
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetically recyclable silica-coated ferrite magnetite-K10 montmorillonite nanocatalyst and its applications in O, N, and S-acylation reaction under solvent-free conditions
Nitin Tandon,
Shripad M. Patil ,
Runjhun Tandon* and
Pushpendra Kumar
,
Runjhun Tandon* and
Pushpendra Kumar
Department of Chemistry, School of Chemical Engineering and Physical Sciences, Lovely Professional University, Phagwara-144411, India. E-mail: runjhun.19532@lpu.co.in
First published on 16th June 2021
Abstract
Novel silica-coated ferrite nanoparticles supported with montmorillonite (K10) have been prepared successfully by using a simple impregnation method. Further, these nanoparticles were characterized by using different analytical methods like FT-IR, PXRD, EDS, and FE-SEM techniques. In addition, these nanoparticles have been explored for their catalytic activity for the O, N, and S-acylation reactions under solvent-free conditions which gave moderate to excellent yields in a much shorter reaction time. Moreover, these nanoparticles could easily be separated out from the reaction medium after the reaction completion by using an external magnetic field and have been re-used for 10 cycles without any significant loss of the catalytic activity.
1. Introduction
Nanocatalysis is an important area of research for organic synthesis using green methodologies. In recent years, many heterogeneous nano-catalysts like zeolites, metal oxides, clay particles etc. have been widely used in the industrial sector for various organic transformations and proven to be more effective than the homogeneous catalysts in terms of ease of work up after the completion of the reaction, fewer chances of formation of by-products and recyclability of the catalysts.1,2 These nano catalysts provide larger surface area for the reactants to come closer to each other for the reaction thereby decreasing the reaction time for the target transformation. The heterogeneous catalysis involving green chemistry is an integral part of the organic synthesis and sustainable chemistry which prevents the use of toxic volatile solvents, reagents, harmful methods and requires minimum reaction time without production of toxic waste materials.3,4 In addition, these eco-friendly methods using green chemistry are also effective in terms of atom economy.5 Recyclability of the catalyst without any significant loss of the activity and yield is also an important aspect which is associated with the reduction in the effluent load and cost-effectiveness eventually leading to the cleaner reactions with desired output in terms of quality and yield.Many attempts have been made in the past to develop nanoparticles which can participate in green chemistry and can be reused for many cycles without significant loss in their activity.6–9 In this context, ferrite magnetic nanoparticles (MNPs) have occupied an important research area in synthetic and bio medicinal chemistry for the development of the target molecules by coupling reactions.10,11 These MNPs have wide range of their applicability in chemical sciences and engineering.
Design and synthesis of MNPs which are highly suitable for various organic synthetic reactions is an art.12–15 Many attempts have been made by various authors to explore chemical synthesis of the MNPs by using precipitation, reverse micelles, photolysis, sonolysis, borohydride reductions etc. which have been further studied for their morphological, structural and magnetic properties.16 These catalysts have shown effective catalytic properties, high selectivity, easy work up from the reaction, and are recoverable without any loss of catalytic properties.17–20 Magnetic nano-material have also been used as a support for the various functionalized catalyst and can easily be recollected by using an external magnet once the target chemical transformation take place. This magnetic nanocatalyst separation (decantation process) is very effective than the other methods such as centrifugation and filtration.
Acylation of the alcohols, amines and thiols to form the corresponding esters and amides is of significant importance in the organic synthesis.21 Number of attempts have been made for the acylation of alcohol and phenols using acetic anhydride or acetyl chloride in the presence of acid or base catalysts under various reaction conditions.22–30 Recently, Anbu et al. have reported the solvent and catalyst free process for the acylation reaction.31 But, all these reported processes are associated with a number of limitations such as longer reaction time, use of toxic solvents, high reaction temperature and ease of handling. Therefore, there is still a need for the development of new catalysts that can be used under mild reaction conditions to get the good yield of the acetylated products using green chemistry.
Solvent free reactions including use montmorillonite (K10) as catalyst have been used in literature for various functional transformations in good to excellent yields.32–34 K10 supported silica coated nanocatalysts have been reported for acylation reactions of alcohols, amines, thiols and phenols and it is an important strategy in organic synthesis for the multistep preparation of molecules applicable in chemical and pharmaceutical industries.35–38 But the disadvantages of the reported methods with K10 includes the extended reaction time or the use of the toxic solvents which puts an extra burden of effluent treatment.
The present work presents the preparation of the core–shell magnetite-montmorillonite (K10) nanoparticle by using the co-precipitation method. The first step involved the preparation of silica-coated ferrite MNPs using tetraethyl orthosilicate (TEOS) in aqueous solution followed by the reaction with montmorillonite (K10) in ethanol under sonication to get magnetite-montmorillonite (K10) nanocatalyst. These nanocatalysts have further been explored for a simple, green and sustainable method for the acylation of amines, alcohols, phenols, and thiols derivatives by using acetic anhydride as one of the reactants (Scheme 1).
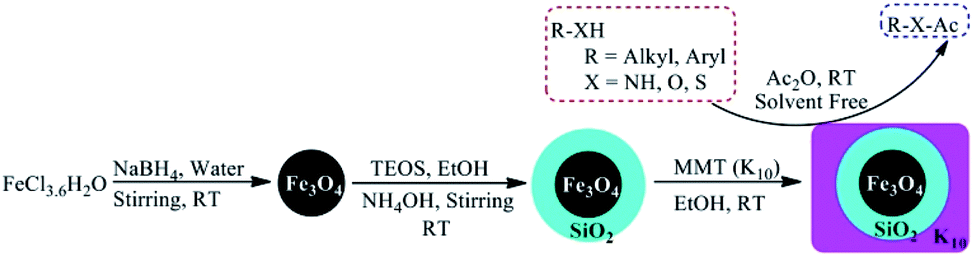 | ||
| Scheme 1 Preparation of Fe3O4@SiO2@K10 and its applications in the acylation of alcohol, amines, and thiols. | ||
2. Results and discussion
2.1. Characterization of silica-coated ferrite magnetite-K10 montmorillonite nanocatalyst
IR spectra of Fe3O4 nanoparticles is depicted in Fig. 1a. Peak at 3219.08 cm−1 was assigned to hydroxyl group due to OH stretching vibrations of the water present on the surface of Fe3O4 nanoparticles. The additional peaks at 1340.07 cm−1 and 1017.83 cm−1 were due to the de-ionized water used as solvent. The IR peaks at 694.49 cm−1 and 451.50 cm−1 were assigned to Fe–O bond vibrations of Fe3O4 nanoparticles.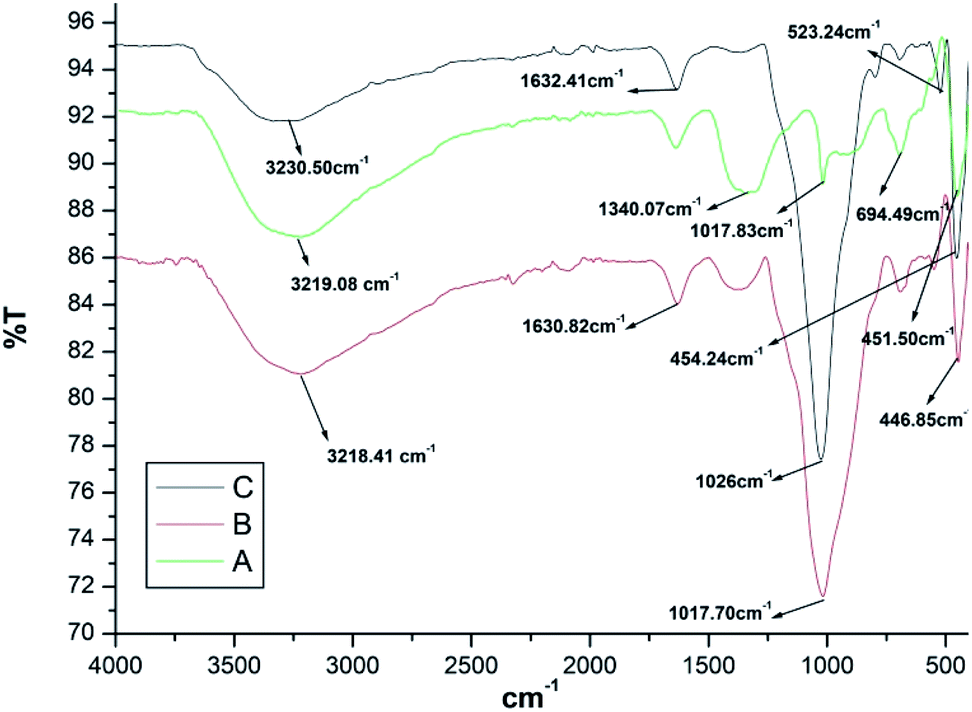 | ||
| Fig. 1 FT-IR spectra of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@K10. | ||
IR spectra of Fe3O4@SiO2 (Fig. 1b) gave two peaks at 1630.82 cm−1 and 1017.70 cm−1 which were assigned to un-symmetrical and symmetrical linear stretching vibrations of Si–O–Si bonding. The peak at 446.85 cm−1 corresponded to bending vibration absorption peak of Si–O–Si.
IR spectrum of Fe3O4@SiO2@K10 is depicted in Fig. 1c which showed additional peak at 523.24 cm−1 due to bending vibration of Al–O–Si bond along with the peak at 454.24 cm−1 which was assigned to bending vibration Si–O–Si bond.
The main characteristic peaks of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@K10 have been summarized in Table 1.
| Compound | Characteristic IR peaks |
|---|---|
| Fe3O4 | 3219.08 cm−1, 1340.07 cm−1, 1017.83 cm−1, 694.49 cm−1 and 451.50 cm−1 |
| Fe3O4@SiO2 | 3218.41 cm−1, 1630.82 cm−1, 1017.70 cm−1 and 446.85 cm−1 |
| Fe3O4@SiO2@K10 | 3230.50 cm−1, 1632.41 cm−1, 1026 cm−1, 523.24 cm−1 and 454.24 cm−1 |
Powder XRD studies of the prepared samples were performed in the range of 2θ value of 20–80° and corresponding patterns are depicted in Fig. 2. In general, presence of multiple peaks in PXRD of all the three samples indicated the polycrystalline nature of samples. The exhaustive evolution of magnetite Fe3O4 along with small trace of Fe2O3 was observed in the PXRD pattern of Fe3O4 (Fig. 2a). Further, no peak corresponding to metallic iron was observed in either of the PXRD of the samples under study. In addition, no additional peak was observed on loading of SiO2 on Fe3O4 due to amorphous nature of the sample (Fig. 2b). However, increase in intensity of 400 peaks (2θ, 44.1) in the PXRD of Fe3O4@SiO2, suggesting improved crystallinity and change in preferred orientation of crystallites. It was also observed that the peaks shifted slightly towards higher angle due to smaller ionic radii of Si2+ as compared to Fe3+ confirming the overloading of SiO2 on Fe3O4. In case of PXRD of Fe3O4@SiO2@K10 (Fig. 2c), two additional intense peaks at 2θ value of 21.20 and 26.30 were attributed to the presence of montmorillonite (K10).39 Also, loading of K10 did not alter the diffraction pattern much, suggesting that metal cations resided only on outer surface of the clay. However, due to marginal shifting in peaks, inclusion of metal cations in the interlayer of clay could not be completely ruled out which also suggested the binding of K10 over Fe3O4@SiO2 surface. The average crystalline size calculated by Scherrer's calculation and was found to be 28 nm.
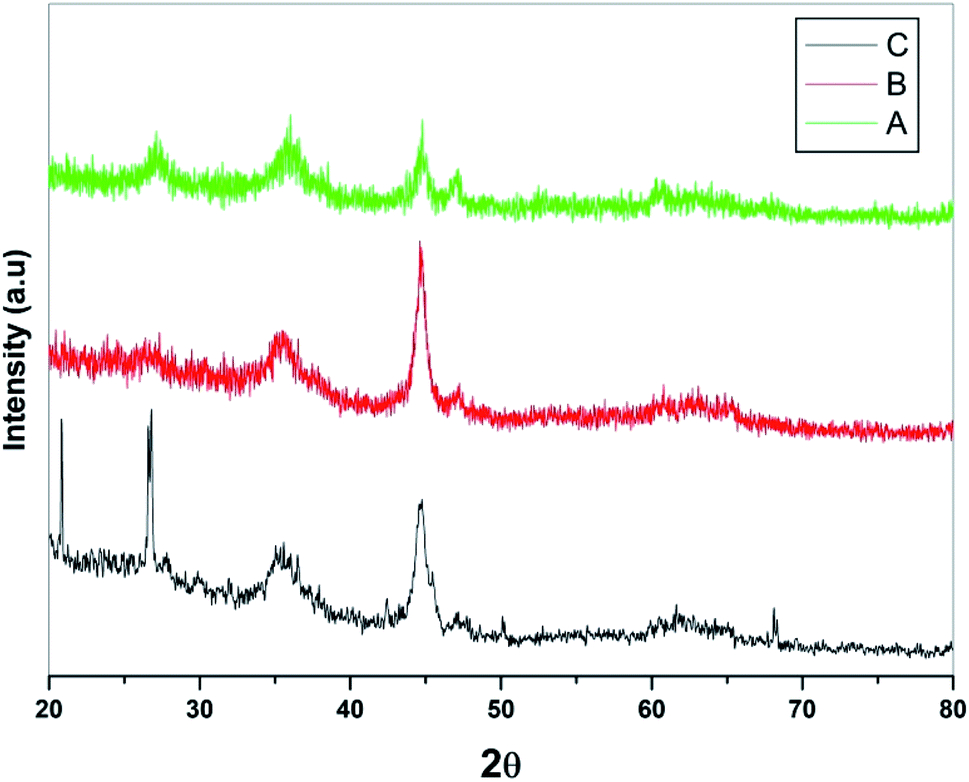 | ||
| Fig. 2 PXRD pattern of (A) Fe3O4, (B) Fe3O4@SiO2, and (C) Fe3O4@SiO2@K10. | ||
The obtained SEM images of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@K10 are depicted in Fig. 3 which suggested that Fe3O4@SiO2 nanoparticles were supported on montmorillonite (K10) surface. According to SEM images, the nanoparticles were spherical in shape. Also, the particle size of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@K10 were calculated as 50, 100, and 100 nm respectively.
 | ||
| Fig. 3 SEM image of (A) Fe3O4 (B) Fe3O4@SiO2 (C) Fe3O4@SiO2@K10 nanocatalyst. | ||
The elemental composition of synthesized material was determined by using EDX detector coupled with SEM. EDX spectra of Fe3O4@SiO2 is shown in Fig. 4A which confirmed the presence of Fe, Si and O in expected ratio of 27.22%, 10.10% and 62.68 at% respectively. Fig. 4b depicts the EDX spectra of Fe3O4@SiO2@K10 which confirmed the presence of Fe, Si, O, Mg, Al, and K in the expected ratio of 8.87%, 32.58, 51.83, 1.15, 5.00 and 0.58 at%, respectively. Although, EDX technique in conjunction with SEM is not very sensitive to detect lower amount of elements but presence of trace amount can be confirmed.
 | ||
| Fig. 4 (a) EDX data for Fe3O4@SiO2, (b) EDX data for Fe3O4@SiO2@K10 nanocatalyst. | ||
In a combined study in the presence of a scanning electron microscope (SEM), wavelength-dispersive X-ray spectroscopy (WDX) can help providing the qualitative information about the distribution of the different chemical elements in the catalyst matrix. Fig. 5 represents the SEM and corresponding WDX images of the prepared catalyst which suggested that Fe metal particles were well dispersed in a composite. The specific area of elemental analysis indicates the uniform and homogeneous distribution of Fe, Si, O, Mg, Al, and K throughout the sample.
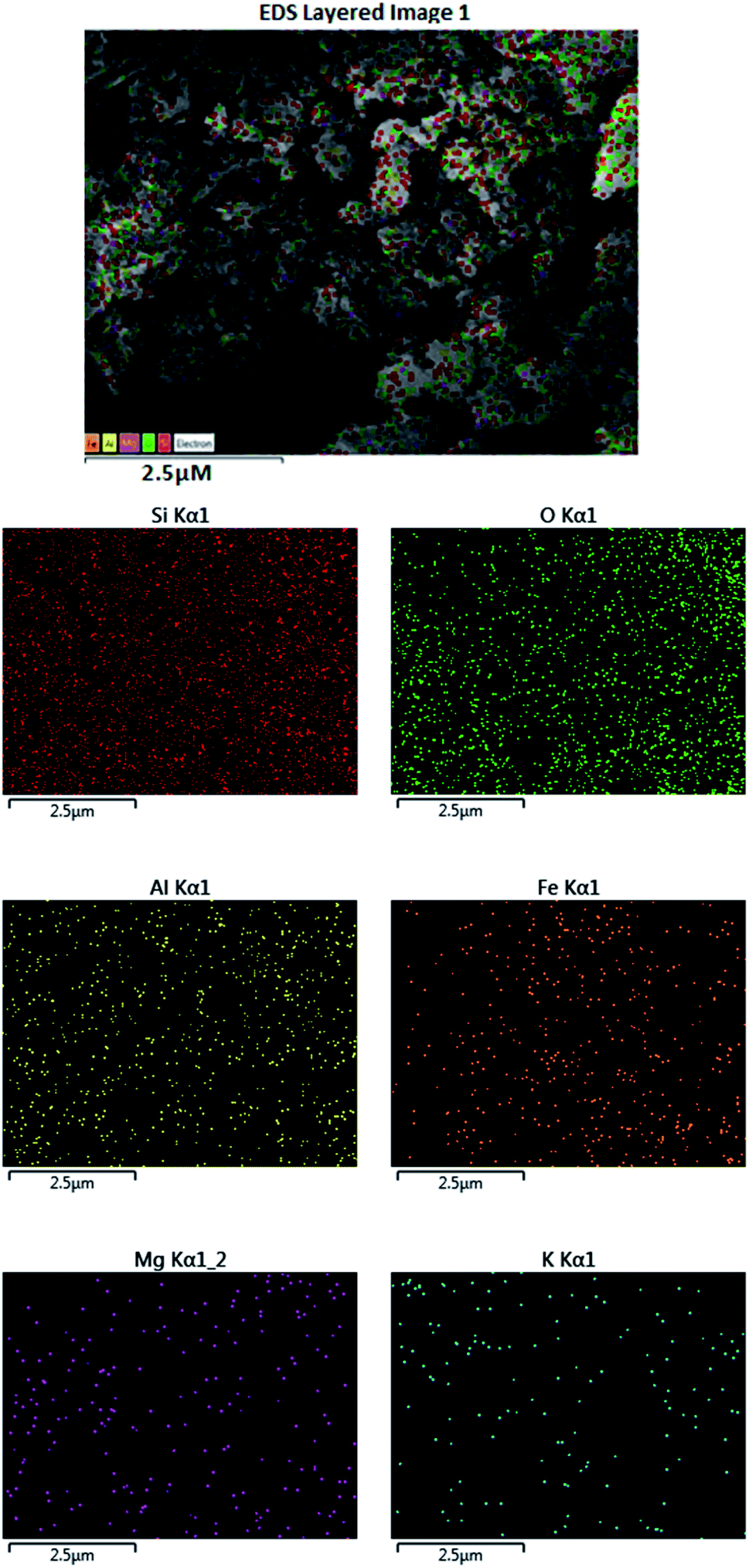 | ||
| Fig. 5 SEM images of Fe3O4@SiO2@K10 showing the presence of Fe, Si, Mg, O, Al, and K atom in the nanocatalyst. | ||
2.2. Catalytic applications of Fe3O4@SiO2@K10 nanoparticles
The catalytic study of synthesized nanoparticles of Fe3O4@SiO2@K10 was studied for the acylation of amines, phenols, alcohols, and thiols in the presence of acetic anhydride (Scheme 1).The reaction of benzyl alcohol with acetic anhydride was used as a model reaction which was carried out at various reaction conditions in the presence of the Fe3O4@SiO2@K10 nanocatalyst to optimize the reaction conditions. Different reactions were carried out in the presence of polar protic solvent (ethanol), polar aprotic solvent (tetrahydrofuran), nonpolar solvent (toluene), solvent with moderate polarity (dichloromethane), in aqueous media (water) and in the absence of any solvent by using 15 mol% of the catalyst. Promising results were obtained when acetonitrile (CH3CN), toluene or dichloromethane (DCM) was used as solvents whereas the less yields were obtained by performing the reaction in ethanol or water. Interestingly, best results were obtained in the absence of the solvent. Further, decreasing the catalyst quantity from 15 to 10 mol% led to decrease in the yield. However, the increase in the quantity to 20 mol% did not change the yield. It was very important to note that the reaction did not take place in the absence of the catalyst under solvent-free conditions. Also, the acylation reaction did not take place in the presence of Fe3O4 nanoparticles or Fe3O4@SiO2 alone confirming the importance of K10 coating (Table 2).
|
||||
|---|---|---|---|---|
| Sr. no. | Solvent | Catalyst (mol%) | Time (min) | Yieldb (%) |
| a Reaction conditions: benzyl alcohol (1 mmol), acetic anhydride (1.5 mmol), room temperature and solvent (5 ml).b Isolated yields. | ||||
| 1 | Ethanol | 15 | 360 | 40 |
| 2 | Water | 15 | 360 | 35 |
| 3 | THF | 15 | 360 | 50 |
| 4 | DCM | 15 | 90 | 75 |
| 5 | Toluene | 15 | 120 | 60 |
| 6 | CH3CN | 15 | 70 | 80 |
| 7 | None | 15 | 20 | 99 |
| 8 | None | 10 | 70 | 88 |
| 9 | None | 20 | 20 | 99 |
| 10 | None | None | 720 | 00 |
| 11 | None | Fe3O4 nanoparticles | 20 | 00 |
| 12 | None | Fe3O4@SiO2 | 20 | 00 |

These optimized reaction conditions were further used for the acylation reactions of different amines, alcohol and thiol derivatives. Table 3 summarizes the reaction output of different aromatic primary aromatic amines with acetic anhydride under the optimized conditions. In general, the presence of the substituents on the aromatic ring did not affect the yield of the reaction much. However, the presence of the electron withdrawing groups (7–10) led to increase in the reaction time to 20 minutes from 10 minutes case of aniline (1). On the other hand, the presence of electron releasing groups at the aromatic ring did not affect the reaction time significantly (2–6). The reaction time was also found to increase in case of reaction with cyclic secondary amine (11).
| Comp. | Substrate | Product | Time (min) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: reagent (1 mmol), catalyst (15 mol%) and acetic anhydride (1.5 mmol), stirring at room temperature under solvent-free conditions.b Isolated yields. | ||||
| 1 | 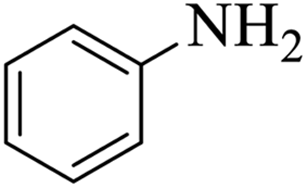 |
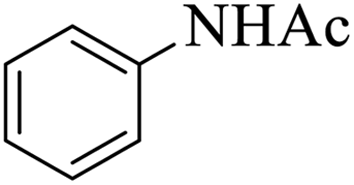 |
10 | 96 |
| 2 |  |
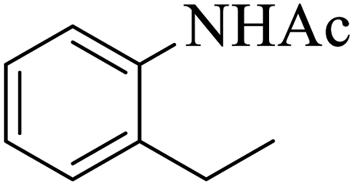 |
12 | 95 |
| 3 | 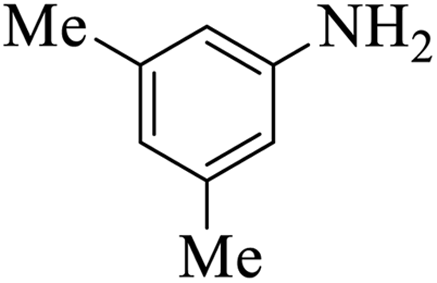 |
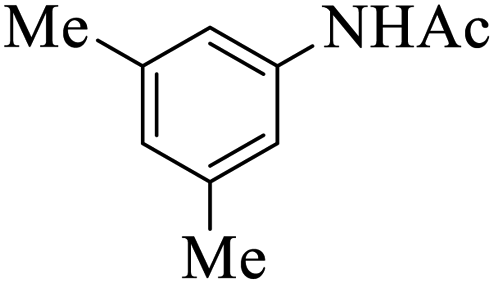 |
12 | 95 |
| 4 | 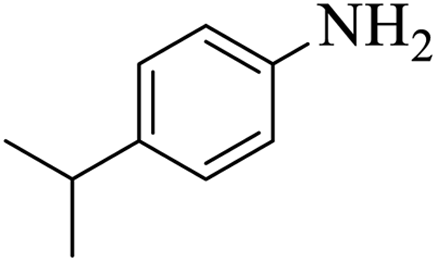 |
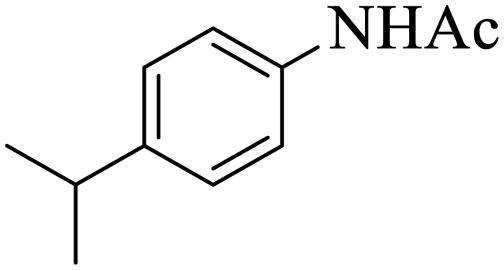 |
15 | 94 |
| 5 | 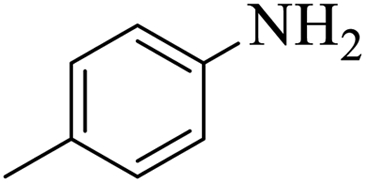 |
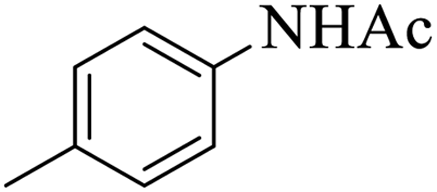 |
10 | 96 |
| 6 | 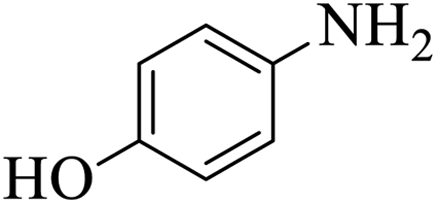 |
 |
12 | 95 |
| 7 | 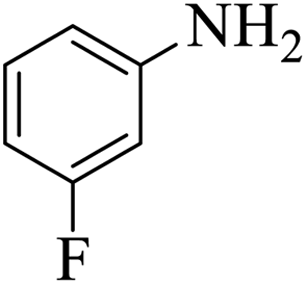 |
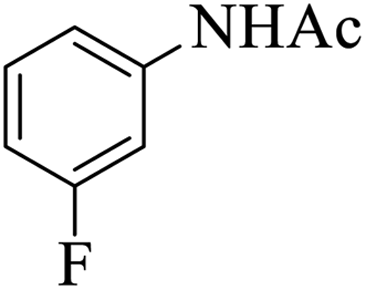 |
20 | 92 |
| 8 | 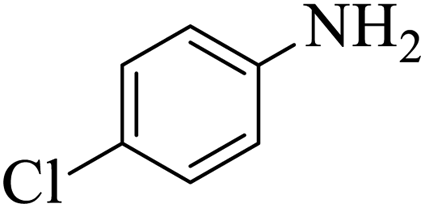 |
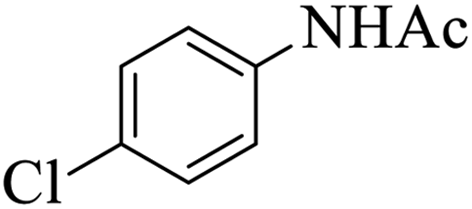 |
20 | 93 |
| 9 | 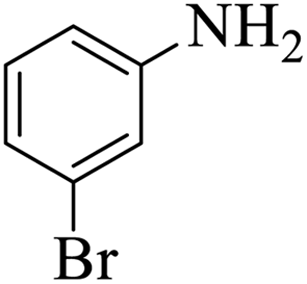 |
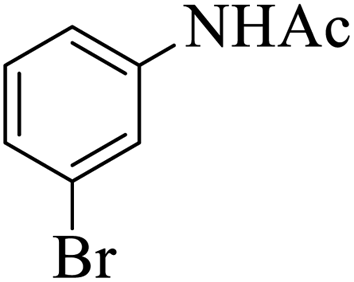 |
25 | 90 |
| 10 | 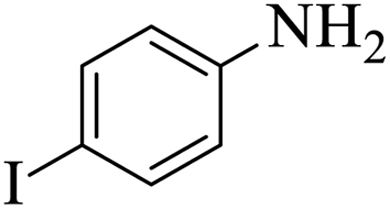 |
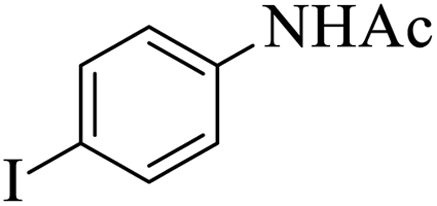 |
25 | 91 |
| 11 | 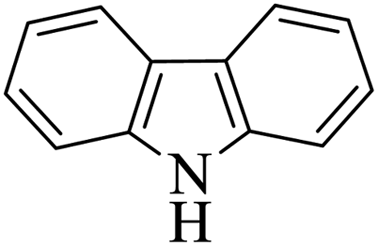 |
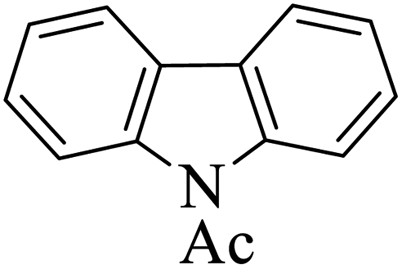 |
20 | 94 |
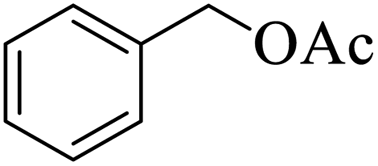
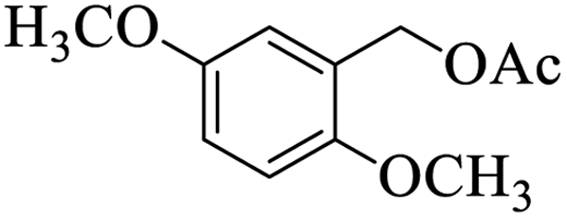
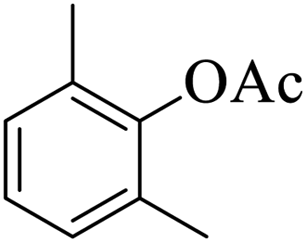
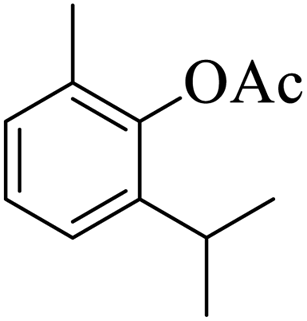
Further, alcohol derivatives involving primary, secondary, tertiary or benzyl alcohols were also explored for their acylation reactions in the presence of the prepared nanocatalyst to give the corresponding acetate products from good to excellent yield in very less reaction time (Table 4). In general, the reaction of alcohol derivatives took longer time than the amine derivatives. The studies suggested that the phenol (12) and benzyl alcohol (16) gave quantitative yield on reaction with acetic anhydride and the reaction was completed in 20 minutes. However, the increase in the steric hindrance (13 versus 12) and (17–20 versus 16) led to decrease in the yield with increase in the reaction time. Further, the reaction gave good results for primary (15) as well as for secondary alcohols (14).
| Comp. | Substrate | Product | Time (min) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: reagent (1 mmol), nanoparticle (15 mol%) and acetic anhydride (1.5 mmol), stirring at room temperature under solvent-free conditions.b Isolated yields. | ||||
| 12 | 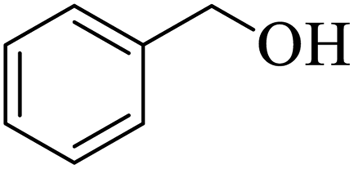 |
 |
20 | 99 |
| 13 | 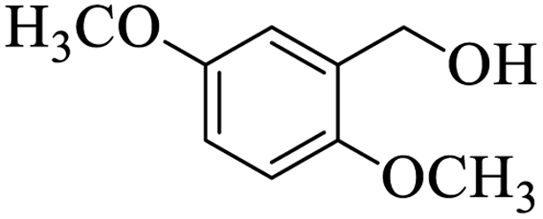 |
 |
20 | 96 |
| 14 | 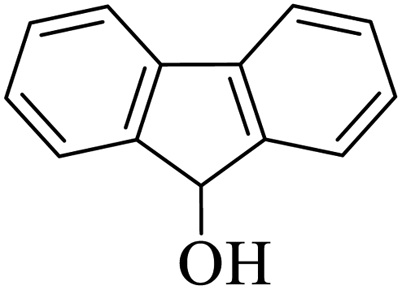 |
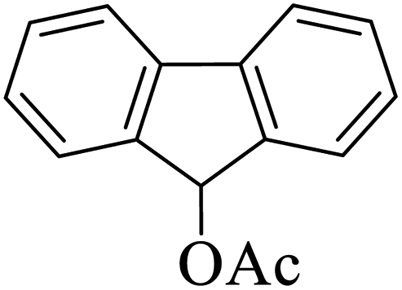 |
30 | 94 |
| 15 | 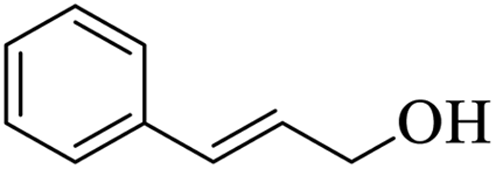 |
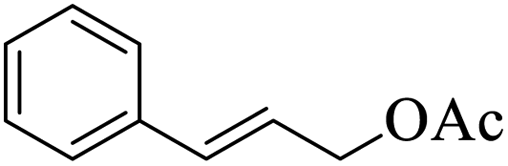 |
20 | 93 |
| 16 |  |
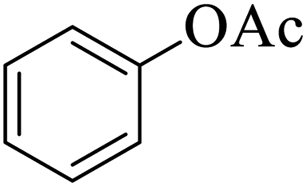 |
20 | 99 |
| 17 | 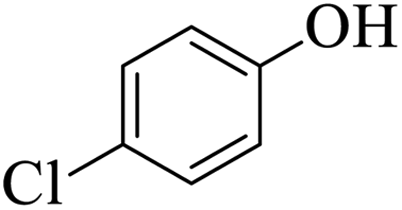 |
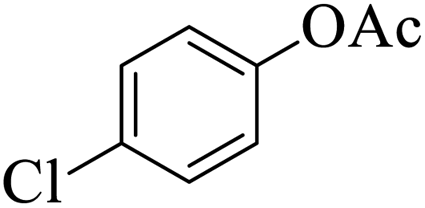 |
10 | 92 |
| 18 | 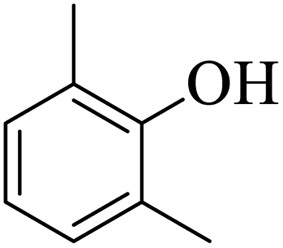 |
 |
15 | 89 |
| 19 | 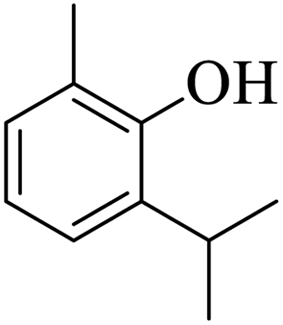 |
 |
15 | 88 |
| 20 | 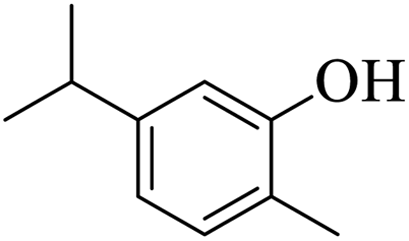 |
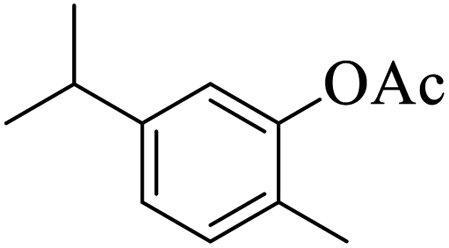 |
15 | 90 |
| 21 | 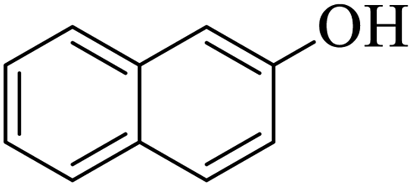 |
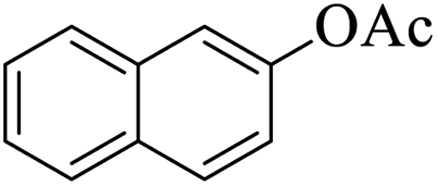 |
10 | 92 |
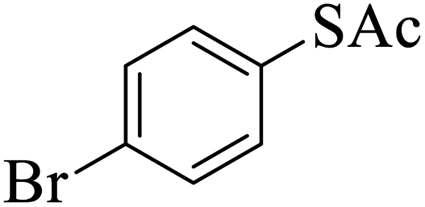
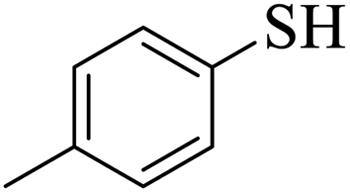
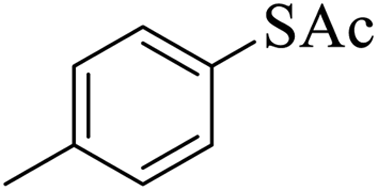
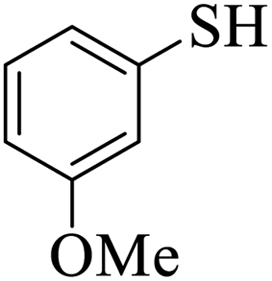
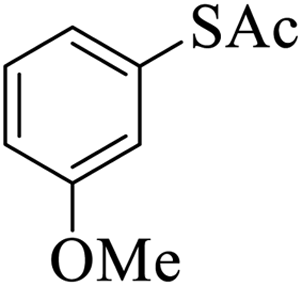
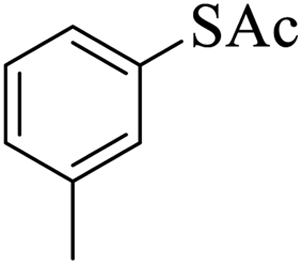
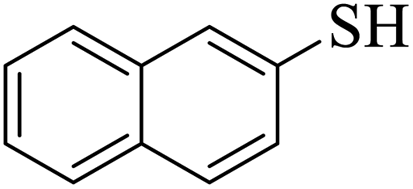
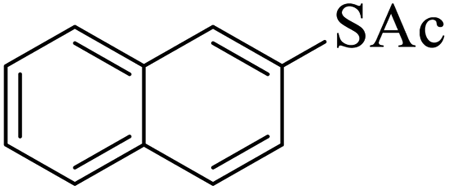
Thiols derivatives were found to be lesser reactive than the amine and alcohol derivatives towards their acylation reactions and thus gave lesser yields and took more time for completion (Table 5). The presence of the electron withdrawing (23–25) or electron releasing groups (entries 26–28) did not affect the reaction output as compared to the parent derivative (22). Interestingly, 2-thionaphthol gave good yield (29).



The possible mechanism of the acylation of alcohols, amines or thiols catalysed by Fe3O4@SiO2@K10 is illustrated in Scheme 2. The reaction is likely to be proceeded via Fe3O4@SiO2@K10 catalyzing the formation of intermediate II which makes carbonyl carbon of acetic anhydride more electrophilic facilitating nucleophilic attack by R–XH(III) to get the intermediate IV. Finally, the rearrangement of intermediate IV resulted in the formation of the acylated product V along with the formation of acetic acid as byproduct. A similar type of mechanism has also been proposed by Safari et al.40
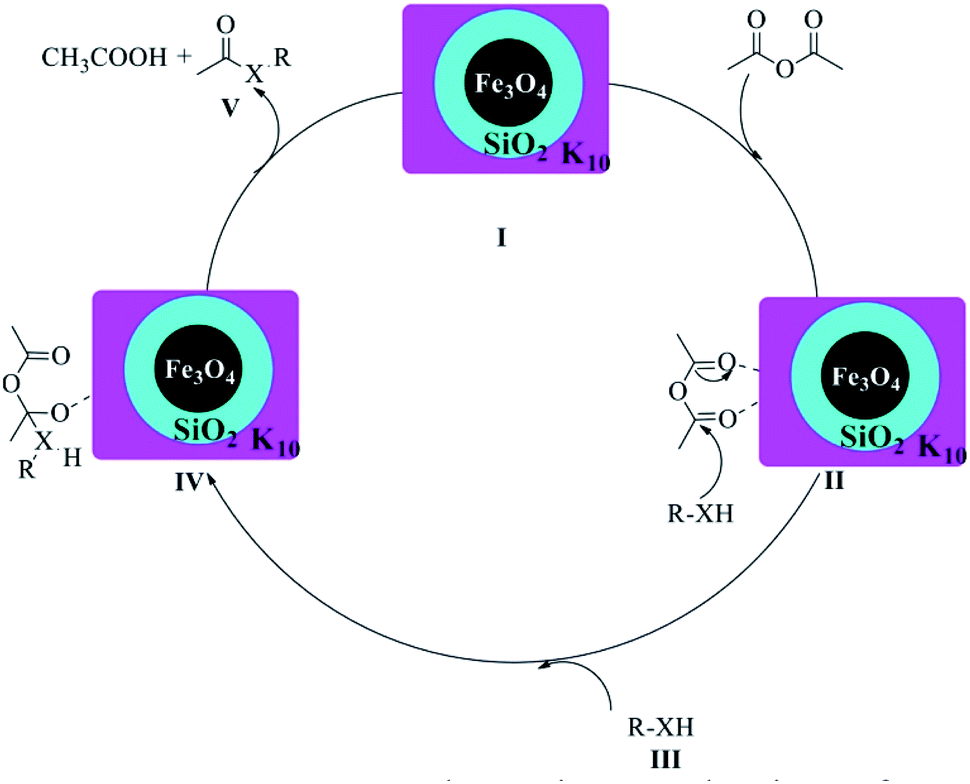 | ||
| Scheme 2 Proposed reaction mechanism of Fe3O4@SiO2@K10 catalyzed under acylation of O, N, and S. | ||
Further, the Fe3O4@SiO2@K10 catalyst was reused for 10 cycles (Fig. 6) for the acylation of benzyl alcohol without any appreciable loss in the yield (Table 6) and the reaction time.
 | ||
| Fig. 6 Recyclability of the Fe3O4@SiO2@K10 nanocatalyst. | ||
| Cycle | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Yield (%) | 99 | 99 | 99 | 99 | 99 | 98 | 98 | 98 | 97 | 97 |
The present work has advantages over the previously reported methods (Table 7) in terms of lesser reaction time, moderate reaction conditions and avoids the use of harmful and toxic solvents. Moreover, the prepared catalyst is easily recoverable from the reaction mass after the completion of the desired transformation and could be used at least for 10 cycles without any significant loss in the catalytic activity.
| Sr. no. | Reaction conditions | Time | Yield (%) | Ref. |
|---|---|---|---|---|
| 1 | Fe3O4@SiO2@K10, RT, solvent-free | 20 min | 99 | Present work |
| 2 | SiO2–ZnCl2, 80 °C, CH3CN | 3 h | 90 | 41 |
| 3 | Borated zirconia, 110 °C, toluene | 15 h | 25 | 42 |
| 4 | Fe3O4@PDA–SO3H, RT, solvent-free | 25 min | 98 | 43 |
| 5 | Silica-sulfamic acid, RT, solvent-free | 15 min | 93 | 44 |
| 6 | Nano γ-Fe2O3, sonication, RT | 1 h | 64 | 45 |
| 7 | CuO–ZnO, 80 °C, CH2Cl2 | 15 min | 92 | 46 |
| 8 | Yttria-zirconia, 110 °C, solvent-free | 6 h | 94 | 47 |
| 9 | Copper zirconium phosphate NPs, 60 °C, solvent-free | 30 min | 91 | 48 |
3. Conclusions
In the present work, we have prepared a novel core–shell Fe3O4@SiO2@K10 nanocatalyst via loading of montmorillonite on the surface of Fe3O4@SiO2 nanoparticles. Furthermore, the prepared catalyst has been explored for the acylation of the various nucleophilic substrates such as alcohols, phenols, amines, and thiols derivatives. This process has many advantages over the previously known methods of acylation in terms of excellent yields, lesser reaction time, ambient reaction conditions and solvent free environment supporting the green chemistry. Also, no significant loss of activity of the catalyst has been observed when used in 10 cycles.4. Experimental
4.1. General methods
All commercial compounds and solvents (Spectrochem, Aldrich, Merck etc.) were used without purification. Melting points were determined in an open capillary and were uncorrected. The prepared nanoparticles of silica-coated ferrite magnetite-K10 montmorillonite nanocatalyst were characterized by using different characterization techniques like Fourier transmission-infrared spectroscopy (FT-IR, Perkin Elmer, Version 10.6.1), powder X-ray diffraction (PXRD Brucker D8 Advances), field emission scanning electron microscope (FE-SEM, JEOL, JSM-7610F plus) and energy-dispersive X-ray spectroscopy (EDS, OXFORD EDS LN2).4.2. Synthesis of Fe3O4 nanoparticles
The synthesis of Fe3O4 nanoparticles was achieved by using simple wet chemical reduction technique.492.7 g of ferric chloride hexahydrate was dissolved in de-ionized water to make the total volume up to 100 ml in a volumetric flask to get 0.1 M of solution. In an another volumetric flask, 9.4 g of sodium borohydride was dissolved in de-ionized water to make total volume up to 100 ml to get 2.5 M of the solution. Added, 40 ml of ferric chloride hexahydrate solution drop wise to the 10 ml of sodium borohydride solution under stirring for about 15 minutes. The completion of the reaction was indicated by the disappearance of the bubbles through the reaction mass and appearance of the black precipitates. The prepared nanoparticles of Fe3O4 were separated through the reaction mass by external magnet and were washed with de-ionized water. Finally the nanoparticles were dried in oven at 60 °C for 12 hours to get 1.65 g of Fe3O4 nanoparticles.
4.3. Synthesis of silica-coated Fe3O4 nanoparticles
The preparation of core–shell Fe3O4@SiO2 nanoparticles was achieved by the simple Stober method by the reaction of Fe3O4 nanoparticles with tetraethyl orthosilicate (TEOS). For this purpose, 1 g of Fe3O4 nanoparticles were dispersed in 100 ml of absolute ethanol and the reaction mass was sonicated for about 15 min. After 15 minutes of sonication, 5 ml of ammonium hydroxide solution (10% w/v) was added to reaction mass followed by drop wise addition of 1.5 ml of tetraethyl orthosilicate with continuous sonication at room temperature for 5 h. After that, the resulting silica-coated nanoparticles Fe3O4@SiO2 were separated from the reaction mass through the external magnet, washed with water and ethanol, then finally dried at 60 °C under vacuum for 12 hours to get 1.3 g of silica-coated nanoparticles Fe3O4@SiO2.4.4. Synthesis of silica-coated ferrite magnetite-K10 montmorillonite nanocatalyst
A suspension of 1 g of Fe3O4@SiO2 nanoparticles in 50 ml of ethanol was kept under sonication for about 30 min and then added 0.5 g of montmorillonite (K10) to the reaction mass under continuous stirring. After that, the pH of the reaction mass was set to 12 by the use of 1 M sodium hydroxide solution and the reaction mass was stirred at room temperature for further 20 hours. Finally, the precipitates formed were separated, washed with de-ionized water and finally dried at 60 °C for 24 hours to get 1.42 g of the target magnetite-montmorillonite (K10).4.5. General procedure for acylation of amines/phenols/alcohols/thiols derivatives
In a typical reaction, 1 mmol of the reactant was reacted with 1.5 mmol of acetic anhydride in the presence of 15 mol% of Fe3O4@SiO2@K10 under vigorous stirring at room temperature. The progress of the reaction was monitored using thin-layer chromatography (TLC). After the completion of the reaction, the reaction mixture was diluted with ethyl acetate (5 ml) and then nanoparticles were separated out by using external magnet. The separated nanoparticles were washed with water and ethanol many times and dried under vacuum. The reaction mixture was washed with saturated NaHCO3 solution and the product was extracted with ethyl acetate, dried over Na2SO4, and the solvent was evaporated under vacuum to get the desired products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to the Central Instrumentation Facility, Department of Chemistry, School of Chemical engineering and physical sciences, Lovely Professional University, Phagwara-144411 Punjab, India for partial support of this work.Notes and references
- A. Vaccari, Catal. Today, 1998, 41, 53 CrossRef CAS.
- K. Tanabe and W. F. Holderich, Appl. Catal., A, 1999, 181, 399 CrossRef CAS.
- V. Polshettiwar and R. S. Varma, Chem. Soc. Rev., 2008, 37, 1546 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- (a) A. E. Diaz-Alvarez, J. Francos, B. Lastra-Barreira, P. Crochet and V. Cadierno, Chem. Commun., 2011, 47, 6208 RSC; (b) A. P. Abbott, D. Bothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9442 CrossRef PubMed.
- P. T. Anastas, L. B. Bartlett, M. M. Kirchhoff and T. C. Williamson, Catal. Today, 2000, 55, 11 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC.
- S. P. Kunde, K. G. Kanade, B. K. Karale, H. N. Akolkar, P. V. Randhavane and S. T. Shinde, Arabian J. Chem., 2017, 43, 7277 CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed.
- M. T. Reetz and M. Maase, Adv. Mater., 1999, 11, 773 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- S. B. Kalidindi and B. R. Jagirdar, ChemSusChem, 2012, 5, 65 CrossRef CAS PubMed.
- P. Chen, X. Zhou, H. Shen, N. M. Andoy, E. Choudhary, K. S. Han and G. Liu, Chem. Soc. Rev., 2010, 39, 4560 RSC.
- A. Schatz, O. Reiser and W. J. Stark, Chem.–Eur. J., 2010, 16, 8950 CrossRef PubMed.
- M. A. Willard, L. K. Kurihara, E. E. Carpenter, S. Calvin and G. Harris, Int. Mater. Rev., 2004, 49, 125 CrossRef CAS.
- (a) R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481 RSC; (b) A. Schatz, T. R. Long, R. N. Grass, W. J. Stark, P. R. Hanson and O. Reiser, Adv. Funct. Mater., 2010, 20, 4323 CrossRef PubMed.
- M. B. Gawande, P. S. Branco, K. Parghi, J. J. Shrikhande, R. K. Pandey, C. A. A. Ghumman, N. Bundaleski, O. M. N. D. Teodoro and R. V. Jayaram, Catal. Sci. Technol., 2011, 1, 1653 RSC.
- (a) M. B. Gawande, S. N. Shelke, A. Rathi, P. S. Branco and R. K. Pandey, Appl. Organomet. Chem., 2012, 26, 395 CrossRef CAS; (b) U. U. Indulkar, S. R. Kale, M. B. Gawande and R. V. Jayaram, Tetrahedron Lett., 2012, 53, 3857 CrossRef CAS; (c) M. B. Gawande, A. K. Rathi, P. S. Branco, T. M. Potewar, A. Velhinho, I. D. Nogueira, A. Tolstogouzov, C. Amjad, A. Ghumman and O. M. N. D. Teodoro, RSC Adv., 2013, 3, 3611 RSC.
- (a) S. Jansat, D. Picurelli, K. Pelzer, K. Philippot, M. Gómez, G. Muller, P. Lecante and B. Chaudret, New J. Chem., 2006, 30, 122 RSC; (b) J. Li, Y. Zhang, D. Han, Q. Gao and C. Li, J. Mol. Catal. A: Chem., 2009, 298, 31 CrossRef CAS.
- J. Otera, Chem. Rev., 1993, 93, 1449 CrossRef CAS.
- A. Orita, C. Tanahashi, A. Kakuda and J. Otera, Angew. Chem., Int. Ed., 2000, 39, 2877 CrossRef CAS PubMed.
- R. Alleti, M. Perambuduru, S. Samanha and V. P. Reddy, J. Mol. Catal. A: Chem., 2005, 226, 57 CrossRef CAS.
- B. Karimi and J. Maleki, J. Org. Chem., 2003, 68, 4951 CrossRef CAS PubMed.
- N. Ghaffari Khaligh, J. Mol. Catal. A: Chem., 2012, 363–364, 90 CrossRef.
- W. Steglich and G. Hofle, Angew. Chem., Int. Ed., 1969, 8, 981 CrossRef CAS.
- E. Vedejs and T. S. Diver, J. Am. Chem. Soc., 1993, 115, 3358 CrossRef CAS.
- E. F. V. Scriven, Chem. Soc. Rev., 1983, 12, 129 RSC.
- S. Tomohumi, O. Kousaburo and O. Takashi, Synthesis, 1999, 7, 1141 Search PubMed.
- R. Ballini, G. Bosica, S. Carloni, L. Ciaralli, R. Maggi and G. Sartori, Tetrahedron Lett., 1998, 39, 6049 CrossRef CAS.
- N. Anbu, N. Nagarjun, M. Jacob, J. M. V. K. Kalaiarasi and A. Dhakshinamoorthy, Chemistry, 2019, 1, 69 CrossRef.
- S. Bonacci, M. Nardi, P. Costanzo, A. D. Nino, M. L. D. Gioia, M. Oliverio and A. Procopio, Catalysis, 2019, 9, 301 Search PubMed.
- S. Bonacci, G. Iriti, S. Mancuso, P. Novelli, R. Paonessa, S. Tallarico and M. Nardi, Catalysis, 2020, 10, 845 CAS.
- A. Procopio, G. D. Luca, M. Nardi, M. Oliverioa and R. Paonessa, Green Chem., 2009, 11, 770 RSC.
- T. W. Green and P. C. M. Wuts, Protective groups in organic synthesis, Wiley, New York, 3rd edn, 1999 Search PubMed.
- J. Otera, Esterification: Methods, Reactions and Applications, Wiley–VCH, 1st edn, 2003 Search PubMed.
- A.-X. Li, T.-S. Li and T.-H. Ding, Chem. Commun., 1997, 1389 RSC.
- T.-S. Li and A.-X. Li, J. Chem. Soc., Perkin Trans. 1, 1998, 1913 RSC.
- J. Julinawati, S. Gea, E. Eddiyanto, B. Wirjosentono and I. Ichwana, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 523, 021023 Search PubMed.
- J. Safari and N.-H. Nasab, Res. Chem. Intermed., 2019, 45, 1025–1038 CrossRef CAS.
- R. Gupta, V. Kumar, M. Gupta, S. Paul and R. Gupta, Indones. J. Chem., 2008, 47, 1739 Search PubMed.
- F. Tamaddon, M. A. Amrollahi and L. Sharafat, Tetrahedron Lett., 2005, 46, 7841 CrossRef CAS.
- H. Veisi, S. Taheri and S. Hemmati, Green Chem., 2016, 18, 6337 RSC.
- K. Niknam and M. Blanco, J. Mol. Catal. A: Chem., 2010, 52, 316 Search PubMed.
- M. A. Bhosale, D. Ummineni, T. Sasaki, D. Nishio-Hamane and B. M. Bhanage, J. Mol. Catal. A: Chem., 2015, 8, 404 Search PubMed.
- J. Albadi, A. Alihosseinzadeh and M. Mardani, Chin. J. Catal., 2015, 36, 308 CrossRef CAS.
- P. Kumar, R. K. Pandey, M. S. Bodas, S. P. Dagade, M. K. Dogare and A. V. Ramaswamy, J. Mol. Catal. A: Chem., 2002, 181, 207 CrossRef CAS.
- A. J. Hajipour and H. Karimi, Chin. J. Catal., 2014, 35, 1982 CrossRef CAS.
- S. H. Chaki, J. M. Tasmira, M. D. Chaudhary, J. P. Tailor and M. P. Dshpandey, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2015, 6, 035009 Search PubMed.
- R. B. Sonawane, N. K. Rasal and S. V. Jagtap, Org. Lett., 2017, 19, 2078 CrossRef CAS PubMed.
- B. P. Fors Karin, D. Qingle, Z. Stephen and L. Buchwald, Tetrahedron, 2009, 65, 6576 CrossRef PubMed.
- G. Zhang, Y. Zhao, L. Xuan and C. Ding, Eur. J. Org. Chem., 2019, 30, 4911 CrossRef.
- M. D. H. Bhuiyan, A. B. Mahon, P. Jensen, J. K. Clegg and A. C. Try, Eur. J. Org. Chem., 2009, 5, 687 CrossRef.
- K. Iida, S. Ishida, T. Watanabe and T. Arai, J. Org. Chem., 2019, 84, 7411 CrossRef CAS PubMed.
- V. Arumugam, W. Kaminsky and D. Nallasamy, Green Chem., 2016, 18, 3295 RSC.
- M. J. Durán-Peña, J. M. Botubol-Ares, J. R. Hanson, R. Hernandez-Galan and I. G. Collado, J. Am. Chem. Soc., 2014, 136, 1062 CrossRef PubMed.
- U. Mandi, A. S. Roy, B. Banerjee and Sk. M. Islam, RSC Adv., 2014, 4, 42670 RSC.
- A. Correa, T. Leon and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062 CrossRef CAS PubMed.
- M. Kumar, S. Bagchi and A. Sharma, New J. Chem., 2015, 39, 8329 RSC.
- R. Das and D. Chakraborty, Synthesis, 2011, 10, 1621 Search PubMed.
- S. M. D. Morais, N. S. Vila-Nova, C. M. L. Bevilaqua, F. C. Rondon, C. H. Lobo, A. A. A. N. Moura, A. D. Sales, A. P. R. Rodrigues, J. R. de Figuereido, C. C. Campello, M. E. Wilson and H. F. de Andrade Jr, Bioorg. Med. Chem., 2014, 22, 6250 CrossRef PubMed.
- H. P. Narkhede, U. B. More, D. S. Dalal and P. P. Mahulikar, Synth. Commun., 2008, 38, 2413 CrossRef CAS.
- K. Kucinski and G. Hreczycho, Org. Process Res. Dev., 2018, 22(4), 489 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |