
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo novel oxetane containing lignans and a new megastigmane from Paronychia arabica and in silico analysis of them as prospective SARS-CoV-2 inhibitors†
Abdelsamed I. Elshamyab,
Tarik A. Mohamedc,
Mahmoud A. A. Ibrahim d,
Mohamed A. M. Atiae,
Tatsuro Yoneyamaa,
Akemi Umeyamaa and
Mohamed-Elamir F. Hegazy*cf
d,
Mohamed A. M. Atiae,
Tatsuro Yoneyamaa,
Akemi Umeyamaa and
Mohamed-Elamir F. Hegazy*cf
aFaculty of Pharmaceutical Sciences, Tokushima Bunri University, Yamashiro-cho, Tokushima, 770-8514, Japan
bChemistry of Natural Compounds Department, National Research Centre, Dokki, Giza, 12622, Egypt
cChemistry of Medicinal Plants Department, National Research Centre, 33 El-Bohouth St., Dokki, Giza, 12622, Egypt. E-mail: me.fathy@nrc.sci.eg; Fax: +20-233370931; Tel: +20-233371635
dComputational Chemistry Laboratory, Chemistry Department, Faculty of Science, Minia University, Minia, 61519, Egypt
eMolecular Genetics and Genome Mapping Laboratory, Genome Mapping Department, Agricultural Genetic Engineering Research Institute (AGERI), Agricultural Research Center (ARC), Giza, 12619, Egypt
fDepartment of Pharmaceutical Biology, Institute of Pharmaceutical and Biomedical Sciences, Johannes Gutenberg University, Staudinger Weg 5, 55128 Mainz, Germany
First published on 4th June 2021
Abstract
The chemical characterization of the extract of the aerial parts of Paronychia arabica afforded two oxetane containing lignans, paronychiarabicine A (1) and B (2), and one new megastigmane, paronychiarabicastigmane A (3), alongside a known lignan (4), eight known phenolic compounds (5–12), one known elemene sesquiterpene (13) and one steroid glycoside (14). The chemical structures of the isolated compounds were constructed based upon the HRMS, 1D, and 2D-NMR results. The absolute configurations were established via NOESY experiments as well as experimental and TDDFT-calculated electronic circular dichroism (ECD). Utilizing molecular docking, the binding scores and modes of compounds 1–3 towards the SARS-CoV-2 main protease (Mpro), papain-like protease (PLpro), and RNA-dependent RNA polymerase (RdRp) were revealed. Compound 3 exhibited a promising docking score (−9.8 kcal mol−1) against SARS-CoV-2 Mpro by forming seven hydrogen bonds inside the active site with the key amino acids. The reactome pathway enrichment analysis revealed a correlation between the inhibition of GSK3 and GSK3B genes (identified as the main targets of megastigmane treatment) and significant inhibition of SARS-CoV-1 viral replication in infected Vero E6 cells. Our results manifest a novel understanding of genes, proteins and corresponding pathways against SARS-CoV-2 infection and could facilitate the identification and characterization of novel therapeutic targets as treatments of SARS-CoV-2 infection.
Introduction
Medicinal plants and their products have been used for the prevention and/or treatment of several diseases.1–3 According to the World Health Organization (WHO), around 80% of people around the worldwide have used herbal medicinal plants, comprising about 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 plant species, as primary health care. According to WHO, around 21000 plant species have potential for being used as medicinal plants.4
000 plant species, as primary health care. According to WHO, around 21000 plant species have potential for being used as medicinal plants.4
The Paronychia genus, one of the genera of the family Caryophyllaceae, includes more than 100 plant species all around the world, especially in warm temperature regions such as Africa, the Mediterranean, North and South America and Eurasia.5 Several traditional uses have been documented for Paronychia plants in different areas all over the world such as for the treatment of prostate, bladder, and abdominal ailments, kidney stones, eczema, as a febrifuge and digestive, diabetes, heart pains, as a gastric analgesic and for ulcers, hypoglycemia, as an aperitif and as a diuretic.6–9
Extracts and isolated compounds from Paronychia plant species have been reported to exhibit significant biological activities such as cytotoxic10 and antioxidant activities.9,11,12 Numerous phytochemical constituents from plants belonging to the Paronychia genus have been documented, such as gypsogenic acid and polygalacic acid-type saponins, oleanane-type glycosides, flavonoids and tocopherols.10,11,13–16
A recent respiratory infectious disease (coronavirus disease (COVID-19)) has been attributed to the novel Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2).17 Up to this date, there are no reports concerning the chemical constituents and/or biological activities of P. arabica although the medicinal importance of these plants has been shown. Continuing our work using natural resources for the isolation and identification of bioactive metabolites,18–23 we described here the isolation and identification of two new oxetane containing lignans 1 and 2, one new megastigmane 3, along with a known lignan (4), eight known phenolic compounds (5–12), a known elemene sesquiterpene (13) and one steroid glycoside (14) (Fig. 1) from the aerial parts.
 | ||
| Fig. 1 Structures of the isolated compounds (1–14). | ||
Over the past decade, the analysis of pathways and networks has provided a deep understanding of the interactions between genetic variations and therapeutic responses to a large number of drugs in terms of their biological framework.24 Meanwhile, pathways are known as groups of biological objects connected to specific functions or targets; biological networks are generally assembled in a systems manner, comprising many pathways concurrently.25 Numerous studies have employed pathway and network-based approaches in targeted therapies to predict drug side effects, explain hazardous toxicity issues and, moreover, to disclose drug resistance mechanisms.26
To better understand the effects of targeted therapies in patients, a package of software tools can be used to visualize patient-specific variations and drug targets followed by developing pathways, which are process-oriented representations of biological reactions or biological networks that predict interactions among genes, proteins and other biological entities.24,27
To combat COVID-19, prevention of SARS-CoV-2 replication could be achieved by targeting the viral main protease (Mpro), papain-like protease (PLpro) and RNA-dependent RNA polymerase (RdRp) enzymes. Therefore, the binding modes and affinities of the isolated compounds were predicted against the three SARS-CoV-2 targets using the molecular docking technique. Furthermore, this study aims to identify targets and pathways enriched in response to megastigmane in terms of SARS-CoV-2 infection.
Results and discussion
The chemical description of the hydro-methanolic extract of P. arabica aerial parts provides fourteen metabolites, comprising two new oxetane containing lignans, paronychiarabicine A (1) and B (2), a new megastigmane, paronychiarabicastigmane A (3) along with eleven known compounds (4–14) (Fig. 1).Paronychiarabicine A (1), a white amorphous powder, exhibited a negative optical rotation in methanol [α] −5.1 (C 0.1, MeOH). The molecular formula of 1 was designated as C30H32O9 from the observed TOF-ESI-MS molecular ion peak at 559.1924 (M + Na)+ (calc.: 559.1944; C30H32O9Na) which displayed 15° of unsaturation. The FT-IR spectrum of 1 revealed the characteristic absorption bands of hydroxyl (at 3346 cm−1) and keto (at 1723 cm−1) functional groups. The 1H NMR spectrum of 1 (Table 1) showed eight aromatic protons at δH 6.42 dd (1H, J = 8.2, 2.0 Hz), 6.46 brs (1H), 6.58 d (1H, J = 1.3 Hz), 6.50 d (1H, J = 1.9 Hz), 6.65 dd (2H, J = 8.2, 2.0 Hz), 6.72 dd (1H, J = 8.2, 2.0 Hz), and 6.82 d (1H, J = 1.9 Hz). Two oxygenated aliphatic methylenes at δH 3.71 ddd (J = 7.5, 3.8, 2.0 Hz), 3.61 ddd (J = 7.5, 3.8, 2.0 Hz) and at δH 3.83 dd (J = 7.3, 1.6 Hz), 4.09 dd (J = 7.3, 1.6 Hz)] as well as one oxygenated methine at δH 5.41 d (J = 6.9 Hz) were assigned. Additionally, three methoxy groups at δH 3.64 (s, 6H) and 3.72 s were characterized. The 13C NMR of 1 (Table 1) displayed thirty carbon resonances that were categorized, depending upon HSQC and DEPT-135, into 11 quaternary, 12 methines, 4 methylenes, and 3 methyls. The quaternary carbons were characterized into a keto group at δC 180.2, six oxygenated aromatic carbons at δC 144.0, 144.9, 146.1, 146.9 and 147.7 (2 × C), and four substituted aromatic carbons at δC 128.6, 130.1, 131.3 and 133.4. The methines were also assigned to eight aromatic carbons at δC 108.9, 112.0, 113.5, 114.7, 114.8, 117.7, 118.3 and 120.9, one oxygenated carbon at δC 87.7, and three aliphatic ones at δC 41.3, 46.5 and 54.0. By the same method, four methylenes were identified at δC 34.2 and 37.5 and two oxygenated at δC 63.6 and 71.5. The three methyls present at δC 54.9 (2 × C) and 55.3 were characterised as for three methoxy groups.
| No. | Paronychiarabicine A (1) | Paronychiarabicine B (2) | ||||
|---|---|---|---|---|---|---|
| δH, mult. (J Hz) | δC | δH mult. (J Hz) | δH | |||
| a s: quaternary, d: methine, t: methylene, q: methyl. | ||||||
| 1 | — | 131.3 | s | — | 132.0 | s |
| 2 | 6.46 br s | 117.7 | d | 6.48 br s | 116.9 | d |
| 3 | — | 128.6 | s | — | 128.8 | s |
| 4 | — | 146.9 | s | — | 146.7 | s |
| 5 | — | 147.7 | s | — | 147.7 | s |
| 6 | 6.58 d (1.3) | 113.5 | d | 6.65 d (2.0) | 112.9 | d |
| 7a | 2.83 dd (14.0, 5.5) | 34.2 | t | 2.76 dd (14.1, 5.4) | 34.0 | t |
| 7b | 2.74 dd (14.0, 5.5) | 2.59 dd (14.1, 5.4) | ||||
| 8 | 2.59 m | 46.5 | d | 2.58 m | 46.4 | d |
| 9 | — | 180.2 | s | — | 180.3 | s |
| 10a | 3.83 dd (7.3, 1.6) | 71.5 | t | 3.84 t (16.9) | 71.5 | t |
| 10b | 4.09 dd (7.3, 1.6) | 4.11 dd (7.6, 1.4) | ||||
| 11 | 2.42 m | 41.3 | d | 2.43 m | 41.1 | d |
| 12 | 2.41 m | 37.5 | t | 2.49 d (1.6), 2.42 d (1.6) | 37.7 | t |
| 13 | — | 130.1 | s | — | 129.9 | s |
| 14 | 6.50 d (1.9) | 112.0 | d | 6.46 d (1.9) | 112.6 | d |
| 15 | — | 144.0 | s | — | 145.0 | s |
| 16 | — | 144.9 | s | — | 144.0 | s |
| 17 | 6.65 dd (8.2, 2.0) | 114.7 | d | 6.66 dd (8.1, 1.9) | 114.7 | d |
| 18 | 6.42 dd (8.0, 2.0) | 120.9 | d | 6.58 d (8.0) | 121.7 | d |
| 1′ | — | 133.4 | s | — | 133.2 | s |
| 2′ | 6.82 d (1.9) | 108.9 | d | 6.85 d (2.0) | 109.1 | d |
| 3′ | — | 147.7 | s | — | 147.6 | s |
| 4′ | — | 146.1 | s | — | 146.2 | s |
| 5′ | 6.65 dd (8.2, 2.0) | 114.8 | d | 6.66 dd (8.1, 1.9) | 114.7 | d |
| 6′ | 6.72 dd (8.2, 2.0) | 118.3 | d | 6.73 dd (8.3, 1.9) | 118.3 | d |
| 7′ | 5.41 d (6.9) | 87.7 | d | 5.39 d (6.4) | 87.7 | d |
| 8′ | 3.34 dd (7.5, 11.4) | 54.0 | d | 3.35 m | 53.9 | d |
| 9′a | 3.71 ddd (7.5, 3.8, 2.0) | 63.6 | t | 3.69 ddd (8.0, 3.9, 2.6) | 63.4 | t |
| 9′b | 3.61 ddd (7.5, 3.8, 2.0) | 3.64 ddd (7.0, 4.3, 1.3) | ||||
| 5-OMe | 3.64 s | 54.9 | q | 3.67 s | 55.0 | q |
| 15-OMe | — | — | 3.71 s | 55.3 | q | |
| 16-OMe | 3.72 s | 55.3 | q | — | — | |
| 3′-OMe | 3.64 s | 54.9 | q | 3.70 s | 55.0 | q |
The analysis of the 1D NMR of 1 (Table 1) as well as the 2D NMR (Fig. 2) displayed three aromatic rings: (i) a tetra-substituted ring (ring A), (ii) a tri-substituted ring (ring B) and (iii) a tri-substituted ring (ring C). The HMBC correlations of H-1 at δH 6.46 br s/C-6 (δC 113.5; J3), H-6 at δH 6.46 br s/C-3 (δC 146.9; J3) and H-6/C-5 (δC 147.7; J2) as well as H-2/C-3 (δC 128.6; quaternary carbon; J2) and H-2/C-1 (δC 131.3; quaternary carbon; J2) established the tetra-substituted ring (ring A). Additionally, the observed strong J3-HMBC correlation of the methyl proton at δH 3.64 s and the oxygenated carbon C-5 at δC 147.7 confirmed that C-5 was methoxylated. Also, the tri-substituted ring (ring B) was constructed depending upon the HMBC correlations of H-14 at 6.50 d (J = 1.9 Hz)/C-16 (δC 144.9; J3), H-14/C-18 (δC 120.9; J3), H-18 at δH 6.42 dd (J = 8.2, 2.0 Hz)/C-16, (H-18 δC 120.9) and H-17 at δH 6.65 dd (2H, J = 8.2, 2.0 Hz)/C-15 (δC 144.0; J3) alongside H-17/C-13 (δC 130.1; quaternary carbon; J3). The methoxylation of C-16 was deduced by the strong J3-HMBC correlation of the methyl proton at δH 3.72 s/C-16.
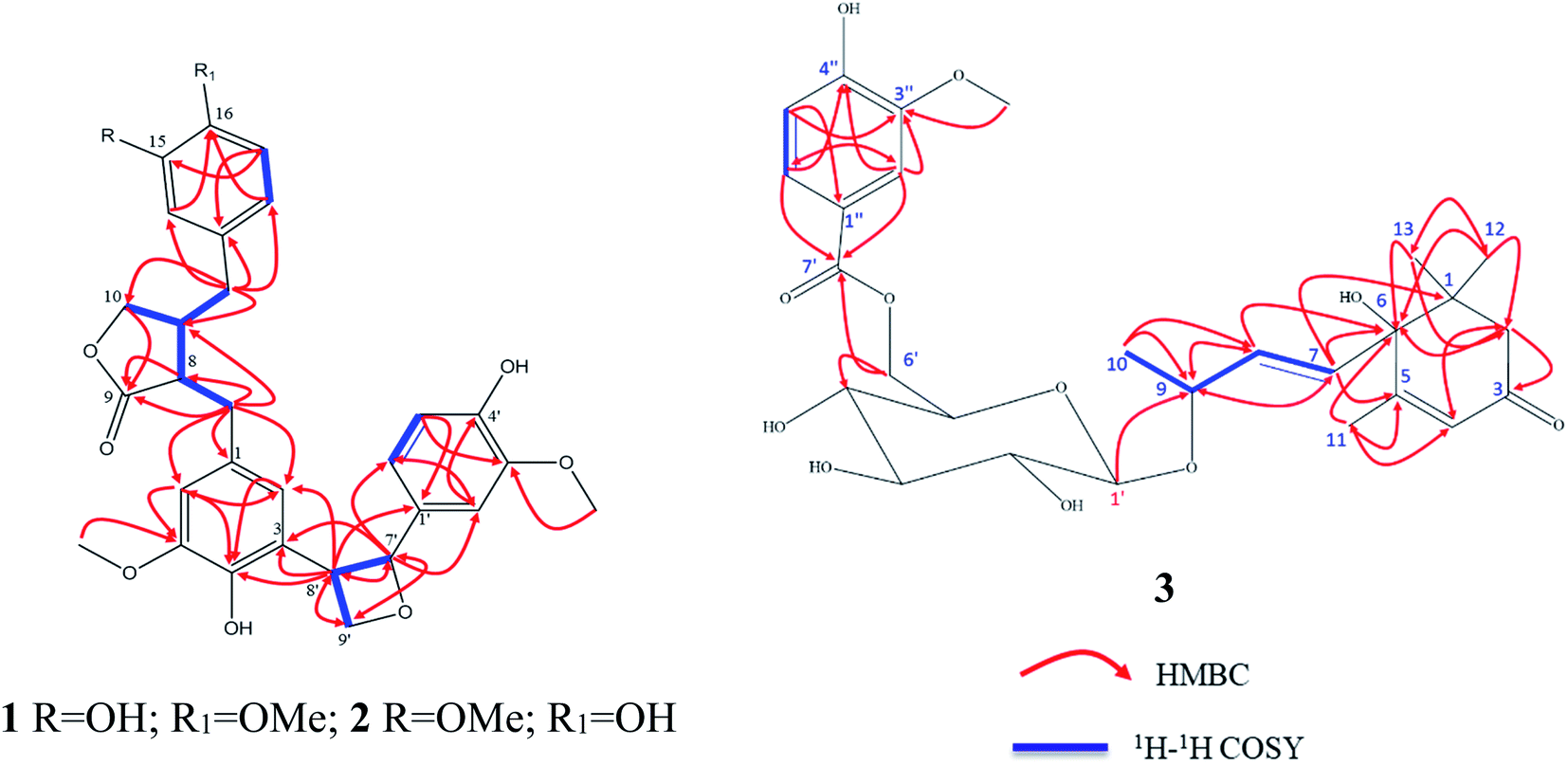 | ||
| Fig. 2 1H–1H COSY and HMBC of 1–3. | ||
The 1H–1H COSY correlations of H-8 at δH 2.59 m/H-11 at δH 2.42 m, H-11/H-10 at δH [3.83 dd (J = 7.3, 1.6 Hz); 4.09 dd (J = 7.3, 1.6 Hz); oxygenated methylene] along with the strong HMBC correlations of H-8/C-9 (1δC 80.2; carbonyl carbon, J2) and H-10/C-9 (J3) were used to deduce the disubstituted dihydrofuranone ring (ring D). Also, the 1H–1H COSY correlations of H-7′ at δH 5.41 d (J = 6.9 Hz)/H-8′ at δH 3.34 t (J = 16.8 Hz) and H–H-8′/H-9′ at δH [3.71 ddd (J = 7.5, 3.8, 2.0, Hz); 3.61 ddd (7.5, 3.8, 2.0)], supported with the HMBC correlations of H-7′/C-8′ (δC 54.0; J2), H-9′/C-7′ (oxygenated methane; δC 87.7; J3) and H-8′/C-9′ (oxygenated methylene; δC 63.6; J2) confirmed the presence of the oxetane ring as ring E.
The 1H–1H COSY correlations of H-11/H-12 at δH 2.41 m (aliphatic methylene) as well as the HMBC correlation of H-12/C-11 (δC 41.3; J2), H-12/C-10 (δC 71.5; J3), H-12/C-13 (δC 130.1; J2), H-12/C-14 (δC 112.0; J3) and H-12/C-18 (δC 120.9; J3) construct the linkage of aromatic ring B and the disubstituted dihydrofuranone ring (ring D) via the methylene carbon C-12.
The 2D NMR spectrum of 1 exhibited clear HMBC correlations of H-7′ (δH 5.41 d, J = 6.9 Hz)/C-1′ (δC 133.4; J2), H-8′ (δH 3.34 m)/C-1′ (J3), H-7′/C-2′ (δC 108.9; J3) and H-7′/C-6′ (δC 118.3; J3). At the same time, HMBC correlations of H-7′/C-3 (δC 128.6; J3), H-8′/C-3 (J2), H-8′/C-2 (δC 117.7; J3) and H-8′/C-4 (δC 146.9; J3) were assigned. These HMBC correlations construct the 3→8′ attachment of ring A by the oxetane moiety (ring E) as well as the 1′→7′ of ring C by the oxetane moiety (ring E).
The absolute configuration of 1 was confirmed from the J coupling constants as well as the NOESY experiments (Fig. 3). Numerous reports have described how the low J coupling constants between both H-7′ and H-8′ (6.9–7.6 Hz) can be used to deduce the trans orientation of diphenyloxetanes.28–30 In 1, the1H NMR exhibited low coupling constant values of 7.1 Hz, confirming the trans orientation of H-7′ and H-8′. The 13C NMR chemical shifts of C-7′ and C-8′ at δC 87.7 and 54.0, respectively, were totally in agreement with a previously published new oxetane containing neolignan that deduced the same configurations.30,31 Also, the NOESY correlations of H-9′b [δH 3.61 ddd (J = 7.5, 3.8, 2.0 Hz)]/H-7′, H-9′a [δH 3.71 ddd (J = 7.5, 3.8, 2.0 Hz)]/H-8′ and H-8′/H-2 [δH 6.46 br s] confirmed the trans orientation of H-7′ and H-8′. Additionally, NOESY correlations of H-2/H-7 [δH 2.83 dd (J = 14.0, 5.5 Hz)], H-7/H-8 [δH 2.59 m], H-8/H-10 [δH 3.83 dd (J = 7.3, 1.6 Hz)], H-7/H-11 [δH 2.42 m] and H-11/H-12 [δH 2.41 m] were assigned.
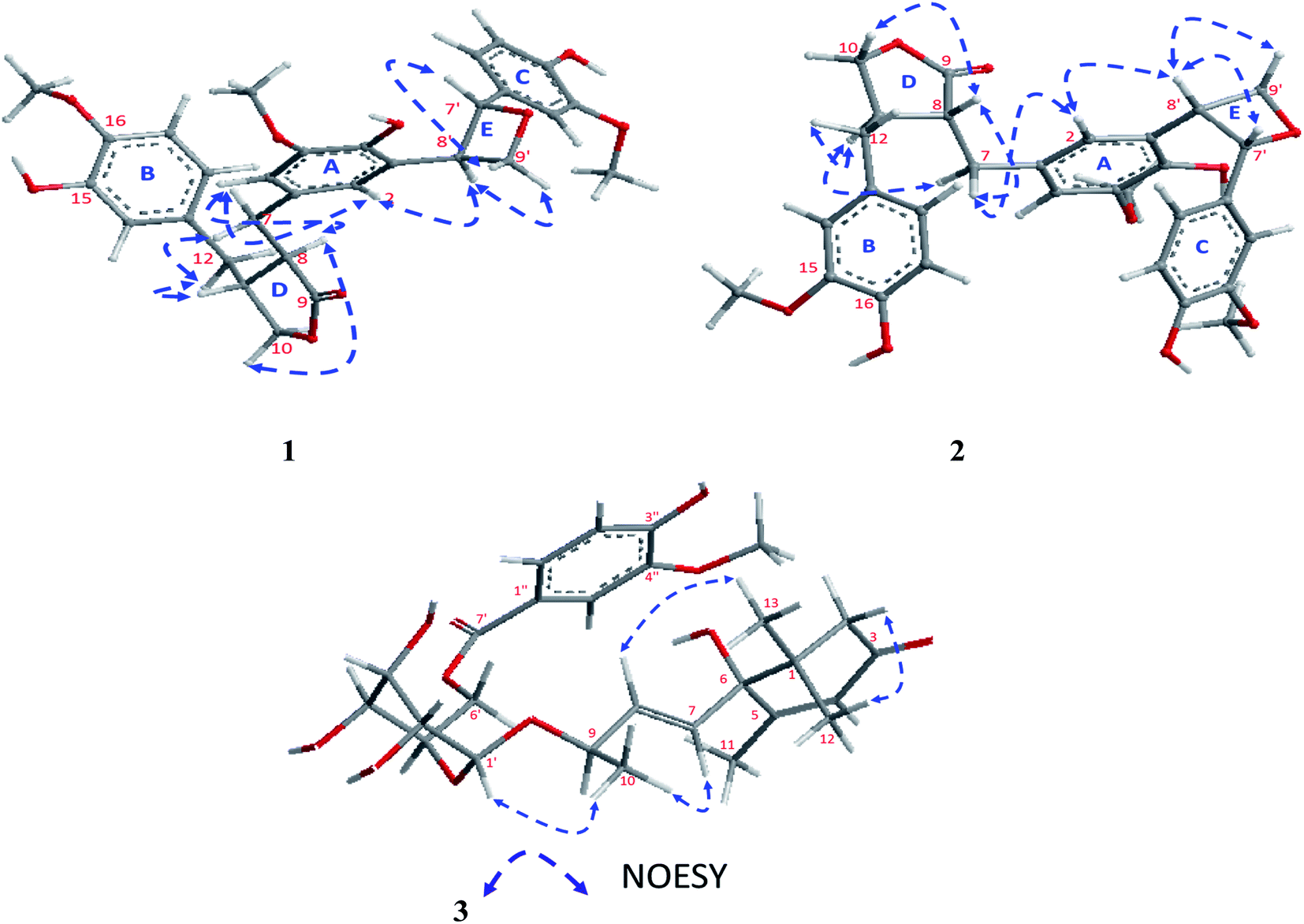 | ||
| Fig. 3 Key NOESY results of 1–3. | ||
To elucidate the absolute configuration of 1, a Boltzmann-weighted TDDFT-ECD spectrum was generated and compared to the experimental one (Fig. 4). The TDDFT-simulated ECD spectrum of 7′R,8′R was in a good agreement with the corresponding experimental ECD spectrum of 1 (Fig. 4). From all of the above-described spectroscopic data, 1 was shown to be a novel oxetane containing lignan, paronychiarabicine A.
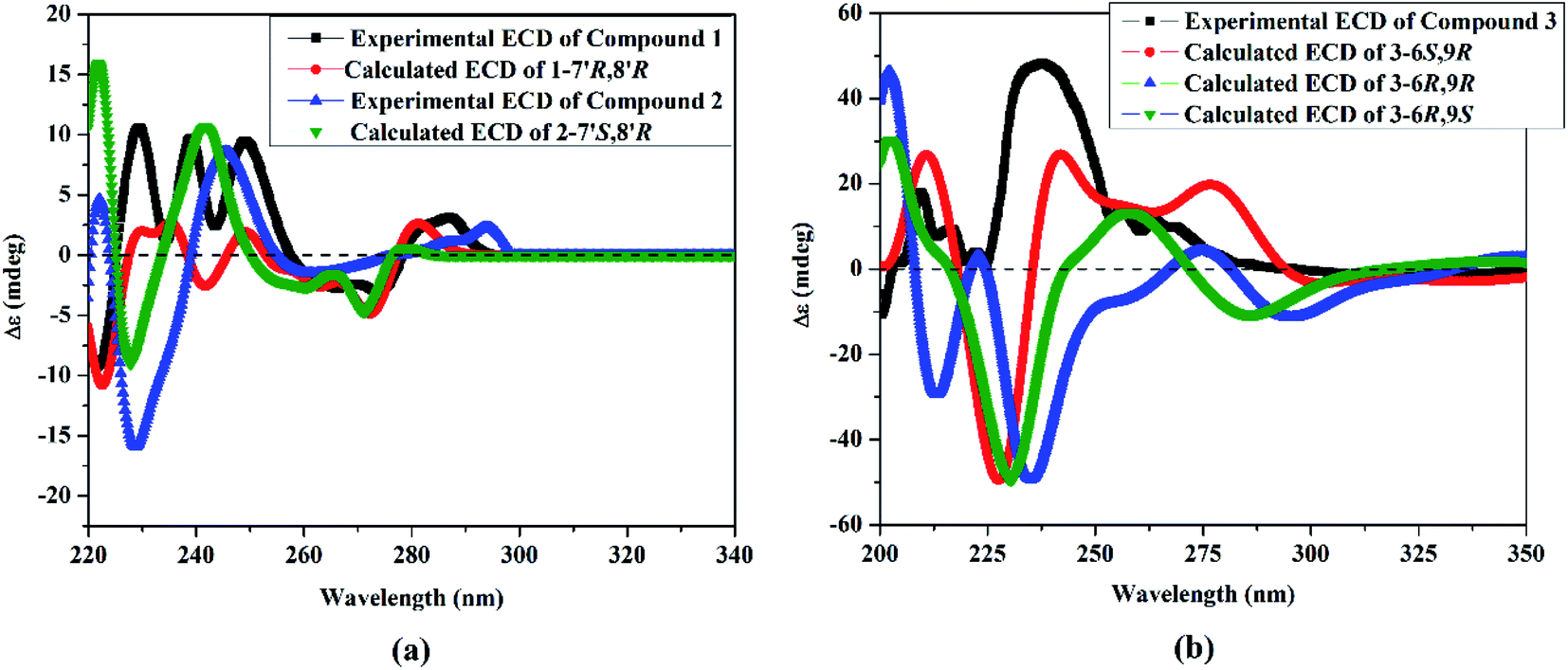 | ||
| Fig. 4 (a) Experimental electronic circular dichroism (ECD) in methanol of 1 (in black) and 2 (in blue), compared with the TDDFT-simulated ECD spectra of 1-7′R,8′R (in red) and 2-7′S,8′R (in green). (b) Experimental ECD in methanol of compound 3 (in black), compared with the TDDFT-simulated ECD spectra of 3-6S,9R, 3-6R,9R and 3-6R,9S. | ||
Paronychiarabicine B (2), a yellow amorphous powder, showed a negative optical rotation in methanol [α] + 6.6 (C 0.1, MeOH). The molecular formula of 2 was established as C30H32O9 from the observed HR-CIMS molecular ion peak at 536.2030 (M)+ (calc.: 536.2046; C30H32O9), which revealed 15° of unsaturation. The FT-IR spectrum of 2 revealed the characteristic absorption bands of hydroxyl (at 3346 cm−1) and keto (at 1723 cm−1) functional groups. Carefully analysis of the 1H and 13C NMR of 2 (Table 1) allowed us to deduce that this compound has same skeleton of 1 except for (i) the down field shift of C-15 by 1 ppm at δC 145, (ii) the up field shift of C-16 by 0.9 ppm at δC 144.0, and (iii) the clear down field shift of H-18 by 0.16 ppm at δH 6.58 d (J = 8.0 Hz) as well as C-18 by 0.6 ppm at δC 121.7. Through the careful analysis of the 2D NMR, the methoxy and hydroxy groups were localized at C-15 and C-16, instead of the opposite in 1, via the HMBC correlations of H-18/C-16 (J3), H-17 [δH 6.66 dd (2H, J = 8.1, 1.9 Hz)]/C-15 (J3) and OMe (δH 3.71 s)/C-15 (J3) (Fig. 2).
The stereochemistry of 2 was established using the coupling constants and NOESY correlations. Regarding to the reports of Saphier et al., 2005, the cis and trans isomers of diphenyloxetanes have different Rf values over a plate of silica gel eluted using n-hexane–EtOAc in which the cis isomer has a lower Rf than that of the trans isomer.29 Also, Saphier et al., 2005, the cis reported that the appearance of H-7′ with a lower coupling constant (<6.9 Hz), as well as the NOE effect of H-8′ due to proximity, allowed the β orientation of H-7′ and H-8′ to be deduced.29 For 2, the Rf (0.62) was found to be lower than that of 1 (0.67) and the coupling constant of H-7′ was assigned at 6.4 Hz (<6.9 Hz), which is in full agreement with the above mentioned reports.28,29 The NOESY correlations of 2 exhibited the same correlations of 1 except for some different correlations that deduced the β orientation of H-7′ and H-8′, such as (i) the absence of correlation of H-9′b [δH 3.64 ddd (J = 7.0, 4.3, 1.3 Hz)]/H-7′ [5.39 d (J = 6.4 Hz)], (ii) the presence of the correlations of H-9′a [δH 3.69 ddd (J = 8.0, 3.9, 2.6 Hz)]/H-8′ (δH 3.35 m), and H-8′/H-2 [δH 6.48 br s] and (iii) the assignment of the correlation of H-8′/H-7′ (Fig. 3). Using the same method, the absolute configuration of 2 was confirmed based upon comparing the Boltzmann-weighted TDDFT-ECD spectra with the experimental one (Fig. 4). The TDDFT-simulated ECD spectrum of the 7′S,8′R isomer was in good agreement with the experimental ECD of 2 (Fig. 4). From all of the mentioned data, 2 was established as a novel oxetane containing lignin, namely paronychiarabicine B.
Paronychiarabicastigmane A (3), a white amorphous powder, exhibited a negative optical orientation ([α] −6.6 (C 0.1, MeOH)). The molecular formula of 3 was determined as C27H36O11 from the two positive mode LR-FAB-MS molecular ion peaks at the m/z 537 (M + 1)+ and 559 (M + Na)+, deduced from the HR-FAB-MS at m/z 559.2157 (M + 1)+, (calc.: 559.2155; C27H36O11Na), and it exhibited 10 degrees of unsaturation. The FT-IR exhibited characteristic absorption bands of hydroxyl and keto groups at 3343 cm−1 and 1725 cm−1, respectively. The 1H NMR of 3 (Table 2) revealed three protons of the 1,3,4-trisubtituted aromatic ring [at δH 6.84 d (J = 8.1 Hz), 7.55 d (J = 8.3 Hz) and 7.58 br s], one anomeric proton of the hexoside moiety [at δH 4.36 d (J = 7.8 Hz)] as well as the hexoside protons [at δH 3.18 m, 3.25 m, 3.28 m, 3.35 m, 3.67 m and 3.87 d (J = 1.8 Hz)], three aliphatic olefinic methine protons [at δH 5.87 d (2H, J = 6.6 Hz) and 5.88 d (J = 2.0 Hz)], one aliphatic methylene proton [at δH 2.16 d (J = 16.8 Hz) and 2.53 d (J = 20.0 Hz)], four methyl protons [at δH 1.05 (s, 6H), 1.31 d (J = 6.4 Hz) and 1.93 d (J = 1.2 Hz)] and one methyl of the methoxy group at δH 3.91 s.
| No. | Paronychiarabicastigmane A (1) | ||||||
|---|---|---|---|---|---|---|---|
| δH, mult. (J Hz) | δC | δH, mult. (J Hz) | δC | ||||
| a s: quaternary, d: methine, t: methylene, q: methyl. | |||||||
| 1 | — | 41.0 | s | 1′′ | — | 122.7 | s |
| 2a | 2.16 d (16.8) | 49.3 | t | 2′′ | 7.58 br s | 112.4 | d |
| 2b | 2.53 d (20.0) | 3′′ | — | 147.2 | s | ||
| 3 | — | 199.8 | s | 4′′ | — | 150.9 | s |
| 4 | 5.88 d (2.0) | 125.8 | d | 5′′ | 6.84 d (8.1) | 114.3 | d |
| 5 | — | 165.9 | s | 6′′ | 7.55 d (8.3) | 123.8 | d |
| 6 | — | 78.6 | s | 3′′-OMe | 3.91 s | 55.0 | q |
| 7 | 5.87 d (6.6) | 130.1 | d | ||||
| 8 | 5.87 d (6.6) | 133.9 | d | ||||
| 9 | 4.43 m | 75.9 | d | ||||
| 10 | 1.31 d (6.4) | 19.8 | q | ||||
| 11 | 1.93 d (1.2) | 18.2 | q | ||||
| 12 | 1.05 s | 22.0 | q | ||||
| 13 | 1.05 s | 22.3 | q | ||||
| 1′ | 4.36 d (7.8) | 101.3 | d | ||||
| 2′ | 3.18 m | 73.8 | d | ||||
| 3′ | 3.25 m | 76.6 | d | ||||
| 4′ | 3.28 m | 70.2 | d | ||||
| 5′ | 3.35 m | 76.7 | d | ||||
| 6′ | 3.67 m | ||||||
| 3.87 d (1.8) | 61.4 | t | |||||
| 7′ | — | 167.0 | s | ||||
Twenty-seven carbon resonances were characterized in the 13C NMR of 3 (Table 2). The complete examination of the 13C-NMR and 1H-NMR revealed the β-glucopyranoside moiety by the six typical substantiated carbons. With more details, these carbons were classified, depending upon the DEPT-135 and HSQC experiments, into 8 quaternary carbons [an α,β-unsaturated keto carbon (at δC 199.8) and an ester keto carbon (at δC 167.0), three substituted aromatic carbons (at δC 122.7, 147.2, 150.9), one aliphatic olefinic carbon (at δC 165.9), one oxygenated carbon (at δC 78.6) and one aliphatic (at δC 41.0)], one anomeric carbon (at δC 101.3) as well as 5 methines of the sugar moiety (at δC 61.4, 70.2, 73.8, 76.6, 76.7), three aliphatic olefinic carbons (at δC 125.8, 130.1, 133.9), three aromatic methines (at δC 112.4, 114.3, 123.8), one oxygenated methine (at δC 75.9), one aliphatic methylene (at δC 49.3), four methyls (at δC 18.2, 19.8, 22.0, 22.3) and one methyl of the methoxy group (at δC 55.0)].
From the careful assignment of the 1D NMR, the structure of 3 was established as a megastigmane skeleton.33 This structure was deduced via 2D NMR using 1H 1H COSY and HMBC (Fig. 2). In HMBC, the correlations of H3-12 [δH 1.05 s]/CH3-13 [δC 22.3, J3], H3-13 [δH 1.05 s]/CH3-12 [δC 22.0, J3], H3-12/CH2-2 [δC 49.3, J3], H3-12/C-6 [δC 78.6, J3], H2-2 [δH 2.16 d (J = 16.8 Hz)] and 2.53 d (J = 20.0 Hz)]/C-6 [J3], H2-2/C-3 [δC 199.8, J2], H2-2/C-4 [δC 125.8, J2], H3-11 [δH 1.93 d (J = 1.2 Hz)]/C-5 [δC 165.9, J2], H3-11/C-4 [J3] and H3-11/C-6 [J3] were used to deduce the megastigmane skeleton. Consequently, the 1H 1H COSY correlations of H-7 [δH 5.87 d (J = 6.6 Hz)]/H-8 [δH 5.87 d (J = 6.6 Hz)], H-8/H-9 [δH 4.43 m], H-9/H3-10 [δH 1.31 d (J = 6.4 Hz)] and the HMBC correlations of H-7/C-6 [δC 78.6, J2], H-7/C-1 [δC 41.0, J3], H-7/C-1 [δC 41.0, J3], H-7/C-5 [J3], H3-10/C-8 [δC 75.9, J2], H3-10/C-8 [δC 133.9, J3] and H-1′ [(δH 4.36 d (J = 7.8 Hz)]/C-9 [δC 75.9, J3] as well were used to elucidate that megastigmane attached to the glucose moiety via C-9. The 1H 1H COSY correlations of H-5′′ [δH 6.84 d (J = 8.1 Hz)]/H-6′′ [δH 7.55 d (J = 8.3 Hz)] together with the HMBC correlations of H-5′′/C-4′′ [δC 150.9, J2], H-5′′/C-3′′ [δC 147.2, J3], H-2′′ [δH 6.84 d (J = 8.1 Hz)]/C-3′′ [J2], H-2′′/C-4′′ [J3], OMe [δH 3.91 s]/C-3′′ [δC 147.2, J3], H-2′′/C-1′′ [δC 122.7, J2], H-2′′/C-6′′ [δC 123.8, J3], H-6′′/C-1′′ [J3], H-2′′/C-7′′ [δC 122.7, ester keto, J3] and H-6′′/C-7′′ [J3] assigned the phenolic moiety as a 3′′-methoxy-4′′-hydroxy-benzen-1′′-carboxylic acid ester. Continuing, the up field shift of the sugar carbon, C-6′ at δC 61.4 as well as the HMBC correlations of H-6′/C-7′ [δC 167.0, J3], H-6′′/C-7′ [J3] and H-2′′/C-7′ [J3] were used to deduce that the attachment of the sugar with the phenolic moiety was via the ester linkage between the sugar carbon H-6′ and the carboxylic acid carbon C-7′. The sugar moiety was determined to be D-glucose by total acid hydrolysis and examinations by TLC with different standard hexosides. The β orientation of the anomeric proton H-1′ was firstly determined by the value of the coupling constant (d, J = 7.8 Hz).32,33
From the NOESY experiment, H-1′-β exhibited a strong correlation with Me-10 [δH 1.31 d (J = 6.4 Hz) which allowed the α orientation of H-9 to be deduced. Additionally, NOESY correlations between H-8 [δH 5.87 d (J = 6.6 Hz)]/Me-13 [δH 1.05 s] and H-2b [δH 2.53 d (J = 20.0 Hz)]/Me-12 [δH 1.05 s] were detected. The R-orientation of C-9 was determined based upon the 13C NMR chemical shift at δC 75.9 (Matsuda et al., 199634 and Wang et al., 201135). To confirm the absolute configuration of 3, the TDDFT-simulated ECD spectra for 3-6S,9R, 3-6R,9R and 3-6R,9S were generated and compared to the experimental spectrum (Fig. 4). The observed positive Cotton effect at 238 nm in the ECD of 3 demonstrated the S-configuration of C-634 as well as the TDDFT-calculated ECD of 3-6S,9R being in a good agreement with the experimental spectrum (Fig. 4). From all of these data, 3 was identified as Paronychiarabicastigmane A.
In addition to compounds 1–3, eleven metabolites were isolated and identified as matairesinol 4′-O-glucoside (4),36 leonuriside A (5),37 arbutine (6),38 isotachioside (7),39 syringin (8),40 dihydrosyringin (9),41 benzyl-O-β-D-glucopyranoside (10),42 benzene-1,2,3,4-tetraol (11),43 2,3-dimethoxyphenol (12),44 8α-(2′-hydroxymethyl-2′-butenoyloxy) derivative of dehydromelitensin (13)45 and daucosterin (14).46
Megastigmane (3) protein targets associated with SARS diseases were predicted using the SwissTargetPrediction-DisGeNET online tools. One hundred and seventeen genes were identified with genes classified using Venn diagram comparison. Based on the PPI network profiles, a STRING web-tool and Cytoscape 3.8.0 for visualization were used to explore the predicted gene targets of megastigmane (3) (Fig. 5). The top 20 genes responding to megastigmane (3) included HRAS, MAPK1, MMP9 and MTOR.
 | ||
| Fig. 5 STRING protein–protein interaction (PPI) network for the top 20 targets for megastigmane (3) as a potent SARS inhibitor. | ||
For further investigation of the megastigmane target–function interactions, pathway enrichment analysis and Boolean Network modeling were carried out. The reactome hierarchy map of megastigmane (3) identified disease pathways which were influenced by the top 20 gene targets responding to megastigmane (3) in terms of SARS-CoV-2 infection (Fig. 6). Out of the top ten biological pathways resulting from the reactome pathway enrichment analysis, four major biological pathways for megastigmane (3) of cytokine signaling in the immune system, signaling by receptor tyrosine kinases, signaling by interleukins and axon guidance with a high significance (FDR <0.00001%) were identified (Table 3).
 | ||
| Fig. 6 (A) The reactome map illustration of disease pathways influenced by the top 20 gene targets responding to megastigmane (3) in terms of SARS-CoV-2 infection. The colours denote over-representation of that pathway in the input dataset. Light grey signifies pathways which are not significantly over-represented. | ||
| Pathway | Pathway size | Number of targets in the pathway | P-value | FDR | Targets in pathway (hit genes) |
|---|---|---|---|---|---|
| Cytokine signaling in the immune system | 780 | 12 | 7.19 × 10−9 | 6.87 × 10−7 | MAP2K1, MMP1, MMP3, PIK3CB, MAPK14, MMP9, MAPK8, PIK3CA, MAPK1, RAF1, HRAS, MCL1 |
| Signaling by receptor tyrosine kinases | 421 | 10 | 4.22 × 10−9 | 6.87 × 10−7 | MAP2K1, PLG, PRKCA, PIK3CB, MAPK14, MMP9, MTOR, PIK3CA, MAPK1, HRAS |
| Signaling by interleukins | 435 | 10 | 5.78 × 10−9 | 6.87 × 10−7 | MAP2K1, MMP1, MMP3, PIK3CB, MAPK14, MMP9, MAPK8, PIK3CA, MAPK1, MCL1 |
| Axon guidance | 492 | 10 | 1.87 × 10−8 | 1.20 × 10−6 | GSK3B, MAP2K1, PRKCA, PIK3CB, MAPK14, MMP9, MAPK8, PIK3CA, MAPK1, HRAS |
| Signaling by NTRK1 (TRKA) | 73 | 6 | 8.48 × 10−9 | 6.87 × 10−7 | MAP2K1, PIK3CB, MAPK14, PIK3CA, MAPK1, HRAS |
| VEGFA–VEGFR2 pathway | 86 | 6 | 2.23 × 10−8 | 1.20 × 10−6 | PRKCA, PIK3CB, MAPK14, MTOR, PIK3CA, HRAS |
| Signaling by NTRKs | 92 | 6 | 3.31 × 10−8 | 1.50 × 10−6 | MAP2K1, PIK3CB, MAPK14, PIK3CA, MAPK1, HRAS |
| Signaling by VEGF | 94 | 6 | 3.76 × 10−8 | 1.50 × 10−6 | PRKCA, PIK3CB, MAPK14, MTOR, PIK3CA, HRAS |
| MAP2K and MAPK activation | 18 | 4 | 6.85 × 10−8 | 2.19 × 10−6 | MAP2K1, MAPK1, RAF1, HRAS |
| Gastrin–CREB signalling pathway via PKC and MAPK | 18 | 4 | 6.85 × 10−8 | 2.19 × 10−6 | MMP3, PRKCA, MAPK1, HRAS |
Investigating the reactome pathway enrichment analysis results highlighted a set of two genes (GSK3 and GSK3B) that represent significant biological targets in the action of megastigmane (3) as a potent SARS-CoV-2 inhibitor. Additionally, the reactome pathway enrichment analysis for SARS-CoV-2 revealed that GSK3B gene interacts with other genes/interactors including AXIN1, CTNNB1, FRAT1, AKT1, APC, GSKIP, PRKACA, MAPT, GYS1 and IPP2 (Fig. 7).
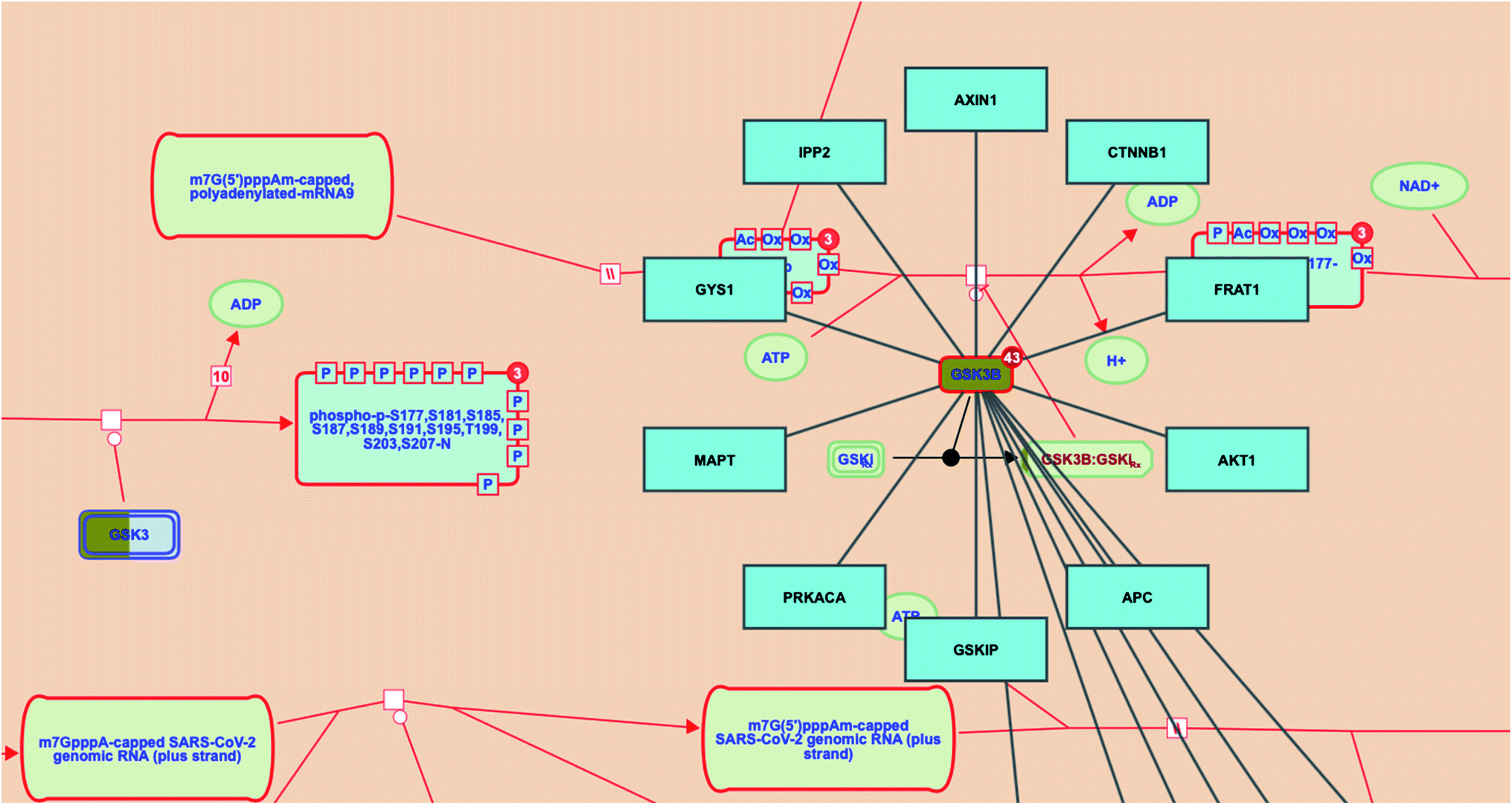 | ||
| Fig. 7 SARS-CoV-2 infection pathway showing the targets (GSK3B; highlighted in dark yellow) in response to megastigmane (3) as a potent SARS-CoV-2 inhibitor. The interactor genes were highlighted in blue. | ||
The SARS-CoV-2 infection pathway
Many additional host factors are involved in the SARS-CoV-2 genome transcription/replication. For instance, the coronavirus N protein plays an important role as a RNA chaperone which enables template switching.47 Importantly, the N protein of SARS-CoV-1 is phosphorylated by the host glycogen synthase kinase 3 (GSK3), and its inhibition was found to significantly inhibit viral replication.48Prospective SARS-CoV-2 inhibitors
To combat COVID-19, the potentials of the isolated compounds 1–3 as SARS-CoV-2 Mpro, PLproand RdRp inhibitors were predicted. The investigated compounds were docked into the active sites of the SARS-CoV-2 targets using AutoDock4.2.6 software.49 The predicted docking scores and the 2D representations of the binding modes of compounds 1–3 inside the active site of SARS-CoV-2 targets are depicted in Fig. 8.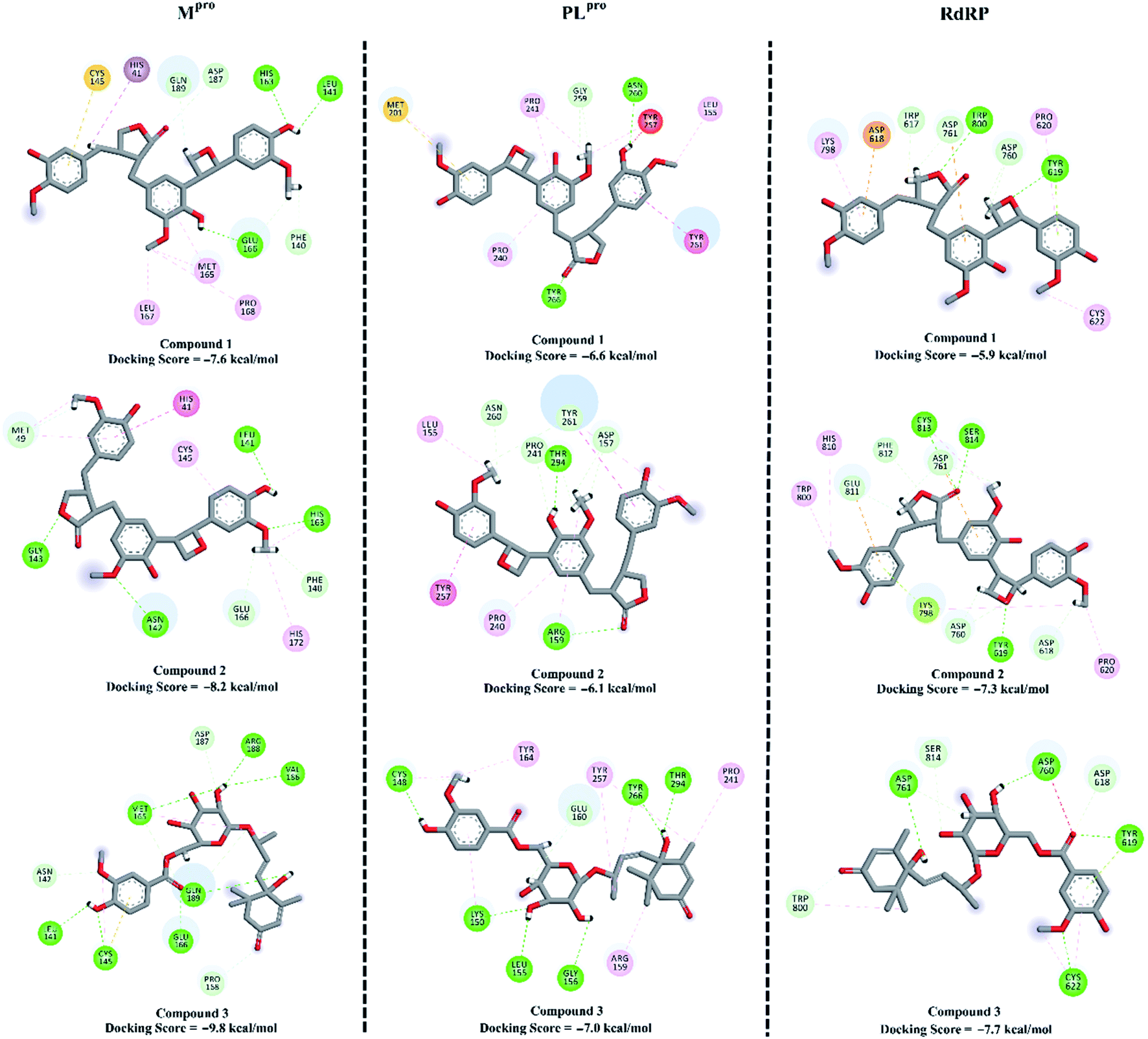 | ||
| Fig. 8 Predicted docking score and binding mode of compounds 1–3 inside the active site of SARS-CoV-2 main protease (Mpro), papain-like protease (PLpro) and RNA-dependent RNA polymerase (RdRp). Interactions: conventional hydrogen bond (green), carbon–hydrogen bond (pale green), pi–sigma and pi–pi (violet), pi–sulfur (yellow), alkyl and pi–alkyl (pale violet), unfavorable donor–donor (red), pi–lone-pair (lemon yellow). | ||
According to the molecular docking calculations, compounds 1–3 demonstrated better binding affinities towards SARS-CoV-2 Mpro compared to those with PLpro and RdRp. Comparing the molecular docking results towards Mpro revealed that compound 3 exhibited an outstanding binding affinity towards Mpro with a docking score of −9.8 kcal mol−1, forming seven hydrogen bonds with the key amino acids inside the active site (Fig. 8). Compounds 1 and 2 showed moderate binding affinities with docking scores of −7.6 and −8.2 kcal mol−1 with Mpro, forming three and four hydrogen bonds, respectively.
Experimental section
General experimental procedures
Optical rotations were measured on a JASCO P-2300 polarimeter (Tokyo, Japan).1H (500 MHz), and 13C NMR (125 MHz) spectra were recorded on a Bruker 500 NMR spectrometer (USA). The chemical shifts were given in δ (ppm), and coupling constants were reported in Hz. HR-MS spectra were obtained on a JEOL JMS-700 instrument (Tokyo, Japan). Column chromatography (CC) was carried out on polyamide 6L and Sephadex LH 20. Precoated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm, Merck, Darmstadt, Germany) and precoated RP-18 F254S plates (Merck, Darmstadt, Germany) were used for TLC analysis. A semi-preparative reversed-phase column (Supelco C18 column 250 × 10 mm, 5 μm) was used for HPLC.Plant material
Aerial parts of P. arabica (L.) DC., 1813 were collected from the Mediterranean coastal belt at Gamsa City, Al-Dakahlia Governorate, Egypt (31°26′35.4′′N 31°33′35.5′′E), during the flowering stage in April–May 2016. The Associate Professor of Taxonomy Dr Ahmed M. Abdel Gawad, Mansoura University, Egypt, collected and authenticated the plant material with a voucher specimen (PARA-016-978).Extraction and purification
Following our previous protocol,50 a black gum (75.6 g) of P. arabica dry material (850 g) was further chromatographed using silica gel column chromatography eluted with a mixture of CHCl3/MeOH step gradient. Eight major fractions (PA 1-8) were afforded after the final collection, according to the TLC profile.The elution of fraction PA-4 (1.18 g) over silica gel CC afforded 4 (68.7 mg) and 14 (11.3 mg) along with the subfraction PA-4A-B. The subfraction PA-4B (106.5 mg) was chromatographed on Sephadex LH-20 eluted with CHCl3/MeOH (1:1) as the solvent system, and afforded 5 (9.8 mg) and 6 (13.6 mg). The subfraction PA-4A (82.6 mg) was eluted by CHCl3/MeOH (1:1) over Sephadex LH-20, and afforded 7 (11.9 mg), 8 (17.2 mg) and 9 (10.5 mg). The fraction PA-5 (1.32 g) was chromatographed on silica gel CC and eluted with a CHCl3/MeOH step gradient, and afforded 10 (18.3 mg), 11 (9.2 mg), and the subfraction PA-5A-B. The chromatography of subfraction PA-5B (43.2 mg) over Sephadex LH-20 using CHCl3/MeOH (1:1) as the elution solvent afforded 12 (15.1 mg). The fraction PA-6 (796.4 mg) was chromatographed over ODS-C18 CC using H2O/MeOH in order of increasing polarity as the eluent, and afforded subfraction PA-6A-C. Subfraction PA-6B (72.1 mg) was subjected to RP-18 HPLC (MeOH–H2O, 3.5:6.5) and afforded 1 (7.3 mg), 2 (5.4 mg) and 13 (16.5 mg). Subfraction PA-6C (19.6 mg) was eluted by CHCl3/MeOH (1:1) over Sephadex LH-20 and afforded 3 (3.4 mg).
Spectroscopic data of compounds 1–3
Acid hydrolysis of 3
Two milligrams of 3 in 1 ml 2% H2SO4 solution was heated under reflux for 2 h, then the reaction mixture was dried followed by dissolving in distilled H2O and neutralizing with NaOH. The neutralized mixture was tested with some sugar moieties such as glucose, rhamnose and others using numerous elution systems over silica gel TLC.32,33,51TDDFT-simulated ECD calculations
To simulate the circular dichroism (ECD) spectra, a conformational analysis was first carried out to generated all possible conformations of compounds 1–3 using Omega2 software.52 The generated conformations within the energy window value of 10 kcal mol−1 were optimized at the B3LYP/6-31G* level of theory, followed by frequency calculations to estimate the Gibbs free energies. Time-dependent density functional theory (TDDFT) calculations with incorporating a polarizable continuum model (PCM) using methanol as a solvent were performed at the B3LYP/6-31+G* level of theory to calculate the first fifty excitation states. SpecDis 1.71 was used to generate ECD spectra for the investigated compounds.53 Gaussian band shapes with a sigma value of 0.20–30 ev were applied for ECD spectra generation. The generated ECD spectra were Boltzmann-averaged. All quantum mechanical calculations were performed using Gaussian09 software.54Molecular docking
The crystal structures of the SARS-CoV-2 main protease (Mpro; PDB code: 6LU7,55 papain-like protease (PLpro; PDB code: 6W9C56 and RNA-dependent RNA polymerase (RdRp; PDB code: 6M7157 were taken as templates for the molecular docking calculations. The docking calculations were carried out using AutoDock4.2.6 software58 according to our previously described protocol, against SARS-CoV-2 targets.49,58,59 The AutoDock protocol was followed to prepare the pdbqt files for the three investigated SARS-CoV-2 targets.60 For the three SARS-CoV-2 targets, the binding site was realized by a docking box around the active site with XYZ dimensions of 60 Å × 60 Å × 60 Å and a spacing value of 0.375 Å. The atomic charges of the isolated compounds were assigned using the Gasteiger method.61 The built-in clustering analysis with 1.0 Å RMSD tolerance was utilized to process the predicted binding positions.
Target prediction and pathway enrichment analysis (PEA)
For the pathway enrichment, we initially predicted all of the biological targets for megastigmane (3) as a SARS-CoV-2 inhibitor using the SwissTargetPredicition online tool (http://www.swisstargetprediction.ch). The DisGeNET database (https://www.disgenet.org) was used to collect the available information on human gene–disease associations (GDAs) for SARS diseases. The protein–protein interaction (PPI) network was developed using the STRING database for the top 20 predicted gene targets.62 To investigate all possible target–function relations based on the network profiles, pathway enrichment analysis was performed for the top 20 SARS diseases-related genes using Cytoscape software V.3.8.2.63 Finally, the Cytoscape-based ReactomeFIViz tool was integrated to visualize and model all possible drug–target interactions.64Conclusion
The chemical characterization of the aerial parts of P. arabica led to the isolation and identification of two novel oxetane containing lignans, paronychiarabicine A (1) and B (2), one new megastigmane, paronychiarabicastigmane A (3), in addition to eleven known metabolites. The absolute configurations of the new compounds were achieved based on NOESY experiments as well as experimental and TDDFT-calculated electronic circular dichroism. Molecular docking calculations demonstrated the competitive binding affinity of the isolated compound 3 as a prospective SARS-CoV-2 Mpro inhibitor.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
Dr Elshamy gratefully acknowledges the subsidy from the Takeda Science Foundation, Japan. Additionally, this work was supported by the Tokushima Bunri University, Japan and the National Research Centre, Egypt. The computational work was completed with resources supported by the Science and Technology Development Fund, STDF, Egypt, grant nos 5480 & 7972 (granted to Dr Mahmoud A. A. Ibrahim). Prof. Mohamed Hegazy acknowledges the financial support from the Alexander von Humboldt Foundation “Georg Foster Research Fellowship for Experienced Researchers”. The National Research Centre, Egypt is gratefully acknowledged.References
- M. K. Cheung, G. G. L. Yue, P. W. Y. Chiu and C. B. San Lau, Front. Pharmacol., 2020, 11, 744 CrossRef CAS PubMed.
- B. Adhikari, B. P. Marasini, B. Rayamajhee, B. R. Bhattarai, G. Lamichhane, K. Khadayat, A. Adhikari, S. Khanal and N. Parajuli, Phytother. Res., 2021, 35, 1298–1312 CrossRef CAS PubMed.
- J. C. Boulos, M. Rahama, M.-E. F. Hegazy and T. Efferth, Cancer Lett., 2019, 459, 248–267 CrossRef CAS PubMed.
- U. Anand, N. Jacobo-Herrera, A. Altemimi and N. Lakhssassi, Metabolites, 2019, 9, 258 CrossRef CAS PubMed.
- D. H. Eroğlu, D. Altay and Ü. Budak, International Journal of Secondary Metabolite, 2017, 4, 103–107 CrossRef.
- F. U. Afifi, B. Al-Khalidi and E. Khalil, J. Ethnopharmacol., 2005, 100, 314–318 CrossRef CAS PubMed.
- S. Bouanani, C. Henchiri, E. Migianu-Griffoni, N. Aouf and M. Lecouvey, J. Ethnopharmacol., 2010, 129, 38–45 CrossRef CAS PubMed.
- M. P. B. De Santayana and E. Morales, J. Ethnopharmacol., 2005, 98, 1–19 (J. Ethnopharmacol., 2005, 98, 1) CrossRef PubMed.
- A. Ferreira, C. Proença, M. L. Serralheiro and M. E. Araújo, J. Ethnopharmacol., 2006, 108, 31–37 CrossRef CAS PubMed.
- D. Gülcemal, M. Masullo, Ö. Alankuş-Çalışkan and S. Piacente, Fitoterapia, 2014, 92, 274–279 CrossRef PubMed.
- S. Sait, S. Hamri-Zeghichi, L. Boulekbache-Makhlouf, K. Madani, P. Rigou, V. Brighenti, F. Pio Prencipe, S. Benvenuti and F. Pellati, J. Pharm. Biomed. Anal., 2015, 111, 231–240 CrossRef CAS PubMed.
- D. M. Zama, Z. Tebibel, W. Benayssa, F. Benayache, S. Benayache and A. Vlietinck, Indian J. Pharmacol., 2007, 39, 145–150 CrossRef.
- S. Avunduk, O. Alankuş-Calişkan, T. Miyamoto, C. Tanaka and M. A. Lacaille-Dubois, Nat. Prod. Commun., 2011, 6, 205–208 CrossRef CAS PubMed.
- S. Avunduk, M. A. Lacaille-Dubois, T. Miyamoto, E. Bedir, S. G. Senol and O. A. Calişkan, J. Nat. Prod., 2007, 70, 1830–1833 CrossRef CAS PubMed.
- A. Braca, A. Bader, T. Siciliano and N. De Tommasi, Magn. Reson. Chem., 2008, 46, 88–93 CrossRef CAS PubMed.
- M. Curini, F. Epifano, L. Menghini and R. Pagiotti, Chem. Nat. Compd., 2004, 40, 190–191 CrossRef CAS.
- N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang, J. Song, X. Zhao, B. Huang, W. Shi, R. Lu, P. Niu, F. Zhan, X. Ma, D. Wang, W. Xu, G. Wu, G. F. Gao, W. Tan, I. China Novel Coronavirus and T. Research, N. Engl. J. Med., 2020, 382, 727–733 CrossRef CAS PubMed.
- A. I. Elshamy, T. A. Mohamed, M. M. Marzouk, T. A. Hussien, A. Umeyama, M. E. F. Hegazy and T. Efferth, Phytochem. Lett., 2018, 24, 105–109 CrossRef CAS.
- A. A. Ahmed, M. E. F. Hegazy, N. M. Hassan, M. Wojcinska, J. Karchesy, P. W. Pare and T. J. Mabry, Phytochemistry, 2006, 67, 1547–1553 CrossRef CAS PubMed.
- M.-E. F. Hegazy, T. A. Mohamed, A. I. Elshamy, A. A. Hassanien, N. S. Abdel-Azim, M. A. Shreadah, I. I. Abdelgawad, E. M. Elkady and P. W. Pare, Nat. Prod. Res., 2016, 30, 340–344 CrossRef CAS PubMed.
- T. A. Mohamed, A. I. Elshamy, A. R. Hamed, K. A. Shams and M. E. F. Hegazy, Phytochem. Lett., 2018, 28, 32–36 CrossRef CAS.
- M.-E. F. Hegazy, T. A. Mohamed, F. F. Abdel-Latif, M. S. Alsaid, A. A. Shahat and P. W. Pare, Phytochem. Lett., 2013, 6, 383–386 CrossRef CAS.
- T. Hirata, A. Takarada, M.-E. F. Hegazy, Y. Sato, A. Matsushima, Y. Kondo, A. Matsuki and H. Hamada, J. Mol. Catal. B: Enzym., 2005, 32, 131–134 CrossRef CAS.
- A. Bezerianos, A. Dragomir and P. Balomenos, in Computational Methods for Processing and Analysis of Biological Pathways, Springer, 2017, pp. 11–46 Search PubMed.
- H. Lee and M. Shin, BioData Min., 2017, 10, 3 CrossRef PubMed.
- P. Y. P. Kao, K. H. Leung, L. W. C. Chan, S. P. Yip and M. K. H. Yap, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 335–353 CrossRef CAS PubMed.
- S. Haider, C. Q. Yao, V. S. Sabine, M. Grzadkowski, V. Stimper, M. H. W. Starmans, J. Wang, F. Nguyen, N. C. Moon and X. Lin, Nat. Commun., 2018, 9, 4746 CrossRef PubMed.
- S. A. G. Fleming and J. J. Gao, Tetrahedron Lett., 1997, 38, 5407–5410 CrossRef CAS.
- S. Saphier, Y. Hu, S. C. Sinha, K. N. Houk and E. Keinan, J. Am. Chem. Soc., 2005, 127, 132–145 CrossRef CAS PubMed.
- S. N. Sulaiman, A. Zahari, S. Y. Liew, M. Litaudon, A. M. Issam, H. A. Wahab and K. Awang, Phytochem. Lett., 2018, 25, 22–26 CrossRef CAS.
- K. Gao, D. Ma, Y. Cheng, X. Tian, Y. Lu, X. Du, H. Tang and J. Chen, J. Agric. Food Chem., 2015, 63, 1067–1075 CrossRef CAS PubMed.
- A. I. Elshamy, T. A. Mohamed, S. L. Al-Rowaily, A. M. Abd-ElGawad, B. A. Dar, A. A. Shahat and M. F. Hegazy, Molecules, 2019, 24, 2412 CrossRef CAS PubMed.
- K. Wei, W. Li, K. Koike, L. Liu, X. Fu, L. Lin, Y. Chen and T. Nikaido, Chem. Pharm. Bull., 2004, 52, 776–779 CrossRef CAS PubMed.
- N. Matsuda and M. Kikuchi, Chem. Pharm. Bull., 1996, 44(9), 1676–1679 CrossRef CAS.
- Y. S. Wang, Z. Liao, Y. Li, R. Huang, H. B. Zhang and J. H. Yang, J. Braz. Chem. Soc., 2011, 22, 2234–2238 CrossRef CAS.
- M. M. A. Rahman, P. M. Dewick, D. E. Jackson and J. A. Lucas, Phytochemistry, 1990, 29, 1971–1980 CrossRef CAS.
- N. P. Thao, B. T. Luyen, J. E. Koo, S. Kim, Y. S. Koh, N. V. Thanh, N. X. Cuong, P. V. Kiem, C. V. Minh and Y. H. Kim, Pharm. Biol., 2016, 54, 588–594 CrossRef PubMed.
- X. Wang, C. Zhang, Y. Peng, H. Zhang, Z. Wang, Y. Gao, Y. Liu and H. Zhang, Food Chem., 2018, 246, 41–47 CrossRef CAS PubMed.
- J. Zhang, S. Yamada, E. Ogihara, M. Kurita, N. Banno, W. Qu, F. Feng and T. Akihisa, Chem. Biodiversity, 2016, 13, 1601–1609 CrossRef CAS PubMed.
- T. T. Guo, X.-F. Tang, J. Chang and Y. Wang, Nat. Prod. Res., 2017, 31, 16–21 CrossRef CAS PubMed.
- H. Zhang, N. Feng, Y. T. Xu, T. X. Li, X. M. Gao, Y. Zhu, Y. S. Song, Y. N. Wang and H. H. Wu, Chem. Biodiversity, 2017, 14, e1600437 CrossRef PubMed.
- Y. Yang, Y. B. Yang, W. Q. Lu, Z. J. Wu and W. S. Chen, Chem. Nat. Compd., 2017, 53, 417–421 CrossRef CAS.
- Y. J. Kuo, S. Y. Hwang, M. D. Wu, C. C. Liao, Y. H. Liang, Y. H. Kuo and H. O. Ho, Chem. Pharm. Bull., 2008, 56, 585–588 CrossRef CAS PubMed.
- A. Pauli and K. H. Kubeczka, Nat. Prod. Commun., 2010, 5, 1387–1394 CrossRef CAS PubMed.
- S. H. García B., F. Navarro, J. Pedro and D. Lazari, Phytochemistry, 1996, 41, 1113–1117 CrossRef.
- R. Vitek, L. M. R. de Novais, H. F. V. Torquato, E. J. Paredes-Gamero, M. G. de Carvalho, P. T. de Sousa Jr, M. J. Jacinto and V. C. da Silva, Nat. Prod. Res., 2017, 31, 1930–1934 CrossRef CAS PubMed.
- S. Zúñiga, J. L. Cruz, I. Sola, P. A. Mateos-Gómez, L. Palacio and L. Enjuanes, J. Virol., 2010, 84, 2169–2175 CrossRef PubMed.
- C.-H. Wu, S.-H. Yeh, Y.-G. Tsay, Y.-H. Shieh, C.-L. Kao, Y.-S. Chen, S.-H. Wang, T.-J. Kuo, D.-S. Chen and P.-J. Chen, J. Biol. Chem., 2009, 284, 5229–5239 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- A. I. Elshamy, T. A. Mohamed, M. Suenaga, M. Noji, A. Umeyama, T. Efferth and M.-E. F. Hegazy, Phytochem. Lett., 2019, 34, 74–78 CrossRef CAS.
- C. A. Seo, E.-K. Ahn, J.-S. Kang, J.-H. Leeb, J. S. Oh and S. Hong, Phytochem. Lett., 2017, 20, 93–97 CrossRef CAS.
- P. C. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, J. Chem. Inf. Model., 2010, 50, 572–584 CrossRef CAS PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision E01, Gaussian Inc., Wallingford CT, USA, 2009 Search PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- J. Osipiuk, S. A. Azizi and S. Dvorkin, Nat. Commun., 2021, 12, 743 CrossRef CAS PubMed.
- Y. Gao, L. Yan, Y. Huang, F. Liu, Y. Zhao, L. Cao, T. Wang, Q. Sun, Z. Ming, L. Zhang, J. Ge, L. Zheng, Y. Zhang, H. Wang, Y. Zhu, C. Zhu, T. Hu, T. Hua, B. Zhang, X. Yang, J. Li, H. Yang, Z. Liu, W. Xu, L. W. Guddat, Q. Wang, Z. Lou and Z. Rao, Science, 2020, 368, 779–782 CrossRef CAS PubMed.
- T. A. Mohamed, A. I. Elshamy, M. A. Ibrahim, A. Zellagui, M. F. Moustafa, A. H. Abdelrahman, S. Ohta, P. W. Pare, M.-E. F. Hegazy, T. A. Mohamed, A. I. Elshamy, M. A. Ibrahim, A. Zellagui, M. F. Moustafa, A. H. Abdelrahman, S. Ohta, P. W. Pare and M.-E. F. Hegazy, RSC Adv., 2020, 10, 34541–34548 RSC.
- M. A. A. Ibrahim, A. H. M. Abdelrahman, T. A. Hussien, E. A. A. Badr, T. A. Mohamed, H. R. El-Seedi, P. W. Pare, T. Efferth and M.-E. F. Hegazy, Nucleic Acids Res., 2020, 126, 104046 CAS.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS PubMed.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- H. V. Cook, N. T. Doncheva, D. Szklarczyk, C. Von Mering and L. J. Jensen, Viruses, 2018, 10, 519 CrossRef PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- A. S. Blucher, S. K. McWeeney, L. Stein and G. Wu, F1000Researh, 2019, 8, 908 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02486h |
| This journal is © The Royal Society of Chemistry 2021 |