
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRigid 3D-spiro chromanone as a crux for efficient antimicrobial agents: synthesis, biological and computational evaluation†
F. H. Elghareeba,
E. M. Kandila,
M. Abou-Elzahaba,
M. Abdelmoteleb b and
M. A. Abozeid*a
b and
M. A. Abozeid*a
aDepartment of Chemistry, Faculty of Science, Mansoura University, Mansoura-35516, Egypt. E-mail: dr.abozeid.chem@gmail.com; mabozeid@mans.edu.eg
bDepartment of Botany, Faculty of Science, Mansoura University, Mansoura-35516, Egypt
First published on 16th June 2021
Abstract
The development of new and effective antimicrobial agents with novel chemical skeletons and working mechanisms is highly desirable due to the increased number of resistant microbes. Different new compounds based upon a 3D-spiro chromanone scaffold such as Mannich bases 2 and 3 in addition to azo dye 4 were synthesized. Besides, the condensation reactions of the hydrazide-spiro chromanone 8 with different ketonic reagents led to the synthesis of pyrazoles (9 & 10) and anils (11 & 13). Moreover, the methoxyl substituted spiro chromanone 14 was condensed with different hydrazines and hydrazides to give the corresponding hydrazones 15–18 in up to 85% yields. The condensation of the hydrazone 18 with salicylaldehyde yielded coumarinyl spiro chromanone 19 in an excellent yield, whereas its reaction with benzaldehyde followed by hydrazine afforded aminopyrazole derivative 21 in 82% yield. The antimicrobial evaluation suggested that hydrazide 8 has a substantial activity against different microbes (S. aureus: D = 22 mm, MIC = 1.64 μM; E. coli: D = 19 mm, MIC = 1.64 μM; C. albicans: D = 20 mm, MIC = 6.57 μM). Moreover, promising antimicrobial activities were observed for azo dye 4 (D = 13–19 mm, MIC = 5.95–11.89 μM), hydrazone 17 (D = 17–23 mm, MIC = 1.88–3.75 μM), and aminopyrazole 21 (D = 14–19 mm, MIC = 2.24–8.98 μM). The molecular docking revealed that compounds 4, 8, 17, and 21 had good to high binding affinities with different microbial targets such as penicillin-binding proteins (−7.4 to −9.9 kcal), DNA gyrase (−7.8 to −9.0 kcal), lanosterol 14-alpha demethylase (−8.2 to −11.2 kcal), and exo-beta-1,3-glucanase (−8.2 to −11.9 kcal). The QSAR analysis ascertained a good correlation between the antimicrobial activity of 3D-spiro chromanone derivatives and their structural and/or physicochemical parameters.
Introduction
Chromone derivatives constitute one of the main families of heterocyclic compounds.1 Particularly, 4-chromanones have attracted significant attention because of their wide natural occurrence such as sterubin,2 morellin,3 and blennolide4 (Fig. 1A). They are the precursors for the synthesis of many naturally occurring compounds such as ripariochromene, hematoxylin, and brazilin.5 In addition, many synthetic chromanone derivatives revealed diversified biological effects such as anti-HIV,6 antibiotic,7 antidiabetic,8 antifungal,9 antiarrhythmic,10 and antitumor11 as well as different agonist and antagonist activities (Fig. 1A). As a result, the chromanone nucleus became a privileged scaffold in drug design and tuning the substitution patterns appeared to be efficient structural screws to build up new biologically active leads.12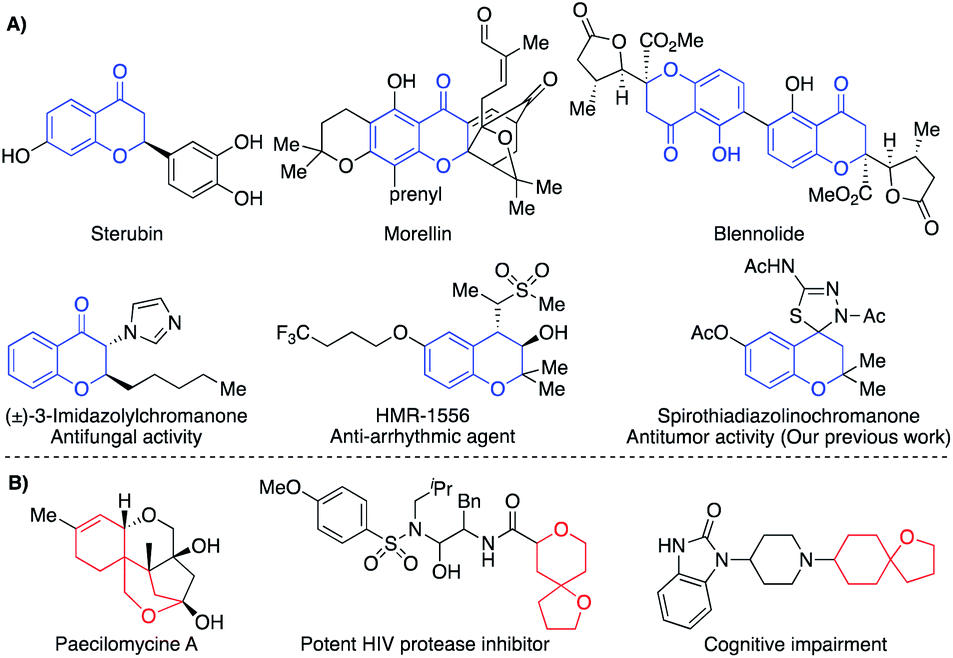 | ||
| Fig. 1 (A) Representative examples of naturally and biologically active chromanones. (B) Representative examples of natural and bioactive oxaspirocyclic derivatives. | ||
On the other hand, spirocyclic compounds represent a privileged class of cyclic organic compounds that comprise rings fused through a tetrahedral central atom.13 The restricted rotation around the spiro-linked atom accommodates the ring planes to be perpendicularly oriented. Based upon the inherent rigidity, higher stability, and precise three-dimensionality, spirocyclic compounds appear as an attractive platform for different applications such as synthesis of chiral ligands14a and organocatalysts14b,c as well as the design of organic optoelectronics.15 Furthermore, spiro compounds were found in a plethora of naturally occurring compounds that manifested diverse biological activities.16 Currently, spiro compounds became a catchy scaffold in the modern drug design because of their intrinsic rigidity and precise 3D-dimensionality which maximize H-bonding, π–π stacking, and hydrophobic interactions.17 These skeletal features could improve drug-receptor binding17 as well as the ADMET score.18 Within the spirocyclic compounds, the oxygen-bearing counterparts constitute a distinguishable subclass due to their wide natural occurrence and notable biological activities in addition to the aforementioned skeletal features for spiro skeletons (Fig. 1B).19
Antimicrobial resistance (AMR) is the ability of microorganisms to grow in the presence of drugs that have been used once against them.20a–c AMR is considered one of the most serious health that leads to persistent infections with high risk of disease transmission. Moreover, increasing the number of multidrug-resistant microbes depicts a cloudy future for the health care systems leading to an urgent need for new antimicrobial agents with novel chemical skeletons as well as working mechanisms.20a–c In our group, we designed different classes of heterocyclic compounds with potent antimicrobial and antiquorum-sensing (QS) activities.20d–g In continuation of our research interest in the synthesis of biologically interesting heterocyclic compounds,20d–i herein we have designed a novel series of spirocyclic chromanone derivatives and evaluated them for their antimicrobial activities with the aid of computational analysis. The construction of a hybrid skeleton combining both chromanone and oxaspiro[5.5]undecane motifs has been the crux of the newly synthesized compounds.
Results and discussion
Chemistry
Mannich reaction of the spirocyclic chromanone 121 with formalin and morpholine afforded the corresponding 8-morpholinomethyl spirocyclic derivative 2 in a good yield (Scheme 1). Similarly, bis-Mannich base 3 was obtained in 56% yield via reaction of spiro chromanone 1 with formalin and piperazine. Moreover, the reaction spiro chromanone 1 with benzene diazonium chloride produced 8-phenyldiazenyl spiro chromanone 4 in 74% yield. The chemical structures of the new compounds 2–4 were secured based on different spectral analyses as shown in the Experimental part.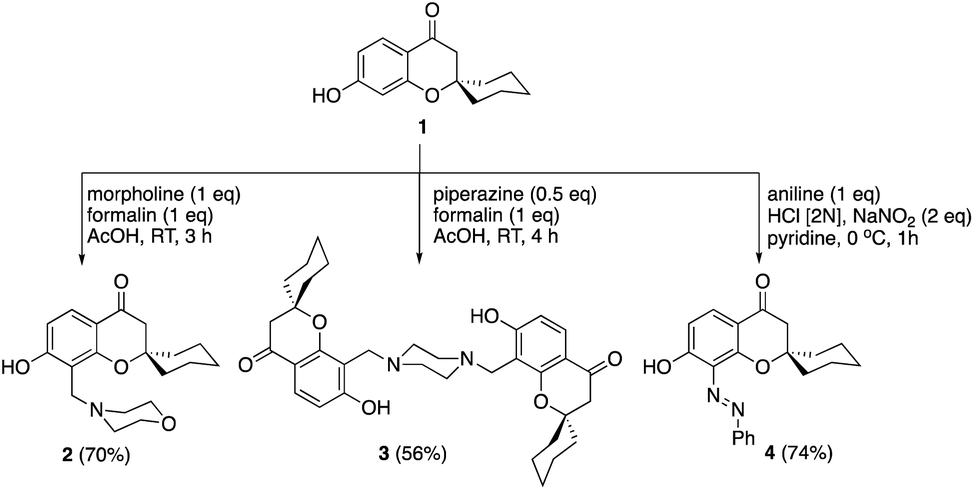 | ||
| Scheme 1 Synthesis of Mannich bases (2 & 3) and azo dye 4. | ||
The chemistry of spiro chromanone 1 was further extended by clipping of ethyl acetate moiety on the pendant phenolic function as in ester 5 (Scheme 2). Treatment of ester 5 with polyphosphoric acid failed to give the desired furanone 6, and instead, the carboxylic acid 7 was obtained in 94% yield. In addition, the hydrazide 8 was synthesized from the ester 5 in an excellent yield by the reaction with hydrazine hydrate in ethanol at room temperature. The absence of ethoxyl group signals in the 1H-NMR spectrum of hydrazide 8 in addition to the appearance of NH and NH2 functionalities at 3303 and 3214 cm−1 in the IR spectrum confirmed its chemical structure.
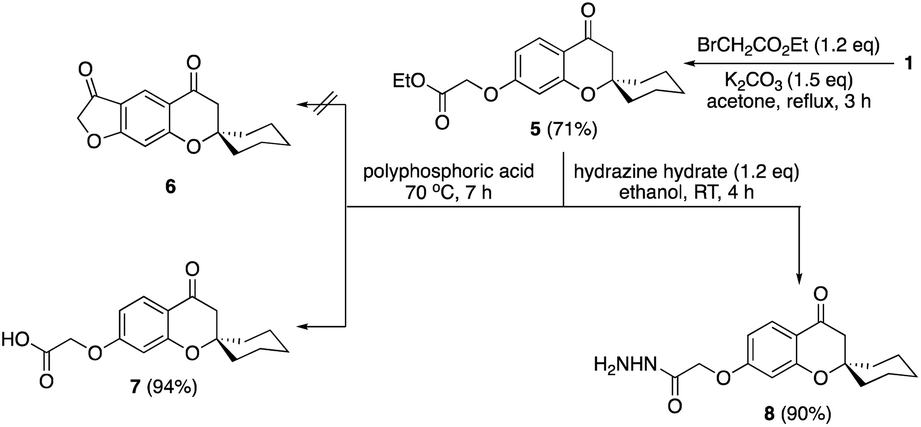 | ||
| Scheme 2 Synthesis of carboxylic acid 7 and hydrazide 8. | ||
The condensation of the hydrazide 8 with ethyl acetoacetate (EAA) in the presence of a catalytic amount of acetic acid in refluxing ethanol gave the pyrazolone 9 in 59% yield (Scheme 3). Similarly, the reaction of 8 with acetylacetone afforded dihydropyrazole 10 in 74% yield. The condensation of hydrazide 8 with benzaldehyde afforded the corresponding hydrazone 11 as an E/Z-mixture in a very good yield. Additionally, the isatin analogue 13 was obtained by refluxing ethanolic solution of the hydrazide 8 and isatin in presence of few drops of acetic acid.
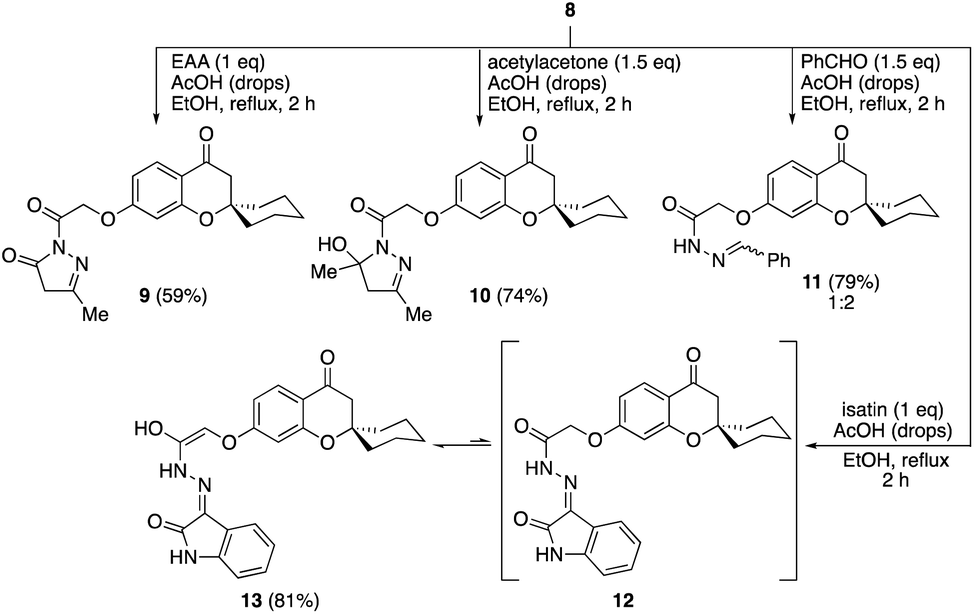 | ||
| Scheme 3 Reaction of hydrazide 8 with ethyl acetoacetate, acetylacetone, benzaldehyde, and isatin. | ||
The condensation of the methyl protected spiro chromanone 1422 with phenylhydrazine afforded the corresponding hydrazone 15 in 56% yield (Scheme 4). Moreover, the acid-mediated synthesis of the bis-hydrazone 16 was accomplished by reaction of 14 with hydrazine hydrate in refluxing ethanol. Reacting precursor 14 with the previously synthesized hydrazide 8 gave hydrazone 17 clipped with two spiro chromanone motifs in 79% yield. Similarly, cyanoacetic acid hydrazide successfully was condensed with the spiro chromanone 14 under similar conditions to produce the cyanoacetyl hydrazone 18 in 85% yield (Scheme 4). The reaction of hydrazone 18 with salicylaldehyde in the presence of a catalytic amount of piperidine gave coumarinyl substituted spiro chromanone 19 in 90% yield. Moreover, the basic condensation of cyanoacetyl hydrazone 18 with benzaldehyde afforded benzylidene 20 which gave aminopyrazole 21 via reaction with hydrazine hydrate in 82% yield. All the compounds were well characterized by spectroscopic methods (ESI material 1†).
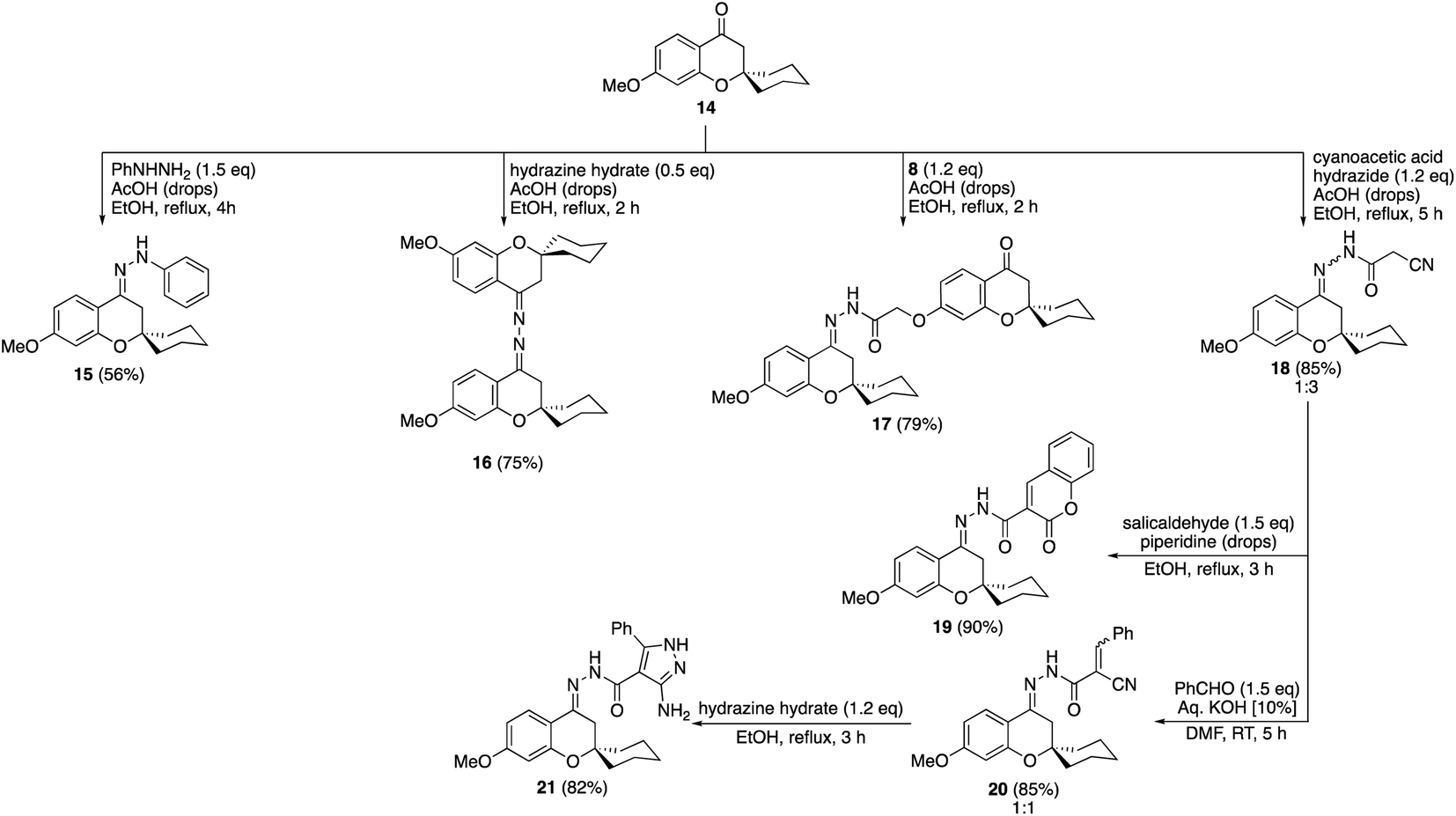 | ||
| Scheme 4 Synthesis of compounds 15–21 via condensation reactions of methyl protected spiro chromanone 14. | ||
Prediction of biological activities and physicochemical parameters
Biological activities of the synthesized derivatives were predicted using PASS online software via comparing the descriptors derived from their structures with descriptors of biologically active drugs in the database.20d–f,23 Results illustrated that some compounds might act as promising anti-infective drugs including antibacterial, antibiotic, antifungal, and antiviral activities (ESI material 2†). Other activities such as anti-inflammatory and anti-cancer in addition to membrane integrity agonists were also anticipated. Moreover, the pharmacokinetics and drug-likeness prediction were calculated using an in silico ADMET (ESI material 3†).24 Compounds 8 and 18 might have better interaction abilities as they have eight rotatable bonds and ten hydrogen bond acceptors. Most of the synthesized derivatives gave log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values < 5 suggesting good cell membrane permeabilities. The compounds with good water solubility will have a good delivery in small doses. All compounds had a bioactivity score of 0.55 indicating higher biological activities in the next steps of drug discovery. Toxicity assessment by AdmetSAR-2 online predictor suggested that the derivatives were likely have no toxicity risk.
P values < 5 suggesting good cell membrane permeabilities. The compounds with good water solubility will have a good delivery in small doses. All compounds had a bioactivity score of 0.55 indicating higher biological activities in the next steps of drug discovery. Toxicity assessment by AdmetSAR-2 online predictor suggested that the derivatives were likely have no toxicity risk.
Antimicrobial activity and toxicity
The antibacterial activities of the synthesized compounds were evaluated against Gram-positive bacteria such as Staphylococcus aureus and methicillin-resistant Staphylococcus aureus (MRSA) in addition to Gram-negative bacteria such as Escherichia coli using agar-diffusion protocol compared with ciprofloxacin as reference antibiotic.25,26 Similarly, the antifungal activity was assessed against Candida albicans compared with clotrimazole as a standard antifungal drug. The results were reported as the average diameter of inhibition zones (D) in millimeters. Compared to the inhibition zones for both standard drugs, the activity indices were calculated according to the equation: activity index (%) = (diameter of the inhibition zone of the tested compound/diameter of the inhibition zone of the reference drug) × 100. The minimal inhibitory concentrations (MICs) for the synthesized compounds that revealed inhibition zones with diameters of ≥10 mm were determined using the two-fold dilution method.27 The diameter of inhibition zones, activity indices, and MICs were summarized in Table 1.| Compound | S. aureusa | E. coli | C. albicans | IC50e (mM) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Db (mm) | AIc (%) | MICd (μM) | Db (mm) | AIc (%) | MICd (μM) | Db (mm) | AIc (%) | MICd (μM) | ||
| a Compounds 2–21 revealed weak or no activity against methicillin-resistant Staphylococcus aureus (MRSA) compared to ciprofloxacin.b D: diameter of inhibition zone.c AI: activity index (%) = (diameter of the inhibition zone of the tested compound/diameter of the inhibition zone of the reference drug) × 100.d MIC: Minimum Inhibitory Concentration.e Cytotoxicity against hepatocellular carcinoma (HePG-2), IC50 is the concentration required to inhibit 50% of cell growth, values were represented as mean ± SD of triplicate readings.f NA: no activity. | ||||||||||
| 2 | 10 | 41.7 | 96.56 | 5 | 19.2 | — | 9 | 33.3 | — | 23.86 ± 1.9 |
| 3 | NAf | — | — | NAf | — | — | 2 | 7.4 | — | 65.63 ± 3.7 |
| 4 | 18 | 75 | 11.89 | 13 | 50 | 5.95 | 19 | 70.4 | 11.89 | 7.31 ± 0.5 |
| 5 | 8 | 33.3 | — | 2 | 7.7 | — | 12 | 44.4 | 100.51 | 41.22 ± 2.8 |
| 7 | 4 | 16.7 | — | NAf | — | — | 5 | 18.5 | — | 32.74 ± 2.5 |
| 8 | 22 | 91.7 | 1.64 | 19 | 73.1 | 1.64 | 20 | 74.1 | 6.57 | 23.86 ± 1.9 |
| 9 | 16 | 66.7 | 10.80 | 11 | 42.3 | 10.80 | 15 | 55.5 | 21.60 | 8.90 ± 0.7 |
| 10 | 15 | 62.5 | 20.70 | 8 | 30.8 | — | 13 | 48.1 | 41.40 | 19.04 ± 1.5 |
| 11 | 11 | 45.8 | 40.77 | 4 | 15.4 | — | 7 | 25.9 | — | 57.73 ± 3.4 |
| 13 | NAf | — | — | NAf | — | — | NAf | — | — | 81.08 ± 4.2 |
| 15 | 15 | 62.5 | 11.89 | 10 | 38.5 | 23.78 | 14 | 51.8 | 23.78 | 10.53 ± 0.9 |
| 16 | NAf | — | — | NAf | — | — | NAf | — | — | 48.58 ± 3.0 |
| 17 | 20 | 83.3 | 1.88 | 17 | 65.4 | 1.88 | 23 | 85.2 | 3.75 | 54.67 ± 3.3 |
| 18 | 5 | 20.8 | — | 2 | 7.7 | — | 10 | 37 | 97.74 | 62.49 ± 3.5 |
| 19 | 7 | 29.2 | — | NAf | — | — | 4 | 14.8 | — | 29.15 ± 2.2 |
| 20 | 2 | 8.3 | — | NAf | — | — | 3 | 11.1 | — | 76.57 ± 3.9 |
| 21 | 19 | 79.2 | 4.49 | 14 | 53.8 | 2.24 | 18 | 66.7 | 8.98 | 14.92 ± 1.2 |
| Ciprofloxacin | 24 | 100 | 1.51 | 26 | 100 | 1.51 | NAf | — | — | — |
| Clotrimazole | NAf | — | — | NAf | — | — | 27 | 100 | 2.90 | — |
However different compounds were active against all tested microbes, MRSA revealed high antimicrobial resistance (ESI material 1†). In the case of S. aureus, hydrazide 8 showed the best antibacterial activity with 91.7% AI and 0.5 μg mL−1 MIC. Besides, azo dye 4, hydrazone 17, and aminopyrazole 21 revealed noticeable activities with MICs (1–4 μg mL−1). In the case of E. coli, hydrazide 8 was the only strong candidate with 73.1% AI and 0.5 μg mL−1 MIC. In the case of C. albicans, azo dye 4, hydrazide 8, and hydrazone 17 were the best compounds with an activity index of more than 70% and minimal inhibitory concentration of 2–4 μg mL−1. Other compounds showed low to moderate activities against S. aureus, E. coli, and C. albicans.
The presence of hydrazide functionality was essential to impart strong antimicrobial activity to compound 8 compared to less active analogues 5, 7, and 9–11 as seen in the previously published literature.28 The combination of spiro chromanone and isatin moieties was very deleterious to give the inactive compound 13. Phenylazo was more fruitful substituent than morpholinomethyl as clearly observed by comparing the antimicrobial activities of compounds 2 and 4. Symmetrical bis-compounds bearing two spiro chromanone moieties such as 3 and 16 were proven to be inactive as antimicrobial. Unsymmetrical bis-compound bearing two spiro chromanone motifs 17 showed very good antibacterial and antifungal activities. Comparing the structures of compounds 18–21 highlighted the role of aminopyrazole moiety in compound 21 to have a selective antibacterial activity for S. aureus.29
The cytotoxicity of the synthesized compounds was evaluated against hepatocellular carcinoma (HePG-2) using MTT assay. The values of IC50 which is the required concentration to suppress 50% of the cell growth were presented in Table 1. Mostly, IC50 of the compounds 4, 8, 17, and 21 were higher than their MICs against Staphylococcus aureus, Escherichia coli, and Candida albicans. This observation indicated that the antimicrobial activity is not resulted from the general toxicity, but it could be rationalized to the selective inhibitory action against the tested microbes.30 Thus, compounds 4, 8, 17, and 21 could be promising antimicrobial candidates.
Molecular docking
The drug–target interactions were investigated using molecular docking analysis in order to clarify the exact binding sites and to understand the mode of action for the best biologically active compounds.31 Penicillin-binding protein from S. aureus (PDB ID: 1VQQ)32 and DNA gyrase from E. coli (PDB ID: 5MMN)33 were used as biological targets to simulate the mechanism of antibacterial activities, while lanosterol 14-alpha demethylase (PDB ID: CYP51)34 and exo-beta-1,3-glucanase (PDB ID: 3N9K)35 from C. albicans were the fungal targets. Protein–ligand docking scores were presented in Table 2. Compounds 4, 8, 17, and 21 revealed good to high binding affinities scores against the four targets. In the case of S. aureus penicillin-binding protein (1VQQ), compounds 4, 8, 17, and 21 with binding affinities from −7.4 to −9.9 kcal mol−1 revealed polar interactions with ASP-320 and LYS-153; GLN-325; GLU-161; ASP-323 and LYS-153, respectively (Fig. 2). The binding affinity of compounds 4, 8, 17, and 21 (−7.8 to −9.0 kcal mol−1) with E. coli DNA gyrase (5MMN) was rationalized to the polar interactions with amino acid residues: ARG-90, GLY-91 and THR-179; ASP-87; THR-179 and GLY-115; THR-179 and ASN-60, respectively (Fig. 3). In addition, compounds 4, 8, 17 and 21 revealed enhanced binding affinities (−8.2 to −11.2 kcal mol−1) with lanosterol 14-alpha demethylase (5TZ1), and this was explained via polar interactions with TYR-90; ARG-339; THR-269; TYR-90 and ARG-339 residues as shown in Fig. 4. In the case of exo-beta-1,3-glucanase (3N9K), compounds 4, 8, 17, and 21 also generated polar interactions with HIS-225 and HIS-252; HIS-134, ASN-145, TYR-254 and SER-291; ASN-145 and TRP-276; ASN-145 and GLU-191, respectively, which were in agreement with the binding affinities (−8.2 to −11.6 kcal mol−1) (Fig. 5). The promising antimicrobial activities of compounds 4, 8, 17, and 21 might be rationalized to abilities to inhibit the biosynthesis of fungal sterols and cell wall branched β-1,3-D-glucans.| Compound | Binding affinity (kcal mol−1) | |||
|---|---|---|---|---|
| 1VQQ | 5MMN | 5TZ1 | 3N9K | |
| 1 | −7 | −5.4 | −7.2 | −8.4 |
| 2 | −7.5 | −6.7 | −8.2 | −7.0 |
| 3 | −9.5 | −8.7 | −8.8 | −7.5 |
| 4 | −8.4 | −8.1 | −9.4 | −8.5 |
| 5 | −6.8 | −7.6 | −7.7 | −8.7 |
| 7 | −6.9 | −7.2 | −8.0 | −9.2 |
| 8 | −7.4 | −8.0 | −8.2 | −8.2 |
| 9 | −8.7 | −9.0 | −9.0 | −10.0 |
| 10 | −8.8 | −8.5 | −9.0 | −9.9 |
| 11 | −8.8 | −7.8 | −9.1 | −9.1 |
| 12 | −9.8 | −9.0 | −10.6 | −10.5 |
| 13 | −9.8 | −8.9 | −10.7 | −10.7 |
| 14 | −6.6 | −6.6 | −7.2 | −8.5 |
| 15 | −7.7 | −8.0 | −9.1 | −9.5 |
| 16 | −8.5 | −7.4 | −10.1 | −10.2 |
| 17 | −9.9 | −9.0 | −10.7 | −11.9 |
| 18 | −7.8 | −7.5 | −8.2 | −9.6 |
| 19 | −9.5 | −8.3 | −10.7 | −11.1 |
| 20 | −8.4 | −8.1 | −10.8 | −10.1 |
| 21 | −8.8 | −7.8 | −11.2 | −11.6 |
| Ciprofloxacin | −6.9 | −7.0 | −8.0 | −8.1 |
| Clotrimazole | −7.3 | −6.1 | −8.0 | −5.7 |
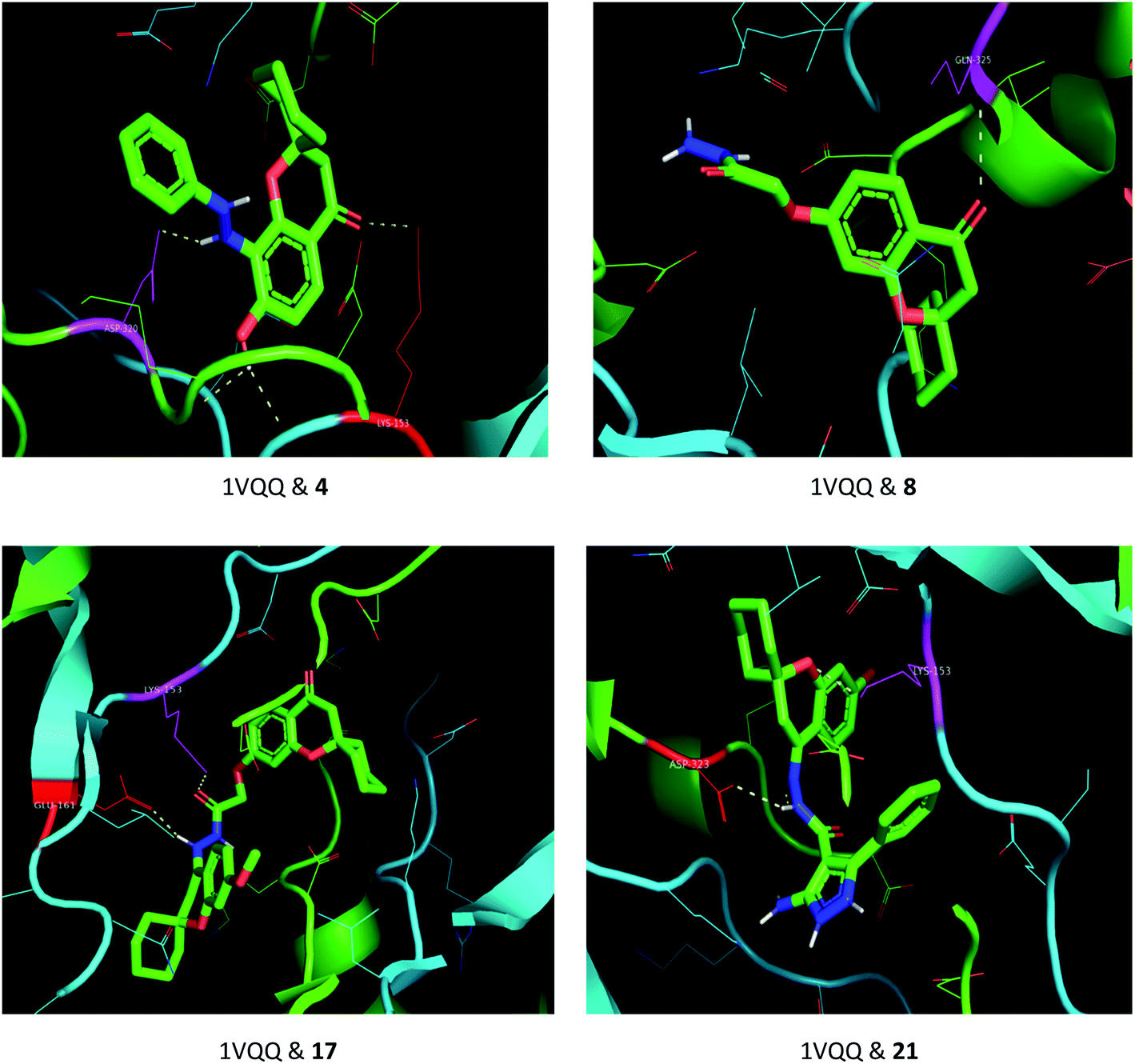 | ||
| Fig. 2 Binding modes between S. aureus penicillin-binding protein (1VQQ) and compounds 4, 8, 17 and 21. | ||
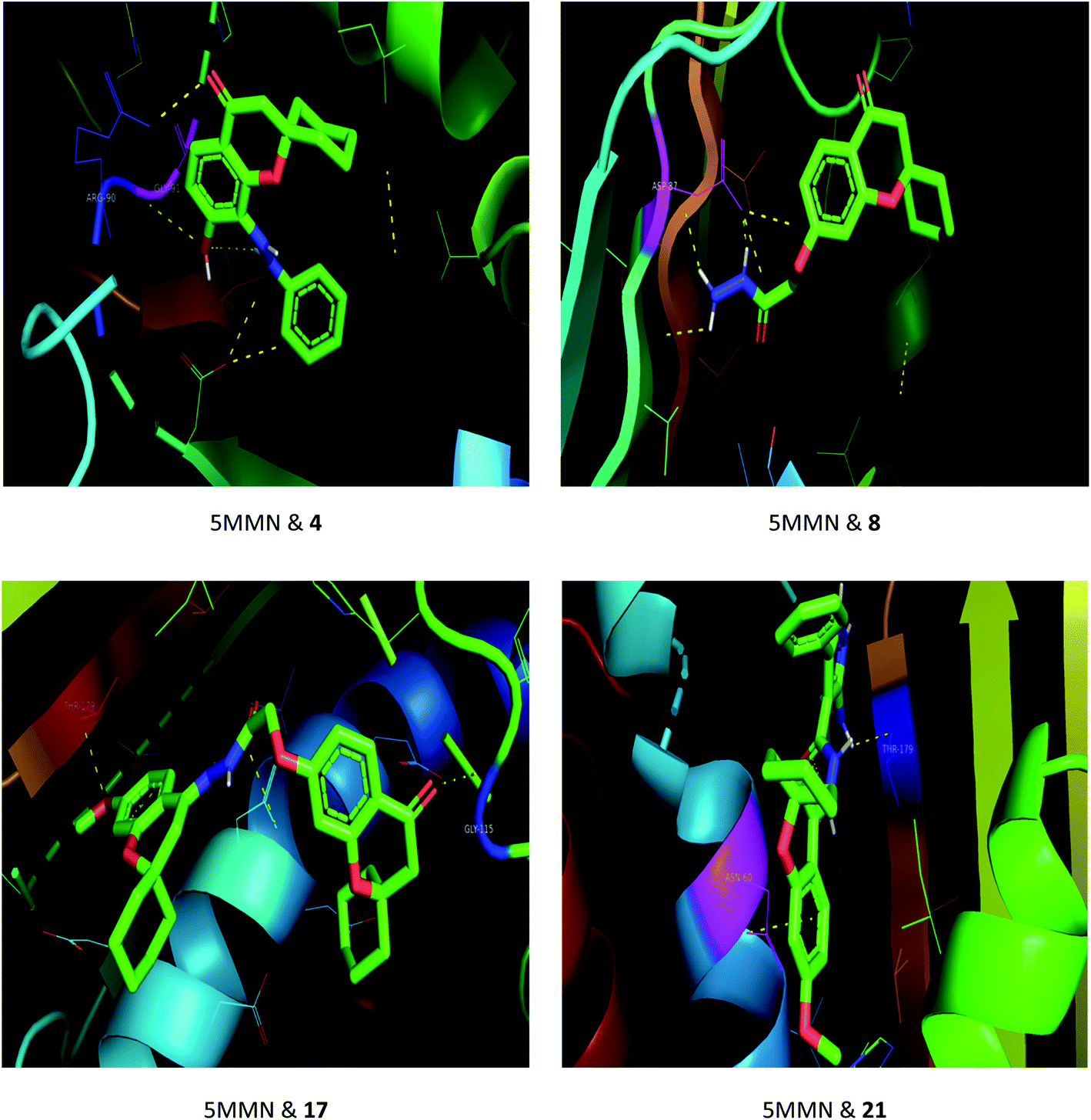 | ||
| Fig. 3 Binding modes between E. coli DNA gyrase (5MMN) and compounds 4, 8, 17 and 21. | ||
 | ||
| Fig. 4 Binding modes between lanosterol 14-alpha demethylase (5TZ1) and compounds 4, 8, 17 and 21. | ||
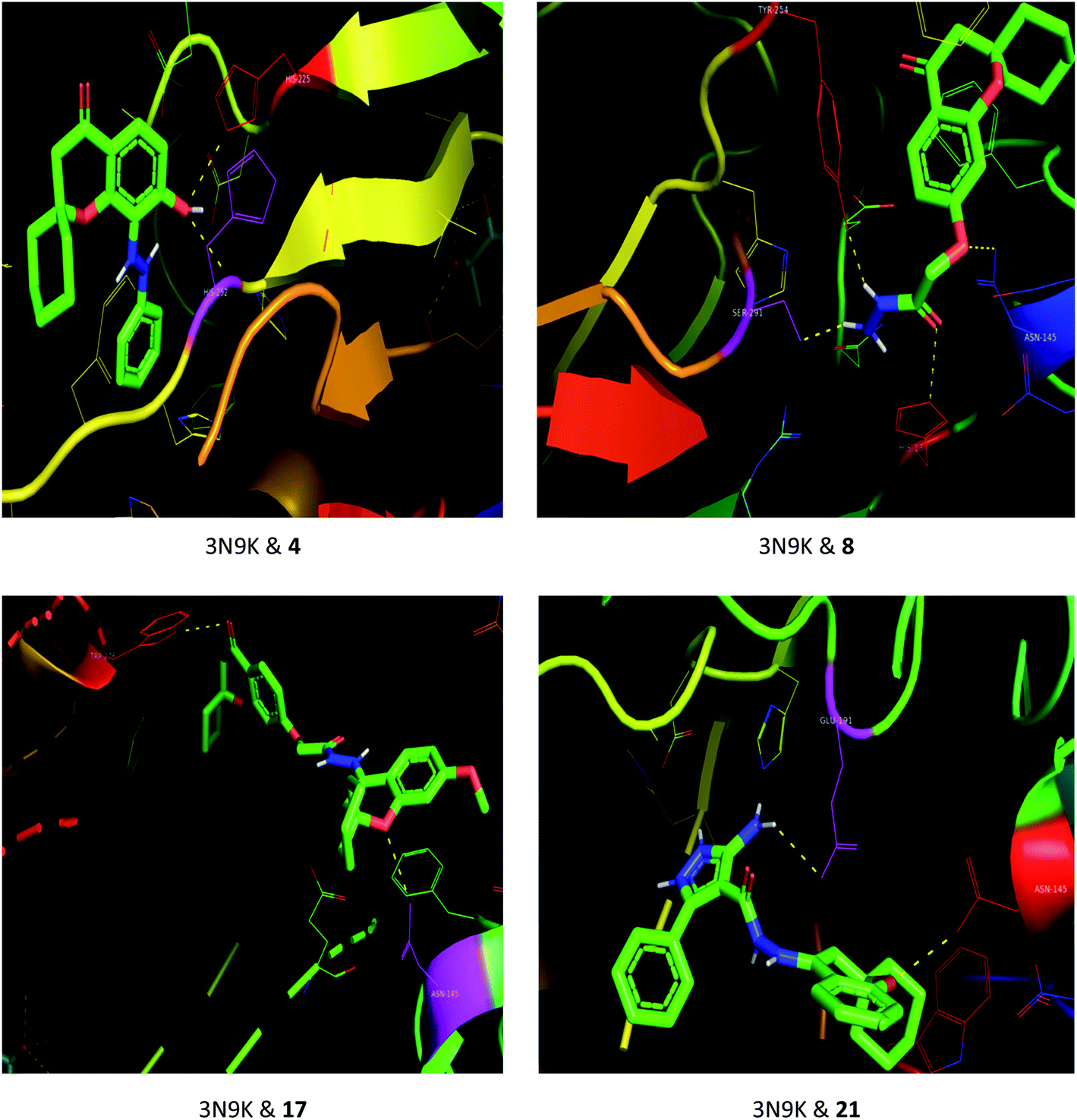 | ||
| Fig. 5 Binding modes between exo-beta-1,3-glucanase (3N9K) and compounds 4, 8, 17 and 21. | ||
Quantitative structure–activity relationship (QSAR)
The QSAR analysis36 was conducted in order to interpret the observed antimicrobial activity based on the structural characteristics of the synthesized spiro chromanone derivatives. The number of calculated descriptors through the PaDEL-Descriptor was 770.37 Then, the dataset of descriptors was imported into QSARINS.38 To mitigate redundancy in the dataset, descriptors with 95% pairwise correlations and 80% sharing the same values were deleted. The whole dataset consisting of all compounds was divided into training and test sets using a random selection of 25% of the total compounds. The selected descriptors were used as independent variables while antibacterial and antifungal activities (diameter of inhibition zone) were used as response variables.The activity trend against S. aureus was evaluated using the following QSAR equation [R2: 0.5279, R2 adj: 0.4421, R2 − R2 adj: 0.0858, RMSE tr: 5.4764, MAE tr: 4.8993, CCC tr: 0.691, s: 6.1782, F: 6.1508, Q2LOO: 0.2923 (R2 = coefficient of determination; R2 adj = adjusted coefficient of determination; RMSE tr = root mean square error for training set; CCC tr = concordance correlation coefficient for training set; s = standard error of estimate; F = Fischer's value; Q2LOO = cross-validation coefficient/Leave-One-Out)]. The activity against E. coli was evaluated using this QSAR equation [R2: 0.5449, R2 adj: 0.469, R2 − R2 adj: 0.0759, RMSE tr: 5.3517, MAE tr: 4.2506, CCC tr: 0.7054, s: 5.9834, F: 7.1839, Q2LOO: 0.354]. Both models were internally and externally validated and selected based on the best fitting (highest R2), and robustness (highest Q2LOO) values. Similarly, the activity against C. albicans was described by following equation [R2: 0.5815, R2 adj: 0.5118, R2 − R2 adj: 0.0697, RMSE tr: 5.3328, MAE tr: 4.3787, CCC tr: 0.7354, s: 5.9622, F: 8.3374, Q2LOO: 0.4029]. High values of R2 and Q2LOO for the equation illustrated good fitting and stability of the QSAR model. Internal and external predictive ability in addition to randomization of the developed models was ascertained.
The scatter plots of the experimental antibacterial and antifungal activities (exp. endpoint) versus calculated activities by QSAR models [pred. by model eq. & pred. by Leave-One-Out (LOO)] were drawn and depicted in Fig. 6. These plots showed that compound 8 had a strong agreement between the experimental antimicrobial activity and those predicted by QSAR models and lied close to ciprofloxacin and clotrimazole along the regression line. In addition, compounds 4, 17, and 21 were the nearest to ciprofloxacin and clotrimazole compared to the other compounds. Therefore, the predictive ability of the QSAR model supported the experimental antibacterial and antifungal activities for these compounds.
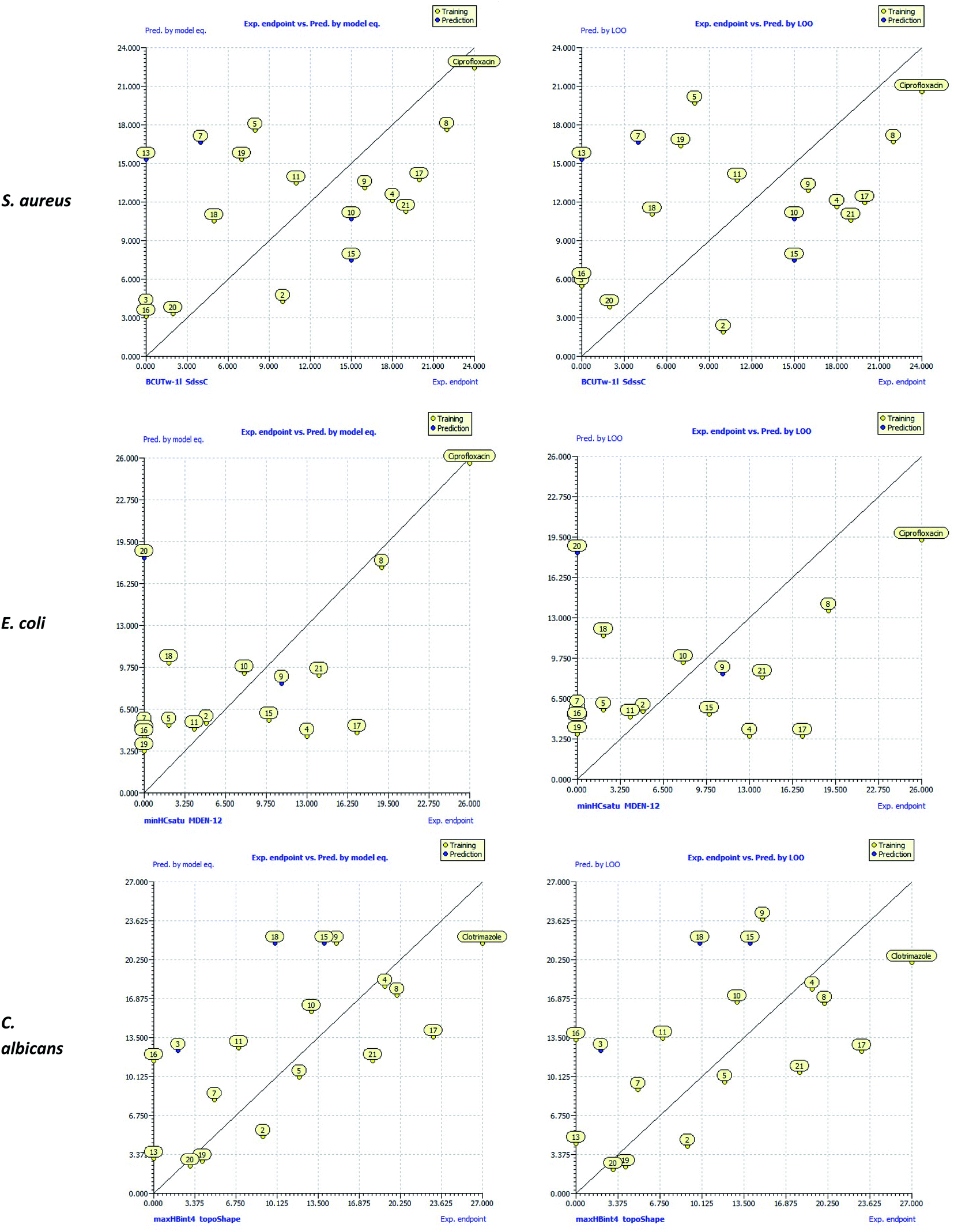 | ||
| Fig. 6 Scatter plots of the experimental antibacterial and antifungal activities (exp. endpoint) versus the calculated activities by QSAR models [pred. by model eq. & pred. by Leave-One-Out (LOO)]. | ||
Conclusion
Novel 3D-spiro chromanone derivatives such as Mannich bases (2 & 3), azo dye 4, pyrazolines (9 & 10), and hydrazones (11 & 13) were efficiently synthesized by adapting the versatile 7-hydroxyspiro[chromane-2,1′-cyclohexan]-4-one (1) as a precursor. Moreover, the condensation of methoxylspirochromanone 14 with different hydrazines and hydrazides gave the corresponding hydrazones 15–18 in yields up to 85%. The coumarinyl spiro chromanone 19 was furnished by the reaction of the hydrazone 18 with salicylaldehyde, whereas the aminopyrazole 21 was synthesized indirectly from the same precursor via the reaction with benzaldehyde followed by hydrazine. The antimicrobial evaluation against different microbes revealed the potent activity of hydrazide 8 (S. aureus: D = 22 mm, MIC = 1.64 μM; E. coli: D = 19 mm, MIC = 1.64 μM; C. albicans: D = 20 mm, MIC = 6.57 μM). Additionally, promising activities of azo dye 4 (D = 13–19 mm, MIC = 5.95–11.89 μM), hydrazone 17 (D = 17–23 mm, MIC = 1.88–3.75 μM), and aminopyrazole 21 (D = 14–19 mm, MIC = 2.24–8.98 μM) were observed. These noticeable antimicrobial activities were compatible with the good to high binding affinities (−7.4 to −11.9 kcal) towards different targets as seen in molecular docking analysis. In addition, QSAR studies highlighted the role of structural and/or physicochemical parameters of 3D-spriochromanones to predict antimicrobial activity. Further studies synthesis of more effective 3D-spiro chromanone derivatives and screening against more challenging microbes such as Methicillin-resistant Staphylococcus aureus (MRSA) in addition to enzyme inhibitory studies are currently underway and will be reported in the due time.Experimental section
General
Melting points for all new compounds were measured using a Gallenkamp apparatus. The IR spectra for all newly synthesized compounds were recorded using KBr method on a Mattson 5000 FT-IR spectrophotometer (Faculty of Pharmacy, Mansoura Uni., Egypt) or Thermo Fisher Nicolete iS 10, USA (Faculty of Science, Mansoura Uni., Egypt). The 1H- and 13C-NMR spectra were measured on Bruker Avance III 400 MHz (Faculty of Pharmacy, Mansoura Uni., Egypt), and/or JEOL ECA II 500 MHz (NMR Unit, Faculty of Science, Mansoura Uni., Egypt). The signal multiplicities were reported as: s = singlet, brs = broad singlet, d = doublet, dd = doublet of doublets, and m = multiplet.13C NMR spectra were measured on JEOL ECA II 125 MHz (NMR Unit, Faculty of Science, Mansoura Uni, Egypt). CDCl3, DMSO-d6 were used as NMR solvents, and the value of chemical shifts (δ) was in ppm unit. The EI-Mass analyses were performed on Thermo Fisher scientific DSQ II GC/MS with Kratos MS-70 ev spectrometer (Faculty of Pharmacy, Al-Azhar Uni., Egypt). The LC-MS analysis was performed on LCMS-2020-Single Quadrupole mass spectrometer from Shimadzu (Faculty of Pharmacy, Al-Azhar Uni., Egypt). High resolution mass spectrometry (HRMS) measurements were carried out using Thermo Fisher Scientific Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (National Research Centre, Egypt). Elemental analyses (C, H, and N) were performed at the microanalytical unit, Cairo Uni., Egypt. Reaction progress and product mixtures were routinely monitored by thin layer chromatography (TLC) on silica gel plates. Antibacterial and antifungal screenings were performed at the Faculty of Pharmacy, Mansoura Uni., Egypt.Synthesis
:ethyl acetate = 3:1) to give 2 as colorless crystals. Yield (0.93 g, 70%); m.p. = 130–131 °C; IR (KBr): ν/cm−1 = 3430 (OH), 2860–2939 (CHAliphatic), 1670 (CO); 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 1.23–1.71 (m, 10H, (CH2)5), 2.57 (s, 2H, CH2), 2.41–2.51 (m, 4H, N(CH2)2), 3.48–3.57 (m, 4H, O(CH2)2), 3.62 (s, 2H, CH2), 6.42 (d, 1H, Ar–H, J = 8.80 Hz), 7.48 (d, 1H, Ar–H, J = 8.80 Hz), 9.72 (brs, 1H, OH); MS (EI, 70 eV): m/z (%) = 331 [M+, 27.7%], 298 [100.0%], 248 [89.7%], 229 [75.8%], 206 [57.4%], 180 [69.7%], 99 [67.4%]; anal. calcd for C19H25NO4 (331.18): C, 68.86; H, 7.60; N, 4.23%. Found: C, 68.92; H, 7.51; N, 4.29%.:ethyl acetate = 3:1) to give 5 as a colorless powder. Yield (0.45 g, 71%); m.p. 73–74 °C; IR (KBr): ν/cm−1 = 2863–2942 (CHAliphatic), 1764 (CO), 1669 (CO); 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 0.98 (t, 3H, CH3, J = 6.80 Hz), 1.03–1.62 (m, 10H, (CH2)5), 3.11 (s, 2H, CH2), 3.94 (q, 2H, CH2, J = 6.80 Hz), 4.63 (s, 2H, CH2), 6.27–6.37 (m, 2H, Ar–H), 7.41 (d, 1H, Ar–H, J = 8.50 Hz); 13C NMR (CDCl3, 125 MHz) δ (ppm): 14.1, 21.4, 25.1, 34.8, 47.8, 61.6, 65.2, 80.5, 102.3, 109.0, 115.4, 128.4, 161.4, 164.1, 168.1, 191.1; MS (EI, 70 eV): m/z (%) = 318 [M+, 6.1%], 297 [46.1%], 287 [100.0%], 240 [58.1%], 187 [88.1%], 160 [37.3%], 123 [63.1%], 46 [82.1%]; LCMS: m/z: 319 [(M + H)+]; HRMS (ESI): m/z calcd for C18H23O5 [(M + H)+] 319.1540, found 319.1528; anal. calcd for C18H22O5 (318.15): C, 67.91; H, 6.97%. Found: C, 67.85; H, 6.99%.:ethyl acetate 3:1) to yield 7 as a colorless powder. Yield (0.82 g, 94%), m.p. 140–141 °C; IR (KBr): ν/cm−1 = 3420–3217 (COOH), 2866–2938 (CHAliphatic), 1767 (CO), 1641 (CO); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.46–1.82 (m, 10H, (CH2)5), 2.67 (s, 2H, CH2), 4.76 (s, 2H, CH2), 6.46 (d, 1H, Ar–H, J = 3.0 Hz), 6.57 (dd, 1H, Ar–H, J = 2.00, 8.00 Hz), 7.62 (d, 1H, Ar–H, J = 8.00 Hz), 13.14 (brs, 1H, COOH); 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 21.0, 24.6, 33.9, 47.0, 64.7, 80.2, 102.1, 109.3, 114.5, 127.5, 160.8, 164.2, 169.6, 190.3; MS (EI, 70 eV): m/z (%) = 290 [M+, 10.6%], 143 [42.8%], 115 [87.3%], 91 [28.7%], 76 [53.2%], 43 [100.0%]; LCMS: m/z: 291 [(M + H)+]; HRMS (ESI): m/z calcd for C17H19O5 [(M + H)+] 291.1227, found 291.1215; anal. calcd for C16H18O5 (290.12): C, 66.20; H, 6.25%. Found: C, 66.02; H, 6.48%.:ethyl acetate 3:1) to give 9 as colorless crystals. Yield (0.44 g, 59%); m.p. 288 °C; IR (KBr): ν/cm−1 = 2858–2931 (CHAliphatic), 1689 (CO), 1665 (CO); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.41–1.81 (m, 10H, (CH2)5), 1.93 (s, 3H, CH3), 2.64 (s, 2H, CH2), 3.54 (s, 2H, CH2), 4.52 (s, 2H, CH2), 6.60 (d, 1H, Ar–H, J = 2.50 Hz), 6.72 (dd, 1H, Ar–H, J = 2.50, 8.50 Hz), 7.62 (d, 1H, Ar–H, J = 8.50 Hz); MS (EI, 70 eV): m/z (%) = 370 [M+, 23.4%], 352 [31.7%], 207 [40.8%], 161 [60.4%], 94 [80.7%], 59 [100.0%], 46 [56.6%]; anal. calcd for C20H22N2O5 (370.15): C, 64.85; H, 5.99; N, 7.56%. Found: C, 64.74; H, 6.04; N, 7.61%.:ethyl acetate 3:1) yielding 10 as colorless crystals. Yield (0.57 g, 74%); m.p. 222–223 °C; IR (KBr): ν/cm−1 = 3446–3351 (OH), 2859–2947 (CHAliphatic), 1672 (CO), 1655 (CO); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.45–1.57 and 1.76–1.84 (m, 10H, (CH2)5), 1.73 (s, 3H, CH3), 1.98 (s, 3H, CH3), 2.65 (s, 2H, CH2), 2.79 (d, 1H, CH, J = 18.50 Hz), 2.91 (d, 1H, CH, J = 18.50 Hz), 4.96 (s, 2H, CH2), 6.38 (d, 1H, Ar–H, J = 2.50 Hz), 6.54 (dd, 1H, Ar–H, J = 3.00, 9.00 Hz), 7.61 (d, 1H, Ar–H, J = 9.00 Hz); MS (EI, 70 eV): m/z (%) = 386 [M+, 16.1%], 368 [58.6%], 292 [49.2%], 228 [28.5%], 167 [41.2%], 85 [100.0%], 45 [32.6%]; anal. calcd for C21H26N2O5 (386.18): C, 65.27; H, 6.78; N, 7.25%. Found: C, 65.37; H, 6.61; N, 7.32%.:2). Yield (0.62 g, 79%); m.p. 128–129 °C; IR (KBr): ν/cm−1 = 3450 (NH), 2854–2928 (CHAliphatic), 1678 (CO), 1639 (CO); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.43–1.85 (m, 10H, (CH2)5, major and minor), 2.66 (s, 2H, CH2, major isomer), 2.68 (s, 2H, CH2, minor isomer), 4.75 (s, 2H, CH2, minor isomer), 5.24 (s, 2H, CH2, major isomer), 6.48 (d, 1H, Ar–H, J = 2.50 Hz, major isomer), 6.56 (d, 1H, Ar–H, J = 2.00 Hz, minor isomer), 6.60 (dd, 1H, Ar–H, J = 2.50, 8.50 Hz, major isomer), 6.66 (dd, 1H, Ar–H, J = 2.50, 9.00 Hz, minor isomer), 7.4–7.46 (m, 5H, Ar–H, E and Z), 7.63 (d, 1H, Ar–H, J = 9.00 Hz, major isomer), 7.66 (d, 1H, Ar–H, J = 7.00 Hz, minor isomer), 7.68–7.72 (m, 3H, Ar–H, major and minor), 8.00 (s, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, major isomer), 8.30 (s, 1H, CHN, minor isomer), 11.59 (brs, 1H, NH, minor isomer), 11.66 (brs, 1H, NH, major isomer); 13C NMR (CDCl3, 125 MHz) δ (ppm): 21.4, 21.5, 25.06, 25.12, 34.7, 34.8, 47.7, 47.8, 65.3, 67.0, 80.5, 80.8, 102.4, 102.5, 109.0, 109.3, 115.3, 115.9, 127.3, 127.9, 128.3, 128.69, 128.72, 128.8, 130.7, 130.9, 133.0, 133.1, 145.8, 149.8, 161.4, 161.6, 163.0, 163.5, 164.7, 169.6, 191.2, 191.3; MS (EI, 70 eV): m/z (%) = 392 [M+, 58.2%], 361 [85.3%], 356 [100.0%], 328 [83.6%], 183 [62.8%], 134 [44.8%]; LCMS: m/z: 393 [(M + H)+]; HRMS (ESI): m/z calcd for C23H25N2O4 [(M + H)+] 393.1809, found 393.1796; anal. calcd for C23H24N2O4 (392.17): C, 70.39; H, 6.16; N, 7.14%. Found: C, 70.28; H, 6.22; N, 7.24%.
N, major isomer), 8.30 (s, 1H, CHN, minor isomer), 11.59 (brs, 1H, NH, minor isomer), 11.66 (brs, 1H, NH, major isomer); 13C NMR (CDCl3, 125 MHz) δ (ppm): 21.4, 21.5, 25.06, 25.12, 34.7, 34.8, 47.7, 47.8, 65.3, 67.0, 80.5, 80.8, 102.4, 102.5, 109.0, 109.3, 115.3, 115.9, 127.3, 127.9, 128.3, 128.69, 128.72, 128.8, 130.7, 130.9, 133.0, 133.1, 145.8, 149.8, 161.4, 161.6, 163.0, 163.5, 164.7, 169.6, 191.2, 191.3; MS (EI, 70 eV): m/z (%) = 392 [M+, 58.2%], 361 [85.3%], 356 [100.0%], 328 [83.6%], 183 [62.8%], 134 [44.8%]; LCMS: m/z: 393 [(M + H)+]; HRMS (ESI): m/z calcd for C23H25N2O4 [(M + H)+] 393.1809, found 393.1796; anal. calcd for C23H24N2O4 (392.17): C, 70.39; H, 6.16; N, 7.14%. Found: C, 70.28; H, 6.22; N, 7.24%.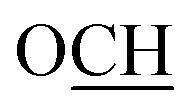 C(OH)NH), 5.40 (brs, 1H, OH or NH), 6.64–6.69 (m, 2H, Ar–H), 6.94 (d, 1H, Ar–H, J = 7.50 Hz), 7.08–7.11 (m, 1H, Ar–H), 7.37–7.40 (m, 1H, Ar–H), 7.56–7.68 (m, 2H, Ar–H), 11.27 (brs, 1H, OH or NH), 13.66 (brs, 1H, OH or NH); 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 21.0, 24.6, 33.9, 47.0, 67.0, 80.3, 102.6, 109.3, 111.2, 115.0, 119.6, 121.0, 122.7, 127.8, 132.0, 138.7, 142.6, 160.8, 162.5, 190.3; MS (EI, 70 eV): m/z (%) = 433 [M+, 17.7], 369 [24.7%], 330 [53.7%], 300 [59.3%], 264 [66.5%], 219 [79.3%], 133 [72.3%], 83 [52.6], 42 [100.0%]; LCMS: m/z: 434 [(M + H)+]; HRMS (ESI): m/z calcd for C24H24N3O5 [(M + H)+] 434.1711, found 434.1691; anal. calcd for C24H23N3O5 (433.16): C, 66.50; H, 5.35; N, 9.69%. Found: C, 66.61; H, 5.23; N, 9.78%.N); 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 1.46–1.81 (m, 10H, (CH2)5), 2.74 (s, 2H, CH2), 3.75 (s, 3H, OCH3), 6.42 (d, 1H, Ar–H, J = 2.00 Hz), 6.55 (dd, 1H, Ar–H, J = 2.40, 8.40 Hz), 6.73 (m, 1H, Ar–H), 7.2–7.21 (m, 4H, Ar), 7.86 (d, 1H, Ar–H, J = 8.80 Hz), 9.25 (brs, 1H, NH); MS (EI, 70 eV): m/z (%) = 336 [M+, 11.3], 334 [36.8%], 300 [75.1%], 260 [54.5%], 217 [51.6%], 155 [100.0%], 153 [64.1%], 119 [53.2%], 112 [39.7%], 99 [68.2%], 64 [56.6%]; anal. calcd for C21H24N2O2 (336.18): C, 74.97; H, 7.19; N, 8.33%. Found: C, 74.85; H, 7.29; N, 8.22%.N); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.41–1.78 (m, 20H, two (CH2)5), 2.95 (s, 2H, 2 CH2), 3.76 (s, 3H, 2 OCH3), 6.45 (d, 2H, Ar–H, J = 2.00 Hz), 6.56 (dd, 2H, Ar–H, J = 3.00, 9.00 Hz), 7.93 (d, 2H, Ar–H, J = 8.50 Hz); 13C NMR (CDCl3, 125 MHz) δ (ppm): 21.7, 25.5, 35.0, 35.9, 55.4, 77.4, 101.5, 108.6, 113.8, 126.2, 155.2, 157.1, 162.9; MS (EI, 70 eV): m/z (%) = 488 [M+, 31.6%], 478 [66.8%], 430 [71.4%], 396 [48.2%], 332 [100.0%], 250 [62.4%], 202 [59.8%], 158 [62.7%]; LCMS: m/z: 489 [(M + H)+]; HRMS (ESI): m/z calcd for C30H36N2O4 [(M + H)+] 489.2748, found 489.2759; anal. calcd for C30H36N2O4 (488.27): C, 73.74; H, 7.43; N, 5.73%. Found: C, 73.59; H, 7.60; N, 5.84%.N); (DMSO-d6, 400 MHz) δ (ppm): 1.21–1.84 (m, 20H, two (CH2)5), 2.84 (s, 2H, CH2), 2.96 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 4.71 (s, 2H, CH2), 6.48 (d, 1H, Ar–H, J = 2.00 Hz), 6.65–7.62 (m, 4H, Ar–H), 7.92 (d, 1H, Ar–H, J = 8.50 Hz), 13.34 (brs, 1H, NH); MS (EI, 70 eV): m/z (%) = 532 [M+, 9.5%], 483 [34.7%], 393 [69.3%], 350 [27.6%], 347 [100.0%], 302 [34.5%], 267 [60.1%], 222 [29.3%], 124 [21.2%]; anal. calcd for C31H36N2O6 (532.26): C, 69.91; H, 6.81; N, 5.26%. Found: C, 70.12; H, 6.89; N, 4.99%.:3). Yield (1.39 g, 85%); m.p. 256–257 °C; IR (KBr): ν/cm−1 = 3192 (NH), 2855–2927 (CHAliphatic), 2260 (C
C(OH)NH), 5.40 (brs, 1H, OH or NH), 6.64–6.69 (m, 2H, Ar–H), 6.94 (d, 1H, Ar–H, J = 7.50 Hz), 7.08–7.11 (m, 1H, Ar–H), 7.37–7.40 (m, 1H, Ar–H), 7.56–7.68 (m, 2H, Ar–H), 11.27 (brs, 1H, OH or NH), 13.66 (brs, 1H, OH or NH); 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 21.0, 24.6, 33.9, 47.0, 67.0, 80.3, 102.6, 109.3, 111.2, 115.0, 119.6, 121.0, 122.7, 127.8, 132.0, 138.7, 142.6, 160.8, 162.5, 190.3; MS (EI, 70 eV): m/z (%) = 433 [M+, 17.7], 369 [24.7%], 330 [53.7%], 300 [59.3%], 264 [66.5%], 219 [79.3%], 133 [72.3%], 83 [52.6], 42 [100.0%]; LCMS: m/z: 434 [(M + H)+]; HRMS (ESI): m/z calcd for C24H24N3O5 [(M + H)+] 434.1711, found 434.1691; anal. calcd for C24H23N3O5 (433.16): C, 66.50; H, 5.35; N, 9.69%. Found: C, 66.61; H, 5.23; N, 9.78%.N); 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 1.46–1.81 (m, 10H, (CH2)5), 2.74 (s, 2H, CH2), 3.75 (s, 3H, OCH3), 6.42 (d, 1H, Ar–H, J = 2.00 Hz), 6.55 (dd, 1H, Ar–H, J = 2.40, 8.40 Hz), 6.73 (m, 1H, Ar–H), 7.2–7.21 (m, 4H, Ar), 7.86 (d, 1H, Ar–H, J = 8.80 Hz), 9.25 (brs, 1H, NH); MS (EI, 70 eV): m/z (%) = 336 [M+, 11.3], 334 [36.8%], 300 [75.1%], 260 [54.5%], 217 [51.6%], 155 [100.0%], 153 [64.1%], 119 [53.2%], 112 [39.7%], 99 [68.2%], 64 [56.6%]; anal. calcd for C21H24N2O2 (336.18): C, 74.97; H, 7.19; N, 8.33%. Found: C, 74.85; H, 7.29; N, 8.22%.N); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.41–1.78 (m, 20H, two (CH2)5), 2.95 (s, 2H, 2 CH2), 3.76 (s, 3H, 2 OCH3), 6.45 (d, 2H, Ar–H, J = 2.00 Hz), 6.56 (dd, 2H, Ar–H, J = 3.00, 9.00 Hz), 7.93 (d, 2H, Ar–H, J = 8.50 Hz); 13C NMR (CDCl3, 125 MHz) δ (ppm): 21.7, 25.5, 35.0, 35.9, 55.4, 77.4, 101.5, 108.6, 113.8, 126.2, 155.2, 157.1, 162.9; MS (EI, 70 eV): m/z (%) = 488 [M+, 31.6%], 478 [66.8%], 430 [71.4%], 396 [48.2%], 332 [100.0%], 250 [62.4%], 202 [59.8%], 158 [62.7%]; LCMS: m/z: 489 [(M + H)+]; HRMS (ESI): m/z calcd for C30H36N2O4 [(M + H)+] 489.2748, found 489.2759; anal. calcd for C30H36N2O4 (488.27): C, 73.74; H, 7.43; N, 5.73%. Found: C, 73.59; H, 7.60; N, 5.84%.N); (DMSO-d6, 400 MHz) δ (ppm): 1.21–1.84 (m, 20H, two (CH2)5), 2.84 (s, 2H, CH2), 2.96 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 4.71 (s, 2H, CH2), 6.48 (d, 1H, Ar–H, J = 2.00 Hz), 6.65–7.62 (m, 4H, Ar–H), 7.92 (d, 1H, Ar–H, J = 8.50 Hz), 13.34 (brs, 1H, NH); MS (EI, 70 eV): m/z (%) = 532 [M+, 9.5%], 483 [34.7%], 393 [69.3%], 350 [27.6%], 347 [100.0%], 302 [34.5%], 267 [60.1%], 222 [29.3%], 124 [21.2%]; anal. calcd for C31H36N2O6 (532.26): C, 69.91; H, 6.81; N, 5.26%. Found: C, 70.12; H, 6.89; N, 4.99%.:3). Yield (1.39 g, 85%); m.p. 256–257 °C; IR (KBr): ν/cm−1 = 3192 (NH), 2855–2927 (CHAliphatic), 2260 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) 1675 (CO); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.35–1.77 (m, 10H, (CH2)5, major and minor), 2.70 (s, 2H, CH2, minor isomer), 2.76 (s, 2H, CH2, major isomer), 3.73 (s, 3H, OCH3, major isomer), 3.74 (s, 3H, OCH3, minor isomer), 3.83 (s, 2H, CH2, minor isomer), 4.21 (s, 2H, CH2, major isomer), 6.40 (d, 1H, Ar–H, J = 2.50 Hz, major isomer), 6.42 (d, 1H, Ar–H, J = 2.50 Hz, minor isomer), 6.52 (dd, 1H, Ar–H, J = 2.00, 8.50 Hz, major isomer), 6.55 (dd, 1H, Ar–H, J = 2.50, 9.00 Hz, minor isomer), 7.77 (d, 1H, Ar–H, J = 8 Hz, major isomer), 7.81 (d, 1H, Ar–H, J = 9 Hz, minor isomer), 10.65 (brs, 1H, NH, major isomer), 11.01 (brs, 1H, NH, minor isomer); 13C NMR (DMSO-d6, 125 MHz): 21.08, 21.15, 24.7, 24.8, 24.9, 33.9, 34.3, 34.4, 35.0, 40.0, 55.3, 76.5, 76.7, 101.4, 101.6, 108.6, 112.4, 116.1, 116.3, 125.5, 125.6, 143.3, 147.7, 155.9, 156.0, 159.0, 162.1, 162.4, 165.5; MS (EI, 70 eV): m/z (%) = 327 [M+, 17.1%], 304 [46.4%], 295 [95.8%], 221 [40.9%], 220 [100.0%], 180 [30.7%], 151 [20.7%], 101 [26.1%], 78 [43.6]; LCMS: m/z: 328 [(M + H)+]; HRMS (ESI): m/z calcd for C18H22N3O3 [(M + H)+] 328.1656, found 328.1644; anal. calcd for C18H21N3O3 (327.16): C, 66.04; H, 6.47; N, 12.84%. Found: C, 66.21; H, 6.31; N, 13.01%.:1). Yield (1.42 g, 85%); m.p. 162–163 °C; IR (KBr): ν/cm−1 = 3435 (NH), 2855–2928 (CHAliphatic), 2213 (CN), 1657 (CO), 1602 (CN); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.46–1.80 (m, 10H, (CH2)5, E and Z), 2.78 (s, 2H, CH2, E or Z), 2.85 (s, 2H, CH2, E or Z), 3.73 (s, 3H, OCH3, E or Z), 3.76 (s, 3H, OCH3, E or Z), 6.38–6.58 (m, 2H, Ar–H, E and Z), 7.59–7.61 (m, 3H, Ar–H, E and Z), 7.83–8.02 (m, 3H, Ar–H, E and Z), 8.18 (s, 1H, CH = CCO, E and Z), 10.88 (brs, 1H, NH, E or Z), 11.28 (brs, 1H, NH, E or Z); MS (EI, 70 eV): m/z (%) = 415 [M+, 19.5%], 387 [100.0%], 322 [53.2%], 309 [58.4%], 282 [57.7%], 210 [45.5%], 119 [42.6%], 69 [52.8%]; anal. calcd for C25H25N3O3 (415.19): C, 72.27; H, 6.07; N, 10.11%. Found: C, 72.18; H, 6.15; N, 10.13%.N); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.23–1.78 (m, 10H, (CH2)5), 2.69 (s, 2H, CH2), 3.78 (s, 3H, OCH3), 6.03 (brs, 2H, NH2), 6.26–6.32 (m, 2H, Ar–H), 6.54–6.59 (m, 1H, Ar–H), 7.49–7.78 (m, 5H, Ar–H), 11.07 (brs, 1H, NH), 11.72 (brs, 1H, NH); MS (EI, 70 eV): m/z (%) = 445 [M+, 26.4%], 330 [78.7%], 235 [26.2%], 133 [39.9%], 90 [62.1%], 77 [100.0%], 63 [86.1%], 50 [38.5%]; anal. calcd for C25H27N5O3 (445.21): C, 67.40; H, 6.11; N, 15.72%. Found: C, 67.48; H, 6.18; N, 15.64%.
N) 1675 (CO); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.35–1.77 (m, 10H, (CH2)5, major and minor), 2.70 (s, 2H, CH2, minor isomer), 2.76 (s, 2H, CH2, major isomer), 3.73 (s, 3H, OCH3, major isomer), 3.74 (s, 3H, OCH3, minor isomer), 3.83 (s, 2H, CH2, minor isomer), 4.21 (s, 2H, CH2, major isomer), 6.40 (d, 1H, Ar–H, J = 2.50 Hz, major isomer), 6.42 (d, 1H, Ar–H, J = 2.50 Hz, minor isomer), 6.52 (dd, 1H, Ar–H, J = 2.00, 8.50 Hz, major isomer), 6.55 (dd, 1H, Ar–H, J = 2.50, 9.00 Hz, minor isomer), 7.77 (d, 1H, Ar–H, J = 8 Hz, major isomer), 7.81 (d, 1H, Ar–H, J = 9 Hz, minor isomer), 10.65 (brs, 1H, NH, major isomer), 11.01 (brs, 1H, NH, minor isomer); 13C NMR (DMSO-d6, 125 MHz): 21.08, 21.15, 24.7, 24.8, 24.9, 33.9, 34.3, 34.4, 35.0, 40.0, 55.3, 76.5, 76.7, 101.4, 101.6, 108.6, 112.4, 116.1, 116.3, 125.5, 125.6, 143.3, 147.7, 155.9, 156.0, 159.0, 162.1, 162.4, 165.5; MS (EI, 70 eV): m/z (%) = 327 [M+, 17.1%], 304 [46.4%], 295 [95.8%], 221 [40.9%], 220 [100.0%], 180 [30.7%], 151 [20.7%], 101 [26.1%], 78 [43.6]; LCMS: m/z: 328 [(M + H)+]; HRMS (ESI): m/z calcd for C18H22N3O3 [(M + H)+] 328.1656, found 328.1644; anal. calcd for C18H21N3O3 (327.16): C, 66.04; H, 6.47; N, 12.84%. Found: C, 66.21; H, 6.31; N, 13.01%.:1). Yield (1.42 g, 85%); m.p. 162–163 °C; IR (KBr): ν/cm−1 = 3435 (NH), 2855–2928 (CHAliphatic), 2213 (CN), 1657 (CO), 1602 (CN); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.46–1.80 (m, 10H, (CH2)5, E and Z), 2.78 (s, 2H, CH2, E or Z), 2.85 (s, 2H, CH2, E or Z), 3.73 (s, 3H, OCH3, E or Z), 3.76 (s, 3H, OCH3, E or Z), 6.38–6.58 (m, 2H, Ar–H, E and Z), 7.59–7.61 (m, 3H, Ar–H, E and Z), 7.83–8.02 (m, 3H, Ar–H, E and Z), 8.18 (s, 1H, CH = CCO, E and Z), 10.88 (brs, 1H, NH, E or Z), 11.28 (brs, 1H, NH, E or Z); MS (EI, 70 eV): m/z (%) = 415 [M+, 19.5%], 387 [100.0%], 322 [53.2%], 309 [58.4%], 282 [57.7%], 210 [45.5%], 119 [42.6%], 69 [52.8%]; anal. calcd for C25H25N3O3 (415.19): C, 72.27; H, 6.07; N, 10.11%. Found: C, 72.18; H, 6.15; N, 10.13%.N); 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.23–1.78 (m, 10H, (CH2)5), 2.69 (s, 2H, CH2), 3.78 (s, 3H, OCH3), 6.03 (brs, 2H, NH2), 6.26–6.32 (m, 2H, Ar–H), 6.54–6.59 (m, 1H, Ar–H), 7.49–7.78 (m, 5H, Ar–H), 11.07 (brs, 1H, NH), 11.72 (brs, 1H, NH); MS (EI, 70 eV): m/z (%) = 445 [M+, 26.4%], 330 [78.7%], 235 [26.2%], 133 [39.9%], 90 [62.1%], 77 [100.0%], 63 [86.1%], 50 [38.5%]; anal. calcd for C25H27N5O3 (445.21): C, 67.40; H, 6.11; N, 15.72%. Found: C, 67.48; H, 6.18; N, 15.64%.Antimicrobial evaluation
The antimicrobial activity of the newly synthesized compounds was evaluated against Staphylococcus aureus (ATCC® 6535), Escherichia coli (ATCC® 8739), and Candida albicans (ATCC® 10231). In addition, methicillin-resistant Staphylococcus aureus (MRSA) was local isolates from the patients of Mansoura University Hospitals, Egypt. The samples were dissolved in dimethyl sulfoxide (DMSO) and solutions of 1 mg mL−1 concentrations were prepared separately. Whatman filter paper discs with a size of 5 mm were cut and then sterilized in the autoclave. After that, the discs were soaked in solutions of the synthesized compounds. To Petri dishes containing agar media [agar (20 g) + beef extract (3 g) + peptone (5 g)] seeded with Staphylococcus aureus, Escherichia coli, and Candida albicans, the previously soaked discs were placed. After incubation of the Petri dishes at 36 °C for 24 h, the inhibition done diameters were measured in millimeter (mm). For each compound, the zone diameter was calculated as the average of three trials. The antimicrobial activities of ciprofloxacin and clotrimazole was assessed as reference drugs under the same conditions. Control experiments using DMSO under the same conditions were run parallel to samples of the synthesized compounds.39 The activity indices were calculated using the following equation:
The minimal inhibitory concentrations (MICs) for compounds with inhibitions zones of ≥10 mm diameters were determined at concentration of 64 μg mL−1. The two-fold dilutions of the solutions were done (64, 32, 16, …, 0.5 μg mL−1). The suspensions of different microbes at concentrations of 106 (CFU mL−1, colony forming unit per mL) were added to 96-well microliter plate followed by incubation at 36 °C. After incubation for 24 h, MICs were determined as the lowest concentrations that inhibited visible growth of the microorganisms (turbidity).40,41
Evaluation of cytotoxicity42
The cytotoxicity was tested against hepatocellular carcinoma (HePG-2) cell line (ATCC® via VACSERA company for biological products, Cairo, Egypt). The reagents 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), RPMI-1640 medium, and dimethyl sulfoxide (DMSO) were purchased from Sigma Chemical Company of St. Louis, USA. Fetal bovine serum was purchased from GIBCO, UK. MTT assay depends upon the conversion of a yellow 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) into a purple formazan by the mitochondrial succinate dehydrogenase in viable cells. HePG-2 cell line was cultured in RPMI-1640 medium containing 10% fetal bovine serum and 100 units per mL 1% antibiotic penicillin/streptomycin at 37 °C in a 5% carbon dioxide incubator. After seeding HePG-2 cell line in a 96-well plate (density: 1.0 × 104 cells per well) at 37 °C FOR 48 h under 5% carbon dioxide, the cells were treated with different concentration of the synthesized compounds 2–21 and followed by an incubation period for 24 h. After that, 20 μL of pre-prepared MTT soln (5 mg mL−1) was added onto each well followed by a second incubation for 4 h. In order to dissolve the generated purple formazan, 100 μL DMSO was added to each well. The spectrophotometric absorbance for each well was measured at 570 nm using microplate reader (ELx800, USA). All tests were carried out three time. The percentage of cell viability was calculated as [A570nm of treated sample/A570nm of untreated sample × 100]. After plotting the relationship between viable cell percentage and compound concentration, the survival curve for HePG-2 was obtained. The IC50 value which is the required concentration to inhibit a 50% of cell growth was calculated by the nonlinear regression analysis by using Graph pad prism software and shown as mean ± standard deviation (SD).Computational evaluation
Molecular docking
To predict the antimicrobial mechanisms and the most suitable targets for the synthesized compounds, two antibacterial and two antifungal targets related to antimicrobial activities were screened. The crystal structures S. aureus penicillin-binding protein (PDB ID: 1VQQ), E. coli DNA gyrase (PDB ID: 5MMN), C. albicans lanosterol 14-alpha demethylase (PDB ID: 5TZ1), and exo-beta-1,3-glucanase (PDB ID: 3N9K) were selected and fetched from Protein Data Bank (www.rcsb.org/pdb). AutoDock 4.2 molecular dockings were performed using PYRX software to predict the binding pose with active sites of the protein targets.43 Water molecules were removed, and hydrogen atoms were added before docking to obtain the correct ionization and tautomeric states for the amino acids. Binding energies of protein–ligand interactions were recorded, and molecular docking was visualized using PYMOL software (version 2.2.3).Quantitative structure–activity relationship (QSAR)
The dataset containing synthesized compounds was used for making QSAR models. The 2D-structures of compounds were made with MavinSketch and converted into optimized MDL*.mol using same tool. The properties were calculated PaDEL-Descriptor (29).37 QSARINS v2.0 was used for creating QSAR models with genetic algorithm-variable subset selection (GA-VSS) method (30).38 The dataset was randomly divided into training and test sets (25% of the whole dataset). External validation was carried out in accordance with the reported criteria.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We would like to deeply thank Prof. Dr Yehia Osman and Dr Sally Elsheikh form the Department of Botany, Faculty of Science, Mansura University, Egypt for their assistance to evaluate the antimicrobial activity of our compounds against Methicillin-resistant Staphylococcus aureus (MRSA).References
- G. P. Ellis, Chromenes, chromanones and chromones, John Willy & Sons, New York, 1977 Search PubMed.
- W. Fischer, A. Currais, Z. Liang, A. Pinto and P. Maher, Redox Biol., 2019, 21, 101089 CrossRef CAS PubMed.
- Y. Sukpondma, V. Rukachaisirikul and S. Phongpaichit, Chem. Pharm. Bull., 2005, 53, 850–852 CrossRef CAS PubMed.
- G. Valdomir, S. Senthilkumar, D. Ganapathy, Y. Zhang and L. F. Tietze, Chem. - Eur. J., 2018, 24, 8760–8763 CrossRef CAS PubMed.
- Y. Gao, Q. Ren, H. Wu, M. Li and J. Wang, Chem. Commun., 2010, 46, 9232–9234 RSC.
- Z. Yang, K. Wu, Y. Xu, X. Xia, X. Wang, M. Ge and L. Shao, J. Antibiot., 2020, 73, 194–199 CrossRef CAS PubMed.
- F. Cottiglia, B. Dhanapal, O. Sticher and J. Heilmann, J. Nat. Prod., 2004, 67, 537–541 CrossRef CAS PubMed.
- M. Ceylan-Ünlüsoy, E. J. Verspohl and R. Ertan, J. Enzyme Inhib. Med. Chem., 2010, 25, 784–789 CrossRef PubMed.
- S. Emami, T. Banipoulad, H. Irannejad, A. Foroumadi, M. Falahati, M. Ashrafi-Khozani and S. Sharifynia, J. Enzyme Inhib. Med. Chem., 2014, 29, 263–271 CrossRef CAS PubMed.
- L. Du, M. Li, Q. Yang, Y. Tang, Q. You and L. Xia, Bioorg. Med. Chem. Lett., 2009, 19, 1477–1480 CrossRef CAS PubMed.
- S. I. El-Desoky, F. A. Badria, M. A. Abozeid, E. M. Kandeel and A. H. Abdel-Rahman, Med. Chem. Res., 2013, 22, 2105–2114 CrossRef CAS.
- (a) H. Chen, J. Xie, D. Xing, J. Wang, J. Tang, Z. Yi, F. Xia, W.-W. Qiu and F. Yang, Org. Biomol. Chem., 2019, 17, 1062–1066 RSC; (b) L. Feng, M. M. Maddox, Md. Z. Alam, L. S. Tsutsumi, G. Narula, D. F. Bruhn, X. Wu, S. Sandhaus, R. B. Lee, C. J. Simmons, Y.-C. Tse-Dinh, J. G. Hurdle, R. E. Lee and D. Sun, J. Med. Chem., 2014, 57, 8398–8420 CrossRef CAS PubMed; (c) N. G. Ghatpande, J. S. Jadhav, R. V. Kaproormath, M. E. Soliman and M. M. Shaikh, Bioorg. Med. Chem., 2020, 28, 115813 CrossRef CAS PubMed.
- R. Rios, Chem. Soc. Rev., 2012, 41, 1060–1074 RSC.
- (a) K. Ding, Z. Han and Z. Wang, Chem.–Asian J., 2009, 4, 32–41 CrossRef CAS PubMed; (b) F. Xu, D. Huang, C. Han, W. Shen, X. Lin and Y. Wang, J. Org. Chem., 2010, 75, 8677–8680 CrossRef CAS PubMed; (c) Y.-H. Yuan, X. Han, F.-P. Zhu, J.-M. Tian, F.-M. Zhang, X.-M. Zhang, Y.-Q. Tu, S.-H. Wang and X. Guo, Nat. Commun., 2019, 10, 3394 CrossRef PubMed.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed.
- E. Chupakhin, O. Babich, A. Prosekov, L. Asyakina and M. Krasavin, Molecules, 2019, 24, 4165 CrossRef CAS PubMed.
- (a) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed; (b) Y.-J. Zheng and C. M. Tice, Expet Opin. Drug Discov., 2016, 11, 831–834 CrossRef PubMed; (c) P. Saraswat, G. Jeyabalan, M. Z. Hassan, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016, 46, 1643–1664 CrossRef CAS; (d) A. Ding, M. Meazza, H. Guo, J. W. Yang and R. Rios, Chem. Soc. Rev., 2018, 47, 5946–5996 RSC.
- L. Guan, H. Yang, Y. Cai, L. Sun, P. Di, W. Li, G. Liu and Y. Tang, RSC Med. Chem., 2019, 10, 148–157 RSC.
- (a) G. Mehta, R. Samineni, P. Srihari, R. G. Reddy and S. Chakravarty, Org. Biomol. Chem., 2012, 10, 6830–6833 RSC; (b) N. Abe, R. Onoda, K. Shirahata, T. Kato, M. C. Woods and Y. Kitahara, Tetrahedron Lett., 1968, 9, 369–373 CrossRef; (c) A. K. Ghosh, K. Krishnan, D. E. Walters, W. Cho, Y. Koo, J. Trevino, L. Holland and J. Buthod, Bioorg. Med. Chem. Lett., 1998, 8, 979–982 CrossRef CAS.
- (a) F. Micoli, F. Bagnoli, R. Rappuoli and D. Serruto, Nat. Rev. Microbiol., 2021, 19, 287–302 CrossRef CAS PubMed; (b) M. Gajdács, E. Urbán, A. Stájer and Z. Baráth, Eur. J. Investig. Health Psychol. Educ., 2021, 11, 71–82 CrossRef; (c) M. Lomazzi, M. Moore, Ap. Johnson, M. Balasegaram and B. Borisch, BMC Publ. Health, 2019, 19, 858 CrossRef PubMed; (d) S. I. El-Desoky, E. M. Keshk, A. A. El-Sawi, M. A. Abozeid, L. A. Abouzeid and A. H. Abdel-Rahman, Saudi Pharmaceut. J., 2018, 26, 852–859 CrossRef PubMed; (e) M. A. Abozeid, M. R. El-Kholany, A. H. Abdel-Rahman and S. I. El-Desoky, Int. J. Mod. Org. Chem., 2018, 5, 1–11 CAS; (f) M. A. Abozeid, M. R. El-Kholany, L. A. Abouzeid, A. H. Abdel-Rahman and S. I. El-Desoky, J. Heterocycl. Chem., 2019, 56, 2922–2933 CrossRef CAS; (g) S. I. El-Desoky, A. A. El-Sawi, M. A. Abozeid, M. Abdelmoteleb, M. Shaaban, E. M. Keshk and A. H. Abdel-Rahman, Med. Chem. Res., 2019, 28, 1601–1617 CrossRef; (h) M. A. Abozeid, A. A. El-Sawi, M. Abdelmoteleb, H. Awad, M. M. Abdel-Aziz, A. H. Abdel-Rahman and S. I. El-Desoky, RSC Adv., 2020, 10, 42998–43009 RSC; (i) M. A. Abozeid, A. A. El-Sawi, M. R. Elmorsy, M. Abdelmoteleb, A. H. Abdel-Rahman and S. I. El-Desoky, RSC Adv., 2019, 9, 27996–28005 RSC.
- M. Mujahid, R. G. Gonnade, P. Yogeeswari, D. Sriram and M. Muthukrishnan, Bioorg. Med. Chem. Lett., 2013, 23, 1416–1419 CrossRef CAS.
- S. Y. Dike, J. R. Merchant and N. Y. Spare, Tetrahedron, 1991, 47, 4775–4786 CrossRef CAS.
- S. Parasuraman, J. Pharmacol. Pharmacother., 2011, 2, 52–53 CrossRef CAS PubMed.
- (a) A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef; (b) H. Yang, C. Lou, L. Sun, J. Li, Y. Cai, Z. Wang and Y. Tang, Bioinformatics, 2018, 35, 1067–1069 CrossRef.
- R. D. Pearson, R. T. Steigbigel, H. T. Davis and S. W. Chapman, Antimicrob. Agents Chemother., 1980, 18, 699–708 CrossRef CAS.
- R. J. Holt, J. Clin. Pathol., 1975, 28, 767–774 CrossRef CAS PubMed.
- J. M. Andrews, J. Antimicrob. Chemother., 2001, 48, 5–16 CrossRef CAS PubMed.
- V. Judge, B. Narasimhan, M. Ahuja, D. Sriram, P. Yogeeswari, E. De Clercq, C. Pannecouque and J. Balzarini, Med. Chem. Res., 2012, 21, 1451–1470 CrossRef CAS.
- H. F. Rizk, M. A. EI-Badawi, S. A. Ibrahim and M. A. EI-Borai, Chin. J. Chem., 2011, 29, 1451–1459 CrossRef CAS.
- H. K. Gençer, U. A. Çevik, S. Levent, B. N. Sağlık, B. Korkut, Y. Özkay, S. Ilgın and Y. Öztürk, Molecules, 2017, 22, 507 CrossRef.
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Nat. Rev. Drug Discovery, 2004, 3, 935–949 CrossRef CAS.
- M. Mohamadzadeh, M. Zarei and M. Vessal, Bioorg. Chem., 2020, 95, 103515 CrossRef CAS PubMed.
- B. Fois, Ž. Skok, T. Tomašič, J. Ilaš, N. Zidar, A. Zega, L. P. Mašič, P. Szili, G. Draskovits, Á. Nyerges, C. Pál and D. Kikelj, ChemMedChem, 2020, 15, 265–269 CrossRef CAS.
- E. R. Trösken, M. Adamska, M. Arand, J. A. Zarn, C. Patten, W. Völkel and W. K. Lutz, Toxicology, 2006, 228, 24–32 CrossRef.
- K. Y. Lum, S. T. Tay, C. F. Le, V. S. Lee, N. H. Sabri, R. D. Velayuthan, H. Hassan and S. D. Sekaran, Sci. Rep., 2015, 5, 9657 CrossRef CAS PubMed.
- (a) E. N. Muratov, J. Bajorath, R. P. Sheridan, I. V. Tetko, D. Filimonov, V. Poroikov, T. I. Oprea, I. I. Baskin, A. Varnek, A. Roitberg, O. Isayev, S. Curtalolo, D. Fourches, Y. Cohen, A. Aspuru-Guzik, D. A. Winkler, D. Agrafiotis, A. Cherkasov and A. Tropsha, Chem. Soc. Rev., 2020, 49, 3525–3564 RSC; (b) Danishuddin and A. U. Khan, Drug Discov. Today, 2016, 21, 1291–1302 CrossRef CAS PubMed.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- P. Gramatica, N. Chirico, E. Papa, S. Cassani and S. Kovarich, J. Comput. Chem., 2013, 34, 2121–2132 CrossRef CAS.
- J. B. Patel, M. P. Weinstein, G. M. Eliopoulos, S. G. Jenkins, J. S. Lewis II, B. Limbago, A. J. Mathers, T. Mazzulli, R. Patel, S. S. Richter, M. Satlin, J. M. Swenson, M. M. Traczewski, J. D. Turnidge, B. L. Zimmer, Performance standards for antimicrobial susceptibility testing, CLSI supplement M100, Clinical and Laboratory Standards Institute, Wayne, PA, 27th edn, 2017. Search PubMed.
- J. H. Rex, B. D. Alexander, D. Andes, B. Arthington-Skaggs, S. D. Brown, V. Chaturvedi, M. A. Ghannoum, A. Espinel-Ingroff, C. C. Knapp, L. Ostrosky-Zeichner, M. A. Pfaller, D. J. Sheehan, T. J. Walsh, Reference method for Broth dilution antifungal susceptibility testing of yeasts; approved standard-third edition. CLSI document M27-A3, Clinical and Laboratory Standards Institute, 2008. Search PubMed.
- J. H. Rex, B. D. Alexander, D. Andes, B. Arthington-Skaggs, S. D. Brown, V. Chaturveli, A. Espinel-Ingroff, M. A. Ghannoum, C. C. Knapp, M. R. Motyl, L. Ostrosky-Zeichner, M. Pfaller, D. J. Sheehan, T. J. Walsh, Reference method for Broth dilution antifungal susceptibility testing of filamentous fungi; approved standard-second edition. CLSI document M38-A2, Clinical and Laboratory Standards Institute, 2008. Search PubMed.
- (a) T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS; (b) F. Denizot and R. Lang, J. Immunol. Methods, 1986, 22, 271–277 CrossRef.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03497a |
| This journal is © The Royal Society of Chemistry 2021 |