
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly sensitive non-enzymatic hydrogen peroxide monitoring platform based on nanoporous gold via a modified solid-phase reaction method†
Zhipeng Yang‡
a,
Jun Li‡a,
Panmei Liua,
An Zhanga,
Jing Wanga,
Yuan Huang a,
Jiangyong Wangb and
Zumin Wang*a
a,
Jiangyong Wangb and
Zumin Wang*a
aState Key Laboratory of Hydraulic Engineering Simulation and Safety, School of Materials Science and Engineering, Tianjin University, 300350 Tianjin, People's Republic of China. E-mail: z.wang@tju.edu.cn
bDepartment of Physics, Shantou University, 515063 Shantou, People's Republic of China
First published on 16th November 2021
Abstract
In this work, nanoporous gold (NPG) fabricated using a modified solid-phase reaction method was developed as an electrocatalyst for the nonenzymatic detection of hydrogen peroxide (H2O2). The NPG morphology and structure were characterized by scanning electron microscopy and high-resolution transmission electron microscopy. The fabricated NPG exhibited a nanoporous framework with numerous structural defects. The NPG-based amperometric H2O2 sensor had a good selectivity, reproducibility, and low detection limit (0.3 μM) under near physiological conditions (pH = 7.4). The sensitivities of this sensor over concentration ranges of 0.002–5 mM and 5–37.5 mM were 159 and 64 μA mM−1 cm−2, respectively. These results indicate that the developed NPG is a promising material for the electrochemical sensing of H2O2.
1. Introduction
Hydrogen peroxide (H2O2) is widely used in several fields such as food, pharmaceutical, and biological applications.1–3 This excessive use of H2O2 can have adverse effects on human health and safety. Thus, analytical determination of H2O2 is essential. Currently, there are several available H2O2-detection techniques such as fluorometry,4–6 chromatography,7,8 spectrophotometry,9,10 chemiluminescence,11 electrochemistry,12 titrimetry,13 and electrochemical methods.14 The electrochemical methods are among the most effective H2O2-detection techniques due to their high sensitivity, fast response, and operational simplicity. These methods detect H2O2 through concentration-dependent electrical signals generated when the H2O2-containing samples are exposed to an electrocatalyst. Two types of electrochemical sensors (enzyme-based and non-enzymatic sensors) have been developed for H2O2 detection. Enzyme-based sensors exhibit serious disadvantages15–19 such as poor reproducibility, sensitivity to pH, low stability, and time-consuming immobilization procedures. Thus, the development of effective and stable non-enzymatic electrode materials is essential.Various electrode materials have been reported as suitable scaffolds for H2O2 detection.20–29 Among them, nanoporous gold (NPG), which has a three-dimensional interconnected porous nanostructure, has attracted significant interest due to its high specific surface area, high electrical conductivity, excellent catalytic activity and good biocompatibility. NPG was recently prepared using a solid-phase reaction method (SPRM) based on a metal-induced crystallization (MIC) process, which exhibited enhanced electro-catalytic properties.30–34 In this study, NPG was designed and manufactured using a modified SPRM method. A triple-layer precursor, i.e., a-Ge (amorphous Ge, top)/Au (middle)/a-Ge (bottom) film precursor, was used to accelerate the MIC process. Using this triple-layer structure, NPGs with nanopores smaller than those prepared using the previously reported double-layer Au (bottom)/a-Ge (top) precursors were obtained. The morphological results indicated that NPGs with smaller grains and nanopores could be obtained using this modified SPRM method. The electrochemical performance of the prepared NPGs during the catalytic reduction of H2O2 was systematically investigated. The results indicated that the obtained NPGs are promising materials for non-enzymatic electrochemical H2O2 sensors.
2. Experimental
2.1. Preparation of the catalyst
The bilayer precursor structure obtained in a previous study consisted of amorphous Ge (a-Ge, top)/Au (bottom).19 Here, a thin a-Ge film was inserted between the previous Au (bottom) layer and the substrate, resulting in a triple-layer film precursor (a-Ge (top)/Au (middle)/a-Ge (bottom)). This precursor thin film was deposited onto Si(100) wafers (covered with 50 nm-thick amorphous SiO2) using magnetron sputtering at 120 °C in a high-vacuum system (base pressure ≤1.0 × 10−5 Pa). Substrates were thoroughly washed before deposition using acetone, ethanol, and ultrapure water. Before sputtering, they were cleaned under Ar+ atmosphere in a vacuum chamber for 5 min. A pure gold target and a pure germanium target were used for direct-current sputtering Au sublayer and radio frequency sputtering Ge sublayer, respectively. A 4 nm-thick a-Ge sublayer was first obtained on the substrate at a rate of 1 nm min−1. Next, while still under vacuum, a 20 nm Au sublayer was deposited on top of the bottom Ge layer. The direct-current sputtering power was 16 W and the deposition rate was 3.3 nm min−1. A 40 nm a-Ge layer was finally deposited on top of the Au film. The deposition rate was 11.6 nm min−1. The substrates were maintained at 120 °C during the entire deposition process to encourage the MIC process of the deposited Au and Ge films, in which the mutual transport of Au and Ge atoms led to the formation of network-like nanostructures. Samples were continuously rotated at 20 rpm during the sputtering deposition process to ensure sample uniformity. The as-sputtered samples were then immersed in a hydrogen peroxide solution (30 vol%) for 5 minutes at 25 °C, resulting in a selective removal of Ge from the precursor. Finally, the samples were thoroughly rinsed and dried using ultrapure water and a stream of nitrogen, respectively.2.2. Characterization of NPGs
Scanning electron microscopy (SEM) with energy-dispersive spectroscopy (EDS) measurements were carried out using a Hitachi S-4800 microscope operated at 15 kV. More detailed high resolution transmission electron (HRTEM) and selected-area electron diffraction (SAED) pattern were characterized by a JEM-2100F microscope (200 kV). The average grain size was determined based on measurements of 300 individual grains using SEM micrographs.2.3. Electrochemical detection of H2O2
Electrochemical measurements were performed with an electrochemical workstation (CHI 660E) at 25 °C. A three-electrode system composed of a 3 mm-diameter NPG-modified glassy carbon electrode (NPG/GCE), saturated calomel electrode (SCE), and platinum wire as the working, reference, and counter electrodes, respectively, was used for the measurements. The working electrode was prepared by the procedure described in Section 2.1, using glassy carbon electrodes as the substrates. For comparison, a polycrystalline gold-modified electrode (Poly-Au/GCE) was prepared using the same magnetron sputtering parameters. Before the fabrication, the bare glassy carbon electrodes were polished to a mirror-like state using Al2O3 suspensions (0.5 and 0.05 μm) and rinsed with pure ethanol and distilled water. They were then dried using nitrogen steam. All experiments were performed at room temperature. Before each electrochemical test, all electrolytes were deoxygenated using nitrogen bubbling (purity ≥99.999 vol%) for at least 30 min.The electrode surface was initially cleaned using ultrapure water and subsequently subjected to 20 cycles of cyclic voltammetry (CV) between −0.8 and 0.8 V in 0.1 M KOH solution. The scan rate was 50 mV s−1. During this process, the NPG/GCE was activated due to OH− penetration in NPG. The CV measurements were conducted in N2-saturated phosphate-buffered solutions (PBS, 0.1 M, pH = 7.4) without and with H2O2 (10 mM) at a scan rate of 50 mV s−1. The effect of the scan rate on the reduction-peak currents was also examined in detail. The amperometric (I–t) measurement at −0.8 V was used to assess the sensitivity of the electrode material toward H2O2 in a continuously stirred PBS solution (pH = 7.4), while recording the change in the current with the increase in the H2O2 concentration. The electrode selectivity was assessed by conducting the detection in a solution containing interferents (i.e., ascorbic acid (AA), uric acid (UA), and glucose (GLU)).
3. Results and discussion
Fig. 1a–d shows SEM and TEM micrographs of NPG prepared using the triple-layer a-Ge (thick, top)/Au (middle)/a-Ge (thin, bottom) film precursor. The NPG thin film exhibited a typical bicontinuous porous structure. The sizes of the NPG grains and nanopores (approximately 14 nm, see the inset in Fig. 1a) were smaller than those of NPG (approximately 26 nm) obtained using a bilayer precursor.35 This can be attributed to the thermodynamic instability of the Au/a-Ge interface of the triple-layer precursor structure.36,37 This instability was caused by placing a gold layer between two amorphous germanium layers, and it increased the driving force of the MIC reaction, accelerating its rate. In addition, the composition of the triple-layer precursor film, as analysed by EDS, is 62 at% Ge and 38 at% Au.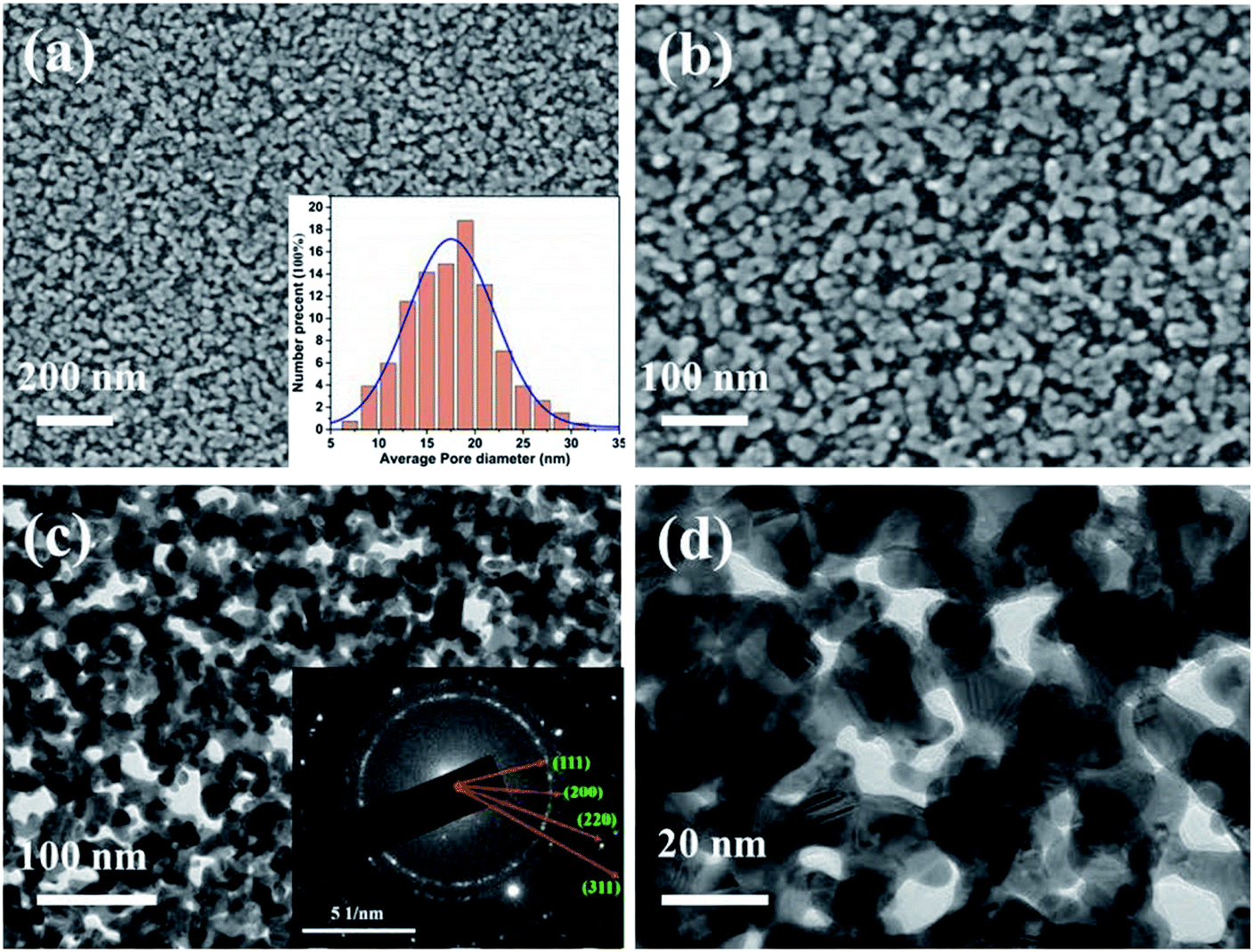 | ||
| Fig. 1 Microstructure of the NPG thin film prepared by the modified solid-phase reaction method: (a, b) SEM and (c, d) TEM images. The inset in (a) and (c) are the average nanopore size and the corresponding SAED pattern of the NPG thin film, respectively. | ||
At the beginning of the metal-induced crystallization process, the amorphous germanium atoms above and below the gold sublayer rapidly diffused along the Au grain boundaries in the gold sublayer. Next, they began to nucleate and produce high density Ge nanocrystalline grains at the gold grain boundaries, resulting in trapping the nanoscale crystalline Ge (c-Ge) in the original Au sublayer. The continuation of the MIC process generated a large stress gradient inside the multilayer film.31 The atoms in the Au sublayer then further diffused to occupy the a-Ge position, promoting the formation of Au nanostructures in the original a-Ge position. As described in Section 2.1, the bottom Ge film must be sufficiently thin (4 nm) to ensure the connection between the gold sublayer and substrate when the MIC process is completed. Subsequently, the germanium was selectively etched from the precursor. As a result, the positions that were occupied by the crystalline germanium in the Au sublayer were exposed, revealing large number of nanopores. Thus, nanoporous gold with the desired morphology was obtained. Due to the good selective etching effect of hydrogen peroxide, the NPG film prepared by the solid-phase reaction method has extremely high purity without residual germanium.33,34 The inset in Fig. 1c shows the SAED pattern of NPG thin film, which indicates the nanocrystalline nature of the obtained NPG thin film.38
Fig. 2a–c show the HRTEM results, revealing that the NPG material contained various typical structural defects such as grain boundaries (Fig. 2a), twins (Fig. 2b) and stacking faults (Fig. 2c). These defects, which are associated with the presence of a high concentration of low-coordination atoms, can effectively work as catalytic active sites in several catalytic reactions.39–43 In addition, the open nanoporous structure with small grains and nanopores creates a large electrochemically active surface area (about 0.33 cm2). Therefore, the as-prepared NPG thin film was expected to show good electrochemical performance during H2O2 detection.
 | ||
| Fig. 2 Representative HRTEM images of the NPG thin film prepared by the modified solid-phase reaction method, showing surface defects: (a) stacking faults, (b) twins, (c) grain boundaries and (d) stepped face. | ||
Before the electrocatalytic measurements, the electrochemical impedance spectroscopy (EIS) was first tested using the method listed in the ESI methods and the result is shown in Fig. S1.† It is found that the diameter of semicircular part of the curve corresponding to the electron transfer resistance (Rct) of the redox reaction is lower (about 65 Ω), suggesting its good conductivity.
After EIS measurements, the electrocatalytic performances were measured using CV. Fig. 3a and b show a comparison between the CV curves of Poly-Au/GCE and NPG/GCE, respectively, in N2-saturated PBS solution (0.1 M, pH = 7.4) without and with H2O2 (10 mM). The Poly-Au/GCE showed low electrocatalytic activity, generating a weak reduction current and onset potential (approximately: −0.2 V) after the addition of H2O2 (10 mM). In contrast, NPG/GCE generated a strong current response to H2O2 at the same potential and a higher positive onset potential at approximately 0 V. The reduction peak of H2O2 occurred at −0.83 V with a peak current four times larger than that obtained from Poly-Au/GCE. The reduction of H2O2 on the surface of NPG/GCE follows the electroreduction mechanism described in eqn (1)–(3):44,45
| H2O2 + e− → OHad + OH− | (1) |
| OHad + e− → OH− | (2) |
| OH− + H+ → H2O | (3) |
 | ||
| Fig. 3 Cyclic voltammograms of (a) Poly-Au/GCE and (b) NPG/GCE in PBS solutions (0.1 M, pH = 7.4) without and with H2O2 (10 mM), the scan rate was 50 mV s−1, (c) cyclic voltammograms of NPG/GCE in a PBS solution (0.1 M, pH = 7.4) with H2O2 (10 mM) at varying scan rates, and (d) cathodic peak current vs. scan rate. | ||
The results obtained in this study support this reaction mechanism because the onset potential of H2O2 reduction (Fig. 3) was almost equal to the adsorption potential of OH−.46 The significant electroreduction of H2O2 by NPG/GCE can be ascribed to the fast interfacial electron transfer on the nanoporous gold nanostructures, which reduces the overpotential and increases the response current.47 Fig. 3c illustrates the CV curves obtained using an NPG/GCE electrode with H2O2 (10 mM) at various scan rates (10–80 mV s−1). Fig. 3d indicates a liner relationship (R2 = 0.996) between the cathodic peak current and the square root of the scan rate, signifying that a classical diffusion-controlled electrochemical process occurred on the NPG/GCE electrode surface.
The pH effect on the NPG/GCE performance during the electroreduction of H2O2 was studied by conducting CVs of NPG/GCE under various pH conditions (Fig. 4). Due to the instability of H2O2 under alkaline conditions, H2O2 reduction was not tested at higher pH values. The reduction peak current increased with pH (from 6.2 to 7.8), and the most positive peak potential occurred at −0.83 V at pH = 7.4. Therefore, a pH of 7.4 was selected to detect H2O2 in the following experiment.
 | ||
| Fig. 4 Peak currents and peak potentials in H2O2 (10 mM) at different pH values. | ||
The practical use of NPG/GCE as a sensing electrode was assessed by examining its electrochemical response to H2O2. This was conducted by monitoring the change in the current with increasing hydrogen peroxide concentration in a 0.1 M PBS solution at a potential of −0.8 V. Fig. 5 shows the amperometric response of NPG/GCE, which indicates that the response current stabilizes within 2 s after each H2O2 addition, suggesting that the electrochemical sensor exhibited rapid diffusion and electron-exchange performance. In addition, the amperometric response exhibited a step-like increase with the H2O2 concentration, indicating high sensitivity. Therefore, the correlation between the concentration of the added H2O2 and the corresponding current response can be used as a calibration curve for H2O2 detection (Fig. 6). A well-defined linear current response was observed with the increase in the H2O2 concentration in two distinct regions: 2 μM to 5 mM and 5–37.5 mM. The calibration linear regression equations of these two regions are are described with the eqn (4) and (5):
| I (μA) = −0.0523 [H2O2] (μM) − 112.422 (2 μM to 5 mM) | (4) |
| I (μA) = −0.0206 [H2O2] (μM) − 286.974 (5 mM to 37.5 mM) | (5) |
 | ||
| Fig. 5 Amperometric recording obtained at the NPG/GCE electrode for different H2O2 concentrations (applied potential: −0.8 V). The inset shows a magnified amperometric response in a lower H2O2 concentration range. | ||
 | ||
| Fig. 6 Calibration curves of the NPG/GCE electrode used in H2O2 detection. | ||
The sensor sensitivity values for the first and second linear regions were estimated to be approximately 159 and 64 μA mM−1 cm−2, respectively. The limit of detection (LOD) were found to be 0.3 μM with a signal to noise ratio of 3 (S/N = 3). A comparison between the sensitivity parameters of NPG/GCE and other Au-based non-enzymatic electrochemical sensor materials is summarized in Table 1. The NPG electrode developed in this work exhibited a much wider detection range and a higher sensitivity for the detection of concentrated H2O2 than those of the previously reported electrodes.48–53 This excellent electrocatalytic activity of NPG/GCE can be ascribed to the large active surface area and plenty of active sites generated due to the solid phase reaction method used here (refer to Fig. 2 and the discussion in Sec. 2.1).
| Electrode materials | Sensitivity (μA mM−1 cm−2) | LOD (μM) | Linear range (μM) | Working potential | Ref. |
|---|---|---|---|---|---|
| a LOD = 3(RSD/slope).b Graphene quantum dots/gold electrode.c Au–polydopamine–graphene quantum dots.d Gold nanograss–nanocube assemblies. | |||||
| GQD/Au electrodeb | 51 | 0.7 | 2–8000 | −0.4 | 48 |
| Au-PDA-GQDc | 48 | 0.006 | 0.1–40 | −0.1 | 49 |
| 8 | 40–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
||||
| Au NG@NCd | 101 | 29 | 50–30000 |
−0.5 | 50 |
| Au nanowires | 185 | 111 | 500–50000 |
−0.7 | 51 |
| Au NP-silica sol–gel | 30 | 0.003 | 2.5–45 | −0.5 | 52 |
| Nanoporous gold | 21 | 3.26 | 5–28000 |
−0.4 | 53 |
| NPG | 159 | 0.3 | 2–5000 | −0.8 | This work |
| 64 | 5000–35000 |
||||
The repeatability of the electrochemical sensor was examined by subjecting the NPG/GCE electrode to 5 repeated CV experiments in a PBS (0.1 M, pH = 7.4) solution with H2O2 (10 mM). The relative standard deviation (RSD) of the reduction peak current over five independent tests was only 1% (Fig. 7a). In addition, five NPG/GCE were fabricated for H2O2 detection under the same conditions. Fig. 7b shows their reduction-peak-current values. The RSD among these values was calculated to be 3%, indicating a good reproducibility of the sensor.
 | ||
| Fig. 7 (a) The repeatability of five experiments using the same NPG/GCE electrode, (b) reproducibility investigation using five NPG/GCE electrodes under the same preparation parameters. In each case, H2O2 (10 mM) was added. | ||
Moreover, the presence of some possible co-existing species in a practical environment may cause interference to current response of the electrodes. Therefore, selectivity is also a key indicator for the actual application of the sensors. Herein, the amperometric response of the NPG/GCE was monitored during the successive injection of interfering substances in the PBS solution (0.1 M). Fig. 8 indicates that there was only a weak response to the addition of multiple interferents (AA, 4 mM; UA, 4 mM; and GLU, 4 mM). However, after the addition of H2O2 (0.2 mM), a very clear current response was obtained. Thus, the effect of interfering species on the sensor response during H2O2 detection was negligible. Therefore, the developed NPG/GCE sensor exhibited excellent selectivity and anti-interference performance during H2O2 detection.
 | ||
| Fig. 8 Effects of AA (4 mM), UA (4 mM), GLU (4 mM) on the amperometric responses of the NPG/GCE electrode in a N2-saturated PBS solution (0.1 M, pH = 7.4). | ||
It is well-known that H2O2 is often used as a stabilizer or a preservative in milk, but excess addition of H2O2 is very harmful to our health. As a result, it is extraordinarily important to monitor the concentration of H2O2. For verifying the feasibility of the prepared electrode, the sensor performance of NPG electrode in real sample (the milk sample is selected here) was tested according to the method in the ESI method.† The recoveries of H2O2 in milk sample are calculated using the corresponding linear regression equation in the insert of Fig. 6. The results are summarized in Table S1 (ESI),† which shows that the recoveries of milk sample with 1 mM, 2 mM and 5 mM H2O2 is 101.0%, 96.5% and 98.4%, respectively. The results further indicate the prepared NPG electrode has good potential to monitor H2O2.
4. Conclusions
A modified solid-phase reaction method was developed based on the MIC process to form NPG thin films using a Ge (thin, bottom)/Au/Ge (thick, top) triple-layer precursor. The NPG thin film prepared by this method exhibited nanoscale grains (approximately 14 nm) smaller than those of the double-layer Au (bottom)/Ge (top) precursor. The as-developed NPG thin film exhibited outstanding electrochemical properties such as high sensitivity, wide linear range, and good selectivity for the electrochemical detection of H2O2 under near-physiological conditions. The excellent electrochemical performance of the sensor can be ascribed to its large active surface area and unique structure, which includes numerous defects generated during the preparation process. These results confirm that the developed NPG thin film can be used as a suitable platform for non-enzymatic electrochemical detection of H2O2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51971153) and the National Key Research and Development Program of China (Grant No. 2017YFE0302600).Notes and references
- Z. Yun, H. J. Gao, X. Chen, Z. S. Z. Chen, Z. K. Zhang, T. T. Li, H. X. Qu and Y. M. Jiang, Food Chem., 2021, 336, 1–10 CrossRef PubMed
.
- K. M. Kersten, M. E. Breen, A. K. Mapp and A. J. Matzger, Chem. Commun., 2018, 54, 9286–9289 RSC
- A. C. del Álamo, C. González, M. I. Pariente, R. Molina and F. Martínez, Chem. Eng. J., 2020, 401, 1–34 CrossRef
- S. J. Lao, H. Y. Qin, L. Q. Ye, B. H. Liu and Z. P. Li, J. Power Sources, 2010, 195, 4135–4138 CrossRef CAS
- G. Gnana kumar, G. Amala and S. M. Gowtham, RSC Adv., 2017, 7, 36949–36976 RSC
- S. A. Kitte, M. N. Zafar, Y. T. Zholudov, X. G. Ma, A. Nsabimana, W. Zhang and G. B. Xu, Anal. Chem., 2018, 90, 8680–8685 CrossRef CAS PubMed
- P. P. Liu, X. B. Ge, R. Y. Wang, H. Y. Ma and Y. Ding, Langmuir, 2009, 25, 561–567 CrossRef CAS PubMed
- J. Z. Li and P. K. Dasgupta, Anal. Chem., 2000, 72, 5338–5347 CrossRef CAS PubMed
- M. Mazloum-Ardakani, H. Rajabi, B. B. F. Mirjalili, H. Beitollahi and A. Akbari, J. Solid State Electrochem., 2010, 14, 2285–2292 CrossRef CAS
- W. Luo, M. E. Abbas, L. H. Zhu, K. J. Deng and H. Q. Tang, Anal. Chim. Acta, 2008, 629, 1–5 CrossRef CAS
- H. Liu, Y. N. Ding, B. C. Yang, Z. X. Liu, Q. Y. Liu and X. Zhang, Sens. Actuators B Chem., 2018, 271, 336–345 CrossRef CAS
- D. Han, J. Zhang, Z. Weng, D. Kong, Y. Tao, F. Ding, D. Ruan and Q.-H. Yang, Mater. Today Energy, 2019, 11, 30–45 CrossRef CAS
- J. He, P. Wu, L. Lu, H. Li, H. Ji, M. He, Q. Jia, M. Hua, W. Zhu and H. Li, ACS Appl. Mater. Interfaces, 2019, 11, 36666–36675 CrossRef CAS
- J. Wang, H. Gao, F. Sun and C. Xu, Sens. Actuators, B, 2014, 191, 612–618 CrossRef CAS
- S. Y. Gu, C. T. Hsieh, B. C. Mallick, C. C. Fu, R. S. Juang, Y. A. Gandomi and K. D. Kihm, Appl. Surf. Sci., 2020, 528, 1–7 CrossRef
- P. Paulraj, A. Umar, K. Rajendran, A. Manikandan, R. Kumar, E. Manikandan, K. Pandian, M. H. Mahnashi, M. A. Alsaiari, A. A. Ibrahim, N. Bouropoulos and S. Baskoutas, Electrochim. Acta, 2020, 363, 1–12 CrossRef
- M. Niamlaem, C. Boonyuen, W. Sangthong, J. Limtrakul, D. Zigah, A. Kuhn and C. Warakulwit, Carbon, 2020, 170, 154–164 CrossRef CAS
- M. Asif, A. Aziz, M. Azeem, Z. Y. Wang, G. Ashraf, F. Xiao, X. D. Chen and H. F. Liu, Adv. Colloid Interface Sci., 2018, 262, 21–38 CrossRef CAS PubMed
- Z. Rahmati, M. Roushani and H. Hosseini, Sens. Actuators, B, 2020, 324, 1–12 CrossRef
- T. Xiao, J. Huang, D. Wang, T. Meng and X. Yang, Talanta, 2020, 206, 1–19 CrossRef PubMed
- S. Li, X. Liu, H. Chai and Y. Huang, Trends Anal. Chem., 2018, 105, 391–403 CrossRef CAS
- R. Zhang and W. Chen, Biosens. Bioelectron., 2017, 89, 249–268 CrossRef CAS PubMed
- Y. Li, J. Zhang, H. Zhu, F. Yang and X. Yang, Electrochim. Acta, 2010, 55, 5123–5128 CrossRef CAS
- E. Arkan, R. Saber, Z. Karimi and M. Shamsipur, Anal. Chim. Acta, 2015, 874, 66–74 CrossRef CAS PubMed
- N. G. Bastús, J. Comenge and V. C. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed
- K. Dhara and D. R. Mahapatra, J. Mater. Sci., 2019, 54, 12319–12357 CrossRef CAS
- I. S. Kucherenko, O. O. Soldatkin, D. Y. Kucherenko, O. V. Soldatkina and S. V. Dzyadevych, Nanoscale Adv., 2019, 1, 4560–4577 RSC
- G. Maduraiveeran and W. Jin, Trends Environ. Anal. Chem., 2017, 13, 10–23 CrossRef CAS
- S. Su, J. Chao, D. Pan, L. Wang and C. Fan, Electroanalysis, 2015, 27, 1062–1072 CrossRef CAS
- Z. Wang, L. Gu, F. Phillipp, J. Y. Wang, L. P. Jeurgens and E. J. Mittemeijer, Adv. Mater., 2011, 23, 854–859 CrossRef CAS PubMed
- Z. M. Wang, J. Y. Wang, L. P. H. Jeurgens, F. Phillipp and E. J. Mittemeijer, Acta Mater., 2008, 56, 5047–5057 CrossRef CAS
- A. Zhang, Z. Yang, Y. Chen, P. Schützendübe, Y. Huang, Y. Liu and Z. Wang, J. Electrochem. Soc., 2020, 167, 1–9 Search PubMed
- A. Zhang, Y. Chen, Z. Yang, S. Ma, Y. Huang, G. Richter, P. Schützendübe, C. Zhong and Z. Wang, ACS Appl. Energy Mater., 2019, 3, 336–343 CrossRef
- A. Zhang, Z. Yang, Y. Chen, L. Yin, P. Schützendübe, Y. Huang, Y. Liu and Z. Wang, J. Electrochem. Soc., 2019, 166, 650–655 CrossRef
- A. Zhang, J. Wang, P. Schützendübe, H. Liang, Y. Huang and Z. Wang, Nanotechnology, 2019, 30, 1–9 Search PubMed
- J. Wang, L. Han, X. Li, D. Liu, L. Luo, Y. Huang, Y. Liu and Z. Wang, J. Mater. Sci. Technol., 2021, 65, 202–209 CrossRef
- Z. M. Wang, J. Y. Wang, L. P. H. Jeurgens and E. J. Mittemeijer, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 1–15 Search PubMed
- J. Du, C. Li, Z. Wang and Y. Huang, J. Mater. Sci. Technol., 2021, 65, 18–28 CrossRef
- K. Yao, C. Zhao, N. Wang, W. Lu, H. Wang, S. Zhao and J. Wang, CrystEngComm, 2018, 20, 6328–6337 RSC
- T. Fujita, P. Guan, K. McKenna, X. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. Chen, Nat. Mater., 2012, 11, 775–780 CrossRef CAS PubMed
- S. Sreedhala, S. Maheshwari, K. J. Betsy and C. P. Vinod, Appl. Catal., A, 2016, 524, 1–7 CrossRef CAS
- M. Krajčí, S. Kameoka and A.-P. Tsai, J. Chem. Phys., 2016, 145, 1–6 CrossRef PubMed
- J. Zhang and C. M. Li, Chem. Soc. Rev., 2012, 41, 7016–7031 RSC
- J. Zhang and M. Oyama, Electrochim. Acta, 2004, 50, 85–90 CrossRef CAS
- R. Zeis, T. Lei, K. Sieradzki, J. Snyder and J. Erlebacher, J. Catal., 2008, 253, 132–138 CrossRef CAS
- Y. Ding and M. W. Chen, MRS Bull., 2009, 34, 569–576 CrossRef CAS
- N. S. K. Gowthaman, B. Sinduja and S. A. John, RSC Adv., 2016, 6, 63433–63444 RSC
- Y. Zhang, C. Y. Wu, X. J. Zhou, X. C. Wu, Y. Q. Yang, H. X. Wu, S. W. Guo and J. Y. Zhang, Nanoscale, 2013, 5, 1816–1819 RSC
- Y. Zhu, S. Lu, A. Gowri Manohari, X. Dong, F. Chen, W. Xu, Z. Shi and C. Xu, J. Electroanal. Chem., 2017, 796, 75–81 CrossRef CAS
- M. Dervisevic, Q. Shi, M. Alba, B. Prieto-Simon, W. Cheng and N. H. Voelcker, J. Colloid Interface Sci., 2020, 575, 24–34 CrossRef CAS PubMed
- E. Dervisevic, M. Dervisevic, Y. Wang, L. F. Malaver-Ortega, W. Cheng, K. L. Tuck, N. H. Voelcker and V. J. Cadarso, Electroanalysis, 2020, 32, 1850–1858 CrossRef CAS
- G. Maduraiveeran and R. Ramaraj, J. Electroanal. Chem., 2007, 608, 52–58 CrossRef CAS
- F. H. Meng, X. L. Yan, J. G. Liu, J. Gu and Z. G. Zou, Electrochim. Acta, 2011, 56, 4657–4662 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03184h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |