
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of a potent macrocyclic peptide inhibitor targeting the PD-1/PD-L1 protein–protein interaction†
Qi Miao‡
 a,
Wanheng Zhang‡a,
Kuojun Zhanga,
He Lib,
Jidong Zhu*b and
Sheng Jiang*a
a,
Wanheng Zhang‡a,
Kuojun Zhanga,
He Lib,
Jidong Zhu*b and
Sheng Jiang*a
aState Key Laboratory of Natural Medicines, School of Pharmacy, School of Engineering, China Pharmaceutical University, Nanjing, 210009, China. E-mail: jiangsh9@gmail.com
bInterdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, China. E-mail: zhujd@sioc.ac.cn
First published on 1st July 2021
Abstract
We report optimization by rational design of JMPDP-027, a potent cyclic peptide that interferes with the PD-1/PD-L1 protein–protein interaction. JMPDP-027 shows a potent restoring ability towards T-cells with an EC50 of 5.9 nM that is comparable to that of the anti-PD-1 monoclonal antibody pembrolizumab. In addition, JMPDP-027 shows not only high resistance to enzymatic hydrolysis in human serum but also no observable toxicity and potent in vivo anticancer activity comparable to that of the mouse PD-L1 antibody in a colon carcinoma (CT26) model. Cyclic peptide antagonists of this sort may provide novel drug candidates for cancer immunotherapy.
Introduction
Immuno-oncology therapy has been recognized as a most efficient and highly selective method for the treatment of cancer. Compared with positive immune checkpoint blockage, antagonizing PD-1/PD-L1 interactions often is beneficial because the PD-1/PD-L1 pathway works as a “normalizer” in immunotherapy.1,2 To date, seven antagonists targeting PD-1 and three targeting PD-L1, all antibodies, have appeared on the market for the treatment of cancer. Besides, small molecules and peptides possess innate advantages, such as minimal immunogenicity, lower manufacturing cost and various modes of administration, and have drawn as much attention as monoclonal antibodies in both the research and the industrial communities.3–6One of the main problems involved with peptides as antagonists would be in vivo stability. Liu and Gao et al., reported a novel D-peptide antagonist targeting the PD-1/PD-L1 pathway. The introduction of D-amino acids led to significant increase of stability in serum. In the meantime, the resulting peptide DPPA had potent tumor growth inhibitory effects in vivo.7 Compared with linear peptides, cyclic peptides possess innate advantages, including restricted conformation, rearrangement of functional groups and resistance to degradation.8 Ma et al. designed and synthesized several cyclic peptides as PD-1/PD-L1 antagonists based on molecular dynamics (MD) simulation. They found that the entire conformation of a linear peptide is stabilized when two cysteine residues form a disulfide bridge. The antagonistic activity of the resulting cyclic peptide was about 10-times higher than that of the linear peptide precursor.9 Recently, Wan and Bu et al. reported a series of cyclic peptides as PD-L1 inhibitors based on structural modification of gramicidin S. The resulting compounds showed strong PD-L1 inhibitory ability and immune activation ability both in vivo and in vitro.10
The Bristol-Myers Squibb Company (BMS) has developed a series of macrocyclic peptides for the treatment of PD-1/PD-L1 related diseases.11,12 Subsequently, Nimmagadda et al. designed and synthesized [68Ga]WL12, a cyclic peptide-68Ga conjugate which can image PD-L1 expression in cancer.13 Most recently, the same group reported their investigations on [64Cu]WL12 as a positive emission tomography (PET) probe to measure PD-L1 occupancy for evaluating therapeutic efficacy.14 The WL12 peptide was chosen from the BMS patent, which further showed that this family of peptides indeed targets the PD-1/PD-L1 interaction. In 2017, Holak et al.15 reported two co-crystal structures of PD-L1 with macrocyclic peptides, peptide-57 and peptide-71, which were chosen from patent WO2014151634.11 Although the two cyclic peptides demonstrated potent inhibitory activity against the PD-1/PD-L1 interaction (IC50 = 9 nM for peptide-57 and 7 nM for peptide-71), some common problems related to peptides still existed, leaving room for further optimization.8 Our mutation strategy, inspired by the research of Bristol-Myers Squibb Company, is to seek for potential close contact between PD-L1 and peptides with assistance of computer-aided drug discovery (CADD) technique to obtain more potent PD-L1 antagonists.
The superimposed structures of peptide-57/PD-L1 and peptide-71/PD-L1 revealed that this series of cyclic peptides occupied essentially the same area and did not induce significant conformational change in PD-L1.15 While the core areas of peptide-57 and peptide-71 were similar, the sequence of peptide-57 from Asn3 to Leu6 occupies an extra area (Fig. S3†).11,15 Moreover, in this sequence, Pro4 in peptide-57 is a “hot spot” according to the results of virtual alanine screening (Table S1†).16–18 The interaction between peptide-57 and PD-L1 are all noncovalent interaction. The most dominant interactions between peptide-57 and PD-L1 involved the peptide-57 side chains of Trp8, Trp10, His5 and Phe1 and the side chains of NMeNle11, NMeNle12 and Pro4 also contributed to binding via van der Waals interactions. In addition, the side chains of Arg13, Cys14, Gly15, Asn3 and Leu6 directly interact with solvent. While NMeAla2, Ser7 and Ser9 appear only to maintain the conformation of peptide-57 and fail to contribute to PD-L1 binding. Based on these findings, we presumed peptide-57 may have the potential for better efficacy.
Results and discussion
Upon conformational analysis, we observed that the side chain of Phe1 in peptide-57 has a relatively smaller Cβ angle compared to other benzyl-like groups (99.5° for Phe1; 115.9° for Trp8; 105.6° for Trp10 in peptide-57; Cβ angles for peptide-71 were recorded in Fig. S5†). We assumed that this rigid conformation in peptide-57 may come from conflicts in Phe1, NMeNle12, Cys14–Gly15 and the hydrophilic part of Arg13. The extension of the Arg13 side chain is close to N-methyl group of NMeNle12, which further affects the side chain of NMeNle12 and eventually, the phenyl group in Phe1 (Fig. S6†). So we hypothesized that mutation of Arg13 to a hydrophobic amino acid could relieve the rigid conformation. In the MD simulation, when Arg13 was mutated to alanine, the angle of Phe1 recovered to the normal angle range but the flexibility of the overall conformation increased with the RMSD varying from 1.4 to 1.7 (Fig. S8†). To maintain the length and space of the side chain the same as Arg13, leucine was chosen to replace Arg13. MD simulation showed both unrestricted rotation of Phe1 as well as relatively stable conformation with the RMSD varying from 1.1 to 1.4 (Fig. S7 and S8†). Ser7 in peptide-57 did not form any interaction with PD-L1 due to the unsuitable distance between Ser7 in peptide-57 and Glu71 in PD-L1. Hence, HoSer may satisfy the extra H-bond formation (all the unnatural amino acids mentioned were defined in Fig. S2†). According to MD simulation results, both the distance and angle suffice to form a hydrogen bond but with the overall flexibility increased. We supposed the substitution of HoSer by Hyp could maintain the condition to form a hydrogen bond and lock the conformation. The following MD simulation indicated that the –OH group in Ser7Hyp forms a hydrogen bond (145.9°, 2.01 Å) with the carbonyl group in side chain of Glu71 as expected (Fig. 2). The resulting compound (2) shows inhibitory ability comparable to that of peptide-57 (IC50 = 46.2 nM for peptide-57 and 40.1 nM for (2), Fig. 6) which suggests that Arg13Leu and Ser7Hyp are acceptable. | ||
| Fig. 1 The structures of cyclic peptide 1–6 and peptide-71. | ||
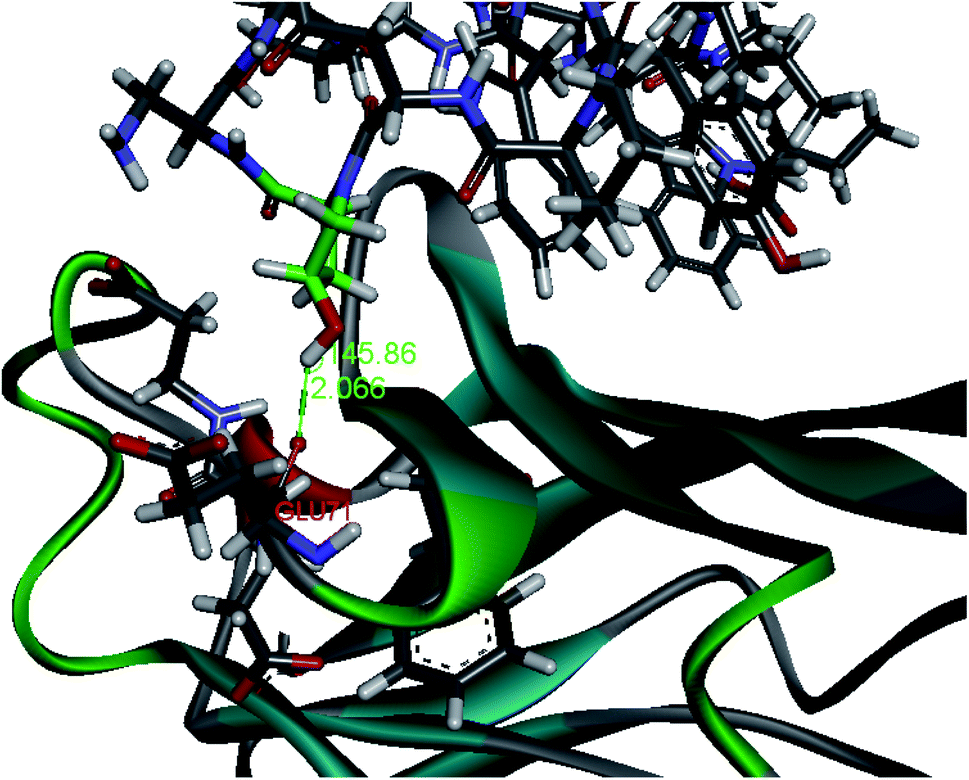 | ||
| Fig. 2 The average conformation in MD simulation of peptide-57 Ser7Hyp. In MD simulations, hydroxyl group in peptide-57 Ser7Hyp could form a hydrogen bond with backbone carbonyl group of Glu71 in PD-L1. The angle of the hydrogen bond is 145.86° and the length is 2.066 Å. | ||
The conformational changes of Phe1 make new contact with Ile54 and Ser117 in PD-L1. It was suggested that a Phe1Tyr mutation would function as an H-bond donor with the Ser117 side chain of PD-L1 (Fig. 3a). Ser9 has no interaction with PD-L1 presumably because the side chain length of Ser9 is too short to reach the nearest PD-L1 residue, Asp61. Considering the presence of the carboxyl group, we further replaced the hydroxyl group with an amino group for potential formation of a salt bridge (Fig. 3b). Trp10 forms a π–π stacking with Arg113 of PD-L1, but due to the direction of its nitrogen, the indole NH cannot form additional interaction with Arg113. Accordingly, we substituted a –CH2COOH group at the nitrogen in the indole. The newly introduced carboxyl group could form a salt bridge with Arg113 (Fig. 3c). In Pro4, although the residue was predicted as a “hot spot”, the pyrrolidine ring did not fully occupy the pocket formed by backbone of Phe67 and Val68 in PD-L1. When we replaced Pro4 with different proline derivatives, it seemed that both Hyp and Oic were preferable. The cyclohexyl group in Oic could well anchor in the shallow cleft formed by Glu71, Val68 and His69 in PD-L1. The hydroxyl group in Hyp could form a H-bond with Val68 in PD-L1. The aforementioned close contacts were all confirmed by MD simulation (Fig. 3d and e). And the resulting compound 3 demonstrated a satisfactory IC50 value of 20.8 nM (Fig. 6).
 | ||
| Fig. 3 Predicted interactions of mutated amino acid. Figures at the left column are the wild type residues in peptide-57 and the right column is the corresponding mutated residues in JMPDP-027 (6). (a) Phe1Tyr. In MD simulation, the Tyr mutation could help form another hydrogen bond between hydroxyl group in Tyr and hydroxyl group of Ser117 in PD-L1. The angle and distance of the hydrogen bond are 157.41° and 1.761 Å. (b) Ser9Dab. In MD simulation, the Dab mutation could help form a salt bridge between amino group in Dab and side chain of Asp31 in PD-L1. The distance between two function groups is 4.002 Å. (c) Trp10Trp(Nin-CH2COOH). In MD simulation, this mutation could help form another salt bridge between carboxyl group in Trp(Nin-CH2COOH) and Arg113 in PD-L1. The distance between two function groups is 4.604 Å. (d) Pro4Hyp. In MD simulation, the Hyp mutation could help form another hydrogen bond between hydroxyl group in Hyp and backbone carbonyl group in Glu71 in PD-L1. The angle and distance of the hydrogen bond are 145.86° and 2.066 Å. (e) Pro4Oic. The Oic mutation could form additional hydrophobic interaction with Phe67, Glu71, Val68 and His69. | ||
To avoid natural amino acid sequence (Tyr1–Asn3, His5–Leu6) in compound 3, we sought for further optimization. In the co-crystal structure of peptide-57 and PD-L1, the imidazole group of His5 in peptide-57 supplied basicity and a suitable conformation for the NH in imidazole to interact with Asp73 in PD-L1. Accordingly, we modified histidine to one of several aliphatic amines. We first chose L-Orn due to the same chain length between nitrogen and the α-C. However, MD simulation showed that the side chain of L-Orn pointed to the solvent area due to the excessive length of carbon chain. L-Dap was supposed to be a better choice of amino acid and this was confirmed in subsequent MD simulations (Fig. 4). Another modification was focused on NMeAla2. Replacement of NMeAla2 with L-proline resulted in compound 4 with inhibitory activity, IC50 = 18.6 nM, comparable to that of compound 3. We then further analyzed the conformation of NMeAla2. The dihedral angles of NMeAla2 are ϕ = −102.8° 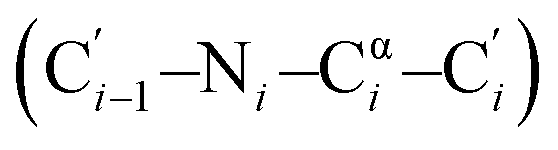 and ψ = 40.1°
and ψ = 40.1°  . According to the literature,19 the dihedral angles of NMeAla2 were more similar to those of HoPro (Fig. 5). The mutation of Pro4HoPro led to the discovery of JMPDP-019 (5) and JMPDP-027 (6) (Fig. 1). These two cyclic peptides inhibit the PD-1/PD-L1 interaction with IC50 values of 14.6 and 13.4 nM, respectively (Fig. 6).
. According to the literature,19 the dihedral angles of NMeAla2 were more similar to those of HoPro (Fig. 5). The mutation of Pro4HoPro led to the discovery of JMPDP-019 (5) and JMPDP-027 (6) (Fig. 1). These two cyclic peptides inhibit the PD-1/PD-L1 interaction with IC50 values of 14.6 and 13.4 nM, respectively (Fig. 6).
 | ||
| Fig. 4 Conformation of peptide-57 and MD simulations of His5Orn and His5Dap. | ||
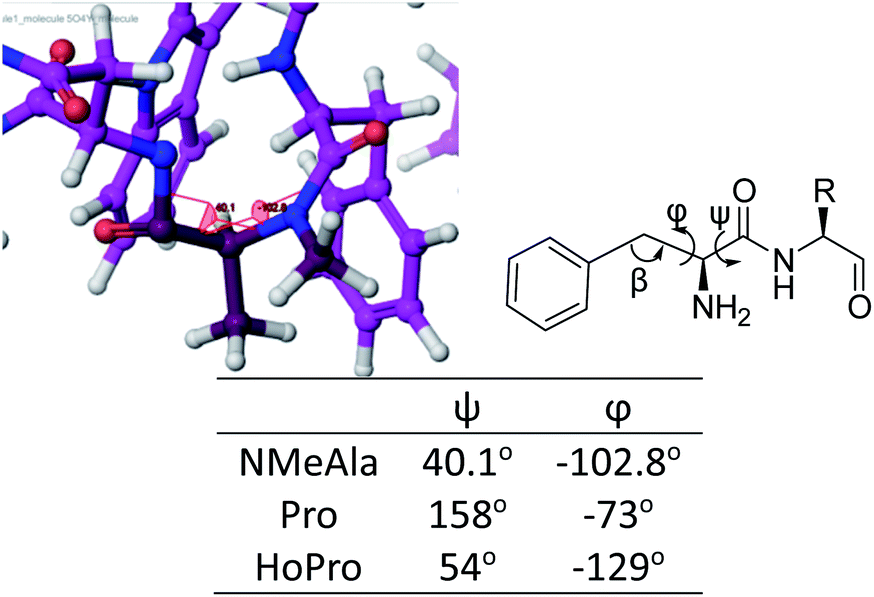 | ||
| Fig. 5 ϕ and ψ angles of NMeAla2 and angles of Pro and HoPro reported in literature.19 | ||
 | ||
| Fig. 6 PD-1/PD-L1 TR-FRET assay: IC50 values of compound 1–6. Error bars were omitted here for clarity, detailed information could be found in ESI.† | ||
To test whether the cyclic peptides are capable of inhibiting the PD-1/PD-L1 interaction in the cellular context, we employed T-cell-like Jurkat cells carrying a reporter luciferase gene under the control of the NFAT promoter and overexpressing PD-1. These cells were contacted with the surrogate of the antigen presenting cells, a CHO cell line that overexpresses a T-cell receptor (TCR) ligand and PD-L1. In this model, the anti-PD-1 monoclonal antibody, pembrolizumab showed a dose-dependent restoring ability with an EC50 of 1.2 nM (all the Relative Light Unit (RLU) data of tested compounds have been normalized to pembrolizumab at a concentration of 5 μM). Compounds 5 and 6 showed similar activity to that of mAbs with EC50 values of 10.6 and 5.9 nM, respectively, while the lead compound (1) showed an EC50 value of 189.5 nM (RLUmax = 8950). The data showed that these cyclic peptides could function as PD-L1 antagonists at the sub-nanomolar level in cell tests (Fig. 7).
 | ||
| Fig. 7 Jurkat T cell/PD-L1-aAPC assay: EC50 values of compound 1, 5, 6 and hPD-1 antibody. Error bars were omitted here for clarity, detailed information could be found in ESI.† | ||
Inspired by these in vitro results, we studied the therapeutic efficacy in vivo of compound 6. First, a test of enzymatic stability in human serum was carried out. Reverse phase HPLC (RP-HPLC) was used to monitor time-dependent peptide hydrolysis. Our measurement showed that compound 6 was stable in 10% human serum after 18 h incubation (Fig. S14†). MTT test indicated that compound 6 did not directly induce cell death of CT26 at the concentration of 50 μM (Fig. S10†).
Subsequently, tumor growth experiments in mice were performed. 15 Balb/c mice were injected with 5 × 105 CT26 cells subcutaneously. After four days, when the tumor volume reached 50 to 100 mm3, the mice were divided randomly into 3 groups with five mice in each group. Compound 6 (4 mg kg−1) was injected intraperitoneally once every day for 14 days. Normal saline and anti-mouse PD-L1 antibodies (10F.9G2, BioXcell) at 10 mg kg−1 were injected separately as blank controls and positive controls. The tumor diameter was measured with a caliper daily, and the volume was calculated with the formula, 1/2 × a × b2. As shown in Fig. 8, Compound 6 significantly inhibits the growth of CT26 tumor in Balb/c mice (with P < 0.01 compared to the negative control), and no obvious bodyweight loss was detected (Fig. 8). In further investigations, we first evaluated the expressions of interferon-γ (IFN-γ) expression in tumor tissue. Obvious upregulation of IFN-γ expression could be seen (Fig. 9), suggesting that 6 could activate cytotoxic T cells thus realizing a therapeutic effect.
 | ||
| Fig. 8 Dosing regimen, tumor growth inhibition investigation and mice weight change of compound 6 and antibody-treated group. | ||
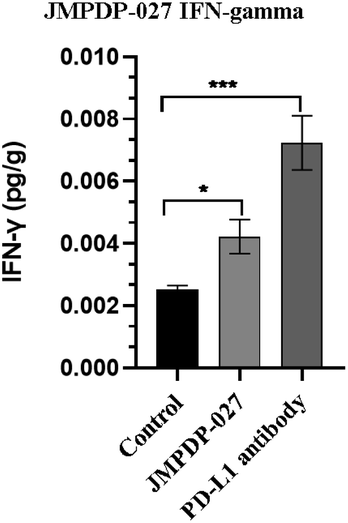 | ||
| Fig. 9 ELISA results of mouse IFN-γ in tumor tissue. | ||
T cell subsets in tumor tissue were further analyzed by flow cytometry experiments. Compared with control group, the 6-treated group showed obvious increase of ratio of CD3+CD8+/CD3+CD4+ (Fig. 10). In the meantime, the ratio of CD3+CD4+Foxp3+ T cells (Treg cells) in tumor tissue decrease sharply compared with control group. While the CD3+CD8+IFN-γ+ T cells (activated cytotoxic T cells) also increased compared with control group (Fig. 10c). This series of changes in T cell subsets indicated activation of immune system.
 | ||
| Fig. 10 Flow cytometry analysis of T cell subsets in tumor tissues. (a) T cell subsets in tumor tissues of control group; (b) T cell subsets in tumor tissues of compound 6-treated group; (c) ratio of different T cell subsets in the two groups. | ||
Tumor tissue biopsies provided another evidences for immunotherapy efficacy of compound 6. Immunohistochemical staining results showed that after treatment, the infiltration of CD8+ T cells in tumor tissues increased sharply. Meanwhile, H&E staining results showed obvious remission of malignancy in the group treated with compound 6 (Table 1). These results further showed that JMPDP-027 (6) could increase the antigenicity of PD-L1-expressing tumors and rescue the immune response against PD-L1-expressed tumors.
| Control | Compound 6 | Control | Compound 6 | ||
|---|---|---|---|---|---|
| H&E |  |
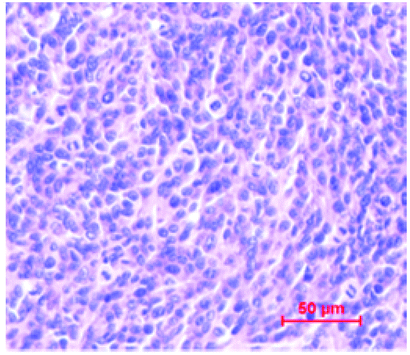 |
IHC (CD8) |  |
 |
Conclusions
In summary, we report the successful design of JMPDP-027 (6) via rational design starting from peptide-57 (1). This lead drug candidate JMPDP-027 antagonizes the PD-1/PD-L1 interaction with subnanomolar inhibitory efficacy at a cellular level. More importantly, JMPDP-027 shows excellent in vivo anticancer activity in the CT26 mouse model. Our results indicate that JMPDP-027 is a new class of antagonist targeting the PD-1/PD-L1 interaction and has potential for clinical immunotherapy.Materials and methods
Cell lines
CT-26 cells were obtained from the American Type Culture Collection (ATCC). Cells were maintained at low passage number and were cultured in RPMI 1640 or DMEM supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin, as specified by ATCC and grown at 37 °C and 5% CO2. Cells were conformed to be mycoplasma free in prior to use.Peptide synthesis
All reagents and solvents were purchased from Bide Pharmatech or Energy Chemical and used without further purification. Rink amide AM resin was obtained from GL Biochem (Shanghai) Ltd. Linear peptides with chloroacetic acid at N-terminus were obtained using rink amide AM resin (loading: 0.782 mmol g−1). Fmoc deprotection was achieved using 20% piperidine in DMF for 30 min. Coupling procedure was performed with solution of corresponding Fmoc-AA (4 eq.), HATU (4 eq.), HOAt (4 eq.) and DIPEA (6 eq.) in DMF (5 mL/0.1 mmol) for 1 hour at rt with the exception of amino acids following N-methylated residues. For amino acid after N-methylated residues the coupling procedure was repeated twice. Each coupling time is 45 min. N-Terminal chloroacetic acid was also coupled with the above method. Cleavage and deprotection of the linear peptides from the resin was accomplished with the mixture of TFA/thioanisole/EDT/anisole (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:3:2, 5 mL mmol−1 of resin) after 3 h of revolving. The crude peptide was precipitated with ice-cold diethyl ether and centrifuged (3000 rpm, 5 min). Peptides were dried in stream of nitrogen.
:1 v/v, 1 mg mL−1) and mixture was stirred for 24 h. The solution was evaporated under vacuum and lyophilized to obtain solid residue.
:5:3:2, 5 mL mmol−1 of resin) after 3 h of revolving. The crude peptide was precipitated with ice-cold diethyl ether and centrifuged (3000 rpm, 5 min). Peptides were dried in stream of nitrogen.
:1 v/v, 1 mg mL−1) and mixture was stirred for 24 h. The solution was evaporated under vacuum and lyophilized to obtain solid residue.Peptide purification
Peptides were purified using preparative HPLC (Shimadzu, SPD-20A UV/VIS detector, LC-20AR LC pump) with a C18 column (Agilent Eclipse XDB-C18, 9.4 × 250 mm, 5 μm) with 10% MeCN (aq.)/acetonitrile (0.1% TFA) eluent system. Gradient: 0–3 min, 0% B; 3–13 min, 0–100% B to obtain a crude product of peptides and gradient: 0–3 min, 0–40% B; 3–30 min, 40–100% B for further purification.Analytical HPLC was done using three methods. Method 1: column: Agilent Eclipse XDB-C18, 4.6 × 250 mm, 5 μm; wavelength: 254 nm; flow: 1 mL min−1; column temperature: 25 °C; eluent system: A: 10% MeCN (aq.)/B: acetonitrile (0.1% TFA) gradient: 0–3 min, 0–30% B; 3–60 min, 30–100% B. Method 2: column: Agilent Eclipse XDB-C18, 4.6 × 250 mm, 5 μm; wavelength: 254 nm; flow: 1 mL min−1; column temperature: 25 °C; eluent system: A: 10% MeOH (aq.)/B: methanol (0.1% TFA); gradient: 0–3 min, 0% B; 3–60 min, 0–100% B. Method 3: column: GL sciences, InertSustain C18, 4.6 × 250 mm, 5 μm; wavelength: 220 nm; flow: 1 mL min−1; column temperature: 45 °C; eluent system: A: 10% MeCN (aq., 0.1% TFA)/B: acetonitrile (0.1% TFA) gradient: 0–5 min, 0–30% B; 5–30 min, 30–80% B.
Protocal for PD-1/PD-L1 blockade bioassay
TR-FRET assay was performed according to the protocol provided by CisBio.20Cell assay21 consists of two genetically engineered cell lines: PD-1 Effector Cells (Jurkat T cells expressing human PD-1 and a luciferase reporter driven by an NFAT response element), and PD-L1 aAPC/CHO-K1 Cells (CHO-K1 cells expressing human PD-L1 and an engineered cell surface protein designed to activate cognate TCRs in an antigen-independent manner). The both cell lines were cultured and propagated following the manufacturer's instructions. APC cells were seeded at 40000 cells per well in 96-well, white, flat-bottom assay plates (Corning) and incubate the cells overnight 16 hours in a 37 °C, 5% CO2 incubator. On the day of the assay serial dilutions of tested macrocyclic peptides and anti-PD-1 antibodies pembrolizumab were prepared in assay buffer (RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 1% Fetal Bovine Serum (BioWest)) and added to the wells containing APC cells. Since stock solutions of peptides were prepared in DMSO, care was taken to keep the concentration of DMSO constant and below observable influence on the test results. Immediately after the addition of tested compounds, 50000 PD-1 Effector Cells were added in the same assay buffer. The cells were incubated for 6 hours in a 37 °C, 5% CO2 incubator and equilibrated for 20 min at room temperature. Bio-Glo reagent (Promega) was added and the luminescence was determined after further 20 min incubation and relative light units (RLU) were normalized to PD-1 antibody controls. Fit curves and determine the half maximal effective concentrations (EC50) value of response using GraphPad Prism® software.
MD simulation and conformation analysis
All the simulation was performed through Accerly Discovery Studio 2016. All the complexes were first applied CHARMm force field and further solvated via solvation module in Discovery Studio in prior to MD simulation. Complexes were solvated with periodic boundary conditions: the cell shape was orthorhombic and the minimum distance from periodic boundary was 7 Å. Counter-ions (Na+ and Cl−) were also added and the salt concentration was set to 0.145 M. MD simulations were performed with simulation-dynamics–standard dynamics cascade module. Detailed settings are recorded in ESI.† Structure/conformation analysis and MD simulations analysis were performed with Accerly Discovery Studio 2016 and Schrödinger Maestro 10.5.Stability evaluation in human serum
The enzymatic hydrolysis stability of compound 1 and compound 6 was detected by a human serum degradation assay. Compounds were dissolved in phosphate buffer solution to a concentration of 0.5 mM. 10% human serum was added into peptide solutions. At certain time point [0 min, 60 min, 120 min, 240 min, 480 min, and overnight (approx. 18 h)], 20 μL of each reaction was sampled and immediately quenched by a mixed solution (90 μL acetonitrile + 90 μL 0.5% acetic acid in H2O) to stop the degradation for the time point. Samples were mixed with 300 μL H2O before analysed by analysis HPLC. Stability of peptides were measured by the following equation:where, St is the peak area of peptides at certain time point and S0 is the peak area of peptides at 0 min. 10% human serum and corresponding peptides were used as blank control and standard, respectively.
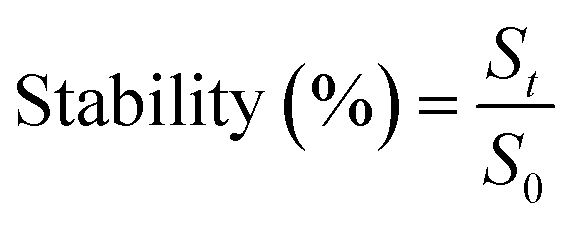
Tumor growth experiments in mice
The experimental procedures, animal use and care protocols were conducted according to the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of China Pharmaceutical University following the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care (Application No. 2020-10-011). Five-week-old male and female Balb/c mice were purchased from College of Veterinary Medicine, Yangzhou University (Yangzhou, Jiangsu, China) and housed in a temperature and humidity controlled room on a 12 h light–dark cycle. Approximately 5 × 105 CT26 cells were subcutaneously injected into the right flank. The mice were randomly divided into three different groups (5 mice per group, 50% female, 50% male). The mice were intraperitoneally injected with 4 mg kg−1 peptide daily after the tumor size reached 50–100 mm3. The anti-mouse PD-L1 antibody (10F.9G2, BioXcell) was administrated as a positive control at a dose of 10 mg kg−1 every two days. The tumor size was assessed with a caliper and calculated with the formula 0.5 × length × width × width. ELISA kits of IFNγ (70-EK280/3-96) in tumors as per the company's instructions (MultiSciences).Flow cytometry analysis
Mice were sacrificed and tumors were harvested. 0.3 mg tumor tissue was isolated and added into a solution of collagenase (400 units per mL), DNase 1 (100 μg mL−1), and hyaluronidase (0.04 units per mL). The mixture was shaked under 37 °C for 1 h. After, the mixture was passed through a 70 μm nylon cell strainer. The sample was washed 3 times with PBS (with 2% FBS) and then resuspended in 1 × 106 cells per mL for flow cytometry analysis. Antibody (AF488-anti mouse CD45, PE/Cy7 anti-mouse CD3, APC anti-mouse CD4, BV510-anti mouse CD8, PE anti-mouse FOXP3 and PE anti-mouse IFN-γ, all purchased from BioLegend) was added following the antibody manufacturer's recommendations and incubated for 30 min on ice. Cells were analyzed on BD FACS Celesta, and data were analyzed using FlowJo 10. Cellular events were first gated by forward and then by side scatter characteristics. The cell counts for the CD45+, CD45+CD3+, CD45+CD3+CD4+, CD45+CD3+CD4+FOXP3+ CD45+CD3+CD8+ and CD45+CD3+CD8+IFN-γ+ T lymphocytes were assessed. For quantification, the proportion of each immune subpopulation was determined by flow cytometry. This number was then multiplied by the total number of cells in the tumor, which had been determined before flow cytometry by counting with a hemocytometer. To determine cell density, cell counts were normalized to tumor weight (cell number per gram tumor).MTT cell viability assay
CT26 cells were cultured in 96 well cell culture plates at a density of 1 × 104 cells per well and incubated with compounds dissolved in 100 μL of serum free DMEM for 24 hour in a CO2 incubator (Make-Healforce) under 37 °C and relative humidity of 90%. This was followed by addition of dimethyl sulfoxide (DMSO) and recording absorbance at 570 nM using a UV-Vis multiwell plate reader (Make-TECAN). In the control group cells were left untreated and only 100 μL of serum free DMEM was added. The cell viability was expressed as a percentage of the control by the following equation:| Cell viability (%) = (Nt/Nc) × 100% |
H&E staining
Tumor sample was immersed in 4% paraformaldehyde for 4 h, and transferred to 70% ethanol. Individual lobes of tumor sample were placed in processing cassettes, dehydrated through a serial alcohol gradient, and embedded in paraffin wax blocks. 5 μm-thick tissue sections were dewaxed in xylene, rehydrated through decreasing concentrations of ethanol, washed in PBS and then stained with hematoxylin and eosin (H&E). After staining, sections were dehydrated through increasing concentrations of ethanol and xylene. Images were acquired on a fluorescent inverted microscope (Nikon Eclipse Ts2R).Immunohistochemistry staining
Isolated tumors were fixed in 4% paraformaldehyde for 24 h at room temperature, dehydrated, and embedded in paraffin wax. Before staining with antibody, tissues were transferred to slides and deparaffinized. The GB13429-anti-mouse CD8 antibody (ServiceBio, China) was added on the slides and incubated at 4 °C overnight for staining. After washing with PBS three times, tissues were incubated with DAPI. Immunofluorescence images were acquired on a fluorescent inverted microscope (Nikon Eclipse Ts2R).Statistics analysis
All the statistical analysis of the samples was conducted by one-way ANOVA. P < 0.05 was regarded as statistically significant (* <0.05, ** <0.01, *** <0.001, **** <0.0001). All data reported are means ± standard deviations, unless otherwise noted.Author contributions
Funding acquisition, project administration, validation: Sheng Jiang.Design of research: Sheng Jiang.
Synthesis, purification and MD simulation: Qi Miao.
Biological evaluation: Wanheng Zhang, He Li and Jidong Zhu.
Data curation: Qi Miao, Wanheng Zhang, Kuojun Zhang, Jidong Zhu and Sheng Jiang.
Writing of original draft: Qi Miao, Wanheng Zhang and Sheng Jiang.
Conflicts of interest
The authors declare the following competing financial interest:Chinese patent application filing is in process. Patent application number 202010447004.X.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81773559), and the Double First-Class University Project (CPU2018GY03).Notes and references
- W. Gu, L. Wang, Y. Wu and J.-P. Liu, Clin. Exp. Pharmacol. Physiol., 2019, 46, 105–115 CrossRef CAS PubMed
.
- M. F. Sanmamed and L. Chen, Cell, 2018, 175, 313–326 CrossRef CAS PubMed
- T. Wang, X. Wu, C. Guo, K. Zhang, J. Xu, Z. Li and S. Jiang, J. Med. Chem., 2019, 62, 1715–1730 CrossRef CAS PubMed
- T. Chen, Q. Li, Z. Liu, Y. Chen, F. Feng and H. Sun, Eur. J. Med. Chem., 2019, 161, 378–398 CrossRef CAS PubMed
- J. Yang and L. Hu, Med. Res. Rev., 2019, 39, 265–301 CrossRef CAS PubMed
- K. Guzik, M. Tomala, D. Muszak, M. Konieczny, A. Hec, U. Błaszkiewicz, M. Pustuła, R. Butera, A. Dömling and T. A. Holak, Molecules, 2019, 24, 2071 CrossRef CAS PubMed
- H.-N. Chang, B.-Y. Liu, Y.-K. Qi, Y. Zhou, Y.-P. Chen, K.-M. Pan, W.-W. Li, X.-M. Zhou, W.-W. Ma, C.-Y. Fu, Y.-M. Qi, L. Liu and Y.-F. Gao, Angew. Chem., Int. Ed., 2015, 54, 11760–11764 CrossRef CAS PubMed
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed
- K. Zhou, J. Lu, X. Yin, H. Xu, L. Li and B. Ma, Biophys. Chem., 2019, 253, 106213 CrossRef CAS PubMed
- H. Sun, D. Chen, S. Zhan, W. Wu, H. Xu, C. Luo, H. Su, Y. Feng, W. Shao, A. Wan, B. Zhou, G. Wan and X. Bu, J. Med. Chem., 2020, 63, 11286–11301 CrossRef CAS PubMed
- Bristol-Myers Squibb Company, Wipo Pat., WO2014151634, 2014 Search PubMed
- Bristol-Myers Squibb Company, Wipo Pat., WO2016039749, 2016 Search PubMed
- R. A. De Silva, D. Kumar, A. Lisok, S. Chatterjee, B. Wharram, K. Venkateswara Rao, R. Mease, R. F. Dannals, M. G. Pomper and S. Nimmagadda, Mol. Pharm., 2018, 15, 3946–3952 CrossRef CAS PubMed
- D. Kumar, A. Lisok, E. Dahmane, M. McCoy, S. Shelake, S. Chatterjee, V. Allaj, P. Sysa-Shah, B. Wharram, W. G. Lesniak, E. Tully, E. Gabrielson, E. M. Jaffee, J. T. Poirier, C. M. Rudin, J. V. Gobburu, M. G. Pomper and S. Nimmagadda, J. Clin. Invest., 2019, 129, 616–630 CrossRef PubMed
- K. Magiera-Mularz, L. Skalniak, K. M. Zak, B. Musielak, E. Rudzinska-Szostak, L. Berlicki, J. Kocik, P. Grudnik, D. Sala, T. Zarganes-Tzitzikas, S. Shaabani, A. Domling, G. Dubin and T. A. Holak, Angew. Chem., Int. Ed., 2017, 56, 13732–13735 CrossRef CAS PubMed
- L. Deng, Q. C. Zhang, Z. Chen, Y. Meng, J. Guan and S. Zhou, Nucleic Acids Res., 2014, 42, W290–W295 CrossRef CAS PubMed
- D. E. Kim, D. Chivian and D. Baker, Nucleic Acids Res., 2004, 32, W526–W531 CrossRef CAS PubMed
- X. Zhu and J. C. Mitchell, Proteins: Struct., Funct., Bioinf., 2011, 79, 2671–2683 CrossRef CAS PubMed
- J. Egli, T. Schnitzer, J. C. B. Dietschreit, C. Ochsenfeld and H. Wennemers, Org. Lett., 2020, 22, 348–351 CrossRef CAS PubMed
- http://www.bpsbioscience.com, catalog #72038.
- Z.-J. J. Cheng, N. Karassina, J. Grailer, J. Hartnett, F. Fan and M. Cong, Cancer Res., 2015, 75, 5440–5440 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03118j |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |