
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of GO/SiO2/PEA as a new solid base catalyst for the green synthesis of some spirooxindole derivatives†
Mahla Toorbaf and
Leila Moradi *
*
Department of Organic Chemistry, Faculty of Chemistry, University of Kashan, P.O. Box 8731753153, Kashan, I. R. Iran. E-mail: l_moradi@kashanu.ac.ir
First published on 21st June 2021
Abstract
Efficient and green one pot multi component synthesis of some spirooxindole derivatives in the presence of graphene oxide functionalized with 2-(1-piperazinyl) ethylamine (GO/SiO2/PEA) as a solid base catalyst was studied. GO/SiO2/PEA has been obtained through a two step reaction and characterized by Fourier transform infrared spectroscopy (FTIR), field emission scanning electron microscopy (FE-SEM), energy-dispersive X-ray spectroscopy (EDX), thermo gravimetric analysis (TGA), Raman spectroscopy and X-ray diffraction (XRD). Green reaction conditions, short reaction times, reusable catalyst and a high to excellent yield of products are some of the advantageous of the presented method.
Introduction
Multicomponent reactions (MCRs) allow quick and easy access to a large group of organic compounds.1–3 Three or more reactants reacted to form the products consisting of all raw materials.4 Because these reactions do not require intermediate separation, their efficiency increases dramatically. Simple conditions, reduction of byproducts, easy separation, energy saving and short reaction times are the benefits of these reactions.5Recently, green approaches in designing the synthetic methods have attracted considerable attention. Green chemistry means the design of chemical products and processes that reduce or eliminate the use and production of harmful substances to human health and the environment. The goals of this approach are the implementation and sustainable development of chemistry and technology in the chemical industry, academia and government.6 Green chemistry is usually presented as a set of twelve principles suggested by Anastas and Warner, including guidelines for the chemists that work on the synthesis of materials, new chemicals, and new technological processes.7,8
Spiroxindoles as a kind of polycyclic compounds, play an important role in the medical fields. Different derivatives of spiroxindoles are seriously on the agenda of scientists. On the other hand, with the change of precursors and methods of preparation of these compounds, new features of them are revealed in the medical and pharmaceutical fields. The antibacterial, anticancer, antitumour, antifungal and antimicrobial activities of these molecules have been demonstrated9–12 therefore; it is desirable to develop simple and highly efficient methods for the preparation of spiroxindoles. Some of catalysts for synthesis of these compounds are include: piperidin13, Et3N,14 MNPs-guanidne,15 Na2CaP2O7,16 L-proline,17 meglumine,18 SnO2 (ref. 19) and CuO nanoparticles20 and also 4-dimethylaminopyridine.21
In recent years, supported catalysts as green and reusable catalysts have received much attention in the design of chemical syntheses. Graphene oxide (GO) and reduced graphene oxide has been used as catalyst support due to high surface area, easily available, low cast and high activity.22–24 Graphene is a two-dimensional sheet of carbon atoms in a hexagonal configuration in which atoms are bonded to the sp2 hybrid. This structure is the newest member of the multi dimensional graphite carbon family.25,26 Graphene oxide is a nanocarbon material with a honeycomb-like carbon plate which has epoxide, hydroxyl and carboxyl functional groups on its surfaces.
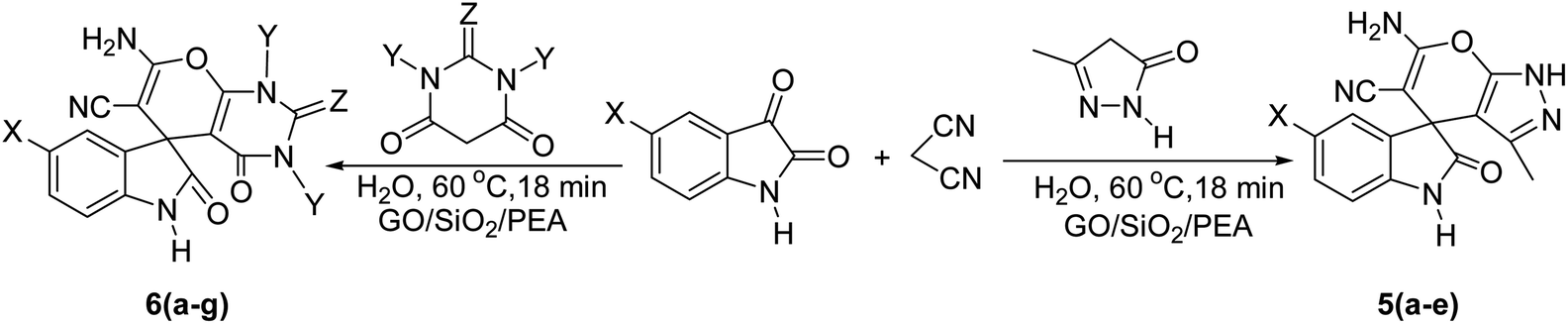
In presented method, we try to design a new heterogeneous catalyst based on graphene oxide. In fact, an amino group has been chemically attached on graphene oxide surfaces through SiO2 linker. Obtained basic catalyst was used in green synthesis of some spirooxindoles (Scheme 1). Results show the high efficiency of catalyst as well as short reaction times with high to excellent yield of products.
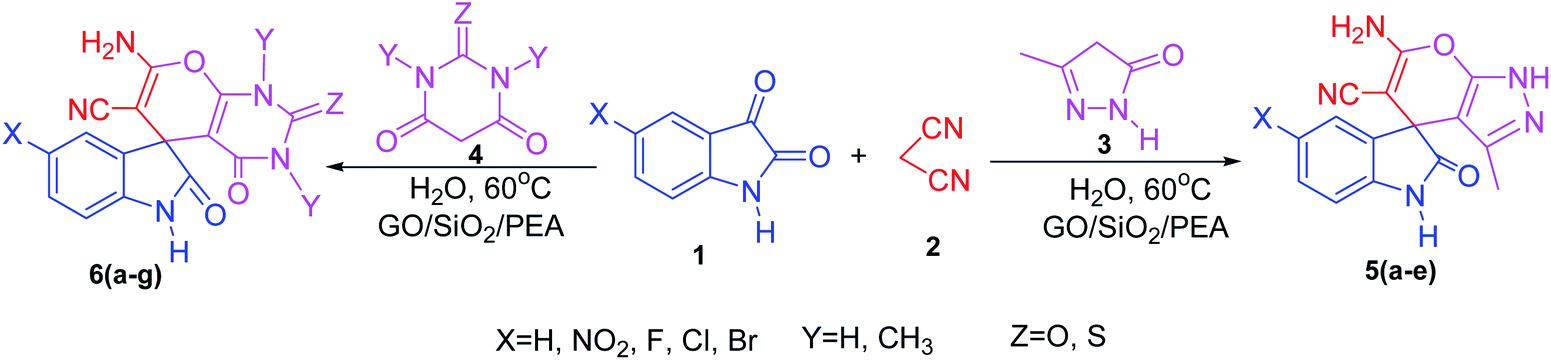 | ||
| Scheme 1 Synthesis of spirooxindoles in the presence of GO/SiO2/PEA. | ||
Experimental section
Materials and methods
Chemical reagents were purchased from the Merck and Aldrich Company. FT-IR spectra were obtained using KBr pellets on a Perkin Elmer 781 spectrophotometer and on an impact 400 Nicolet FTIR spectrophotometer. 1H NMR and 13C NMR spectra were measured with DMSO-d6 solution and tetramethylsilane as internal standard on a Bruker DRX-400 spectrometer. Thermo gravimetric analysis (TGA) was performed on a mettle TA4000 system TG-50 at a heating rate of 10 K min−1 under N2 atmosphere. Raman spectroscopy was provided using FRA/106/S. To investigate the morphology of the MWCNTs and prepared catalyst, FE-SEM images and EDX analysis visualized by a Sigma ZEISS, Oxford Instruments Field Emission Scanning Electron Microscope. TEM images was provided using a Philips CM 120, Netherlands microscope with an accelerating voltage of 150 kV.Preparation of graphene oxide (GO)
GO was synthesized from graphite using the Hummers method.27 Briefly in a typical procedure, 1 g graphite, 15 ml H3PO4 and 135 ml H2SO4 were mixed. 6.6 g KMnO4 was slowly added while stirring, and the rate of addition was controlled to prevent the mixture temperature exceeding from 5 °C. Then, to remove the excess amounts of KMnO4, 3.5 ml of H2O2 was slowly added and stirred for 10 minutes. After left the mixture to cool down, 25 ml of HCl and 15 ml of (deionized water) DI water was added and centrifuged. The supernatant was decanted and the residuals was then rewashed again with HCl and DI water for 3 times and centrifuged. Obtained washed GO was dried at 90 °C for 24 hours to produce the powder of GO.Preparation of graphene oxide functionalized with 2-(1-piperazinyl) ethylamine (GO/SiO2/PEA)
In a balloon equipped with a magnetic stirrer and a nitrogen gas conduction system, 1 g of graphene oxide, 100 ml of dry toluene and 1 ml of (3-chloropropyl) triethoxyethane were mixed and refluxed at 60 °C for 18 h. After the completion of this step, the content of the balloon was filtered and obtained solid (GO/CPTES) washed with EtOH and dried at 80 °C for 16 hours.28 For preparation of GO/SiO2/PEA, 0.5 g of GO/CPTES was dispersed in 15 ml of dry toluene for 10 min by ultrasonication. Then 0.5 ml of 2-(1-piperazinyl) ethylamine (PEA) was added to the mixture and refluxed for 28 hours. After the time had elapsed, the mixture was filtered and the final product was washed with ethanol and dried at 80 °C for 12 h (Scheme 2).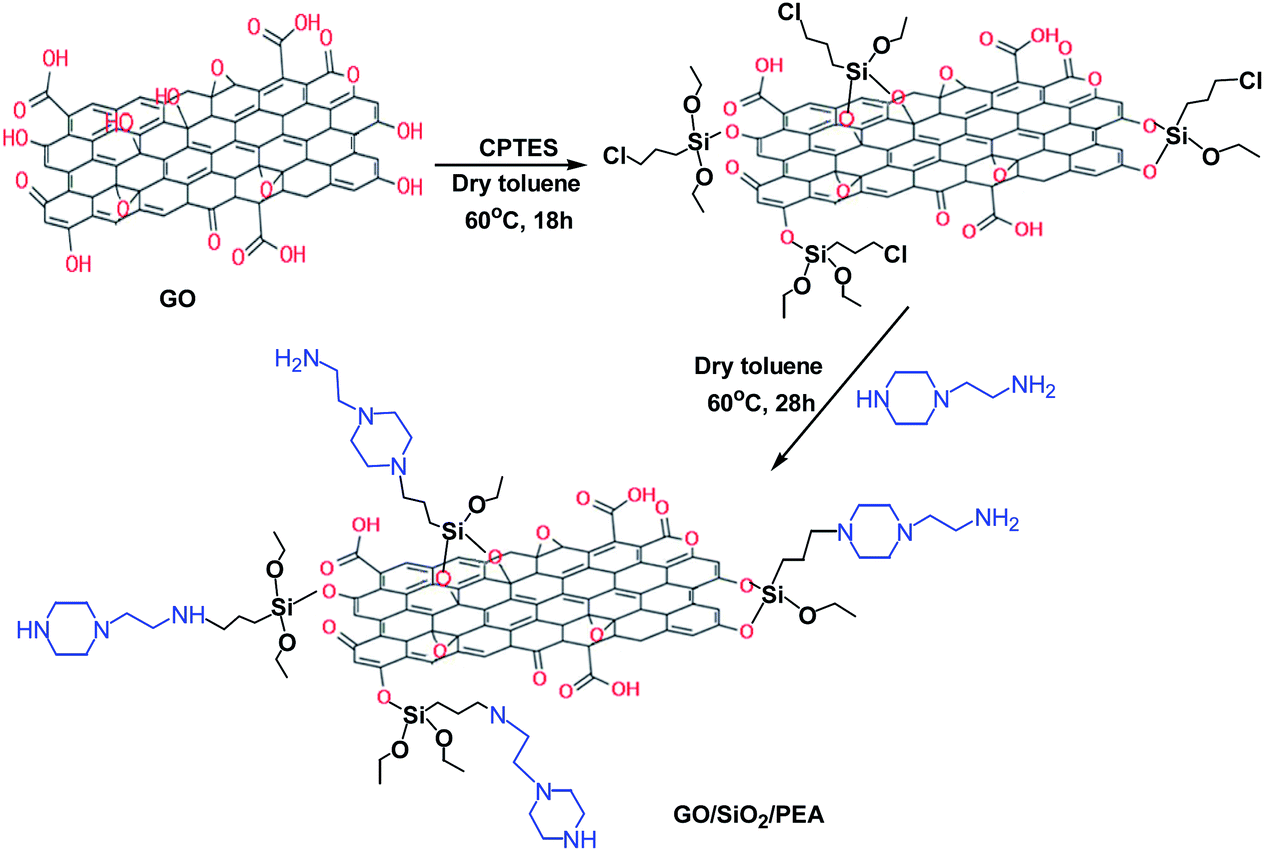 | ||
| Scheme 2 Preparation of GO/SiO2/PEA catalyst. | ||
General procedure for the preparation of spiro derivatives
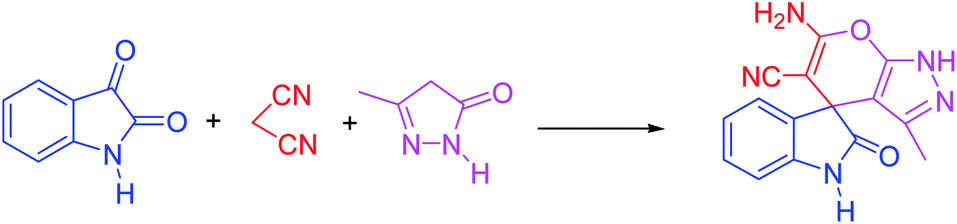
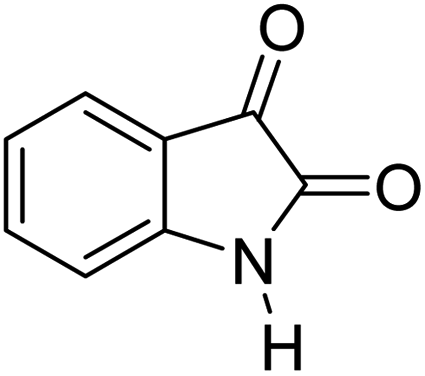
For the synthesis of spiro [indulin-3,4′-pyrano[2,3-c]pyrazole]-5′-carbonitrile derivatives (5a–e), a mixture of ethyl acetoacetate (1 mmol) and hydrazine hydrate (1 mmol) were stirred by magnetic stirrer in 5 ml of water for 15 minutes at 60 °C. After the formation of 3-methyl-2-pyrazoline-5-one as a white precipitate, isatin (1 mmol) and malononitrile (1 mmol) and also catalyst (0.015 g) were added and reaction was continued to complete (followed by thin-layer chromatography). After the reaction was completed, the precipitate was dissolved in ethanol and the catalyst was separated by filtration. Evaporation of solvent from the filtrate, resulted the crud product and for further purification, recrystallized from ethanol (Scheme 1).In order to the synthesis of 7-amino-2,4-dihydro [indoline-3,5-pyrano(2,3-d)pyrimidine]-6-carbonitriles (6a–g), a mixture of isatin (1 mmol), malononitrile (1 mmol) and barbituric acid derivative (1 mmol) in the presence of 0.015 g of catalyst and 5 ml of water was stirred by magnetic stirrer at 60 °C for 18 min. After the completion of reaction was complete, the separation and purification of products was the same as previous synthetic method.
Spectral data
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1711 (CO), 1496 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.51 (3H, s, CH3), 6.88 (1H, d, J = 7.0 Hz, ArH), 6.96 (2H, m, ArH), 7.22 (2H, s, NH2), 7.24 (1H, s, ArH), 10.59 (1H, s, NH), 12.27 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 12.5, 47.1, 58.5, 111.1, 112.5, 116.2, 122.1, 125.6, 127.7, 140.7, 162.6, 166.9, 175.3 ppm. Anal. calcd for C15H11N5O2: C, 61.43; H, 3.78; N, 23.88%. Found: C, 61.36; H, 3.75; N, 23.82%.N), 1720 (CO), 1498 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.57 (3H, s, CH3), 7.12 (1H, d, J = 7.0 Hz, ArH), 7.41 (2H, s, NH2), 7.90 (1H, s, ArH), 8.21 (1H, d, J = 8.0 Hz, ArH), 11.36 (1H, s, NH), 12.40 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 9.8, 48.3, 55.4, 95.2, 110.8, 119.8, 121.6, 127.3, 134.6, 136.5, 144.5, 148.9, 156.7, 164.1, 178.9 ppm; anal. calcd for C15H10N6O4: C, 53.25; H, 2.96; N, 24.85%. Found: C, 53.22; H, 3.12; N, 24.79%.N), 1706 (CO), 1490 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.56 (3H, s, CH3), 6.90 (1H, s, ArH), 6.97 (1H, d, J = 6.0 Hz, ArH), 7.07 (1H, d, J = 7.0 Hz, ArH), 7.28 (2H, s, NH2), 10.62 (1H, s, NH), 12.31 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 9.4, 48.3, 55.3, 95.4, 111.0, 116.2, 119.1, 134.9, 135.2, 138.3, 147.6, 155.8, 157.8, 163.3, 178.2 ppm; anal. calcd for C15H10FN5O2: C, 57.82; H, 3.21; N, 22.49%. Found: C, 57.79; H, 3.23; N, 22.51%.N), 1714 (CO), 1487 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.57 (3H, s, CH3), 6.90 (1H, d, J = 6.0 Hz, ArH), 7.12 (1H, d, J = 7.0 Hz, ArH), 7.29 (1H, s, ArH), 7.29 (2H, s, NH2), 10.74 (1H, s, NH), 12.33 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 9.4, 48.1, 55.1, 95.2, 112.0, 119.1, 125.1, 127.1, 129.5, 135.6, 141.3, 155.7, 163.3, 177.9 ppm; anal. calcd for C15H10ClN5O2: C, 54.88; H, 3.05; N, 21.34%. Found: C, 54.89; H, 3.09; N, 21.41%.N), 1713 (CO), 1498 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.56 (3H, s, CH3), 6.86 (1H, d, J = 6.0 Hz, ArH), 7.22 (1H, s, ArH), 7.30 (2H, s, NH2), 7.41 (1H, d, J = 7.0 Hz, ArH), 10.76 (1H, s, NH), 12.34 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 9.4, 48.0, 55.0, 95.2, 112.2, 114.7, 119.5, 128.0, 132.2, 135.2, 135.5, 141.3, 155.6, 163.1, 178.2 ppm; anal. calcd for C15H10BrN5O2: C, 48.41; H, 2.71; N, 18.82%. Found: C, 48.44; H, 2.65; N, 18.74%.N), 1679 (CO amide), 1249 (CO) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 6.77(1H, d, J = 6.0 Hz, ArH), 6.88 (1H, t, J = 6.0 Hz ArH), 7.16 (2H, t, J = 7.0 Hz, ArH), 7.45 (2H s, NH2), 10.56 (1H, s, NH), 12.51 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δc: 45.1, 57.5, 85.2, 112.4, 119.6, 123.7, 124.3, 129.1, 133.7, 142.6, 163.9, 166.1, 167.9, 176.6, 183.2 ppm; anal. calcd for C15H9N5O2S: C, 45.79; H, 2.29; N, 17.81%. Found: C, 45.81; H, 2.27; N, 17.92%.N), 1684 (CO amide), 1258 (CO), 1485 (CC), 1184–1341 (C–O) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 6.76 (1H, t, J = 6.0 Hz ArH), 6.95 (1H, t, J = 6.0 Hz, ArH), 7.18 (1H, d, J = 7.0 Hz, ArH), 7.44 (2H s, NH2), 10.53 (1H, s, NH), 12.40 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 47.1, 57.0, 91.2, 110.0, 112.3, 115.1, 116.8, 134.7, 138.4, 152.9, 157.1, 158.2, 159.2, 174.1, 177.4 ppm; anal. calcd for C15H8FN5O3S: C, 50.42; H, 2.24; N, 19.61%. Found: C, 50.44; H, 2.22; N, 19.59%.N), 1681 (CO amide), 1222 (CO), 1476 (CC), 1302–1172 (C–O) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 6.77 (1H, d, J = 6.0 Hz, ArH), 7.19 (1H, d, J = 7.0 Hz, ArH), 7.36 (1H, s, ArH), 7.45 (2H s, NH2), 10.63 (1H, s, NH), 12.39 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 48.3, 56.8, 91.0, 110.8, 116.9, 124.4, 126.1, 128.6, 135.2, 141.1, 153.3, 158.3, 160.1, 174.3, 177.1 ppm; anal. calcd for C15H8ClN5O3S: C, 48.15; H, 2.14; N, 18.72%. Found: C, 48.14; H, 2.18; N, 18.81%.N), 1691 (CO amide), 1213 (CO), 1339–1169 (C–O) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 6.74 (1H, d, J = 6.0 Hz, ArH), 7.32 (1H, t, J = 7.0 Hz, ArH), 7.50 (1H, s, ArH), 7.50 (2H s, NH2), 10.68 (1H, s, NH), 12.54 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 47.3, 57.3, 91.5, 111.7, 114.1, 117.3, 127.5, 131.8, 137.1, 142.5, 153.8, 158.8, 160.0, 174.8, 177.5 ppm; anal. calcd for C15H8BrN5O3S: C, 43.06; H, 1.91; N, 16.74%. Found: C, 43.11; H, 1.89; N, 16.69%.N), 1681 (CO amide), 1245 (CO), 1499 (CC), 1302–1172 (C–O) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 3.02 (3H, s, CH3), 3.35 (3H, s, CH3), 6.79 (1H, d, J = 6.0 Hz, ArH), 7.18 (1H, d, J = 7.0 Hz, ArH), 7.27 (1H s, ArH), 7.64 (2H, s, NH2), 10.63 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δc: 28.2, 29.7, 48.1, 56.9, 86.1, 113.5, 117.2, 118.0, 126.4, 130.6, 136.3, 140.6, 151.1, 152.6, 157.9, 165.9, 178.1 ppm; anal. calcd for C17H12ClN5O4: C, 31.10; H, 3.11; N, 18.13%. Found: C, 31.12; H, 3.09; N, 18.15%.N), 1681 (CO amide), 1288 (CO), 1486 (CC) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 3.01 (3H, s, CH3), 3.46 (3H, s, CH3), 6.77 (1H, m, ArH), 6.97 (1H, t, J = 6.0 Hz ArH), 7.07 (1H, s, ArH), 7.62 (2H, s, NH2), 10.52 (1H, s, NH), 12.50 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 28.2, 30.1, 47.8, 56.9, 85.4, 87.1, 112.3, 117.2, 132.6, 136.9, 137.5, 142.3, 150.1, 153.1, 159.1, 161.0, 178.3 ppm; anal. calcd for C17H12FN5O4: C, 32.49; H, 3.24; N, 18.95%. Found: C, 32.46; H, 3.28; N, 18.91%.N), 1693 (CO amide), 1652 (CO) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 3.01 (3H, s, CH3), 3.71 (3H, s, CH3), 6.95 (1H, d, J = 6.0 Hz ArH). 7.28 (2H, t, J = 7.0 Hz, ArH), 7.43 (1H, t, J = 7.0 Hz, ArH), 7.13 (2H, s, NH2) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 27.4, 29.6, 48.0, 57.5, 87.3, 109.6, 117.0, 121.8, 123.9, 128.4, 133.5, 142.2, 150.0, 152.1, 158.3, 159.2, 177.5 ppm; anal. calcd for C17H13N5O4: C, 34.16; H, 3.70; N, 19.94%. Found: C, 43.14; H, 3.73; N, 19.97%.
N), 1711 (CO), 1496 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.51 (3H, s, CH3), 6.88 (1H, d, J = 7.0 Hz, ArH), 6.96 (2H, m, ArH), 7.22 (2H, s, NH2), 7.24 (1H, s, ArH), 10.59 (1H, s, NH), 12.27 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 12.5, 47.1, 58.5, 111.1, 112.5, 116.2, 122.1, 125.6, 127.7, 140.7, 162.6, 166.9, 175.3 ppm. Anal. calcd for C15H11N5O2: C, 61.43; H, 3.78; N, 23.88%. Found: C, 61.36; H, 3.75; N, 23.82%.N), 1720 (CO), 1498 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.57 (3H, s, CH3), 7.12 (1H, d, J = 7.0 Hz, ArH), 7.41 (2H, s, NH2), 7.90 (1H, s, ArH), 8.21 (1H, d, J = 8.0 Hz, ArH), 11.36 (1H, s, NH), 12.40 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 9.8, 48.3, 55.4, 95.2, 110.8, 119.8, 121.6, 127.3, 134.6, 136.5, 144.5, 148.9, 156.7, 164.1, 178.9 ppm; anal. calcd for C15H10N6O4: C, 53.25; H, 2.96; N, 24.85%. Found: C, 53.22; H, 3.12; N, 24.79%.N), 1706 (CO), 1490 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.56 (3H, s, CH3), 6.90 (1H, s, ArH), 6.97 (1H, d, J = 6.0 Hz, ArH), 7.07 (1H, d, J = 7.0 Hz, ArH), 7.28 (2H, s, NH2), 10.62 (1H, s, NH), 12.31 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 9.4, 48.3, 55.3, 95.4, 111.0, 116.2, 119.1, 134.9, 135.2, 138.3, 147.6, 155.8, 157.8, 163.3, 178.2 ppm; anal. calcd for C15H10FN5O2: C, 57.82; H, 3.21; N, 22.49%. Found: C, 57.79; H, 3.23; N, 22.51%.N), 1714 (CO), 1487 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.57 (3H, s, CH3), 6.90 (1H, d, J = 6.0 Hz, ArH), 7.12 (1H, d, J = 7.0 Hz, ArH), 7.29 (1H, s, ArH), 7.29 (2H, s, NH2), 10.74 (1H, s, NH), 12.33 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 9.4, 48.1, 55.1, 95.2, 112.0, 119.1, 125.1, 127.1, 129.5, 135.6, 141.3, 155.7, 163.3, 177.9 ppm; anal. calcd for C15H10ClN5O2: C, 54.88; H, 3.05; N, 21.34%. Found: C, 54.89; H, 3.09; N, 21.41%.N), 1713 (CO), 1498 (C–H, Ar) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 1.56 (3H, s, CH3), 6.86 (1H, d, J = 6.0 Hz, ArH), 7.22 (1H, s, ArH), 7.30 (2H, s, NH2), 7.41 (1H, d, J = 7.0 Hz, ArH), 10.76 (1H, s, NH), 12.34 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 9.4, 48.0, 55.0, 95.2, 112.2, 114.7, 119.5, 128.0, 132.2, 135.2, 135.5, 141.3, 155.6, 163.1, 178.2 ppm; anal. calcd for C15H10BrN5O2: C, 48.41; H, 2.71; N, 18.82%. Found: C, 48.44; H, 2.65; N, 18.74%.N), 1679 (CO amide), 1249 (CO) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 6.77(1H, d, J = 6.0 Hz, ArH), 6.88 (1H, t, J = 6.0 Hz ArH), 7.16 (2H, t, J = 7.0 Hz, ArH), 7.45 (2H s, NH2), 10.56 (1H, s, NH), 12.51 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δc: 45.1, 57.5, 85.2, 112.4, 119.6, 123.7, 124.3, 129.1, 133.7, 142.6, 163.9, 166.1, 167.9, 176.6, 183.2 ppm; anal. calcd for C15H9N5O2S: C, 45.79; H, 2.29; N, 17.81%. Found: C, 45.81; H, 2.27; N, 17.92%.N), 1684 (CO amide), 1258 (CO), 1485 (CC), 1184–1341 (C–O) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 6.76 (1H, t, J = 6.0 Hz ArH), 6.95 (1H, t, J = 6.0 Hz, ArH), 7.18 (1H, d, J = 7.0 Hz, ArH), 7.44 (2H s, NH2), 10.53 (1H, s, NH), 12.40 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 47.1, 57.0, 91.2, 110.0, 112.3, 115.1, 116.8, 134.7, 138.4, 152.9, 157.1, 158.2, 159.2, 174.1, 177.4 ppm; anal. calcd for C15H8FN5O3S: C, 50.42; H, 2.24; N, 19.61%. Found: C, 50.44; H, 2.22; N, 19.59%.N), 1681 (CO amide), 1222 (CO), 1476 (CC), 1302–1172 (C–O) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 6.77 (1H, d, J = 6.0 Hz, ArH), 7.19 (1H, d, J = 7.0 Hz, ArH), 7.36 (1H, s, ArH), 7.45 (2H s, NH2), 10.63 (1H, s, NH), 12.39 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 48.3, 56.8, 91.0, 110.8, 116.9, 124.4, 126.1, 128.6, 135.2, 141.1, 153.3, 158.3, 160.1, 174.3, 177.1 ppm; anal. calcd for C15H8ClN5O3S: C, 48.15; H, 2.14; N, 18.72%. Found: C, 48.14; H, 2.18; N, 18.81%.N), 1691 (CO amide), 1213 (CO), 1339–1169 (C–O) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 6.74 (1H, d, J = 6.0 Hz, ArH), 7.32 (1H, t, J = 7.0 Hz, ArH), 7.50 (1H, s, ArH), 7.50 (2H s, NH2), 10.68 (1H, s, NH), 12.54 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 47.3, 57.3, 91.5, 111.7, 114.1, 117.3, 127.5, 131.8, 137.1, 142.5, 153.8, 158.8, 160.0, 174.8, 177.5 ppm; anal. calcd for C15H8BrN5O3S: C, 43.06; H, 1.91; N, 16.74%. Found: C, 43.11; H, 1.89; N, 16.69%.N), 1681 (CO amide), 1245 (CO), 1499 (CC), 1302–1172 (C–O) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 3.02 (3H, s, CH3), 3.35 (3H, s, CH3), 6.79 (1H, d, J = 6.0 Hz, ArH), 7.18 (1H, d, J = 7.0 Hz, ArH), 7.27 (1H s, ArH), 7.64 (2H, s, NH2), 10.63 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δc: 28.2, 29.7, 48.1, 56.9, 86.1, 113.5, 117.2, 118.0, 126.4, 130.6, 136.3, 140.6, 151.1, 152.6, 157.9, 165.9, 178.1 ppm; anal. calcd for C17H12ClN5O4: C, 31.10; H, 3.11; N, 18.13%. Found: C, 31.12; H, 3.09; N, 18.15%.N), 1681 (CO amide), 1288 (CO), 1486 (CC) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 3.01 (3H, s, CH3), 3.46 (3H, s, CH3), 6.77 (1H, m, ArH), 6.97 (1H, t, J = 6.0 Hz ArH), 7.07 (1H, s, ArH), 7.62 (2H, s, NH2), 10.52 (1H, s, NH), 12.50 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 28.2, 30.1, 47.8, 56.9, 85.4, 87.1, 112.3, 117.2, 132.6, 136.9, 137.5, 142.3, 150.1, 153.1, 159.1, 161.0, 178.3 ppm; anal. calcd for C17H12FN5O4: C, 32.49; H, 3.24; N, 18.95%. Found: C, 32.46; H, 3.28; N, 18.91%.N), 1693 (CO amide), 1652 (CO) cm−1; 1H NMR (400 MHz, DMSO-d6) δH: 3.01 (3H, s, CH3), 3.71 (3H, s, CH3), 6.95 (1H, d, J = 6.0 Hz ArH). 7.28 (2H, t, J = 7.0 Hz, ArH), 7.43 (1H, t, J = 7.0 Hz, ArH), 7.13 (2H, s, NH2) ppm; 13C NMR (100 MHz, DMSO-d6) δC: 27.4, 29.6, 48.0, 57.5, 87.3, 109.6, 117.0, 121.8, 123.9, 128.4, 133.5, 142.2, 150.0, 152.1, 158.3, 159.2, 177.5 ppm; anal. calcd for C17H13N5O4: C, 34.16; H, 3.70; N, 19.94%. Found: C, 43.14; H, 3.73; N, 19.97%.Results and discussions
Characterization of catalyst
We have established that GO/SiO2/PEA is an efficient, reusable and heterogeneous solid base catalyst for the synthesis of spirooxindole derivatives. Presented method has several advantages including excellent yield of products, green reaction conditions, reusability of catalyst and the simple work-up process. Prepared catalyst was characterized using various methods.Fig. 1a and b shows the FTIR spectra of GO and GO/SiO2/PEA. In the FT-IR spectrum, of graphene oxide, the peak at 1578 cm−1 corresponds to carbon–carbon double bonds. The absorption peaks in the 1057, 1678 and 3437 cm−1 are related to the vibration of C–O, carbonyl and hydroxyl groups, respectively. In the FT-IR spectra of GO/SiO2/PEA, vibration peaks corresponding to CO, CC, Si–O–C and N–H are at 1109, 1459, 1628, and 3437 cm−1 demonstrated that the chemically attachment of PEA on GO surfaces was done through SiO2 linker.
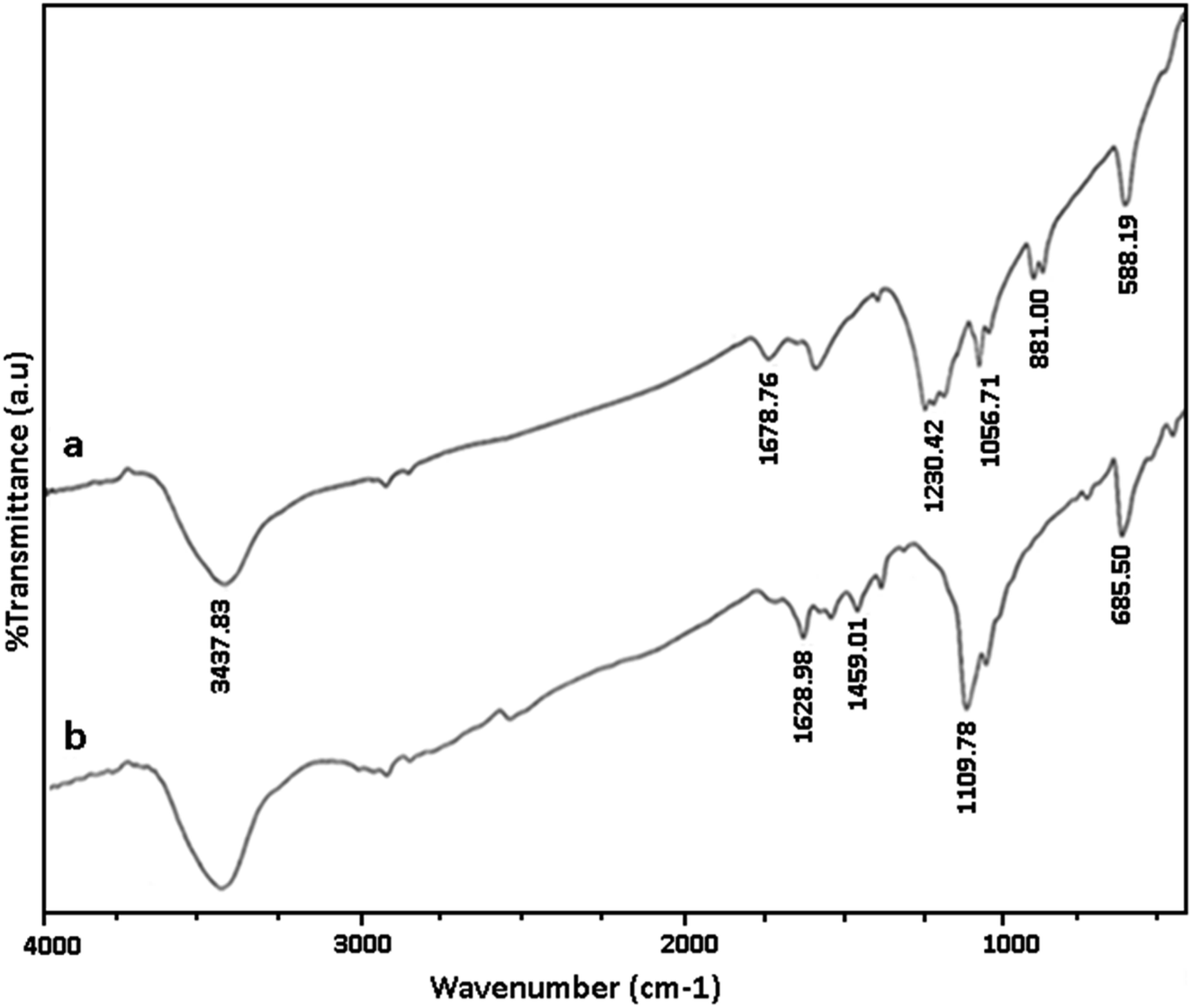 | ||
| Fig. 1 FT-IR of: (a) GO, (b) GO/SiO2/PEA. | ||
Fig. 2 shows the XRD patterns of graphite, GO and GO/SiO2/PEA. The XRD pattern of graphite shows a sharp and long peak at about 26.5° (with the intermolecular distance of 0.34 nm). After the Hummer process and graphene oxide formation, the peak is removed and one peak at about 12.7° was appeared (the interlayer distance become 0.7 nm). In the XRD pattern of the GO/SiO2/PEA, two signals are observed at 2θ = 10.8°, which is due to the structure of the graphene oxide substrate, and the second widespread signal at 2θ = 22.8°, indicates that the prepared catalyst has a layered structure contains amine groups.29
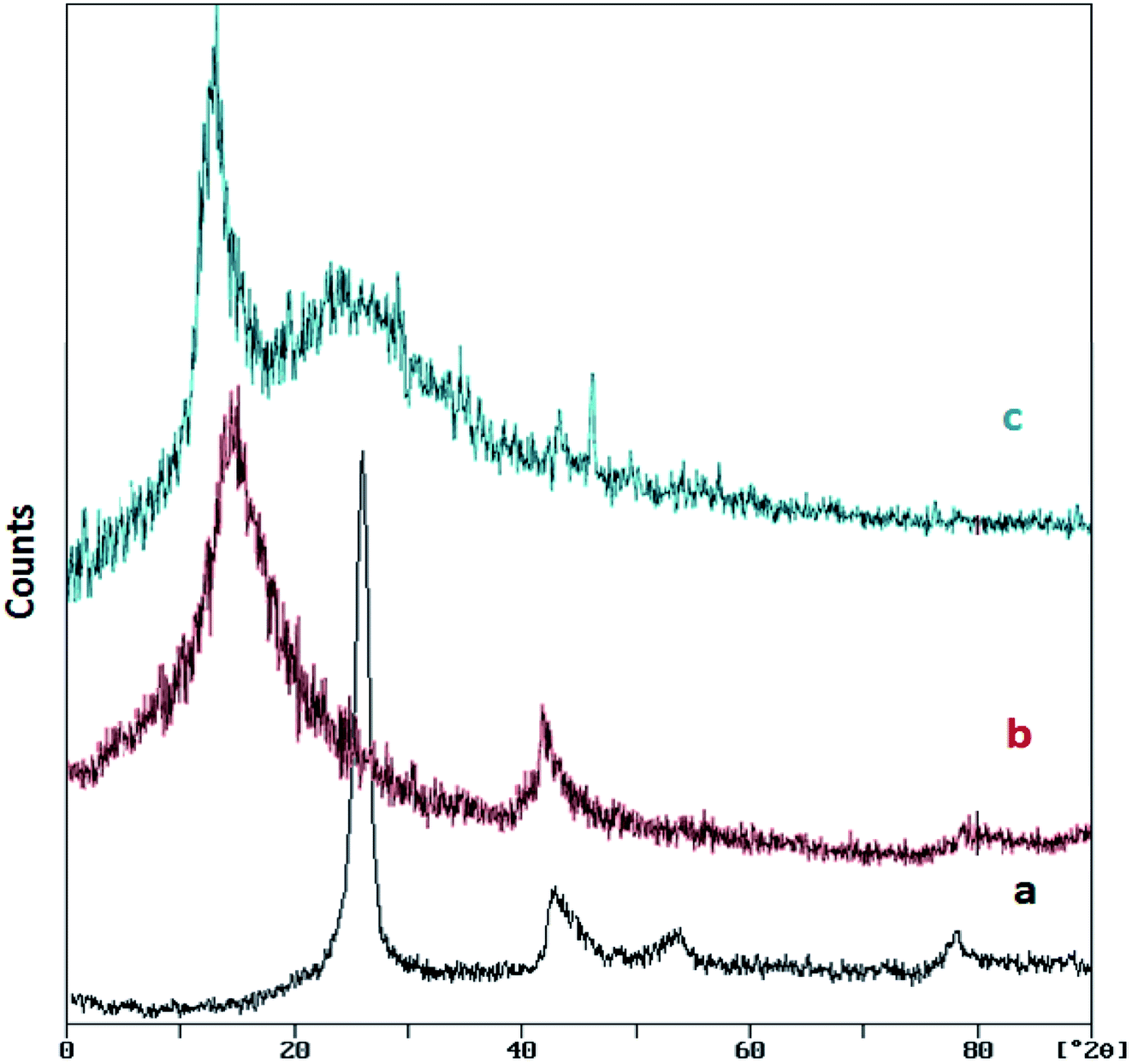 | ||
| Fig. 2 XRD patterns of (a) graphite, (b) GO and (c) GO/SiO2/PEA. | ||
TGA analysis was used to determine the stability of catalyst and percentage of functional groups attached on graphene oxide surfaces. Fig. 3 exhibited the TGA thermograms of GO and GO/SiO2/PEA. As can be seen in Fig. 3a, TGA curve of GO is stable to 220 °C. A major weight loss (about 35%) between 220 °C to 350 °C is due to the decomposition of water and the labile oxygen-containing functional groups on the GO.30 As shown in TGA curve of GO/SiO2/PEA (Fig. 3b), the weight loss about 5% between 20–120 °C is assigned to removal of physically adsorbed water. The second weight loss (about 20%) occur at the temperatures ranging from 120 to 330 °C, and it is attributed to the removal of chemically grafting of SiO2/PEA on to the GO surfaces (Fig. 3b).31
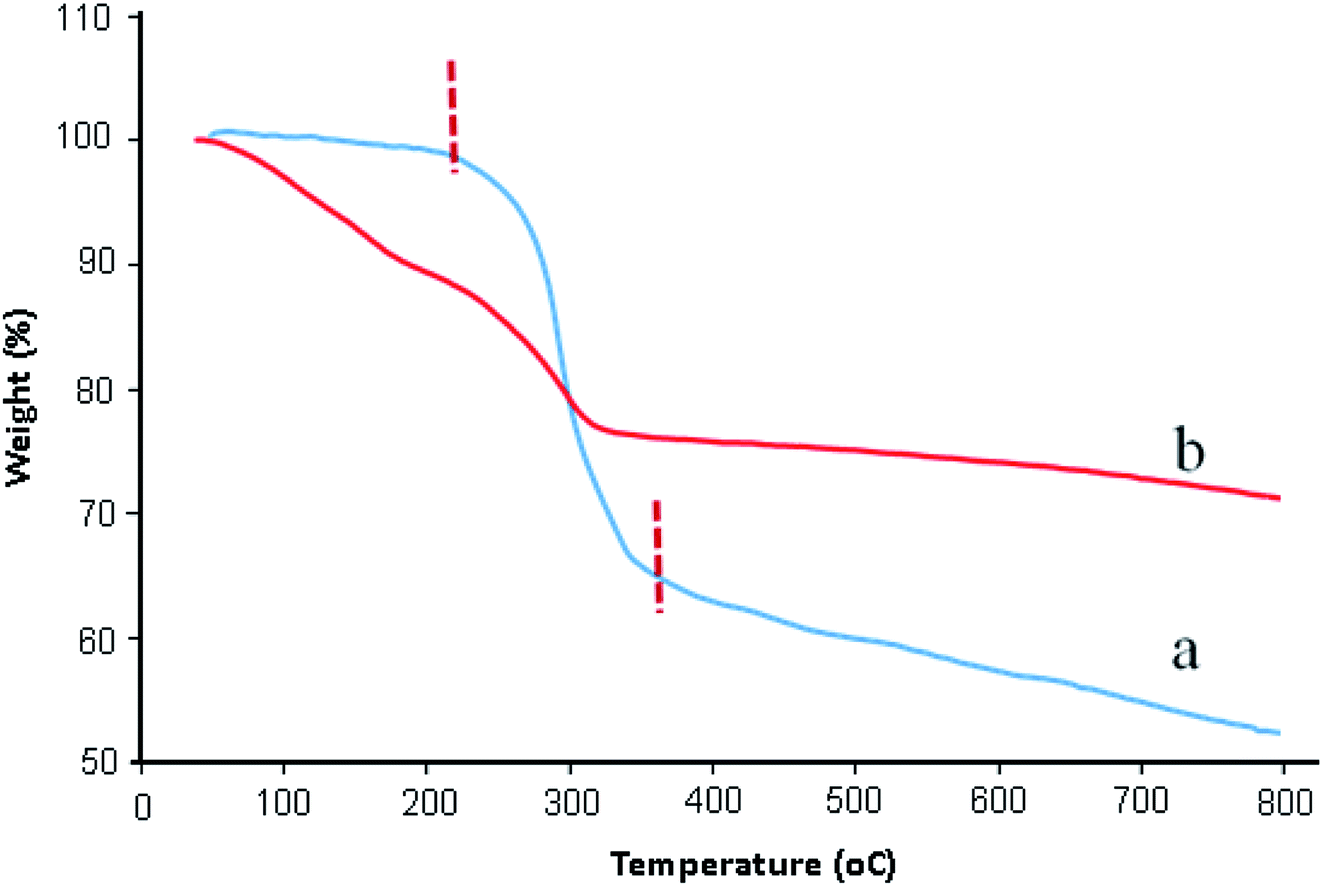 | ||
| Fig. 3 TGA curve of: (a) GO and (b) GO/SiO2/PEA. | ||
In addition, the morphology of prepared catalyst was investigated by the FE-SEM and TEM techniques. As can be seen, the layered, wrinkled and folded structure of catalyst (in Fig. 4a, c and d) proved that SiO2/PEA chemically attached on the GO surfaces.32,33 Also, EDX was used to prove the composition of GO/SiO2/PEA. Results show the presence of nitrogen, silicon, carbon and oxygen in catalyst structure to demonstrate the successfully preparation of GO/SiO2/PEA.
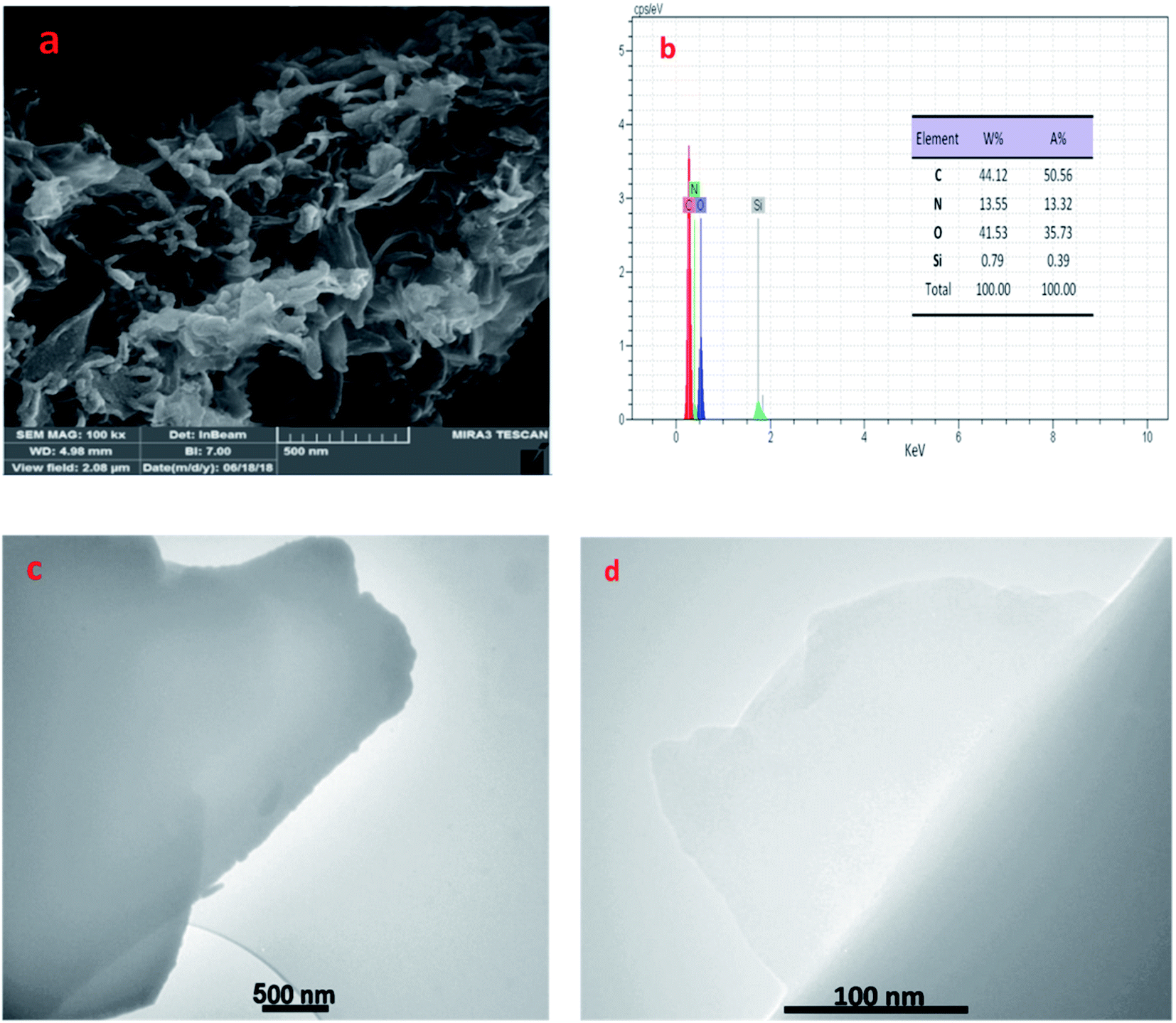 | ||
| Fig. 4 FE-SEM (a), EDX (b) and TEM images (c and d) of GO/SiO2/PEA. | ||
Raman spectroscopy is a nondestructive technique to characterize the structure of carbon based materials. Fig. 5 shows Raman spectra of GO (curve a) and GO/SiO2/PEA (curve b). For carbon materials, the main attributes for a Raman spectra is identified by the D and G bands present around 1345 cm−1 and 1571 cm−1.34,35 The D band, generated by the zone boundary phonons, provides information about the defects and impurities and the G band arises from the doubly degenerate zone centre E2g mode and is present for all carbon based materials.34,36 Fig. 4 also shows that the position of the G band is shifted from 1571 to 1564 cm−1 for GO/SiO2/PEA. This is consistent with the restoration of sp2 graphitic network.37 The intensity ratio IG/ID for both GO and GO/SiO2/PEA are found to be around 0.39 indicating similar degrees of disorder which is understandable considering that functionalization of GO occurs by attachment of SiO2/PEA predominantly on the defect sites where oxidizing groups were already present and hence it does not generally involve conversion of sp2 bonds into sp3.38
 | ||
| Fig. 5 Raman spectra of GO and GO/SiO2/PEA. | ||
After preparation and characterization of catalyst, to optimize the reaction conditions (catalyst amounts, temperature and solvent), the reaction between ethyl acetoacetate (1 mmol), hydrazine hydrate (1 mmol), isatin (1 mmol) and malononitrile (1 mmol) was used as model reaction. Firstly, optimization of solvent and temperature was studied. Based on the results depicted in Table 1, it is founded that water is the best solvent in terms of yield and time of reaction (at 60 °C) compared to other solvents.
|
||||
|---|---|---|---|---|
| Entry | Solvent | T (°C) | Time (min) | Yieldb (%) |
| a Ethyl acetoacetate (1 mmol), hydrazine hydrate (1 mmol), isatin (1 mmol) and malononitrile (1 mmol) in the presence of 0.015 g of catalyst.b Isolated yield. | ||||
| 1 | H2O | r.t. | 25 | 80 |
| 2 | H2O | 60 | 18 | 90 |
| 3 | C2H5OH | r.t | 25 | 72 |
| 4 | C2H5OH | 60 | 25 | 83 |
| 5 | C2H5OH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
r.t. | 25 | 78 |
| 6 | C2H5OH/H2O (1:1) |
60 | 25 | 82 |
| 7 | CH3CN | r.t | 25 | 12 |
| 8 | CH3CN | 60 | 25 | 18 |
Additionally, the optimization of catalyst amount and its effect on the reaction time and yield was investigated. The data in Table 2 show that 0.015 g of catalyst cause to the best yield of product and also the further amount of catalyst did not increase the yield of product.
| Entry | Catalyst (g) | Time (min) | Yield (%) |
|---|---|---|---|
| a (1 mmol) ethyl acetoacetate, (1 mmol) hydrazine hydrate, (1 mmol) isatin, (1 mmol) malononitrile at 60 °C. | |||
| 1 | 0.005 | 30 | 78 |
| 2 | 0.01 | 23 | 84 |
| 3 | 0.015 | 18 | 90 |
| 4 | 0.018 | 18 | 90 |
| 5 | 0.02 | 18 | 90 |
After the optimization of reaction conditions, for evaluation of catalyst efficiency, a variety of spirooxindoles were synthesized at optimum conditions. The results and details were summarized in Table 3. As shown in this table, good to excellent yields were achieved after short reaction times in all cases.
|
|||||
|---|---|---|---|---|---|
| Entry | Carbonyl compound | Isatin | Product | Yield (%) | Mp (°C) |
| 1 | 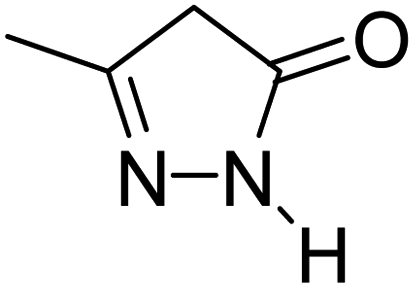 |
 |
 |
90 | 232–234 (ref. 28) |
| 2 |  |
 |
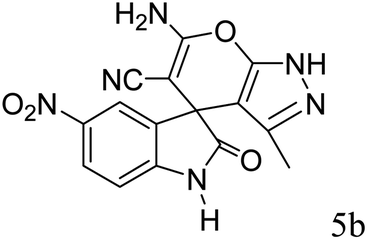 |
92 | 226–228 (ref. 28) |
| 3 | 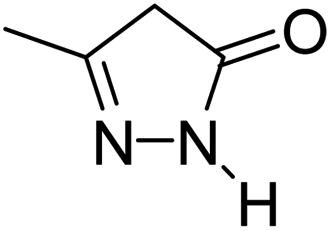 |
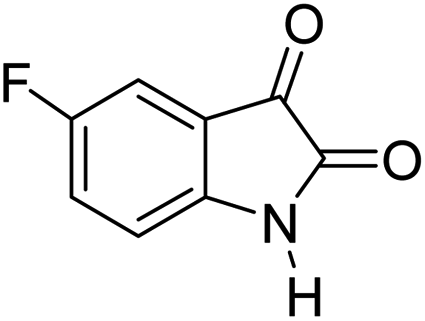 |
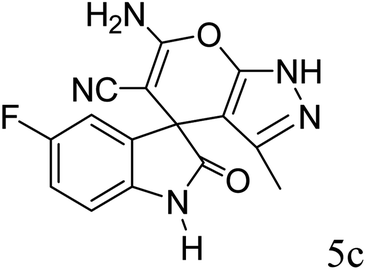 |
90 | 262–265 (ref. 27) |
| 4 |  |
 |
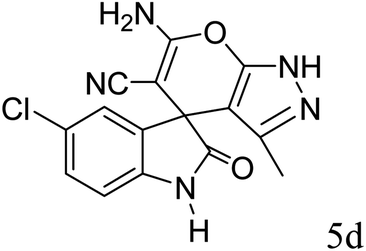 |
89 | 232–234 (ref. 27) |
| 5 |  |
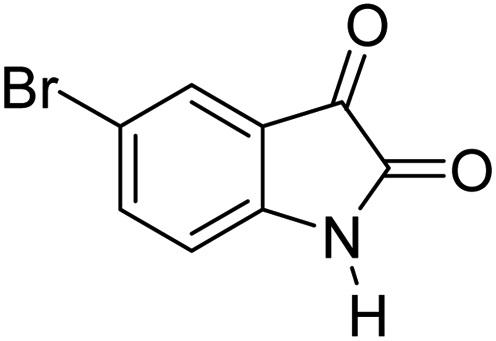 |
 |
90 | 248–250 (ref. 27) |
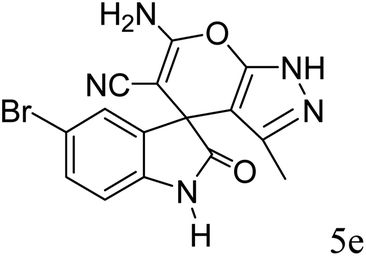
| 6 |  |
 |
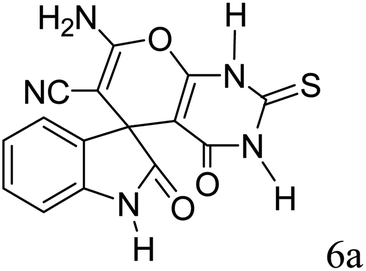 |
90 | 281–283 (ref. 39) |
| 7 |  |
 |
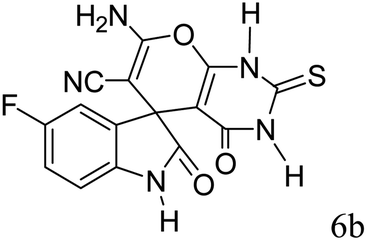 |
93 | 299–301 (ref. 39) |
| 8 |  |
 |
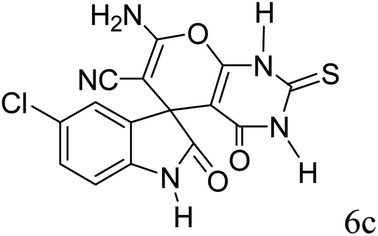 |
92 | 289–291 (ref. 39) |
| 9 |  |
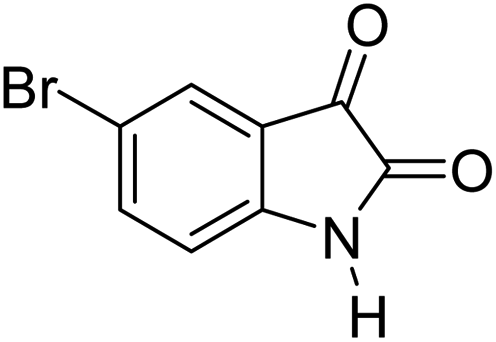 |
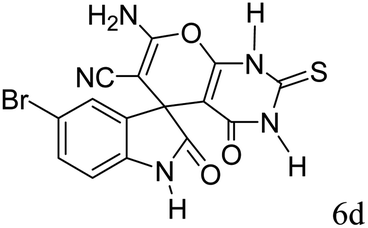 |
91 | 238–240 (ref. 39) |
| 10 | 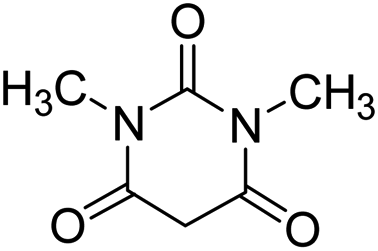 |
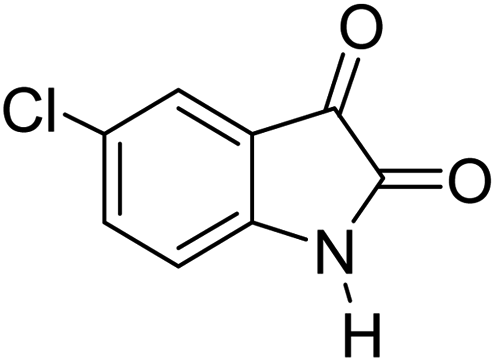 |
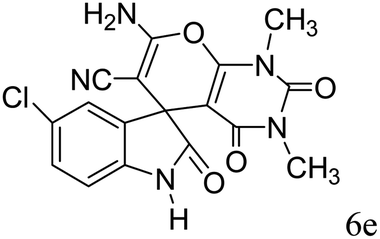 |
89 | 248–250 (ref. 33) |
| 11 |  |
 |
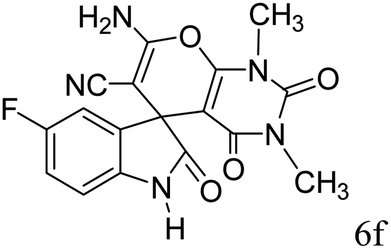 |
90 | 276–278 |
| 12 | 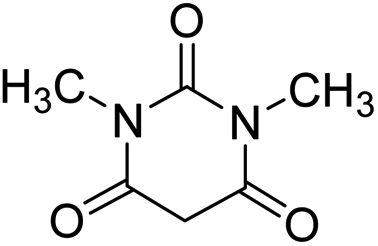 |
 |
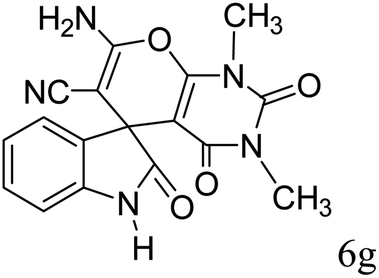 |
90 | 299–301 (ref. 20) |
A proposed mechanism for the synthesis of spirooxindoles in the presence of GO/SiO2/PEA is displayed in Scheme 3. As can be seen, firstly hydrazine hydrate reacts with ethyl acetoacetate to form 3-methyl-2-pyrazoline-5-one (I). On the other hand, Knoevenagel condensation of malononitrile and isatin in the presence of catalyst, lead to formation of intermediate (II). In the next step, Michael addition occurs between intermediate I and II to produce intermediate (III). Finally, the intramolecular cyclization follow by tautomerization of intermediate III resulted the mentioned product.32,33
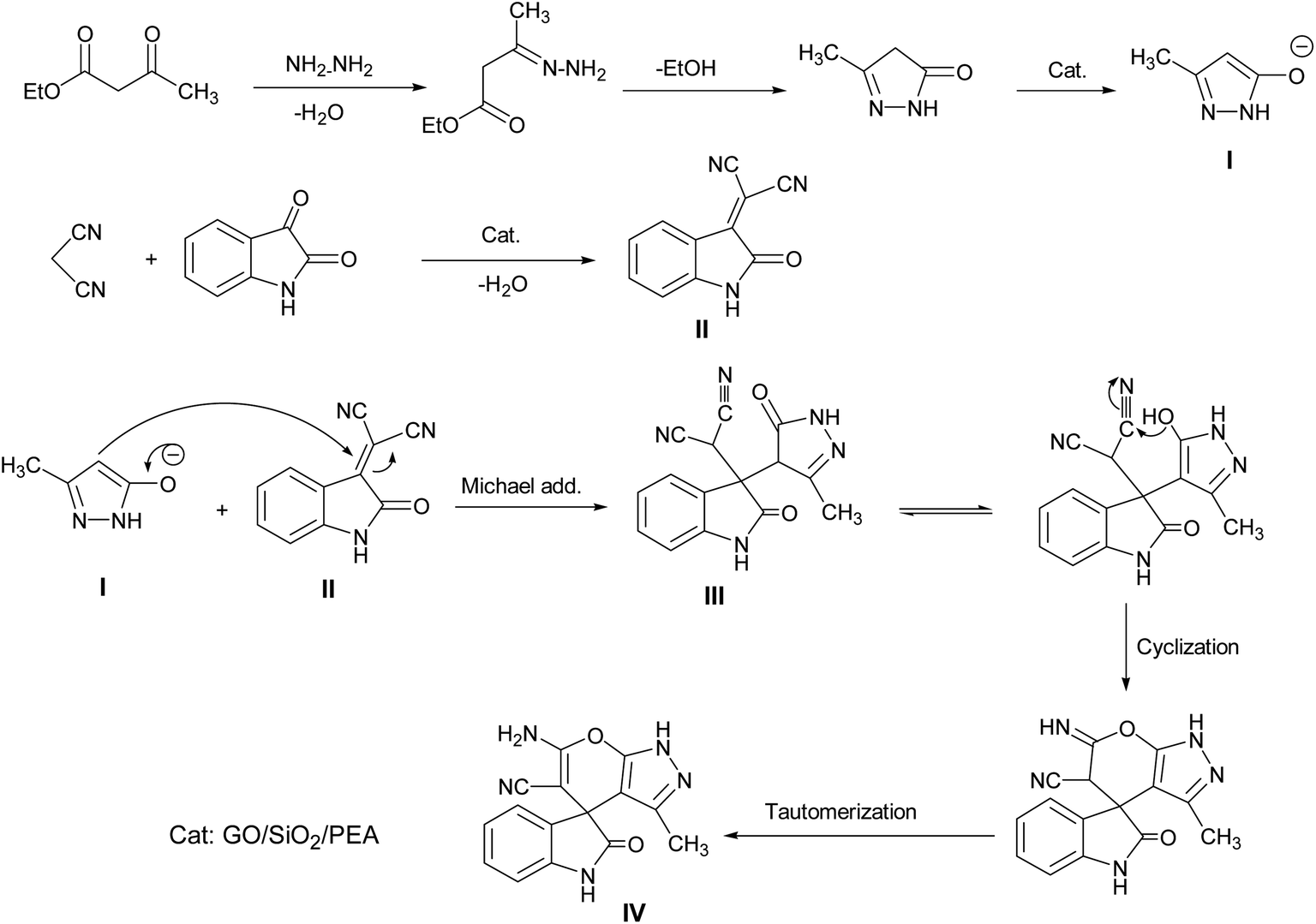 | ||
| Scheme 3 Proposed reaction pathway for the synthesis of spirooxindoles. | ||
In order to study on the performance and recyclability of catalyst, after the completion of sample reaction, catalyst was separated, washed with ethanol and water and dried for using in other similar reaction. Fig. 6 shows that recycled catalyst could be reused five times without considerable decrease in the yield of product to demonstrate the high efficiency and reusability of GO/SiO2/PEA.
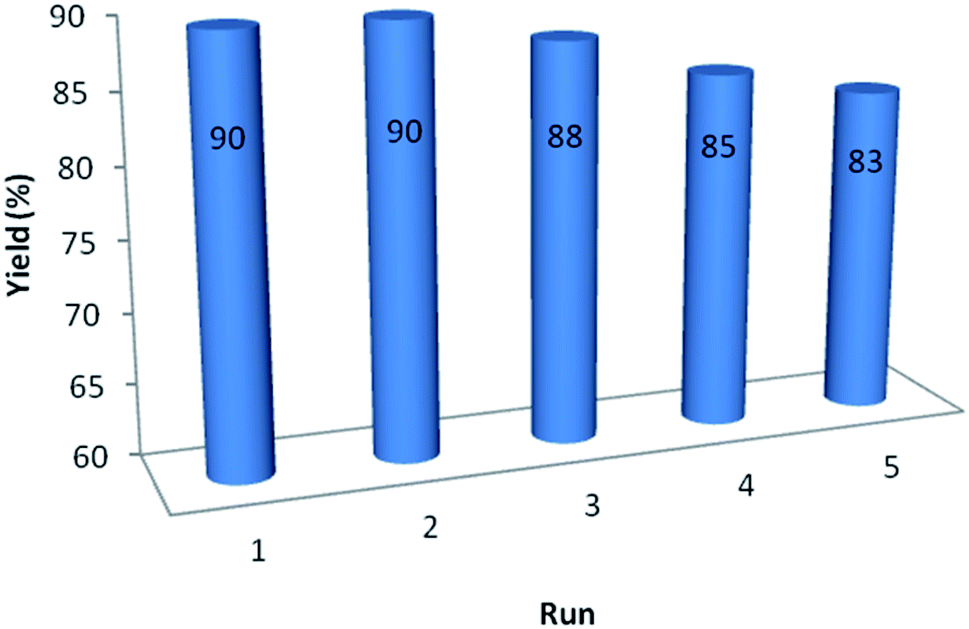 | ||
| Fig. 6 Investigation of catalyst reusability. | ||
Hot filtration test has been done to detect the catalyst nature (homogeneous or heterogeneous). In this way, model reaction was performed under optimized conditions. During the test, catalyst was separated from the reaction mixture by filtration after 9 min (half time of reaction), and the reaction of filtrate was continued for other 9 min. The yield of product (about 40%) shown that after removal of the catalyst, the reaction did not proceed; indicating that no catalytically active species (amino groups) remained in the filtrate and the reaction was performed in the presence of heterogeneous GO/SiO2/PEA catalyst.
The efficiency of GO/SiO2/PEA was compared with some of the other catalysts applied for the synthesis of two kinds of spirooxindole derivatives (5a and 6a). As shown in Table 4, when the reaction was performed in the presence of GO/SiO2/PEA in water, the reaction time was shorter and the product yield was higher than others. Green reaction conditions contain to reusable and efficient catalyst, short reaction time, low temperature as well as easy and clean workup, make this method as a green and eco-friendly protocol.
| Entry | Catalyst | Time | Yield (%) | Reference |
|---|---|---|---|---|
| a Entry 1–10: reaction conditions for the synthesis of 5a and 11–18 for 6a.b Tris-hydroxymethylaminomethane.c The yield of 5a. | ||||
| 1 | Piperidin | 5 h | 89 | 13 |
| 2 | Et3N | 5 h | 80 | 14 |
| 3 | Na2CaP2O7 | 50 min | 78 | 16 |
| 4 | L-Proline | 30 min | 92 | 17 |
| 5 | Carbon–SO3H | 30 min | 90 | 40 |
| 6 | Meglumine | 35 min | 92 | 18 |
| 7 | THAMb in ethanol, r.t. | 4 h | 84 | 41 |
| 8 | SBA-Pr-NH2, r.t., solvent-free | 15 min | 80 | 42 |
| 9 | Chitosan, r.t., (bmim)OH | 210 min | 90 | 43 |
| 10 | EDDF–PEG60, r.t. | 5 h | 87 | 44 |
| 11 | Ionic liquid/K2CO3 in water | 1 h | 90 | 45 |
| 12 | CoFe2O4@SiO2@PUF@Zn(OAc)2 in water | 45 min | 89 | 46 |
| 14 | Fe2O3@SiO2@vitB1Np in water for sonication | 10 min | 85 | 47 |
| 15 | In water at 6 °C | 30 min | 83 | 48 |
| 16 | γ-Fe2O3@HAp-Si(CH2)3SO3H in water at 30 °C | 20 min | 78 | 49 |
| 17 | Et3N microwave irradiation | 2 min | 79 | 50 |
| 18 | GO/SiO2/PEA | 18 min | 90c | This work |
Conclusion
We have established that GO/SiO2/PEA is a robust, reusable and heterogeneous solid catalyst for the synthesis of spirooxindoles in water under thermal conditions. This method has several advantages, including excellent yields of products, green reaction solvent, reusability of catalyst and simple work-up process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research Council of Kashan University should be appreciated for supporting of this work.References
- A. P. Combs, P. A. Tempest, S. D. Brown and T. A. Keating, Acc. Chem. Res., 1996, 29, 123 CrossRef.
- P. R. Faldu, S. K. Dadhaniya, C. J. Patel and G. Vidyasagar, Asian J. Res. Chem., 2011, 4, 1818 Search PubMed; I. Ugi, A. Domling and H. Werner, Endeavour, 1994, 18, 115 CrossRef CAS.
- I. A. Ibarra, A. Islas-Jácome and E. González-Zamora, Org. Biomol. Chem., 2018, 16, 1402 RSC.
- L. Weber, Drug Discovery Today, 2002, 7, 143 CrossRef CAS PubMed.
- D. Cheng, ACS Catal., 2014, 4, 743 CrossRef CAS.
- P. T. Anastas and T. C. Williamson, Green Chem., 1996, 626, 1 CAS.
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford University Press, New York. 2000 Search PubMed.
- Green Chemical Syntheses and Processes, ed. P. T. Anastas, L. G. Heine and T. C. Williamson, American Chemical Society, Washington DC, 2000 Search PubMed.
- B. Bhanupriya, Int. J. Pharmaceut. Sci. Drug Res., 2010, 2, 229 Search PubMed.
- P. Surendra Nath, Acta Pharm., 2005, 551, 27 Search PubMed.
- M. Santos, Trtrahedron, 2014, 52, 9735 CrossRef.
- T. L. Pavlovska, R. G. Redkin, V. V. Lipson and D. V. Atamanuk, Mol. Diversity, 2016, 20, 299–344 CrossRef CAS PubMed.
- S. Ahadi, Z. Yasaei and A. Bazgir, J. Heterocycl. Chem., 2010, 47, 1090 CrossRef CAS.
- C. Wang, Y. H. Jiang and C. G. Yan, Chin. Chem. Lett., 2015, 26, 889 CrossRef CAS.
- F. Alemi, J. S. Ghomi, M. Mahmoudi and H. Shahbazi, RSC Adv., 2016, 6, 74802 RSC.
- B. Maleki, N. Nasiri, R. Tayebee and A. Khojastehnezhad, RSC Adv., 2016, 6, 79128 RSC.
- Y. Jiangxia, Z. Yongbing, S. Tianhua and M. Wengang, J. Chem. Res., 2013, 37, 365 CrossRef.
- R. Y. Guo, Z. M. An, L. P. Mo, S. T. Yang, H. X. Liu, S. X. Wang and Z. H. Zhang, Tetrahedron, 2013, 69, 9931 CrossRef CAS.
- L. Moradi, Z. Ataei and Z. Zahraei, J. Iran. Chem. Soc., 2019, 16, 1273 CrossRef CAS.
- L. Moradi and Z. Ataei, Green Chem. Lett. Rev., 2017, 10, 380 CrossRef CAS.
- J. Feng, K. Ablajan and A. Sali, Tetrahedron, 2014, 70, 484 CrossRef CAS.
- H. Naeimi and M. Farahnak Zarabi, Tetrahedron, 2018, 74, 2314 CrossRef CAS.
- W. Xiao, P. Zhao, S. Deng and N. Zhang, New J. Chem., 2015, 39, 3719 RSC.
- D. Chen, H. Zhang, K. Yang and H. Wang, J. Hazard. Mater., 2016, 310, 179 CrossRef CAS PubMed.
- X. Jia, J. Campos-Delgado, M. Terrones and V. Meunier, Nanoscale, 2011, 3, 86 RSC.
- Q. Wang, X. wang and W. Hu, Chem. Soc. Rev., 2013, 42, 8821 RSC.
- K. Niknam, A. Piran and Z. Karimi, J. Iran. Chem. Soc., 2016, 13, 859 CrossRef CAS.
- S. Ahadi, Z. Yasaei and A. Bazgir, J. Heterocycl. Chem, 2010, 47, 1090 CrossRef CAS.
- P. Zhang, X. Zhang, S. Zhang, X. Lu, Q. Li, Z. Su and G. Wei, J. Mater. Chem. B, 2013, 1, 6525 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- T. A. Pham, N. A. Kumar and Y. T. Jeong, Synth. Met., 2010, 160, 2028 CrossRef CAS.
- M. Gkikas, G. V. Theodosopoulos, B. P. Das, M. Tsianou, H. Iatrou and G. Sakellariou, Eur. Polym. J., 2014, 60, 106 CrossRef CAS.
- S. H. Lee, D. R. Dreyer, J. An, A. Velamakanni, R. D. Piner, S. Park, Y. Zhu, S. O. Kim, C. W. Bielawski and R. S. Ruoff, Macromol. Rapid Commun., 2010, 31, 281 CrossRef CAS PubMed.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS.
- L. Moradia, G. R. Najafi and H. Saeidiroshan, Iran. J. Catalysis, 2015, 5, 357 Search PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- W. Li, X. Z. Tang, H. B. Zhang, Z. G. Jiang, Z. Z. Yu, X. S. Du and Y. W. Mai, Carbon, 2011, 49, 4724 CrossRef CAS.
- S. Chakraborty, S. Saha, V. R. Dhanak, K. Biswas, M. Barbezat, G. P. Terrasid and A. K. Chakraborty, RSC Adv., 2016, 6, 67916 RSC.
- A. Rashidi, Z. Tavakoli, Y. Tarak and S. Khodabakhshi, J. Chin. Chem. Soc., 2016, 63, 399 CrossRef CAS.
- B. M. Rao, G. N. Reddy, T. V. Reddy, B. L. A. Prabhavathi Devi, R. B. N. Prasad, J. S. Yadav and B. V. Subba Reddy, Tetrahedron Lett., 2013, 54, 2466 CrossRef.
- S. S. Khot, P. V. Anbhule, U. V. Desai and P. P. Wadgaonkar, C. R. Chim., 2018, 21, 814 CrossRef CAS.
- G. Mohammadi Ziarani, M. Rahimifard, F. Nouri and A. R. Badiei, J. Serb. Chem. Soc., 2015, 80, 1265 CrossRef.
- P. Rai, M. Srivastava, J. Singh and J. damba Singh, New J. Chem., 2014, 38, 3181 RSC.
- A. Thakur, M. Tripathi and U. C. Rajesh, RSC Adv., 2013, 3, 18142 RSC.
- K. Niknam, M. Khataminejad and F. Zeyaei, Tetrahedron Lett., 2016, 57, 361 CrossRef CAS.
- M. A. Bodaghifard and Z. Mousavi, Appl. Organomet. Chem., 2020, 34, e5859 CrossRef CAS.
- G. S. Nongthombam, L. Makroni, P. D. Shougaijam, R. Noimur, A. Aziz, K. C. Asit and N. Rishanlang, Green Chem., 2016, 18, 4216 RSC.
- L. Q. Zhao, B. Zhou and Y. Q. Li, Heteroat. Chem., 2011, 22, 673 CrossRef CAS.
- L. Jalili-Baleh, N. Mohammadi, M. Khoobi, L. Mamani, A. R. Foroumadi and A. Shafiee, Helv. Chim. Acta, 2013, 96, 1601 CrossRef CAS.
- A. F. Mahmoud, F. F. Abd El-Latif and A. M. Ahmed, Chin. J. Chem., 2010, 28, 91 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02850b |
| This journal is © The Royal Society of Chemistry 2021 |