
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCholine chloride-based deep eutectic solvents as effective electrolytes for dye-sensitized solar cells†
De Nguyenac,
Tuan Van Huynhbc,
Vinh Son Nguyend,
Phuong-Lien Doan Caoce,
Hai Truong Nguyen ce,
Tzu-Chien Weid,
Phuong Hoang Tran*ce and
Phuong Tuyet Nguyen*ac
ce,
Tzu-Chien Weid,
Phuong Hoang Tran*ce and
Phuong Tuyet Nguyen*ac
aFaculty of Chemistry, University of Science, Ho Chi Minh City, Vietnam. E-mail: ntphuong@hcmus.edu.vn
bFaculty of Physics and Engineering Physics, University of Science, Ho Chi Minh City, Vietnam
cVietnam National University Ho Chi Minh City, Vietnam. E-mail: thphuong@hcmus.edu.vn
dDepartment of Chemical Engineering, National Tsing-Hua University, Hsinchu 30013, Taiwan
eDepartment of Organic Chemistry, Faculty of Chemistry, University of Science, Ho Chi Minh City, Vietnam
First published on 17th June 2021
Abstract
Electrolytes for dye-sensitized solar cells remain a challenge for large-scale production and commercialization, hindering the wide application of solar cells. We have developed two new electrolyte-based deep eutectic solvents using a mixture of choline chloride with urea and with ethylene glycol for dye-sensitized solar cells. The prominent features of the two deep eutectic solvent electrolytes are simple preparation for large-scale production with inexpensive, available, and nontoxic starting materials and biodegradability. The solar cell devices proceeded in a safe manner as the two deep eutectic solvents afforded low-cost technology and comparative conversion efficiency to a popular ionic liquid, namely 1-ethyl-3-methylimidazolium tetracyanoborate. Results showed that devices with choline chloride and urea electrolyte exhibited improved open circuit voltage values (VOC), while the ones with choline chloride and ethylene glycol showed an increase in the short circuit current (Isc). Characterization of the devices by electrochemical impedance spectroscopy helped explain the effects of their molecular structures on the enhancement of either VOC or Isc values. These new solvents expand the electrolyte choices for designing dye-sensitized solar cells, especially for the purpose of using low-cost and eco-friendly materials for massive production.
1. Introduction
Dye-sensitized solar cells (DSCs) have attracted numerous scientific and technological interests as efficient and low-cost alternative photovoltaic devices compared to conventional solar cells.1,2 Three important components of DSC include a TiO2 loaded dye sensitizer photo-anode, a platinum cathode, and an electrolyte between the two electrodes.2 A typical DSC electrolyte comprises a coupled redox mediator (e.g. I−/I3−) and electrolyte additives dissolved in liquid or ionic liquid solvents. The first DSCs used liquid electrolytes and achieved high energy conversion efficiencies.2,3 However, the required volatile and hazardous solvents often lead to leakage, which impedes the durability and photovoltaic performance of DSCs and damages the environment.3–6 Such so-called “non-robust” electrolytes also caused a fast thermal degradation of ruthenium dyes,7,8 key components in DSCs, leading to a loss in long-term photovoltaic performance of the devices.8,9 Ionic liquid electrolytes have been thus developed as alternative electrolyte solvents10,11 due to their non-volatility, high conductivity, and thermal stability.12–14 However, the performance of DSCs with ionic liquid electrolyte still faces challenges including (i) high viscosity leading to low ionic diffusion of the mediator and hence low conversion efficiency of the devices;15 (ii) high cost and low purity;10 (iii) and potentially harmful effects from the electrolyte waste to the environment and human health.11,16 Some alternative, low-viscosity ionic liquid electrolytes such as 1-ethyl-3-methylimidazolium dicyanamide,17 imidazolium selenocyanate,18 triethylammonium perfluorocarboxylate,19 and imidazolium tetracyanoborate or imidazolium tricyanomethanide20 have been subsequently studied as promising solvents for high-performance DSCs, but the economic and environmental issues remain. In the search for a more eco-friendly, low-cost, and efficient electrolyte for DSC devices, deep eutectic solvents (DESs) have been considered as potential candidates due to many of their advantages such as high availability and sustainability, simple and inexpensive manufacturing, and biodegradability.21Deep eutectic solvents (DESs) were first described by Abbott and coworkers by mixing the substituted ammonium salts with hydrogen bond donor components.22–24 Up until now, several kinds of DESs have been fabricated by mixing two or three cheap and non-hazardous materials, which through hydrogen bonding interactions, have freezing points lower than the starting components.25 As a result, DESs show better properties illustrating significant advantages for industrial application.26 DESs can be prepared by simply mixing two or three components,27 which are relatively inexpensive and environmentally friendly.28,29 For example, choline chloride, also well-known as vitamin B4 and as an additive in fish feed, is a useful quaternary ammonium salt to prepare DESs.30 In a recent development, DESs have been used in a wide variety of applications, such as biotransformation,31–33 catalysis,34–36 extraction processes,37–39 and material sciences.40
DESs are emerging as potential substitutes for ionic liquids in DSC electrolytes. Jhong et al. reported a eutectic mixture of glycerol and choline iodide as an electrolyte in DSCs with organic dye (D149) and achieved an efficiency of 3.88%.41 Later on, Boldrini and coworkers applied an aqueous choline chloride-based DES as an electrolyte and phenothiazine organic sensitizer in DSC, which reached an efficiency of nearly 2%.42 In 2019, our group mixed a choline chloride: phenol DES with the liquid electrolyte of acetonitrile as a solvent.43 Results showed that even though the DES-added cells were initially less efficient than the DES-free ones, the efficiencies of the two devices became comparable after more than 1000 hours of operation. DES helped stabilize and improve the long-term photovoltaic performance of the DSC devices, particularly the short circuit current. However, further studies are needed to develop more eco-friendly, volatile organic solvent-free electrolytes.
To solve the problem, in this study, we developed two choline chloride-based DES mixtures with urea (DES-CU) and with ethylene glycol (DES-CE), for applications in DSC electrolytes. The electrolytes were prepared with redox mediator I−/I3− and electrolyte additives in the DES without any addition of liquid solvents. The DES cells were characterized and analyzed by the current–voltage (J–V) curve; incident photon to current efficiency (IPCE), and electrochemical impedance spectroscopy (EIS) measurements in comparison with standard cells used the popular ionic liquid – 1-ethyl-3-methylimidazolium tetracyanoborate (EMITCB). The results demonstrated that the DES devices achieved comparative energy conversion efficiency to the standard ionic liquid devices. EIS analysis gave a finding of the DES molecular structures affecting the photovoltaic performance of the devices, especially on the open-circuit voltage (VOC) and short circuit current density (JSC) values.
2. Experimental
A. Preparation of DESs
DESs were prepared according to a previously reported procedure.44 Specifically, DES-CE (choline chloride/ethylene glycol molar ratio of 1/2) and DES-CU (choline chloride/urea molar ratio of 1/2) were prepared by heating the corresponding individual pure components to 80 °C under stirring until a colorless solution was formed.B. DSC fabrication & characterization
The cathode was prepared by drilling two small holes in the 1.5 × 1.5 cm FTO glass, followed by cleaning and platinum paste (Dyesol PT1) deposition process.45
The reference EMITCB ionic liquid electrolyte comprises 1 M 1-methyl-3-propylimidazolium iodide (PMII), 0.2 M iodine (I2), 0.1 M guanidinium thiocyanate (GuSCN), 0.5 M N-methylbenzimidazole (NMB), 0.74 M EMITCB, all from Sigma Aldrich. The DES-based electrolytes were prepared by replacing EMITCB with DES-CE or DES-CU. To optimize the electrolyte composition, different DES/EMITCB volume ratio, including 0.5 (CE0.5, CU0.5), 1 (CE1, CU1), 1.5 (CE1.5, CU1.5), were evaluated.
The electrolyte was then injected through the hole on the platinized cathode. Finally, another thin cover glass slide with the Surlyn film was hot-pressed to enclose the two holes. The complete DSC devices were kept in the dark for further characterization.
J–V curve and IPCE measurement. A black mask with an active area of 0.16 cm2 was attached to the photoanode. The J–V curve of DSC was measured with a computer-controlled digital source meter (Keithley 2400) under the exposure of a standard solar simulator (PECCELL, PEC-L15. AM1.5, Class B) with a light intensity of 1000 W m−2, relative humidity around 30–40% at 20 °C.
IPCE was measured using a PEC-S20 (Peccell Technologies). The light wavelength was set from 300 to 900 nm with an increment of 5 nm.
EIS full cell in the dark. A Schlumberger SI 1255 HF frequency response analyzer integrated with an electrochemical interface SI 1286 was used to measure the EIS of fully assembled DSC cells in dark conditions with a frequency range of 105 Hz to 10−1. The negative potentials of −0.4, −0.5, −0.6, −0.7 V were applied to the TiO2 photo-anode during measurements. The obtained impedance spectra were fitted using the Z-view software to extract impedance parameters.
3. Results and discussion
A. Characterization of DES-CU and DES-CE
For pure choline chloride, vibrational bands at 3476 cm−1 refer to the presence of hydroxyls, while the bands at 3019–2965 cm−1 and 1478–1376 cm−1 indicate the presence of an alkyl group (Fig. S2†). The characteristic C–N vibration can be found at 1135 cm−1. The vibration bands at 1087 and 955 cm−1 are assigned to C–O and C–C–O asymmetric stretching associated with the –CH2CH2–OH group of the choline cation. In the FT-IR spectrum of pure urea, vibrational bands at 3441 and 1567 cm−1 refer to the presence of N–H vibration. The characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration can be found at 1662 cm−1. The vibrational bands at 1457 cm−1 are assigned to C–N stretching of urea. During the formation of DES, the spectra of DES-CU is an overlap with those of both choline chloride and urea, and it illustrated that the structures of choline chloride and urea were not destroyed in DES-CU. Specifically, the small difference in wavenumbers compared to the signals of the N–H and CO groups in urea and DES-CU indicates that hydrogen bonds were formed between choline chloride and urea.
O vibration can be found at 1662 cm−1. The vibrational bands at 1457 cm−1 are assigned to C–N stretching of urea. During the formation of DES, the spectra of DES-CU is an overlap with those of both choline chloride and urea, and it illustrated that the structures of choline chloride and urea were not destroyed in DES-CU. Specifically, the small difference in wavenumbers compared to the signals of the N–H and CO groups in urea and DES-CU indicates that hydrogen bonds were formed between choline chloride and urea.
In the FT-IR spectrum of pure ethylene glycol, vibrational bands at 3315 cm−1 refer to the presence of O–H vibration. The vibration bands at 2930 cm−1 are assigned to –CH2 stretching of ethylene glycol (Fig. S3†). During the formation of DES, the spectrum of DES-CE overlaps with those of both choline chloride and ethylene glycol. It illustrated that the structures of choline chloride and ethylene glycol were not destroyed in DES-CE. Moreover, the absorption bands of DES-CE at 3440 cm−1 could be ascribed to the stretching vibration of the O–H functional group (Fig. S3†). There was a slight difference in the wavenumber compared to the signal of the O–H group in choline chloride as well as ethylene glycol, indicating that hydrogen bond formation was created between the two components of DES.
Thermal gravimetric analysis (TGA) from 50 to 750 °C showed that DES-CU started to decompose at around 150 °C (Fig. S4-A†). The major weight loss occurs in the temperature range from 150 to 350 °C. The decomposition temperature of both choline chloride and urea was found to be higher due to the formation of complexes of DES-CU. It was suggested that the DES obtained by the complexation of a quaternary ammonium salt and a hydrogen bond altered the properties of the choline chloride and urea.46 DES exhibited a lower melting temperature than their individual components due to the weaker interaction between the choline cation and the correspondent hydrogen bond donors. Similarly, TGA showed that DES-CE started to decompose at around 150 °C. The major weight loss occurs between 150 and 300 °C. The decomposition of pure choline chloride and pure ethylene glycol was between 300 to 350 °C and 100 to 200 °C, respectively (Fig. S4-B†).
1H NMR spectrum of DES-CU showed the resonances of choline chloride at 4.03–3.99 ppm (m, 2H) and 3.53–3.50 ppm (m, 2H), which were assigned to the –CH2– group of the choline cation. The signal at 3.23 ppm (s, 9H) was attributed to the –CH3 resonance of the choline cation (Fig. S5†). The 1H NMR spectrum of DES-CE indicated the resonances of choline chloride at 3.64–3.57 ppm (m, 2H), 3.31 ppm (m, 2H), and 3.74 ppm (s, 9H). The signal of ethylene glycol at 2.03 (9 s, 4H) showed –CH2–, the resonance of the ethylene glycol (Fig. S6†). DES with high viscosities and several inter-and intra-dipolar interactions is the main reason for the broadening effect on the lineshape of the NMR spectrum. Additionally, the 1H NMR spectrum shows that the structure of choline chloride and urea are not destroyed in DES-CU.
The glass transition temperature was determined using differential scanning calorimetry (DSC). The glass transition temperature depended strongly on the nature of the two components of DES. DSC spectrum of DES-CU under heating the deep eutectic mixture from −60 °C to 100 °C with a heating rate of 10 °C min−1 showed that there is an obvious exotherm with an onset temperature of −20 °C corresponding to the crystallization of glass (Fig. S7†). The process displayed one sharp melting endotherm with an onset temperature of 12 °C, which was consistent with the melting point. Obviously, DES-CU demonstrated two phase transitions from glass to crystal and crystal to melt during the DSC process. For DES-CE, the DSC spectrum showed that the glass transition temperature of the DES-CE mixture was from −34.62 °C (Fig. S8†).
B. IPCE and J–V characterization
Devices with different electrolytes showed similar IPCE spectra (Fig. 1). All devices had IPCE values of 50–60% at the spectrum band at around 540 nm, which was the characteristic IPCE peak of N719-sensitized cells. The IPCE spectra of CE and CU devices were a little bit lower than that of EMITCB. This might be due to the lower diffusion of I3− in CE and CU electrolytes than in EMITCB. The integrated current density values calculated from IPCE measurements are shown in Table 1. There was a small difference between the current density values from the J–V curves and the IPCE spectra. This could be due to the dissimilarity of light intensity between the two measurement systems. | ||
| Fig. 1 IPCE spectra of DSC devices using EMITCB, CE, CU electrolytes. | ||
| VOC (mV) | JSC (mA cm−2) | FF (%) | PCE (%) | JSC (mA cm−2) from IPCE | |
|---|---|---|---|---|---|
| EMITCB | 689 ± 1 | 11.5 ± 0.3 | 68 ± 2 | 5.4 ± 0.1 | 10.4 ± 0.1 |
| CE0.5 | 658 ± 0 | 11.2 ± 0.3 | 69 ± 1 | 5.1 ± 0.2 | 9.9 ± 0.1 |
| CE1 | 665 ± 9 | 10.0 ± 1.2 | 61 ± 3 | 4.0 ± 0.5 | 9.5 ± 0.1 |
| CE1.5 | 648 ± 3 | 8.5 ± 1.9 | 67 ± 5 | 3.6 ± 0.6 | 9.9 ± 0.1 |
| CU0.5 | 723 ± 2 | 10.3 ± 0.2 | 69 ± 2 | 5.1 ± 0.2 | 9.3 ± 0.2 |
| CU1 | 745 ± 1 | 9.0 ± 0.3 | 64 ± 1 | 4.3 ± 0.2 | 7.6 ± 0.2 |
| CU1.5 | 737 ± 3 | 5.7 ± 1.5 | 73 ± 8 | 3.0 ± 0.4 | 7.5 ± 0.1 |
The shape of J–V curves shows the photovoltaic performance of all devices, demonstrating that DES-CE and DES-CU could function well as solvents in DSC devices.47 The forward and backward J–V curves were identical, indicating no hysteresis in the measurement (Fig. S9†). The JSC of CE0.5 devices was 11.2 ± 0.3 mA cm−2, comparable to 11.5 ± 0.3 mA cm−2 of EMITCB ones. EMITCB was considered a standard DSC ionic liquid solvent for its high JSC, thanks to its low viscosity and high conductivity, 18 cP (25 °C) and 15.1 mS cm−1 (20 °C),48 respectively. In our study, DES-CE had viscosity 37 cP (25 °C)47 and conductivity of 7.61 mS cm−1 (20 °C),49 comparable to those of EMITCB. In contrast, the viscosity of DES-CU was 750 cP (25 °C), much higher than those of DES-CE and EMITCB. Furthermore, it had lower conductivity, 0.199 mS cm−1 (40 °C), than those of the other two solvents.50 These underlied CU0.5's JSC value of 10.3 ± 0.2 mA cm−2, lower than that of CE0.5 (11.2 ± 0.3 mA cm−2). However, the JSC value of CU0.5 was still high compared to its disadvantageous nature, e.g., its low viscosity. For the cells in the same electrolyte group, when increasing the amount of DES-CE by 3 times, JSC dropped from 11.2 ± 0.3 mA cm−2 to 8.5 ± 1.9 mA cm−2 (Table 1). Moreover, it dropped nearly 50% of the initial value in the case of DES-CU.
The overall conversion efficiency of EMITCB was 5.4 ± 1%, the highest among the tested electrolytes. CE0.5 and CU0.5 achieved the same conversion efficiency, 5.1 ± 0.2%, which was a very competitive performance for any electrolyte at the same cost range as of these two. Even though the VOC of CE0.5 devices was about 30 mV, lower than that of EMITCB, CE0.5 still achieved this impressive efficiency by maintaining a JSC as high as that of the standard electrolyte. Although the initial JSC of CU0.5 devices performed with JSC was around 1 mA cm−2, lower than that of CE0.5, it was maintained over a broader potential range (Fig. 2).
 | ||
| Fig. 2 Current–voltage characteristics of DSC devices using different electrolytes with various amounts of DES-CE, DES-CU compared to EMITCB. The light intensity was set at AM1.5, 100 mW cm−2. | ||
The fill factor value was strongly impacted by the series resistance,51 but that was not the case for the observed results. Although the series resistance was around 20 Ω for all devices, the fill factor considerably varied between 60 to 70%.
Regarding the open-circuit potential (VOC), the CU electrolyte group yielded the highest VOC value, while the CE group had the VOC value lower than EMITCB (Fig. 1). Considering CU1, CE1, and EMITCB electrolytes with an equivalent amount of solvents, the VOC measured in CU1 was 745 ± 1 mV, significantly higher than EMITCB and CE1, 689 ± 1 mV and 665 ± 9 mV, respectively. Thus, DES-CU had a positive effect on improving the VOC. We speculated that this effect might come from the restriction of charge leakage from the TiO2–electrolyte interface, and proceeded to investigate with EIS analysis.
C. EIS of full cell in the dark
Under dark conditions with the applied bias voltage, dye-uncovered mesoporous TiO2 electrode donates an electron to I3−, while the reduction of I3− to I− occur at the Pt-FTO surface.52 The charge transfer process at the Pt-FTO and electrolyte interface is described as a charge transfer resistance RCE in parallel with a constant phase element CPECE. The charge transfer process at the TiO2 electrode is a coupled parallel to RPE and CPEPE. The diffusion of I3− in the viscous electrolyte is modeled by a Warburg impedance element. RS is the sum of all contact-resistant distributions (Fig. 3). | ||
| Fig. 3 Nyquist plot of DSC devices using EMITCB, CE1, CU1 electrolytes in dark condition under −0.7 V bias voltage. From left to right, the three arcs are related to the charge transfer process at the counter electrode–electrolyte interface, carrier transport at photoanode TiO2|dye|electrolyte interfaces, and incomplete arc of electrolyte diffusion. The inset shows the enlarged first arcs. The dots are experimental points; lines are fitted experimental data with the equivalent circuit model attached in the figure. | ||
To reveal the influence of DES on the surface of TiO2 and open circuit potential, we conducted EIS under various bias potentials. From this analysis, the CPE related to the charge accumulation on bare TiO2 the surface was extracted and converted to the capacitance value by eqn (1)
 | (1) |
CPE is proportional exponentially to the bias voltage applied in the EIS measurement or the TiO2 Fermi level as shown in eqn (2).53
 | (2) |
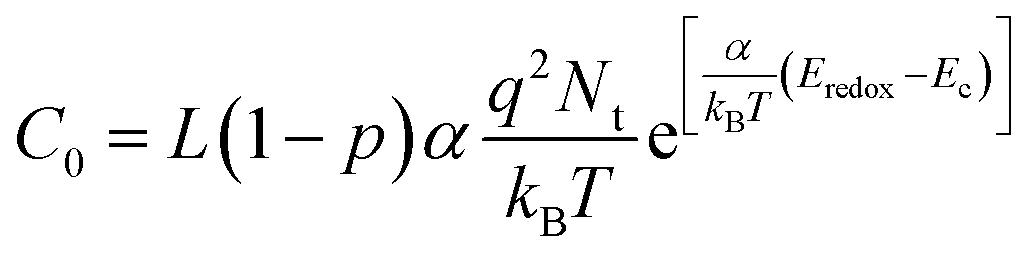 | (3) |
The experimental interface capacitance points were well fitted by the exponential eqn (2) with the shortcut formula as y = a × e(−b × x) (Fig. 4A). Each electrolyte displayed different C curves of which the b values were proportional to the electron trap numbers. The application of a more negative potential on the anode increased the anode's electron density. Consequently, the density of oppositely charged ions at the interface (as shown in C values) and, with it, the probability of electron loss due to the ion exchange continued to surge.
 | ||
| Fig. 4 (A) Interface capacitance of TiO2 interface CPE, and (B) charge transfer resistance of TiO2 interface RPE at various bias potentials under dark conditions. Experimental data (dot) were fitted (line) with R2 = 0.99 and 1 for (A) and (B), respectively. | ||
As can be seen in Fig. 4A, the group of DES-CE had stronger capacitance curves banding upward corresponding to the more negative charge state of the TiO2 electrode. Meanwhile, the DES-CU group curves lay under those of the EMITCB. It is worth noting that opposite trends were observed from the two groups of DES when different amounts of DES were used in the electrolytes. For the electrolytes of DES-CU, the density of oppositely charged ions at the interface was low (small b values), leading to lower electron loss probability and higher VOC. The DES-CE electrolytes displayed the highest b values, meaning the highest electron trap distribution on the anode interface, with little variance. This was consistent with the lowest VOC average with a small variance observed for these electrolytes. We suggested that these results could be explained due to the different molecular structures of the two DES.
Ashish Pandey et al. reported that DES-CE with alcohol functional groups was more dipolar than DES-CU.54 Although the polarity of these two solvents was not quantified, the results showed a correlation with conductivity, since polarity is an essential factor to determine the conductivity. Due to their higher polarity, components in DES-CE were more attracted by the negative charge at the TiO2 interface, resulting in a higher possibility of charge release. For the same reason, DES-CE reached the highest charge density at the lowest potential among other solvents (Fig. 4A). Additionally, although an increasing percentage of DES-CE in the electrolytes diluted the I−/I3− redox couple – the main electron carrier in DSC devices, the electrolyte could reach an even higher charge density at a lower applied voltage (Fig. 4A, arrow sign). However, the trend was observed oppositely when the amount of DES-CU was enhanced in the electrolyte.
The phenomenon observed with DES-CU was similar to the shielding effect, usually observed from conventional nitrogen heterocyclic additives such as N-methylbenzimidazole (NMB) or 4-tert-butylpyridine (4-TBP). NMB or 4-TBP could adsorb onto the TiO2 surface causing the negative shift of the Fermi level of TiO2, as well as preventing the triiodide penetration.55–57 Notably, although NMB was present in all of the electrolytes in our study, its influence on VOC was markedly different from each other. Thus, we speculated that DES-CU, which contains an amine functional group in urea, promoted the activity of NMB. Alternatively, DES-CU might act as an additive in DES electrolyte and help to improve VOC. The effect of choline chloride on VOC values as observed here could be eliminated as it was present both of DESs.
Using EMITCB's VOC (689 mV) as the reference, we calculated the theoretical voltage of DSC devices via the fitted TiO2 interface capacitance equations (Table 2). We assumed that the effect of the series resistance and the charge transfer resistance on TiO2 conduction band shift were negligible compared to the influence of charge distribution and thus normalized the TiO2 charge state of different electrolytes to the same condition. Consequently, the shielding efficiency of individual solvents EMITCB, CE, or CU could vary the activation energy of the charge transfer process at the TiO2 interface and determine the required potential to reach the same charge density with the reference EMITCB.
| EMITCB | CE0.5 | CE1 | CE1.5 | CU0.5 | CU1 | CU1.5 | |
|---|---|---|---|---|---|---|---|
| Calculated potential (mV) | 689 | 657 | 651 | 644 | 725 | 745 | 809 |
| VOC from J–V curves (mV) | 689 ± 1 | 658 ± 0 | 665 ± 9 | 648 ± 3 | 723 ± 2 | 745 ± 1 | 737 ± 3 |
Using the same concept as in the TiO2 interface capacitance inspection, we plotted the TiO2 interface charge transfer resistance as a function of the applied potential and fitted the experimental data by eqn (4) (Fig. 4B).53
 | (4) |
 | (5) |
Our results showed that charge transfer resistance at the TiO2 interface of DES-CU dramatically increased over the low potential area (Fig. 4B). As mentioned above, DES-CU was less polar than DES-CE and thus provided a good insulator environment under the internal electric field of the device. The lines of TiO2 charge transfer resistance lay above the others showed that DES-CU devices displayed higher RPE, indicating that these devices could resist the leakage current better than DES-CE and even EMITCB devices.
4. Conclusions
We prepared two choline chloride-based DESs with ethylene glycol and urea as room-temperature solvents for eco-friendly, inexpensive volatile solvent-free DSC electrolytes and under standard illumination achieved conversion efficiencies as high as 5.1 ± 0.2, comparable to that of the commercial ionic liquid EMITCB. DES-CE displayed higher JSC than that of DES-CU thanks to its lower viscosity and much higher conductivity. In contrast, DES-CU, which contains an amine functional group, showed the shielding effect on TiO2 interface. DES-CU not only played a role as a mediator solvent but also acted as an effective additive in the system that helps improve the VOC. This noteworthy characteristic of DES-CU should be considered when designing other versatile DES-based electrolytes for eco-friendly DSCs with nonvolatile, non-flammable, low-cost, and scalable solvents. This type of DES-electrolyte is promising for large-scale production of DSC. Our work also demonstrated that the capacitance–potential curves from EIS could be used as a simple tool to estimate the theoretical VOC values, with application potential in the high-throughput evaluation of electrolyte systems or additives on DSC devices.Author contributions
De Nguyen: methodology, validation, investigation, writing – original draft. Tuan Van Huynh: methodology, investigation, formal analysis, writing – original draft. Vinh Son Nguyen: investigation, formal analysis. Phuong-Lien Doan Cao: investigation, formal analysis. Hai Truong Nguyen: formal analysis, writing – original draft, writing – review & editing. Tzu-Chien Wei: supervision, writing – review & editing. Phuong Hoang Tran: supervision, conceptualization, formal analysis, data curation, visualization, writing – review & editing. Phuong Tuyet Nguyen: funding acquisition, supervision, conceptualization, formal analysis, data curation, visualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by Vietnam National University HoChiMinh City (VNU-HCM) under grant number VL2020-18-04. The authors thank Dr Trinh-Don Nguyen for critically proofreading the manuscript.References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan and G. Luo, Chem. Rev., 2015, 115, 2136–2173 CrossRef CAS PubMed.
- S. P. Mohanty and P. Bhargava, Electrochim. Acta, 2015, 168, 111–115 CrossRef CAS.
- J. Preat, D. Jacquemin and E. A. Perpète, Energy Environ. Sci., 2010, 3, 891–904 RSC.
- M. I. Asghar, K. Miettunen, J. Halme, P. Vahermaa, M. Toivola, K. Aitola and P. Lund, Energy Environ. Sci., 2010, 3, 418–426 RSC.
- P. T. Nguyen, R. Degn, H. T. Nguyen and T. Lund, Sol. Energy Mater. Sol. Cells, 2009, 93, 1939–1945 CrossRef CAS.
- P. T. Nguyen, A. R. Andersen, E. M. Skou and T. Lund, Sol. Energy Mater. Sol. Cells, 2010, 94, 1582–1590 CrossRef.
- P. T. Nguyen, B. X. T. Lam, A. R. Andersen, P. E. Hansen and T. Lund, Eur. J. Inorg. Chem., 2011, 2011, 2533–2539 CrossRef.
- P. Wang, S. M. Zakeeruddin, J.-E. Moser and M. Grätzel, J. Phys. Chem. B, 2003, 107, 13280–13285 CrossRef CAS.
- R. Kawano, H. Matsui, C. Matsuyama, A. Sato, M. A. B. H. Susan, N. Tanabe and M. Watanabe, J. Photochem. Photobiol., A, 2004, 164, 87–92 CrossRef CAS.
- T. Lund, P. T. Nguyen, H. M. Tran, P. Pechy, S. M. Zakeeruddin and M. Grätzel, Sol. Energy, 2014, 110, 96–104 CrossRef CAS.
- P. T. Nguyen, T. A. P. Phan, N. H. T. Ngo, T. V. Huynh and T. Lund, Solid State Ionics, 2018, 314, 98–102 CrossRef CAS.
- P. T. Nguyen, N. P. D. Nguyen and L. T. Nguyen, J. Phys. Chem. Solids, 2018, 122, 234–238 CrossRef CAS.
- W. Kubo, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, Chem. Commun., 2002, 374–375, 10.1039/B110019J.
- D. Zhao, Y. Liao and Z. Zhang, Clean: Soil, Air, Water, 2007, 35, 42–48 CAS.
- M. Bidikoudi, L. F. Zubeir and P. Falaras, J. Mater. Chem. A, 2014, 2, 15326–15336 RSC.
- C.-T. Li, C.-P. Lee, C.-T. Lee, S.-R. Li, S.-S. Sun and K.-C. Ho, ChemSusChem, 2015, 8, 1244–1253 CrossRef CAS PubMed.
- H. H. Lin, J. D. Peng, V. Suryanarayanan, D. Velayutham and K. C. Ho, J. Power Sources, 2016, 311, 167–174 CrossRef CAS.
- J.-D. Decoppet, S. B. Khan, M. S. A. Al-Ghamdi, B. G. Alhogbi, A. M. Asiri, S. M. Zakeeruddin and M. Grätzel, Energy Technol., 2017, 5, 321–326 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. P. Abbott, J. C. Barron, K. S. Ryder and D. Wilson, Chem.–Eur. J., 2007, 13, 6495–6501 CrossRef CAS PubMed.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- W. Guo, Y. Hou, S. Ren, S. Tian and W. Wu, J. Chem. Eng. Data, 2013, 58, 866–872 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- L. I. N. Tomé, V. Baião, W. da Silva and C. M. A. Brett, Appl. Mater. Today, 2018, 10, 30–50 CrossRef.
- B.-Y. Zhao, P. Xu, F.-X. Yang, H. Wu, M.-H. Zong and W.-Y. Lou, ACS Sustainable Chem. Eng., 2015, 3, 2746–2755 CrossRef CAS.
- S. Khandelwal, Y. K. Tailor and M. Kumar, J. Mol. Liq., 2016, 215, 345–386 CrossRef CAS.
- B. L. Gadilohar and G. S. Shankarling, J. Mol. Liq., 2017, 227, 234–261 CrossRef CAS.
- P. Vitale, V. M. Abbinante, F. M. Perna, A. Salomone, C. Cardellicchio and V. Capriati, Adv. Synth. Catal., 2017, 359, 1049–1057 CrossRef CAS.
- K. O. Wikene, H. V. Rukke, E. Bruzell and H. H. Tonnesen, J. Photochem. Photobiol., B, 2017, 171, 27–33 CrossRef CAS PubMed.
- O. S. Hammond, K. J. Edler, D. T. Bowron and L. Torrente-Murciano, Nat. Commun., 2017, 8, 14150–14157 CrossRef CAS PubMed.
- C. Vidal, L. Merz and J. García-Álvarez, Green Chem., 2015, 17, 3870–3878 RSC.
- W.-H. Zhang, M.-N. Chen, Y. Hao, X. Jiang, X.-L. Zhou and Z.-H. Zhang, J. Mol. Liq., 2019, 278, 124–129 CrossRef CAS.
- M. J. Rodríguez-Álvarez, J. García-Álvarez, M. Uzelac, M. Fairley, C. T. O'Hara and E. Hevia, Chem.–Eur. J., 2018, 24, 1720–1725 CrossRef PubMed.
- J. Chen, M. Liu, Q. Wang, H. Du and L. J. M. Zhang, Molecules, 2016, 21, 1383 CrossRef PubMed.
- A. H. Panhwar, M. Tuzen and T. G. Kazi, Talanta, 2018, 178, 588–593 CrossRef CAS PubMed.
- R. A. Zounr, M. Tuzen, N. Deligonul and M. Y. Khuhawar, Food Chem., 2018, 253, 277–283 CrossRef CAS PubMed.
- D. Carriazo, M. C. Serrano, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, Chem. Soc. Rev., 2012, 41, 4996–5014 RSC.
- H.-R. Jhong, D. S.-H. Wong, C.-C. Wan, Y.-Y. Wang and T.-C. Wei, Electrochem. Commun., 2009, 11, 209–211 CrossRef CAS.
- C. L. Boldrini, N. Manfredi, F. M. Perna, V. Trifiletti, V. Capriati and A. Abbotto, Energy Technol., 2017, 5, 345–353 CrossRef CAS.
- P. T. Nguyen, T.-D. T. Nguyen, V. S. Nguyen, D. T.-X. Dang, H. M. Le, T.-C. Wei and P. H. Tran, J. Mol. Liq., 2019, 277, 157–162 CrossRef CAS.
- G. García, S. Aparicio, R. Ullah and M. Atilhan, Energy Fuels, 2015, 29, 2616–2644 CrossRef.
- J.-L. Lan, C.-C. Wan, T. C. Wei, W.-C. Hsu, C. Peng, Y.-H. Chang and C.-M. Chen, Int. J. Electrochem. Sci., 2011, 6, 1230–1236 CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- C. D'Agostino, R. C. Harris, A. P. Abbott, L. F. Gladden and M. D. Mantle, Phys. Chem. Chem. Phys., 2011, 13, 21383–21391 RSC.
- S. Seki, N. Serizawa, K. Hayamizu, S. Tsuzuki, Y. Umebayashi, K. Takei and H. Miyashiro, J. Electrochem. Soc., 2012, 159, A967–A971 CrossRef CAS.
- A. P. Abbott, R. C. Harris and K. S. Ryder, J. Phys. Chem. B, 2007, 111, 4910–4913 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper and S. Gray, ChemPhysChem, 2006, 7, 803–806 CrossRef CAS PubMed.
- Q. Wang, S. Ito, M. Grätzel, F. Fabregat-Santiago, I. Mora-Seró, J. Bisquert, T. Bessho and H. Imai, J. Phys. Chem. B, 2006, 110, 25210–25221 CrossRef CAS PubMed.
- Q. Wang, J.-E. Moser and M. Grätzel, J. Phys. Chem. B, 2005, 109, 14945–14953 CrossRef CAS PubMed.
- S. R. Raga, E. M. Barea and F. Fabregat-Santiago, J. Phys. Chem. Lett., 2012, 3, 1629–1634 CrossRef CAS PubMed.
- A. Pandey, R. Rai, M. Pal and S. Pandey, Phys. Chem. Chem. Phys., 2014, 16, 1559–1568 RSC.
- T. Stergiopoulos, E. Rozi, C.-S. Karagianni and P. Falaras, Nanoscale Res. Lett., 2011, 6, 307 CrossRef PubMed.
- H. Kusama and H. Arakawa, J. Photochem. Photobiol., A, 2004, 162, 441–448 CrossRef CAS.
- T. A. P. Phan, N. P. Nguyen, L. T. Nguyen, P. H. Nguyen, T. K. Le, T. V. Huynh, T. Lund, D.-H. Tsai, T.-C. Wei and P. T. Nguyen, Appl. Surf. Sci., 2020, 509, 144878 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03273a |
| This journal is © The Royal Society of Chemistry 2021 |