
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGel-polymer electrolytes based on polyurethane ionomers for lithium power sources
I. M. Davletbaeva *a,
A. A. Nizamova,
A. V. Yudinab,
G. R. Baymuratovab,
O. V. Yarmolenkob,
O. O. Sazonov*a and
R. S. Davletbaevc
*a,
A. A. Nizamova,
A. V. Yudinab,
G. R. Baymuratovab,
O. V. Yarmolenkob,
O. O. Sazonov*a and
R. S. Davletbaevc
aKazan National Research Technological University, 68 Karl Marx Str., Kazan, Republic of Tatarstan 420015, Russian Federation. E-mail: davletbaeva09@mail.ru; sazonov.oleg2010@gmail.com
bInstitute of Problems of Chemical Physics of RAS, Acad. Semenov Avenue 1, Chernogolovka, Moscow Region, 142432, Russian Federation
cKazan National Research Technical University Named After A. N. Tupolev – KAI, 10 Karl Marx Str., Kazan, Republic of Tatarstan 420111, Russian Federation
First published on 17th June 2021
Abstract
Polyurethanes based on the aminoethers of ortho-phosphoric acid and polyisocyanates of an aliphatic nature were studied as a substrate for the preparation of new polymer electrolyte. The conductivity of polyurethane ionomer samples obtained using the optimal amount of aliphatic polyisocyanates and after keeping them in a 1 M LiBF4 solution in γ-butyrolactone reaches 0.62 mS cm−1. It has been established that the transport of positively charged ions through the polymer matrix is due to the formation of clusters of phosphate ions and their association into the conducting channels. The introduction of carboxylate ions into the conducting channels by modifying the aminoethers of ortho-phosphoric acid with phthalic anhydride leads to an increase in their size and rise in the mobility of cations. As a result, the conductivity of polyurethane gel electrolytes increased to 2.1 mS cm−1.
1 Introduction
Currently, lithium-ion batteries (LIB) are widely used in electronic devices such as mobile phones, laptops, and digital cameras due to their useful array of characteristics, including low weight, high energy density, minimal memory effect, fast charging, low self-discharge, and environment-friendliness.1–7 The main component of the power source, which determines the energy consumption of the system, is the electrolyte. LIBs use liquid electrolytes, which are solutions of lithium salts in aprotic organic solvents, the ionic conductivity of which at room temperature can reach values of 10−3 to 10−2 S cm−1.8 However, during the electrode processes (intercalation–deintercalation of lithium ions into the anode and cathode materials), the electrolyte decomposes, which ultimately can lead to such undesirable consequences as a decrease in the service life of devices, leakage and ignition.9–13 In order to make LIB's safer, it was decided to replace the liquid electrolyte with a polymer one.14,15 The use of polymer electrolytes (PE), among other reasons, makes it possible to increase their capacity by improving the design and service life.16,17PE are of two types: solid electrolytes and gel electrolytes.18 Many researchers have proven that the use of solid polymer electrolytes (SPE) with their inherent high electrochemical stability and good mechanical properties can solve the problems associated with the use of liquid electrolytes. At the same time, despite the advantages inherent in SPEs, their practical application is limited due to the low ionic conductivity at room temperature.19–25 For example, for a well-studied SPE based on polyethylene oxide (PEO), the ionic conductivity is in the range from 10−8 to 10−4 S cm−1.26,27 The reason for the low level of conductivity in this case is the appearance of obstacles to the transport of lithium ions, which, due to the transition of complexes into a more ordered structure, appear below the crystallization temperature.28–30
To increase the ionic conductivity, it was proposed to plasticize SPE with organic solutions of a lithium salt and ionic liquids.31–39 Retention of lithium ions solutions by a polymer matrix leads to the formation of polymer gel electrolytes (GPE),40 which simplify the technology of manufacturing various electrochemical devices with effective electrode–electrolyte contacts.24–28
At this moment, polymer matrices based on polyethylene oxide (PEO),41–45 polycarbonate (PC),46,47 polyacrylonitrile (PAN),48 poly(methyl methacrylate) (PMMA)49,50 and polyvinylidene fluoride (PVDF)51 are well studied. GPEs obtained from these polymer systems have a fairly high ionic conductivity, reaching about 10−3 S cm−1. However, the low mechanical parameters52–57 of the GPE limit the area of their practical application. Currently, the search continues for polymer systems to create GPE combining high conductivity and the required level of mechanical strength. In this regard, GPEs based on polyurethanes are interesting and promising.58–61 In works,60,61 PEs were obtained, exhibiting high ionic conductivity in the range of 10−4 to 10−3 S cm−1 at room temperature and good mechanical properties. Also, one of the promising directions, in our opinion, could be the development of GPEs based on polyurethanes of ionomeric nature. Polyurethane ionomers refer to ionomers containing from 1 to 15 mol% ionogenic groups in their main chains.62 This feature leads to a high probability of self-organization with formation of various types of nano- and microstructures containing ionic clusters and ion conductive channels.63–68 The volume of the conducting channels is the main factor determining the conductivity of such polymers. Therefore, by influencing this parameter, it is possible to obtain high ionic conductivity.
In works,69,70 polyurethane ionomers (AEPA–PU) based on aminoethers of ortho-phosphoric acid (AEPA) were obtained. It was found that during the preparation of AEPA using triethanolamine as a catalyst and a branching center, a branched compound is formed, in which, due to incomplete etherification of H3PO4 with triethanolamine and polyoxypropylene glycol, phosphate anions are present (Fig. 1).
 | ||
| Fig. 1 AEPA formation. | ||
The aim of this work was synthesis and studying new polyurethane gel electrolytes based on branched aminoethers of ortho-phosphoric acid modified with phthalic anhydride.
2 Experimental
2.1 Materials
Polypropylene glycol (PPG), (Wanol 2310) was purchased from Wanhua Chemical (Beijing, China). Triethanolamine (TEOA), acetone were obtained from Ltd. “Component-reaktiv” (Moscow, Russia). 85% aqueous solution of ortho-phosphoric acid (OPA) was purchased from Ltd “MCD-Chemicals” (Moscow, Russia). Phthalic anhydride was obtained from “Kupavnareaktiv” (Moscow, Russia). Dibutyltindilaurate (DD) was obtained from “Sigma-Aldrich” (United States of America). Aliphatic polyisocyanate (PIC) (Attonate AL 75) obtained from “Attica chemicals” (Lithuania, Siauliai). The liquid electrolyte (1 M LiBF4 solution in γ-butyrolactone) was obtained from “Ekotekh” (Chernogolovka, Moscow region).2.2 Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[H3PO4]:[PPG] = 1:6:6. The calculated amount of OPA and PPG was placed in a round-bottom flask, mixed for two minutes, and then TEOA was added to the reaction system. Within two hours, the reaction mass was stirred at T = 80 °C and a residual pressure of 0.7 kPa.
:[H3PO4]:[PPG] = 1:6:6. The calculated amount of OPA and PPG was placed in a round-bottom flask, mixed for two minutes, and then TEOA was added to the reaction system. Within two hours, the reaction mass was stirred at T = 80 °C and a residual pressure of 0.7 kPa.To obtain AEPA–PA certain amounts of phthalic anhydride was added in the AEPA. The mole ratios between the components were [TEOA]:[H3PO4]:[PPG]:[PA] = 1:6:6:(0.1 ÷ 3.0). The synthesis at T = 90 °C and the residual pressure of 0.7 kPa was carried out until PA completely reacted. The AEPA obtained in this way were named as AEPA-(0.1 ÷ 3.0)PA.
2.3 Preparation of polyurethane gel electrolytes
The prepared polymer films were cut into circular specimens with a 5 mm diameter. Then the specimens were swelled in 1 M LiBF4 in γ-butyrolactone at 40 °C for an hour and used for the ionic conductivity measurement.2.4 The battery assembly
In this work batteries prototypes with a cathode based on the lithium salt of the tetraazapentacene derivative LiOTAP71 were assembled. The electrochemical reduction and oxidation of LiOTAP is shown in Scheme 1. Each molecule of LiOTAP can undergo 8-electron oxidation releasing also eight Li+ cations, which corresponds to the theoretical specific capacity of ∼468 mA h g−1. | ||
| Scheme 1 Electrochemical reduction and oxidation of LiOTAP.71 | ||
The electrochemical performance of LiOTAP was evaluated in the coin-type lithium batteries. The cathode composition comprising of 45 wt% of LiOTAP, 50 wt% of conductive carbon black (Timical Super C65) and 5 wt% of PVDF polymer binder (Kynar Flex HSV 900, Arkema France) was dispersed in N-methylpyrrolidone (NMP, 99%, Aldrich) to obtain ∼12% suspension, which was homogenized mechanically (Isolab homogenizer “Light Duty”) and then by sonication (SONOPULS HD 3200, 100 W).
Cathode slurry was applied onto the current collector (20 μm thick Al-foil) using tape coating (Gelon Lib. coater). The resulting films were annealed at 120 °C in vacuum (15 mbar). The organic cathode (d = 1.6 cm, active weight leading of 3–4 mg) and lithium anode (d = 1.6 cm) were assembled in coin-type lithium batteries using the polymer electrolyte films with liquid electrolyte activations 1 M LiBF4 solution in gamma-butyrolactone as electrolyte. The assembly of the cells was carried out inside MBraun argon glove box.
2.5 Fourier transform infrared spectroscopy analysis (FTIR)
The FTIR spectra were recorded on an InfraLUM FT 08 Fourier transform spectrometer (Lumex, St. Petersburg, Russia) using the attenuated total reflection technique in the spectral range of 3800 to 600 cm−1. The spectral resolution was 2 cm−1 and the number of scans was 16.2.6 Viscosity and density measurements
The dynamic viscosity of the samples was determined at the T = 30 °C and atmospheric pressure on an SVM 3000 Stabinger Viscometer (Anton Paar, Austria), with a systematic error of ±0.35% of the measured value. At the same time, the density of the samples was determined with a systematic error of 0.0005 g cm−3.2.7 Tensile stress–strain measurements
Tensile stress–strain measurements were obtained from the film samples of size 40 mm × 15 mm using Universal Testing Machine Inspekt mini (Hegewald & PeschkeMeß-und Prüftechnik GmbH, Nossen, Germany) at 293 ± 2 K and 1 kN. The crosshead speed was set at 50 mm min−1 and the test continued until sample failure. Minimums of five tests were analyzed for each sample and the average values were reported.2.8 Measurement of temperature dependence of dielectric loss tangent
The temperature dependence of the dielectric loss tangent (DLT) of polymer samples was registered in the temperature range from −120 °C to 160 °C at a frequency of 1 kHz. A facility consisted of the measuring cell that was placed in a Dewar vessel filled with nitrogen and to which an E7-20 RLC-meter and a B7-78 universal voltmeter functioning as a precision thermometer were connected.2.9 Measurements of the surface tension
The droplet counting method was used to determine the surface tension (σ). The basis of the calculations is the equation, where the weight of the drop that comes off the pipette is proportional to the surface tension of the fluid and the radius of the pipette (R): m = 2πRσ/g, where: g – is acceleration of gravity; m – is the drop mass of the test liquid. Following this equation: σ = mg/2πR. Further, according to the obtained results was constructed characteristic curve of the surface tension (σ) from concentration (C).2.10 Thermomechanical analysis (TMA)
The thermomechanical curves of polymer samples were obtained using TMA 402 F (Netzsch, Selb, Germany) thermomechanical analyzer in the compression mode. The sample thickness was 2 mm, and the rate of heating was 3 °C min−1 from 20 °C to 250 °C in the static mode. The load was 2 N.2.11 Mechanical loss tangent measurements (MLT)
The MLT curves of polymer samples were taken using the dynamic mechanical analyser Netzsch DMA 242 (Germany) in the mode of oscillating load. Force and stress–strain correspondence were calibrated using a standard mass. The thickness of the sample was 2 mm. Viscoelastic properties were measured under nitrogen. The samples were heated from 20 °C to 250 °C at the rate of 3 °C min−1 and frequency of 1 Hz. The mechanical loss tangent was defined as the ratio of the viscosity modulus G′′ to the elasticity modulus G′.2.12 Thermal gravimetric analysis (TGA)
TGA was performed using STA-600 TGA-DTA combined thermal analyzer (PerkinElmer, Waltham, MA, USA). The samples (0.1 g) were loaded in alumina pans and heated from 30 to 750 °C at a rate of 5 K min−1 in a nitrogen atmosphere.2.13 Ionic conductivity measurement
Electrochemical impedance spectroscopy (EIS) was used to study the ionic conductivities of prepared GPEs. The electrochemical impedance of GPEs films was measured at ambient temperature in the frequency range from 500 kHz to 1 kHz at an AC signal amplitude of 10 mV on an Elins Z-2000 (Russia) impedance meter using hermetic symmetric cells equipped with stainless steel (SS) blocking electrodes. In each series of experiments, four samples were used in impedance measurements. The bulk resistance (Rb) expressed in ohms were recalculated to specific conductivities (σsp) expressed in S cm−1 for a given specimen thickness (d) and surface area (A) of the specimens from eqn (1):
 | (1) |
The mean value obtained from impedance data for four specimens was taken as the result.
2.14 The electrochemical stability
The GPE electrochemical stability window was voltammetrically recoded with an Elins P-8nano potentiostat in a Li/GPE/SS coin cell, wherein a polymer film activated with liquid electrolyte was placed between the lithium and SS electrodes.2.15 Lithium ion transference number measurement
The transport numbers of Li+ ions were measured by chronoamperometry in Li/GPE/Li cells. The coin-type cells C2032 were assembled in MBraun argon box. The electrodes represented circles of lithium metal foil with diameter of 16 mm between which the polymer electrolyte film of the same size was placed. The cells were preliminarily exposed at room temperature for 1 week to let the formation of a solid-electrolyte layer, similarly in the work.72 After this, the cells were polarized at voltage (ΔV) of 10 mV for 1 h by means of P-2X device (Elins, Russia). The initial current (I0) and the equilibrium (steady-state) current (ISS) were measured. The cell impedance was measured before and after polarization in the frequency range from 1 to 2 × 106 Hz with signal amplitude of 10 mV on the aforementioned device. From impedance hodographs, we calculated the resistance at the electrode/electrolyte interface before (R0) and after polarization (RSS). The transport numbers of Li+ ions (t+), were calculated by the following equation:
 | (2) |
2.16 Charge–discharge characteristics measurement
The electrochemical performance of the batteries was evaluated using battery analyzer BTS-5V10 mA (Neware Technology Ltd.) by performing charge/discharge cycling.The Li/GPE/LiOTAP half-cells were tested in a galvanostatic regime with the current rate of C/10 within the potential range of 0.7–3.5 V.
3 Results and discussion
3.1 AEPA–PU characterization
To obtain polyurethanes, a wide range of isocyanates are used, which are divided into two main groups – isocyanates of aromatic and aliphatic nature. According to preliminary studies, the use of aromatic isocyanates results in the formation of a relatively rigid polymer matrix. As a result, high values of ionic conductivity were not achieved for polymer gel electrolytes obtained from such polyurethanes. In this regard, polyisocyanates of an aliphatic nature (PIC) were used, which are branched due to the biuret bonds of hexamethylene diisocyanate derivatives.Since the content of PIC can have a significant effect on the topological structure of polyurethanes and, accordingly, on their free volume, the effect of the relative PIC content on the properties of AEPA–PU was investigated. It turned out that the PIC content does not affect the characteristics of polymers thermal stability. So, for a number of samples obtained in the range of the ratio [PIC]:[AEPA] = 1:(1 ÷ 1.5) (wt%), the temperature of 5% weight loss is 275 °C, the temperature of 10% weight loss is in the region of 300 °C, and 50% of the weight loss occurs at 335 °C.
According to the physical and mechanical tests (Fig. 2), the maximum strength of AEPA–PU is achieved at a 1.3 ÷ 1.5 mass excess of PIC relative to AEPA. High stress values at deformation of less than 10% are a consequence of the high density of the polymer network nodes of the investigated AEPA–PU. The observed high elastic deformation is a consequence of the flexible-chain polyether component combination into its own microphase. According to the data presented, the smallest possible degree of crosslinking is achieved at the ratio [PIC]:[AEPA] = 1:1 (wt%). With a further decrease in the PIC content in the polymer, crosslinking sites sufficient to obtain a material with stable geometric characteristics can not be formed.
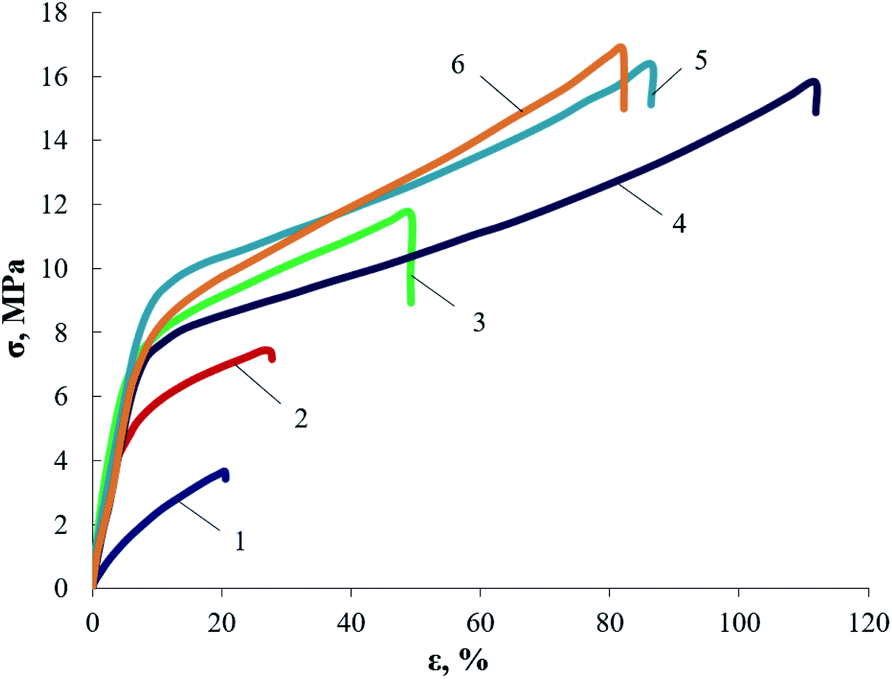 | ||
| Fig. 2 Tensile tests for AEPA–PU, obtained at ratios of [PIC]:[AEPA] = 1 (1), 1.1 (2), 1.2 (3), 1.3 (4), 1.4 (4), 1.5 (6), wt%. | ||
To obtain a heterophase polymer gel electrolyte, dry samples were kept for 1 hour at 40 °C in a 1 M solution of LiBF4 in γ-butyralactone. The choice of this lithium salt solution is related to the fact that it is the most common electrolyte in lithium power sources, therefore, it is better studied.
According to the dependence of the ionic conductivity on the relative mass excess of PIC of polymer gel electrolytes (Fig. 3), the ionic conductivity of the samples increases after their activation with a liquid electrolyte. The highest values of ionic conductivity are observed for the samples obtained at the ratio [PIC]:[AEPA] = 1:1 (wt%). The conductivity of polyurethane ionomer samples obtained using an optimal amount of aliphatic polyisocyanates and after keeping them in a LiBF4 solution in γ-butyrolactone reaches values of 0.62 mS cm−1 (Fig. 3).
 | ||
| Fig. 3 Dependencies of the ionic conductivity for AEPA–PU,obtained at ratios of [PIC]:[AEPA], wt%. T = 20 °C. | ||
The resistance of the GPE obtained in this way were measured in symmetric cells with blocking stainless steel (SS) electrodes. Fig. 4 shows a typical view of the SS/GPE/SS hodographs for each sample.
 | ||
| Fig. 4 Nyquist plots for the SS/GPE/SS cells. GPE based on AEPA–PU, obtained at ratios of [PIC]:[AEPA] = 1 (1), 1.1 (2), 1.2 (3), 1.3 (4), 1.4 (5), 1.5 (6), wt%. | ||
To establish the features of the supramolecular organization of AEPA–PU, measurings in the low-temperature region using the temperature dependences of the dielectric loss tangent (tgδ) were carried out. The curves show that the temperatures of the onset of segmental mobility (α-transition) are in the region of −20 °C (Fig. 5). Such α-transitions are characteristic for AEPA–PU in the entire used range of the ratio [PIC]:[AEPA] (wt%). The demonstration of low-temperature α-transitions confirms the conclusions that the polyoxypropylene component of AEPA–PU is combined into its own microphase. According to the temperature dependences of the dielectric loss tangent, the course of curves at T > 0 °C for the samples obtained at the ratios [PIC]:[AEPA] = 1:1 (wt%) is flatter and less pronounced in comparison with the course of the curves for the samples obtained at a higher the relative content of PIC. The observed difference in the nature of the temperature dependences of tgδ is a consequence of a significant decrease in the density of the polymer network for the samples obtained with the minimum content of PIC.
 | ||
| Fig. 5 Temperature dependencies of the dielectric loss tangent for AEPA–PU, obtained at ratios of [PIC]:[AEPA] = 1 (1), 1.1 (2), 1.2 (3), 1.3 (4), 1.4 (5), wt%. | ||
In the temperature range of 80–100 °C, characteristic changes on the TMA and MLT curves are observed (Fig. 6), by which one can judge the appearance of supramolecular formations in AEPA–PU, caused by the clustering of ionic groups. The temperature of the onset of thermodestructive flow (Tf) for the AEPA–PU sample obtained at [PIC]:[AEPA], (wt%) = 1:1 (with the lowest potentially possible degree of crosslinking) is in the region of 195 °C. For specimens cured at a higher PIC content, Tf reaches 220 °C.
 | ||
| Fig. 6 TMA and MLTcurves for AEPA–PU, obtained at ratios of [PIC]:[AEPA] = 1 (1), 1.1 (2), 1.2 (3), 1.3 (4), 1.4 (4), 1.5 (6), wt%. | ||
Under the conditions of AEPA–PU swelling in liquids of a polar nature, the dissociation of P–OH groups becomes possible with the subsequent combination of clusters into ion-conducting channels (Fig. 7).
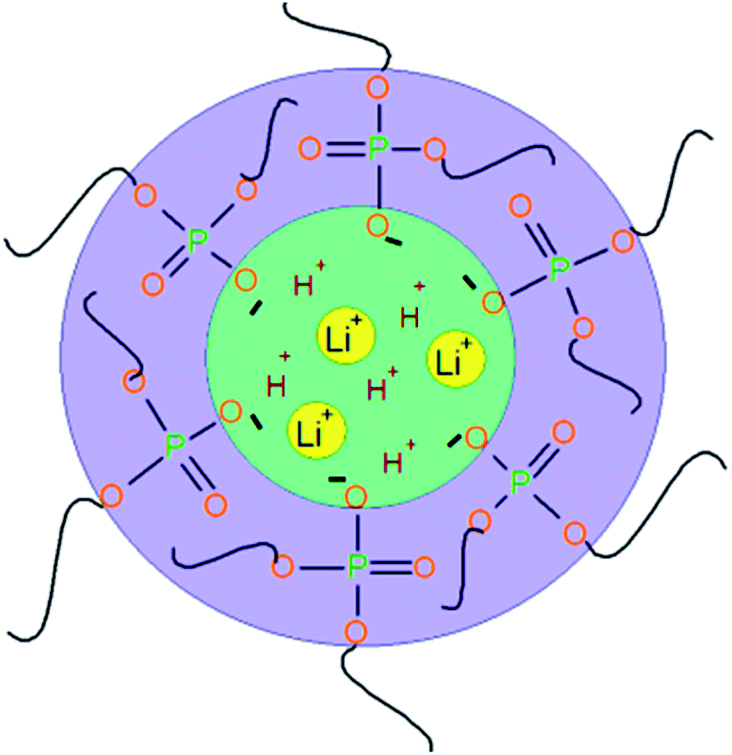 | ||
| Fig. 7 Scheme of ion-conducting channels in polymer gel electrolytes based on AEPA–PU. | ||
The drop in ionic conductivity with an increase in the amount of PIC introduced is explained by an increase in the density of the polymer network and a corresponding decrease in the free volume of AEPA–PU. As a result, the probability of the alignment of ionic clusters into a single conducting channel decreases.
3.2 AEPA–PA and AEPA–PA–PU characterization
To increase the content of ionogenic groups, mobility of protons in the composition of AEPA–PU and to increase the free volume in the polymer matrix due to the creation of steric hindrances in the branched structure of AEPA, phthalic anhydride (PA) was used. The scheme of interaction of AEPA with PA is shown in Fig. 8. | ||
| Fig. 8 Scheme of interaction of AEPA with PA. | ||
The reaction of AEPA with PA is confirmed by the data of FTIR spectroscopy (Fig. 9). The initial AEPA is characterized by the presence of bands at 1720 cm−1 and 1736 cm−1, correlated with the stretching vibrations of phosphates. In the spectra of the product of the AEPA and PA (AEPA–PA) interaction, the bands in the region of 1760 cm−1, caused by the stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in the PA, disappear. In the spectra of AEPA-0.5PA the band 1720 cm−1 disappears while the band 1736 cm−1, caused by stretching vibrations of the CO bond in the composition of the ester group formed here, becomes more pronounced. The intensity of the 1736 cm−1 band increases with an increase in the PA content in the reaction system. The observed changes in the spectra of AEPA, caused by the addition of PA into the composition, indicate the formation of new intermolecular interactions in AEPA–PA.
O bond in the PA, disappear. In the spectra of AEPA-0.5PA the band 1720 cm−1 disappears while the band 1736 cm−1, caused by stretching vibrations of the CO bond in the composition of the ester group formed here, becomes more pronounced. The intensity of the 1736 cm−1 band increases with an increase in the PA content in the reaction system. The observed changes in the spectra of AEPA, caused by the addition of PA into the composition, indicate the formation of new intermolecular interactions in AEPA–PA.
 | ||
| Fig. 9 FTIR spectra of PA (1), AEPA (2), AEPA-0.5PA (3), AEPA-2.3PA (4), AEPA-3.0PA (5). | ||
Since the established maximum conductivity was found for the sample obtained at [PIC]:[AEPA] = 1:1 (wt%), this ratio was used to synthesize the corresponding AEPA–PA–PU. According to the dependence shown in Fig. 10, the introduction of an aromatic carboxyl-containing fragment into the branched structure of AEPA leads to an increase in the conductivity of AEPA–PA–PU in comparison with AEPA–PU by more than 3 times. At the same time, the dependence of the values of conductivity on the content of phthalic anhydride is complex. The view of Nyquist plots for the SS/GPE/SS cells were the same as shown at Fig. 4. The highest values of conductivity are observed for the samples of AEPA-(0.2 ÷ 0.5)PA–PU, obtained at the molar ratio of [PA]:[TEOA] = (0.2 ÷ 0.5):1 in the composition of AEPA–PA, i.e. in the region of relatively low phthalic anhydride content.
 | ||
| Fig. 10 Dependence of the ionic conductivity for AEPA–PA–PU on the molar excess of PA relative to TEOA. T = 20 °C. | ||
Due to the fact that the dependence of the conductivity of AEPA–PA–PU on the content of PA in the composition of AEPA–PA is not additive, it became necessary to study the reasons for the revealed pattern. For these purposes, the dependences of the change in the properties of AEPA–PA and obtained on their basis AEPA–PA–PU on the content of PA in the composition of AEPA–PA were studied.
According to Fig. 11, the dependences of the dynamic viscosity (η) and density (ρ) of AEPA–PA on the PA content are not additive. In the region where an unusual increase in the ionic conductivity of the AEPA–PA–PU samples is observed, there is a sharp increase in the dynamic viscosity and density of AEPA–PA, followed by a slight decrease in their values. Starting from the molar ratio of [PA]:[TEOA] = (0.5 ÷ 3.0):1 in the composition of AEPA–PA, the slope of the dependences becomes smaller.
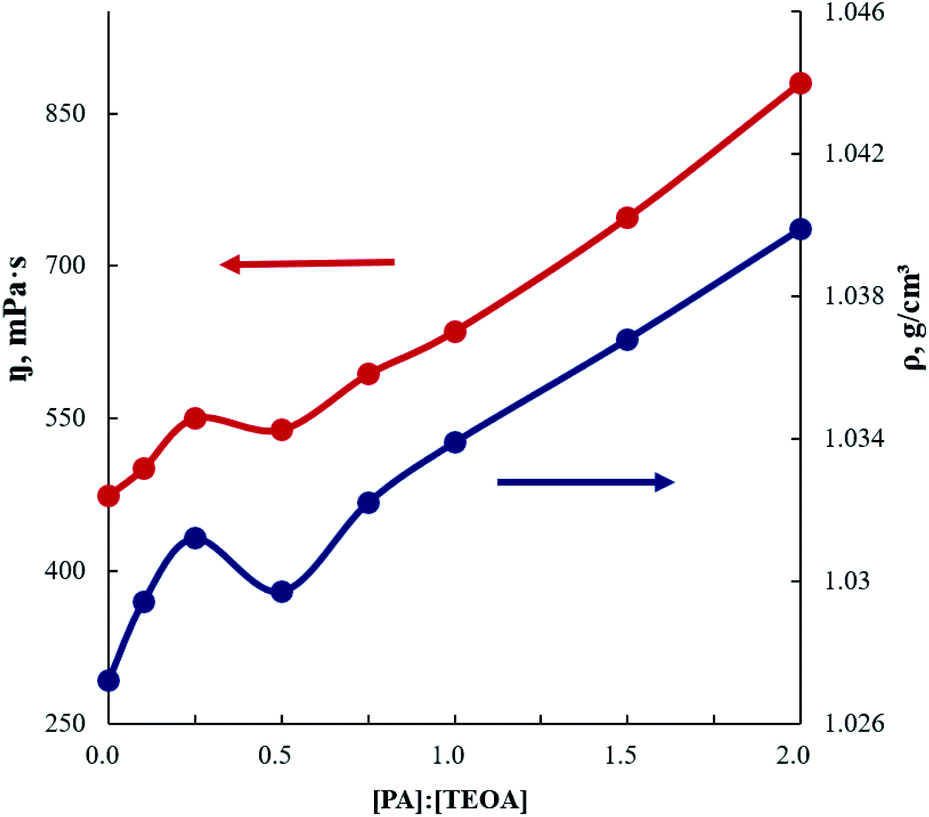 | ||
| Fig. 11 Dependencies of the density and dynamic viscosity for AEPA–PA on the molar excess of PA relative to TEOA. T = 30 °C. | ||
The obtained regularities make it possible to judge that at a small relative content of phthalic anhydride derivatives in the composition of AEPA–PA, cohesive interactions between particles of modified aminoethers of ortho-phosphoric acid are enhanced. The dependences of the AEPA–PA dynamic viscosity and density on molar excess of PA relative to TEOA in the AEPA–PA composition are not additive and has two different areas of manifestation. The peculiarities of the η and ρ values increase are presumably due to the formation of a specific supramolecular structure of AEPA–PA, based on the combination of phosphate ions into clusters (Fig. 7). The introduction of carboxylate anions into such clusters should lead to corresponding changes in the size and packing of these macromolecular formations.
The above reasoning is also supported by the results of measurements of the surface-active properties of AEPA–PA, obtained at the different molar excess of PA relative to TEOA (Fig. 12). It turned out that the nature of the change in the surface tension of AEPA–PA from the molar ratio of [PA]:[TEOA] in the composition of AEPA–PA correlates with the regularities of changes in the ionic conductivity, density, and dynamic viscosity of the obtained compounds. An increase in the surface tension of AEPA–PA in the region of molar ratio [PA]:[TEOA] = (0.1 ÷ 0.6):1 in the composition of AEPA–PA is associated with an increase in the size of cluster structures built on the basis of the corresponding AEPA–PA.
 | ||
| Fig. 12 Surface tension isotherms for AEPA (1)AEPA-0.1PA (2), AEPA-0.25PA (3), AEPA-0.5PA (4), AEPA-1.3PA (5), AEPA-2.25PA (6), AEPA-3.0PA (7). | ||
The fact that the formation of the supramolecular structure of AEPA–PA–PU is due to the peculiarities of the structural organization of the initial AEPA–PA can also be judged by the strength, thermomechanical properties and temperature dependences of the dielectric loss tangent for AEPA–PA–PU obtained at different molar excess of PA relative to TEOA in the AEPA–PA composition.
According to measurements of the temperature dependences of the dielectric loss tangent, in the low-temperature region for the initial AEPA–PU that does not contain PA, an α-transition is observed in the region of −20 °C (Fig. 13). For AEPA–(0.25 ÷ 0.5)PA–PU an abrupt drop in the α-transition temperature to −40 °C occurs. The observed decrease in the temperature of the onset of segmental mobility for AEPA-(0.25 ÷ 0.5)PA–PU is a consequence of the intensification of the processes of release of the polyoxypropylene component into its own microphase.
 | ||
| Fig. 13 Temperature dependencies of the dielectric loss tangent for AEPA–PU (1), AEPA-0.25PA–PU (2), AEPA-0.5PA–PU (3),AEPA-0.9PA–PU (4), AEPA-1.4PA–PU (5), AEPA-1.8PA–PU (6). | ||
A relatively sharp increase in the tgδ values in the temperature range 60–160 °C for AEPA-(0.25 ÷ 0.5)PA–PU samples in comparison with AEPA–PU and AEPA-(0.9 ÷ 1.8)PA–PU (Fig. 13) is due to their looser macromolecular structure. The same conclusion can be drawn from the analysis of the TMA and MLT curves (Fig. 14). Thus, for AEPA–PU a relaxation transition is observed in the range of 80–110 °C, and for AEPA-0.5PA–PU, this transition is shifted to a lower temperature range (60–85 °C).
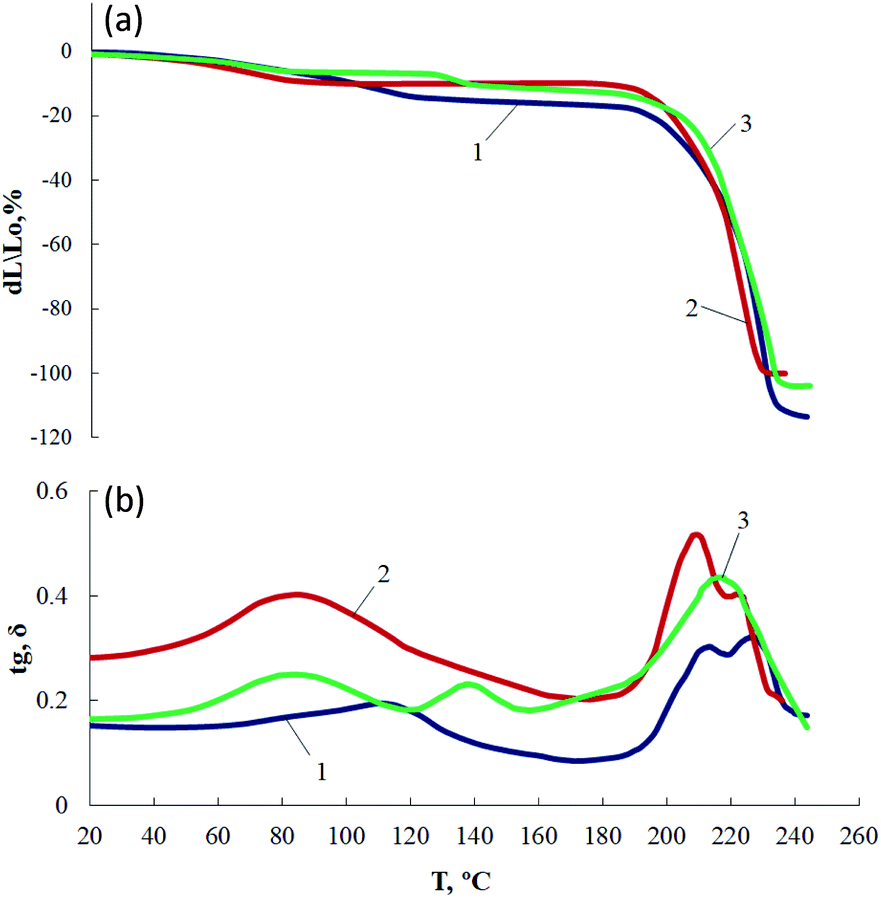 | ||
| Fig. 14 TMA (a) and MLT (b) curves of AEPA–PU (1), AEPA-0.5PA–PU(2), AEPA-0.9PA–PU (3). | ||
An increase in the strength of AEPA–PA–PU with an increase of molar excess of PA relative to TEOA in the AEPA–PA composition confirms the segregation of aromatic ester fragments (Fig. 15). An increase in the values of the relative elongation at break for AEPA-(0.25 ÷ 0.5)PA–PU (Fig. 16) is an additional confirmation of segregation enhancement in these polymers of the flexible polyoxypropylene component into its own microphase.
 | ||
| Fig. 15 Tensile tests of AEPA–PA–PU obtained at the molar ratio of [PA]:[TEOA] = 0 (1); 0.1 (2); 0.2 (3); 0.3 (4); 0.4 (5), 0.5 (6); 0.6 (7); 0.7 (8); 0.8 (9); 0.9 (10); 1.4 (11); 1.8 (12); 2.3 (13); 3.0 (14). | ||
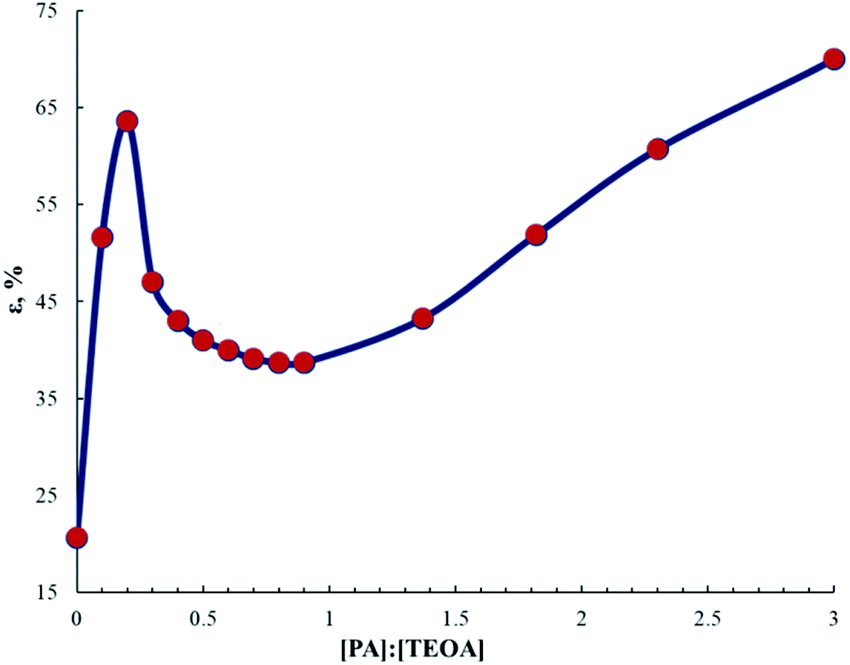 | ||
| Fig. 16 Dependence of elongation at break for AEPA–PA–PU on the molar ratio of [PA]:[TEOA]. | ||
Thus, macromolecular packing centers are formed in AEPA-(0.25 ÷ 0.5)PA–PU, which, as a result of the cooperative effect, lead to significant changes in the processes of supramolecular organization of these polymers. Judging by the noticeable increase in the conductivity of AEPA-(0.25 ÷ 0.5)PA–PU in comparison with AEPA–PU (Fig. 10), the peculiarities of the supramolecular structure of AEPA-(0.25 ÷ 0.5)PA–PU are the reason for the increase in the volume of ion-conducting channels (Fig. 17) and the mobility of cations in the polymer matrix.
 | ||
| Fig. 17 Scheme of ion-conducting channels in polymer gel electrolytes based on AEPA–PA–PU. | ||
At the next stage of the work, the electrochemical stability and lithium ion transference numbers of GPEs obtained on the basis of AEPA–PU, AEPA-0.2PA–PU and AEPA-0.5PA–PU were investigated. Using these polymer electrolytes, prototype batteries with a cathode based on the lithium salt of the tetraazapentacene derivative LiOTAP and lithium as anode were assembled.
Voltammetric studies (Fig. 18) showed that the upper limit of the electrochemical stability window (relative to Li+/Li) of GPE based on AEPA–PU, AEPA-0.2PA–PU and AEPA-0.5PA–PU are equal to 5.37, 5.29 and 3.84 V respectively. It can be seen from Fig. 18 that at the content of PA >0.2 in the polymer, the electrochemical stability of the electrolyte decreases by 1.5 V.
 | ||
| Fig. 18 Voltammograms of Li/GPE/SS cells for electrolytes based on AEPA–PU (1), AEPA-0.2PA–PU (2) and AEPA-0.5PA–PU (3). Potential scan rate 2 mV s−1; potential given vs. Li+/Li pair. | ||
Then the transfer numbers of lithium ions of the polymer electrolytes were measured. Fig. 19 shows typical curves that served the basis for calculating the parameters for eqn (2) by the example GPE based on AEPA–PU.
 | ||
| Fig. 19 Curves that served the basis for calculating transport numbers t+ in GPE based on AEPA–PU: (a) – impedance hodograph of Li/GPE/Li cell before passing electric current (1), after passing the current (2) and (b) – chronoamperogram of Li/GPE/Li cell. | ||
The results of calculating of the transport numbers t+ according to eqn (2) are given in Table 1. It is seen that the Li+ transport number <0.2. Apparently, the main carriers of ionic transport are protons, not lithium ions.
| Polymer matrix of electrolyte | Iss, μA | I0, μA | ΔV, mV | R0, Ω | Rss, Ω | t+ |
|---|---|---|---|---|---|---|
| AEPA–PU | 7.86 | 14.49 | 10.00 | 869.00 | 2659.00 | 0.13 |
| AEPA-0.2PA–PU | 11.00 | 16.00 | 10.00 | 732.00 | 1780.00 | 0.12 |
| AEPA-0.5PA–PU | 12.00 | 16.21 | 10.00 | 613.00 | 812.00 | 0.196 |
According to the charging–discharge characteristics of the batteries (Fig. 20), the maximum discharge capacity of the LiOTAP cathode is 250 mA h g−1 at the second cycle for a prototype battery with GPE based on AEPA-0.2PA–PU.
 | ||
| Fig. 20 The charge–discharge profiles of Li/GPE/LiOTAP cells at C/10 current rate for AEPA–PU (a), AEP-0.2PA–PU (b), AEP-0.5PA–PU (c) for cycles number 2–5. | ||
4. Conclusions
New polyurethane gel electrolytes obtained based on branched aminoethers of ortho-phosphoric acid and aliphatic polyisocyanates have been synthesized and investigated. It is shown that the drop in ionic conductivity with an increase in the amount of introduced polyisocyanate is explained by an increase in the density of the polymer network and a corresponding decrease in the free volume of AEPA–PU. As a result, the likelihood of the alignment of ionic clusters into a single conducting channel, which facilitates the transport of cations through the polymer matrix, decreases. The conductivity of polyurethane ionomer samples obtained using the optimal amount of aliphatic polyisocyanates and after keeping them in 1 M LiBF4 solution in γ-butyrolactone reaches 0.62 mS cm−1.The introduction of carboxylate ions into the conducting channels by modifying the aminoethers of ortho-phosphoric acid with phthalic anhydride leads to an increase in their size and rising in the mobility of positive ions. As a result, the conductivity of polyurethane gel electrolytes increased to 2.1 mS cm−1.
It was found by voltammetry that GPE with maximum conductivity exhibits high electrochemical stability equal to 5.29 V relative to Li+/Li.
Together with the data on ionic conductivity, the results of measurements of the transport number for lithium ions turned out to be interesting, according to which it is in the region of less than 0.2. It was assumed that the increase in conductivity after the modification of AEPA–PU occurs due to a corresponding increase in the number of transfer over lithium cations. However, it turned out that the increase in conductivity is most likely due to an increase in the mobility of protons. Therefore, it can be argued that the conductivity of electrolytes is provided by the joint transfer of both lithium cations and hydrogen, a large proportion of which is probably accounted for by protons.
Despite the small values of the transfer numbers for lithium cations, the maximum discharge capacity of the LiOTAP cathode is 250 mA h g−1 at the second cycle for a prototype battery with GPE based on AEPA-0.2PA–PU. It can be assumed that the high values of the discharge capacity of the cathode are due to the possibility of its operation with both lithium ions and protons.
Thus, we have obtained a promising polymer material for GPE, which can be used in electrochemical devices, which, in turn, requires further research.
Abbreviation
| OPA | ortho-Phosphoric acid |
| TEOA | Triethanolamine |
| PPG | Polypropylene glycol |
| PIC | Aliphatic polyisocyanate |
| PA | Phthalic anhydride |
| PU | Polyurethane |
| PUI | Polyurethane ionomer |
| AEPA | Amino ether of ortho-phosphoric synthesized on the basis of [TEOA]:[H3PO4]:[PPG] = 1:6:6 |
| AEPA-(0.1 ÷ 3.0)PA | Amino ethers of ortho-phosphoric acid synthesized on the basis of [TEOA]:[H3PO4]:[PPG]:[PA] = 1:6:6:(0.1 ÷ 3.0) |
| AEPA-(0.1 ÷ 3.0)PA–PU | Polyurethanes synthesized on the basis of AEPA-(0.1 ÷ 3.0)PA |
Funding
This work was supported by the Russian Science Foundation (grant no. 19-19-00136).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the group of researchers who, within the framework of the project supported by the Ministry of Education and Science of the Russian Federation (project no. AAAA-A19-119071190044-3) (Experiment on Battery prototype testing), developed the cathode material and provided us with the opportunity to use it to assemble the batteries prototypes using the polymer electrolytes we obtained.References
- H. Li, Z. Xu, J. Yang, J. Wang and S. Hirano, Sustainable Energy Fuels, 2020, 4, 5469–5487 RSC.
- Y. Tang, L. Zhang, J. Chen, H. Sun, T. Yang, Q. Liu, Q. Huang, T. Zhu and J. Huang, Energy Environ. Sci., 2021, 14, 602–642 RSC.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- S. Li, S. Q. Zhang, L. Shen, Q. Liu, J. Bin Ma, W. Lv, Y. B. He and Q. H. Yang, Adv. Sci., 2020, 7, 1903088 CrossRef CAS PubMed.
- A. M. Tripathi, W.-N. Su and B. J. Hwang, Chem. Soc. Rev., 2018, 47, 736–851 RSC.
- L. Liu, H. Zhao and Y. Lei, InfoMat, 2019, 1, 74–84 CrossRef CAS.
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC.
- L.-P. Wang, X.-D. Zhang, T.-S. Wang, Y.-X. Yin, J.-L. Shi, C.-R. Wang and Y.-G. Guo, Adv. Energy Mater., 2018, 8, 1801528 CrossRef.
- H. Xu, A. Wang, X. Liu, D. Feng, S. Wang, J. Chen, Y. An and L. Zhang, Polymer, 2018, 146, 249–255 CrossRef CAS.
- G. Feng, Z. Li, L. Mi, J. Zheng, X. Feng and W. Chen, J. Power Sources, 2018, 376, 177–183 CrossRef CAS.
- C. Zuo, M. Yang, Z. Wang, K. Jiang, S. Li, W. Luo, D. He, C. Liu, X. Xie and Z. Xue, J. Mater. Chem. A, 2019, 7, 18871–18879 RSC.
- M. Forsyth, L. Porcarelli, X. Wang, N. Goujon and D. Mecerreyes, Acc. Chem. Res., 2019, 52, 686–694 CrossRef CAS PubMed.
- C. Wang, Y. Yang, X. Liu, H. Zhong, H. Xu, Z. Xu, H. Shao and F. Ding, ACS Appl. Mater. Interfaces, 2017, 9, 13694–13702 CrossRef CAS PubMed.
- Y. Gao, Z. Yan, J. L. Gray, X. He, D. Wang, T. Chen, Q. Huang, Y. C. Li, H. Wang, S. H. Kim, T. E. Mallouk and D. Wang, Nat. Mater., 2019, 18, 384–389 CrossRef CAS PubMed.
- A. Manthiram and X. Y. S. Wang, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- Y. G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D. S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- K. Mishra, N. Yadav and S. A. Hashmi, J. Mater. Chem. A, 2020, 8, 22507–22543 RSC.
- Y. Li, L. Zhang, Z. Sun, G. Gao, S. Lu, M. Zhu, Y. Zhang, Z. Jia, C. Xiao, H. Bu, K. Xi and S. Ding, J. Mater. Chem. A, 2020, 8, 9579–9589 RSC.
- Q. Han, S. Wang, Z. Jiang, X. Hu and H. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 20514–20521 CrossRef CAS PubMed.
- C. Zuo, B. Zhou, Y. H. Jo, S. Li, G. Chen, S. Li, W. Luo, D. He, X. Zhou and Z. Xue, Polym. Chem., 2020, 11, 2732–2739 RSC.
- D. Cao, X. Sun, Q. Li, A. Natan, P. Xiang and H. Zhu, Matter, 2020, 3, 57–94 CrossRef.
- H. Duan, Y. X. Yin, Y. Shi, P. F. Wang, X. D. Zhang, C. P. Yang, J. L. Shi, R. Wen, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2018, 140, 82–85 CrossRef CAS PubMed.
- J. Cheng, G. Hou, Q. Sun, Z. Liang, X. Xu, J. Guo, L. Dai, D. Li, X. Nie, Z. Zeng, P. Si and L. Ci, Solid State Ionics, 2020, 345, 115156 CrossRef CAS.
- S. Choudhury, Z. Tu, A. Nijamudheen, M. J. Zachman, S. Stalin, Y. Deng, Q. Zhao, D. Vu, L. F. Kourkoutis, J. L. Mendoza-Cortes and L. A. Archer, Nat. Commun., 2019, 10, 3091 CrossRef PubMed.
- L. Porcarelli, C. Gerbaldi, F. Bella and J. R. Nair, Sci. Rep., 2016, 6, 19892 CrossRef CAS PubMed.
- I. Shin, J. Nam, K. Lee, E. Kim and T.-H. Kim, Polym. Chem., 2018, 9, 5190–5199 RSC.
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Chem, 2019, 5, 2326–2352 CAS.
- J. Suk, Y. H. Lee, D. Y. Kim, D. W. Kim, S. Y. Cho, J. M. Kim and Y. Kang, J. Power Sources, 2016, 334, 154–161 CrossRef CAS.
- K. Leš and C.-S. Jordan, RSC Adv., 2020, 10, 41296–41304 RSC.
- Y. Ma, J. Ma, J. Chai, Z. Liu, G. Ding, G. Xu, H. Liu, B. Chen, X. Zhou, G. Cui and L. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 41462–41472 CrossRef CAS PubMed.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- J. B. Goodenough and Y. Kim, Mater. Chem., 2010, 22, 587–603 CrossRef CAS.
- S. Khurana and A. Chandra, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 207–218 CrossRef CAS.
- I. Osada, H. de Vries, B. Scrosati and S. Passerini, Angew. Chem., Int. Ed., 2016, 55, 500–513 CrossRef CAS PubMed.
- S. Wang, X. Liu, A. Wang, Z. Wang, J. Chen, Q. Zeng, X. Wang and L. Zhang, Polym. Chem., 2018, 9, 4674–4682 RSC.
- S. Xiong, Y. Liu, P. Jankowski, Q. Liu, F. Nitze, K. Xie, J. Song and A. Matic, Adv. Funct. Mater., 2020, 30, 2001444 CrossRef CAS.
- Q. Liu, Y. Liu, X. Jiao, Z. Song, M. Sadd, X. Xu, A. Matic, S. Xiong and J. Song, Energy Storage Mater., 2019, 23, 105–111 CrossRef.
- J. Shi, Y. Yang and H. Shao, J. Membr. Sci., 2018, 547, 1–10 CrossRef CAS.
- C. Wang, T. Wang, L. Wang, Z. Hu, Z. Cui, J. Li, S. Dong, X. Zhou and G. Cui, Adv. Sci., 2019, 6, 1901036 CrossRef CAS PubMed.
- Z. Li, H.-M. Huang, J.-K. Zhu, J.-F. Wu, H. Yang, L. Wei and X. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 784–791 CrossRef CAS PubMed.
- N. Molinari, J. P. Mailoa and B. Kozinsky, Chem. Mater., 2018, 30, 6298–6306 CrossRef CAS.
- B. Jinisha, K. M. Anilkumar, M. Manoj, V. S. Pradeep and S. Jayalekshmi, Electrochim. Acta, 2017, 235, 210–222 CrossRef CAS.
- Q. Ma, A. Chakrabarti, X. Mei, Z. Yue, H. Dunya, R. Filler and B. K. Mandal, Ionics, 2018, 25, 1633–1643 CrossRef.
- J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS.
- D. Zhang, X. Xu, X. Huang, Z. Shi, Z. Wang, Z. Liu, R. Hu, J. Liu and M. Zhu, J. Mater. Chem. A, 2020, 8, 18043–18054 RSC.
- X. Yu, L. Xue, J. B. Goodenough and A. Manthiram, Adv. Funct. Mater., 2020, 2002144 Search PubMed.
- P. Pal and A. Ghosh, Solid State Ionics, 2018, 319, 117–124 CrossRef CAS.
- W. Zhai, H.-j. Zhu, L. Wang, X.-m. Liu and H. Yang, Electrochim. Acta, 2014, 133, 623–630 CrossRef CAS.
- P. Yao, B. Zhu, H. Zhai, X. Liao, Y. Zhu, W. Xu, Q. Cheng, C. Jayyosi, Z. Li, J. Zhu, K. M. Myers, X. Chen and Y. Yang, Nano Lett., 2018, 18, 6113–6120 CrossRef CAS PubMed.
- W. Xiao, Z. Wang, Y. Zhang, R. Fang, Z. Yuan, C. Miao, X. Yan and Y. Jiang, J. Power Sources, 2018, 382, 128–134 CrossRef CAS.
- H. Zhai, P. Xu, M. Ning, Q. Cheng, J. Mandal and Y. Yang, Nano Lett., 2017, 17, 3182–3187 CrossRef CAS PubMed.
- J. Wan, J. Xie, X. Kong, Z. Liu, K. Liu, F. Shi, A. Pei, H. Chen, W. Chen, J. Chen, X. Zhang, L. Zong, J. Wang, L. Chen, J. Qin and Y. Cui, Nat. Nanotechnol., 2019, 14, 705–711 CrossRef CAS PubMed.
- M. Dirican, C. Yan, P. Zhu and X. Zhang, Mater. Sci. Eng., R, 2019, 136, 27–46 CrossRef.
- K. Dai, C. Ma, Y. Feng, L. Zhou, G. Kuang, Y. Zhang, Y. Lai, X. Cui and W. Wei, J. Mater. Chem. A, 2019, 7, 18547–18557 RSC.
- A. Varzi, R. Raccichini, S. Passerini and B. Scrosati, J. Mater. Chem. A, 2016, 4, 17251–17259 RSC.
- C. S. Wong, K. H. Badri, N. Ataollahi, K. P. Law, M. S. Su’ait and N. I. Hassan, International Journal of Chemical, Nuclear, Metallurgical and Materials Engineering, 2014, 8, 1168 Search PubMed.
- J. S. Parveen and S. S. M. A. Maeed, Int. J. Sci. Eng. Res., 2013, 4, 553 Search PubMed.
- J. Kozakiewicz, J. Przybylski and K. Sylwestrzak, Polym. Adv. Technol., 2016, 27, 258 CrossRef CAS.
- M. H. Ugur, H. Kilic, M. L. Berkem and A. Gungor, Chem. Pap., 2014, 68, 1561 CAS.
- J. Potaufeux, J. Odent, D. Notta-Cuvier, F. Lauro and J. Raquez, Polym. Chem., 2020, 11, 5914–5936 RSC.
- N. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175 CrossRef CAS.
- G. He, Z. Li, J. Zhao, S. Wang, H. Wu, M. D. Guiver and Z. Jiang, Adv. Mater., 2015, 27, 5280 CrossRef CAS PubMed.
- C. C. Yang, R. Narimani, B. J. Frisken and S. Holdcro, J. Membr. Sci., 2014, 469, 251 CrossRef.
- J. R. Rowlett, V. Lilavivat, A. T. Shaver, Y. Chen, A. Daryaei, H. Xu, C. Mittelsteadt, S. Shimpalee, J. S. Riffle and J. E. McGrath, Polymer, 2017, 122, 296 CrossRef CAS.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759 CrossRef CAS PubMed.
- D. Basu, A. Das, K. W. Stockelhuber and S. Wiebner, Designing of Elastomer Nanocomposites, 2016, vol. 275, p. 235 Search PubMed.
- I. M. Davletbaeva, O. O. Sazonov, A. R. Fazlyev, R. S. Davletbaev, S. V. EfimovandV and V. Klochkov, RSC Adv., 2019, 9, 18599–18608 RSC.
- I. M. Davletbaeva, O. O. Sazonov, A. R. Fazlyev, I. N. Zakirov, R. S. Davletbaev, S. V. Efimov and V. V. Klochkov, Polym. Sci., Ser. A, 2020, 20, 337–349 Search PubMed.
- A. A. Slesarenko, I. K. Yakuschenko, V. Ramezankhani, V. Sivasankaran, O. E. Romanyuk, A. V. Mumyatov, A. F. Shestakov, O. V. Yarmolenko, K. J. Stevenson and P. A. Troshin, J. Power Sources, 2019, 435, 226724 CrossRef CAS.
- J. Evans, C. A. Vincet and P. G. Bruce, Polymer, 1987, 28, 2324 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |