
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zinc(II) and cadmium(II) amorphous metal–organic frameworks (aMOFs): study of activation process and high-pressure adsorption of greenhouse gases†
Miroslav Almáši *a,
Nikolas Királya,
Vladimír Zeleňáka,
Mária Vilkováb and
Sandrine Bourrellyc
*a,
Nikolas Királya,
Vladimír Zeleňáka,
Mária Vilkováb and
Sandrine Bourrellyc
aDepartment of Inorganic Chemistry, Faculty of Science, P. J. Šafárik University, Moyzesova 11, SK-041 54 Košice, Slovak Republic. E-mail: miroslav.almasi@upjs.sk
bNMR Laboratory, Faculty of Science, P. J. Šafárik University, Moyzesova 11, SK-041 01 Košice, Slovak Republic
cAix-Marseille University, CNRS, MADIREL, Marseille Cedex 20, F-133 97, France
First published on 4th June 2021
Abstract
Two novel amorphous metal–organic frameworks (aMOFs) with chemical composition {[Zn2(MTA)]·4H2O·3DMF}n (UPJS-13) and {[Cd2(MTA)]·5H2O·4DMF}n (UPJS-14) built from Zn(II) and Cd(II) ions and extended tetrahedral tetraazo-tetracarboxylic acid (H4MTA) as a linker were prepared and characterised. Nitrogen adsorption measurements were performed on as-synthesized (AS), ethanol exchanged (EX) and freeze-dried (FD) materials at different activation temperatures of 60, 80, 100, 120, 150 and 200 °C to obtain the best textural properties. The largest surface areas of 830 m2 g−1 for UPJS-13 (FD) and 1057 m2 g−1 for UPJS-14 (FD) were calculated from the nitrogen adsorption isotherms for freeze-dried materials activated at mild activation temperature (80 °C). Subsequently, the prepared compounds were tested as adsorbents of greenhouse gases, carbon dioxide and methane, measured at high pressures. The maximal adsorption capacities were 30.01 wt% CO2 and 4.84 wt% CH4 for UPJS-13 (FD) and 24.56 wt% CO2 and 6.38 wt% CH4 for UPJS-14 (FD) at 20 bar and 30 °C.
1 Introduction
Metal–organic frameworks (MOFs) are one of the most exciting classes of porous materials discovered in the past few decades. MOFs are crystalline materials constructed from organic molecules (linkers) coordinated to metal ions or clusters to form interesting polymeric frameworks with a larger surface area and the advantage of pore size changing. Due to the porosity and tunable textural properties, MOF materials find applications in gas storage and separation,1,2 heterogeneous catalysis,3,4 drug delivery,5,6 magnetic refrigeration,7,8 sensing9,10 and energy storage.11,12 A small but growing number of noncrystalline MOFs are steadily capturing scientific interest, though it is only recently that well-characterised examples of amorphous metal–organic frameworks (aMOFs) have started to appear.13–15 aMOFs retain the basic building blocks and connectivity of their crystalline counterparts, though they lack any long-range periodic order. aMOFs combine properties that are usually found in crystalline with the unique properties of the amorphous domain such as isotropy, absence of grain boundaries, abundant defects and active sites, and flexibility. Most aMOFs have been prepared by applying stress to a crystalline framework. To date, both static and hydrostatic pressures, heating, mechanical stress (ball milling, grinding), radiation, and also chemical treatment have been applied to crystalline MOFs to induce their collapse and amorphization.16 However, examples of aMOF materials which have been prepared in the amorphous state by direct synthesis are also known. It should be noted that only a minority number of compounds prepared by the mentioned way are known in the literature: a-UiO-66-SO3H (SBET = 11 m2 g−1),17 a-FMM-120 (SBET = 43 m2 g−1),18 CPPs-X (X = 1, 3, 5; SBET = 522–588 m2 g−1),19 aZr-MOF(X) (X = 23–26; SBET = 410–955 m2 g−1),20 NEU-X (X = 2–8; SBET = 10–294 m2 g−1).21–23 As was described above, aMOFs have been mostly prepared in a top-down approach by applying stress and introducing disorder into the parent frameworks. In our work we developed bottom-up synthesis of aMOFs, with a high and permanent porosity. Amorphous MOFs offer many exciting opportunities for practical application, either as novel functional materials themselves or facilitating other processes, though the domain is mostly unexplored.24 Published articles dealing with aMOFs have been demonstrated to be of potential in several fields, e.g. catalysis,25–28 drug delivery,29–33 supercapacitors34–36 and irreversible harmful substance capture.37,38To study the porosity and textural properties of porous materials is necessary to activate them and thus obtain an open structure. A typical activation process is a thermal activation, in which the process of releasing solvent molecules is caused by heat. This activation way often leads to the shrink or collapse of the polymeric frameworks causing a decrease of material's porosity or non-porosity. To reduce the activation temperature, a solvent exchange process is also used as a tool, in which high-boiling solvents are exchanged for low-boiling solvents and the thermal activation of porous material occurs at a lower temperature. The freeze-drying process, also known as lyophilisation or cryodesiccation, is a dehydration/desolvation process performed at low temperature, which involves freezing the material with guest molecules and subsequent sublimating the guests under low temperature and low pressure. Lyophilisation is a commercial process preliminary used in biological, biomedical processing, fine food production and preservation. The freeze-drying process has also found an application in the activation of porous materials.39 Large specific surface area and better textural properties can be obtained by exchanging of high boiling point solvents (N,N′-dimethylformamide (153 °C), N,N′-diethylformamide (176 °C), N,N′-dimethylacetamide (165 °C), dimethylsulfoxide (189 °C) etc.), that are typically located in the pores of as-synthesized material by the solvents with high melting point and high vapour pressure (benzene (5.5 °C, 75 mmHg at 20 °C), cyclohexane (4 °C, 77 mmHg at 20 °C), tert-butyl alcohol (23 °C, 31 mmHg at 20 °C), o-xylene (−23 °C, 7 mmHg at 20 °C) and compressed CO2) followed by evacuation at low temperature. For the organic nature of porous coordination polymers, many MOFs exhibit surface areas that are only a small fraction of predicted/calculated surfaces based on their crystal structures. It was shown that a freeze-drying process could achieve a higher surface area and the desired features of porous materials. For example, freeze-dried compounds [Cu2(MTB)(H2O)2]·6DEF·2H2O and [Cu2(MTC)(H2O)2] ·14DMF·5H2O, which also contain tetrahedral linkers (MTB = methanetetrabenzoate, MTC = methanetetra(biphenyl-p-carboxylate)) exhibited surface areas of 526 m2 g−1 and 791 m2 g−1 for evacuated materials (activation temperature 60 °C) and 1560 m2 g−1, 1020 m2 g−1 for freeze-dried samples, respectively.40 As is obvious from described values, lyophilisation is an efficient process for activating porous materials that can increase the surface area three times compared to standard thermal activation.
Global temperature has been rising since the first industrial revolution in the early 19th century, due to the emissions of large quantities of greenhouse gases. The main greenhouse gases are carbon dioxide and methane, but also nitrogen oxides and water vapour. Carbon dioxide is a major by-product of fossil fuel combustion, electricity generation and other anthropogenic activities. Major advances in carbon capture technology could provide power plants and road transport with an efficient and inexpensive way to remove carbon dioxide from their flue gas emissions, which is necessary to reduce greenhouse gas emissions to slow global warming and climate change. The 2030 Climate and Energy Framework commits all EU member states to a 40% reduction in greenhouse gas emissions over the coming years.41 For this reason, it is necessary to develop new and effective carbon dioxide and methane adsorbents as the main greenhouse gases. Many porous materials such as carbon dioxide adsorbents, e.g. MOFs or porous oxides, are modified with nitrogen-containing amine molecules to capture the CO2 (chemisorption) at low-temperature steam to flush out the CO2 for other uses or to sequester it underground are currently being developed and intensively studied. Examples are tetraamine-appended MOF Mg2(dobpdc) (dobpdc = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate, SBET = 2880 m2 g−1), which can store up 90 wt% CO2 (ref. 42) and amine grafted silica materials.43–45 In order to avoid CO2-intensive energy processes, CO2 molecules must be captured in porous materials by physisorption as a chemisorption process. However, physisorption brings new challenges, because there is a lack of selectivity for CO2 because no chemical bond is formed or when the functional groups are tuned to increase selectivity, the absorption of CO2 is limited.
In the present study, extended tetrahedral nitrogen-rich tetraazo-tetracarboxylic acid (H4MTA) was prepared by seven-step organic synthesis and used as a linker in the preparation of two novel aMOFs containing Zn(II) (UPJS-13) and Cd(II) (UPJS-14) ions. After characterisation, the textural properties of prepared materials were studied by nitrogen adsorption at −196 °C on as-synthesized (AS), ethanol exchanged (EX) and freeze-dried (FD) samples. The specific surface area (SBET) depended on the sample pretreatment (AS < EX < FD) and the activation temperature (60–200 °C). The highest SBET values were obtained for FD materials after activation process at 80 °C: UPJS-13 (EX) 830 m2 g−1 and UPJS-14 (EX) 1057 m2 g−1. After finding the best activation conditions, the materials were tested as adsorbents of greenhouse gases in high-pressure adsorption measurements of carbon dioxide and methane up to 20 bar.
2 Experimental
2.1 Synthetic part
 | ||
| Fig. 1 Synthesis of methanetetrayltetrakis(benzene-4,1-diyl)tetrakis(aza)tetrakis(methan-1-yl-1-yliden)tetrabenzoic acid (H4MTA) with corresponding synthetic conditions. | ||
2.1.2.1 Synthesis of 4-tritylaniline (1). 4-Tritylaniline was prepared by the condensation reaction of triphenylmethanol and aniline. Triphenylmethanol (20 g; 77 mmol) was dissolved in glacial acetic acid (230 cm3) and to this solution aniline (11 cm3; 121 mmol) was added. Finally, the reaction mixture was acidified with concentrated HCl (38.5 cm3) and refluxed at 130 °C for 48 h. Subsequently, the reaction mixture was cooled to ambient temperature and diluted in water (1.5 dm3). The resulting precipitate of 1 was filtered off under reduced pressure using a Buchner funnel. The filter cake was washed several times with water to remove residual acetic and hydrochloric acid. The washed product was placed in an oven and dried at 70 °C overnight to yield 18.16 g of 1 (71% based on triphenylmethanol). 1H NMR (600 MHz, CDCl3) δ (ppm): 5.00 (s, 2H); 6.46 (d, 2H, J = 6.98 Hz); 6.75 (d, 2H, J = 6.98 Hz); 7.09–7.29 (m, 15H) ppm. Elemental analysis for C25H21N1 (335.44 g mol−1) calculated: C, 89.51%; H, 6.31%; N, 4.18%; measured: C, 89.46%; H, 6.21%; N, 4.09. IR (KBr): ν(NH) 3469 (w), 3382 (w); ν(CH)ar. 3082 (w), 3054 (w), 3028 (w); δ(NH) 1621 (m); ν(C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)ar. 1593 (w), 1511 (w); δ(CCH)ar. 1183 (w), 1081 (w), 1033 (w); γ(CCH)ar. 825 (m) cm−1.
C)ar. 1593 (w), 1511 (w); δ(CCH)ar. 1183 (w), 1081 (w), 1033 (w); γ(CCH)ar. 825 (m) cm−1.
2.1.2.2 Synthesis of tetrakis(phenyl)methane (2). The tetrakis(phenyl)methane core was prepared by deamination reaction of 4-tritylaniline. 1 (10 g; 30 mmol) was dispersed in N,N′-dimethylformamide (DMF, 125 cm3) and the solution was acidified by dropwise addition of concentrated sulphuric acid (62.5 cm3). Sulphuric acid protonated the primary amine of 1, causing its complete dissolution in DMF. Subsequently, the reaction mixture was cooled with the solution of ice/sodium chloride to ≈−10 °C, and then isopentyl nitrite (15 cm3; 112.5 mmol) was added dropwise. After the addition of isopentyl nitrite, the solution was stirred for 1 h at ≈−10 °C. Hypophosphorous acid (30 cm3; 0.5 mol) was added dropwise after the mentioned reaction time, which catalysed the azo bond cleavage of the intermediate. During the addition of H3PO2, the release of nitrogen was observed and a precipitate of 2 was formed in the mixture. 2 was filtered off under reduced pressure, washed with DMF, methanol and dried in an oven at 70 °C overnight (yield: 8.83 g, 92.4% based on 1). 1H NMR (600 MHz, CDCl3) δ (ppm): 7.21 (m, 20H) ppm. 13C NMR (151 MHz, CDCl3) δ (ppm): 67.0; 125.8; 127.4; 131.1; 146.7 ppm. Elemental analysis for C25H20 (320.43 g mol−1) calculated: C, 93.71%; H, 6.29%; measured: C, 92.67%; H, 6.19%. IR (KBr): ν(CH)ar. 3080 (w), 3052 (w), 3029 (w), 3011 (w); ν(C
C)ar. 1591 (w), 1490 (m); δ(CCH)ar. 1181 (w), 1080 (w), 1034 (w); γ(CCH)ar. 748 (s), 699 (s) cm−1.
2.1.2.3 Synthesis of tetrakis(p-nitrophenyl)methane (3). Tetrakis(p-nitrophenyl)methane was prepared by nitration reaction of 2 using fuming acid. Fuming acid (nitric acid, 99% HNO3, 12.81 cm3; 0.3 mol) was cooled to ≈−10 °C using H2O/NaCl bath and 2 (2.56 g; 8 mmol) was added to the solution in small portions as the reaction is extremely exothermic. After complete addition of 2, acetic anhydride (4.27 cm3; 45 mmol) was added dropwise and the reaction mixture was stirred for 30 min. The last step was a dilution of the reaction mixture into glacial acetic acid (17.1 cm3). A yellow precipitate of 3 was filtered off, washed with acetic acid, methanol and dried in an oven at 50 °C overnight (yield 1.63 g, 40.8% based on 2). Finally, the product was recrystallised from toluene, resulting in a reaction yield of 30%. 1H NMR (600 MHz, DMSO): δ = 7.61 (8H, d, J = 9.01 Hz); 8.22 (8H, d, J = 9.01 Hz) ppm. Elemental analysis for C25H16N4O8 (500.42 g mol−1) calculated: C, 60.00%; H, 3.22%; N,11.20%; N, 11.20%; measured C, 60.31%; H, 3.18%; N, 11.02%. IR (KBr): ν(CH)ar. 3109 (w), 3079 (w); ν(C
C)ar. 1604 (m), 1589 (m), 1493 (w); νas.(C–NO2) 1520 (s); νs.(C–NO2) 1337 (s); δ(CCH)ar. 1106 (m), 1009 (w); γ(CCH)ar. 837 (s), 743 (s) cm−1.
2.1.2.4 Preparation of tetrakis(p-aminophenyl)methane (4). The aim of this synthetic step was the reduction reaction of nitro to an amino group. The reduction was carried out by two synthetic procedures using different catalysts and reducing agents: hydrazine/RaNi or Pd/C and hydrogen, respectively.
2.1.2.4.1 Synthesis using hydrazine/RaNi. The reaction mixture of 3 (0.3 g; 0.9 mmol), hydrazine monohydrate (N2H4·H2O; 0.4 g; 8 mmol; 50% aqueous solution) and RANEY® nickel (Ra/Ni; 10 g) in THF (50 cm3) was refluxed for 12 hours. The reaction mixture was hot filtered under reduced pressure to remove the heterogeneous catalyst and washed with THF. The filtered solution was evaporated under vacuum until the first visible crystals of the product were formed. The crystallised product 4 was filtered off, slightly washed with ethanol and finally purified from hot ethyl acetate (yield: 0.18 g, 78% based on 3).
2.1.2.4.2 Synthesis using Pd/C and hydrogen. 3 (0.5 g; 0.99 mmol) was dissolved in methanol (20 cm3) and heterogeneous Pd/C catalyst (0.1 g; 0.05 mmol) was added to the solution in small portions. The reaction mixture was stirred vigorously and the hydrogen atmosphere was created as follows: the pressure in the system was reduced by a vacuum pump until boiling of the methanol occurred and hydrogen was introduced into the system. This procedure was repeated four times. Subsequently, the reaction mixture was stirred for 24 h in a reducing atmosphere of hydrogen at room temperature. The product was filtered off, washed with methanol and dried under vacuum. 4 was purified by column chromatography on silica gel using a solvent mixture of DCM
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (7:1, v/v) as mobile phase (yield: 0.27 g, 71% based on 3).
:MeOH (7:1, v/v) as mobile phase (yield: 0.27 g, 71% based on 3).
2.1.2.4.3 1H NMR (600 MHz, DMSO) δ (ppm). 4.84 (4H, s); 6.39 (8H, d, J = 8.4 Hz); 6.67 (8H, d, J = 8.4 Hz). Elemental analysis for C25H24N4 (380.48 g mol−1) calculated: C, 78.92%; H, 6.36%; N, 14.73%; measured: C, 79.13%; H, 6.28%; N, 14.49%. IR (KBr): ν(NH2) 3465 (m), 3382 (m); ν(CH)ar. 3082 (w), 3054 (w), 3027 (w); ν(C
C)ar. 1505 (s), 1593 (m); δ(NH2) 1621 (m); ν(C–N) 1286 (s); δ(CCH)ar. 1182 (m), 1115 (m), 1033 (w); γ(CCH)ar. 825 (w), 749 (m), 701 (s) cm−1.
2.1.2.5 Preparation of ethyl 4-nitrosobenzoate (5). 2.5 g (15 mmol) of ethyl 4-aminobenzoate was dissolved in 50 cm3 of dichloromethane (DCM) and solution of oxone (potassium peroxymonosulfate, KHSO5·0.5KHSO4·0.5K2SO4) 20.3 g (66 mmol) in 200 cm3 of deionised water was added. The reaction mixture was stirred at room temperature for 2 h. After the colour change from colourless to green, the mixture was extracted with CH2Cl2 and washed with H2O several times. The organic layer was further washed with 30 cm3 of 1 M HCl and 40 cm3 of NaHCO3 saturated aqueous solution. The suspension was extracted with CH2Cl2 (4 × 50 cm3) and dried with Na2SO4. The organic layers were collected and the solvent was removed under reduced pressure to form the light-yellow product of 5 (yield 2.03 g, 75% based on ethyl 4-aminobenzoate). 1H NMR (600 MHz, CDCl3) δ (ppm): 1.24 (3H, t, J = 7.1 Hz), 4.14 (2H, q, J = 7.1 Hz), 8.07 (2H, ddd, J = 8.5, 1.6, 0.5 Hz), 8.13 (2H, ddd, J = 8.5, 1.4, 0.5 Hz). Elemental analysis for C9H9NO3 (179.17 g mol−1) calculated: C, 60.33%; H, 5.06%; N, 7.82%; measured: C, 60.54%; H, 4.99%; N, 7.89%. IR (KBr): ν(CH)ar. 3103 (w), 3072 (w), 3050 (w); ν(CH)aliph. 2864 (w), 2901 (w), 2932 (w), 2979 (w); ν(C
O) 1703 (s); ν(C–N) 1529 (w); γ(CCH)ar. 874 (s), 689 (s) cm−1.
2.1.2.6 Preparation of methanetetrayltetrakis(benzene-4,1-diyl)tetrakis(aza))tetrakis (methan-1-yl-1-yliden)tetrabenzoic acid ethyl ester (Et4MTA) (6). Et4MTA was prepared by the condensation reaction of ethyl 4-nitrosobenzoate and tetrakis(p-aminophenyl)methane. Solution of 5 (1.6 g, 9 mmol) in 40 cm3 of acetic acid was added dropwise during 10 min time period in solution of 4 (0.7 g, 1.8 mmol) dissolved in 40 cm3 of glacial acetic acid and 50 cm3 of extra dry methanol under nitrogen. The reaction mixture was stirred at laboratory temperature in an inert atmosphere of nitrogen for 2 days. Formed orange suspension of 6 was filtered off, washed with saturated aqueous solution of NaHCO3, water and methanol (3 × 15 cm3) and dried at 60 °C in an oven overnight (yield: 1.21 g, 64% based on 4). 1H NMR (600 MHz, CDCl3) δ (ppm): 8.19 (8H, d, J = 8.9, H-3′′,5′′), 7.94 (8H, d, J = 8.9, H-2′′,6′′), 7.92 (8H, d, J = 8.8, H-3′,5′), 7.54 (8H, d, J = 8.8, H-2′,6′), 4.42 (8H, q, J = 7.1, H-5), 1.43 (12H, t, J = 7.1, H-6); 13C NMR (151 MHz, CDCl3) δ (ppm): 166.1 (C-4), 155.2 (C-1′′), 151.0 (C-4′), 149.3 (C-1′), 132.5 (C-4′′), 131.8 (C-2′,6′), 130.7 (C-3′′,5′′), 122.9 (C-3′,5′), 122.8 (C-2′′,6′′), 65.6 (C-1), 61.4 (C-5), 14.5 (C-6). Elemental analysis for C61H52N8O8 (1025.11 g mol−1) calculated: C, 71.47%; H, 5.11%; N, 10.93%; measured: C, 71.37%; H, 5.03%; N, 11.18%. IR (KBr): ν(C
O) 1679 (s); ν(CC)ar. 1599 (w); ν(NN) 1494 (w); δ(CCH)ar. 1085 (m), 1009 (m); γ(CCH)ar. 868 (m), 699 (s) cm−1. Assignments of hydrogen and carbon atoms are depicted in Fig. S1 in ESI† and 2D NMR experiments: 1H,1H–COSY; 1H,13C-HSQC; 1H,13C-HMBC spectra of 6 are presented in Fig. S2–S4 in ESI.†
2.1.2.7 Preparation of methanetetrayltetrakis(benzene-4,1-diyl)tetrakis(aza))tetrakis(methan-1-yl-1-yliden)tetrabenzoic acid (H4MTA) (7). The last synthesis step in H4MTA preparation was deesterification reaction of its ethyl ester (6). 6 (270 mg, 0.26 mmol) was suspended and stirred in 150 cm3 solution of 1 M NaOH in methanol and heated at 80 °C for 2 h. After this time the reaction mixture was cooled down to ambient temperature, filtrated and the solvent was removed under vacuum. The crude product was dissolved in water and solution was subsequently acidified with concentrated HCl until pH = 1. Orange solid of 7 was precipitated, filtered off, washed with water and dried in an over at 50 °C overnight (yield: 226 mg, 94% based on 6). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 13.25 (4H, s, OH), 8.14 (8H, d, J = 8.5, H-3′′,5′′), 7.95 (8H, d, J = 8.5, H-2′′,6′′), 7.96 (8H, d, J = 8.8, H-3′,5′), 7.60 (8H, d, J = 8.5, H-2′,6′). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 166.6 (C-4), 154.4 (C-1′′), 150.1 (C-4′), 149.2 (C-1′), 132.9 (C-4′′), 131.5 (C-2′,6′), 130.7 (C-3′′,5′′), 122.8 (C-3′,5′), 122.5 (C-2′′,6′′), 65.2 (C-1). Elemental analysis for C53H36N8O8 (912.9 g mol−1) calculated: C, 69.73%; H, 3.97%; N, 12.27%; measured: C, 69.35%; H, 4.08%; N, 12.18%. IR (KBr): ν(OH)ar. 3420 (w, br); ν(CH)ar. 3106 (w), 3069 (w); ν(CH)aliph. 2982 (w), 2960 (w), 2929 (w); ν(C
O) 1709 (s); ν(CC)ar. 1597 (w); ν(NN) 1521 (w); δ(CCH)ar. 1085 (m), 1009 (m); γ(CCH)ar. 858 (m), 697 (s) cm−1. 2D NMR experiments: 1H,13C-HSQC and 1H,13C-HMBC spectra of 7 are presented in Fig. S5 and S6 in ESI.†
 | ||
| Fig. 2 Prepared orange quasi-crystals of (a) UPJS-13 (AS) and (b) UPJS-14 (AS) in screwcapped vials and after filtration process and 4× zoom. | ||
2.1.4.1 As-synthesized samples (AS). The as-synthesized samples for nitrogen adsorption experiments were prepared by repeating the synthetic procedure described in Section 2.1.3 Synthesis of UPJS-13 and UPJS-14. After collecting of sufficient amount of the sample, the material was stored in the mother liquor. The collected material was further activated under vacuum at selected temperatures described in Section 2.2.6 Nitrogen adsorption measurements.
2.1.4.2 Ethanol exchanged samples (EX). The ethanol exchanged samples were prepared by immersing of the as-synthesized materials in ethanol for 14 days, while ethanol was changed every day. Ethanol exchanged samples were activated under vacuum at selected temperatures described in Section 2.2.6 Nitrogen adsorption measurements.
2.1.4.3 Freeze-dried samples (FD). The freeze-dried samples were prepared by immersing the as-synthesized materials in benzene for 14 days, while benzene was changed every day. Subsequently, the suspension of the sample in benzene was frozen at −20 °C. After five freeze–thaw cycles, the benzene exchanged material in sample cells were placed under vacuum using a turbomolecular pump at −20 °C for 24 h, and further heated under vacuum at selected temperatures described in Section 2.2.6 Nitrogen adsorption measurements. Schematic representation of the described freeze-drying process is shown in Fig. 3.
 | ||
| Fig. 3 Scheme of the freeze-drying process. | ||
2.2 Methods and characterisation
:100 (sample: KBr). Potassium bromide was before measurements dried in an oven at 500 °C for 4 h and cooled in a desiccator. | ||
| Fig. 4 (a) 1H (600 MHz) and (b) 13C (151 MHz) NMR spectra of H4MTA in DMSO-d6 with assignments of hydrogen and carbon atoms. | ||
O)) for calibration of 13C NMR scale.The gas adsorption measurements at 30 °C and pressures up to 20 bar were made with carbon dioxide and methane using a homemade high-throughput instrument.50 Gas adsorption was measured via a manometric gas dosing system on both samples in parallel. The amounts of gas adsorbed were calculated by an equation of state using the Reference Fluid Thermodynamic and Transport Properties (REFPROP) software package 8.0 of the National Institute of Standards and Technology (NIST).51 Approximately 100 mg of sample was used, and the sample was thermally activated individually in situ under a primary vacuum, at a given final temperature overnight. Methane was of 99.9995% purity (N55), carbon dioxide was of 99.995% purity (N45) and gases were obtained from Air Liquide. The weight of the samples used in the high-pressure adsorption experiments was approximately 100 mg and before measurements, the freeze-dried samples were activated at 80 °C under vacuum for 12 h.
3 Results and discussion
3.1 Synthesis and NMR spectroscopy
The H4MTA ligand used in the preparation of aMOFs in the present study was synthesized by the seven-step organic synthesis (see Fig. 1). First, a tetraphenylmethane core was synthesized by the condensation reaction of triphenylmethanol and aniline and the prepared product was subsequently deaminated. Further reactions led to the introduction of nitro groups using fuming acid, which were subsequently reduced to amine groups. Described reactions led to the preparation of the first essential component required for the construction of H4MTA. The second basic component, ethyl 4-nitrosobenzoate, was prepared by transforming the nitro group to the nitroso group using oxone. The resulting H4MTA acid was prepared by the condensation reaction of ethyl 4-nitrosobenzoate with tetrakis-(p-aminophenyl)methane in anhydrous methanol and prepared ethyl ester was further deesterified in a concentrated solution of sodium hydroxide in methanol, followed by acidifying the solution to pH = 1.NMR spectroscopy was used for molecular structure determination of all prepared organic compounds (see 2.1.2 Synthesis of H4MTA). The procedure to assign the NMR data of derivative 6 is described as an example. Compound 6 consists of twelve carbons. The carbonyl carbon is easily detectable in 13C NMR spectrum because of the chemical shift 166.2 ppm. The carbon peak at 166.2 ppm showed long-range couplings to signals of proton at 4.42 ppm (q, J = 7.1 Hz), protons at 8.19 ppm (d, J = 8.8 Hz) and protons at 7.94 ppm (d, J = 8.8 Hz). The proton signal at 4.42 ppm was easily assigned as H-5 because of the splitting pattern as quartet and chemical shift of corresponding carbon peak at 61.4 ppm in the HSQC spectra. The chemical shift for H-6, methyl group, is shown as a triplet of splitting pattern at the lowest frequency at 1.43 ppm. The protons at 8.19 ppm were H-3′′,5′′. They correlated with 130.7 ppm of 13C peak at HSQC spectrum and were coupled to both C-2′′,6′′ and C-1′′ in the HMBC spectrum. The chemical shift for H-2′,6′ (7.54 ppm (d, J = 8.8 Hz)) was assigned by HMBC correlation with quaternary carbon C-1 (65.6 ppm). Protons H-3′,5′ (7.92 ppm (d, J = 8.8 Hz)) were determined from the COSY experiment. The C-4′ is long-range coupled to H-2′,6′. The chemical shift for C-1′ was assigned by HMBC correlation with H-3′,5′.
The successful synthesis of the resulting tetrahedral tetraazo-tetracarboxylic acid was confirmed by 1H and 13C NMR measurements and corresponding spectra with assignments of hydrogen and carbon atoms are depicted in Fig. 4 and 2D NMR 1H,13C-HSQC and 1H,13C-HMBC spectra are presented in Fig. S5 and S6 in ESI.† Derivative 7 has a carboxyl group adjacent to carbonyl carbon atom C-4. Carbonyl carbon C-4 was found to correlate with protons H-3′′,5′′ in HMBC spectrum. The chemical shift of C-3′′,5′′ was observed at 130.7 ppm. The protons H-2′′,6′′ were observed at 7.96 ppm, whereas the corresponding carbon appeared at 122.5 ppm. The protons H-2′′,6′′ exhibit long-range coupling to the quaternary carbon C-4′′ at 132.9 ppm and the protons H-3′′,5′′ are long-range coupled to C-1''. The 1H doublet at 7.59 ppm exhibits a long-range coupling to C-1 (65.2 ppm) in the HMBC spectrum suggesting this signal corresponds to H-2′,6′. In the HMBC spectrum H-2′,6′ also correlate with the carbon at 150.1 ppm, which is C-4′. This implies that the chemical shift for H-3′,5′ is 7.94 ppm. C-1′ with the chemical shift at 149.2 ppm displays a long-range coupling to the protons H-3′,5′. Measured NMR spectra correlate well with a molecular structure of desired tetrahedral linker.
H4MTA was used for the preparation of UPJS-13 (AS) and UPJS-14 (AS) using zinc(II) and cadmium(II) nitrates at different synthetic conditions. From the structural analysis point of view, our primary goal was to prepare single crystals of studied materials suitable for single-crystal X-ray diffraction experiments. We performed many syntheses (specific synthetic conditions are summarized in Table S1 in ESI†), in which we changed the synthetic conditions: from the molar ratio of reactants, H4MTB in the form of carboxylic acid or sodium salt, through reaction time, reaction temperature, different solvents and its volume ratios. The mentioned variable conditions led to the formation of predominantly powdered products, which were not suitable for single-crystal X-ray diffraction experiments. Although aMOFs UPJS-13 and UPJS-14 were prepared in the form of crystals (see Fig. 2) which at first sight appeared to be single crystals, the results of structural measurements (SXRD and PXRD) showed their amorphous character. The best synthetic conditions for the preparation of presented aMOF compounds were found in the mixture of solvent DMF/H2O at 80 °C. The solvent molecules, DMF and water located in the cavities of the prepared compounds were exchanged with ethanol (UPJS-13 (EX) and UPJS-14 (EX)). A third variant of the materials was prepared by the freeze-drying process using benzene (UPJS-13 (FD) and (UPJS-14 (FD)). All described aMOFs materials were subsequently studied by nitrogen adsorption to find the best textural properties and after finding the best activation conditions, the selected compounds were tested as adsorbents for greenhouse gases, carbon dioxide and methane, by high-pressure adsorption measurements.
3.2 Single crystal and powder X-ray diffraction experiments
Since the materials were prepared in the form of crystals (see Fig. 2), compounds were first subjected to single-crystal X-ray diffraction analysis to solve their crystal structures. Because the samples did not provide a diffraction image, even after selecting other crystals, the materials were subjected to powder X-ray diffraction (PXRD) experiments. These measurements aimed to confirm the amorphous composition of the whole bulk of prepared materials and confirm their amorphous character. Measured PXRD patterns of materials are shown in Fig. 5 and confirmed their disordered character. Aperiodic arrangements of atoms result in their PXRD patterns being dominated by broad “humps” caused by diffuse scattering and thus they are mostly indistinguishable from one another. | ||
| Fig. 5 Powder X-ray diffraction patterns of UPJS-13 (AS) and UPJS-14 (AS). | ||
3.3 Infrared spectroscopy
The infrared spectra of as-synthesized, ethanol exchanged and freeze-dried samples are shown in Fig. 6 and the assignment of absorption bands is summarised in Table 1. The IR spectra of both as-synthesized samples (see Fig. 6, black lines) exhibit broad band centred at about 3400 cm−1 which can be attributed to the OH stretching vibrations (ν(OH)) of water molecules, used in the synthesis. The presence of MTA linker in the prepared materials is evident from the intensive adsorption bands at 1603 cm−1 and 1378 cm−1 which can be assigned to antisymmetric and symmetric stretching vibrations of coordinated carboxylate groups (νas(COO−) and vs(COO−)). The presence of MTA in prepared materials demonstrated absorption band at 1563 cm−1 for UPJS-13 (AS) and 1543 cm−1 for UPJS-14 (AS) which can be attributed to the NN stretching vibrations of an azo bond. The stretching vibration of aromatic CH (ν(CH)ar) groups of MTA was found at 3053 cm−1 for UPJS-13 (AS) and 3047 cm−1 for UPJS-14 (AS). The stretching vibrations of aliphatic CH groups (ν(CH)aliph) of DMF molecules adsorbed in the porous system of samples were observed in the region under 3000 cm−1 (2926 and 2853 cm−1 for UPJS-14 (AS); 2929 and 2850 cm−1 for UPJS-14 (AS)). The presence of DMF is also evident from the strong absorption band of carbonyl group ν(CO) located at 1656 cm−1 and the ν(C–N) vibration at 1253 cm−1 for both compounds. In the IR spectra of ethanol exchanged samples (see Fig. 6, red lines) absorption bands belonging to the MTA linker are still present, but the characteristic vibration ν(CO) around 1650 cm−1 for DMF molecules is no longer present. Described observation confirms the successful exchange of DMF molecules for ethanol. Ethanol molecules in the IR spectra showed a shift in ν(OH) vibration to lower wavelength values (3277 cm−1 for UPJS-13 (EX) and 3260 cm−1 for UPJS-14 (EX)) and also new aliphatic ν(CH)aliph vibrations appeared under 2930 cm−1. In the IR spectra of freeze-dried compounds (see Fig. 6, blue lines), no characteristic absorption bands originated from DMF or ethanol are present, which confirm the successful activation of UPJS-13 and UPJS-14 materials.
 | ||
| Fig. 6 Infrared spectra of as-synthesized (AS), ethanol exchanged (EX) and freeze-dried (FD) materials of (a) UPJS-13 and (b) UPJS-14. | ||
| UPJS-13 (AS) | UPJS-13 (EX) | UPJS-13 (FD) | UPJS-14 (AS) | UPJS-14 (EX) | UPJS-14 (FD) | |
|---|---|---|---|---|---|---|
| a w – weak, m – medium, s – strong, br – broad, ar – aromatic, aliph – aliphatic, s – symmetric, as – antisymmetric. | ||||||
| ν(OH) | 3440 (w, br) | 3277 (w, br) | — | 3407 (w, br) | 3260 (w, br) | — |
| ν(CH)ar | 3053 (w) | 3053 (w) | — | 3047 (w) | 3047 (w) | — |
| ν(CH)aliph | 2926 (w) | 2926 (w) | — | 2929 (w) | 2929 (w) | — |
| 2853 (w) | 2853 (w) | 2850 (w) | 2850 (w) | |||
| ν(CO) |
1656 (s) | — | — | 1656 (s) | — | — |
| ν(NN) |
1563 (w) | 1563 (w) | 1563 (w) | 1543 (w) | 1543 (w) | 1543 (w) |
| νas(COO−) | 1603 (s) | 1603 (s) | 1603 (s) | 1603 (s) | 1603 (s) | 1603 (s) |
| ν(CC)ar | 1490 (m) | 1490 (m) | 1490 (m) | 1493 (m) | 1493 (m) | 1493 (m) |
| νas(COO−) | 1378 (s) | 1378 (s) | 1378 (s) | 1378 (s) | 1378 (s) | 1378 (s) |
| ν(CN) | 1253 (m) | — | — | 1253 (m) | — | — |
| δ(COO−) | 785 (s) | 785 (s) | 785 (s) | 787 (s) | 787 (s) | 787 (s) |
3.4 Thermal stability study
The thermal behaviour and stability of as-synthesized, ethanol exchanged and freeze-dried compounds were investigated using thermogravimetry and obtained TG curves are shown in Fig. 7. The AS samples (see black curves in Fig. 7) are thermally stable after heating to 80 °C, above this temperature in the temperature range 80–380 °C desolvation processes take place in two overlapping decomposition steps. In mentioned range total mass loss of 21.6 wt% for UPJS-13 (AS) and 32.0 wt% for UPJS-14 (AS) was observed and correspond to release of solvent molecules (3 × DMF, 4 × H2O, clcd. mass loss 21.9 wt% for UPJS-13 (AS); 4 × DMF, 5 × H2O, clcd. mass loss 31.8 wt% for UPJS-14 (AS)). Further heating led to the decomposition of frameworks, which occurs in 380–530 °C. Final thermal decomposition products were ZnO (observed residual mass 12.6 wt%, clcd. residual mass 12.2 wt% for UPJS-13 (AS)) and CdO (observed residual mass 17.4 wt%, clcd. residual mass 16.9 wt% for UPJS-14 (AS)). The desolvation process in ethanol exchanged samples takes place at a lower temperature (see red curves in Fig. 7), the release of ethanol molecules starts at the beginning of heating and complete desolvation occurs about 180 °C. The desolvated forms of UPJS-13 (EX) and UPJS-14 (EX) are thermally stable from 180–420 °C as is seen from the plateau on TG curves. The decomposition of polymeric frameworks takes place in two overlapping decomposition steps, similarly to the as-synthesized samples. As can be seen from obtained TG curves for EX materials, the solvent exchange process significantly impacts the compounds' activation temperature. The TG curves of the freeze-dried samples (see green curves in Fig. 7) confirm the successful activation of the compounds, as no weight loss is observed in the temperature range 30–420 °C. The decomposition of UPJS-13 (FD) and UPJS-14 (FD) frameworks, [Zn2(MTA)] and [Cd2(MTA)], takes place in temperature range 420–530 °C with residual masses 15.5 wt% and 33.9 wt% corresponding to the formation of ZnO and CdO as final decomposition products (clcd. residual masses 15.7 wt% for ZnO and 34.4 wt% for CdO). | ||
| Fig. 7 Thermogravimetric curves of as-synthesized, ethanol exchanged and freeze-dried samples of (a) UPJS-13 and (b) UPJS-14 measured in an air atmosphere and temperature range of 30–800 °C. | ||
3.5 Solid-state NMR spectroscopy
The formation of interactions/bonds between the building blocks in the prepared aMOF materials was also studied by solid-state NMR (ssNMR) spectroscopy. A comparison of the 13C CP/MAS NMR spectra of the linker (H4MTA) and the freeze-dried materials UPJS-13 and UPJS-14 are shown in Fig. 8. The 13C NMR spectra of H4MTA measured in the liquid (see Fig. 4) and solid phases (see Fig. 8) show differences in the chemical shifts of the individual signals, which are mostly shifted in the solid phase to lower values. Changes in the position of the NMR signals can be explained by the formation of intermolecular interactions52 between H4MTA molecules in the solid-state. However, it should be noted that the particle size of the material used for measurement can also affect the position of the signals in the resulting NMR spectrum.53 Significant differences were observed for the carbonyl carbon (C4) signal, which shifts 2.6 ppm lower from the original value (166.7 ppm) in the liquid phase. This observation can be explained by the formation of dimers between the carboxyl groups of H4MTA molecules with each other.54 Significant changes in chemical shifts occur after coordination of the MTA linker to Zn(II) and Cd(II) ions, when significant changes in the signal positions of the carbon atoms C4 and C4′′ were observed (see Fig. 8). Since Zn(II) and Cd(II) ions are diamagnetic metals, they do not show so significant changes in chemical shifts after coordination of linker molecules compared to the paramagnetic ions.55–58 Nevertheless, based on the described changes of chemical shifts in the 13C CP/MAS NMR spectra of H4MTA and prepared compounds, the formation of a coordination bond between the building blocks can be confirmed. | ||
| Fig. 8 Assignment of the signals to the carbon atoms in H4MTA and activated forms of UPJS-13 and UPJS-14 in 13C CP/MAS NMR spectra. | ||
3.6 Gas adsorption
 | ||
| Fig. 9 Nitrogen adsorption isotherms of (a) as-synthesized, (b) ethanol exchanged and (c) freeze-dried samples of UPJS-13 and UPJS-14 after activation at 60, 80, 100, 120, 150 and 200 °C. | ||
| Sample | SBET surface area (m2 g−1) | |||||
|---|---|---|---|---|---|---|
| Activation temperature (°C) | ||||||
| 60 | 80 | 100 | 120 | 150 | 200 | |
| As-synthesized | ||||||
| UPJS-13 (AS) | 154 | 349 | 415 | 404 | 448 | 375 |
| UPJS-14 (AS) | 254 | 405 | 629 | 656 | 621 | 613 |
|
||||||
| Exchanged | ||||||
| UPJS-13 (EX) | 447 | 721 | 210 | 237 | 280 | 225 |
| UPJS-14 (EX) | 681 | 767 | 880 | 862 | 869 | 851 |
|
||||||
| Freeze dried | ||||||
| UPJS-13 (FD) | 648 | 830 | 769 | 623 | 618 | 615 |
| UPJS-14 (FD) | 754 | 1057 | 1052 | 1024 | 1013 | 989 |
 | ||
| Fig. 10 Calculated BET surface areas from nitrogen isotherms of activated materials at 60, 80, 100, 120, 150 and 200 °C for (a) UPJS-13 and (b) UPJS-14. | ||
For as-synthesized materials containing DMF/water molecules in the pores, it is characteristic that as the activation temperature increases, the surface area gradually increases up to 100 °C (see black curves in Fig. 10a). Above this temperature, there is no more pronounced increase in SBET and the values are almost constant. This is caused by the gradual removal of solvent molecules from the pore system of materials. The calculated surface area at 100 °C were 415 m2 g−1 for UPJS-13 (AS) and 629 m2 g−1 for UPJS-14 (AS). After activating samples at 200 °C, a slight decrease in surface area was observed (see Fig. 9, Table 2).
For UPJS-13 (EX), the trend in surface area values at selected activation temperature is different. The maximum SBET of 721 m2 g−1 was reached at 80 °C and by subsequent heating at 100 °C decreases to the value of 210 m2 g−1. The further increase in temperature does not have a significant effect on the textural properties (see red curves in Fig. 10), probably due to the collapse of the polymer framework. Because ethanol has a low boiling point, its release at 100 °C occurs all at once, which lead to pore perforation associated with the destruction of the framework. The sample UPJS-14 (EX) showed similar behaviour compared to UPJS-14 (AS) and gradually increased the SBET with increasing temperature up to 80 °C and the maximal surface area after activation at 100 °C was 880 m2 g−1.
The best textural properties were observed for materials after the freeze-drying process (see green curves in Fig. 10). The highest values of surface areas were calculated after activation of the samples at 80 °C (see green curves in Fig. 10, Table 2) and corresponding values were 830 m2 g−1 (pore volume, Vp = 0.57 cm3 g−1; pore diameter, d = 0.73 nm calculated by DFT) for UPJS-13 (FD) and 1057 m2 g−1 (pore volume, Vp = 0.72 cm3 g−1; pore diameter, d = 0.87 nm calculated by DFT) for UPJS-14 (FD). Gradual heating to 120 °C leads to a slight decrease in SBET values and further heating did not affect the values of surface area for both compounds.
It is to note, that the observed SBET values for UPJS-13 and UPJS-14 are higher compared with MOFs constructed from shorter tetrahedral linker, methanetetrabenzoate (MTB) containing Ca(II) (126 m2 g−1),59 Zn(II) (248 m2 g−1),46 Co(II) (356 m2 g−1),60 Ba(II) (358 m2 g−1),59 Cu(II) (526 m2 g−1)40 and Ni(II) (700 m2 g−1)61 ions. A further comparison could be performed with a copper(II) MOF [Cu2(MTC)(H2O)2]·14DMF·5H2O,40 which contains similar extended tetrahedral linker MTC (methanetetra(biphenyl-p-carboxylate) to MTA described in the present study. The methanol exchanged and vacuum dried compounds, both activated at 60 °C exhibited surface area of 791 m2 g−1 and its freeze-dried form 1020 m2 g−1. The zinc(II) analogue with formulae [Zn2(MTC)(H2O)2] may also be mentioned, which exhibited after dichloromethane exchange a surface area of 1170 m2 g−162 and calculated SBET is similar to UPJS-14 (FD). The highest reported surface area was published for zirconium(IV) MOF containing MTC linker with a composition of [Zr6(OH)16(MTC)2] and SBET = 3411 m2 g−1.63,64,74
In summary, it can be concluded that UPJS-14 material showed better textural properties compared to UPJS-13, regardless of the post-synthetic treatment and activation conditions. The highest values of surface areas of prepared materials were observed for samples after the freeze-drying process, followed by ethanol-exchanged and as-synthesized samples. Compound UPJS-14 (FD) activated at 80 °C exhibited large surface area compared with other reported MOF compounds containing similar extended tetrahedral linker.
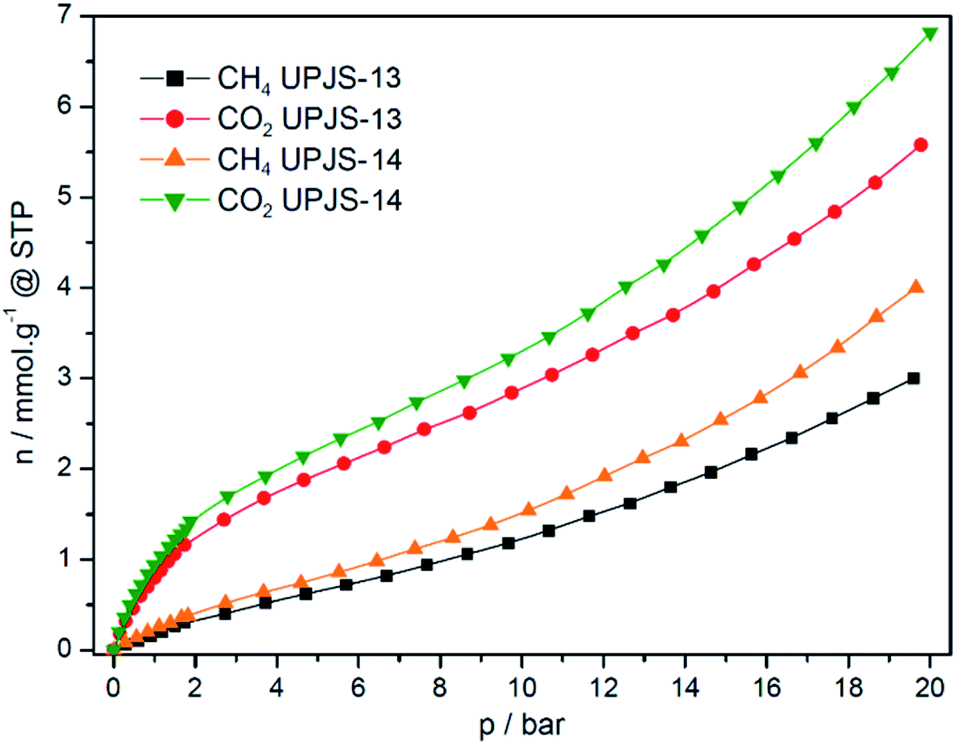 | ||
| Fig. 11 Methane and carbon dioxide high-pressure adsorption isotherms of UPJS-13 and UPJS-14 measured at 30 °C and pressure up to 20 bar. | ||
| Bar | CO2 | CH4 | |||
|---|---|---|---|---|---|
| mmol g−1 | wt% | mmol g−1 | wt% | ||
| UPJS-13 (FD) | 1 | 0.94 | 4.14 | 0.22 | 0.35 |
| 20 | 6.82 | 30.1 | 2.21 | 1.4 | |
| UPJS-14 (FD) | 1 | 0.80 | 3.52 | 0.36 | 0.58 |
| 20 | 5.58 | 24.56 | 3.98 | 6.38 | |
aMOFs storage capacities of carbon dioxide at 1 bar and 30 °C were 0.94 mmol g−1 (4.14 wt%) for UPJS-13 FD and 0.8 mmol g−1 (3.52 wt%) for UPJS-14 FD. With increasing pressure, an increase in the amount of adsorbed carbon dioxide was observed. The maximal adsorption uptake and corresponding adsorption capacities were 6.82 mmol g−1 (30.01 wt%) for UPJS-13 FD and 5.58 mmol g−1 (24.56 wt%) for UPJS-14 FD at 30 °C and 20 bar. Both compounds show high CO2 storage capacities, however, the UPJS-13 FD framework showed a higher affinity for CO2 compared to UPJS-14 FD, although it has a smaller surface area. Due to the presence of an electron-deficient carbon atom, carbon dioxide is expected to interact with protic electronegative functional groups, leading to strong chemisorption. This simple interaction has so far led to a number of nanoporous polymers in which nitrogen-rich functions are incorporated, such as triazine,65,66 tetrazole,67 imidazole,68,69 imide70 and amines.42,71 Increased CO2 adsorption capacity in UPJS-13 (FD) and UPJS-14 (FD) can be explained by an interaction between CO2 molecules and adsorption sites represented by a lone pair on the azo functional groups located in the molecular structure of the MTA linker. In the case of azo group, the interaction with CO2 was also confirmed by computational studies, in which the relative binding affinity of CO2 to the –NN– group using trans-azobenzene as a model system were performed. Calculations have shown that the lowest energy structure of the CO2/trans-azobenzene complex indicates that carbon dioxide preferentially binds to the azo group of trans-azobenzene by physisorption with a binding energy of 17.0 kJ mol−1. CO2 also has a favourable interaction with the aromatic ring, including a large stabilizing energy, 14.4 kJ mol−1.72 It is known that MOFs with coordinatively unsaturated sites (CUSs) offer interesting possibilities for tuning the affinity of these materials towards certain adsorbates, potentially increasing their selectivity and storage capacity.73 In MOFs with CUSs, at least one available coordination site of the metal centre is occupied by an atom belonging to a solvent molecule. The solvent molecules can be removed through the activation process, leaving the metal atom with an open coordination site. Examples are Zn(II) and Cd(II) compounds, which have been constructed with methanetetrabenzoate (MTB, short tetrahedral carboxylate linker compared to MTA) with compositions: [Zn2(MTB)(H2O)2]·3H2O·3DMF46 and [Cd4(MTB)2(DMF)4]·4DMF·4H2O.64,74 The framework of zinc form imitates the PtS structure and contains paddle-wheel cluster (2 × CUSs, bold text in the formula) and the cadmium framework mimics the fluorite (CaF2) structure, which contains a tetranuclear cluster containing four terminally coordinated solvent molecules (4 × CUSs, bold text in the formula). We can only speculate, that the increased affinity of CO2 for the presented aMOFs is the presence of CUSs on Zn(II) and Cd(II) ions in the frameworks of UPJS-13 and UPJS-14.
According to the literature data, the high-pressure CO2 adsorption measurements were performed for two compounds containing MTB linker. Nickel(II) MOF (SBET = 700 m2 g−1) adsorbed 18.05 wt% (4.1 mmol g−1) of CO2 (ref. 61) and lead(II) compound (SBET = 980 m2 g−1) 19.81 wt% (4.5 mmol g−1) of CO2 (ref. 75 and 76) at 30 °C and 21 bar. The highest to date CO2 storage capacities were reported for the following unmodified MOFs: UiO(dpdc) (dpdc = 2,2′-bipyridine-5,5′-dimethyl, SBET = 2646 m2 g−1)77 18.11 mmol g−1 (79.7 wt%) at 30 °C and 20 bar; NU-111 (Cu(4-PmBC), 4-PmBC = 4-(pyrimidin-5-yl)benzoate, SBET = 4932 m2 g−1)78 14.04 mmol g−1 (61.8 wt%) at 25 °C and 30 bar; DGC-MIL-101 (DGC = dry gel conversion, Cr(BDC), BDC = benzene-1,4-dicarboxylate, SBET = 4198 m2 g−1)79 13.59 mmol g−1 (59.8 wt%) at 25 °C and 40 bar and DMOF (Zn2(BDC)2(DABCO), DABCO = 1,4-diazabicyclo[2.2.2]octane, SBET = 1980 m2 g−1)80 8.66 mmol g−1 (38.1 wt%) at 25 °C and 20 bar.
Methane is the main component of natural gas and the second most abundant greenhouse gas in Earth's atmosphere. CH4 is a non-polar molecule with zero quadrupole moment and has a low volumetric energy density, which poses a challenge for efficient storage to reduce its concentration in the atmosphere or for energy application. For this reason, methane was also used as an adsorbate for high-pressure adsorption and measured isotherms are presented in Fig. 11. Materials adsorb methane with a maximal storage capacity of 0.22 mmol g−1 (0.35 wt%) for UPJS-13 (FD) and 0.36 mmol g−1 (0.58 wt%) for UPJS-14 (FD) at 30 °C and 1 bar and 3.02 mmol g−1 (4.84 wt%) for UPJS-13 (FD) and 3.98 mmol g−1 (6.38 wt%) for UPJS-14 (FD) at 30 °C and 20 bar. It could be noted that the adsorbed amounts are approximately four/five times lower compared to carbon dioxide. Contrary, the values of stored CH4 are relatively high and can be explained by the pore size of aMOF materials. Several theoretical studies have shown that the optimum pore size for methane storage is 8 Å, which corresponds to distance between pore walls of about twice larger, than the molecule diameter.81,82 As it was described above, the effective pore size of UPJS-13 (FD) and UPJS-14 (FD) based on nitrogen adsorption are 7.3 and 8.7 Å; thus it could be an explanation for increased CH4 adsorption.
A comparison of the adsorption capacity with compounds containing shorter tetrahedral carboxylate linker (MTB) has shown that the CH4 adsorption capacities were reported for Ni(II)61 and Pb(II)75 compounds with a methane storage capacity of 3.29 wt% (2.05 mmol g−1) and 3.05 wt% (1.9 mmol g−1) at 30 °C and 21 bar, respectively. The highest documented CH4 capacities were reported for NU-125 (Cu(BTTI), BTTI = 5,5′,5′′-(4,4′,4′′-(benzene-1,3,5-triyl)tris(1H1,2,3-triazole-4,1-diyl))triisophthalate, SBET = 3120 m2 g−1)83 which can store 11.47 mmol g−1 (18.40 wt%) at 25 °C and 20 bar and BUT-22 (Al3(O)(OH)(H2O)2(PPTTA)3/2, PPTTA = (1,4-phenylenebis(pyridine-4,2,6-triyl))tetrabenzoate, SBET = 4380 m2 g−1)84 with storage capacity of 9.9 mmol g−1 (15.88 wt%) at 23 °C and 20 bar. Other MOFs, such as [Co(BDP)] (BDP = 1,4-benzenedipyrazolate, SLANG = 2911 m2 g−1)85 and commercial product, Basolite A520 (aluminum fumarate, SBET = 1300 m2 g−1)86 have also shown promise as methane sorbents with methane storage capacity of 6.5 mmol g−1 (10.43 wt%) at 25 °C and 20 bar and 3.80 mmol g−1 (6.10 wt%) at room temperature and pressure 20 bar, respectively.
4 Conclusion
The present study dealt with the preparation, characterisation and application of two novel aMOF compounds as adsorbents of greenhouse gases, carbon dioxide and methane. In the first phase, the extended nitrogen-rich tetraazo-tetracarboxylic acid, H4MTB was prepared by a seven-step organic synthesis. Spectroscopic and NMR measurements confirmed its successful preparation. The next step was the preparation of aMOF compounds in combination with H4MTB and zinc(II) or cadmium(II) cations under the formation of as-synthesized materials, UPJS-13 (AS) and UPJS-14 (AS). In order to facilitate the activation of the compounds, the solvent molecules located in the cavities of the as-synthesized materials were exchanged with ethanol (UPJS-13 (EX) and UPJS-14 (EX)). Another variant of the compounds was prepared by a freeze-drying process using benzene (UPJS-13 (FD) and UPJS-14 (FD)). The compounds were characterised by elemental analysis, ICP-MS, infrared spectroscopy, solid-state NMR spectroscopy and PXRD. All prepared aMOFs were studied by nitrogen adsorption to find the best activation conditions. The compounds showed the highest surface areas after the freeze-drying process and at an activation temperature of 80 °C (SBET = 830 m2 g−1 for UPJS-13 (EX) and SBET = 1057 m2 g−1 for UPJS-14 (EX)). aMOF materials were subsequently tested as greenhouse gases adsorbents, carbon dioxide and methane, by high-pressure adsorption measurements at 30 °C and pressure up to 20 bar. The maximal gas storage capacities were 30.01 wt% for UPJS-13 FD, 24.56 wt% for UPJS-14 FD of CO2 and 4.84 wt% for UPJS-13 FD and 6.38 wt% for UPJS-14 FD of CH4 at 30 °C and 20 bar. The increased adsorption capacity of carbon dioxide can be explained by the interaction of CO2 molecules with azo groups located in the structure of MTA ligand, or by interaction with CUSs. And the increased adsorption capacity of methane can be explained by the suitable pore size of the prepared materials.Author contributions
Miroslav Almáši – conceptualization, data curation, formal analysis, investigation, supervision, validation, visualization, writing - original draft; writing - review & editing, Nikolas Király – data curation, formal analysis, investigation, validation, visualization, writing - original draft; writing – review & editing, Vladimír Zeleňák – funding acquisition, investigation, validation, Mária Vilková – data curation, formal analysis, investigation, validation, Sandrine Bourrelly – data curation, investigation, validation.Conflicts of interest
No potential conflict of interest was reported by the authors.Acknowledgements
This work was supported by the Scientific Grant Agency of the Slovak Republic (VEGA) project no. 1/0865/21 and from P. J. Safarik University project no. VVGS-2020-1667.References
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. Mizrahi Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed.
- M. Fakhraei Ghazvini, M. Vahedi, S. Najafi Nobar and F. Sabouri, J. Environ. Chem. Eng., 2021, 9, 104790 CrossRef CAS.
- T. A. Goetjen, J. Liu, Y. Wu, J. Sui, X. Zhang, J. T. Hupp and O. K. Farha, Chem. Commun., 2020, 56, 10409–10418 RSC.
- H. Konnerth, B. M. Matsagar, S. S. Chen, M. H. G. Prechtl, F.-K. Shieh and K. C.-W. Wu, Coord. Chem. Rev., 2020, 416, 213319 CrossRef CAS.
- J. Cao, X. Li and H. Tian, Curr. Med. Chem., 2020, 27, 5949–5969 CrossRef CAS PubMed.
- Y. Wang, J. Yan, N. Wen, H. Xiong, S. Cai, Q. He, Y. Hu, D. Peng, Z. Liu and Y. Liu, Biomaterials, 2020, 230, 119619 CrossRef CAS PubMed.
- S. Kim, K. Son and H. Oh, Korean J. Mater. Res., 2020, 30, 136–141 CrossRef.
- V. Zeleňák, M. Almáši, A. Zeleňáková, P. Hrubovčák, R. Tarasenko, S. Bourelly and P. Llewellyn, Sci. Rep., 2019, 9, 15572 CrossRef PubMed.
- L. Wang, Sens. Actuators, A, 2020, 307, 111984 CrossRef CAS.
- H.-Y. Li, S.-N. Zhao, S.-Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364–6401 RSC.
- D. Capková, M. Almáši, T. Kazda, O. Čech, N. Király, P. Čudek, A. S. Fedorková and V. Hornebecq, Electrochim. Acta, 2020, 354, 136640 CrossRef.
- J. Cai, Y. Song, X. Chen, Z. Sun, Y. Yi, J. Sun and Q. Zhang, J. Mater. Chem. A, 2020, 8, 1757–1766 RSC.
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef.
- H. Ohtsu, T. D. Bennett, T. Kojima, D. A. Keen, Y. Niwa and M. Kawano, Chem. Commun., 2017, 53, 7060–7063 RSC.
- A. Halder and D. Ghoshal, CrystEngComm, 2018, 20, 1322–1345 RSC.
- J. Fonseca, T. Gong, L. Jiao and H. L. Jiang, J. Mater. Chem. A, 2021, 9, 10562–10611 RSC.
- Z. Hu, J. Lin, N. Ogiwara, A. Rodriguez, Y. Peng, Y. Wang, S. Horike and D. Zhao, CrystEngComm, 2016, 18, 2803–2807 RSC.
- T. Zhang, J. Wang, W. Zhang, C. Yang, L. Zhang, W. Zhu, J. Sun, G. Li, T. Li and J. Wang, J. Mater. Chem. A, 2019, 7, 2845–2854 RSC.
- R. Z. Zhang, S. Quan, M. Xia, Q. Wang, W. Zhang and J. M. Yang, J. Colloid Interface Sci., 2018, 525, 54–61 CrossRef CAS PubMed.
- M. Marmier, M. D. Wise, J. J. Holstein, P. Pattison, K. Schenk, E. Solari, R. Scopelliti and K. Severin, Inorg. Chem., 2016, 55, 4006–4015 CrossRef CAS PubMed.
- J. Fonseca and S. Choi, Microporous Mesoporous Mater., 2021, 310, 1–10 CrossRef.
- J. Fonseca and S. Choi, Dalton Trans., 2021, 50, 3145–3154 RSC.
- J. Fonseca and S. Choi, Catal.: Sci. Technol., 2020, 10, 8265–8282 RSC.
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555–1562 CrossRef CAS PubMed.
- X. Zhang, H. Li, X. Lv, J. Xu, Y. Wang, C. He, N. Liu, Y. Yang and Y. Wang, Chem.–Eur. J., 2018, 24, 8822–8832 CrossRef CAS PubMed.
- P. Ma, F. Meng, N. Wang, J. Zhang, J. Xie and B. Dai, ChemistrySelect, 2018, 3, 10694–10700 CrossRef CAS.
- B. Li and H. C. Zeng, Chem. Mater., 2019, 31, 5320–5330 CrossRef CAS.
- H. Zhou, M. Zheng, H. Tang, B. Xu, Y. Tang and H. Pang, Small, 2019, 16, 1904252 CrossRef PubMed.
- C. Orellana-Tavra, E. F. Baxter, T. Tian, T. D. Bennett, N. K. Slater, A. K. Cheetham and D. Fairen-Jimenez, Chem. Commun., 2015, 51, 13878–13881 RSC.
- X. Wu, H. Yue, Y. Zhang, X. Gao, X. Li, L. Wang, Y. Cao, M. Hou, H. An, L. Zhang, S. Li, J. Ma, H. Lin, Y. Fu, H. Gu, W. Lou, W. Wei, R. N. Zare and J. Ge, Nat. Commun., 2019, 10, 5165 CrossRef PubMed.
- K. Suresh and A. J. Matzger, Angew. Chem., Int. Ed., 2019, 58, 16790–16794 CrossRef CAS PubMed.
- Y. Liu, S. Lv, D. Liu and F. Song, Acta Biomater., 2020, 116, 16–31 CrossRef CAS PubMed.
- C. Orellana-Tavra, M. Köppen, A. Li, N. Stock and D. Fairen-Jimenez, ACS Appl. Mater. Interfaces, 2020, 12, 5633–5641 CrossRef CAS PubMed.
- F. Yang, W. Li and B. Tang, J. Alloys Compd., 2018, 733, 8–14 CrossRef CAS.
- W. Niu and Y. Yang, ACS Appl. Energy Mater., 2018, 1, 2440–2445 CrossRef CAS.
- J.-W. Xiu, G.-E. Wang, M.-S. Yao, C.-C. Yang, C.-H. Lin and G. Xu, Chem. Commun., 2017, 53, 2479–2482 RSC.
- T. D. Bennett, P. J. Saines, D. A. Keen, J.-C. Tan and A. K. Cheetham, Chem.–Eur. J., 2013, 19, 7049–7055 CrossRef CAS PubMed.
- T. Zhang, J. Wang, W. Zhang, C. Yang, L. Zhang, W. Zhu, J. Sun, G. Li, T. Li and J. Wang, J. Mater. Chem. A, 2019, 7, 2845–2854 RSC.
- H. Zhang, Ice templating and freeze-drying for porous materials and their applications, Wiley-VCH, Weinheim, Germany, 2018 Search PubMed.
- L. Ma, A. Jin, Z. Xie and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 9905–9908 CrossRef CAS PubMed.
- https://ec.europa.eu/clima/policies/strategies/progress_en, Climate Action – European Commission, 2017.
- E. J. Kim, R. L. Siegelman, H. Z. Jiang, A. C. Forse, J.-H. Lee, J. D. Martell, P. J. Milner, J. M. Falkowski, J. B. Neaton, J. A. Reimer, S. C. Weston and J. R. Long, Science, 2020, 369, 392–396 CrossRef CAS PubMed.
- V. Zeleňák, M. Skřínska, F. R. Siperstein and A. Patti, Appl. Surf. Sci., 2019, 476, 886–896 CrossRef.
- J.-T. Anyanwu, Y. Wang and R. T. Yang, Ind. Eng. Chem. Res., 2019, 59, 7072–7079 CrossRef.
- P. Zhao, G. Zhang and L. Hao, Adsorption, 2020, 26, 749–764 CrossRef CAS.
- M. Almáši, V. Zeleňák, A. Zukal, J. Kuchár and J. Čejka, Dalton Trans., 2016, 45, 1233–1242 RSC.
- C. Klumpen, M. Breunig, T. Homburg, N. Stock and J. Senker, Chem. Mater., 2016, 28, 5461–5470 CrossRef CAS.
- S. Shukla, A. Mahata, B. Pathak and B. Lochab, RSC Adv., 2015, 5, 78071–78080 RSC.
- J. Gonda, S. Fazekašová, M. Martinková, T. Mitríková, D. Roman and M. B. Pilátová, Org. Biomol. Chem., 2019, 17, 3361–3373 RSC.
- A. D. Wiersum, C. Giovannangeli, D. Vincent, E. Bloch, H. Reinsch, N. Stock, J. S. Lee, J.-S. Chang and P. L. Llewellyn, ACS Comb. Sci., 2013, 15, 111–119 CrossRef CAS PubMed.
- E. W. Lemmon and H. M. McLinden, MO reference fluid thermodynamic and transport properties; REFPROP 8.0, National Institute of Standards and Technology, Gaithersburg MD 2007 Search PubMed.
- Y. Xu, P. M. J. Szell, V. Kumar and D. L. Bryce, Coord. Chem. Rev., 2020, 411, 213237 CrossRef CAS.
- S. Naoki, N. Akira and K. Keisaku, Bull. Chem. Soc. Jpn., 1995, 68, 2151–2157 CrossRef.
- B. H. Meier, F. Graf and R. R. Ernst, J. Chem. Phys., 1982, 76, 767 CrossRef CAS.
- F. Gul-E-Noor, B. Jee, A. Poppl, M. Hartmann, D. Himsl and M. Bertmer, Phys. Chem. Chem. Phys., 2011, 13, 7783–7788 RSC.
- J. Tang, S. Li, Y. Chu, Y. Xiao, J. Xu and F. Deng, J. Phys. Chem. C, 2019, 123, 24062–24070 CrossRef CAS.
- V. Martins, J. Xu, X. Wang, K. Chen, I. Hung, Z. Gan, C. Gervais, C. Bonhomme, S. Jiang, A. Zheng, B. E. G. Lucier and Y. Huang, J. Am. Chem. Soc., 2020, 142, 14877–14889 CrossRef CAS PubMed.
- H. C. Hoffmann, M. Debowski, P. Müller, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Materials, 2012, 5, 2537–2572 CrossRef CAS.
- M. Almáši, V. Zeleňák, R. Gyepes, Ľ. Zauška and S. Bourrelly, RSC Adv., 2020, 10, 32323–32334 RSC.
- Y. E. Cheon and M. P. Suh, Chem. Commun., 2009, 2296 RSC.
- M. Almáši, V. Zeleňák, M. Opanasenko and J. Čejka, Dalton Trans., 2014, 43, 3730 RSC.
- L. Wen, P. Cheng and W. Lin, Chem. Commun., 2012, 48, 2846 RSC.
- M. Zhang, Y.-P. Chen, M. Bosch, T. Gentle, K. Wang, D. Feng, Z. U. Wang and H.-C. Zhou, Angew. Chem., Int. Ed., 2013, 53, 815–818 CrossRef PubMed.
- Y. Wang, Q. Liu, Q. Zhang, B. Peng and H. Deng, Angew. Chem., Int. Ed., 2018, 57, 7120–7125 CrossRef CAS PubMed.
- P. Das and S. K. Mandal, ACS Appl. Mater. Interfaces, 2020, 12, 37137–37146 CrossRef CAS PubMed.
- A. Mukhtar, S. Ullah, A. Inayat, S. Saqib, N. B. Mellon, M. A. Assiri, A. G. Al-Sehemi, M. B. Khan Niazi, Z. Jahan, M. A. Bustam and M. Ibrahim, Energy, 2021, 216, 119230 CrossRef CAS.
- R. Zhang, D.-X. Meng, F.-Y. Ge, J.-H. Huang, L.-F. Wang, Y.-K. Xv, X.-G. Liu, M.-M. Meng, H. Yan, Z.-Z. Lu, H.-G. Zheng and W. Huang, Dalton Trans., 2020, 49, 2145–2150 RSC.
- S. S. Dhankhar and C. M. Nagaraja, New J. Chem., 2020, 44, 9090–9096 RSC.
- W. R. Webb, M. E. Potter, D. J. Stewart, S. J. Elliott, P. J. Sazio, Z. Zhang, H. K. Luo, J. Teng, L. Zhang, C. Ivaldi, I. Miletto, E. Gianotti and R. Raja, Chem.–Eur. J., 2020, 26, 13606–13610 CrossRef CAS PubMed.
- L. Wang, L. Yang, L. Gong, R. Krishna, Z. Gao, Y. Tao, W. Yin, Z. Xu and F. Luo, Chem. Eng. J., 2020, 383, 123117 CrossRef CAS.
- S. Salehi, M. Anbia and F. Razavi, Environ. Prog. Sustainable Energy, 2019, 39, 13302 CrossRef.
- H. A. Patel, S. Hyun Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- Ü. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- H. Chun, D. Kim, D. N. Dybtsev and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 971–974 CrossRef CAS PubMed.
- M. Almáši, V. Zeleňák, R. Gyepes, S. Bourrelly, M. V. Opanasenko, P. L. Llewellyn and J. Čejka, Inorg. Chem., 2018, 57, 1774–1786 CrossRef PubMed.
- M. Almáši, V. Zeleňák, M. V. Opanasenko and J. Čejka, Catal. Lett., 2018, 148, 2263–2273 CrossRef.
- L. Li, S. Tang, C. Wang, X. Lv, M. Jiang, H. Wu and X. Zhao, Chem. Commun., 2014, 50, 2304 RSC.
- J. Luo, J. Wang, G. Li, Q. Huo and Y. Liu, Chem. Commun., 2013, 49, 11433 RSC.
- J. Kim, Y.-R. Lee and W.-S. Ahn, Chem. Commun., 2013, 49, 7647 RSC.
- N. C. Burtch, H. Jasuja, D. Dubbeldam and K. S. Walton, J. Am. Chem. Soc., 2013, 135, 7172–7180 CrossRef CAS PubMed.
- J. Alcañiz-Monge, M. A. De La Casa-Lillo, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 1997, 35, 291–297 CrossRef.
- R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2011, 112, 703–723 CrossRef PubMed.
- B. Wang, X. Zhang, H. Huang, Z. Zhang, T. Yildirim, W. Zhou, S. Xiang and B. Chen, Nano Res., 2020, 14, 507–511 CrossRef.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357–361 CrossRef CAS PubMed.
- M. Gaab, N. Trukhan, S. Maurer, R. Gummaraju and U. Müller, Microporous Mesoporous Mater., 2012, 157, 131–136 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02938j |
| This journal is © The Royal Society of Chemistry 2021 |