DOI:
10.1039/D1RA03827C
(Paper)
RSC Adv., 2021,
11, 23574-23588
Design and fabrication of photoactive imidazole-based poly(ether-imide)s and a polyimide/HBP-modified SiO2 composite: toward high heat-resistance, antimicrobial activity and removal of heavy metal ions
Received
16th May 2021
, Accepted 15th June 2021
First published on 5th July 2021
Abstract
This article describes the synthesis and properties of novel imidazole-based aromatic polyimides (PIs) containing bulky groups from direct polycondensation of two diamines with 3,3′,4,4′-benzophenone tetracarboxylic dianhydride (BTDA) and (hexafluoroisopropylidene)diphthalic anhydride (6FDA). The structure–property relationship of the prepared samples was fully determined via FT-IR, 1H and 13C NMR and elemental analysis (CHN) techniques. The inherent viscosity values of the polyimides ranged from 0.51 to 0.73 dL g−1. These PIs showed glass transition temperatures ranging from 273 to 306 °C, and 10% mass loss temperatures within the range of 478–504 °C in a N2 atmosphere. High transparency with a UV-visible absorption cut-off wavelength was found to range between 285 and 300 nm. Good antimicrobial activity can be correlated with the presence of xanthene and imidazole units into the main structure of PIs. Next, SiO2 nanoparticles as inorganic nanoparticles were added to one of the synthesized polyimides (BTDA-PIb), causing changes in the attributes of both the nanoparticles and PI. The data obtained from examining the properties of the prepared BTDA-PIb/HBP@SiO2 demonstrated increased heat resistance, photoluminescence intensity, and antimicrobial inhibition compared to pure PI. Also, in this article, the polymeric samples as adsorbents were evaluated for extraction of heavy metal ions (Hg2+ and Co2+) from water sources.
Introduction
Nowadays, polymer nanocomposites (PNCs) have received extensive attention in the field of science and technology, owing to the combination of properties of inorganic nanomaterials and polymeric matrices.1–3 Nano-silica (SiO2) has been widely applied in polymer composites as an amplifier thanks to enjoying several desirable properties including environmental compatibility, affordability, non-toxicity, excellent thermal stability, and capability of enhancing the mechanical properties of the polymeric matrix; hence PNCs based on SiO2 nanofillers have sparked huge interest in recent years.4–6 Nonetheless, monotonous distribution and dispersion of nanofillers all over the polymer matrix and adequate interaction between the two corresponding phases (matrix and filler) is one of the main requirements when preparing polymeric nanocomposites. Modifying nanoparticles (NPs) by silane coupling agents is a beneficial procedure for controlling the good dispersion of NPs in the polymeric phase.7,8 Hyperbranched polymers (HBPs) are highly branched macromolecules with a 3D spherical structure and abundant agglomeration of active terminal groups that can link nano-surfaces (i.e. GO, SiO2, Al2O3). The modifying of HBPs NPs can prevent the agglomeration of the nanofiller and increment their surface association for organic solvent and polymer matrices.9–11
One of the most progressive categories of great performance polymeric materials is aromatic polyimides (PIs), owing to their prominent properties such as robust chemical resistance, outstanding thermal stability, relatively small dielectric permittivity, prominent mechanical fortification, supreme electrical attributes, etc.12–15 Also, as matrices, they are often considered the appropriate choice for preparing polymeric nanocomposites.16–19 PIs have engendered significant consideration for diverse applications such as functional membranes, gas separation, aerospace industry, and interlayer dielectrics (ILD) in electronic devices.20–22 The rigid structure characteristics of PIs have a close relationship with the formation of charge transfer complexes (CTCs) in polymer chains; as such, the application and processability of traditional aromatic PIs are very limited.23 Hence, structural modifications to polyimides for overcoming these drawbacks have been widely investigated. One impressive procedure is inserting bulky pendant groups in the main backbone of polymers. These groups prevent from packing of the polymer chains and CTC formation as a result solubility in common solvent improves dramatically.24,25 Other methods that have been developed in this field include introducing electron-donating moieties,26 heterocyclic segments,27 aliphatic moiety,28 flexible linkages,29 and spiro structure into the polyimide chains.30,31
Ring components containing heteroatoms have been extensively incorporated into the backbone of thermostable polymers such as polyisocyanurate, polyureas, polyesters, polyimides, and polyamides, which progress the solubility of these polymers in organic solvents and enhance their chemical plus thermal resistance.32–36 Imidazole and xanthene rings can be classified as one of the most prominent heterocyclic scaffolds in many fields including medicine and organic chemistry.37,38 The polar imidazole ring has two nitrogens, which easily dissolve in a polar solvent and aqueous solutions. These compounds and their derivatives with their unique properties such as stability to hydrogenation, consistency in harsh bases and acids, high thermal and chemical stability,39 and structural stiffness have claimed a central position in pharmacological activities and chemistry. Further, diverse pharmacological activities including antibacterial, antifungal, anti-HIV, and anticancer activity have been reported by previous studies.40,41 In recent years, numerous studies have been performed to covalently bond the imidazole monomers as an antimicrobial agent to the polymer chain. Covalent bonding can enhance the antimicrobial activity when using the polymer.42 Xanthene derivatives are the 6-membered oxygen heterocycles, which make part of the backbone of many natural products. They have an important role in medicinal chemistry due to their biological properties.43 Fluorescent organic compounds are well-known as a means to generate light with a range of color profiles. The compounds containing xanthene or imidazole due to having optical properties have been of special interest for material scientists and applied as agents for bio-imaging.33,44
Herein, we report the synthesis of a new series of imidazole-based diamines containing bulky aromatic pendant groups to prepare thermostable and processable polyimides. The UV-vis spectra, crystallinity, solubility, antimicrobial activity, and thermal properties of synthesized polyimides have been characterized in detail. Additionally, a nanocomposite of linking between a selective PI (BTDA-PIb) and hyperbranched aromatic polyamide grafted SiO2 NPs (20 wt%) were synthesized. SiO2 NPs were grafted onto NH2-terminated hyperbranched aromatic polyamide via a 2-step path for creating potent interfacial interactions with the PI matrix. Field energy-dispersive X-ray (EDX), emission scanning electron microscopy (FE-SEM), X-ray diffraction (XRD), thermogravimetric analysis (TGA), and Fourier transform infrared spectroscopy (FT-IR) were used to identify of the structure and properties of the modified SiO2 NPs, nanocomposite, and polymers. Finally, the properties of the neat PI and relevant nanocomposite were compared with each other. Next, the application of PIs and nanocomposite as adsorbent was investigated for eliminating two heavy metal ions Hg2+ and Co2+ from aqueous solutions.
Experimental
Reagents
All chemicals and solvents were procured from Merck Chemical Co. (Germany) and Sigma-Aldrich. Terephthalaldehyde, sulfuric acid (H2SO4), benzil, 4,4′-dimethoxybenzil, ammonium ethanoate, palladium on carbon (Pd/C), hydrazine monohydrate, acetic anhydride (Ac2O), 3,3′,4,4′-benzophenone tetracarboxylic dianhydride (BTDA), and 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (6FDA), 3-triethoxysilylpropylamine (APTES), 3,5-diaminobenzoic acid (DABz), and triphenyl phosphite (TPP) were applied as obtained. N,N-Dimethylacetamide (DMAc), N,N-dimethylformamide (DMF), dimethylsulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), and pyridine (Py) were purified by distillation before usage. SiO2 nanoparticles (SiO2 NP) with an average diameter of 10 nm were purchased from Degussa (Germany). 6-(4-Nitrophenoxy)naphthalen-2-ol (NOH) was prepared in our research laboratory.34
Instrumentation and characterizations
13C-NMR and 1H-NMR spectra were recorded on a Bruker 400 NMR spectrometer Bruker Avance DRX (Germany) appliance with tetramethylsilane (TMS) as an interior modulus and DMSO-d6 as solvent. The atomic absorption spectrophotometer (BRAICWFX-130 AA) was used to determine metal cation concentration in the liquid phase. Better dispersion of NPs was performed on Branson S3200 (50 kHz, 150 W) sonicating bath. To record FE-SEM the Hitachi S4160 instrument was applied. X-ray diffraction evaluation was accomplished on Philips X-ray diffractometer using Ni-filtered Cu Kα irradiance at 35.4 kV and 28 mA at 25 °C. The inherent viscosity of dried polymer samples was evaluated by Ubbelohde suspended-level viscometer at ambient temperature in NMP at a concentration of 0.5 g dL−1. FT-IR spectra were performed at 25 °C from 400 to 4000 cm−1, on Bruker Tensor 27 spectrometer. Determination of elemental concentrations (hydrogen, carbon, and nitrogen) for the samples prepared was carried out by CHN-600 Leco elemental analyzer. The Barnstead Electrothermal engineering LTD 9200 device was used to measure the Melting point (uncorrected) of synthesized samples. Perkin-Elmer LS-50B spectrophotometers (Norwalk, CT, USA) and Cecil 5503 (Cecil Instruments, Cambridge, UK) were used for obtaining fluorescence emission and Ultraviolet-Visible spectra of dilute monomers (2 × 10−5 M in DMSO) and polymers (0.20 g dL−1 in DMSO), respectively. Solubility testing of all components was performed through dissolving 0.1 g of each of the samples in 1 mL of solvent. Thermogravimetric analysis (TGA) was carried out to determine the amount of the heat resistance of polymeric samples on a DuPont Instruments (TGA 951) analyzer under N2 atmosphere at a heating rate of 10 °C min−1 within the range of 30–700 °C. To characterize the glass transition temperature (Tg) values of the polymeric components from obtained Differential scanning calorimetry (DSC) curves on a Perkin Elmer pyris 6 DSC at 10 °C min−1 heating rate from 25 to 350 °C under N2 atmosphere was used.
Synthesis of monomers
Synthesis of 4-(3,11-bis(4-nitrophenoxy)-14H-dibenzo[a,j]xanthen-14-yl)benzaldehyde (NXB). NOH (1.124 g, 4 mmol) and terephthalaldehyde (0.268 g, 2 mmol) were dissolved in dilute acetic acid (HOAc) (10 mL) in a 100-mL tow-necked flask equipped with a magnetic stirrer bar and condenser. Then, H2SO4 was transferred to the solution as a catalyst and placed at 80 °C. After passing 3 h, the mixture was cooled and precipitated in distilled water. The formed product was accumulated by filtration and dried in an oven at 90 °C. Yield: 97% (1.28 g). Mp: 269–274 °C. FT-IR (KBr, cm−1): 3076 (aromatic C–H), 2921, 2850 (aliphatic C–H), 2727 (aldehyde C–H), 1699 (aldehyde C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1587 (CC), 1514, 1342 (NO2), 1240 (C–O–C) stretching. 1H-NMR (400 MHz, DMSOd6, δ, ppm): 6.90 (s, 1H, CH), 7.09 (d, J = 9.13 Hz, 4H, Ar–H), 7.51 (dd, J1 = 9.04 Hz, J2 = 1.43, 2H, Ar–H), 7.59 (d, J = 8.9 Hz, 2H, Ar–H), 7.67 (d, J = 7.93 Hz, 2H, Ar–H), 7.70 (d, J = 1.7 Hz, 2H, Ar–H), 7.87 (d, J = 6.61 Hz, 2H, Ar–H), 7.91 (d, J = 7.2 Hz, 2H, Ar–H), 8.15 (d, J = 8.9 Hz, 4H Ar–H), 8.83 (d, J = 9.28 Hz, 2H, Ar–H), 9.72 (s, 1H, CH aldehyde). 13C NMR (100 MHz, DMSO-d6, δ in ppm): 36.88 (CH xanthene), 116.82, 117.51, 117.83, 118.82, 121.12, 126.07, 128.28, 128.43, 128.58, 128.92, 129.86, 131.70, 134.50, 142.287 (C–NO2), 147.89, 151.17, 151.58, 162.73, 192.11 (CH aldehyde).
O), 1587 (CC), 1514, 1342 (NO2), 1240 (C–O–C) stretching. 1H-NMR (400 MHz, DMSOd6, δ, ppm): 6.90 (s, 1H, CH), 7.09 (d, J = 9.13 Hz, 4H, Ar–H), 7.51 (dd, J1 = 9.04 Hz, J2 = 1.43, 2H, Ar–H), 7.59 (d, J = 8.9 Hz, 2H, Ar–H), 7.67 (d, J = 7.93 Hz, 2H, Ar–H), 7.70 (d, J = 1.7 Hz, 2H, Ar–H), 7.87 (d, J = 6.61 Hz, 2H, Ar–H), 7.91 (d, J = 7.2 Hz, 2H, Ar–H), 8.15 (d, J = 8.9 Hz, 4H Ar–H), 8.83 (d, J = 9.28 Hz, 2H, Ar–H), 9.72 (s, 1H, CH aldehyde). 13C NMR (100 MHz, DMSO-d6, δ in ppm): 36.88 (CH xanthene), 116.82, 117.51, 117.83, 118.82, 121.12, 126.07, 128.28, 128.43, 128.58, 128.92, 129.86, 131.70, 134.50, 142.287 (C–NO2), 147.89, 151.17, 151.58, 162.73, 192.11 (CH aldehyde).Anal. calcd for C40H24N2O8 (660 g mol−1): C, 72.72%; H, 3.63%; N, 4.24%; found: C, 72.56%; H, 3.71%; N, 4.25%.
Synthesis of 2-(4-(3,11-bis(4-nitrophenoxy)-14H-dibenzo[a,j]xanthen-14-yl)phenyl)-4,5-diphenyl-1H-imidazole (NXIa). A mixture of compounds (3.3 g, 5 mmol) NXB, (1.05 g, 5 mmol) benzil, and (2.69 g, 35 mmol) ammonium ethanoate was placed into a 250-mL tow-necked flask equipped with a reflux condenser and a mechanical stirrer, and dissolved in 10 mL dilute acetic acid (HOAc). The suspension was kept at 120 °C stirring for 12 h. Then the system was cooled down and obtained yellow precipitate gathered by filtration and washed with distilled ethanol and water, and dried in an oven at 90 °C to provide the raw produce. Yield: 93% (3.95 g). Mp: 228–232 °C. FT-IR (KBr, cm−1): 3410 (imidazole N–H), 3068 (aromatic C–H), 2925, 2852 (aliphatic C–H), 1587 (CC), 1515, 1342 (NO2), 1242 (C–O–C) stretching. 1H-NMR (400 MHz, DMSOd6, δ, ppm): 6.82 (s, 1H, CH), 7.14 (d, J = 9.16 Hz, 4H, Ar–H), 7.23 (t, J = 6.88 Hz, 2H, Ar–H), 7.43 (m, 4H, Ar–H), 7.46 (d, J = 8.0 Hz, 2H, Ar–H), 7.52 (dd, J1 = 8.9 Hz, J2 = 2.26, 2H, Ar–H), 7.61 (d, J = 8.86 Hz, 4H, Ar–H), 7.75 (d, 2H, J = 2.08 Hz, Ar–H), 7.78 (d, J = 8.0 Hz, 2H, Ar–H), 7.88 (d, J = 8.2 Hz, 2H, Ar–H), 7.93 (d, J = 9.11 Hz, 2H, Ar–H), 8.20 (d, J = 9.15 Hz, 4H, Ar–H), 8.85 (d, J = 9.30 Hz, 2H, Ar–H), 12.51 (s, 1H, NH imidazole). 13C NMR (100 MHz, DMSO-d6, δ in ppm): 36.60 (CH xanthene), 111.31, 117.37, 117.53, 117.73, 117.88, 118.83, 121.00, 125.45, 125.99, 126.15, 126.96, 128.10, 128.27, 128.54, 128.68, 129.46, 131.73, 142.29, 145.00 (C–NO2), 145.28, 147.84, 151.08, 162.67, 162.85 (CN).Anal. calcd for C54H34N4O7 (850 g mol−1): C, 76.23%; H, 4.00%; N, 6.58%; found: C, 76.19%; H, 4.03%; N, 6.56%.
Synthesis of 2-(4-(3,11-bis(4-nitrophenoxy)-14H-dibenzo[a,j]xanthen-14-yl)phenyl)-4,5-bis(4-methoxy phenyl)-1H-imidazole (NXIb). Similarly, NXIb was synthesized from the compound NXB (3.3 g, 5 mmol) and 4,4′-dimethoxybenzil (1.35 g, 5 mmol) in a like manner to NXIa. Yield: 91% (4.14 g), mp: 222–224 °C. FT-IR (KBr, cm−1): 3404 (imidazole N–H); 3072 (aromatic C–H); 2927, 2840 (aliphatic C–H), 1589 (CC); 1514, 1342 (NO2), 1244 (C–O–C) stretching. 1H-NMR (400 MHz, DMSOd6, δ, ppm): 3.72 (s, 6H, CH methoxy group), 6.81 (s, 1H, CH), 6.89 (d, J = 8.2 Hz, 4H, Ar–H), 7.13 (d, J = 8.0 Hz, 2H, Ar–H), 7.36 (d, J = 8.31 Hz, 2H, Ar–H), 7.49 (d, J = 7.63 Hz, 2H, Ar–H), 7.51 (d, J = 7.82 Hz, 2H, Ar–H), 7.60 (d, J = 8.86 Hz, 2H, Ar–H), 7.73 (s, 2H, Ar–H), 7.77 (d, J = 7.93 Hz, 2H, Ar–H), 7.85 (d, J = 8.87 Hz, 2H, ArH), 7.88 (d, J = 8.46 Hz, 2H, Ar–H), 7.91 (d, J = 9.01 Hz, 2H, Ar–H), 8.19 (d, J = 9.02 Hz, 4H, Ar–H), 8.84 (d, J = 9.12 Hz, 2H, Ar–H), 12.49 (s, 1H, NH imidazole). 13C NMR (100 MHz, DMSO-d6, δ in ppm): 36.68 (CH xanthene), 55.92, 55.77 (OCH3), 11.70, 113.82, 114.77, 117.36, 117.51, 117.91, 118.83, 120.99, 125.46, 126.14, 128.27, 128.55, 128.67, 128.94, 131.74, 132.01, 142.30, 144.24 (C–NO2), 145.23, 147.85, 151.08, 158.39, 162.86, 164.75 (CN).Anal. calcd for C56H38N4O9 (910 g mol−1): C, 73.84%; H, 4.17%; N, 6.15%; found: C, 73.79%; H, 4.20%; N, 6.16%.
Synthesis of 4,4′-((14-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenyl)-14H-dibenzo[a,j]xanthene-3,11-diyl)bis(oxy))dianiline (IXDAa). Amount 4.25 g, (5 mmol) NXIa, and 0.1 g palladium on carbon Pd/C (10%) were put in a 250-mL two-necked flask equipped by a mechanical stirrer, a condenser containing 50 mL ethanol. After stirring the mixture at 70 °C for 30 min, 16 mL of a mixture of hydrazine monohydrate/ethanol (6/10, v/v) was transferred dropwise in 1 h. The reaction system was continued for 4 h. The completed reaction was filtered while hot to remove the solid Pd/C. After the solvent evaporated, the resulting white crystals were poured into distilled water, filtered off, and dried in an oven at 90 °C. Yield: 94% (3.71 g), mp: 248–251 °C. FT-IR (KBr, cm−1): 3267–3622 (NH2 and imidazole N–H); 3047 (aromatic C–H); 2925, 2856 (aliphatic C–H), 1596 (CC); 1234 (C–O–C) stretching. 1H-NMR (400 MHz, DMSOd6, δ, ppm): 5.04 (s, 4H, NH2), 6.62 (d, J = 7.7 Hz, 4H, Ar–H), 6.69 (s, 1H, CH), 6.83 (d, J = 8.7 Hz, 4H, Ar–H), 7.18 (d, J = 2.5 Hz, 2H, Ar–H), 7.25 (t, J = 7.7 Hz, 2H, Ar–H), 7.25 (dd, J1 = 6.6 Hz, J2 = 2.5, 2H, Ar–H), 7.38 (d, J = 8.0 Hz, 4H, Ar–H), 7.41 (d, J = 7.75 Hz, 2H, Ar–H), 7.47 (t, J = 7.9 Hz, 4H, Ar–H), 7.69 (d, J = 8.3 Hz, 2H, Ar–H), 7.76 (d, J = 9.0 Hz, 2H, Ar–H), 7.84 (d, J = 8.3 Hz, 2H, Ar–H), 865 (d, J = 9.4 Hz, 2H, Ar–H), 12.39 (s, 1H, NH imidazole). 13C NMR (100 MHz, DMSO-d6, δ in ppm): 36.42 (CH xanthene), 111.87, 114.95, 117.08, 118.29, 119.22, 120.99, 125.36, 126.54, 126.97, 127.97, 128.09, 128.19, 128.34, 128.60, 131.00, 131.64, 135.06, 145.10, 145.48 (C–NH2), 145.60, 146.81, 155.98, 156.52, 157.91 (CN).Anal. calcd for C54H38N4O3 (790 g mol−1): C, 82.02%; H, 4.81%; N, 7.08%; found: C, 81.98%; H, 4.87%; N, 7.09%.
Synthesis of 4,4′-((14-(4-(4,5-bis(4-methoxyphenyl)-1H-imidazol-2-yl)phenyl)-14H-dibenzo[a,j]xanthene-3,11-diyl)bis(oxy))dianiline (IXDAb). Similarly, IXDAb was synthesized from the compound NXIb (4.55 g, 5 mmol) in a like manner to IXDAa. Yield: 96% (4.08 g), mp: 239–241 °C. FT-IR (KBr, cm−1): 3261–3635 (NH2 and imidazole N–H); 3037 (aromatic C–H); 2927, 2837 (aliphatic C–H), 1596 (CC); 1232 (C–O–C) stretching. 1H-NMR (400 MHz, DMSOd6, δ, ppm): 3.69 (s, 3H, CH methoxy group), 3.74 (s, 3H, CH methoxy group), 5.06 (s, 4H, NH2), 6.66 (d, J = 9.6 Hz, 4H, Ar–H), 6.69 (s, 1H, CH), 6.84 (d, J = 8.5 Hz, 4H, Ar–H), 6.86 (d, J = 8.2 Hz, 2H, Ar–H), 6.96 (d, J = 7.3 Hz, 2H, Ar–H), 7.21 (s, 2H, Ar–H), 7.36 (d, J = 7.8 Hz, 2H, Ar–H), 7.38 (d, J = 8.2 Hz, 2H, Ar–H), 7.42 (d, J = 8 Hz, 2H, Ar–H), 7.48 (d, J = 8.4 Hz, 2H, Ar–H), 7.70 (d, J = 8.2 Hz, 2H, Ar–H), 7.74 (d, J = 9.2 Hz, 2H, Ar–H), 7.86 (d, J = 8.0 Hz, 2H, Ar–H), 8.66 (d, J = 9.3 Hz, 2H, Ar–H), 12.36 (s, 1H, NH imidazole). 13C NMR (100 MHz, DMSO-d6, δ in ppm): 36.76 (CH xanthene), 55.92, 55.10 (OCH3), 111.96, 113.57, 114.06, 115.04, 117.12, 118.33, 119.27, 121.05, 123.52, 125.27, 126.60, 127.08, 127.51, 127.80, 127.82, 127.96, 128.15, 128.76, 129.67, 131.67, 136.417, 144.47 (C–NH2), 145.36, 145.62, 146.85, 156.02, 156.82, 157.88, 158.72 (CN).Anal. calcd for C56H42N4O5 (850 g mol−1): C, 79.06%; H, 4.94%; N, 6.59%; found: C, 79.02%; H, 4.99%; N, 6.57%.
Synthesis of polyimides
The below total manner was applied for the provision of four series of PIs from the newly synthesized diamines with the corresponding dianhydrides. IXDAa or IXDAb (1 mmol) and 5 mL dry NMP as a solvent were transferred into a 100-mL tow-necked flask. The mixture was stirred at ambient temperature for 30 min, then 1 mmol of dianhydride was moved to the mixture and stirred at 25 °C for 24 h under the N2 atmosphere and in this stage poly(amic acid) (PAA) solution was formed. After completing the first step a total of 3 mL of the mixture Ac2O (1.8 mL) and Py (1.2 mL) was added to the solution and stirred at 25 °C for 1 h under N2 gas, then, Then the mixture was stirred at 130 °C for 12 h and the chemical imidization of PAA was carried out. In the end step, the system was cooled and the obtained viscous solution was precipitated in 150 mL methanol. The product was filtered and washed with hot water. Subsequently, to prepare a purer product and eliminate the low molecular weight oligomers, the precipitate was washed via refluxing methanol for 24 h in a Soxhlet device.
BTDA-PIa. To prepare this polymer, the reaction between of the IXDAa and BTDA was applied. Yield = 89% and ηinh = 0.73 dL g−1. FT-IR (KBr, cm−1): 3134–3701 (N–H imidazole), 2927, 2856 (aliphatic C–H), 1773 and 1722 (CO imide), 1598 (CN), 1504 (CC), 1371 (C–N), 1236 (C–O–C). 1H-NMR (400 MHz, DMSOd6, δ, ppm): δ: 6.77–8.79 (m, 39H, Ar–H), 12.55 (s, 1H, N–H, imidazole ring).Anal. calcd for (C71H40N4O8)n (1076 g mol−1): C, 79.18%; H, 3.71%; N, 5.20%; found: C, 79.14%; H, 3.77%; N, 5.21%.
6FDA-PIa. To prepare this polymer, the reaction between of the IXDAa and 6FDA was applied. Yield = 94% and ηinh = 0.62 dL g−1. FT-IR (KBr, cm−1): 3137–3693 (N–H imidazole), 3058 (aromatic C–H), 2927, 2856 (aliphatic C–H), 1769 and 1728 (CO imide), 1602 (CN), 1504 (CC), 1371 (C–N), 1240 (C–O–C). 1H-NMR (400 MHz, DMSOd6, δ, ppm): δ: 6.78–8.78 (m, 39H, Ar–H), 12.51 (s, 1H, N–H, imidazole ring).Anal. calcd for (C73H40F6N4O7)n (1198 g mol−1): C, 73.12%; H, 3.39%; N, 4.67%; found: C, 73.09%; H, 3.44%; N, 4.65%.
BTDA-PIb. To prepare this polymer, the reaction between of the IXDAb and BTDA was applied. Yield = 84% and ηinh = 0.63 dL g−1. FT-IR (KBr, cm−1): 3186–3681 (N–H imidazole), 3059 (aromatic C–H), 2925, 2844 (aliphatic C–H), 1774 and 1722 (CO imide), 1604 (CN), 1504 (CC), 1371 (C–N), 1240 (C–O–C). 1H-NMR (400 MHz, DMSOd6, δ, ppm): δ: 3.72 (s, 6H, OCH3), 6.88–8.79 (m, 37H, Ar–H), 12.40 (s, 1H, N–H, imidazole ring).Anal. calcd for (C73H44N4O10)n (1136 g mol−1): C, 77.11%; H, 3.87%; N, 4.92%; found: C, 77.08%; H, 3.90%; N, 4.93%.
6FDA-PIb. To prepare this polymer, the reaction between of the IXDAb and 6FDA was applied. Yield = 91% and ηinh = 0.51 dL g−1. FT-IR (KBr, cm−1): 3186–3689 (N–H imidazole), 3066 (aromatic C–H), 2925, 2854 (aliphatic C–H), 1776 and 1728 (CO imide), 1612 (CN), 1506 (CC), 1375 (C–N), 1245 (C–O–C). 1H-NMR (400 MHz, DMSOd6, δ, ppm): δ: 3.70 (s, 6H, OCH3), 6.87–8.78 (m, 37H, Ar–H), 12.35 (s, 1H, N–H, imidazole ring).Anal. calcd for (C75H44F6N4O9)n (1258 g mol−1): C, 71.54%; H, 3.49%; N, 4.45%; found: C, 71.53%; H, 3.53%; N, 4.42%.
Synthesis of amine functionalized SiO2 nanoparticles (SiO2–NH2 NPs)
In a 100 mL tow-necked flask, ethanol (20 mL) was added to the SiO2 NPs (0.5 g), then the mixture reaction was sonicated by a 150-W water ultrasonic bath for 30 minutes. In the following, APTES (2 mL) was added dropwise to the system within 30 min under N2 gas and stirred for 24 h at room temperature. Finally, the formed product was separated by centrifugation and washed with ethanol, which wiped the excess silane coupling agent out.
Preparation of SiO2–NH2 grafted hyperbranched aromatic polyamide (SiO2–NH2@HBP)
At first, SiO2–NH2 (0.4 g) and DABz (0.4 g) were dissolved in NMP (10 mL), then a mixture of TPP (1.28 mL) and Py (1.38 mL) was added to it. The reaction mixture proceeds for 12 h at 110 °C with the protection of N2 gas. After cooling, the obtaining solution was cast into 100 mL of methanol to produce purple color sediment. The solid product was gathered by filtration and washed with chilled methanol.
Preparation of polyimide/SiO2–NH2@HBP nanocomposite (BTDA-PIb/HBP@SiO2)
A typical synthesis method for preparation polymeric nanocomposite is as follows: 0.2 g of BTDA-PIb was moved into a flask containing 5 mL of dry DMF. After the complete dissolution of the polymer, 20 wt% (based on polymer content) of SiO2–NH2@HBP was added to it and sonicated for 2 h. Then the temperature was adjusted to 80 °C and stirred for 12 h under N2 gas. After cooling, the viscous polymer solution was poured into methanol and precipitated. In the end, the obtained product was centrifuged and dried in a vacuum oven at 60 °C.
Antimicrobial activity by agar well diffusion procedure
In this study, the disk diffusion procedure was conducted for the initial screening of antimicrobial activities of polyimides and polymeric nanocomposite using two fungi Aspergillus niger and Aspergillus oryzae, plus one Gram-positive bacteria Staphylococcus aureus, and one Gram-negative bacteria Pseudomonas aeruginosa. In the first step, bacterial and fungi cultures for antimicrobial activity were prepared by picking colony from 18-h-old nutrient agar and potato dextrose agar (PDA) plates, and the density of the organism suspensions was adjusted to the equal of the 0.5 McFarland standard (1.5 × 108 CFU mL−1) with the help of normal saline (sterilized). Then nutrient agar plates were inoculated by spreading 0.5 mL of each bacterial suspension (107 to 108 CFU mL−1) and potato dextrose agar (PDA) plates were inoculated by spreading 0.5 mL of each fungus suspension (107 to 108 CFU mL−1) over the entire agar surface. The growth rate of bacterium and fungus was reported about 0.1 mL. In this method, DMF in an amount related to the quantity present in bioassays was applied as a negative control and also as positive controls, gentamicin (antibacterial agent) and fluconazole (antifungal agent) were used. Initially, the solutions of dried polymeric samples were prepared in DMF solvent with a concentration 100 mg mL−1. Then, 100 μL of each dilution was impregnated into wells (diameter = 6 mm) created by a sterile cork borer and placed on potato dextrose agar (PDA) plus nutrient agar. Afterward, incubation of nutrient agar and the PDA plates was done at 37 °C and 25 °C, respectively. The diameter of the inhibition zone (DIZ) of nutrient agar and the PDA plates were measured after 24 h and 72 h, respectively.
Adsorption studies
The solid–liquid extraction of Co2+ as its chloride salts and Hg2+ as its nitrate salt was carried out from aqueous the solutions at pH = 7–8. The solution pH of Co2+ was regulated by HCl (0.1 M) and NaOH (0.1 M) and solution pH of Hg2+ was regulated by HNO3 (0.1 M) and NaOH (0.1 M). For extraction of Co2+ and Hg2+ from aqueous solution, initially, 0.05 g from each one of powder samples was shaken with of 25 mL metal ion solution with an initial concentration of 20 ppm for 48 h at ambient temperature. Then, the liquid phase was separated from the solid phase through centrifugation. Distribution coefficient (kd) for measuring the sorption efficiency of absorbing also the removal percentage were calculated using equations given in following:| | |
% removal = (Co − Ce)/Co
| (1) |
| | |
Kd = ((Co − Ce) × V)/(Co × W)
| (2) |
where Co and Ce are the amounts of concentrations of the Co2+ and Hg2+ ions in the initial and in the liquid phase after adsorption, (mg mL−1), V is liquid phase volume (mL) and W is the weight of the sorbent (g).
Result and discussion
Synthesis and characterization of monomers
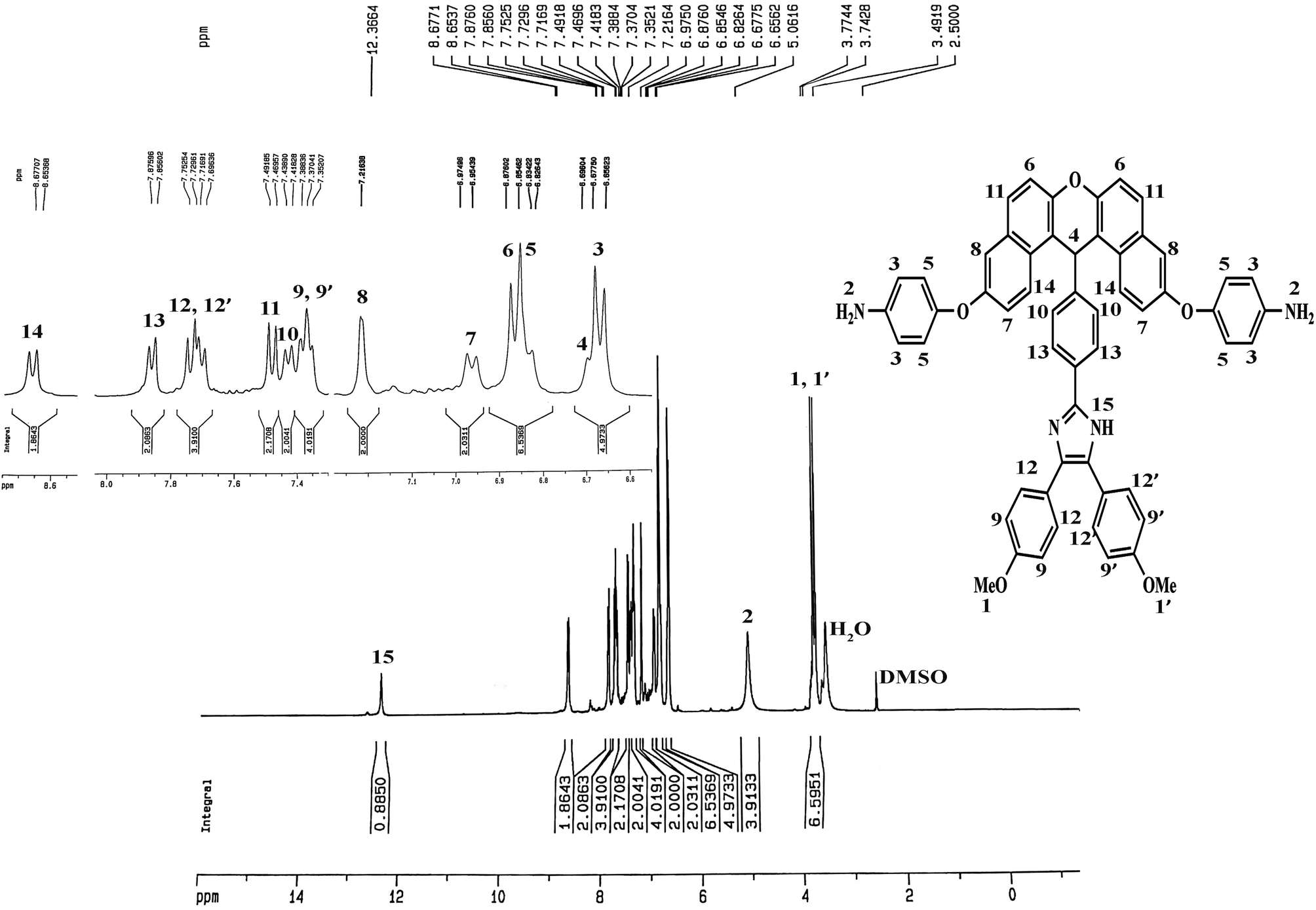
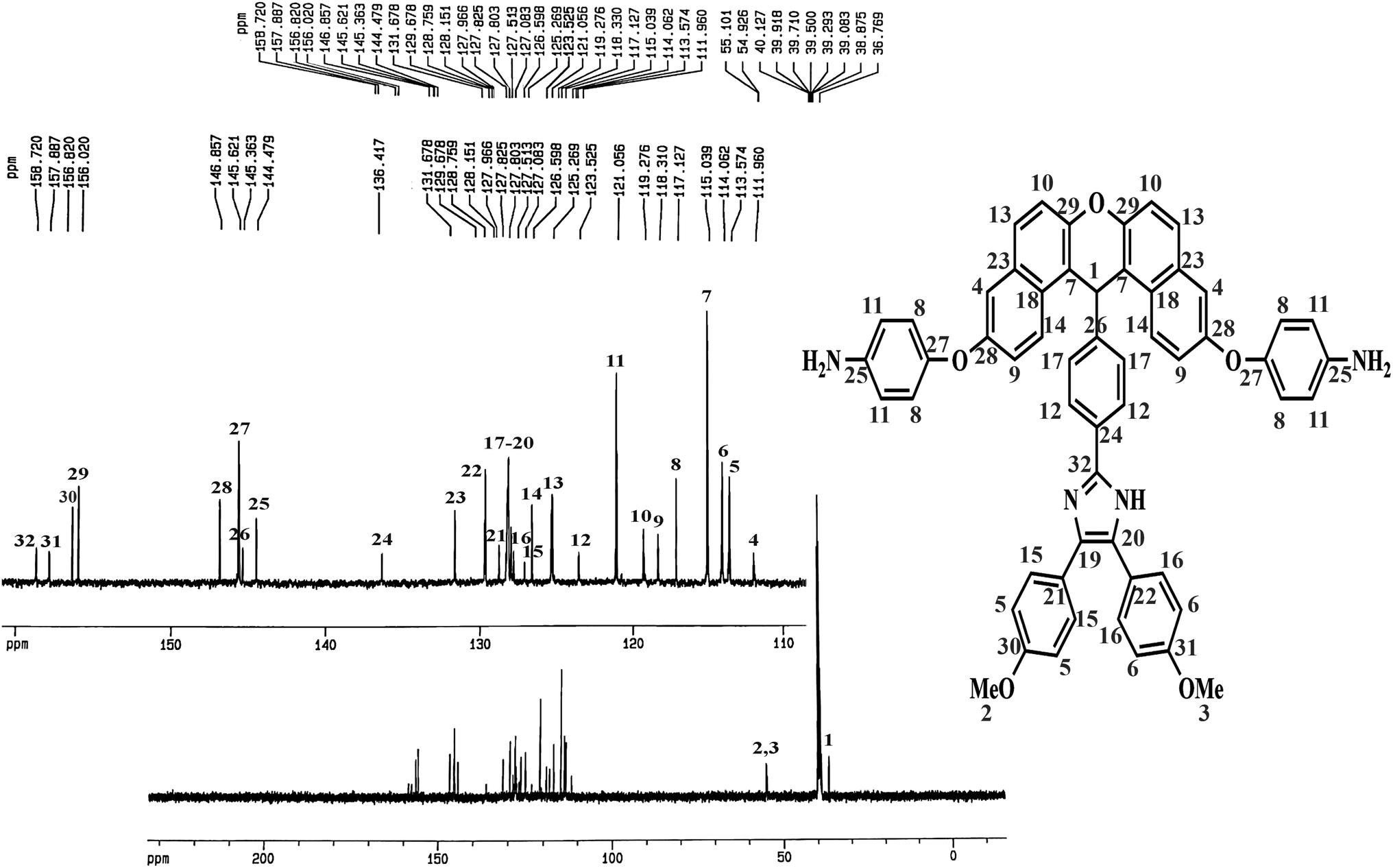
The method of preparing two novel xanthene-based diamines with pendant groups and containing imidazole ring and other monomers is illustrated in Scheme 1. In the first step, the xanthene-based product (NXB) was prepared via the reaction between (NOH) and terephthalaldehyde in the presence of H2SO4 and HOAc as catalyst and solvent, respectively. Next, (NXB) intermediate reacted with 4,4′-dimethoxybenzil or benzil in the presence of ammonium ethanoate and HOAc, whereby a product containing imidazole ring was produced (NXIs). The end step involved hydrogenation of synthesized dinitro compounds (NXIs) using Pd/C and hydrazine monohydrate in ethanol as a solvent to prepared diamines. All reaction steps were controlled by a thin layer chromatography (TLC). FT-IR, 13C-NMR, and 1H-NMR spectroscopies as well as elemental analysis were utilized for identifying the monomers. A comparison of the FT-IR spectra of compounds NXIa and IXDAa is shown in Fig. 1. In Fig. 1a, the peaks at 3410, 1340, and 1515 cm−1 correspond to the imidazole ring N–H, symmetric and asymmetric –NO2 stretching vibrations, respectively. Hydrogenation of the dinitro (NXIa) caused the disappearance of the –NO2 peak and the manifestation of NH2 stretching vibration peaks, occurring at 3373 and 3431 cm−1. Additionally, the C–O–C stretching absorption peak was observed near 1242 cm−1 (Fig. 1a) and 1234 cm−1 (Fig. 1b), which well justifies the presence of ether linkage in the main structure of these compounds. In the 1H-NMR spectra of IXDAb (Fig. 2), the signals in 3.74 and 3.77 ppm correspond to the protons of the methoxy groups, with the particular resonance of the primary amine protons centered at 5.06 ppm. Also, two signals at 6.69 and 12.36 ppm are attributed to the C–H xanthene and N–H imidazole ring, with aromatic rings hydrogen atoms observed from 6.65 to 8.67 ppm. Furthermore, 13C NMR spectra of IXDAb has also been introduced in Fig. 3. As shown in Fig. 3, 32 carbon signals were observed at 36.76 to 158.72 ppm, with the signal at 36.76 ppm related to xanthene CH unite and two signals at 54.92 and 55.10 ppm related to –OCH3 groups. All data suggest that these compounds have fully compatible with the proposed structures.
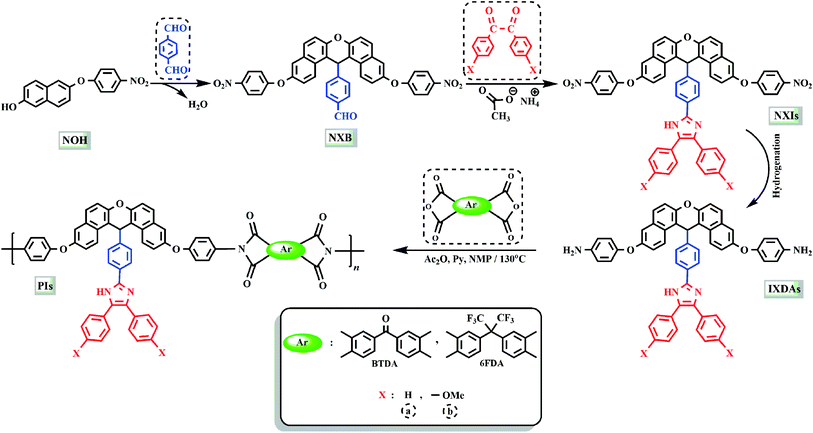 |
| | Scheme 1 Preparation route of monomers and PIs. | |
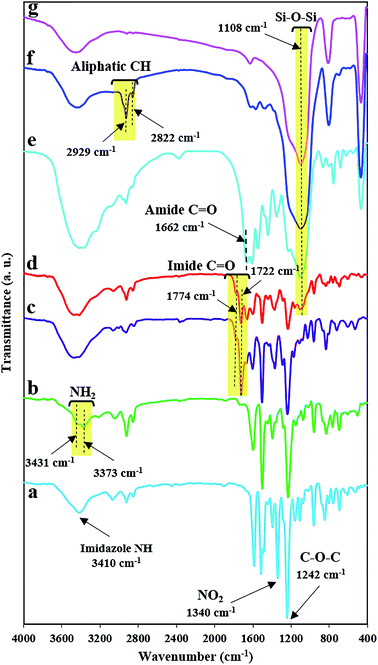 |
| | Fig. 1 The FT-IR spectra of a NXIa (a), IXDAa (b), BTDA-PIb (c), BTDA-PIb/HBP@SiO2 (d), SiO2–NH2@HBP (e), SiO2–NH2 (f) and SiO2 (g). | |
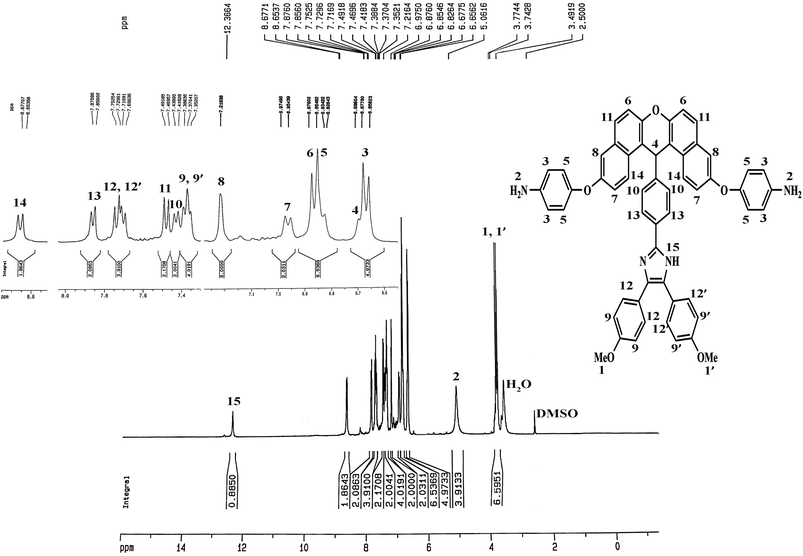 |
| | Fig. 2 1H-NMR spectrum of DAXb. | |
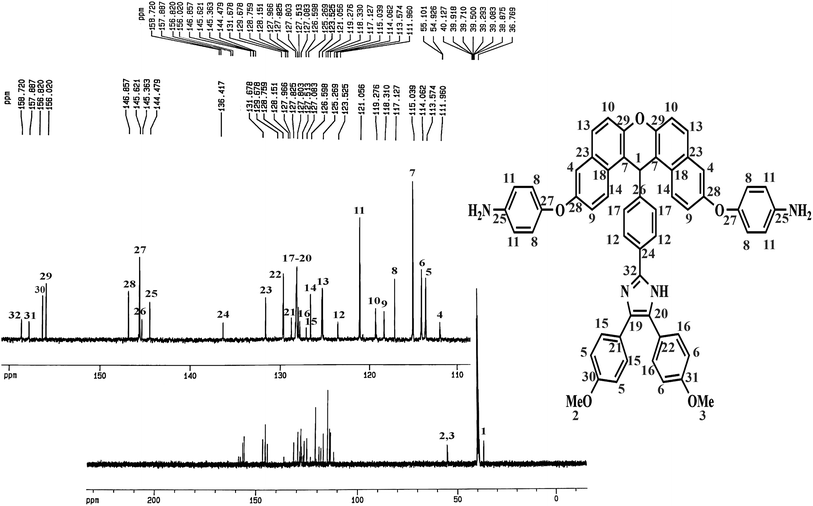 |
| | Fig. 3 13C-NMR spectrum of DAXb. | |
Synthesis of the PIs
Here, a two-stage polymerization procedure was used to synthesize four new PIs from two commercially dianhydrides (BTDA and 6FDA) and two synthesized diamines (IXDAa and IXDAb), as outlined in Scheme 1. Initially, polycondensation of the diamines and the dianhydrides was carried out at 25 °C whereby solvable polyamic acid (PAA) precursor was prepared. Next, the PIs were synthesized via dehydration and polymerization of PAA at elevated temperatures. According to Table 1, the inherent viscosity values of polymers ranged from 0.51 to 0.73 dL g−1, with isolate yield >84%. Identifications including elemental analysis and 1H-NMR and FT-IR spectra were applied to verify the chemical structure of PIs. In Fig. 1c. specific N–H and CN vibration bands of the imidazole ring could be seen at the wavenumber of 3460 and 1604 cm−1, respectively. Specifically, the absorption bands at 1774 and 1722 cm−1 in the spectra of BTDA-PIb verified the stretching of asymmetric and symmetric imide CO. The absorption of the imide ring deformation and C–N stretching bands occurred at 721 cm−1 and 1371 cm−1. The 1H-NMR spectrum of 6FDA-PIa demonstrates the specified resonance of imidazole NH at 12.51 ppm and xanthene CH at 6.78 ppm, where the protons related to aromatic units emerged within the range of 7.19–8.78 ppm (Fig. 4). The chemical structure of 6FDA-PIa was confirmed based on the presence of resonance signals corresponding to IXDAa and 6FDA in the 1H-NMR spectrum.
Table 1 The solubility of synthesized componentsa
| Sample |
Yield (%) |
ηinhb (dL g−1) |
Solvent |
| NMP |
DMAc |
DMSO |
DMF |
THF |
Py |
CHCl3 |
 soluble at room temperature; soluble at room temperature;  soluble on heating at 60 °C; soluble on heating at 60 °C;  partially soluble on heating at 60 °C; partially soluble on heating at 60 °C;  insoluble on heating at 60 °C. Measured at a polymer concentration of 0.5 g dL−1 in NMP at 25 °C. insoluble on heating at 60 °C. Measured at a polymer concentration of 0.5 g dL−1 in NMP at 25 °C. |
| IXDAa |
96 |
— |
 |
 |
 |
 |
 |
 |
 |
| IXDAb |
94 |
— |
 |
 |
 |
 |
 |
 |
 |
| BTDA-PIa |
88 |
0.73 |
 |
 |
 |
 |
 |
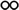 |
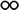 |
| 6FDA-PIa |
94 |
0.62 |
 |
 |
 |
 |
 |
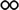 |
 |
| BTDA-PIb |
83 |
0.63 |
 |
 |
 |
 |
 |
 |
 |
| 6FDA-PIb |
91 |
0.51 |
 |
 |
 |
 |
 |
 |
 |
| BTDA-PIb/HBP@SiO2 |
— |
— |
 |
 |
 |
 |
 |
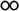 |
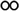 |
 |
| | Fig. 4 1H-NMR spectrum of 6FDA-PIa. | |
Preparation and characterization of BTDA-PIb/HBP@SiO2
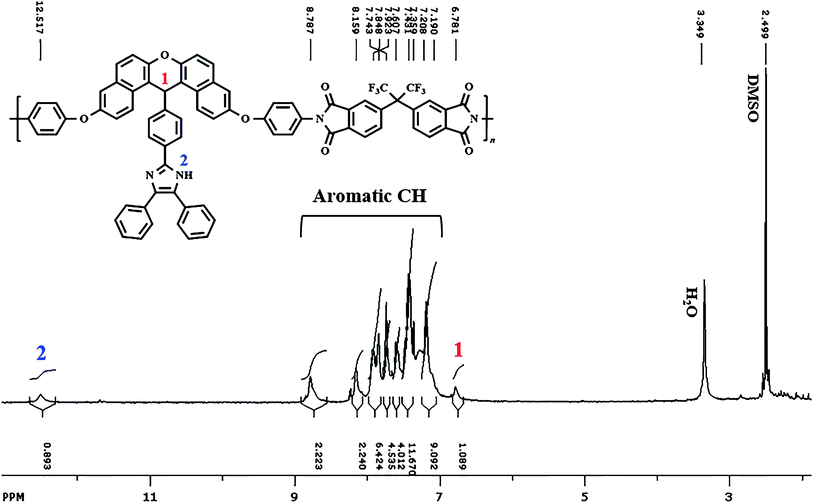
Scheme 2 indicates the general route to prepare the BTDA-PIb/HBP@SiO2. Silanization and linking of HBP onto modified-SiO2 NPs stages were performed to modify SiO2 NPs, which would prevent the aggregation of SiO2 NPs in the PI matrix in the final step. In the second step, the presence of NH2 groups on the surface of SiO2–NH2 NPs as initiator sites caused DABz grafted onto modified NPs surface, and SiO2–NH2@HBP NPs were synthesized. The terminal groups of the bonded SiO2–NH2@HBP NPs chains are amino groups, which lead to good adaptability between functionalized SiO2 and polyimide via the formation of hydrogen bonds.
 |
| | Scheme 2 Grafting of BTDA-PIb onto SiO2–NH2@HBP nanoparticles. | |
FT-IR was used to verify the attendance of some functional groups in SiO2 NPs, SiO2–NH2 NPs, SiO2–NH2@HBP, and BTDA-PIb/HBP@SiO2. As shown in Fig. 1g, asymmetric bending and stretching vibrations of the Si–O–Si bonds were centered at 470 and 1108 cm−1, respectively. Also, the absorption peak at 3442 cm−1 is assigned to the –OH groups of SiO2. The emergence of the new peaks at 2822 and 2929 cm−1 in the FT-IR spectrum of SiO2–NH2 NPs as compared with the FT-IR spectrum of SiO2 NPs, which can be related to the stretching vibration of (–CH2) groups, confirmed the presence of silane agent in SiO2–NH2 NPs. The FT-IR spectrum of SiO2–NH2@HBP showed amide CO bonds and aromatic CC stretching at 1662 cm−1 and 1598 cm−1, respectively (Fig. 1e), verifying hyperbranched polyamide was successfully grafted onto SiO2 NPs surface. For BTDA-PIb/HBP@SiO2, the emergence of relatively broad-stretching vibration band at around 1108 cm−1 confirmed the presence of SiO2–NH2@HBP in the polymeric matrix (Fig. 1d).
The crystallization performance of the resulting samples (SiO2 NPs, SiO2–NH2@HBP, BTDA-PIb/HBP@SiO2, and BTDA-PIb) was evaluated by X-ray diffraction (XRD) with the corresponding patterns illustrated in Fig. 5. Fig. 5 shows the BTDA-PIb had an amorphous pattern and it could be related to disturbed the packing of polymers chains by the imidazole-based pendant groups. Due to the amorphous structure of SiO2 NPs, SiO2–NH2@HBP NPs will also have a non-crystalline structure. The BTDA-PIb/HBP@SiO2 (Fig. 5) showed an amorphous nature similar to PI's. The BTDA-PIb/HBP@SiO2 (Fig. 5) showed an amorphous nature similar to BTDA-PIb with a wide peak at 2θ = 21.47°.
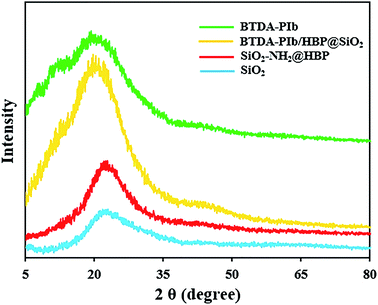 |
| | Fig. 5 XRD patterns of SiO2, SiO2–NH2@HBP, BTDA-PIb/HBP@SiO2 and BTDA-PIb. | |
The surface morphology of the synthesized components was observed by FE-SEM. The SEM images of the BTDA-PIb, SiO2–NH2@HBP and BTDA-PIb/HBP@SiO2 are shown in Fig. 6 with the same magnifications. As Fig. 6(a, a′) shows, the prepared BTDA-PIb indicated a uniform surface with a relatively flat structure. According to Fig. 6(c, c′), substantial difference can be seen in the surface morphology of SiO2–NH2@HBP NPs grafted onto PI matrix as compared with neat BTDA-PIb, indicating SiO2–NH2@HBP NPs have adhered well and uniformly to the functional segments of the PI phase. The outcome of FE-SEM demonstrated PI was successfully grafted onto SiO2 NPs.
 |
| | Fig. 6 FE-SEM images of BTDA-PIb (a, a′), SiO2–NH2@HBP (b, b′) and BTDA-PIb/HBP@SiO2 (c, c′). | |
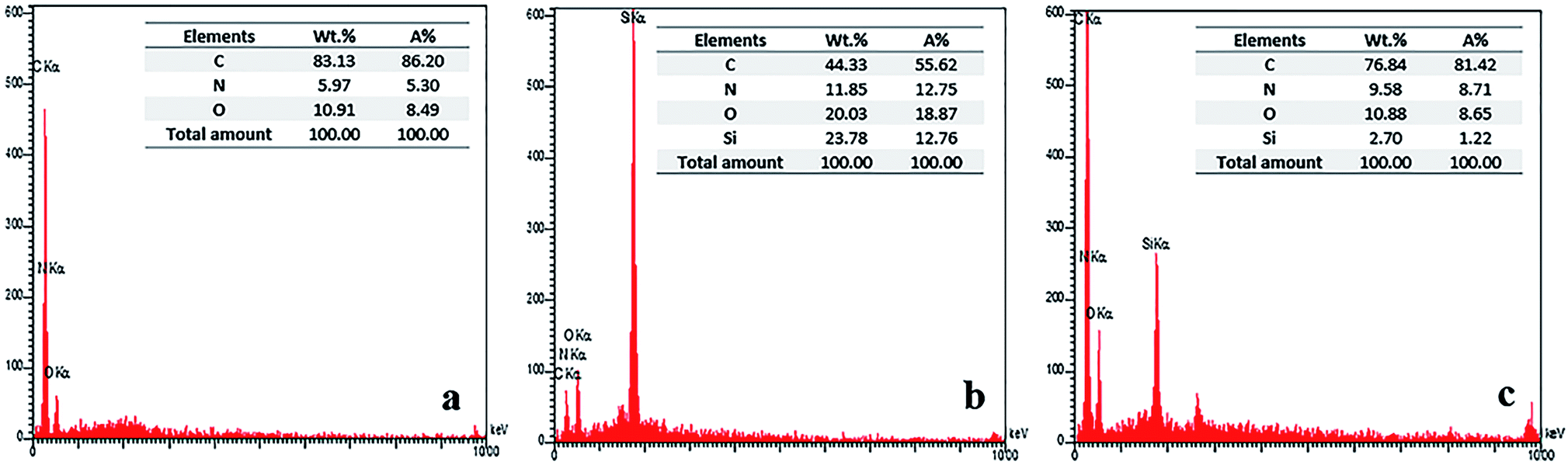
The mapping of C, O, Si, N elements determined by EDS is given in Fig. 7. In the EDS analysis of SiO2–NH2@HBP NPs (Fig. 7b), the emergence of X-ray signals at 0.29 and 0.4 keV which relate to C and N elementals respectively, are indicating that DABz and APTES have been successfully grafted into SiO2 NPs surface. According to EDS mapping of BTDA-PIb/HBP@SiO2 (20 wt%) in Fig. 7c, the presence of a new X-ray signal at 1.90 keV, which corresponding to Si element, is confirmed the successful incorporation of SiO2–NH2@HBP NPs into PI matrix.
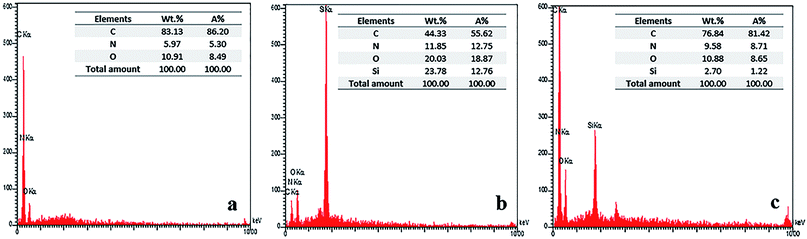 |
| | Fig. 7 EDS analysis of BTDA-PIb (a), SiO2–NH2@HBP (b) and BTDA-PIb/HBP@SiO2 (c). | |
Solubility test
Generally, polyimide chain packing capability and interaction of polymer chains (CTC) affect their solubility in various organic solvents. In this paper, the solubility behavior of the PIs and other components was investigated at 5 mg mL−1 (sample/solvent) and 25 or 60 °C, with the results listed in Table 1. As can be observed, all PIs have shown good solubility in polar aprotic solvents such as DMAc, DMF, DMSO, and NMP at ambient temperature, which can be attributed to dipole–dipole interactions of polar segments (imidazole ring, ether linkage, and carbonyl groups) in the main chain structure of polymers with polar amide solvents. In contrast, the nanocomposite was partially soluble in those solvents at 60 °C. Bending created between stiff phenyl rings by the ether bonds in these polymers was enhanced their flexibility and followed that improved their solubility. Insertion of bulky pendant groups (4,4′-dimethoxybenzil and benzil) in the main chain structure of PIs would disturb the interaction of polymer chains (CTC); consequently, the solvent molecules will be capable to diffuse more easily in between polymer chains. The presence of the methoxy (–OMe) groups in the group (b) polyimides (BTDA-PIb, 6FDA-PIb) caused these PIs to have better solubility in low boiling solvents such as Py and THF at 60 °C. Also, 6FDA-based PIs displayed better solubility than BTDA-based PIs in THF and CHCl3, due to the reduction of the rigidity of polymer structure and the creation of free volume in between polymers chains by replacing two bulky (–CF3) groups instead of the carbonyl group in PIs derived from BTDA.
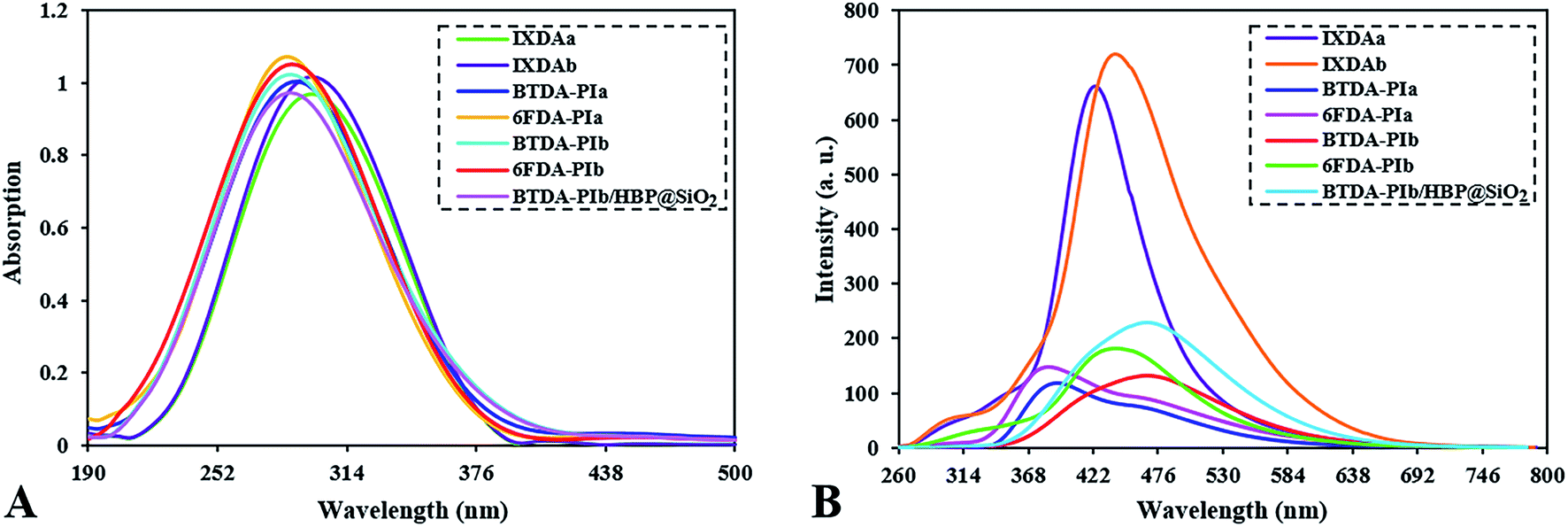
Optical properties
The prepared polyimides (BTDA-PIa, 6FDA-PIa, BTDA-PIb, 6FDA-PIb) in this work show high-quality optical properties due to the presence of xanthene and imidazole group, and also owning pendant part which causes more free volume in the polymer chain and reduces the formation of charge–transfer complex (CTC). The ultraviolet-visible and fluorescence spectroscopy of each prepared sample are depicted in Fig. 8. Also, the corresponding data are shown in Table 2. DMSO solution consisting of 0.2 g dL−1 nanocomposite, 0.2 g dL−1 PIs, and 2 × 10−5 M DA was used to determine the UV-visible spectrum and spectrofluorometric. The π → π* transition of the conjugated aromatic rings in the studied compounds creates maxima (λmax) in the range of 285 to 300 nm as shown in Fig. 8A. According to Fig. 8B, the emission spectrum of PIs, IXDAs, and nanocomposites solution are calculated at 384–471 nm. Considering Table 2, the reported quantum efficiencies for PIs, IXDAs, and nanocomposites are 9–15%, 33 and 35%, and 18%, respectively, extracted via the dilute solution of DMSO nanocomposite and PIs with a concentration of 0.2 g dL−1. Here applied standard was considered a quinine solution with a concentration of 0.1 N in H2SO4 (Φf = 0.53). Typically, polymeric compounds have a longer conjugate system than diamine monomers, which prolongs the entrapment process of photon wavelength. According to this point as well as considering Fig. 8B, PIs and BTDA-PIb/HBP@SiO2 have lower fluorescence intensities than IXDAs. The difference in fluorescence intensity between 6-FDA-derived polyimides and BTDA-derived polyimides may be due to differences in the structure of the dianhydrides. The benzophenone carbonyl groups in the BTDA structure are stronger electro-withdrawing than –CF3 groups in 6FDA-based PIs. Thus polyimides based on 6-FDA have more quantum yields (Φf) than the BTDA (Fig. 8B). According Table 2 and Fig. 8B, the derived-(BTDA-PIb) polymeric nanocomposite exhibited emission at λem = 471 nm, absorption at λab = 290 nm. It has more fluorescence intensity compared to pure BTDA-PIb that showed a green shift with Φf = 15%.
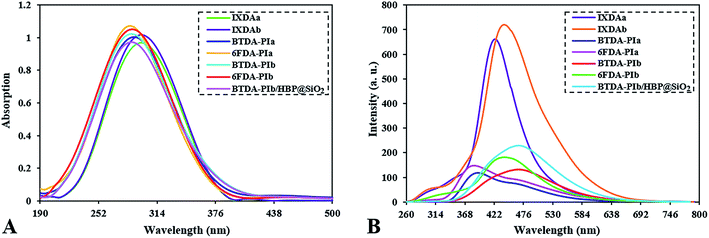 |
| | Fig. 8 UV-Vis (A) and fluorescence (B) spectra of diamines, PIs and BTDA-PIb/HBP@SiO2. | |
Table 2 Optical properties of PIs, diamines and nanocomposite
| Sample |
λabsa (nm) |
λema (nm) |
Φfb (%) |
| UV-visible absorption and fluorescence emission data of the compounds in solution. Fluorescence quantum yield relative to 10−5 M quinine sulfate in 1 N H2SO4(aq) (Φf = 0.55) as a standard. |
| IXDAa |
295 |
430 |
33 |
| IXDAb |
300 |
448 |
35 |
| BTDA-PIa |
295 |
394 |
7 |
| 6FDA-PIa |
285 |
384 |
9 |
| BTDA-PIb |
285 |
473 |
10 |
| 6FDA-PIb |
290 |
444 |
12 |
| BTDA-PIb/HBP@SiO2 |
290 |
471 |
15 |
Thermal properties
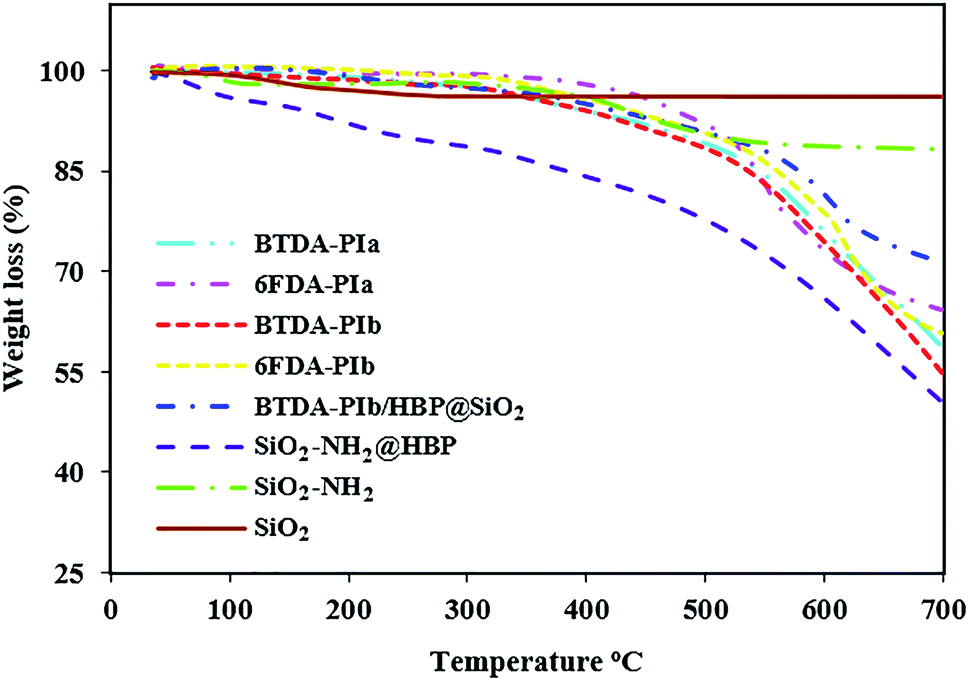
According to differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) techniques, the thermal performance of the resulting polymeric compounds was examined, with the relevant data summarized in Table 3. As shown in Table 3, the glass transition temperature (Tg) of the PIs was found at 273–306 °C, extracted through DSC with a heating rate of 10 °C min−1 under N2 gas. The non-crystalline nature of the PIs can be attributed to the insertion of the bulky pendant group in the main chain of these polymers which increased the rotational barrier and weakening of polymer chain packaging, in turn enhancing the Tg values of these PIs. The presence of –OMe groups in the main structure of group b PIs (BTDA-PIb, 6FDA-PIb) causes slightly looser polymer chains in these polymers than group a PIs (BTDA-PIa, 6FDA-PIa). As a result, these polyimides demonstrated relatively lower Tg values when compared with PIs without methoxy groups. The Tg values of 6FDA-based PIs were higher than those of BTDA-based PIs, which can be related to greater steric limitation of CF3 groups in 6FDA imparted by the sp3-swivel segment between phenylene rings.45 The char yield at 700 °C and temperature at 10% wt. loss (Td,10) was determined by TGA curves (Fig. 9). The Td,10 values in nitrogen atmospheres were overstepped from 478 °C and the char yield was found within the range of 39.5–45.5%, which offered excellent thermal consistency of PIs. It confirmed that introducing bulky pendant groups into the structure of these PIs did not have a negative effect on the amount of the inherent thermal resistance of PIs. The amount of the minimum concentration of oxygen required for the combustion of a polymeric material is known as the limiting oxygen index (LOI), which will be counted by equation LOI = 17.5 + 0.4CR. Where CR = char yield. Remarkably, the LOI of polyimides and nanocomposite were above 26%, which corresponds to the attributes of non-flammable polymer. As listed in Table 3, with the enhancing of dianhydride structural rigidity, the Tg value enhanced too. The increasing order of thermal resistance was as follows: PIs-6FDA > PIs-BTDA. The lower Td,10 values of BTDA-PIs compared to 6FDA-PIs originated from the employment of –CF3 groups in the main structure of 6FDA. In addition, SiO2, SiO2–NH2, SiO2–NH2@HBP, and BTDA-PIb/HBP@SiO2 were characterized using the TGA (Fig. 9). Both modified nanoparticles exhibited an initial mass loss below 90 °C, which is assigned to desorption of absorbed humidity. In the next stage, due to decomposition of organic section SiO2–NH2 and SiO2–NH2@HBP NPs, both indicated 12% and 44% of decomposition, respectively. At the same time, no decomposition was seen in unmodified SiO2 NPs. Higher values of T10 and CR were perceived on BTDA-PIb/HBP@SiO2 compared to neat PI, which can be related to the presence of the inorganic materials and the formation of interactions between two phases.
Table 3 Thermal characteristic data of PAs and nanocomposite
| Sample |
Td10a (°C) |
Tgb (°C) |
CYc (wt%) |
LOId (%) |
| Thermal decomposition at 10% weight loss was recorded by T-GA in N2. Glass transition temperature was recorded at a heating rate of 10 °C min−1 in N2. CY, char yield; weight percentage of material left at 700 °C in N2. Limiting oxygen index percentage evaluated char yield at 700 °C. |
| BTDA-PIa |
486 |
278 |
58 |
40.7 |
| 6FDA-PIa |
503 |
306 |
64 |
43.1 |
| BTDA-PIb |
478 |
273 |
55 |
39.5 |
| 6FDA-PIb |
504 |
301 |
60 |
41.5 |
| BTDA-PIb/HBP@SiO2 |
502 |
— |
70 |
45.5 |
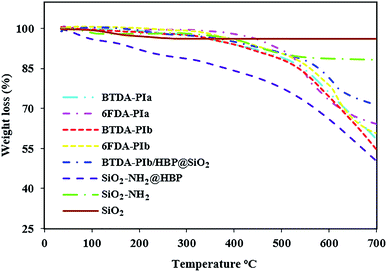 |
| | Fig. 9 TGA curves of PIs, SiO2, SiO2–NH2, SiO2–NH2@HBP, BTDA-PIb/HBP@SiO2. | |
Antimicrobial activities
All polymer samples were evaluated against two bacterial strains and two fungal strains, and their activities were analyzed by IZD through the well diffusion method. The corresponding data from antimicrobial tests are summarized in Table 4. As shown in Table 4, all tested polymers demonstrated good inhibition activities, which is related to the presence of the xanthene group and the imidazole ring in their backbone. The results also showed that the value of the inhibitory effect of the compounds on microbial growth was different due to differences in the structure of the dihydride and diamine used in the main structure of the polyimides. In comparison to the standard drugs applied, antimicrobial activity results showed that components BTDA-PIb and 6FDA-PIb substituted with methoxy groups had less inhibition against antibacterial and antifungal agents as compared to BTDA-PIa and 6FDA-PIa, which is attributed to the effect of increasing electron density in a molecule by –OCH3 as an electron donating group. Relatively higher antimicrobial potency of 6FDA-drived PIs than BTDA-derived PIs may be correlated to the presence electro-withdrawing and hydrophobicity nature of groups –CF3 in compared to the carbonyl group.46 Further, SiO2 NPs act as antimicrobial agents due to their high surface area, porous structure, and adsorption attributes.6 As a result, based on the data listed in Table 4, polymeric nanocomposite demonstrated relatively more antimicrobial activity than native BTDA-PIb.
Table 4 Antimicrobial activity of polyamides and nanocomposite solutions with (100 mg mL−1) concentrationa
| Sample |
Antibacterial activity DIZ |
Antifungal activity DIZ |
| Staphylococcus aureus |
Pseudomonas aeruginosa |
Aspergillus oryzae |
Aspergillus niger |
| DIZ (mm) = The diameter of the inhibition zone. Each experiment has been conducted three times at least. Error values were within ±1 mm. NA = not active. |
| BTDA-PIa |
12 |
11 |
11 |
12 |
| 6FDA-PIa |
13 |
12 |
13 |
12 |
| BTDA-PIb |
10 |
10 |
9 |
9 |
| 6FDA-PIb |
11 |
11 |
12 |
10 |
| BTDA-PIb/HBP@SiO2 |
13 |
12 |
11 |
12 |
| Gentamycin |
15 |
16 |
— |
— |
| Fluconazole |
— |
— |
16 |
16 |
| DMF |
NA |
NA |
NA |
NA |
Adsorption of Co2+ and Hg2+ ions
A group of trace elements such as mercury, chromium, iron, cobalt, cadmium, arsenic, etc. is classified as heavy metal ions. They have undesirable properties including toxicity and carcinogenicity. Thus, they can cause diseases via consuming heavy-metal contaminated water and their accumulation in tissues.47 Extraction of these pollutants from drinking water sources through adsorption is one of the best technical and economical solutions owing to its high efficiency, low cost, and ease of operation.48 As the prepared polymeric compounds are insoluble in water therefore in this work, synthesized polyimides and nanocomposite were used for eliminating Co2+ and Hg2+ ions from wastewater through adsorption. Polymeric samples contain various functional groups, including imidazole NH, ether linkage, and carbonyl. Indeed, these groups acted as hosts for the target metal ions, and as a result, these ions embedded in the polymer phase by physical interaction (Scheme 3). Furthermore, to further elucidate the adsorption mechanism of Co2+ and Hg2+ onto polymers, one of the polyimides (BTDA-PIb) was analyzed by using SEM (Fig. 10). SEM images after adsorption are given in Fig. 10B and C, which showed surface morphology of BTDA-PIb was considerably changed after adsorption and seems the surface and porosity of the PI have been agglomerated with Co2+ and Hg2+. These results indicating that presence of functional groups in the main chain structure of polyimide can be involved in the adsorption process of ions. The important factors on the adsorption of heavy metal ions from aqueous solutions and their effect enhancement efficiency in adsorbing are, contact time, pH range, concentration, Lewis hard–soft acid–base principle, amount of ion exchanger, sizes of cavities, and radii of metal ions.49 The solid–liquid extraction for the elimination of individual metal cations from aqueous solution was carried out in pH = 7–8. In adsorption studies, pH is a critical parameter because it influences the chemistry of active binding sites on the adsorbent. In the acidic region protonation of more nitrogen in the imidazole rings is happens and also excess H+ made the PI surface positively charged, which produced strong electrostatic repulsion attraction with the cationic species. Moreover, in high pH the competition interaction between metal ions and hydroxyl ions may be cause of the lower adsorption. The related adsorption data, as listed in Table 5, showed the order of Co2+ > Hg2+, which can be justified based on an inverse relationship between the radius of metal ions and the amount of adsorption. Cobalt(II) with a smaller radius than mercury(II) can be better absorbed into the free space between the polymer chains. As expected, the adsorption values for 6-FDA-derived PIs were lower than those of BTDA-derived PIs due to the replacement of the two hydrophobic –CF3 groups with the CO group. As shown in Table 5, the largest absorption percentage for both heavy metal ions have been related to the BTDA-PIb/HBP@SiO2 compound. Indeed, the existence of modified nanoparticles with a high surface area in the polymeric nanocomposite would cause higher-efficiency sorption than neat polymer.50
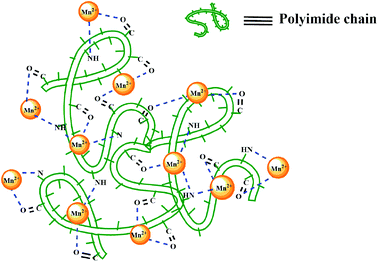 |
| | Scheme 3 Possible mechanism of adsorption of metal ions by PIs. | |
 |
| | Fig. 10 SEM images of BTDA-PIb (A), BTDA-PIb/Hg2+ (B), and BTDA-PIb/Co2+ (C). | |
Table 5 The removal percentage and distribution coefficient parameters for Hg2+ and Co2+ solutions (20 ppm), on the sorbents
| Sample |
Hg2+ content solution (ppm) |
Co2+ content solution (ppm) |
| Final (Ce) |
Removal (%) |
Kd (mL g−1) |
Final (Ce) |
Removal (%) |
Kd (mL g−1) |
| BTDA-PIa |
13.96 |
30.20 |
151.00 |
11.27 |
43.63 |
203.25 |
| 6FDA-PIa |
14.76 |
26.19 |
131.00 |
12.58 |
37.10 |
170.50 |
| BTDA-PIb |
13.38 |
33.06 |
165.50 |
10.94 |
45.27 |
196.5 |
| 6FDA-PIb |
14.29 |
28.91 |
142.75 |
12.01 |
39.91 |
184.25 |
| BTDA-PIb/HBP@SiO2 |
7.26 |
63.70 |
318.50 |
4.48 |
77.58 |
388.00 |
Conclusion
We successfully prepared heat-resistant PIs (6FDA-based PIs and BTDA-based PIs) containing imidazole and xanthene segments via two-step thermic imidization using NMP as a solvent. The improved solubility of polyimides in common organic solvents owing to the attendance of voluminous pendant groups and ether linkage in PIs backbone. The thermal properties of the PIs showed a significant 10% wt. loss (Td,10) of >478 °C and char yield (CR) between 39.5 and 45.5%. 6FDA-PIs revealed greater thermal stability compared to BTDA-PIs, with respect to the presence of polar –CF3 groups in their main structure. Meanwhile, it was well investigated that incorporation of xanthene and imidazole as antimicrobial agents in the PIs backbone induced good inhibition against fungus, Gram-negative and germ-positive bacteria. Further, the presence of these groups in PIs structure conferred fluorescence properties into them, showing fluorescence emission upon irradiation with Φf ranging from 9 to 15%. These polymers could also be a good candidate for preparing polymeric nanocomposite. To achieve uniform dispersion of SiO2 NPs in the PI phase, we carried out hyperbranched aromatic polyamide linking of SiO2 NPs. Finally, all properties of nanocomposite were examined and its effects in BTDA-PIb matrix were investigated. The photoluminescence intensity, heat resistance, and antimicrobial inhibition properties of the BTDA-PIb/HBP@SiO2 indicated an obvious enhancement compared to the pure PI. Further, Hg2+ and Co2+ ions removal from water was tested by PIs and polymeric nanocomposite as chelating agents, with the results indicating nanocomposite could be a better option for extracting related ions from aqueous solution than neat PI.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the University of Tabriz for the financial supports for this research, the Research Center for pharmaceutical nanotechnology (RCPN) of the Tabriz University of Medical Science, and the University of Kurdistan (UOK), Sanandaj (Iran) for the financial supports for this research.
References
- Y. Ahmadian, A. Bakravi, H. Hashemi and H. Namazi, Polym. Bull., 2019, 76, 1967–1983 CrossRef CAS.
- M. Pooresmaeil and H. Namazi, Polym. Adv. Technol., 2019, 30, 447–456 CrossRef CAS.
- H. Namazi, A. Dadkhah and M. Mosadegh, J. Polym. Environ., 2012, 20, 794–800 CrossRef CAS.
- X. Wang, L. Wang, Q. Su and J. Zheng, Compos. Sci. Technol., 2013, 89, 52–60 CrossRef CAS.
- H. Namazi and H. Ahmadi, J. Power Sources, 2011, 196, 2573–2583 CrossRef CAS.
- R. Venkatesan and N. Rajeswari, Silicon, 2019, 11, 2233–2239 CrossRef CAS.
- R. Alizadeh, M. Ghaemy, M. Bazzar and F. Hashemi Nasr, J. Appl. Polym. Sci., 2014, 131, 1–14 CrossRef.
- S. A. Tharakan and S. Muthusamy, RSC Adv., 2021, 11, 16645–16660 RSC.
- M. Suraj Belgaonkar and B. Kandasubramanian, Eur. Polym. J., 2021, 147, 110301 CrossRef CAS.
- Y. Yu, M. Z. Rong and M. Q. Zhang, Polymer, 2010, 51, 492–499 CrossRef CAS.
- R. Francis, N. Joy, E. P. Aparna and R. Vijayan, Polym. Rev., 2014, 54, 268–347 CrossRef CAS.
- L. Li, Y. Xu, J. Che and Z. Ye, Polym. Adv. Technol., 2019, 30, 120–127 CrossRef CAS.
- L. Zhou, Y. Li, Z. Wang, M. Zhang, X. Wang, H. Niu and D. Wu, RSC Adv., 2019, 9, 7314–7320 RSC.
- Y. Zhao, T. Feng, G. Li, F. Liu, X. Dai, Z. Dong and X. Qiu, RSC Adv., 2016, 6, 42482–42494 RSC.
- C. K. Chen, Y. C. Lin, L. C. Hsu, J. C. Ho, M. Ueda and W. C. Chen, ACS Sustain. Chem. Eng., 2021, 9, 3278–3288 CrossRef CAS.
- B. Feng, K. Xu and A. Huang, RSC Adv., 2017, 7, 2211–2217 RSC.
- S. N. Hu, Y. Lin and G. Z. Wu, Chinese J. Polym. Sci., 2020, 38, 100–108 CrossRef CAS.
- K. M. Jeong, Y. H. Li, H. G. Lee and C. S. Ha, Polym. Adv. Technol., 2016, 27, 1345–1350 CrossRef CAS.
- O. Baykara, Ş. G. İrim, A. A. Wis, M. A. Keskin, G. Ozkoc, A. Avcı and M. Doğru, Polym. Adv. Technol., 2020, 31, 2466–2479 CrossRef CAS.
- R. Sulub-Sulub, M. I. Loriá-Bastarrachea, J. L. Santiago-Garciá and M. Aguilar-Vega, RSC Adv., 2018, 8, 31881–31888 RSC.
- I. Gouzman, E. Grossman, R. Verker, N. Atar, A. Bolker and N. Eliaz, Adv. Mater., 2019, 31, 1–15 CrossRef PubMed.
- S. Chisca, V. E. Musteata, I. Sava and M. Bruma, Eur. Polym. J., 2011, 47, 1186–1197 CrossRef CAS.
- H. T. Zuo, F. Gan, J. Dong, P. Zhang, X. Zhao and Q. H. Zhang, Chinese J. Polym. Sci., 2021, 39, 455–464 CrossRef CAS.
- M. Shabanian, H. Moghanian, M. Khaleghi, M. Hajibeygi, H. A. Khonakdar and H. Vahabi, RSC Adv., 2016, 6, 112568–112575 RSC.
- S. M. Amininasab, S. Esmaili and Z. Shami, High Perform. Polym., 2020, 11–12 Search PubMed.
- L. Shi, H. Ye, W. Liu, G. Tian, S. Qi and D. Wu, J. Mater. Chem. C, 2013, 1, 7387–7399 RSC.
- Z. Li, Y. Wang and S. Zhu, J. Phys. Conf. Ser., 2021, 1732, 012126 CrossRef CAS.
- Y. Zhuang, J. G. Seong and Y. M. Lee, Prog. Polym. Sci., 2019, 92, 35–88 CrossRef CAS.
- R. Iqbal, M. K. Khosa, M. A. Jamal, S. Ilyas, M. T. Hussain and M. Hamid, Polym. Adv. Technol., 2016, 27, 221–227 CrossRef CAS.
- Y. Wu, Z. Chen, J. Ji, Y. Zhou, H. Huang, S. Liu and J. Zhao, Eur. Polym. J., 2020, 132, 109742 CrossRef CAS.
- B. Deng, S. Zhang, C. Liu, W. Li, X. Zhang, H. Wei and C. Gong, RSC Adv., 2018, 8, 194–205 RSC.
- M. Taghavi, R. Alizadeh and M. Ghaemy, RSC Adv., 2015, 5, 9581–9590 RSC.
- S. M. A. Nasab and M. Ghaemy, J. Polym. Res., 2011, 18, 1575–1586 CrossRef CAS.
- D. Khaki, S. M. Amininasab and H. Namazi, New J. Chem., 2020, 44, 17346–17359 RSC.
- J. Li, G. Zhang, Y. Yao, Z. Jing, L. Zhou and Z. Ma, RSC Adv., 2016, 6, 60094–60100 RSC.
- D. Khaki, H. Namazi and S. M. Amininasab, React. Funct. Polym., 2021, 158, 104780–104792 CrossRef CAS.
- M. L. da Silva, R. R. Teixeira, L. de Azevedo Santos, F. T. Martins and T. C. Ramalho, Arab. J. Chem., 2020, 13, 974–987 CrossRef CAS.
- A. Siwach and P. K. Verma, BMC Chem., 2021, 15, 1–69 CrossRef.
- C. Yin, J. Dong, D. Zhang, J. Lin and Q. Zhang, Eur. Polym. J., 2015, 67, 88–98 CrossRef CAS.
- N. Rani, A. Sharma and R. Singh, Mini-Reviews Med. Chem., 2013, 13, 1812–1835 CrossRef CAS.
- S. M. Amininasab, A. Rashidi, M. Taghavi and Z. Shami, Chinese J. Polym. Sci., 2016, 34, 766–776 CrossRef CAS.
- S. M. Amininasab, S. Esmaili, M. Taghavi and Z. Shami, J. Fluor. Chem., 2016, 192, 48–57 CrossRef CAS.
- D. Kumar, P. Sharma, H. Singh, K. Nepali, G. K. Gupta, S. K. Jain and F. Ntie-Kang, RSC Adv., 2017, 7, 36977–36999 RSC.
- H. Ding, L. Peng, G. Yuan and L. Zhou, Dye. Pigment., 2020, 176, 3–8 CrossRef.
- Z. Mi, Z. Liu, C. Tian, X. Zhao, H. Zhou, D. Wang and C. Chen, J. Polym. Sci. Part A Polym. Chem., 2017, 55, 3253–3265 CrossRef CAS.
- M. Fioroni, K. Burger, A. E. Mark and D. Roccatano, J. Phys. Chem. B, 2003, 107, 4855–4861 CrossRef CAS.
- Z. Lin, Y. Hu, Y. Yuan, B. Hu and B. Wang, Ecotoxicol. Environ. Saf., 2021, 208, 111451 CrossRef CAS.
- X. Liu, H. Pang, X. Liu, Q. Li, N. Zhang, L. Mao, M. Qiu, B. Hu, H. Yang and X. Wang, Innovation, 2021, 2, 4855–4861 Search PubMed.
- B. Wang, Y. Li, J. Zheng, Y. Hu, X. Wang and B. Hu, Chemosphere, 2020, 254, 126898 CrossRef CAS PubMed.
- G. Zhao, X. Huang, Z. Tang, Q. Huang, F. Niu and X. Wang, Polym. Chem., 2018, 9, 3562–3582 RSC.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Hassan Namazi
a,
Hassan Namazi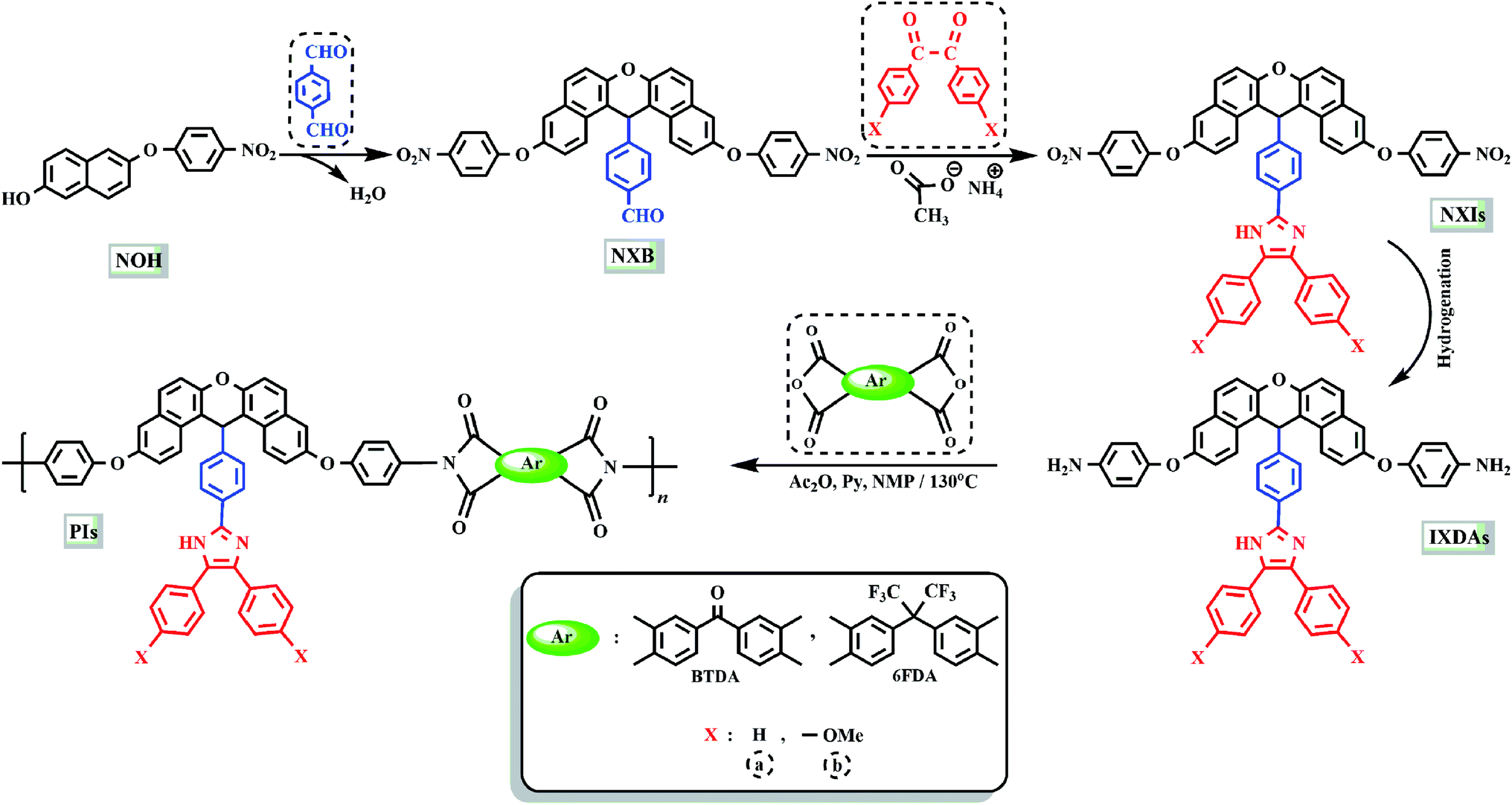

 soluble at room temperature;
soluble at room temperature;  soluble on heating at 60 °C;
soluble on heating at 60 °C;  partially soluble on heating at 60 °C;
partially soluble on heating at 60 °C;  insoluble on heating at 60 °C.b Measured at a polymer concentration of 0.5 g dL−1 in NMP at 25 °C.
insoluble on heating at 60 °C.b Measured at a polymer concentration of 0.5 g dL−1 in NMP at 25 °C.