ther., 2012, 18, 53–61, DOI:10.1007/s10989-011-9278-4.
- P. K. Kanchi and A. K. Dasmahapatra, Enhancing the binding of the β-sheet breaker peptide LPFFD to the amyloid-β fibrils by aromatic modifications: a molecular dynamics simulation study, Comput. Biol. Chem., 2021, 92, 107471, DOI:10.1016/j.compbiolchem.2021.107471 . Epub 2021 Mar. 3. PMID: 33706107..
- B. M. Austen, K. E. Paleologou, S. A. Ali, M. M. Qureshi, D. Allsop and O. M. El-Agnaf, Designing peptide inhibitors for oligomerization and toxicity of Alzheimer's beta-amyloid peptide, Biochemistry, 2008, 47(7), 1984–1992, DOI:10.1021/bi701415b . Epub 2008 Jan. 12. PMID: 18189413..
- M. Taylor, S. Moore, J. Mayes, E. Parkin, M. Beeg, M. Canovi, M. Gobbi, D. M. Mann and D. Allsop, Development of a proteolytically stable retro-inverso peptide inhibitor of beta-amyloid oligomerization as a potential novel treatment for Alzheimer's disease, Biochemistry, 2010, 49(15), 3261–3272, DOI:10.1021/bi100144m . PMID: 20230062..
- C. Giordano, A. Masi, A. Pizzini, A. Sansone, V. Consalvi, R. Chiaraluce and G. Lucente, Synthesis and activity of fibrillogenesis peptide inhibitors related to the 17–21 beta-amyloid sequence, Eur. J. Med. Chem., 2009, 44(1), 179–189, DOI:10.1016/j.ejmech.2008.03.036 . Epub 2008 Apr. 8. PMID: 18501995..
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, 3D structure of Alzheimer's amyloid-beta (1–42) fibrils, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(48), 17342–17347, DOI:10.1073/pnas.0506723102 . Epub 2005 Nov. 17. PMID: 16293696; PMCID: PMC1297669..
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fändrich, Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer's brain tissue, Nat. Commun., 2019, 10(1), 4760, DOI:10.1038/s41467-019-12683-8 . PMID: 31664019; PMCID: PMC6820800..
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall, S. Linse and R. G. Griffin, Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils, J. Am. Chem. Soc., 2016, 138(30), 9663–9674, DOI:10.1021/jacs.6b05129 . Epub 2016 Jul. 14. PMID: 27355699; PMCID: PMC5389415..
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schröder, Fibril structure of amyloid-β (1–42) by cryo-electron microscopy, Science, 2017, 358(6359), 116–119, DOI:10.1126/science.aao2825 . Epub 2017 Sep. 7. PMID: 28882996; PMCID: PMC6080689..
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease, Nat. Struct. Mol. Biol., 2015, 22(6), 499–505, DOI:10.1038/nsmb.2991 . Epub 2015 May 4. PMID: 25938662; PMCID: PMC4476499..
- P. H. Nguyen, A. Ramamoorthy, B. R. Sahoo, J. Zheng, P. Faller, J. E. Straub, L. Dominguez, J. E. Shea, N. V. Dokholyan, A. De Simone, B. Ma, R. Nussinov, S. Najafi, S. T. Ngo, A. Loquet, M. Chiricotto, P. Ganguly, J. McCarty, M. S. Li, C. Hall, Y. Wang, Y. Miller, S. Melchionna, B. Habenstein, S. Timr, J. Chen, B. Hnath, B. Strodel, R. Kayed, S. Lesné, G. Wei, F. Sterpone, A. J. Doig and P. Derreumaux, Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer's Disease, Parkinson's Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis, Chem. Rev., 2021, 121(4), 2545–2647, DOI:10.1021/acs.chemrev.0c01122 . Epub 2021 Feb. 5. PMID: 33543942..
- A. Acharya, J. Stockmann, L. Beyer, T. Rudack, A. Nabers, J. C. Gumbart, K. Gerwert and V. S. Batista, The Effect of (−)-Epigallocatechin-3-Gallate on the Amyloid-β Secondary Structure, Biophys. J., 2020, 119(2), 349–359, DOI:10.1016/j.bpj.2020.05.033 . Epub 2020 Jun. 10. PMID: 32579965; PMCID: PMC7376235..
- A. Jarmuła, J. Ludwiczak and D. Stępkowski, β-Sheet breakers with consecutive phenylalanines: insights into mechanism of dissolution of β-amyloid fibrils, Proteins, 2021, 89(7), 762–780, DOI:10.1002/prot.26057 . Epub 2021 Feb. 18. PMID: 33550630..
- M. Cheon, M. Kang and I. Chang, Polymorphism of fibrillar structures depending on the size of assembled Aβ17-42 peptides, Sci. Rep., 2016, 6, 38196, DOI:10.1038/srep38196 . PMID: 27901087; PMCID: PMC5128875..
- A. Kahler, H. Sticht and A. H. Horn, Conformational stability of fibrillar amyloid-beta oligomers via protofilament pair formation – a systematic computational study, PLoS One, 2013, 8(7), e70521, DOI:10.1371/journal.pone.0070521 . PMID: 23936224; PMCID: PMC3729696..
- A. Kaur, A. Kaur, D. Goyal and B. Goyal, How Does the Mono-Triazole Derivative Modulate Aβ42 Aggregation and Disrupt a Protofibril Structure: Insights from Molecular Dynamics Simulations, ACS Omega, 2020, 5(25), 15606–15619 CrossRef CAS PubMed.
- S. Gupta and A. K. Dasmahapatra, Destabilization potential of phenolics on Aβ fibrils: mechanistic insights from molecular dynamics simulation, Phys. Chem. Chem. Phys., 2020, 22(35), 19643–19658 RSC.
- J. A. Lemkul and D. R. Bevan, Destabilizing Alzheimer's Abeta(42) protofibrils with morin: mechanistic insights from molecular dynamics simulations, Biochemistry, 2010, 49(18), 3935–3946, DOI:10.1021/bi1000855 . PMID: 20369844..
- S. Gupta and A. K. Dasmahapatra, Caffeine destabilizes preformed Aβ protofilaments: insights from all atom molecular dynamics simulations, Phys. Chem. Chem. Phys., 2019, 21(39), 22067–22080, 10.1039/c9cp04162a . PMID: 31565708..
- W. J. Du, J. J. Guo, M. T. Gao, S. Q. Hu, X. Y. Dong, Y. F. Han, F. F. Liu, S. Jiang and Y. Sun, Brazilin inhibits amyloid β-protein fibrillogenesis, remodels amyloid fibrils and reduces amyloid cytotoxicity, Sci. Rep., 2015, 5, 7992, DOI:10.1038/srep07992 . PMID: 25613018; PMCID: PMC4303869..
- H. S. Kundaikar and M. S. Degani, Insights into the Interaction Mechanism of Ligands with Aβ42 Based on Molecular Dynamics Simulations and Mechanics: Implications of Role of Common Binding Site in Drug Design for Alzheimer's Disease, Chem. Biol. Drug Des., 2015, 86(4), 805–812, DOI:10.1111/cbdd.12555 . Epub 2015 Mar. 28. PMID: 25763767..
- R. K. Saini, S. Shuaib, D. Goyal and B. Goyal, Insights into the inhibitory mechanism of a resveratrol and clioquinol hybrid against Aβ42 aggregation and protofibril destabilization: a molecular dynamics simulation study, J. Biomol. Struct. Dyn., 2019, 37(12), 3183–3197, DOI:10.1080/07391102.2018.1511475 . Epub 2018 Dec. 24. PMID: 30582723..
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713, DOI:10.1021/acs.jctc.5b00255 . Epub 2015 Jul. 23. PMID: 26574453; PMCID: PMC4821407..
- W. J. Allen, T. E. Balius, S. Mukherjee, S. R. Brozell, D. T. Moustakas, P. T. Lang, D. A. Case, I. D. Kuntz and R. C. Rizzo, DOCK 6: impact of new features and current docking performance, J. Comput. Chem., 2015, 36(15), 1132–1156, DOI:10.1002/jcc.23905 . PMID: 25914306; PMCID: PMC4469538..
- D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao and P. A. Kollman, AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- D. R. Roe and T. E. Cheatham III, Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data, J. Comput. Chem., 2018, 39(25), 2110–2117, DOI:10.1002/jcc.25382 . Epub 2018 Oct. 3. PMID: 30368859; PMCID: PMC7313716..
- B. R. Miller III, T. D. McGee Jr, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, MMPBSA.py: An Efficient Program for End-State Free Energy Calculations, J. Chem. Theory Comput., 2012, 8(9), 3314–3321, DOI:10.1021/ct300418h . Epub 2012 Aug. 16. PMID: 26605738..
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions, Protein Eng., 1995, 8(2), 127–134, DOI:10.1093/protein/8.2.127 . PMID: 7630882..
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, PLIP: fully automated protein-ligand interaction profiler, Nucleic Acids Res., 2015, 43(W1), W443–W447, DOI:10.1093/nar/gkv315 . Epub 2015 Apr. 14. PMID: 25873628; PMCID: PMC4489249..
- B. Han, Y. Liu, S. W. Ginzinger and D. S. Wishart, SHIFTX2: significantly improved protein chemical shift prediction, J. Biomol. NMR, 2011, 50(1), 43–57, DOI:10.1007/s10858-011-9478-4 . Epub 2011 Mar. 30. PMID: 21448735; PMCID: PMC3085061..
- F. Azam, N. H. Alabdullah, H. M. Ehmedat, A. R. Abulifa, I. Taban and S. Upadhyayula, NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer's disease: an investigation by docking, molecular dynamics, and DFT studies, J. Biomol. Struct. Dyn., 2018, 36(8), 2099–2117, DOI:10.1080/07391102.2017.1338164 . Epub 2017 Jun. 15. PMID: 28571516..
- E. Terzi, G. Hölzemann and J. Seelig, Reversible random coil-beta-sheet transition of the Alzheimer beta-amyloid fragment (25–35), Biochemistry, 1994, 33(6), 1345–1350, DOI:10.1021/bi00172a009 . PMID: 8312252..
- S. Shuaib, S. S. Narang, D. Goyal and B. Goyal, Computational design and evaluation of β-sheet breaker peptides for destabilizing Alzheimer's amyloid-β42 protofibrils, J. Cell. Biochem., 2019, 120(10), 17935–17950, DOI:10.1002/jcb.29061 . Epub 2019 Jun. 4. PMID: 31162715..
- S. Hou, R. X. Gu and D. Q. Wei, Inhibition of β-Amyloid Channels with a Drug Candidate wgx-50 Revealed by Molecular Dynamics Simulations, J. Chem. Inf. Model., 2017, 57(11), 2811–2821, DOI:10.1021/acs.jcim.7b00452 . Epub 2017 Nov. 15. PMID: 29099594..
- Y. Sun, W. Xi and G. Wei, Atomic-level study of the effects of O4 molecules on the structural properties of protofibrillar Aβ trimer: β-sheet stabilization, salt bridge protection, and binding mechanism, J. Phys. Chem. B, 2015, 119(7), 2786–2794, DOI:10.1021/jp508122t . Epub 2015 Feb. 9. PMID: 25608630..
- M. S. Dutta and S. Basu, Identifying the key residues instrumental in imparting stability to amyloid beta protofibrils – a comparative study using MD simulations of 17–42 residues, J. Biomol. Struct. Dyn., 2020, 1–26, DOI:10.1080/07391102.2019.1711192 . Epub ahead of print. PMID: 31900057..
- K. E. Marshall, K. L. Morris, D. Charlton, N. O'Reilly, L. Lewis, H. Walden and L. C. Serpell, Hydrophobic, aromatic, and electrostatic interactions play a central role in amyloid fibril formation and stability, Biochemistry, 2011, 50(12), 2061–2071, DOI:10.1021/bi101936c . Epub 2011 Feb. 22. PMID: 21288003..
- H. J. Dyson, P. E. Wright and H. A. Scheraga, The role of hydrophobic interactions in initiation and propagation of protein folding, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(35), 13057–13061, DOI:10.1073/pnas.0605504103 . Epub 2006 Aug. 17. PMID: 16916929; PMCID: PMC1559752..
- S. S. Barale, R. S. Parulekar, P. M. Fandilolu, M. J. Dhanavade and K. D. Sonawane, Molecular Insights into Destabilization of Alzheimer's Aβ Protofibril by Arginine Containing Short Peptides: A Molecular Modeling Approach, ACS Omega, 2019, 4(1), 892–903 CrossRef CAS.
- N. Agrawal and A. A. Skelton, 12-Crown-4 Ether Disrupts the Patient Brain-Derived Amyloid-β-Fibril Trimer: Insight from All-Atom Molecular Dynamics Simulations, ACS Chem. Neurosci., 2016, 7, 1433–1441, DOI:10.1021/acschemneuro.6b00185.
- R. K. Saini, S. Shuaib and B. Goyal, Molecular insights into Aβ42 protofibril destabilization with a fluorinated compound D744: a molecular dynamics simulation study, J. Mol. Recognit., 2017, 30(12) DOI:10.1002/jmr.2656 , Epub 2017 Aug. 29, PMID: 28850770.
Footnote |
| † Electronic supplementary information (ESI) available: Details about interactions between protein and peptides, MM-GBSA free of binding and comparison of the experimental structure with respect to simulated structure. See DOI: 10.1039/d1ra03609b |
| This journal is © The Royal Society of Chemistry 2021 |