DOI:
10.1039/D1RA02245H
(Paper)
RSC Adv., 2021,
11, 20123-20136
Effect of OH substitution in 3-benzylchroman-4-ones: crystallographic, CSD, DFT, FTIR, Hirshfeld surface, and energy framework analysis†
Received
21st March 2021
, Accepted 22nd May 2021
First published on 4th June 2021
Abstract
3-Benzylchroman-4-ones (homoisoflavanones) are oxygen-containing heterocycles with a sixteen-carbon skeleton. They belong to the class of naturally occurring polyphenolic flavonoids with limited occurrence in nature and possess anti-inflammatory, antibacterial, antihistaminic, antimutagenic, antiviral, and angioprotective properties. Recently, we reported the synthesis and anticancer activity studies of fifteen 3-benzylchroman-4-one molecules, and most of them were proven to be effective against BT549 and HeLa cells. In this work, we report the single-crystal X-ray crystallographic studies of two molecules 3-[(2-hydroxyphenyl)methyl]-3,4-dihydro-2H-1-benzopyran-4-one and 3-[(2,4-dimethoxyphenyl)methyl]-3,4-dihydro-2H-1-benzopyran-4-one. The single crystals were grown using a novel laser-induced crystallization technique. We observed that the 3-benzylchroman-4-one derivative bearing OH substitution at the 2′ position adopted different conformation due to formation of dimers through O–H⋯O, and C–H⋯O intermolecular hydrogen bondings. The role of OH substitution in the aforementioned conformational changes was evaluated using density functional theory (DFT), Hirshfeld surface, energy framework and FTIR spectroscopy analysis. In addition, we have carried out a Cambridge Structural Database (CSD) study to understand the conformational changes using five analogue structures. X-ray crystallographic, computational, and spectroscopic studies of 3-benzylchroman-4-ones provided an insight into the role of substitution at benzyl moieties in stabilizing the three-dimensional (3D) structures.
1. Introduction
Homoisoflavanones belong to the class of naturally occurring polyphenolic flavonoids. Homoisoflavanones have been extracted from several flowering plants such as Eucomis, Muscari, and Bellevalia in the family of Hyacinthaceae, Liliaceae, Agavaceae, Fabaceae, and Polygonaceae.1,2 3-Benzylchroman-4-ones are a family of homoisoflavanones, which have the chemical structure of a oxygen-containing heterocyclic ring, and two phenyl rings with a sixteen-carbon skeleton. They possess anti-inflammatory, antibacterial, antihistaminic, antimutagenic, antiviral, and angioprotective properties.3–5 For example, the caesalpinanone and 6-O-methylcaesalpinianone isolated from Fabaceae inhibited glutathione S-transferase and were shown to have antioxidant, anti-inflammatory effects.6 Sappanone A inhibited cisplatin-induced kidney injury,7 and homoisopogon A (1) exhibited potent cytotoxicity against human lung cells.8
In a recent work, we have synthesized 15 derivatives of 3-benzylchroman-4-one and identified them as a potential anticancer inhibitor against BT549 and HeLa cells.9 We have also identified that they have an affinity with p53 protein. To date, crystal structure of five 3-benzylchroman-4-one derivatives have been reported in the literature.9–12 In this work, we report the single-crystal X-ray crystallographic studies of two 3-benzylchroman-4-one derivatives named as 3-[(2-hydroxyphenyl)methyl]-3,4-dihydro-2H-1-benzopyran-4-one (HIF-4) and 3-[(2,4-dimethoxyphenyl)methyl]-3,4-dihydro-2H-1-benzopyran-4-one (HIF-13). The results show that the conformation of HIF-13 is similar to its analogue structures. However, the conformation of HIF-4 is strikingly deviant from all other previously reported structures, including HIF-13. A better understanding of the conformational changes has been acquired through DFT calculations on HIF-4 and HIF-13 and FTIR spectroscopy on HIF-4. The CSD study was used to understand the conformational changes of various 3-benzylchroman-4-one derivatives. Besides Hirshfeld surface and energy frameworks studies were also conducted for seven 3-benzylchroman-4-one structures. The theoretical and experimental studies revealed the mechanism behind the deviation of HIF-4 structure compared to other 3-benzylchroman-4-one derivatives.
2. Materials and methods
2.1 Synthesis and characterization
The HIF-4 and HIF-13 compounds were synthesized and characterized, as reported in earlier articles. The synthesis part is outlined in Scheme 1A. The molecular structure of derivatives HIF-4 and HIF-13 were confirmed using elemental analysis, NMR spectroscopy, and mass spectrometry analysis.9–12 Briefly, the synthetic pathway for the preparation of homoisoflavanone involved the synthesis of chalcone followed by the reduction of chalcone to dihydrochalcone and its conversion to homoisoflavanone. A mixture of 2′-hydroxyacetophenone and the corresponding benzaldehyde derivative in 20% KOH/EtOH was added to a well-closed glass container and heated on an oil bath at 132 °C for three to four minutes. Then the reaction mixture was cooled and poured into ice-cold water. Diluted hydrochloric acid was added, and the reaction mixture was kept in the refrigerator overnight. The separated solid, 2′-hydroxy chalcone, was filtered and recrystallized in methanol. 2′-Hydroxy chalcone, saturated ammonium formate solution [methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (1:1)], and 10% Pd/C were refluxed for 90 minutes. The reaction mixture was filtered. The product which remained in the filtrate was isolated in good yield by dispersing the residue in water, extracting it with ethyl acetate, and drying over anhydrous Na2SO4 to obtain 2′-hydroxydihydrochalcone. 2′-Hydroxydihydrochalcone was dissolved in ethanol and refluxed with paraformaldehyde and 50% aqueous diethylamine for 9 hours. Ethanol was distilled off, and the residue was taken up in ethyl acetate. Ethyl acetate was distilled off, and the oily residue was column chromatographed over silica using pet ether:ethyl acetate (7:3) as eluent to get the 3-(4-methoxybenzyl)-2,3-dihydro-4H-chroman-4-one in 60–70% yield.
:THF (1:1)], and 10% Pd/C were refluxed for 90 minutes. The reaction mixture was filtered. The product which remained in the filtrate was isolated in good yield by dispersing the residue in water, extracting it with ethyl acetate, and drying over anhydrous Na2SO4 to obtain 2′-hydroxydihydrochalcone. 2′-Hydroxydihydrochalcone was dissolved in ethanol and refluxed with paraformaldehyde and 50% aqueous diethylamine for 9 hours. Ethanol was distilled off, and the residue was taken up in ethyl acetate. Ethyl acetate was distilled off, and the oily residue was column chromatographed over silica using pet ether:ethyl acetate (7:3) as eluent to get the 3-(4-methoxybenzyl)-2,3-dihydro-4H-chroman-4-one in 60–70% yield.
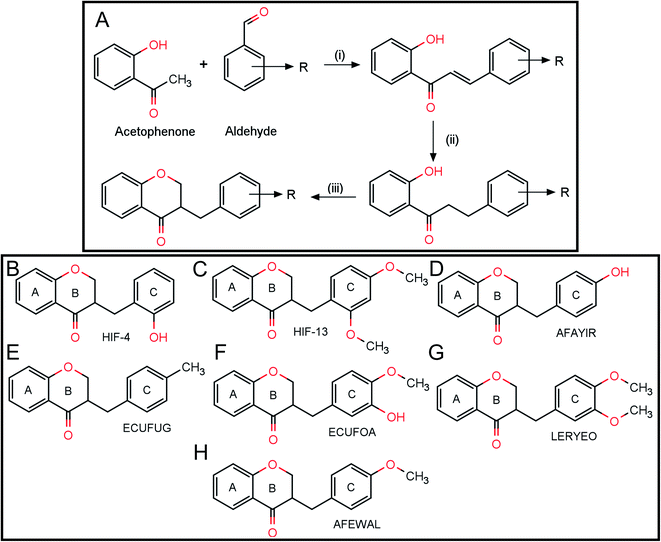 |
| | Scheme 1 (A) Scheme for the synthesis of homoisoflavanone. (B) Chemical structures of HIF-4 and (C) HIF-13. The chemical structures of analogue structures retrieved from the CSD database with refcodes AFAYIR (D), ECUFUG (E), ECUFOA (F), LERYEO (G), and AFEWAL (H). | |
Reagents and conditions: (i) 20% KOH/EtOH heated on an oil bath at 132 °C, 3–4 minutes (ii) 10% Pd–C, HCOONH4, MeOH–THF (1:1), reflux, 90 minutes; (iii) 50% v/v aqueous diethylamine, (HCHO)n, EtOH, reflux, 9 hours (Scheme 1A). Here R= 2-hydroxy in HIF-4 (Scheme 1B), and R = 2,4-dimethoxy in HIF-13 (Scheme 1C).
2.2 Laser-induced crystallization
Our initial attempts to obtain the crystals of HIF-4 using conventional crystallization methods failed. Thus, the single crystals of HIF-4 (Scheme 1B) and HIF-13 (Scheme 1C) were obtained using the laser-induced crystallization technique as described.13–15 Fig. 1 shows the schematic diagram of the laser-induced crystallization setup used in this study. The HIF-4 and HIF-13 compounds were dissolved in an equal volume of ethanol and methanol (1:1) mixture followed by heating it to 40 °C. Each dissolved solution was taken into a coverslip (50 µl) separately, and the nucleant material coir was added to the crystallization solutions individually as described.13 The crystallization solution was placed on an inverted microscope (M). A continuous-wave laser beam (λ = 1064 nm) was used as a source of excitation with an output power of 60 mW. A manual beam expander (BE) to a size of 9 mm was used to expand the laser beam. The expanded laser beam, a dichroic mirror (M) with high reflectivity at 1064 nm, was used via a 1:1 telescopic arrangement to the back aperture of the microscope objective (10×, NA = 0.3). A CCD camera was accomplished to visualize and record the crystallization process (Nikon DS-Fi1c, Japan) attached to the microscope at 50 interlaced frames per second. A linear x–y translational stage was used to control the sample focusing (Fig. 1).13 When the laser light was focused on the nucleant material coir, bubble formation was observed, followed by the Brownian motion in the solution. Tiny crystals started to appear within three minutes of laser exposure (shown as insets in Fig. 1). These tiny crystals were used as seed and transferred into a 5 mL beaker containing crystallization solutions for further growth by slow evaporation. The crystal growth was monitored every 12 hours using a microscope. Diffraction quality crystals with a size of 0.35 × 0.28 × 0.19 mm3 were grown within 32 hours. A similar crystallization experiment was conducted for the HIF-13 molecule, and we took the crystal size of 0.30 × 0.25 × 0.18 mm3 for X-ray diffraction studies.
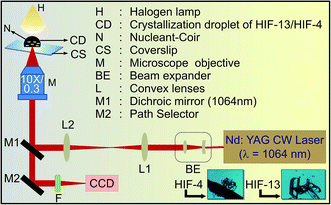 |
| | Fig. 1 Schematic diagram of laser-induced crystallization setup. Components involved in the setup are marked, and the HIF-4 and HIF-13 crystals grown using the laser-induced crystallization technique were also shown at the bottom. | |
2.3 Single crystal data collection, single crystal structure determination, refinement, and analysis
Single-crystal X-ray diffraction experiments were carried out for the compounds HIF-4 and HIF-13 (Scheme 1B and C). The preliminary cell determination and the 3D data collection for HIF-4 and HIF-13 were carried out in a Rigaku Saturn 72+ single-crystal X-ray diffractometer using graphite monochromatized MoKα radiation (λ = 0.71075 Å). The cell parameters were refined by the least-squares method in the θ range of 2–31° for HIF-4 and 3–25° for HIF-13. A complete data set was processed using CrystalClear software.16 Structure solution by direct methods using the SHELXS9717 exposed the positions of all non-hydrogen atoms of HIF-4 and HIF-13. The initial structures were refined by the least-squares method until convergence. Carbon-bound H atoms were placed geometrically, with C–H = 0.93 Å, and 0.96 Å (methyl) forced to ride on their parent atoms with Uiso(H) = 1.2Ueq(C) or Uiso(H) = 1.5Ueq(Cmethyl). The hydroxy H atom was located in a difference Fourier map and refined with Uiso(H) = 1.2Ueq(O). The final refinement converged to an R-value of 0.065 for HIF-4 and 0.053 for HIF-13. The Δρmax, Δρmin (e Å−3) being 0.16 and −0.19 for HIF-4 and 0.18 and −0.18 for HIF-13. These calculations were carried out using the packages SHELXL18 and WinGX.19 ORTEP-3 was used to prepare the thermal ellipsoid diagrams of HIF-4 and HIF-13 (Fig. 2).20 Tabulation of atomic and thermal parameters was done using the software CIFTAB.18 The characterization of rings was done from puckering parameters' values,21 and symmetry parameters were obtained using PARST97.22 Molecular packing diagrams were drawn using Mercury.23 The relevant crystallographic data of title compounds were deposited at Cambridge Crystallographic Data Centre (CCDC) with CCDC no. 1834972 (HIF-4), and 1834992 (HIF-13).† Crystal data and structure refinement details were summarized in Table 1. Hydrogen bonds were listed in Tables 2 and 3 for HIF-4, and HIF-13, respectively. The atomic coordinates of all the non-hydrogen atoms with their equivalent isotropic, anisotropic displacements parameters, the positional and isotropic displacement of the hydrogen atoms for HIF-4, HIF-13 were given in ESI data (Tables S1–S6†).
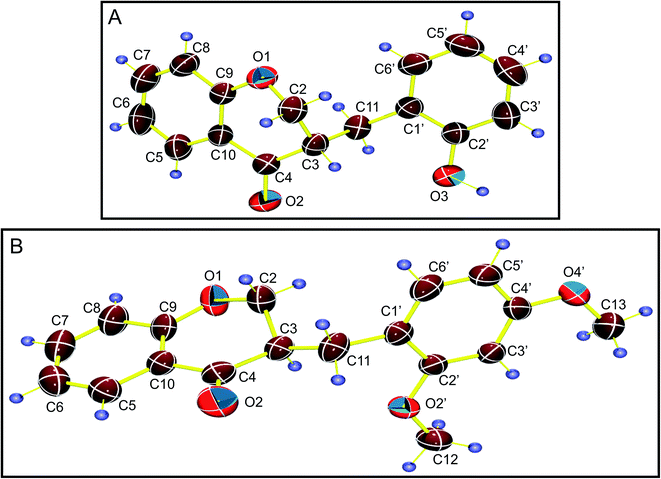 |
| | Fig. 2 The thermal ellipsoid diagrams of HIF-4 (A) and HIF-13 (B) drawn with a 50% probability level with atom numbering. | |
Table 1 Crystal data and structure refinement details for HIF-4 and HIF-13
| Identification code |
HIF-4 |
HIF-13 |
| Empirical formula |
C16H14O3 |
C18H18O4 |
| Formula weight |
254.27 |
298.34 |
| Temperature/K |
293(2) |
293(2) |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/n |
| a/Å |
12.0638(14) |
7.385(7) |
| b/Å |
8.9307(9) |
27.33(3) |
| c/Å |
12.9341(14) |
8.364(8) |
| α/° |
90 |
90 |
| β/° |
108.465(12) |
115.630(11) |
| γ/° |
90 |
90 |
| Volume/Å3 |
1321.8(3) |
1522(3) |
| Z |
4 |
4 |
| ρcalc./g cm−3 |
1.278 |
1.302 |
| µ/mm−1 |
0.088 |
0.091 |
| F(000) |
536.0 |
632.0 |
| Crystal size/mm3 |
0.350 × 0.280 × 0.190 |
0.300 × 0.250 × 0.180 |
| Radiation |
MoKα (λ = 0.71073) |
MoKα (λ = 0.71075) |
| 2θ range for data collection/° |
5.642 to 62.356 |
6.168 to 50.778 |
| Index ranges |
−17 ≤ h ≤ 16, −12 ≤ k ≤ 12, −17 ≤ l ≤ 18 |
−8 ≤ h ≤ 8, −31 ≤ k ≤ 32, −10 ≤ l ≤ 9 |
| Reflections collected |
17821 |
11860 |
| Independent reflections |
3945 [Rint = 0.0520, Rsigma = 0.0457] |
2768 [Rint = 0.0759, Rsigma = 0.0775] |
| Data/restraints/parameters |
3945/0/176 |
2768/0/201 |
| Goodness-of-fit on F2 |
1.029 |
0.925 |
| Final R indexes [I ≥ 2σ(I)] |
R1 = 0.0647, wR2 = 0.1302 |
R1 = 0.0533, wR2 = 0.1090 |
| Final R indexes [all data] |
R1 = 0.1217, wR2 = 0.1632 |
R1 = 0.1171, wR2 = 0.1405 |
| Largest diff. peak/hole/e Å−3 |
0.16/−0.19 |
0.18/−0.18 |
| CCDC deposition no. |
1834972 |
1834992 |
2.4 Cambridge structural database (CSD)
CSD provides the opportunity to study the conformational behavior in analogue structures. We investigated similarities and differences of HIF-4 and HIF-13 with respect to analogue structures retrieved from the CSD. Five analogue structures possessing the 3-benzylchroman-4-one skeleton (CSD refcodes: AFAYIR (rac-3-(4-hydroxybenzyl)chroman-4-one),10 ECUFOA (3-(3-hydroxy-4-methoxybenzyl)chroman-4-one), ECUFUG (3-(4-methylbenzyl)chroman-4-one),9 AFEWAL (3-(4-methoxybenzyl)-2,3-dihydro-4H-chromen-4-one),12 and LERYEO (3-(3,4-dimethoxybenzyl)chroman-4-one))11 were extracted from the CSD.
2.5 Hirshfeld surface and energy frameworks
The Hirshfeld surface (HS) analysis, including two-dimensional (2D) fingerprint (FP) plots and electrostatic energy frameworks of HIF-4 and HIF-13, along with other five other analogue structures, were generated using the CrystalExplorer (version 17.5), and corresponding CIF files were used as the input files.24,25
2.6 Density functional theory (DFT)
The DFT calculations on HIF-4 and HIF-13 were performed using the hybrid functional Becke 3 parameter combined with the Lee–Yang–Parr correlation functional (B3LYP) with a Gaussian type basis 6-31+G(d,p). The B3LYP method has been proven as reliable and relatively accurate for smaller molecular systems. In this work, geometry optimization was performed on possible isomers of HIF-4 and HIF-13 molecules. Following it, optimization was also performed on homodimer of all low energy structures. All the calculations were performed under the methanol solvent environment to follow the crystallization solvent condition. Gaussian 09 (Rev. D) program suite was used for all the calculations.26 The ball-and-stick model figures were made using PyMOL (https://www.pymol.org).27
2.7 FTIR spectroscopy
To understand the hydrogen bonding of HIF-4, infrared absorption spectra of the HIF-4 sample were performed before (semi-crystalline/amorphous) and after crystallization using JASCO FT/IR 6300 spectrometer.
3. Results and discussion
3.1 Conformation analysis of HIF-4 and HIF-13
The C2 and C3 atoms of HIF-4 significantly deviate from the mean plane formed by other atoms in the ten membered chromanone ring as 0.400(2) and −0.340(2), respectively. The pyranone ring of HIF-4 adopts slightly distorted envelope conformation with the puckering parameters q2 = 0.4154 (19) Å, q3 = −0.2622 (18) Å, QT = 0.4912 (19) Å, and φ = 258.9(2)°. Fig. 3A shows the packing of the HIF-4 molecule viewed along the a-axis. In the crystal, the molecules are stabilized by strong O3–H3O⋯O2 (symmetry 1 − x, 2 − y, 1 − z), and C5′–H5′⋯O1 (symmetry code: 1 − x, ½ + y, ½ − z) hydrogen bonds (Table 2 and Fig. 3A). Also, C2–H2A⋯π (C1′–C6′) (Fig. 3B) interaction with H⋯Cg – 2.84 Å [symmetry code 1 − x, ½ + y, ½ − z] plays a role in stabilizing the crystal structure (Fig. 3B). There are no π–π stacking interactions present in the structure.
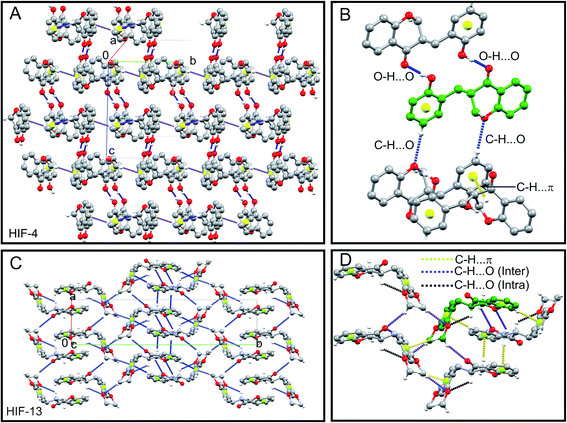 |
| | Fig. 3 3D molecular packing of HIF-4 and HIF-13. (A) Molecular packing of HIF-4 viewed along the a-axis. (B) The close view of hydrogen bond networking of HIF-4. The O–H⋯O, C–H⋯O dimer formation of HIF-4 along with the C–H⋯π interactions are shown. (C) Molecular interactions of HIF-13 viewed along the c-axis. (D) Close view of the C–H⋯O hydrogen bonds and C–H⋯π networking of HIF-13. The atoms are shown in the ball-and-stick model. Carbon, oxygen, and hydrogen atoms are coloured as grey, red, and white, respectively. For clarity purposes, hydrogen atoms, which are not involved in interactions, are omitted. All the interactions are shown in dotted lines. The centroid of phenyl rings involved in C–H⋯π interactions are shown in the yellow sphere. The carbon atoms of the parent molecule in B and D are shown in green colour. | |
Table 2 Hydrogen bonds for HIF-4a
| D–H⋯A |
d(D–H)/Å |
d(H–A)/Å |
d(D–A)/Å |
D–H–A/° |
| Symmetry transformation used to generate equivalent atoms: #11 − X, 2 − Y, 1 − Z, #21 − X, 1/2 + Y, 1/2 − Z, #31 −X, 1/2 + Y, 1/2 − Z. |
| O3–O3H⋯O2#1 |
0.94(3) |
1.83(3) |
2.768(2) |
174(2) |
| C5′–H5′⋯O1#2 |
0.93(4) |
2.671(3) |
3.492(2) |
148(2) |
| C2–H2A⋯Cg#3 |
0.97 |
2.84 |
3.494(2) |
125 |
The pyranone ring in HIF-13 (ring B in Scheme 1C) is not planar, as seen in HIF-4 and previously observed.9–12 The carbon C2 has the largest offset from the plane (−0.354(3)) followed by the C3 atom (−0.314(2)) defined by the adjacent benzene ring (ring A in Scheme 1C). The pyronone ring of HIF-13 adopts a distorted envelope conformation with ring puckering parameters q2 = 0.354 (3) Å, q3 = 0.274(3) Å, QT = 0.447(3) Å, and φ = 84.1(4)°. The packing of the HIF-13 is stabilized by a 3D network of weak intramolecular C3–H3⋯O2′, and C12–H12C⋯O2, C13–H13B⋯O2′ intermolecular interactions, which serve to link inversion-related sheets (Fig. 3C). It should be noted that HIF-13 is the only structure that forms an intramolecular C–H⋯O hydrogen bond comparison with HIF-4 and the other five analogue structures retrieved from CSD, as discussed in Section 2.4. In addition to C–H⋯O hydrogen bonds, there are three C2–H2B⋯π (C5–C10), C8–H8⋯π (C1′–C6′), C12–H12A⋯π (C1′–C6′) interactions stabilize the 3D structure of HIF-13 (Table 3 and Fig. 3D).
Table 3 Hydrogen bonds for HIF-13a
| D–H⋯A |
d(D–H)/Å |
d(H–A)/Å |
d(D–A)/Å |
D–H–A/° |
| Symmetry transformation used to generate equivalent atoms: #1X − 1, Y, Z, #2X − 1/2, Y − 1/2, Z + 1/2, #32 − X − 2, 1 − Y, 1 − Z, #41 − X, 1 − Y, 1 − Z, #5X − 1/2, 1/2 − Y, Z − 1/2. |
| C3–H3⋯O2′ |
0.98 |
2.58 |
3.144(4) |
116.8 |
| C12–H12C⋯O2#1 |
0.96 |
2.67 |
3.564(4) |
155.1 |
| C13–H13B⋯O2′#2 |
0.96 |
2.70 |
3.369(4) |
127.1 |
| C2–H2B⋯Cg#3 |
0.97 |
2.72 |
3.668(5) |
167.0 |
| C8–H8⋯Cg#4 |
0.93 |
2.76 |
3.623(5) |
156.0 |
| C12–H12A⋯Cg#5 |
0.96 |
3.00 |
3.903(5) |
158.0 |
3.2 CSD studies of 3-benzylchroman-4-ones
The geometric parameters of HIF-4 and HIF-13 are comparable with the analogue structures AFAYIR,10 ECUFOA,9 ECUFUG,9 AFEWAL,12 and LERYEO11 reported earlier. The bond lengths of HIF-4 and HIF-13 pyranone rings are compared with the five analogue structures, and the results are shown in Fig. 4A. The results show that except C2–C3, all other bond lengths are similar (Fig. 4 and Table 4). The average C2–C3 bond length is 1.433 Å, much smaller than HIF-4 and HIF-13 because this bond is often disordered in most of the reported structures. Out of five analogue structures, three of them (AFAYIR, AFEWAL, EUCFOA) have either C2 or C3 atoms disordered in O1–C2, C2–C3 bonds. In the case of AFAYIR, AFEWAL, both C3 and C4 atoms are disordered in the C3–C4 bond. Hence, the C2–C3 bond disorder is observed frequently; the respective average bond length has a considerable deviation from HIF-4 and HIF-13 (Table 4). Similarly, the C3–C11 and C11–C1′ bond lengths of HIF-4 and HIF-13 are comparable with the analogue structures. While C11–C1′ bond length of HIF-4 (1.506 Å) and HIF-13 (1.501 Å) is closer to the average bond length (1.505 Å), the C3–C11 bond length of HIF-4 (1.550 Å) is longer than HIF-13 (1.520 Å), and the average of analogue structures (1.507 Å) (Fig. 4A).
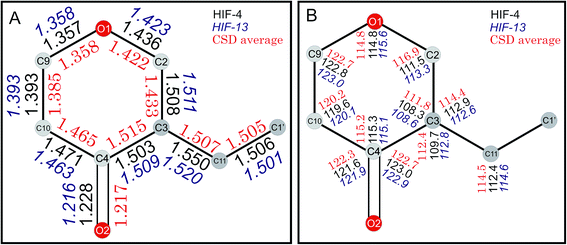 |
| | Fig. 4 CSD survey of HIF-4 and HIF-13. (A) Bond lengths of HIF-4, HIF-13, and the average structures. (B) Bond angles of HIF-4, HIF-13, and the average structures. The values of HIF-4 (black), HIF-13 (blue and italics), and the CSD average (red) are marked. | |
Table 4 Selected bond lengths (Å) of the pyranone ring of HIF structures and analogue structures retrieved from CSD. The bond lengths calculated for the disordered atoms are indicated in * mark. For example, O1–C2B*, in which the C2 atom is disordered
| Bond |
HIF-4 |
HIF-13 |
AFAYIR |
EUCFUG |
AFEWAL |
ECUFOA |
LERYEO |
Average |
| O1–C2 |
1.436(2) |
1.423(3) |
1.388 |
1.444 |
1.437 |
1.423 |
1.418 |
1.422(22) |
| O1–C2B* |
— |
— |
1.536 |
— |
1.487 |
1.502 |
— |
1.508(25) |
| C2–C3 |
1.508(2) |
1.511(4) |
1.393 |
1.500 |
1.342 |
1.434 |
1.496 |
1.433(68) |
| C2–C3B* |
— |
— |
1.254 |
— |
1.218 |
1.273 |
— |
1.248(28) |
| C3–C4 |
1.503(2) |
1.509(3) |
1.512 |
1.507 |
1.530 |
1.514 |
1.510 |
1.515(9) |
| C4–C3B* |
— |
— |
1.512 |
— |
1.543 |
— |
— |
1.528(22) |
| C4–C10 |
1.471(2) |
1.463(4) |
1.457 |
1.474 |
1.466 |
1.460 |
1.468 |
1.465(7) |
| C10–C9 |
1.393(2) |
1.393(4) |
1.388 |
1.395 |
1.379 |
1.373 |
1.389 |
1.385(9) |
| O1–C9 |
1.357(2) |
1.358(3) |
1.359 |
1.357 |
1.354 |
1.363 |
1.355 |
1.358(4) |
| O2–C4 |
1.228(2) |
1.216(3) |
1.218 |
1.216 |
1.212 |
1.227 |
1.213 |
1.217(6) |
| C3–C11 |
1.550(2) |
1.520(3) |
1.507 |
1.533 |
1.502 |
1.486 |
1.507 |
1.507(17) |
| C3B–C11* |
— |
— |
1.458 |
— |
1.522 |
— |
— |
1.490(45) |
| C11–C1′ |
1.506(2) |
1.501(3) |
1.510 |
1.502 |
1.500 |
1.503 |
1.509 |
1.505(4) |
The bond angles of pyranone rings of HIF-4 and HIF-13, along with the average bond angles of analogue structures, are shown in Fig. 4B, and the details are summarized in Table 5. Almost all bond angles of HIF-4 and HIF-13 have good agreement with the average bond angle values. The O1–C2–C3 of HIF-4 (111.5°) and HIF-13 (113.3°) are slightly lower than the average value (116.9°), and it may be due to the disordered behaviour observed in the analogue structures. The O1–C2–C3 and C11–C3–C2 bond angles are slightly higher than the normal for the disordered structures AFAYIR, AFEWAL, and ECUFOA (Table 5 and Fig. 4B).
Table 5 Selected bond angles of HIF structures and analogue structures retrieved from CSD. The bond angles that correspond to either one or two disordered atoms are marked as *. For example, in C9–O1–C2B, the C2 atom is disordered, and C4–C3B–C2B, the C3 and C2 atoms are disordered
| Bond angle |
HIF-4 |
HIF-13 |
AFAYIR |
ECUFUG |
AFEWAL |
ECUFOA |
LERYEO |
Average |
| C9–O1–C2 |
114.8(1) |
115.6(2) |
114.6 |
114.4 |
115.2 |
114.6 |
115.0 |
114.8(0.3) |
| C9–O1–C2B* |
— |
— |
115.3 |
— |
112.8 |
113.0 |
— |
113.7(1.4) |
| O1–C2–C3 |
111.5(1) |
113.3(2) |
119.9 |
112.3 |
124.0 |
115.2 |
112.9 |
116.9(5.0) |
| O1–C2B–C3 |
— |
— |
127.2 |
— |
126.3 |
120.5 |
— |
124.7(3.6) |
| C4–C3–C2 |
108.3(1) |
108.6(2) |
113.1 |
109.4 |
116.7 |
111.4 |
108.4 |
111.8(3.3) |
| C4–C3B–C2B* |
— |
— |
123.0 |
— |
117.0 |
116.0 |
— |
118.7(3.8) |
| C10–C4–C3 |
115.3(2) |
115.1(2) |
116.4 |
115.4 |
114.3 |
115.8 |
114.3 |
115.2(0.9) |
| C10–C4–C3B* |
— |
— |
114.1 |
— |
114.5 |
— |
— |
114.3(0.3) |
| C9–C10–C4 |
119.6(2) |
120.1(2) |
120.4 |
120.2 |
120.3 |
119.9 |
120.1 |
120.2(0.2) |
| O1–C9–C10 |
122.8(2) |
123.0(2) |
122.6 |
122.5 |
122.5 |
122.8 |
122.9 |
122.7(0.2) |
| C11–C3–C2 |
112.9(2) |
112.6(2) |
109.5 |
112.1 |
112.8 |
123.4 |
114.3 |
114.4(5.3) |
| C11–C3B–C2B* |
— |
— |
116.1 |
— |
115.1 |
129.0 |
— |
120.1(7.8) |
| C11–C3–C4 |
109.7(1) |
112.8(2) |
113.2 |
112.9 |
109.4 |
114.2 |
112.3 |
112.4(1.8) |
| C11–C3B–C4 |
— |
— |
116.0 |
— |
111.2 |
— |
— |
113.6(3.4) |
| C1′–C11–C3 |
112.4(1) |
114.6(2) |
113.4 |
113.7 |
115.0 |
116.3 |
114.0 |
114.5(1.2) |
| C1′–C11–C3B* |
— |
— |
121.6 |
— |
117.0 |
— |
— |
119.3(3.3) |
| O2–C4–C3 |
123.0(2) |
122.9(2) |
123.5 |
122.9 |
122.6 |
121.6 |
122.9 |
122.7(0.7) |
| O2–C4–C3B* |
— |
— |
118.3 |
— |
121.3 |
— |
— |
119.8(2.1) |
| O2–C4–C10 |
121.6(2) |
121.9(3) |
122.2 |
121.7 |
122.3 |
122.6 |
122.8 |
122.3(0.4) |
The selected six torsion angles around the pyranone ring and phenyl ring C1′–C6′ of HIF-4, HIF-13, and their analogue structures are summarized in Table 6. The C2–C3–C11–C1′ torsion angle adopts either −synclinal or +synclinal conformation (Table 6), and HIF-13 has the maximum (69.6(3)°). The C3–C11–C1′–C2′ torsion angle adopts +synclinal (from +30° to +90°) conformation, except ECUFUG (97.7°), and AFEWAL (94.6°), which adopt +anticlinal conformation. The C3–C11–C1′–C6′ (−anticlinal) and C4–C3–C11–C1′ (±antiperiplanar) torsion angles of all seven structures adopt similar conformations. The torsion angle O1–C2–C3–C11 adopts two different orientations. The HIF-13 and its five analogue structures adopt antiperiplanar (±150° to ±180°) conformation, whereas HIF-4 (61.7(2)°) adopt +synclinal conformation. Similarly, O2–C4–C3–C11 of HIF-4 (88.9°) also adopts +synclinal conformation, and all other structures adopt synperiplanar (±0° to ±30°) conformation. Thus, it is clear that HIF-4 adopts an entirely different conformation than HIF-13 and five analogue structures extracted from CSD.
Table 6 Selected torsion angles of HIF structures and analogue structures retrieved from CSD. The * mark represents the calculated torsion angles contains disordered atoms. For example, torsion angle O1–C2B–C3B–C11* has two (C2B and C3B) disordered atoms
| Torsion angle |
HIF-4 |
HIF-13 |
AFAYIR |
ECUFUG |
AFEWAL |
ECUFOA |
LERYEO |
| O1–C2–C3–C11 |
61.7(2) |
−177.5(2) |
−171.8 |
176.9 |
−177.0 |
167.4 |
−174.6 |
| O1–C2B–C3B–C11* |
— |
— |
−174.9 |
— |
172.4 |
152.5 |
— |
| C2–C3–C11–C1′ |
60.4(2) |
69.6(3) |
61.2 |
−66.2 |
−50.8 |
65.5 |
54.3 |
| C2B–C3B–C11–C1′* |
— |
— |
−26.7 |
— |
50.0 |
−12.3 |
— |
| C3–C11–C1′–C2′ |
81.4(2) |
74.7(3) |
87.3 |
97.7 |
94.6 |
74.8 |
70.3 |
| C3B–C11–C1′–C2′* |
— |
— |
62.7 |
— |
66.3 |
— |
— |
| C3–C11–C1′–C6′ |
−98.3(2) |
−106.1(3) |
−117.9 |
−81.3 |
−114.2 |
−104.5 |
−108.8 |
| C3B–C11–C1′–C6′* |
— |
— |
−93.3 |
— |
−85.9 |
— |
— |
| O2–C4–C3–C11 |
88.9(2) |
18.5(3) |
17.3 |
−24.2 |
26.0 |
9.71 |
16.4 |
| O2–C4–C3B–C11* |
— |
— |
−33.4 |
— |
−27.5 |
— |
— |
| C4–C3–C11–C1′ |
−178.8(1) |
−167.1(2) |
−174.2 |
169.8 |
170.3 |
178.9 |
178.3 |
3.3 Conformational flexibilities of 3-benzylchroman-4-ones
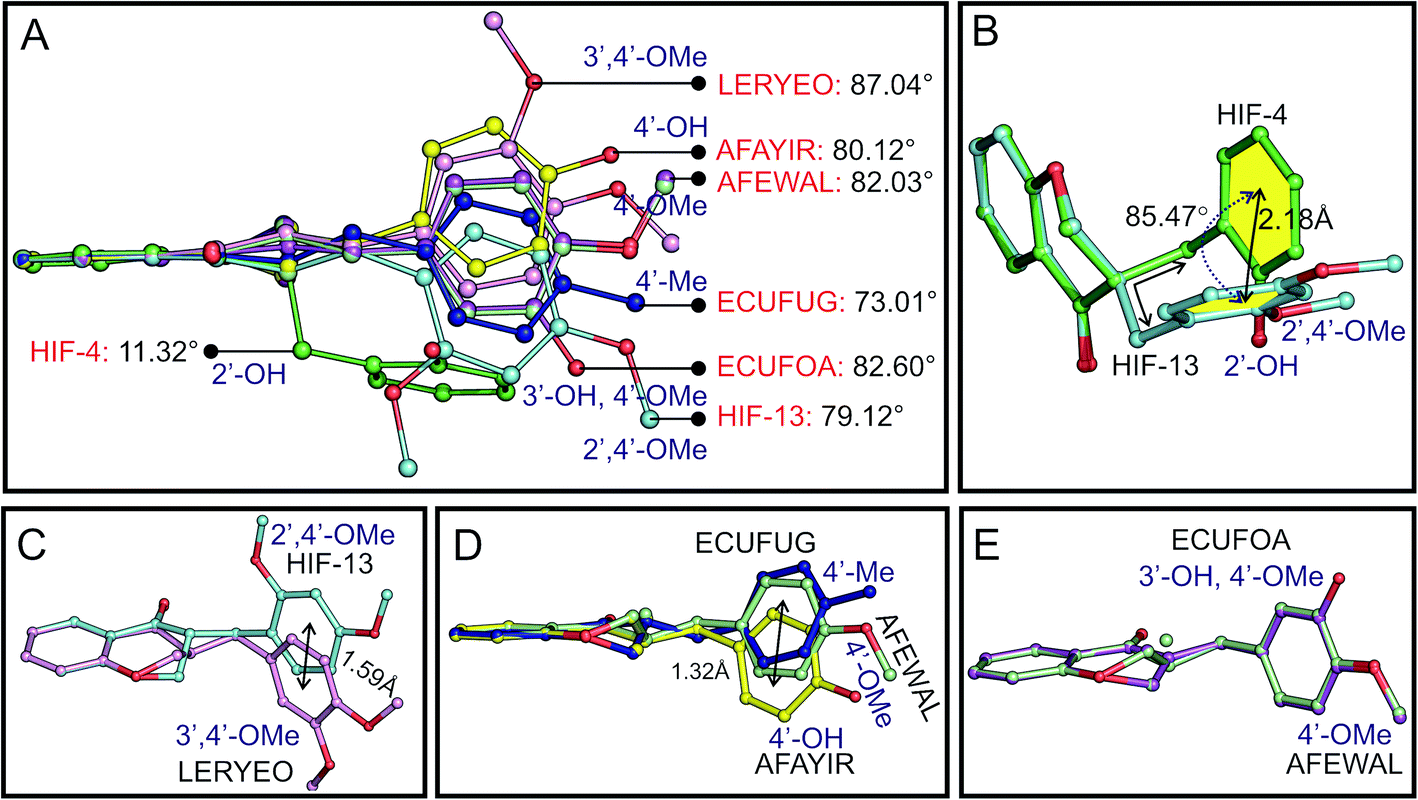
The dihedral angle between the planes of phenyl rings C5–C10 (ring A in Scheme 1B) and C1′–C6′ (ring C in Scheme 1B) can be used to systematically explain the conformation of the molecules. The angle is found to be 11.32° for HIF-4 and 79.12° for HIF-13. This corresponding angle is 87.04° for LERYEO, 80.12° for AFAYIR, 82.03° for AFEWAL, 73.01° for ECUFUG, and 82.60° for ECUFOA (Fig. 5A).
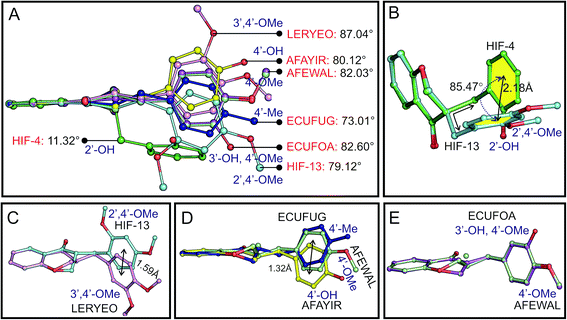 |
| | Fig. 5 Conformational analysis of 3-benzylchroman-4-ones. (A) Superposition of seven 3-benzylchroman-4-one structures based on the similar atoms of chromanone moiety (except C2 atom). The dihedral angle between the planes of two phenyl rings of seven 3-benzylchroman-4-one structures and the corresponding substitution are marked. (B) Comparison of HIF-4 and HIF-13. The angle between the two phenyl rings of HIF-4 and HIF-13 and the distance between the centroid of the two phenyl rings are shown. (C) Comparison of LERYEO (OMe at 3′, 4′ positions) and HIF-13 (OMe at 2′, 4′ positions). (D) Comparison between AFAYIR, AFEWAL, and EUCFUG. (E) Comparison of ECUFOA and AFEWAL. | |
Superposition of the HIF-4 and HIF-13 structures with ten common non-hydrogen atoms of the chromanone moiety (except C2 atom) yielded the RMSD of 0.173 Å. At the same time, the phenyl ring C of these two structures deviates from each other with 2.18 Å and rotates with an angle of 85.47° (Fig. 5B). The C11 atom, which connects the pyranone ring B, and the phenyl ring C, is axially attached to the C3 atom in HIF-4. In contrast, the C11 atom in HIF-13 is equatorially connected to the C3 atom. The puckering of the C2 atom from the mean plane through the other five atoms of the pyranone ring may also play a significant role in the phenyl ring movement. For example, HIF-13 has OMe moiety at 2′ and 4′, and LERYEO has OMe moiety at 3′ and 4′ positions. The C2 and C3 atoms of HIF-13 and LERYEO adopt an opposite position. Due to this fact, the phenyl rings deviate from each other around 1.59 Å from their centroid (Fig. 5C).
The EUCFUG (4′-CH3), AFEWAL (4′-OMe), and AFAYIR (4′-OH) structures have a single substitution at the 4′ position and have disordered C2/C3 atoms. Thus the AFEWAL (4′-OMe) is positioned between EUCFUG and AFAYIR, and the phenyl rings of the two end structures (EUCFUG and AFAYIR) are moved around 1.32 Å distance (Fig. 5D). Interestingly the ECUFOA and AFEWAL have OMe at 4′ position of phenyl ring C, and both adopt similar conformation, though ECUFOA has an additional substation at 3′ (OH). HIF-4, ECUFOA, and AFAYIR have a hydroxyl group in the phenyl ring C in 2′, 3′, and 4′ positions, respectively. Though all three structures have the same moiety, ECUFOA and AFAYIR have almost similar dihedral angles (Fig. 5A), and thus both adopt similar conformation. However, HIF-4 has a dihedral angle of 11.32° between the planes, and it adopts a different conformation. Thus, the CSD structural analysis shows that (i) irrespective of any moieties present at the 2′, 3′ and 4′ positions in phenyl ring C, all structures adopt similar conformation, except HIF-4, which has OH at 2′ position; (ii) substitution at 4′ position makes the phenyl ring C near perpendicular concerning the chromanone moiety, (iii) substitution at 3′ position does not bring any significant conformational changes, and (iv) OH substitution at 2′ position makes the phenyl ring C near parallel to chromanone moiety. Thus HIF-4 adopts an entirely different conformation compared to all other molecules.
3.4 Hirshfeld surface studies of 3-benzylchroman-4-ones
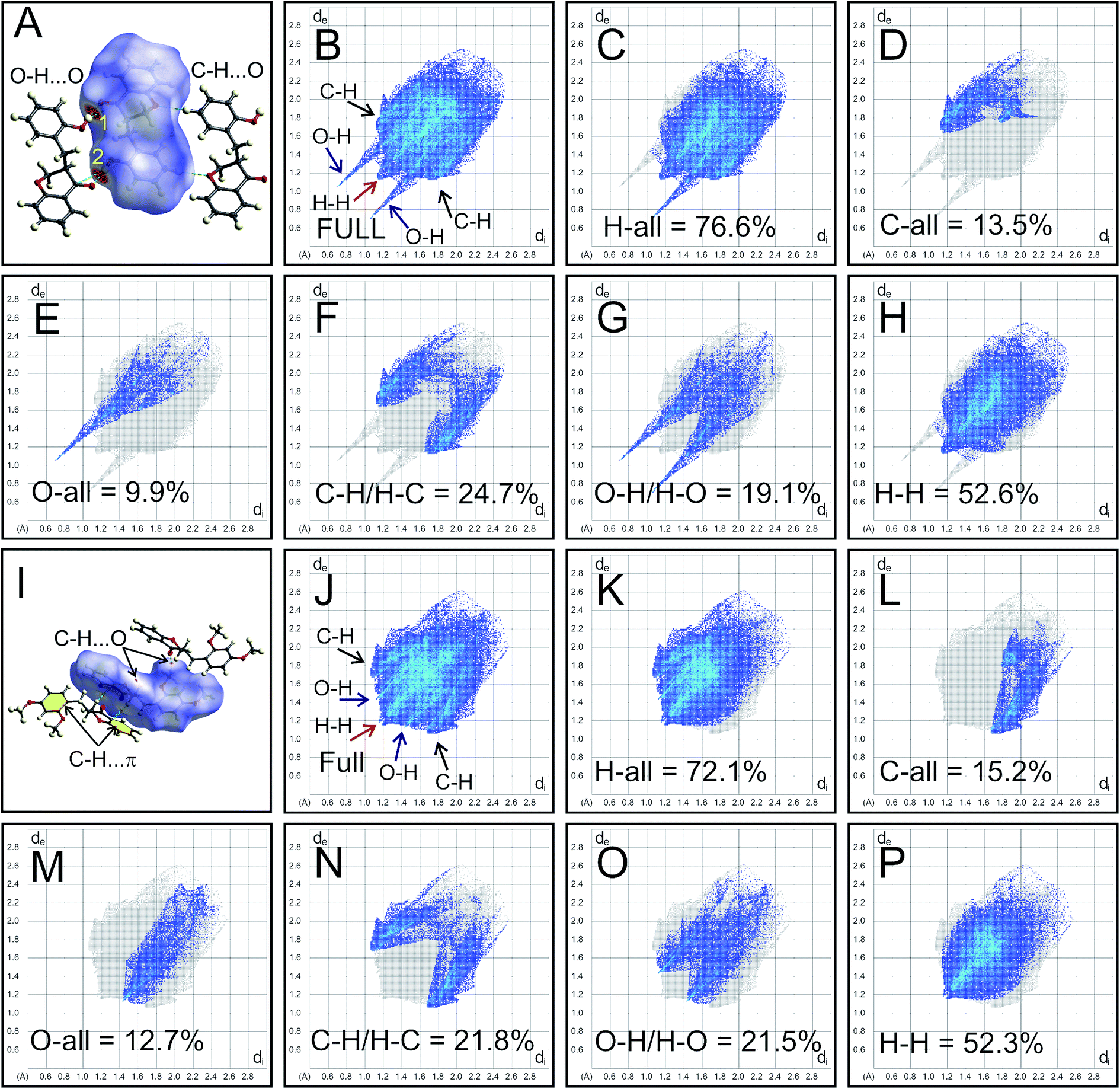
The intermolecular interactions of HIF-4 and HIF-13 were further studied with Hirshfeld surface (HS) analysis, including 2D fingerprint (FP) plots along with other five other analogue structures.24 Fig. 6 shows the HS map and 2D-FP plots of HIF-4 and HIF-13. In the HS of HIF-4, two bright red spots and two less intense red spots were observed. The label 1 and 2 denote the strong O–H⋯O (O3–O3H⋯O2) hydrogen bond, which makes a dimer (Fig. 6A). The less intense red spots represent the weak C5′–H5′⋯O1 hydrogen bond, which also forms another dimer. The full 2D-FP plot of HIF-4 (Fig. 6B), as well as the finger plots illustrating the different contacts of the HIF-4, H-all (76.6%; Fig. 6C), C-all (13.5%; Fig. 6D), O-all (9.9%; Fig. 6E), C–H/H–C (24.7%; Fig. 6F), O–H/H–O (19.1; Fig. 6G), and H–H (52.6%; Fig. 6H), are shown. According to divided fingerprints, the highest contribution of contacts received by the HIF-4 is H–H (52.6%). In the fingerprint plots, the H-all and O-all contacts develop as sharp spikes, indicating a dimer formation in the crystal structure through O3–O3H⋯O2 hydrogen bonds between two adjacent molecules of HIF-4 (Fig. 6B). The C–H contribution is slightly higher than the O–H contribution, which may be due to the combination of weak C–H⋯O, and C–H⋯π interactions.
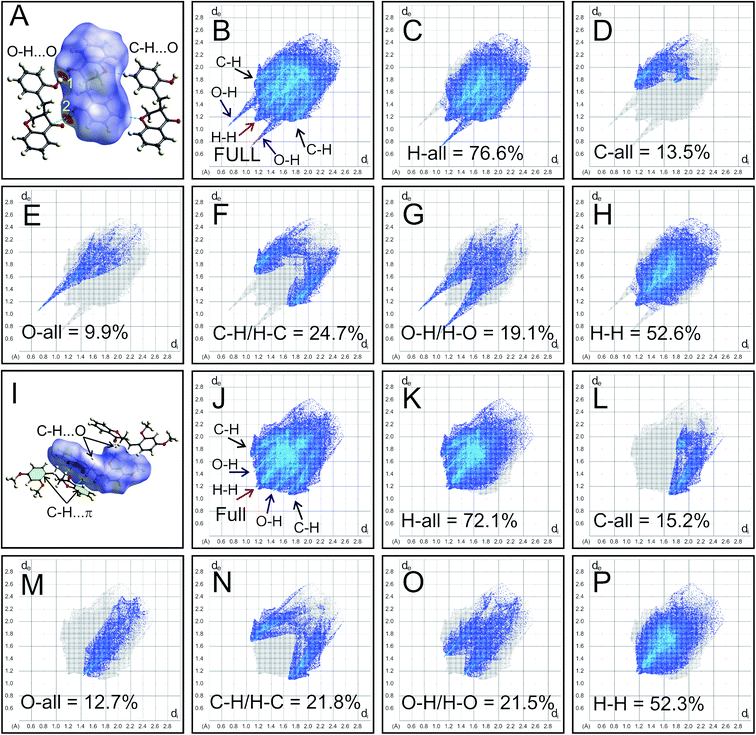 |
| | Fig. 6 HS analysis of HIF-4 and HIF-13. (A) HS for HIF-4 mapped with dnorm. Labels 1 and 2 represent the O–H⋯O dimer. Finger plots for HIF-4 full (B) and the specific pairs of atom-types H-all (C), C-all (D), O-all (E), C–H/H–C (F), O–H/H–O (G), and H–H (H) for HIF-4. (I) HS for HIF-13 mapped with dnorm. C–H⋯O, and C–H⋯π interactions are marked, and the phenyl rings involved in the C–H⋯π interactions are shown in yellow. Finger plots for HIF-13 full (J) and the specific pairs of atom-types H-all (K), C-all (L), O-all (M), C–H/H–C (N), O–H/H–O (O), and H–H (P) for HIF-13 are shown. | |
The C–H⋯O and C–H⋯π (marked as yellow in Fig. 6I) interactions of HIF-13 appear as small and pale-red areas (Fig. 6I). The full 2D-FP plot (Fig. 6J), as well as the finger plots illustrating the different contacts of the HIF-13, H-all (72.1%; Fig. 6K), C-all (15.2%; Fig. 6L), O-all (12.7%; Fig. 6M), C–H/H–C (21.8%; Fig. 6N), O–H/H–O (21.5%; Fig. 6O), and H–H (52.3%; Fig. 6P), are shown. In comparison, HIF-4 (52.6%) and HIF-13 (52.3%) have nearly similar H–H contributions. The histogram of the percentage of oxygen, carbon, and hydrogen intermolecular contributions of all seven 3-benzylchroman-4-one structures shows that LERYEO and ECUFOA have the most considerable oxygen contribution (12.6%), and ECUFOG (8.5%) has the lowest contribution (ESI Fig. S1H†). In terms of carbon, HIF-4 (13.5%) and HIF-13 (13.2%) have the highest contribution than analogue structures. In general, hydrogen contributes around 77% on average to make intermolecular interactions for all 3-benzylchroman-4-one structures. In which, ECUFOA (75.6%), HIF-13 (75.8%) are the lowest, and ECUFUG (79.1%) contributes higher than all other structures (ESI Fig. S1H†). Out of seven structures, HIF-4, ECUFOA, and AFAYIR have OH substitution at 2′, 3′, and 4′ positions in the phenyl ring C. The 2D-FP plot of the analogue structures shows that the AFAYIR and ECUOFA contribution of O–H is almost similar to HIF-4 (ESI Fig. S1A–G†). Then what makes the HIF-4 different than other structures? To understand the fact, along with Hirshfeld surface, the O–H⋯O intermolecular interactions are scrutinized further for HIF-4, ECUFOA, and AFAYIR structures (ESI Fig. S1I–K†). HIF-4 is the only structure that forms a dimer via O–H⋯O hydrogen bonds among all three structures. In terms of distance (hydrogen-acceptor: 1.83 Å) and the angle (174(2)°), HIF-4 is stronger than the other two structures (ESI Fig. S1I†). Though AFAYIR also forms a strong O–H⋯O hydrogen bond, the hydrogen bond pattern is different than HIF-4 (ESI Fig. S1J†). In addition, the oxygen atom in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of the pyranone ring act as an acceptor atom for both HIF-4 and AFAYIR structures. In ECUCOA, the OMe moiety in phenyl ring C acts as an acceptor, and the pyranone ring is not involved in the O–H⋯O hydrogen bond interactions. The hydrogen bond strength of the ECUFOA is comparatively weaker than HIF-4 and AFAYIR (ESI Fig. S1K†). Thus, it is evident that the intermolecular interactions, especially O–H⋯O dimer formation in HIF-4, play a significant role in adopting different conformation due to the OH substitution at 2′ position in phenyl ring C.
O moiety of the pyranone ring act as an acceptor atom for both HIF-4 and AFAYIR structures. In ECUCOA, the OMe moiety in phenyl ring C acts as an acceptor, and the pyranone ring is not involved in the O–H⋯O hydrogen bond interactions. The hydrogen bond strength of the ECUFOA is comparatively weaker than HIF-4 and AFAYIR (ESI Fig. S1K†). Thus, it is evident that the intermolecular interactions, especially O–H⋯O dimer formation in HIF-4, play a significant role in adopting different conformation due to the OH substitution at 2′ position in phenyl ring C.
3.5 Energy framework analysis of 3-benzylchroman-4-ones
Energy framework analysis helps to understand the crystal packing and visualize the interaction topologies based on electrostatic, polarization, dispersion, and exchange-repulsion.28 The interaction energies (kJ mol−1) calculated from energy framework calculations using CrystalExplorer are summarized in ESI Table S7† for all seven HIF-structures. HIF-4 (−100.5 kJ mol−1) has the highest electrostatic energy than all other structures, followed by AFAYIR (−56.8 kJ mol−1) and ECUFOA (−38.0 kJ mol−1). It may be because all three structures have OH moiety present in the phenyl ring C. It should be noted that the HIF-4 and AFAYIR structures have only OH substitution at the phenyl ring C, and their electrostatic energies are higher than any other structures. Interestingly AFAYIR (−61.7 kJ mol−1) has the highest dispersion energy, and HIF-4 has the lowest (−31.2 kJ mol−1). In terms of total energy, HIF-4 is once again dominating with −85.5 kJ mol−1 followed by AFAYIR with −54.3 kJ mol−1. The AFEWAL (OMe at 4′ position) and ECUFUG (CH3 at 4′ position) structures have the lowest total energy, which is 50% lower than the HIF-4 structure.
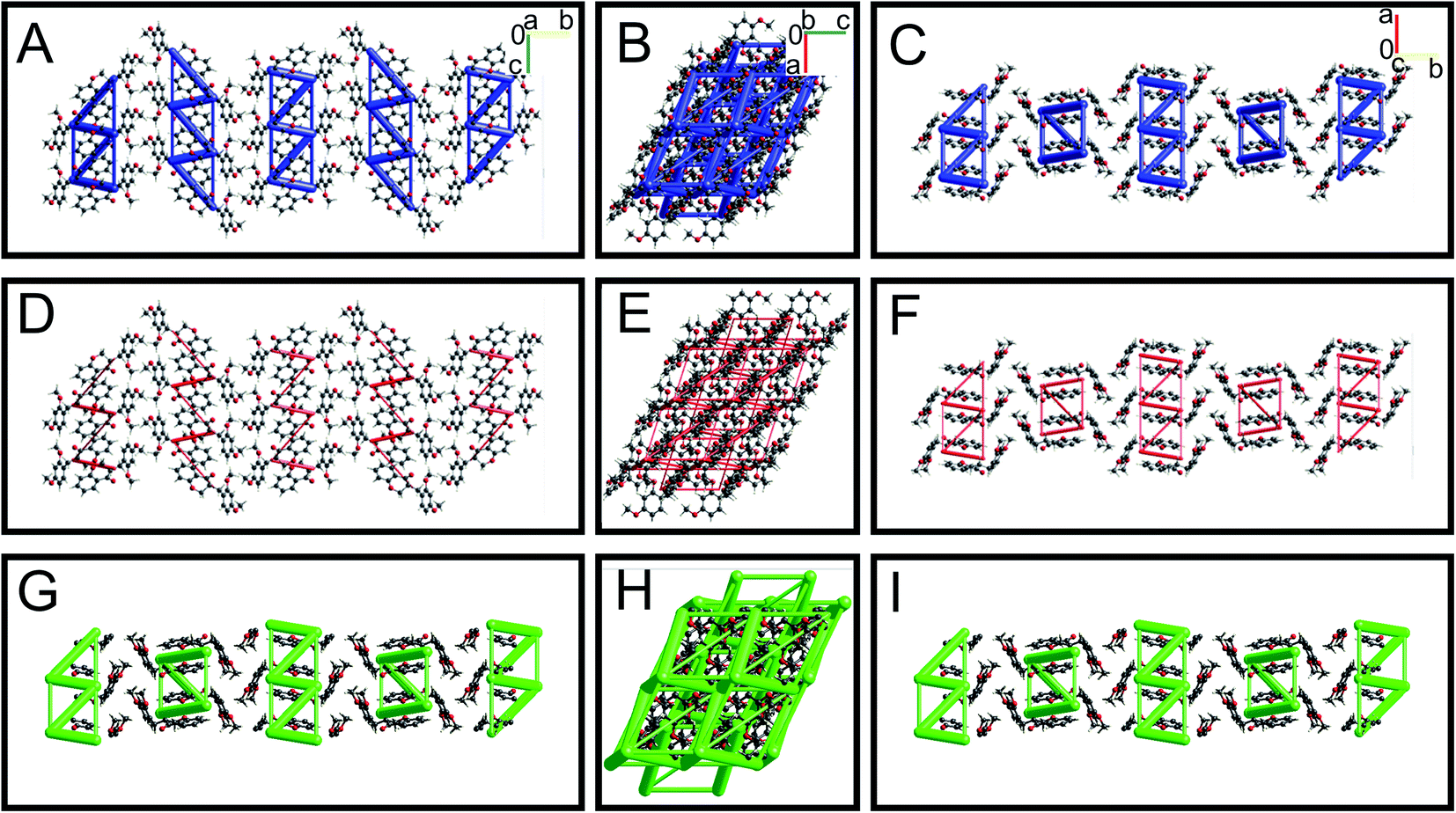
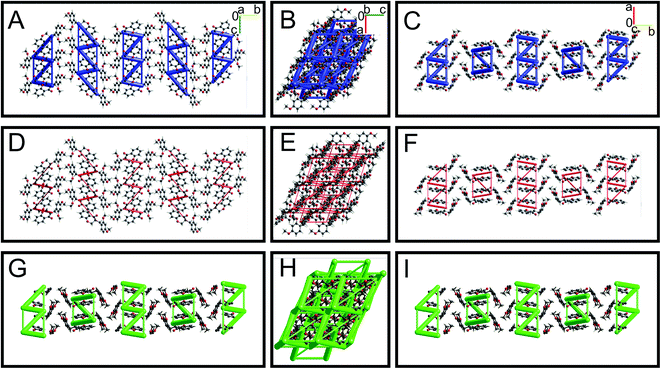
The energy framework interaction plots for HIF-4 and HIF-13 are depicted in Fig. 7 and 8, respectively. The energy frameworks for Etot (Fig. 7A–C), Eele (Fig. 7D–E), and Edisp (Fig. 7G–I) for HIF-4 are shown in blue, red, and green colour cylinders, respectively. Similarly, Etot (Fig. 8A–C), Eele (Fig. 8D–E), and Edisp (Fig. 8G–I) for HIF-13 are also shown. The energy frameworks for LERYEO, AFAYIR, AFEWAL, ECUFOA, and ECUFUG are shown in the ESI Fig. S2–S6.† The results show that HIF-4 forms a parallel network mainly through OH atoms (Fig. 7), and the HIF-13 forms a zig-zag energy pattern formed between the two molecules of HIF-13 (Fig. 8). In HIF-4, the electrostatic terms dominate the energy pattern due to the OH substation at the 2′ position. In HIF-13, the energy pattern is dominated by the dispersion energies due to the OMe substitutions at 2′ and 4′ positions in phenyl ring C. The remaining structures LERYEO (ESI Fig. S2†), AFAYIR (ESI Fig. S3†), AFEWAL (ESI Fig. S4†), EUCFOA (ESI Fig. S5†), and ECUFUG (ESI Fig. S6†) form parallel zig-zag sheet energy networks mainly through C3–C11 and C11–C1′ bonds, and are comparable to each other. However, as discussed earlier, the network pattern of HIF-4 is unique and different than all other structures due to highly dominated electrostatic energies.
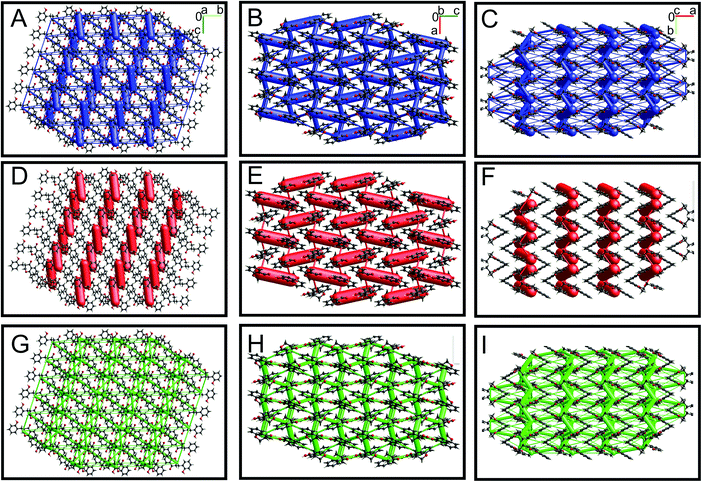 |
| | Fig. 7 Energy frameworks analysis for HIF-4. The molecular arrangement of HIF-4 is viewed along with the a (A, D, and G), b (B, E, and H), and c (C, F, and I) directions. The models are shown in ball-and-stick. The total interaction energies (A–C), electrostatic terms (D–F), and dispersion energy terms (G–I) are shown in blue, red, and green, respectively. The energy framework interactions are calculated using B3LYP/6-31G(d,p) electron-density functions. The energies between molecular pairs are represented as cylinders with a scale size of 150 by joining the centroids of pairs of molecules. | |
 |
| | Fig. 8 Energy framework analysis for HIF-13. The molecular arrangement of HIF-13 is viewed along with the a (A, D, and G), b (B, E, and H), and c (C, F, and I) directions. The models are shown in ball and stick. The total interaction energies (A–C), electrostatic terms (D–F), and dispersion energy terms (G–I) are shown in blue, red, and green, respectively. | |
3.6 DFT studies of HIF-4 and HIF-13
The structures of HIF-4 and HIF-13 molecules were further investigated using DFT calculations. Although there are several geometries possible, Fig. 9A shows the top five low energy models of HIF-4. Three isomers, isomer-1 (0.0 kJ mol−1), isomer-2 (2.7 kJ mol−1), and isomer-3 (10.0 kJ mol−1) adopt different conformation than HIF-4 crystallographic structure. Isomer-1 appeared as the most stable conformation with O–H⋯O (1.703 Å) intramolecular hydrogen bond. The isomer-2 of HIF-4 is similar to isomer-1, except the phenyl ring is slightly oriented away with a higher O–H⋯O (1.732 Å) hydrogen bond length. The isomer-3 adopts a similar conformation as HIF-13. The isomer-4 (10.92 kJ mol−1) and isomer-5 (16.96 kJ mol−1) adopt similar conformation like HIF-4. The only structural difference between isomer-4 and isomer-5 is in the orientation of the hydrogen atom in 2′-OH (Fig. 9A).
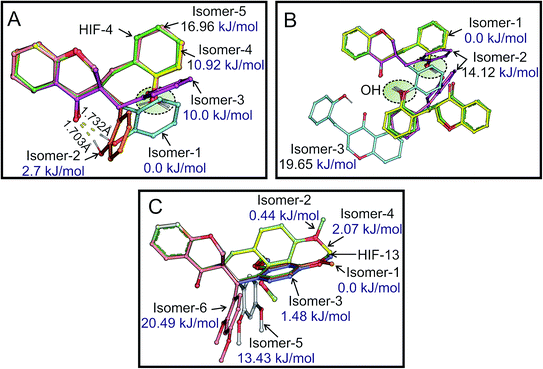 |
| | Fig. 9 DFT models of HIF-4 and HIF-13. (A) Isomers of HIF-4 superimposed with HIF-4 crystal structure based on chroman-4-one moiety. The five isomers obtained from DFT studies were shown and coloured differently, and the respective energy values are marked. The intramolecular O–H⋯O hydrogen bonds are shown in yellow dashed lines, and the hydrogen bond distances are marked. (B) Three possible homodimer structures of HIF-4 calculated using DFT studies superimposed with the HIF-4 dimer crystal structure shown along with the energy values. The orientation of OH molecules is highlighted in the green circle. (C) Six possible isomers of HIF-13 were calculated from DFT studies along with the crystal structure of HIF-13. | |
The formation of a molecular dimer is a critical process to start the nucleation of the molecular system towards the crystal. Because of this, optimizations of homodimers of monomers were performed in the methanol environment. Fig. 9B shows the energy-optimized structures of HIF-4 dimers with their relative energies. The intermolecular O–H⋯O (O3–O3H⋯O2) hydrogen bond stabilizes the dimer structure of isomer-1 with the distance of 1.765 Å and adopts the most stable conformation. Interestingly this lowest energy dimer structure is similar to the crystal structure of HIF-4. In the dimer of isomer-2, the intermolecular O–H⋯O (O3–O3H⋯O2) hydrogen bond is formed with the length of 1.833 Å. In isomer-3, the intermolecular O–H⋯O (O3–O3H⋯O2) hydrogen bond was intact with a distance of 1.699 Å. In addition, a weak C5–H5⋯O3 (2.582 Å) intermolecular hydrogen bond was also observed. Except for one of the monomers of isomer-3, the orientation of polar hydrogen O3H is similar for all the isomers and closely resembles the HIF-4 dimer structure. Though the isomer-2 (14.12 kJ mol−1) and isomer-3 (19.65 kJ mol−1) form stronger O–H⋯O hydrogen bonds, their energies do not favour the formation of 3D networks. Thus, the DFT studies support the crystal structure of HIF-4.
In the case of HIF-13, six low-energy optimized structures are produced (Fig. 9C). Isomer-1, isomer-2 (0.44 kJ mol−1), and isomer-3 (1.48 kJ mol−1) are the most stable models. Therefore, in the solution phase at room temperature, all the three structures with energy ≤2 kJ mol−1 are possible and can form dimers. Both isomer-1 (0.0 kJ mol−1) and isomer-3 (1.48 kJ mol−1) are similar to HIF-13 and are superimposable. Except that the C13 atom of isomer-1 (0.0 kJ mol−1), which occupy an opposite position in comparison with HIF-13 and isomer-3. The isomer-2 (0.44 kJ mol−1), and isomer-4 (2.07 kJ mol−1) adopt a conformation similar to HIF-4, while isomer-5 (13.43 kJ mol−1), and isomer-6 (20.49 kJ mol−1) adopt entirely different conformation. Thus, the DFT study suggests that the two different conformations of HIF-4 and HIF-13 are possible. The substitution position, dimer formation, and electrostatic energies of substituted atoms play a significant role in determining the conformation of these molecules.
3.7 FTIR spectroscopy studies of HIF-4
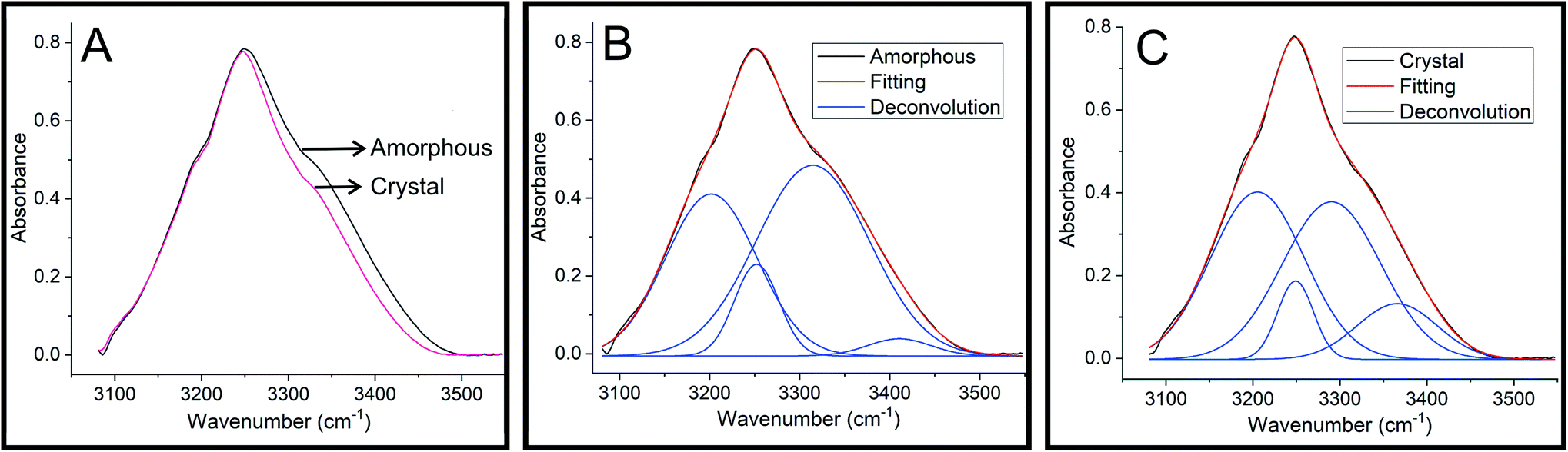
Infrared spectroscopy has been widely used to understand the influence of hydrogen bonding on O–H stretching frequencies.29,30 It has been observed that the O–H stretching frequency shows red-shift when involved in a hydrogen bond formation.30 To understand the hydrogen bonding of HIF-4, FTIR spectra were recorded on semi-crystalline/amorphous and crystal. Fig. 10A shows the overlaid spectra in the O–H stretching region. It is clear from the figure that the O–H vibrations in the crystalline HIF-4 are shifted to the lower frequency side and become slightly narrower. These O–H bands can be deconvoluted into four peaks each at 3201.7, 3252.0, 3314.5 and 3410.6 cm−1 for amorphous (Fig. 10B) and 3205.0, 3248.9, 3290.3 and 3365.7 cm−1 for crystalline HIF-4 (Fig. 10C). The results obtained by FTIR study on HIF-4 suggest that, both inter and intra molecular hydrogen bonding exist in amorphous or semi crystalline powder sample of HIF-4. Thus, there is band spread at the higher frequency edge and is contributed by the effect of intra-molecular hydrogen bonding on O–H stretching. It is assumed that the intermolecular hydrogen bonds are shorter and therefore have lower O–H stretching frequencies than intramolecular hydrogen bonds.31 The intramolecular hydrogen bonding contribution to O–H stretching has vanished when crystal is formed (Fig. 10C).
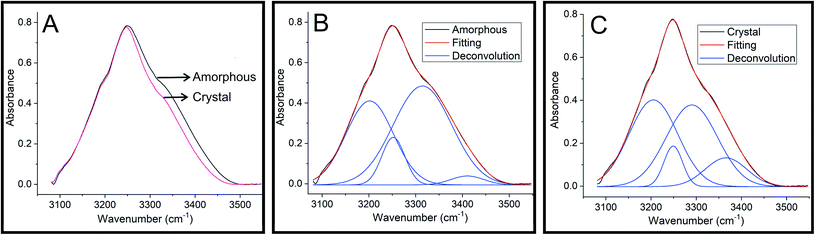 |
| | Fig. 10 FTIR spectra of HIF-4. (A) FTIR spectra of HIF-4 in the O–H stretching region. Deconvolution of the O–H stretching bands of amorphous (B) and crystal (C) of HIF-4. | |
4. Conclusions
Efforts to obtain the crystals of HIF-4 using conventional crystallization techniques are not successful. Thus, we have used the laser-induced crystallization technique to grow the microcrystals. Later those microcrystals were used as a seed to develop good quality crystals for single-crystal X-ray diffraction analysis. Therefore laser-induced crystallization technique may be explored futher to crystallize organic, inorganic, and biological molecules.13 Single crystal X-ray crystallographic studies revealed that the 3-benzylchroman-4-one derivatives tend to crystallize in a particular conformation. However, the HIF-4, a molecule bearing OH at 2′ position in phenyl ring C, adopted a different conformation. DFT, FTIR, Hirshfeld, energy frameworks, and CSD studies are also used to analyze the conformational changes of 3-benzylchroman-4-ones. All these studies complemented each other to understand the effect of substitutions at 2′, 3′; and 4′ positions in phenyl ring C, and role of hydrogen bonds in conformation of 3-benzylchroman-4-ones. In HIF-13, OMe moiety is present at the 2′ and 3′ positions, and it adopts a similar conformation as other 3-benzylchroman-4-ones. Some of the reported structures, AFAYIR, ECUFOA have OH substitution at phenyl ring C in 3′ or 4′ position, and they also adopt similar conformation like HIF-13. Whereas the CSD, energy frameworks, DFT studies revealed that OH substitution at 2′ position activates the dimer formation via strong O–H⋯O hydrogen bonds, which facilitate the conformational changes in HIF-4. Our earlier studies have reported homoisoflavanones as an effective inhibitor for cancer exclusively for tumor suppressor protein p53.9 Therefore the new reported molecules with different conformations may be an excellent candidate to be explored further. Further experimental studies may be carried out to understand the anticancer properties of deviated HIF-4 structure, which may provide insight into anticancer drug molecule development.
Conflicts of interest
Authors declare no conflict of interest.
Acknowledgements
Abdul Ajees Abdul Salam acknowledges the MAHE intramural research grant (MAHE/DREG/PhD/IMF/2019). We gratefully acknowledge support for this work from the Department of Biotechnology (BT/PR6413/MED/14/802/2005) and the Department of Science and Technology's FIST program (SR/FST/PSI-174/2012).
References
- S. N. López, M. G. Sierra, S. J. Gattuso, R. L. Furlán and S. A. Zacchino, Phytochemistry, 2006, 67, 2152–2158 CrossRef PubMed.
- A. Ata, E. M. Gale and R. Samarasekera, Phytochem. Lett., 2009, 2, 106–109 CrossRef CAS.
- K. Du Toit, E. E. Elgorashi, S. F. Malan, S. E. Drewes, J. Van Staden, N. R. Crouch and D. A. Mulholland, Bioorg. Med. Chem., 2005, 13, 2561–2568 CrossRef CAS PubMed.
- F. Fusi, A. Ferrara, C. Koorbanally, N. R. Crouch, D. A. Mulholland and G. Sgaragli, J. Pharm. Pharmacol., 2008, 60, 489–497 CrossRef CAS PubMed.
- A. Gaspar, M. J. Matos, J. Garrido, E. Uriarte and F. Borges, Chem. Rev., 2014, 114, 4960–4992 CrossRef CAS PubMed.
- S. Hur, Y. S. Lee, H. Yoo, J. H. Yang and T. Y. Kim, J. Dermatol. Sci., 2010, 59, 163–169 CrossRef CAS PubMed.
- L. Kang, H. Zhao, C. Chen, X. Zhang, M. Xu and H. Duan, Int. Immunopharmacol., 2016, 38, 246–251 CrossRef CAS PubMed.
- N. H. Dang, N. D. Chung, H. M. Tuan, N. T. Hiep and N. T. Dat, Chem. Pharm. Bull., 2017, 65, 204–207 CrossRef CAS PubMed.
- L. Simon, A. A. Abdul Salam, S. Madan Kumar, T. Shilpa, K. K. Srinivasan and K. Byrappa, Bioorg. Med. Chem. Lett., 2017, 27, 5284–5290 CrossRef CAS PubMed.
- S. Shalini, C. R. Girija, L. Simon, K. K. Srinivasan and T. V. Venkatesha, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o1011–o1012 CrossRef CAS PubMed.
- S. Shalini, C. R. Girija, L. Simon, K. K. Srinivasan, T. V. Venkatesha and M. M. Jotani, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o241 CrossRef CAS PubMed.
- L. Simon, S. Shalini, C. R. Girija, K. K. Srinivasan and T. V. Venkatesha, Eur. Sci. J., 2013, 9, 366–373 Search PubMed.
- S. Thippeshappa, S. D. George, A. Bankapur, S. Chidangil, D. Mathur and A. A. Abdul Salam, Sci. Rep., 2018, 8, 16018 CrossRef PubMed.
- T. Shilpa, S. D. George, A. Bankapur, S. Chidangil, A. K. Dharmadhikari, D. Mathur, S. Madan Kumar, K. Byrappa and A. A. Abdul Salam, Photochem. Photobiol. Sci., 2017, 16, 870–882 CrossRef CAS PubMed.
- T. Shilpa, S. G. Bhat, V. R. Rodrigues, S. George, A. K. Dharmadhikari, C. Santhosh and A. A. Ajees, Proc. Indian Natl. Sci. Acad., 2015, 81, 517–523 Search PubMed.
- CrystalClear Software User’s Guide, Molecular Structure Corporation, Rigaku Corporation, 1999, pp. 1718–1725 Search PubMed.
- G. M. Sheldrick, SHELXS-97 and SHELXL-97, Program for Crystal Structure Solution and Refinement, University of Gottingen, Gottingen, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van De Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D., Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- B. S. P. User's guide and W. L. DeLano, in Molecular Graphic System, 2004, DeLano Scientific LLC, San Carlos, California, USA Search PubMed.
- M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 CAS.
- S. H. Kim, C. M. Lee and K. Kafle, Korean J. Chem. Eng., 2013, 30, 2127–2141 CrossRef CAS.
- C. M. Lee, J. D. Kubicki, B. Fan, L. Zhong, M. C. Jarvis and S. H. Kim, J. Phys. Chem. B, 2015, 119, 15138–15149 CrossRef CAS PubMed.
- N. V. Ivanova, E. A. Korolenko, E. V. Korolik and R. G. Zhbankov, J. Appl. Spectrosc., 1989, 51, 847–851 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1834972 and 1834992. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02245h |
| ‡ Current address: Department of Chemistry and Molecular Biology, University of Gothenburg, Medicinaregatan 9C, 413 90, Göteborg, Sweden. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Shilpa T.ae,
Madan Kumar S.‡
b,
Aseefhali Bankapurc,
Rajeev K. Sinha
*a,
Shilpa T.ae,
Madan Kumar S.‡
b,
Aseefhali Bankapurc,
Rajeev K. Sinha