
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural characterisation of amyloid-like fibrils formed by an amyloidogenic peptide segment of β-lactoglobulin†
Vasantha Gowda a,
Michal Bilerb,
Andrei Filippovcd,
Malisa V. Mantonicoa,
Eirini Ornithopouloua,
Mathieu Linaresbef,
Oleg N. Antzutkincg and
Christofer Lendel*a
a,
Michal Bilerb,
Andrei Filippovcd,
Malisa V. Mantonicoa,
Eirini Ornithopouloua,
Mathieu Linaresbef,
Oleg N. Antzutkincg and
Christofer Lendel*a
aDept. of Chemistry, KTH Royal Institute of Technology, Stockholm, Sweden. E-mail: lendel@kth.se
bDept. of Theoretical Chemistry, KTH Royal Institute of Technology, Stockholm, Sweden
cChemistry of Interfaces, Luleå University of Technology, Sweden
dDept. Medical and Biological Physics, Kazan State Medical University, 420012, Kazan, Russia
eLaboratory of Organic Electronics and Group of Scientific Visualization, ITN, Linköping University, 60174 Norrköping, Sweden
fSwedish e-Science Research Centre (SeRC), Linköping University, 60174 Norrköping, Sweden
gDept. of Physics, University of Warwick, Coventry, UK
First published on 17th August 2021
Abstract
Protein nanofibrils (PNFs) represent a promising class of biobased nanomaterials for biomedical and materials science applications. In the design of such materials, a fundamental understanding of the structure–function relationship at both molecular and nanoscale levels is essential. Here we report investigations of the nanoscale morphology and molecular arrangement of amyloid-like PNFs of a synthetic peptide fragment consisting of residues 11–20 of the protein β-lactoglobulin (β-LG11–20), an important model system for PNF materials. Nanoscale fibril morphology was analysed by atomic force microscopy (AFM) that indicates the presence of polymorphic self-assembly of protofilaments. However, observation of a single set of 13C and 15N resonances in the solid-state NMR spectra for the β-LG11–20 fibrils suggests that the observed polymorphism originates from the assembly of protofilaments at the nanoscale but not from the molecular structure. The secondary structure and inter-residue proximities in the β-LG11–20 fibrils were probed using NMR experiments of the peptide with 13C- and 15N-labelled amino acid residues at selected positions. We can conclude that the peptides form parallel β-sheets, but the NMR data was inconclusive regarding inter-sheet packing. Molecular dynamics simulations confirm the stability of parallel β-sheets and suggest two preferred modes of packing. Comparison of molecular dynamics models with NMR data and calculated chemical shifts indicates that both packing models are possible.
Introduction
Amyloid fibrils are filamentous protein or peptide aggregates with a β-sheet secondary structure in which the β-strands run perpendicular to the fibril axis (cross-β structure).1 Apart from their vital role in the pathogenesis of certain diseases,2–4 including Alzheimer's disease, Parkinson's disease, and type II diabetes,5 amyloid-like protein nanofibrils (PNFs) have also gained interest as functional nanomaterials and innovative biomaterials.6–10 Over the years, great efforts have been made to generate PNFs from readily available sources such as whey, eggs, and legume products.11,12 The increased interest in PNF materials is linked to their tuneable mechanical properties, high chemical and thermodynamic stabilities as well as inherent biocompatibility and biodegradability.13–17 Protein based nanofibrils have found many applications,18–20 among others, in thin films, foams, composites and gels e.g., in biomedical applications such as drug delivery systems, biosensors and scaffolds for tissue regeneration.9,21–24Our understanding of the nature of fibrillar aggregates has largely evolved in recent years, particularly, through the structure determination of their elementary β-strand building blocks and the characterisation of their assembly into cross-β steric zippers.25 However, a thorough understanding of the hierarchical assembly of these individual structural elements into mature fibrils requires both a complete knowledge of the cross-β structures at the atomic level and a characterisation of the nanoscale morphology. Often, PNFs display polymorphism, which further complicates their structure elucidation. In particular, the structure–function relationship of polymorphic forms of nanofibrils is of interest not only for disease related studies but also for the possibility to control the nanomechanical properties of the functional materials. Polymorphism in cross-β fibrils can occur at different structural levels due to (i) different number of protofilaments forming the fibril, (ii) relative arrangements of protofilaments in mature fibrils, and (iii) variations in the protofilament substructure.26 Molecular structures of fibrils formed by a given protein/peptide can vary depending on the aggregation conditions, and multiple distinct structures (polymorphs) often coexist under a single set of conditions. It is challenging to establish which of the distinct morphologies, observed for example in AFM images, also contain distinct molecular structures as the polymorphic samples can, in some cases, produce uniform spectroscopic signatures (e.g., chemical shifts), indicating that the fibril morphologies present in such a sample consist of similarly structured building blocks or protomers. Conversely, although a uniform spectral characteristic does not always guarantee a monomorphic sample, the observation of spectroscopic heterogeneity implies to the presence of several polymorphs in the sample. To our favour, magic-angle-spinning (MAS) solid-state nuclear magnetic resonance (ss-NMR) methods have the capability of providing detailed structural information, sufficient for the development of full molecular models of fibrils from proteins or peptides.27–29
The β-lactoglobulin (β-LG) is the main protein component of the whey fraction of bovine milk. It is one of the most frequently used model systems for exploring PNFs in materials applications.30 In many ways, whey PNFs closely resemble lightweight and elastic properties of silk fibers,31 and they possess functional properties as foaming and emulsifying agents, hydrogel formation, and increasing viscosity.32,33 Also, whey PNFs, have become a promising candidate in the field of food research for being biocompatible nanomaterial.34,35 In contrast to the random aggregates that are formed when whey protein isolate (WPI) is heated at a pH value close to the isoelectric point, fibrils are formed when WPI is heated at pH 2, 80–90 °C and low ionic strength.36 The single fibrils derived from β-LG typically have a length between 1 and 10 μm, a thickness of about 2–4 nm, with a periodicity of 30–40 nm.37 Although WPI is a mixture of different proteins, previous studies38,39 have shown that PNFs formed under the applied conditions are built from β-LG -derived peptides. To explain the molecular basis of fibril formation of β-LG proteins, which represents about 60% of the protein content in WPI, it has been proposed that a critical early step is the partial unfolding of the protein, resulting in flexible conformers that expose aggregation-prone regions of the sequence.38,40–42 It is also known that hydrolysis of the polypeptide chain into shorter segments plays a central role in the fibrillation process38 (Scheme 1). In the current study we investigate PNFs formed by a short peptide segment derived from β-LG, particularly, the residues 11–20 of the β-LG protein (β-LG11–20). The segment is the central part of the major PNF-forming segment in β-LG (i.e., residues 1–32/33, Scheme 1).38,39 It has also been shown to form amyloid-like fibrils on its own in presence of 3–5 M urea at 37 °C and pH 7.0.43
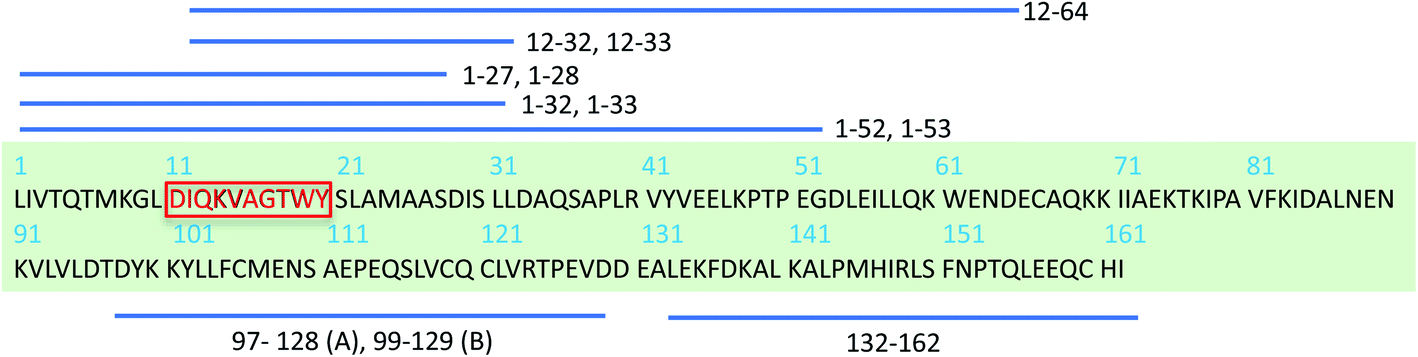 | ||
| Scheme 1 Schematic display of β-LG full sequence and peptides derived from β-LG identified in PNFs by mass spectrometry.38,39 Peptides fragments that differ by one residue are depicted using one stripped line. A and B represents bovine β-LG variants.38 The red-highlighted peptide segment is chosen to study here. | ||
Here, we use an approach to seed fibril growth of β-LG11–20 monomers by β-LG PNF seeds. It is well-known that fibrils with a given structure can be amplified by seeded growth protocols.43–46 Typically, the seeds represent 5–10% of the total peptide or protein in the solution, producing amplification factors of 10 or more in each round of seeded growth. It has been observed that seeding with structurally homogeneous fibrils produces morphologically homogeneous fibrils at the molecular level.45 The AFM images of the nanofibrils formed by the β-LG11–20 peptide display different morphologies. Based on statistical analysis of the AFM images in combination with ss-NMR and molecular dynamics (MD) simulations we are addressing the question whether the polymorphism originate from the molecular structure or from the assembly of protofilaments at the nanoscale. We present a first structural description for amyloid-like fibrils from a β-LG derived segment.
Material and methods
Materials
Bovine β-LG, fluorenylmethoxycarbonyl (F-moc) protected amino acid residues, and all other chemicals used for solid-phase peptide synthesis (SPPS) were purchased from Sigma-Aldrich. Unlabelled β-LG11–20 peptide (sequence Ac-DIQKVAGTWY–NH2) was purchased from AlexoTech AB (Umeå, Sweden). The labelled peptides were synthesised using standard F-moc protocols with H-benzotriazol-1-yl-tetramethyluronium hexafluorophosphate activation on an automated peptide synthesiser (Applied Biosystems model 433A). The F-moc N-protected C-terminal amino-acid residue (Y20) was anchored via carboxyl group to an amino resin (rink amide) to yield amide-linked peptide that will ultimately produce a C-terminal amide peptide. Two uniformly 13C- and 15N-labelled peptides were prepared by placing residues at positions (i) I2 and T8, and (ii) V5, A6, G7. After loading the first amino acid (here, Y10), the desired peptide sequence was assembled in a linear fashion from the C-terminus to the N-terminus (the C–N strategy) by repetitive cycles of Nα deprotection and amino acid coupling reactions. Before starting the solid phase synthesis, the resin was swollen in DCM for 30 min. A large excess of the activated amino acid was used (typically 2–10 times excess compared to the resin functionality). Hexafluorophosphate H-benzotriazol-1-yl-tetramethyluronium (HBTU) coupling reagent was used for the in situ activation of carboxylic functional groups of the N-protected amino acids. Removal of the F-moc protecting group from the N-terminus of the peptidyl-resin was achieved by treatment with 20% piperidine in DMF. The N-terminal acetylation of the peptide was performed with 10% acetic anhydride in DMF. Trifluoroacetic acid was used for the simultaneous cleavage of the peptide from the resin and removal of sidechain protecting groups in ice-cold diethyl ether with a standard mixture of scavengers of free radicals (phenol and sulphur-containing compounds) to avoid spurious reactions and by-products.The peptides were purified via reversed-phase high performance liquid chromatography (HPLC) using semi-preparative columns, which had been pre-equilibrated in 20% acetonitrile, a flowrate of 3 mL min−1 at 50 °C. The peptides were dissolved in 20% acetonitrile in 0.1% TFA (0.5 mg mL−1). Elution was carried out by running a linear gradient of 20–55% acetonitrile in 0.1% aq. TFA over 30 minutes with Sepax polyRP column (5 μm, 300 Å, 4.6 × 150 mm). The eluent was monitored at 214 and 254 nm using a UV absorbance detector. The recovered fractions at a retention time of 14.5–16.5 min were lyophilised to get the purified peptides in powder form. The peptide identities were confirmed by MALDI-TOF. The peptide was mixed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with matrix solution (150 mg mL−1 DHB in 1:1 water:acetonitrile), deposited on a gold-plated MALDI target, air dried, and subjected to MALDI-TOF analysis.
:1 with matrix solution (150 mg mL−1 DHB in 1:1 water:acetonitrile), deposited on a gold-plated MALDI target, air dried, and subjected to MALDI-TOF analysis.
Fibrillation of pure β-lactoglobulin
β-LG fibrils were prepared using the protocol from Akkerman C. et al.38 About 120 mg of β-LG was dissolved in 4 mL MilliQ H2O. The pH was adjusted to 2 using one drop of 3 M HCl and fine adjusted with 0.1 M HCl. The solution was centrifuged for 30 min at 1500 × g and filtrated (0.45 μm) to remove undissolved protein. The protein solution was incubated at 90 °C for 2 days. The fibrils were purified either by spin-filtration or dialysis (both with 100 kDa cut off) and then lyophilised.Preparation of β-lactoglobulin seeds
The lyophilised β-LG fibrils were dissolved in 10 mM HCl to a final concentration of 30 mg mL−1 and diluted to a concentration of 1.0 mg mL−1 using MilliQ water. The sample was sonicated 2 × 2 minutes in water bath. The solution was diluted to 0.05 mg mL−1, lyophilised, and stored at −20 °C.Seeding of peptide solution with β-LG and 1st generation PNF seeds
4 mg of β-LG11–20 peptide was dissolved in 4 mL 10 mM HCl containing 5% β-LG seeds. The solution was centrifuged at 15655 × g and the supernatant solution was incubated at 50 °C and 500 rpm for 48 h in a Thermomixer (Thermo Scientific). A lower temperature than for the β-LG fibrillation was used to avoid hydrolysis. The incubated solution was kept at 50 °C for two more days. The fibrils were collected by centrifugal spin filtration using 10 kDa cut-off filter.
Preparations of 2nd generation isotopically labelled PNFs
Peptide PNF seeds were obtained by sonication of the 1st generation fibrils (unlabelled), lyophilised, and were stored under −20 °C. About 2 mg of β-LG11–20 peptide (isotopically labelled) was dissolved in 2 mL 10 mM HCl solution. The 2nd generation fibrils were obtained by seeding the above peptide solution with ∼5% of peptide seeds. The solution was incubated for two days at 50 °C at 500 rpm in the Thermomixer. The solution was left without agitation for two more days. The fibrils were collected by spin filtration using 10 kDa cut-off filter.Thioflavin T (ThT) fluorescence
ThT fluorescence of protein/fibril samples were measured on a Cary Eclipse Spectrofluorometer (Varian) using a quartz cuvette according to Nilsson.47 About 50 μL of protein/fibril solutions were mixed with 150 μL of ThT solution (0.005 mg mL−1). The emission spectrum from 460 to 600 nm was obtained at an excitation wavelength of 440 nm; the excitation and emission slits were 5 and 10 nm, respectively. 10 mM HCl solution was deducted as a background.Atomic force microscopy (AFM)
PNF morphology was investigated using a Dimension FastScan atomic force microscope (Bruker) operating in tapping mode. The peptide PNFs were diluted between 1:10 and 1:100 in 10 mM HCl and applied on a freshly cleaved mica surface. Periodicity and height profiles of the fibrils were measured from AFM images using Gwyddion software.48 Between 50 and 100 fibrils were measured for each fibril morphology.
NMR spectroscopy
All 1H and 13C NMR spectra were recorded on a Bruker AV III (500 MHz, B0 = 11.4 T) spectrometer operating at 500.13 MHz and 125.76 MHz for 1H and 13C, respectively.Solution-state NMR measurements
The solution-state NMR spectral measurements for the synthetic peptides were measured in H2O + 10% D2O solutions. Suppression of water signals during 1H acquisitions were achieved by the WET (water suppression enhanced through T1 effects) pulse sequence. The spectral parameters for 1D (1-dimensional) 1H/13C experiments were as follows: π/2 pulse lengths 7.0/11.5 μs, recycle delay 2.5 s, number of transients 16/10240. Both 1H and 13C spectra were referenced to TSP (0.0 ppm). For two-dimensional 1H–1H TOCSY and 1H–13C HSQC experiments the following parameters were used: 2048 data points in the 1H dimension and 256 increments in the indirect (F1) dimension (F1 incremental delays for TOCSY (1H) and HSQC (13C) were 153.81 and 37.86 μs, respectively); TOCSY spin-lock (mixing) delay was 80 ms.
Solid-state NMR measurements
The short-peptide PNFs were spin-filtered using 10 kDa membrane, pelleted fibrils were resuspended in deionized water and then lyophilised. Typical ss-NMR samples contained 2–3 mg of peptide fibrillar material. Lyophilisation of fibril samples allowed more material to be placed into the rotor, resulting in higher signal-to-noise ratios in the solid-state NMR spectra. The magic angle for the rotor was adjusted using powder KBr (79Br NMR). The 13C CP-MAS NMR chemical shifts were externally referenced to the downfield peak of powder adamantane (38.48 ppm with respect to TMS, 0 ppm). However, for easy comparison with Wishart reference chemical shifts,49 the chemical shifts are recalibrated for DSS as secondary reference using the formula50| δneat TMSMAS = δDSS in waterinternal − 2.67 = δsolid adamantaneMAS + 38.48 |
1H–13C cross-polarisation (CP) was optimised with following parameters: the MAS rate was 9000 Hz, 1H 90-degree pulse 3.0 μs at 100 W (ω1(1H) = 83.3 kHz), 13C 90-degree pulse 3.0 μs at 131.83 W, relaxation delay was 2.5 s, CP contact time was 700–1500 μs, 1H–13C CP nutation frequency ω1(1H) = 40.3 kHz (50 W), broadband spinal64 decoupling at protons was ω1(1H) = 83.3 kHz.
To obtain interatomic correlations that extend to longer length scales, additional 2D 13C–13C DARR (Dipolar Assisted Rotational Resonance) spectra were measured at 298 K. Mixing time was 250–500 ms and the MAS spinning rate was 12 kHz. The spectral widths of both the direct (F2) and indirect (F1) dimensions were 24 kHz. A total of 256 t1 slices and 80 scans per slice were measured, resulting in an experimental time of 15 hours.
TALOS+ secondary structure prediction
Experimentally derived 13C and 15N chemical shifts were used for TALOS+ prediction. Based on secondary chemical shifts and sequence information, TALOS+ empirically predicts backbone torsion angles and a measure of rigidity expressed as RCI S2 value. Dihedral angles were predicted for all but first and last residues in the sequence. Resulting from the analysis were 8 pairs of dihedral angles ϕ and ψ, which were classified as strong predictions. Residues were designated as part of a β-strand if TALOS+ predicted the secondary structure with a probability higher than 85%.Molecular dynamics (MD) simulations
Classical MD simulations were carried out using Amber16 package51 with the protein and solvent molecules described using ff14SB52 force field and the TIP3P53 water model, respectively. Topology and initial coordinates of a linear form of the peptide (β-LG11–20, Ac-DIQKVAGTWY–NH2, from N-terminus to C-terminus) was created using the LEaP program. Note that this peptide also has modifications of the N-terminus (acetylation) and C-terminus (amidation). After neutralising the system using chloride ions, the MD simulations ran at 300 K using Langevin dynamics and constant pressure with an average pressure of 1 atm and with isotropic position scaling using the Berendsen barostat.54 The simulations ran for up to 80 ns (unless stated otherwise) with a time step of 2 fs per step. The trajectories were recorded every 5000 step and the cut-off were set to 12 Å for non-bonded interactions. All analyses of our MD trajectories were performed using CPPTRAJ.55Building different systems/arrangements for MD
First, we built smaller systems containing one to up to four peptides, and we ran the simulations up to 80 ns to identify possible inter/intra-molecular interactions. From these, we observed that the peptides tend to form π-stacking interaction between individual tryptophan moieties, and individual tyrosine moieties. Moreover, the system is stabilised by hydrogen bonding interactions of the neighbouring peptides (part between W and Y). The rest of the peptide forms β-sheet alignment. Based on these observations, we manually replicated this 4-peptide system in one of the axes resulting in the 20-peptide system, still making sure the previously observed interactions are still held in place. From here, we extended the system to a two-layer assembly, where one layer is formed from 20 peptides in the π-stacking and H-bonding fashion as described above (see all considered possibilities in Fig. S7 in the ESI†). Such arrangements ran for 30 ns.DFT calculations
Both geometry optimisation and NMR calculations of the monomer β-LG11–20 were performed using Gaussian 16, Revision A.03 (ref. 56) program. For geometry optimisation of the monomer, B3LYP57 exchange correlation functional was used together with the D3 dispersion correction58 and the 6-31G(d,p) basis set. In order to capture the solvation effects, a solvation model based on density (SMD) was adopted using water as solvent. To ensure well-converged local minimum geometry, extremely tight convergence criteria were used along with an ultrafine integration grid. The 13C NMR shielding constants were calculated based on the gauge-independent atomic orbitals (GIAO) approach. The NMR shielding constants were calculated at the DFT level employing a hybrid functional PBE0 (with 25% exact exchange).59 Molecular dynamics ensembles of peptides, both in parallel and anti-parallel conformations, were also assessed based on their back-calculated Cα, Cβ, and C′ chemical shifts in comparison with NMR experiments. However, due to large ensemble size, the DFT/NMR shifts were averaged over a small molecular ensemble containing four molecules from the final snapshots after 30 ns MD simulations, which were selected from the centre part of the β-sheets.Results and discussions
Fibril formation of the β-LG11–20 peptide
The β-LG11–20 was chosen as a simplistic model for studying β-LG PNF structure as it is a part of one of the larger peptide fragments β-LG12-32 that was identified as part of the β-LG fibrils core (Scheme 1).38,39 The shorter peptide β-LG11–20 is also interesting as it has two aromatic residues, one tryptophan and one tyrosine, which could drive amyloid assembly and stabilise the resulting fibrils.60The fibrillation process was monitored using thioflavin T (ThT) fluorescence. ThT fluorescence is frequently used to detect amyloid-like fibrils, because of strongly increased fluorescence of ThT when binding to the β-sheet structure in the fibrils. A clear increase of the ThT fluorescence intensity of the β-LG11–20 samples was observed when incubated up to 96 h, as displayed in Fig. 1. The results indicate that the fibril formation typically reaches a stationary phase within 96 hours of thermal treatment.
 | ||
| Fig. 1 ThT fluorescence spectra of β-LG11–20 peptide solution (1 mg mL−1) with 5% pure β-LG PNF seeds, incubated at 50 °C and pH 2 for 0, 24, 36, 48, and 96 hours, respectively. | ||
AFM analysis of fibril morphologies
We studied the β-LG11–20 fibrils using AFM to characterise the fibrils' morphology and organisation of the underlying protofilaments. As shown in Fig. 2A, the β-LG11–20 fibrils are composed of multiple strands with ‘twisted nanotube morphologies’,61 having characteristic periodicity and height values. PNFs that formed mesh-like (Fig. 2B) and web-like (Fig. 2C) structures in areas with a high fibril density were observed. Further details of the fibrils were obtained from statistical analysis of high-resolution images of selected regions. The statistical abundance of fibrils with varying periodicity are depicted as histogram bars in Fig. 2D, while the corresponding heights of various fibrillar filaments are shown in Fig. 2E. There is a linear correspondence between the periodicity and height profiles of the fibrils (Fig. 2E) indicating that the morphological differences may be related to different number of protofilaments. In the investigated samples, fibril lengths up to ca. 10 μm were observed. Most of the fibrils in Fig. 2 comprised of twisted protofilaments with periodicities varying between 30 and 60 nm and maximum heights being from 3 to 6 nm. From here, statistically, there is a dominant fibril morphology with periodicity ranging between 30–40 nm with corresponding heights between 2.5–3.6 nm. It is worth mentioning that the periodicity and heights of the single fibrils of β-LG11–20 are comparable with fibrils from pure β-LG and WPI prepared under similar conditions with the periodicity and the height being 35–40 nm and 3–4 nm, respectively.36,37 Hence, it is possible that the use of β-LG seeds during the fibrillation process has templated a cross-β structure in the peptide similar to the core structure of the fibrils formed from the full-length protein.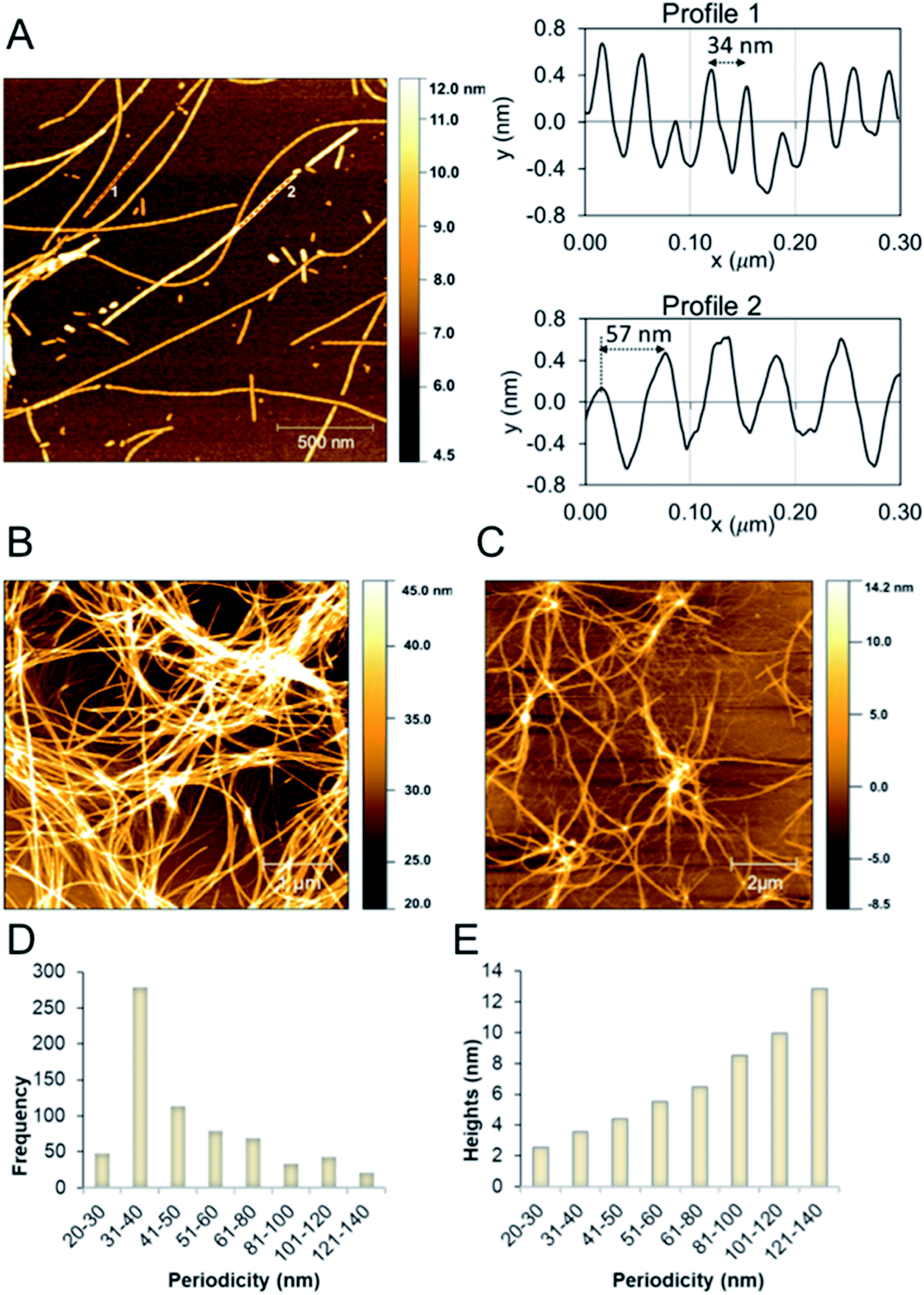 | ||
| Fig. 2 (A) AFM height images of β-LG11–20 fibrils formed at 50 °C and pH 2 (after 96 hours). Images were collected in air. The right side shows the profiles with different periodicity that are highlighted in the left side image. (B and C) AFM height images of β-LG11–20 displaying a mesh-like (B) and web-like (C) structure in areas with a high fibril density. (D and E) Statistical AFM analysis of fibril morphologies. The periodicity distribution showing that the majority of formed fibrils have periodicity of 35 ± 5 nm (D) and the correlation between periodicity and fibril heights (E). | ||
Solution-structure of monomeric β-LG11–20 peptide
Monomeric β-LG11–20 peptide in solution was investigated by solution-state NMR. The 1H and 13C NMR chemical shift assignment was achieved by the 1H–1H TOCSY and the 1H–13C HSQC, respectively, and are shown in Tables 1 and S1 (ESI†). The 1H spectrum for the unlabelled β-LG11–20 peptides is depicted in Fig. S1 (ESI†) and 13C spectra for the peptides with uniform 13C/15N labelling of selected residues are depicted in Fig. S2 (ESI†). The 1HN, 1Hα, 13C′, 13Cα and 13Cβ chemical shifts are very similar to the random coil values49 indicating that the peptide does not adopt any defined secondary structure in solution (Fig. 3).| C' | Cα | Cβ | Cγ | Cγ1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M(aq) | M(s) | Fib(s) | M(aq) | M(s) | Fib(s) | M(aq) | M(s) | Fib(s) | M(aq) | M(s) | Fib(s) | M(aq) | M(s) | Fib(s) | |
| D | 53.81 | 53.1 | 53.5 | 42.41 | 45.2 | 45.2 | |||||||||
| I | 176.61 | 175.0 | 175.1 | 62.21 | 61.6 | 61.3 | 39.11 | 42.1 | 42.6 | 27.51 | 28.37 | 28.8 | 17.9 | 17.67 | 18.5 |
| Q | 56.41 | 57.0 | 56.5 | 30.31 | 33.4 | 33.4 | 36.91 | ||||||||
| K | 54.11 | 55.3 | 55.3 | 33.41 | 36.6 | 37.4 | 25.01 | ||||||||
| V | 176.31 | 176.1 | 175.6 | 62.41 | 61.2 | 60.8 | 33.31 | 34.9 | 35.8 | 21.51 | 23.37 | 23.9 | 20.9 | 22.27 | 22.9 |
| A | 178.41 | 177.7 | 176.8 | 53.11 | 50.7 | 51.0 | 19.61 | 21.9 | 22.2 | ||||||
| G | 174.51 | 174.0 | 173.2 | 45.71 | 45.9 | 46.2 | |||||||||
| T | 174.41 | 172.7 | 172.3 | 62.61 | 62.1 | 61.8 | 70.31 | 71.6 | 72.0 | 21.81 | 22.77 | 23.2 | |
||
| W | 58.01 | 58.5 | 58.4 | 30.11 | 34.2 | 34.3 | |||||||||
| Y | 57.71 | 58.7 | 58.2 | 39.31 | 43.0 | 42.8 | |||||||||
| Cδ | N | ||||
|---|---|---|---|---|---|
| M(aq) | M(s) | Fib(s) | M(s) | Fib(s) | |
| D | |||||
| I | 13.1 | 14.37 | 15.1 | 122.9 | 123.4 |
| Q | |||||
| K | 29.4 | ||||
| V | 126.0 | 124.8 | |||
| A | 128.5 | 129.5 | |||
| G | 108.3 | 111.4 | |||
| T | 116.3 | 116.6 | |||
| W | |||||
| Y | |||||
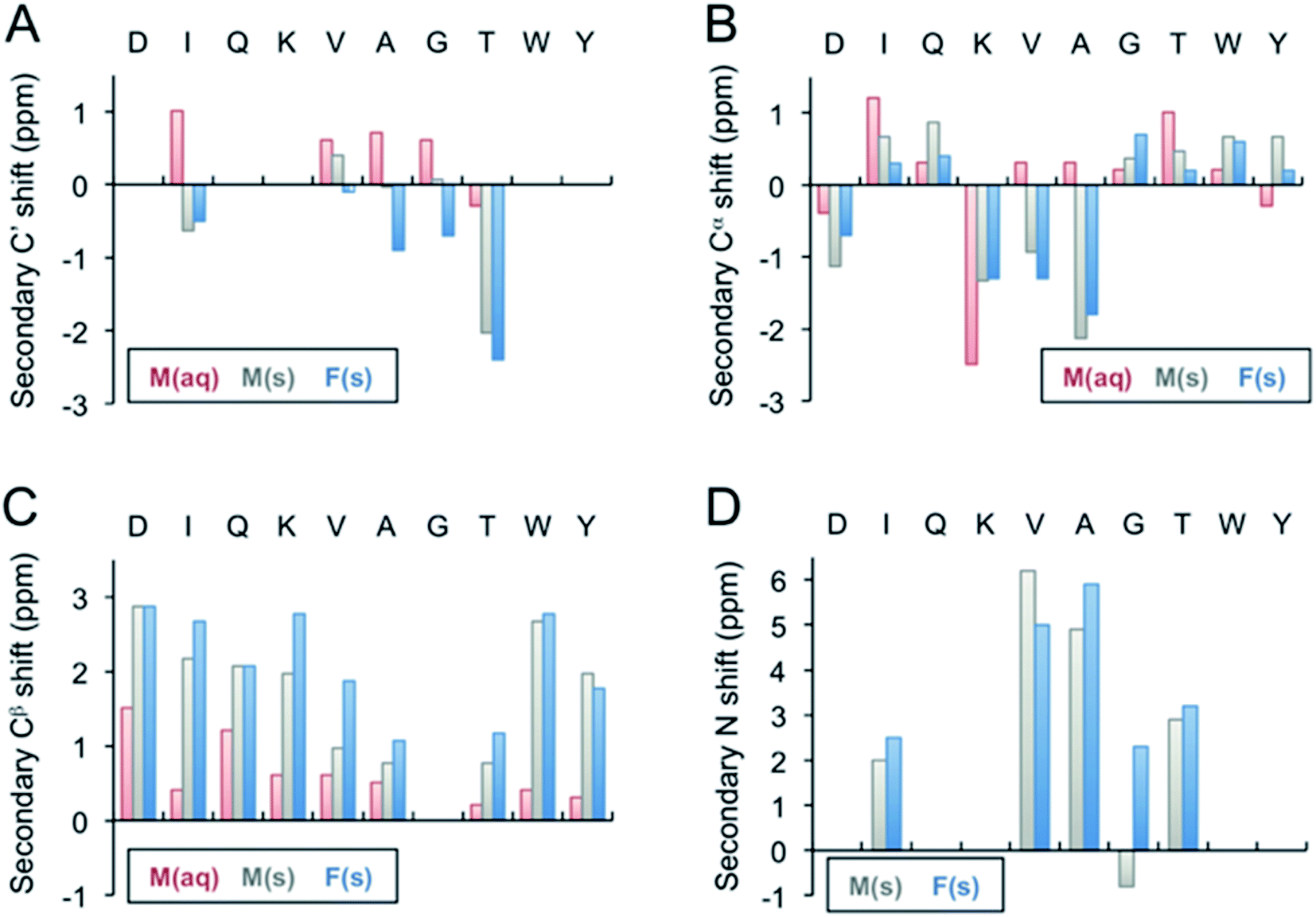 | ||
| Fig. 3 Secondary 13C′ (A), 13Cα (B), 13Cβ (C), and 15N (D) chemical shifts of the β-LG11–20 fibrils. The secondary shifts were calculated as Δδ = δ(exp) − δ(X), where δ(exp) is the experimentally observed chemical shift of either the monomer values in liquid, M(aq), monomer in solid state, M(s), or for the fibrils, F(s), whereas δ(X) represents the Wishart49 random coil values in ppm. The secondary 13C′ and 15N shifts for D1, Q3, K4, W9, Y10 could not be calculated as the residues were not labelled in any of the samples. | ||
Solid-state NMR analysis of unlabelled β-LG11–20 peptide and fibrils
Two of the main challenges in ss-NMR involve broad resonance lines and low natural abundance of the preferred 13C and 15N isotopes. The level of sample homogeneity is directly reflected in the ss-NMR line width, and thus has a consequent impact on the structural analysis. Local structural disorders in rigid solids results in broad 13C NMR resonance lines. However, hydration of the samples by addition of small amounts (a few μL) of water may increase the molecular motions, reducing inhomogeneous broadening through motional averaging.The 13C CP-MAS spectrum of lyophilised unlabelled β-LG11–20 peptide monomers (i.e., before fibril formation) and fibrils are depicted in Fig. S3A and B (ESI†). The lyophilised samples result in broad lines for both monomer and the fibrils, having line widths up to 4 ppm. A large number of spectral accumulations were required to get a decent signal to noise ratio, which makes the site-specific assignments a challenging task. The broad features of the resonance lines make it difficult to draw any conclusion of the fibril morphologies and potential structural differences between fibrils with different morphologies. However, there is a significant decrease (a factor of 4–5) in the 13C resonance line widths for the hydrated fibrils (see Fig. S3C in the ESI†).
Solid-state NMR analysis of selectively labelled β-LG11–20 peptide and fibrils
Site-specific isotopic enrichment of the β-LG11–20 peptide fibrils allows site-specific studies and was employed to enhance signal intensity. The 13C and 15N chemical shifts are sensitive to variations in local molecular conformation and structural environment. It has been established that β-strand secondary structure results in simultaneous ‘shielding effects’ on the chemical shifts for the carbonyls and Cα resonances and ‘de-shielding effects’ on the Cβ resonances, relative to the same amino acid residue within a random-coil peptide in aqueous solution.49 The chemical shifts for C', Cα, and Cβ are the most sensitive to secondary structure and relative to random coil values, the C'and Cα move down-field in α-helices and up-field for β-strand and vice versa for Cβ.We prepared two separate β-LG11–20 samples labelled uniformly with 13C and 15N at (underscored) residues D![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) QVAG
QVAG![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) WY and
WY and 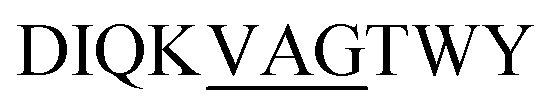 to make spectral assignments, and to characterise selectively the intermolecular packing of adjacent β-sheets in the fibrils. The 1D 13C CP-MAS NMR spectra of the IT and VAG labelled β-LG11–20 peptides are depicted in Fig. S4 (ESI†). The corresponding 2D 13C–13C DARR spectra of IT and VAG labelled β-LG11–20 peptides are depicted in Fig. S5 (ESI†). The 13C resonance line widths for the labelled non-fibrillar peptides are between 1.5 and 2.5 ppm, while the fibrils have around 40% smaller linewidths. This supports a more well-defined structure in the fibrils compared to the monomer although parts of the effect may be due to different degrees of sample hydration.
to make spectral assignments, and to characterise selectively the intermolecular packing of adjacent β-sheets in the fibrils. The 1D 13C CP-MAS NMR spectra of the IT and VAG labelled β-LG11–20 peptides are depicted in Fig. S4 (ESI†). The corresponding 2D 13C–13C DARR spectra of IT and VAG labelled β-LG11–20 peptides are depicted in Fig. S5 (ESI†). The 13C resonance line widths for the labelled non-fibrillar peptides are between 1.5 and 2.5 ppm, while the fibrils have around 40% smaller linewidths. This supports a more well-defined structure in the fibrils compared to the monomer although parts of the effect may be due to different degrees of sample hydration.
For the monomeric lyophilised peptide, intra and inter-residue cross peaks are distinct and show no overlap except for the Cα of I2, (V5) and T8 residues. A DARR mixing time of 250 and 500 ms was used to assign intra- and inter-residue backbone and sidechain cross peaks, respectively. As expected, no inter-residue cross peaks between the far apart residues I2 and T8 were observed. On the other hand, at 500 ms mixing time, A6 shows both i − 1 and i + 1 cross peak. In addition, i + 2 cross peaks were observed between V5 and G7.
Fibril samples with selective labelling were prepared from the respective peptide monomers. The resulting fibrils from the individual monomers were named after the labelling scheme viz. Fib-IT and Fib-VAG. 2D 13C–13C CP-DARR experiments with a mixing time of 500 ms were recorded for both fibril samples. All DARR spectra contain peaks corresponding to a single spin system for each amino acid residue, indicating that all residues exist in one unique structure. With the assignment of the labelled residues available, additional peak assignments in the 1D 13C spectrum of the unlabelled fibrils were made. Secondary C', Cα, Cβ and N chemical shifts for the monomer in solution-state (Liq.), monomer in solid-state (M-s) and fibrils (Fib) were calculated according to Wishart et al.49 (Fig. 3). The C' and some of the Cα chemical shifts in the fibrils shifted upfield, while Cβ and N shifted downfield relative to the random coil values. The positive Cα secondary shifts (shifted downfield) are small and therefore it can be concluded that the fibrils contain the expected β-sheet structure. Notably, the secondary shifts of the monomeric peptide obtained by solid-state NMR are, with a few exceptions, similar to those in the fibrils and it can be speculated that the peptide adopts an extended structure with some resemblance of a β-strand while packing in a solid-state powder. The monomeric peptide in solution, however, display smaller secondary shifts, which is in agreement with a more random coil-like structure.
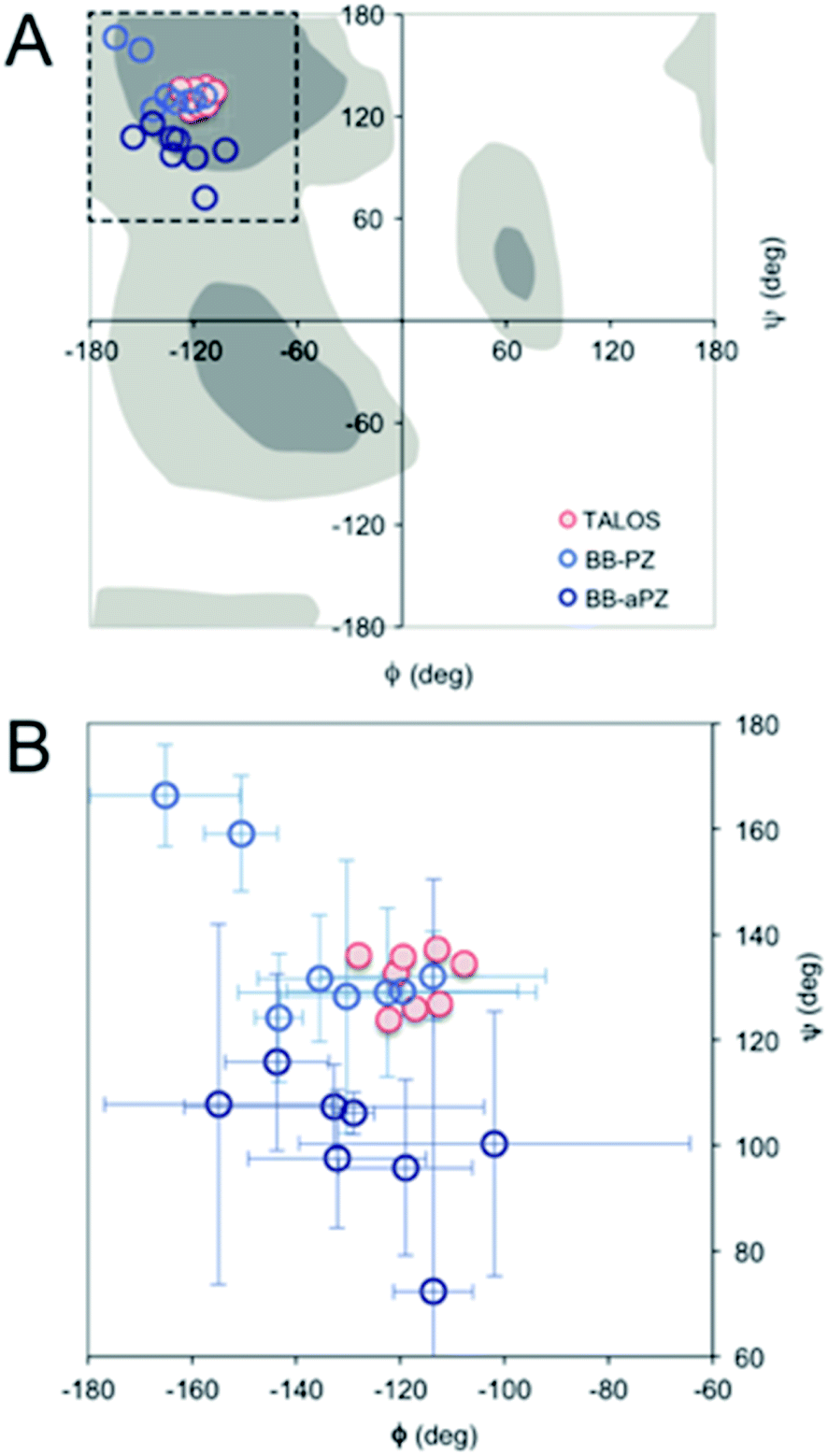 | ||
| Fig. 4 (A) Ramachandran plot showing the ϕ and ψ values predicted by TALOS+ (red) and the values obtained from the two best MD models (blue). (B) A magnification with error bars (±1 STD) included. | ||
For a more detailed analysis of secondary structure, the experimentally derived C′, Cα, Cβ and N chemical shifts were used for a TALOS+ prediction of backbone torsion angles ϕ and ψ (Fig. 4 and Table S2 in the ESI†). Satisfactory convergence is obtained for eight of the ten residues, for which the program can predict ϕ and ψ. All eight residues (the termini excluded) show a high degree of β-sheet secondary structure. Sites for which TALOS+ predicted torsion angles typical of β-strand region of the Ramachandran plot, i.e., the torsion angles with estimated uncertainties of approximately ±10° are all between ≈−110° and −130° for ϕ, and ≈120° and 140° for ψ.
Solid-state NMR analysis of β-LG11–20 fibrils – quaternary structure
Next, the arrangement of the strands within the β-sheets was examined. The labelling scheme of the Fib-IT sample was selected to reveal dipolar interactions between the I2 and T8 residues in potential anti-parallel β-sheets. However, 13C–13C cross-peaks in the 13C 2D DARR experiments result from only the intra-residue resonances but no inter-residue cross-peaks between residues I2 and T8, suggesting that the β-LG11–20 peptide form parallel β-sheets in the fibrils. To further corroborate this conclusion, a fibril sample with 50% mixture of the monomers Fib-VAG-IT was prepared and analysed for long range inter-molecular correlations (Fig. 5). The 13C–13C CP-DARR spectrum contains five C' resonances between ∼169 and 174 ppm associated with different Cα chemical shifts (between 43 and 59 ppm). All five C' have dipolar couplings that extend to their side chain carbons (intra-residual) as well as some inter-residue dipolar couplings between VAG in the Fib-VAG-IT. Most importantly, there are inter-molecular cross-peaks between G7 and T8 residues. The cross-peaks were symmetrical about the diagonal peaks. Inter-strand dipolar couplings between G7C'–T8Cα, T8C′–G7Cα and G7Cα–T8Cα were observed. No intermolecular cross-peak involving I2 was observed, confirming the presence of a parallel β-sheet arrangement of the fibrils.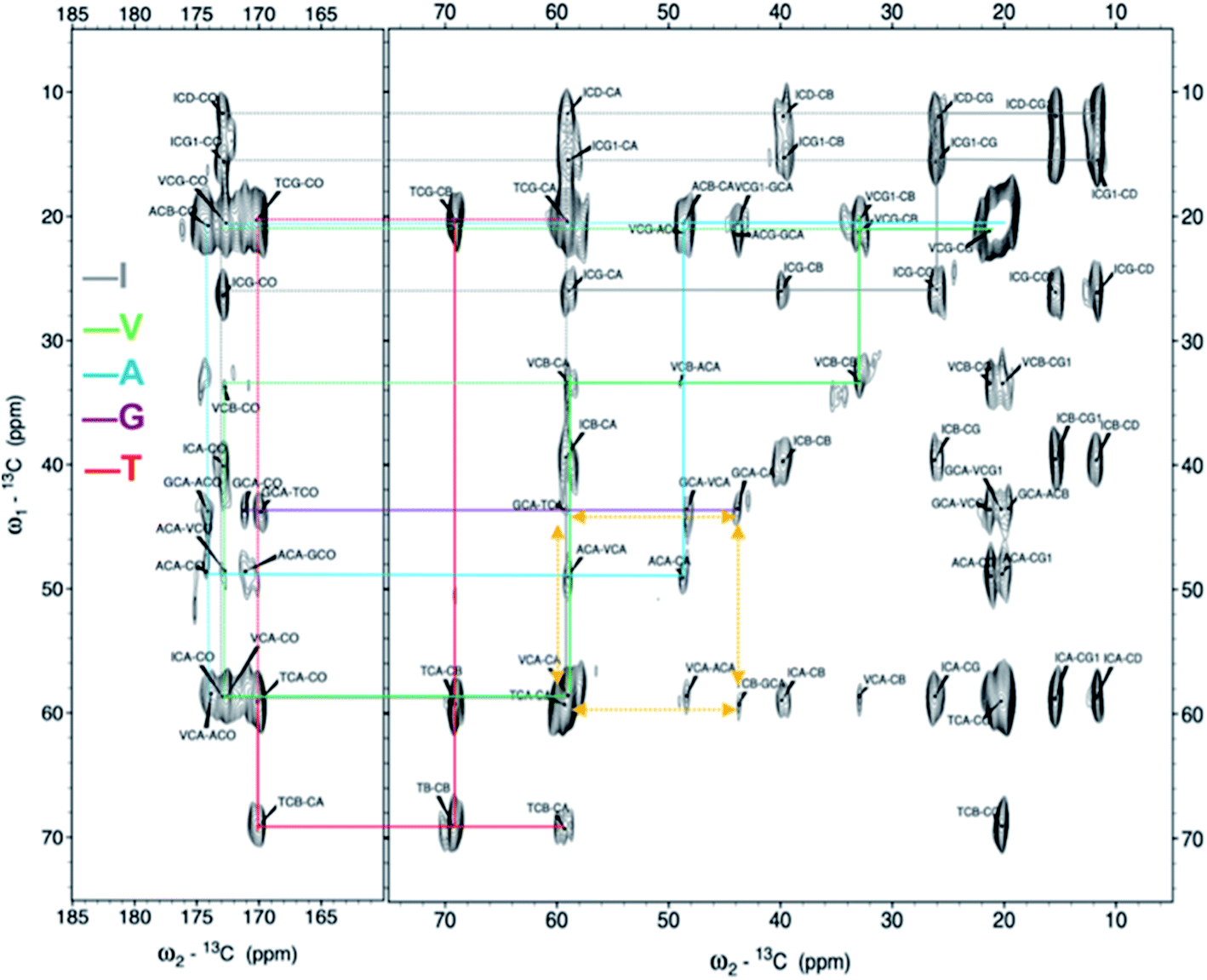 | ||
| Fig. 5 2D 13C–13C solid-state DARR NMR spectra of the mixed (50:50%) fibrils IT-VAG. The lines indicate the assigned spin systems. The yellow arrows highlight inter-molecular cross peaks. | ||
Next, we evaluated how the β-sheets pack against each other, although no unambiguous intermolecular cross peaks could be detected in the NMR data to guide this analysis. The parallel β-sheets formed by the peptide display two distinct faces. On the first face (A), the sidechains of I2, K4, A6, T8 and Y10 are displayed while the other face (B) are made up form D1, Q3, V5, (G7) and W9 (see Fig. S6B in the ESI†). Based on knowledge from previous studies of amyloid structures originating form short peptides, we hypothesize that the parallel β-sheets would pack against each other to form a “steric zipper”.62–64 With the two distinct faces, the sheets can pack in six different modes (Fig. S6, ESI†). From inspection, the variants involving a buried positively charged lysine side chain seems less likely. Hence, a zipper involving the face B residues would be preferred. This is indeed in agreement with the absence of cross-peaks between I2 and T8 side chains in the NMR spectra as these would not face each other in this arrangement. The B–B zipper packing would potentially involve burying of the Trp side chain.
Molecular dynamics simulations
As the NMR data could not unambiguously reveal the packing of the sheets against each other to form steric zipper structures previously described for other peptide fibrils,65–67 we decided to use molecular dynamics simulation to explore the fibril formation of the β-LG11–20 peptide. One of the first simulations involved a small β-sheet structure consisting of four peptide units in a parallel arrangement. The initial structure was built, based on previous observations, with the π-stacking interactions between W10 moieties, and Y9 moieties. Each peptide unit was placed in a position to form hydrogen bonds with the neighbouring peptide unit (Fig. 6A). During our MD simulation, the system remained very close to the initial arrangement (keeping its π–π stacking and H-bonding interactions); moreover, the peptides formed a β-sheet alignment. Simulations of a longer β-sheet structure with 20 peptide units showed that despite the partly broken π–π stacking interactions between the all Y9 and W10 moieties after 13 ns of the MD simulation, the hydrogen bonding interactions in the β-sheet were still preserved, supporting the previous experimental results of the peptide to form stable parallel β-sheet structures (Fig. 6B).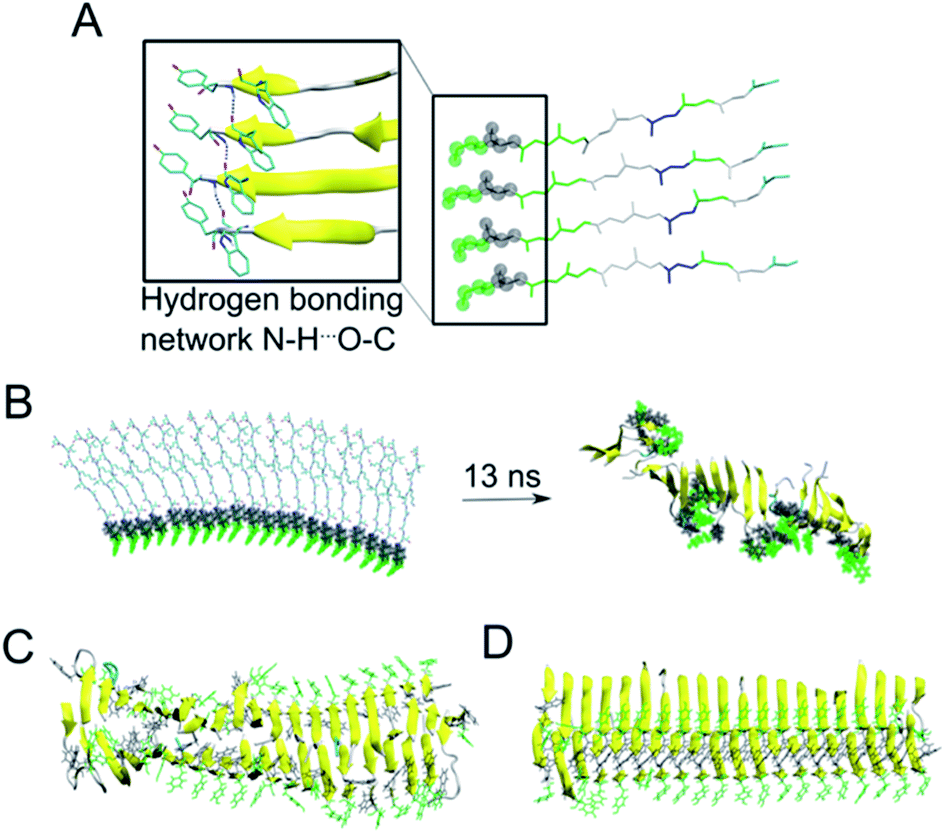 | ||
| Fig. 6 (A) A representative structure of four peptides in parallel alignment. For simplicity, only the protein backbone is shown, and the tyrosine and tryptophane moieties are shown in green and grey spheres, respectively. The structure is (i) stabilised by the π–π stacking interactions involving Tyr and Trp moieties, (ii) stabilised by hydrogen bonding interactions within the neighbouring peptides, and (iii) the peptides form β-sheet secondary structure during our MD simulations (as represented by the arrows). (B) In a similar manner to panel A, a 20-peptide sheet was built to study a stability of this more complex structure. Even after ca. 13 ns of MD simulation, the end point of our simulation showed that the π–π stacking and H-bonding interactions are not preserved among all Tyr and Trp moieties (highlighted in green and grey), despite the ability to preserve most of the β-sheets (yellow). (C) MD simulation (30 ns) of 40 peptides in the anti-parallel B–B zipper mode packing (stabilised by 194 ± 9 H-bonding interactions). (D) MD simulation (30 ns) of 40 peptides in the parallel B–B zipper mode packing (stabilised by 192 ± 9 H-bonding interactions). Panels C and D show the most stable arrangements of a 40-peptide double sheets; the non-stable packing variants are shown in Fig. S7 in the ESI.† | ||
Subsequently, we explored the relative stability of different steric zipper arrangements by performing 30 ns MD simulations of two parallel β-sheets, each containing 20 peptides. We considered six different arrangements: three parallel orientations (tyrosines of each β-sheets being on opposite sides of each β-sheet) and (i) all lysines' side chains residues point inward and are packed against each other (AA-PZ), (ii) all lysines' side chains residues point outward (BB-PZ), (iii) lysines' side chains of one β-sheets point inwards and the same side chains of the other β-sheets point outward (AB-PZ); and another three antiparallel arrangements (with tyrosines of both β-sheets being on the same side) with the same lysines' arrangement as described earlier i.e., (iv) all lysines inwards (AA-aPZ), (v) outwards (BB-aPZ) or (vi) one inward and another outward (AB-aPZ). All six packing modes (see Fig. S6 in the ESI†) were explored, and all arrangements with the positively charged lysine side chain packed against the neighbouring sheet (arrangements, where lysine side chains point inwards) were found to be unstable during our MD simulations (Fig. S7 in the ESI†) as the systems failed in holding a perfect β-sheet structure, and some of the π–π stacking and hydrogen bonding interactions broke resulting in the lower stabilisation of the studied systems. The two variants with the B-faces packed against each other, either in a parallel manner (BB-PZ) or anti-parallel manner (BB-aPZ), were both found to be very stable within the time of our simulations with preserved π–π stacking and hydrogen-bonded β-sheets (Fig. 6C and D).
Looking to the two MD models closely, we find that the intra β-sheets Cα distance between G7-T8 residues in both parallel- and anti-parallel zipper arrangement are short (<7 Å) and could potentially be detected by the 13C–13C DARR NMR experiment. On the other hand, intra- or inter-β-sheet Cα distances between I2–T8 in both parallel- and anti-parallel zipper arrangements are at least 11 Å, which are not possible to observe in the current 13C–13C DARR experimental setup. As the side chains of both I2 and T8 are located in the face opposite in B–B zipper arrangements, the distances between sidechain atoms are even longer than for Cα contacts.
DFT/PBE0 calculations of 13C chemical shifts
We used DFT approach and calculated the 13C chemical shifts of the two most probable MD models. The computed 13C chemical shifts for the monomer (β-LG11–20) are compared with the experimentally observed values in liquid-state, whereas those calculated for selected molecular ensembles (average of four central peptide units) obtained from our MD simulations are compared with the MAS NMR results for the fibrils. There is a linear correlation between the calculated shielding values and the experimental chemical shifts for Cα and side chain carbons for both monomer and fibrils (Fig. 7). The 13C′ shift correlation is not as reliable as they are only few data set points available and the C′ shifts span over a small range of values (data not shown). Based on the comparison, one can conclude that both structural models are in agreement with the NMR data although the parallel packing (BB-PZ) seems to be slightly better (R2 close to unity). | ||
| Fig. 7 Experimental vs. DFT/PBE0 calculated 13C Cα and Cβ chemical shifts for monomer (A) in liquid-state, parallel BB-PZ (B), and anti-parallel BB-aPZ (C) molecular ensembles β-sheets. The calculated values for panel B and C were obtained from snapshots taken at the end point of our 30 ns of MD simulations, and the experimental values are taken from solid-state MAS NMR. | ||
Conclusions
We have explored the structure of the nanofibrils formed by a 10-residue peptide derived from β-lactoglobulin (β-LG11–20). The presented results show that the peptide forms micrometre-long straight, amyloid-like fibrils. Although the fibrils appear to be polymorphic when examined by AFM, the further observation of a single set of NMR resonances indicates the presence of a single molecular structure. Hence, the polymorphism observed at the nanoscale originates from packing differences of protofilaments. Secondary chemical shifts derived from peptides with site-specific labelling confirm β-sheet secondary structure. The intermolecular contacts observed in the NMR data also suggest parallel arrangements of the β-sheets, but they are inconclusive about the packing of the sheets against each other. We used MD simulations to evaluate the relative stability of different packing models and found that packing most probably involves the face of the peptide that consists of D1, Q3, V5, G7, W9 as it avoids the electrostatic repulsion coming from the two lysine residues (K4) in close proximity. We suggested two possible packing modes: parallel and anti-parallel. Although the energetics is slightly more favourable for the anti-parallel mode, the parallel mode displays slightly better agreement with secondary structure content. Calculated 13C chemical shifts from the parallel model is also in better agreement with the experimental values than the anti-parallel model.In comparison with existing structures of short peptides in amyloid-like structures, we find that parallel arrangement of the sheets is rare, at least if the strands are organised into parallel β-sheets. Only one such model was found in the PDB database (PDB ID 6ODG), confirming that parallel packing is possible. However, the majority of these structures are determined by X-ray diffraction experiments of amyloid-like crystals. Such crystals may have different preferences in the packing mode than amyloid fibrils formed in solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Carl Tryggers stiftelse (CTS16:273 and CTS18:810). The Kempe Foundation in memory of J. C. and Seth M. Kempe is acknowledged for a grant from which an automated peptide synthesiser (Applied Biosystems model 433A) was purchased. M. L. thanks SeRC (Swedish e-Science Research Center) for funding. The molecular dynamics simulations and DFT calculations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC.References
- M. Sunde and C. C. Blake, Q. Rev. Biophys., 1998, 31, 1–39 CrossRef CAS PubMed.
- J. D. Sipe, Annu. Rev. Biochem., 1992, 61, 947–975 CrossRef CAS PubMed.
- N. Agrawal and A. A. Skelton, Protein J., 2019, 38, 425–434 CrossRef CAS PubMed.
- V. N. Uversky and A. L. Fink, Protein Misfolding, Aggregation, and Conformational Diseases: Part B: Molecular Mechanisms of Conformational Diseases, Springer US, Boston, MA, 2007, pp. 61–110 Search PubMed.
- J. W. Höppener, B. Ahrén and C. J. Lips, N. Engl. J. Med., 2000, 343, 411–419 CrossRef.
- S. Mankar, A. Anoop, S. Sen and S. K. Maji, Nano Rev., 2011, 2, 6032 CrossRef.
- C. Li and R. Mezzenga, Nanoscale, 2013, 5, 6207–6218 RSC.
- T. P. J. Knowles and M. J. Buehler, Nat. Nanotechnol., 2011, 6, 469–479 CrossRef CAS PubMed.
- S. F. Oliveira, G. Bisker, N. A. Bakh, S. L. Gibbs, M. P. Landry and M. S. Strano, Carbon, 2015, 95, 767–779 CrossRef CAS.
- S. Ling, C. Li, J. Adamcik, Z. Shao, X. Chen and R. Mezzenga, Adv. Mater., 2014, 26, 4569–4574 CrossRef CAS.
- M. A. Lambrecht, K. J. A. Jansens, I. Rombouts, K. Brijs, F. Rousseau, J. Schymkowitz and J. A. Delcour, Compr. Rev. Food Sci. Food Saf., 2019, 18, 1277–1291 CrossRef CAS PubMed.
- X. Ye, C. Lendel, M. Langton and R. T. Olsson, Protein nanofibrils: Preparation, properties, and possible applications in industrial nanomaterials, in Industrial Applications of Nanomaterials, Elsevier, 2019, pp. 29–63 Search PubMed.
- D. Cho, A. N. Netravali and Y. L. Joo, Polym. Degrad. Stab., 2012, 97, 747–754 CrossRef CAS.
- S. T. Sullivan, C. Tang, A. Kennedy, S. Talwar and S. A. Khan, Food Hydrocolloids, 2014, 35, 36–50 CrossRef CAS.
- J. F. Smith, T. P. J. Knowles, C. M. Dobson, C. E. Macphee and M. E. Welland, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15806–15811 CrossRef CAS PubMed.
- T. P. Knowles, A. W. Fitzpatrick, S. Meehan, H. R. Mott, M. Vendruscolo, C. M. Dobson and M. E. Welland, Science, 2007, 318, 1900–1903 CrossRef CAS PubMed.
- U. Slotta, S. Hess, K. Spiess, T. Stromer, L. Serpell and T. Scheibel, Macromol. Biosci., 2007, 7, 183–188 CrossRef CAS PubMed.
- C. Akkermans, A. J. Van der Goot, P. Venema, H. Gruppen, J. M. Vereijken, E. Van der Linden and R. M. Boom, J. Agric. Food Chem., 2007, 55, 9877–9882 CrossRef CAS.
- J. S. Mounsey, B. T. O'Kennedy, M. A. Fenelon and A. Brodkorb, Food Hydrocolloids, 2008, 22, 65–73 CrossRef CAS.
- J. Weiss, P. Takhistov and D. J. McClements, J. Food Sci., 2006, 71, R107–R116 CrossRef CAS.
- W. Zhang, X. Yu, Y. Li, Z. Su, K. D. Jandt and G. Wei, Prog. Polym. Sci., 2018, 80, 94–124 CrossRef CAS.
- A. Antonucci, J. Kupis-Rozmysłowicz and A. A. Boghossian, ACS Appl. Mater. Interfaces, 2017, 9, 11321–11331 CrossRef CAS PubMed.
- X. Yu, Z. Wang, Z. Su and G. Wei, J. Mater. Chem. B, 2017, 5, 1130–1142 RSC.
- L. Sasso and J. A. Gerrard, in Micro and Nanofabrication Using Self-Assembled Biological Nanostructures, Elsevier, 2015, pp. 1–20 Search PubMed.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. Ø. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS PubMed.
- M. Fändrich, S. Nyström, K. P. R. Nilsson, A. Böckmann, H. LeVine and P. Hammarström, J. Intern. Med., 2018, 283, 218–237 CrossRef.
- G. Comellas and C. M. Rienstra, Annu. Rev. Biophys., 2013, 42, 515–536 CrossRef CAS PubMed.
- R. Tycko, Annu. Rev. Phys. Chem., 2011, 62, 279–299 CrossRef CAS PubMed.
- S. E. Ashbrook, J. M. Griffin and K. E. Johnston, Annu. Rev. Anal. Chem., 2018, 11, 485–508 CrossRef PubMed.
- Q. Wang, H. Yu, B. Tian, B. Jiang, J. Xu, D. Li, Z. Feng and C. Liu, Coatings, 2019, 9, 583 CrossRef CAS.
- A. Kamada, N. Mittal, L. D. Söderberg, T. Ingverud, W. Ohm, S. V. Roth, F. Lundell and C. Lendel, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1232–1237 CrossRef CAS PubMed.
- R. A. Mantovani, G. de Figueiredo Furtado, F. M. Netto and R. L. Cunha, J. Food Eng., 2018, 223, 99–108 CrossRef CAS.
- S. K. Ng, K. L. Nyam, I. A. Nehdi, G. H. Chong, O. M. Lai and C. P. Tan, Food Sci. Biotechnol., 2016, 25, 15–21 CrossRef CAS PubMed.
- A. Kroes-Nijboer, P. Venema and E. van der Linden, Food Funct., 2012, 3, 221–227 RSC.
- Y. Hu, C. He, M. W. Woo, H. Xiong, J. Hu and Q. Zhao, Food Funct., 2019, 10, 8106–8115 RSC.
- S. Ikeda and V. J. Morris, Biomacromolecules, 2002, 3, 382–389 CrossRef CAS.
- D. Oboroceanu, L. Wang, A. Brodkorb, E. Magner and M. A. E. Auty, J. Agric. Food Chem., 2010, 58, 3667–3673 CrossRef CAS.
- C. Akkermans, P. Venema, A. J. van der Goot, H. Gruppen, E. J. Bakx, R. M. Boom and E. van der Linden, Biomacromolecules, 2008, 9, 1474–1479 CrossRef CAS PubMed.
- X. Ye, M. S. Hedenqvist, M. Langton and C. Lendel, RSC Adv., 2018, 8, 6915–6924 RSC.
- S. G. Bolder, H. Hendrickx, L. M. C. Sagis and E. van der Linden, J. Agric. Food Chem., 2006, 54, 4229–4234 CrossRef CAS.
- L. N. Arnaudov and R. de Vries, in Food Colloids: Interactions, Microstructure and Processing, ed. E. Dickinson, Royal Society of Chemistry, Cambridge, 2007, pp. 237–246 Search PubMed.
- L. N. Arnaudov, R. de Vries, H. Ippel and C. P. M. van Mierlo, Biomacromolecules, 2003, 4, 1614–1622 CrossRef CAS PubMed.
- D. Hamada, T. Tanaka, G. G. Tartaglia, A. Pawar, M. Vendruscolo, M. Kawamura, A. Tamura, N. Tanaka and C. M. Dobson, J. Mol. Biol., 2009, 386, 878–890 CrossRef CAS PubMed.
- J. T. Jarrett and P. T. Lansbury, Cell, 1993, 73, 1055–1058 CrossRef CAS PubMed.
- W. Qiang, W.-M. Yau and R. Tycko, J. Am. Chem. Soc., 2011, 133, 4018–4029 CrossRef CAS PubMed.
- S. G. Bolder, L. M. C. Sagis, P. Venema and E. van der Linden, J. Agric. Food Chem., 2007, 55, 5661–5669 CrossRef CAS PubMed.
- M. R. Nilsson, Methods, 2004, 34, 151–160 CrossRef CAS PubMed.
- D. Nečas and P. Klapetek, Open Phys., 2012, 10, 181–188 Search PubMed.
- D. S. Wishart, C. G. Bigam, J. Yao, F. Abildgaard, H. J. Dyson, E. Oldfield, J. L. Markley and B. D. Sykes, J. Biomol. NMR, 1995, 6, 135–140 CrossRef CAS.
- C. R. Morcombe and K. W. Zilm, J. Magn. Reson., 2003, 162, 479–486 CrossRef CAS.
- D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao and P. A. Kollman, Amber 2016, University of California, San Francisco, 2016 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. J. Berendsen, J. V. Postma, W. F. van Gunsteren, A. R. H. J. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji and X. Li, Gaussian 16 Rev. A.03, Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- E. Gazit, FASEB J., 2002, 16, 77–83 CrossRef CAS PubMed.
- M. Žganec and E. Žerovnik, Biochim. Biophys. Acta, 2015, 1840, 2944–2952 CrossRef PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS PubMed.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Nature, 2017, 547, 185–190 CrossRef CAS PubMed.
- M. A. Wälti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Böckmann, P. Güntert, B. H. Meier and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef.
- A. W. P. Fitzpatrick, G. T. Debelouchina, M. J. Bayro, D. K. Clare, M. A. Caporini, V. S. Bajaj, C. P. Jaroniec, L. Wang, V. Ladizhansky, S. A. Müller, C. E. MacPhee, C. A. Waudby, H. R. Mott, A. De Simone, T. P. J. Knowles, H. R. Saibil, M. Vendruscolo, E. V. Orlova, R. G. Griffin and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5468–5473 CrossRef CAS PubMed.
- C. P. Jaroniec, C. E. MacPhee, V. S. Bajaj, M. T. McMahon, C. M. Dobson and R. G. Griffin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 711–716 CrossRef CAS PubMed.
- G. T. Debelouchina, M. J. Bayro, A. W. Fitzpatrick, V. Ladizhansky, M. T. Colvin, M. A. Caporini, C. P. Jaroniec, V. S. Bajaj, M. Rosay, C. E. Macphee, M. Vendruscolo, W. E. Maas, C. M. Dobson and R. G. Griffin, J. Am. Chem. Soc., 2013, 135, 19237–19247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03575d |
| This journal is © The Royal Society of Chemistry 2021 |