
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydromagnesite sheets impregnated with cobalt–ferrite magnetic nanoparticles as heterogeneous catalytic system for the synthesis of imidazo[1,2-a]pyridine scaffolds†
Deepika Geedkara,
Ashok Kumar a,
Kranti Kumarb and
Pratibha Sharma*a
a,
Kranti Kumarb and
Pratibha Sharma*a
aSchool of Chemical Sciences, Devi Ahilya University, Indore-452001, MP, India. E-mail: drpratibhasharma@yahoo.com
bUGC-DAE, Consortium for Scientific Research, Devi Ahilya University, Indore-452001, MP, India
First published on 1st July 2021
Abstract
This paper manifests an A3-coupling strategy assisted by novel hydromagnesite sheets impregnated with cobalt ferrite (CoFe2O4-HMS) magnetic nanoparticles (MNPs) as an environmentally benign nanocomposite to synthesize imidazo[1,2-a]pyridine scaffolds under ultrasonication. The synthesis of these biologically active derivatives was achieved through A3-coupling employing 2-aminopyridines derivatives, pertinent aryl aldehydes, and phenylacetylene in the presence of polyethylene glycol 400 (PEG 400) as a green solvent under aerobic conditions. Based on its high product yield (up to 94%) in a short reaction time, with a modest catalyst loading, excellent catalyst, and solvent recyclability without substantial loss of operation (up to five synthetic cycles), as demonstrated by the high ecological compatibility and sustainability factors, this strategy follows the principles of green chemistry. The synthesized nanocomposite was characterized via several spectroanalytical techniques, including PXRD, FE-SEM, HR-TEM, EDAX, ICP-AES, FT-IR, Raman spectroscopy, CO2-TPD, TGA-DTA-DTG analyses, magnetic studies, and nitrogen porosimetry. Furthermore, the structures of synthesized compounds were confirmed based on FT-IR, 1H NMR, 13C NMR, mass spectroscopy, and elemental analysis data.
1. Introduction
Multicomponent coupling reaction strategies embracing green chemistry protocols have blossomed as sustainable pathways for achieving the synthesis of numerous heterocyclic scaffolds. Particularly, nitrogen-containing fused frameworks are the most prevalent structural motifs found in pharmaceutical chemistry as biologically active synthetic equivalents and natural products.1 Therefore, the development of efficient and environmentally sustainable protocols for the synthesis of new series of highly functionalized imidazo-fused pyridine scaffolds has attracted considerable attention on account of their synthetic and effective biological significance. Their chemistry has gained substantial interest in recent years owing to their contribution in various medicinal applications as potent antiviral,2 anxiolytic,3 anticancer,4 antibacterial,5 and anti-inflammatory agents.6 Several biologically active representatives such as zolpidem, alpidem, olprinone, zolimidine, and miroprofen contain an imidazo[1,2-a]pyridine (I–V) nucleus (Fig. 1).7 The presence of the imidazo[1,2-a]pyridine nucleus in the framework has potential relevance in optoelectronics, dyes, and sensing materials, which has resulted in the development of many novel strategies for their preparation.8–10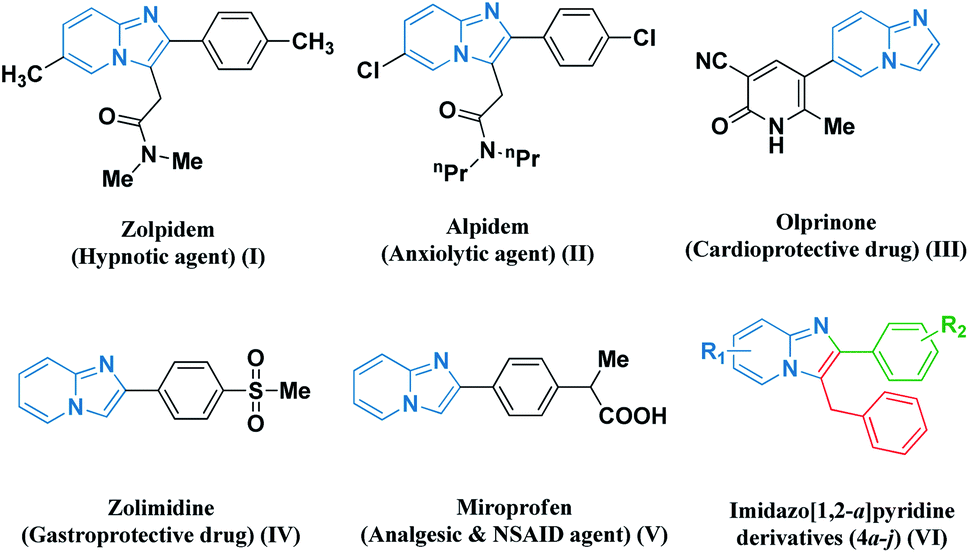 | ||
| Fig. 1 Some representative examples of biologically active imidazo fused pyridines (I–V) and our designed molecule (VI). | ||
Based on these observations, it is considered to be worthwhile to develop robust, economical, and environment-friendly catalytic frameworks for the synthesis of pharmacologically significant imidazo-fused pyridine derivatives (VI). A systemic perusal of the literature revealed that there are recent reports on several transition-metal-catalyzed techniques for the synthesis of imidazole-fused heterocyclic core nuclei. These catalytic strategies include the use of CuSO4·5H2O,11 MNP@BiimCu,12 CuO/CuAl2O4,13 Fe3O4@SiO2 NPs,14 CuI/L-proline,15 Cu–NPs16 and nano-copper/nano-ZnAl2O4 under an N2 atmosphere,17 PW-CIS500,18 and CoFe2O4@CNT-Cu19 towards the green synthesis of amalgamated heterocyclic moieties. However, these reported methods have some bottlenecks including the use of hazardous and expensive catalysts, extensive moisture-sensitive metallic reagents, harsh reaction conditions, prolonged reaction time, tedious work-up processes, use of volatile organic solvents and non-recyclable and large catalytic loading, leading to the generation of an enormous amount of waste. Hence, considering the diverse significance of imidazo-fused pyridine scaffolds, we endeavoured to develop hydromagnesite sheets impregnated with cobalt–ferrite magnetic nanoparticles derived as an alternative strategy employing 2-aminopyridine derivatives, pertinent aryl aldehydes, and phenyl acetylene as precursors. To the best of our knowledge, the synthesis of amalgamated imidazo-pyridine frameworks derived from hydromagnesite sheets impregnated with cobalt–ferrite magnetic nanoparticles has not been reported to date.
Furthermore, ultrasonic irradiation has attracted significant attention from scientific and industrial researchers as an intuitive, green technique in many chemical processes.20,21 Currently, the use of ultrasonic irradiation as an alternative source of energy for organic reactions results in a shorter reaction time with green and mild reaction conditions, minimum chemical waste in high isolated yields, and improvement in the eco-sustainability and selectivity of the system. This acceleration of reactions under ultrasonic conditions can be attributed to the acoustic cavitations arising from the high pressures and energy produced instantly. Hydromagnesite sheets (HMS) have attracted significant attention on account of their specific applications in many areas such as the sequestration of CO2 gas, corrosion prevention, fire retardants, RNA chromatography, photoluminescence, versatile magnesium oxide precursor, and endothermic coolant in yellow smoke formulations for hand-held signals. However, despite the diverse applications of HMS, its use as a catalyst has not been substantially explored.22 Thus, based on the literature,22,23 the crystal structure models of HMS and CoFe2O4-HMS are shown in Scheme 2.
Interestingly, magnetic nanocomposites are considered efficient and recoverable catalysts on account of their unique properties and remarkable catalytic potential, simple preparation, non-corrosive nature, non-toxicity, physiochemical stability, and environmental compatibility. Moreover, the incorporation of support media has become important in enhancing the surface area and overcoming the problems associated with the aggregation of nanocomposites. Therefore, considering these fascinating features, we synthesized hydromagnesite sheets impregnated with cobalt–ferrite magnetic nanoparticles. The magnetic separation of a synthesized catalyst is an intriguing alternative to filtration or centrifugation, given that it reduces the loss of the catalyst and improves the recovery and reusability of catalysts. Moreover, the eventual substitution of conventional hazardous organic solvents by safe and environmentally benign media is also substantially relevant given that organic solvents account for more than 80% of the overall waste products of a process.24,25 Therefore, to circumvent all these bottlenecks, herein, we report a green and efficient protocol for the synthesis of amalgamated imidazo-pyridine derivatives in high to excellent yields considering recyclable PEG 400 as the reaction medium under aerobic conditions.
Furthermore, despite the multifarious properties associated with heterogeneous nanocomposite catalysts and pharmacologically significant imidazo-fused pyridine scaffolds, and in continuation to our work towards nanocatalysis-oriented synthesis,25,26 we demonstrate the unprecedented ultrasonic-assisted environmentally friendly synthesis of highly functionalized imidazo[1,2-a]pyridines in the presence of hydromagnesite sheets impregnated with cobalt–ferrite magnetic nanoparticles (CoFe2O4-HMS) as a recyclable magnetic nano-catalyst composite under mild and green reaction conditions via A3-coupling followed by 5-exo-dig cyclization. To the best of our knowledge, this is the first report to date on the use of hydromagnesite sheets impregnated with cobalt–ferrite magnetic nanoparticles (CoFe2O4-HMS) as a heterogeneous nanocatalyst for the synthesis of imidazo[1,2-a]pyridines scaffolds.
2. Results and discussion
2.1. Optimization of reaction conditions
To optimize the reaction conditions, a model condensation reaction of 2-aminopyridine (1a), benzaldehyde (2a), and phenyl acetylene (3) was carried out in the presence of various solvents under ultrasonic irradiation. In the presence of organic solvents such as ethanol, 1,4-dioxane, THF (tetrahydrofuran), DMF (dimethylformamide), and DMSO (dimethylsulfoxide) the reactions proceeded smoothly to afford the product (4a) in quantitative yields (Table 1, entries 1–5). The same reaction was attempted in aqueous medium and green solvents including water, glycerine, EG (ethylene glycol), DEG (diethylene glycol), and PEG 400 (polyethylene glycol 400) (entries 6–14). It was observed that the reaction did not proceed with a substantial yield in water as the solvent (Table 1, entry 6). It can be inferred from the data presented in Table 1 that the product yields with EG and DEG were 76% and 68%, respectively, even after a reaction time of one hour (Table 1, entries 8 and 9, respectively). Intriguingly, PEG 400 was considered to be the best among the solvents used, affording the product yield of 89% (Table 1, entry 10). The reaction was extended to one hour but no further enhancement in the yield of the product was observed (Table 1, entry 11). Furthermore, a high catalytic loading (8 mol%) under similar conditions did not result in a substantial improvement in the yield (Table 1, entry 12). After the solvent optimization, the reaction was also performed in CoCl2·6H2O as the catalyst and in bare CoFe2O4 MNPs, but the yield of the product was not appreciable (Table 1, entries 13 and 14, respectively). After ascertaining the optimal reaction conditions, 2-aminopyridine derivatives, pertinent aromatic aldehydes and, phenyl acetylene were employed to observe the extent and prevalence of this reaction in the presence of PEG 400 (Scheme 1). The results are summarized in Table 1. The correlation between catalytic activity and reaction efficiency for the synthesis of the desired product (4a) is illustrated in Fig. S1 of the ESI.†| Entry | Condition | Solvent | Timeb (min) | Yieldc (%) |
|---|---|---|---|---|
| a Reaction conditions: 2-aminopyridine (1 mmol) (1a), benzaldehyde (1 mmol) (2a) and phenyl acetylene (1 mmol) (3).b All reactions were monitored by TLC.c Isolated yield. | ||||
| 1 | CoFe2O4-HMS (7 mol%) | 1,4 dioxane | 25 | 58 |
| 2 | CoFe2O4-HMS (7 mol%) | DMSO | 25 | 61 |
| 3 | CoFe2O4-HMS (7 mol%) | THF | 25 | 42 |
| 4 | CoFe2O4-HMS (7 mol%) | DMF | 25 | 56 |
| 5 | CoFe2O4-HMS (7 mol%) | EtOH | 25 | 54 |
| 6 | CoFe2O4-HMS (7 mol%) | H2O | 25 | 68 |
| 7 | CoFe2O4-HMS (7 mol%) | Glycerine | 25 | 79 |
| 8 | CoFe2O4-HMS (7 mol%) | EG | 25 | 76 |
| 9 | CoFe2O4-HMS (7 mol%) | DEG | 60 | 68 |
| 10 | CoFe2O4-HMS (7 mol%) | PEG 400 | 25 | 89 |
| 11 | CoFe2O4-HMS (7 mol%) | PEG 400 | 60 | 89 |
| 12 | CoFe2O4-HMS (8 mol%) | PEG 400 | 25 | 86 |
| 13 | CoFe2O4 MNPs (7 mol%) | PEG 400 | 30 | 69 |
| 14 | CoCl2·6H2O (7 mol%) | PEG 400 | 30 | 66 |
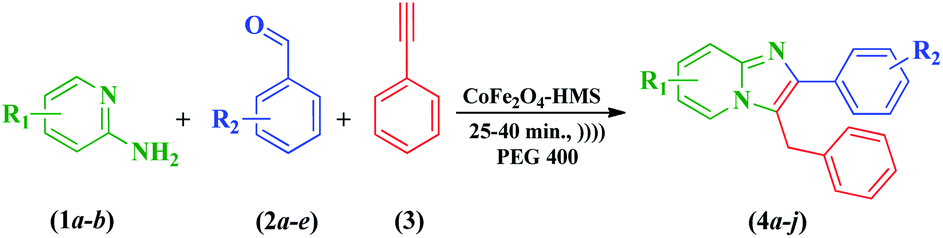 | ||
| Scheme 1 Synthesis of imidazo[1,2-a]pyridine derivatives (4a–j). | ||
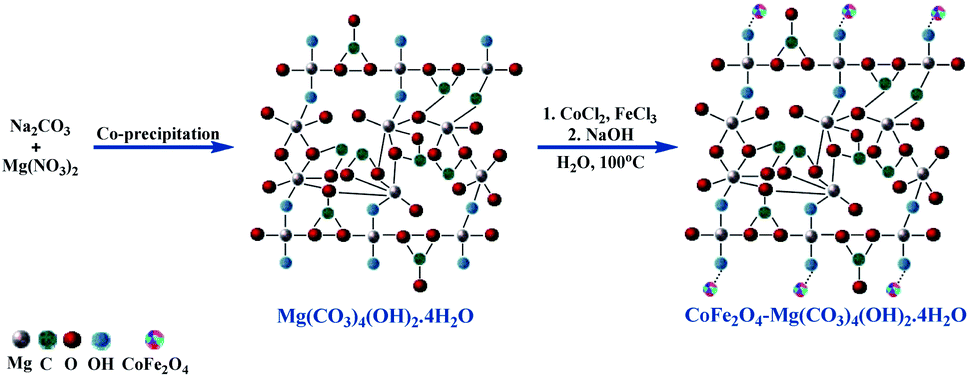 | ||
| Scheme 2 Synthesis of hydromagnesite sheets impregnated with cobalt–ferrite magnetic nanoparticles (CoFe2O4-HMS). | ||
Furthermore, the effects of various substituents were determined in a similar fashion, yielding the corresponding anticipated products (4a–j) in high to excellent yields (78–94%). The nitro group at the ortho position of the benzene ring gave an appreciable yield of 82% (Table 2, entry 4), whereas substitution with chloro and hydroxyl groups at the para position resulted in an improvement in the yield (Table 2, entries 2 and 3, respectively). It was inferred that the electron-withdrawing and electron-donating substituents at the para positions intensified the rate of reaction, resulting in better yields in comparison to the ortho position (Table 2, entries 2–5). Also, the presence of di-substituted aryl aldehydes resulted in the desired products in satisfactory yields (Table 2, entries 5 and 10). In contrast, the presence of strong electron-donating groups such as methyl at the para position resulted in an enhancement in the yield from 87% to 94% (Table 2, entries 6–10).
| Entry | R1 (amino pyridine) | R2 (aryl aldehydes) | Productb | Time (min) | Yieldc (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 2-aminopyridine derivatives (1 mmol) (1a–b), pertinent aryl aldehyde (1 mmol) (2a–e), phenyl acetylene (1 mmol) (3) and a catalytic amount (7 mol%) of CoFe2O4-HMS in the presence of PEG 400.b All reactions were monitored by TLC.c Isolated yield. | |||||
| 1 | H | H | 4a | 25 | 89 |
| 2 | H | 4-Cl | 4b | 30 | 86 |
| 3 | H | 4-OH | 4c | 35 | 84 |
| 4 | H | 2-NO2 | 4d | 40 | 82 |
| 5 | H | 2-OMe, 4-OH | 4e | 30 | 78 |
| 6 | 4-Me | H | 4f | 30 | 91 |
| 7 | 4-Me | 4-Cl | 4g | 25 | 94 |
| 8 | 4-Me | 4-OH | 4h | 30 | 88 |
| 9 | 4-Me | 2-NO2 | 4i | 35 | 90 |
| 10 | 4-Me | 2-OMe, 4-OH | 4j | 40 | 87 |
To analyze the role of ultrasonic irradiation, the reactions were performed in the presence of the same amount of CoFe2O4-HMS in PEG 400 medium, under different conditions, together with stirring and refluxing. The obtained results were compared with the results from sonication and summarised in Table 3. This revealed that the reactions under ultrasonic irritation led to comparatively higher yields and shorter reaction times compared to both stirring and refluxing conditions. It is assumed that the efficiency with the use of ultrasound irritation is due to the cavitation phenomenon. The chemical and physical effects of ultrasound are primarily due to acoustic cavitations, which involve design, escalation and collapse of the cavity. A substantial concentration of energy is instigated by an alteration in the temperature of the bubble of kinetic energy of the fluid due to the collapse of the bubble in the fluid. The high temperatures and pressures arising from cavitations produce a set of ultrasonic applications consisting of accelerating the rate of the reaction, amending the reaction pathway, and enhancing chemical reactivity, and thus have significant use in the synthesis of organic compounds.20,25 To assist the sustainability of the protocol regarding the green chemistry approach, the methodology was assessed based on the atom economy,27 carbon efficiency, reaction mass efficiency,28 eco-scale score,29 and E-factor values.30 The reaction of 2-amino-4methyl pyridine (1b), 4-chlorobenzaldehyde (2b) and phenyl acetylene (3) were considered for the estimation of these parameters. The atom economy of 93.60% and 74.07% carbon efficiency with an eco-scale factor of 75 demonstrated the feasibility of this methodology. The estimate of green chemistry metrics is summarized in the ESI.† The E-factor, mass intensity and reaction mass efficiency for the model reaction are 0.0639%, 1.0639% and 90.26%, respectively. These criteria support that this method is a clean and green synthetic approach for the synthesis of amalgamated imidazo-pyridine heterocyclic scaffolds.
| Entry | R1 (amino pyridine) | R2 (aryl aldehydes) | Time (min) | Yieldb (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Sc | Rd | Ue | Sc | Rd | Ue | |||
| a Reaction conditions: 2-aminopyridine derivatives (1 mmol) (1a–b), pertinent aryl aldehyde (1 mmol) (2a–e), phenyl acetylene (1 mmol) (3) and a catalytic amount (7 mol%) of CoFe2O4-HMS in the presence of PEG 400.b Yields refer to isolated products.c Stirring condition.d Refluxing condition.e Ultrasonic condition. | ||||||||
| 1 | H | H | 45 | 40 | 25 | 85 | 76 | 89 |
| 2 | H | 4-Cl | 55 | 45 | 30 | 74 | 69 | 86 |
| 3 | H | 4-OH | 65 | 50 | 35 | 80 | 73 | 84 |
| 4 | H | 2-NO2 | 75 | 55 | 40 | 79 | 67 | 82 |
| 5 | H | 2-OMe, 4-OH | 55 | 40 | 30 | 71 | 64 | 78 |
| 6 | 4-Me | H | 50 | 45 | 30 | 85 | 79 | 91 |
| 7 | 4-Me | 4-Cl | 55 | 40 | 25 | 88 | 81 | 94 |
| 8 | 4-Me | 4-OH | 60 | 45 | 30 | 72 | 63 | 88 |
| 9 | 4-Me | 2-NO2 | 55 | 40 | 35 | 83 | 75 | 90 |
| 10 | 4-Me | 2-OMe, 4-OH | 75 | 60 | 40 | 76 | 66 | 87 |
According to the literature and our results,11,14,22,25,31–35 a plausible reaction mechanism to account for the formation of imidazo[1,2-a]pyridines is presented in Scheme 3. The domino reaction begins with condensation between 2-aminopyridine (1a–b) and the pertinent aryl aldehyde derivatives (2a–e), followed by dehydration, resulting in the formation of a Schiff base (A). In the A3-coupling strategy, where the basic sites of HMS promote the formation of cobalt–ferrite acetylide complex (3)′ by the activation of a proton from phenyl acetylene (3), the cobalt–ferrite acetylide (3)′ attacks the preformed iminium ion (A) to form intermediate (B). Consequently, the latter undergoes a 5-exo dig cyclization step initiated by an attack on the alkyne unit by the pyridine nitrogen, which results in the formation of imidazo-fused pyridine systems (4a–j).
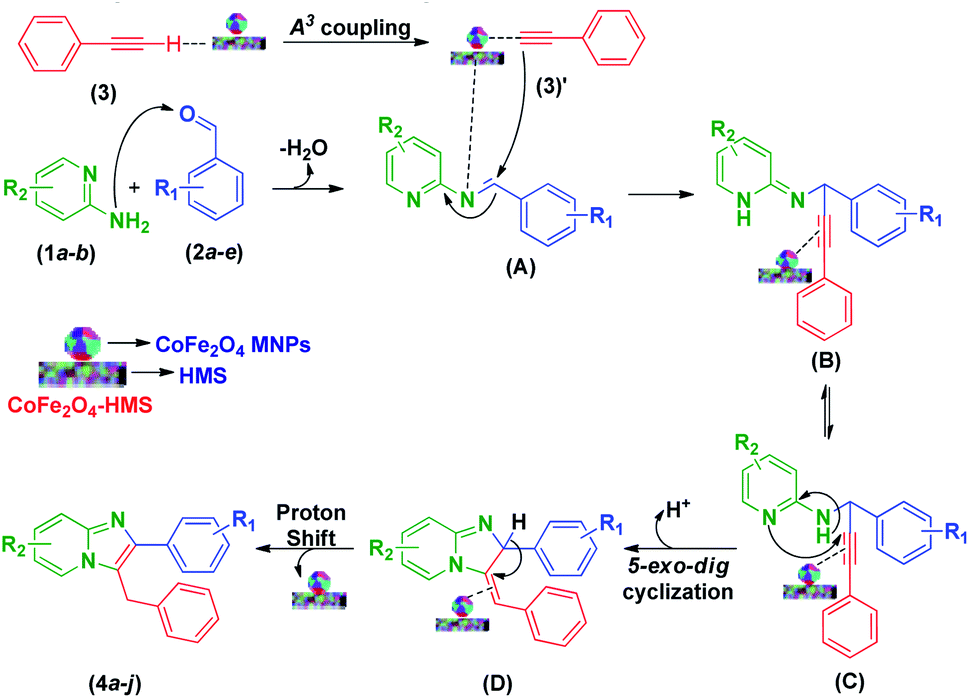 | ||
| Scheme 3 Plausible mechanistic pathway for the formation of multi-substituted imidazo[1,2-a]pyridine scaffolds. | ||
2.2. Recycling and reusability of magnetic nanocomposite and solvent
The reusability and recyclability of a catalytic framework are of great relevance in industrial processes. Thus, to assess the level of reusability and stability of the synthesized magnetic nanocomposite, we executed the reaction of 2-amino-4-methyl pyridine (1b) with 4-chloro benzaldehyde (2b) and phenyl acetylene (3) using PEG 400 as the reaction medium to acquire the desired product (4g). After the completion of the initial reaction, the magnetic nanoparticles were dragged to the inner sidewall of the reaction flask with the use of an external magnet. Then, the isolated catalyst was recovered according to the comprehensive procedure for the recovery of catalyst, as mentioned in the Experimental section.25,36 The magnetically recovered nanocatalyst was further subjected to the model reaction for the synthesis of (4g), (up to five runs) under analogous conditions. The outcomes of the recyclability of the nanocomposite are presented in Fig. S2(i) in the ESI.† It can be observed in Fig. S2(i)† that the recovered CoFe2O4-HMS catalyzed the reaction to give the product in 94%, 93%, 90%, 89%, and 87% yield. The significant yields of the product confirmed that there was no substantial deterioration in the catalytic activity. The morphology and crystalline structure of the recycled catalyst were characterized via FE-SEM and PXRD analyses (Fig. S3 and S4, respectively, in the ESI†). The analysis of the results revealed that there was no significant changes in the morphology and crystalline structure of the recovered catalyst.To investigate the recyclability of the used PEG 400, the solvent was collected after completion of the initial reaction, subjected to the normal workup and dried at 50 °C under vacuum. The recovered solvent was reused in the subsequent synthetic runs (up to five runs) for the model reaction. The results of the solvent recyclability are shown in Fig. S2(ii) in the ESI.† Furthermore, it can be seen from Fig. S2(ii)† that the recovered PEG 400 gave the product in 87%, 85%, 85%, 83%, and 82% yield.25
2.3. Heterogeneous nature of CoFe2O4-HMS magnetic nanocatalyst
The heterogeneity of the CoFe2O4-HMS magnetic nanocatalyst was studied by performing a hot filtration test experiment for the model reaction of 2-amino-4-methyl pyridine (1b) with 4-chlorobenzaldehyde (2b) and phenyl acetylene (3) in the presence of PEG 400 to afford the product (4g). The reaction was stopped after 15 min at around 50% conversion of the starting materials and the catalyst was filtered from the reaction mixture. There was no substantial improvement in the progress of the reaction upon further continuing it with the filtrate for the next 10 min after the removal of the catalyst. Moreover, the conversion remained constant with 57% yield even after a reaction time of 40 min, which revealed that the reaction occurred only due to the presence of the CoFe2O4-HMS MNPs. The filtrate was further analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES). The ICP-AES analysis of the filtrate did not show a detectable amount of cobalt and iron ions, which confirms the heterogeneous nature of the catalyst with its negligible leaching into the reaction mixture. The result of the hot filtration test and the leaching effect of the CoFe2O4-HMS magnetic nanocatalyst is depicted in Fig. S5 of the ESI.†2.4. Comparison of the results of the present protocol with the reported methods
A comparative report of the literature protocols for the synthesis of amalgamated imidazo-pyridine derivatives and our present work is summarized in Table 4. Gratifyingly, in comparison to the reported methodologies, our present approach offers prominent advantages including environmental acceptability, wide substrate scope, operational simplicity, practicability, economic viability, excellent yield of the products in a shorter reaction time, easy work-up, and reusability of both the magnetic nanocatalyst and solvent. Consequently, performing the reaction by utilizing a small amount of catalyst, reusability within multiple runs, and ecofriendly nature of CoFe2O4-HMS are among some of the value-added advantages of this newly synthesized magnetic nano-composite, leading to the highly efficient synthesis of multi-substituted imidazo-fused pyridine derivatives.| Entry | Condition | Solvent | Temperature (°C)/time | Yield (%) | Ref. |
|---|---|---|---|---|---|
| 1 | CuSO4·5H2O (10 mol%)/sodium ascorbate (20 mol%)/SDS (10 mol%) | H2O | 50/6–16 h | 52–89 | 11 |
| 2 | MNP@BiimCu (1.2 mol%)/CTAB (5 mg) | H2O | Reflux/5–8.5 h | 65–95 | 12 |
| 3 | CuO/CuAl2O4 (15 mol%)/D-glucose (30 mol%) | Toluene | Reflux/24 h | 41–89 | 13 |
| 4 | Fe3O4@SiO2 NPs (5 mol%)/K2CO3 | EtOH | Reflux/3–6 h | 77–90 | 14 |
| 5 | CuI (5 mol%)/L-proline | DMSO | 110/3 h | 76–88 | 15 |
| 6 | Cu-NPs (1 mol%)/N2 | — | 120/12–16 h | 14–85 | 16 |
| 7 | Nano-copper/nano-ZnAl2O4/N2 | — | 90/4–6 h | 89–94 | 17 |
| 8 | PW-CIS500 (0.9 mol%) | — | 100/2.5–3.5 h | 82–95 | 18 |
| 9 | CoFe2O4@CNT–Cu (5 mol%) | PEG 400 | 80/3–6 h | 80–96 | 19 |
| 10 | CoFe2O4–HMS (7 mol%) | PEG 400 | )))/25–40 min | 78–94 | Our work |
Furthermore, a comparative account of the synthesized catalyst with some reported catalytic systems for the formation of the model compound (4a) via the A3 coupling strategy followed by a 5-exo-dig cyclization approach is presented in Table S1 of the ESI.†
2.5. Characterization of nanocatalysts
D = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ |
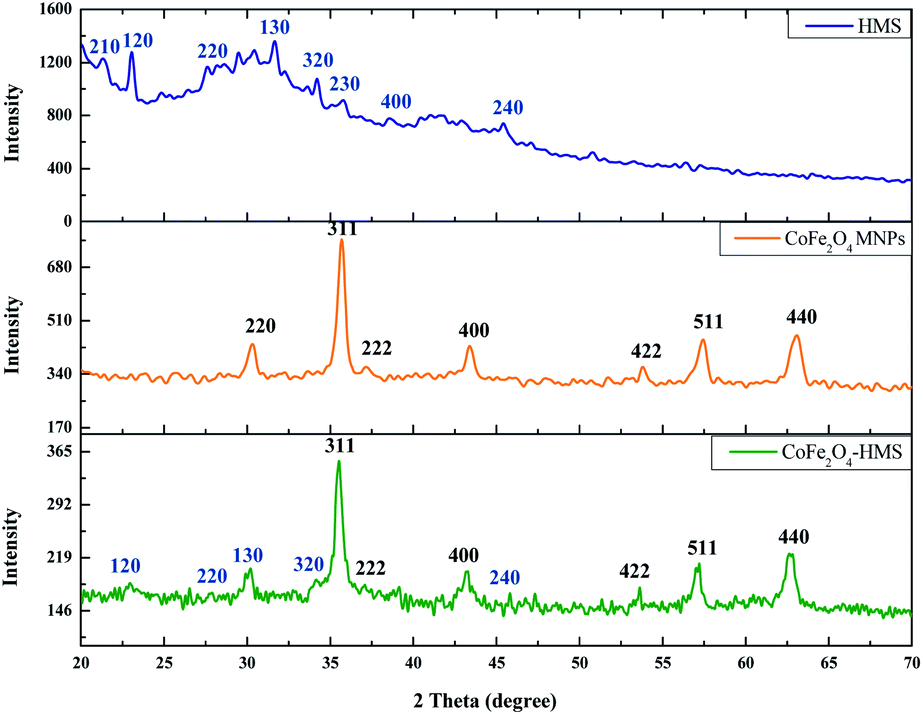 | ||
| Fig. 2 PXRD diffraction pattern of CoFe2O4-HMS, CoFe2O4 MNPs and HMS. | ||
 | ||
| Fig. 3 Field-emission scanning electron microscopy (FE-SEM) images of (A–F) CoFe2O4-HMS and (G and H) CoFe2O4 MNPs and (I and J) HMS at different magnifications. | ||
The powder X-ray diffractograms of CoFe2O4-HMS, CoFe2O4 MNPs and HMS are shown in Fig. 2, showing broadened peaks centred in the 2θ range of 20°–70°. The diffraction pattern of HMS could be indexed to monoclinic symmetry with refined lattice parameters of a-axis of about 10.12 Å, b-axis of 8.93 Å and c-axis of 8.38 Å with β = 114.56°. The major peaks corresponding to the (210), (120), (220), (130), (320), (230), (400) and (240) planes at 2θ positions of 21.33°, 23.04°, 27.60°, 31.66°, 34.24°, 35.77°, 38.57° and 45.44°, respectively, are the characteristic peaks of hydromagnesite sheets according to the standard JCPDS card no. 25-513.38
The peaks centred at 30.33°, 35.71°, 37.20°, 43.40°, 53.80°, 57.44° and 63.08° are attributed to the presence of (220), (311), (222), (400), (422), (511) and (440) crystal planes of inverse spinel CoFe2O4 MNPs, respectively.25,39 These cores can be indexed as inverse spinel and face-centred cubic (fcc) structure (CoFe2O4 MNPs), according to the standard JCPDS card no. 10-0325.40,41 The most intense diffraction peak is at 2θ = 35.678° and has a plane spacing of d = 2.5145 Å, which corresponds to the (311) plane and is characterized by cubic spinel ferrite. This confirmed the formation of a single-phase face-centred cubic spinel structure with no extra peaks corresponding to other crystallographic phases.25,41 The presence of a peak at 30.100° with d-spacing = 2.9664 Å (which corresponds to HMS) and 35.521° with d-spacing = 2.5251 Å (which corresponds to CoFe2O4 MNPs) in the diffraction pattern of the CoFe2O4-HMS composite, indicates the successful formation of the corresponding hybrid material. The sharpness of the peaks indicates the crystalline nature of the cobalt–ferrite nanoparticles and their impregnation in the hydromagnesite sheets.
The chemical purity of the as-synthesized nanocomposite sample was verified via EDAX studies. The characteristic peaks in the EDAX spectrum (Fig. 4) confirm the presence of cobalt (Co), iron (Fe), oxygen (O), magnesium (Mg) and carbon (C) as the only elements in the decorated magnetic nanoparticles. The spectrum revealed that the amount of cobalt and iron stabilized on the HMS was 12.33 at% and 25.57 at%, respectively. The content of cobalt and iron in CoFe2O4-HMS was found to be 23.23 and 46.04 wt%, respectively, which are in accordance with the amount of Co (23.14 wt%) and Fe (45.97 wt%) analyzed by the ICP-AES technique.
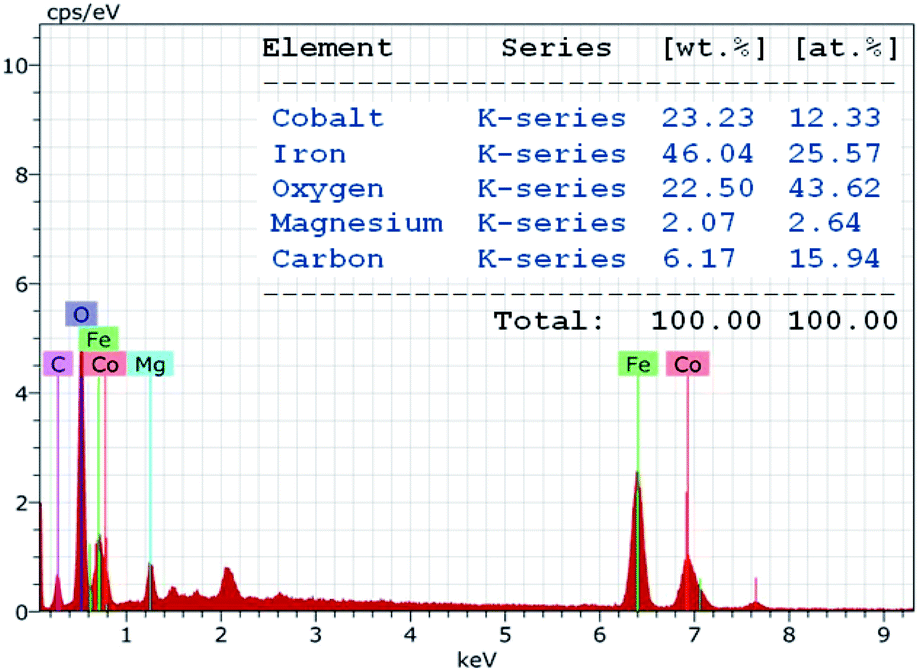 | ||
| Fig. 4 EDAX pattern of CoFe2O4-HMS. | ||
The surface and internal morphologies of the as-synthesized hydromagnesite sheets (HMS) were further determined via high-resolution transmission electronic microscopy (HR-TEM), as shown in Fig. 5. The results revealed the existence of thin and transparent porous flakes with a 2D sheet morphology of HMS at different magnifications. The polycrystalline nature of the Mg (CO3)4(OH)2·4H2O sheets (HMS) was authenticated using selected area electron diffraction (SAED).
 | ||
| Fig. 5 Selected area electron diffraction (SAED) pattern and high-resolution transmission electron microscopy (HR-TEM) images of hydromagnesite sheets (HMS) at different magnifications. | ||
The broad band at around 3600–2500 cm−1 can be ascribed to the O–H stretching vibration of the carboxyl functional groups produced on their surface. The bands at 2981 and 2891 cm−1 are attributed to the stretching vibration of the C–H group. The characteristic peaks of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration are located at 1644 and 1634 cm−1 in the composite. The bands in the range of 1476 to 880 cm−1 are ascribed to the O–H bending of the carboxylic groups, whereas the bands at 1348 and 1251 cm−1 are due to the –CH bending. The band in the spectrum at around 1155–1070 cm−1 shows the presence of C–O group vibrational modes. The metal–oxygen bands with peaks range in the range of ∼723 to ∼534 cm−1 are ascribed to the deformation vibration of the Fe–OH groups. These two bands are usually assigned as the vibration of ions in the crystal lattices. This indicates the presence of continuously distributed ferrite particles. The band at ∼588 cm−1 corresponds to the intrinsic stretching vibrations of the metal at the tetrahedral site (Fe ↔ O), whereas the band at ∼493 is assigned to the octahedral-metal stretching (Co ↔ O).25,39,41 Moreover, the FTIR spectra of CoFe2O4 MNPs and HMS were also recorded and compared with that of the hydromagnesite sheets impregnated with cobalt ferrite, as displayed in Fig. 6. The functional groups with their wavenumber values for the samples are summarized in Table S2, in the ESI.†
O stretching vibration are located at 1644 and 1634 cm−1 in the composite. The bands in the range of 1476 to 880 cm−1 are ascribed to the O–H bending of the carboxylic groups, whereas the bands at 1348 and 1251 cm−1 are due to the –CH bending. The band in the spectrum at around 1155–1070 cm−1 shows the presence of C–O group vibrational modes. The metal–oxygen bands with peaks range in the range of ∼723 to ∼534 cm−1 are ascribed to the deformation vibration of the Fe–OH groups. These two bands are usually assigned as the vibration of ions in the crystal lattices. This indicates the presence of continuously distributed ferrite particles. The band at ∼588 cm−1 corresponds to the intrinsic stretching vibrations of the metal at the tetrahedral site (Fe ↔ O), whereas the band at ∼493 is assigned to the octahedral-metal stretching (Co ↔ O).25,39,41 Moreover, the FTIR spectra of CoFe2O4 MNPs and HMS were also recorded and compared with that of the hydromagnesite sheets impregnated with cobalt ferrite, as displayed in Fig. 6. The functional groups with their wavenumber values for the samples are summarized in Table S2, in the ESI.†
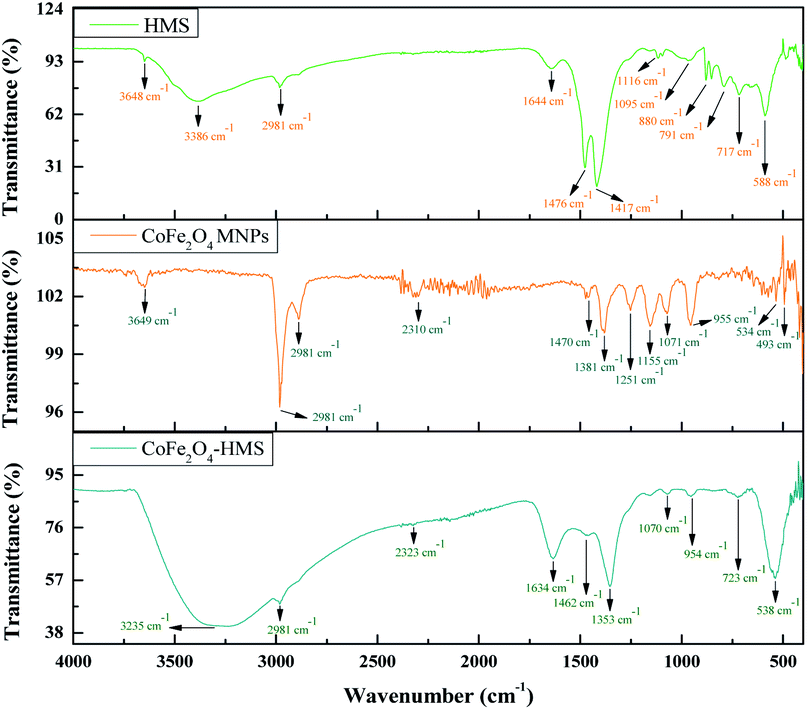 | ||
| Fig. 6 FT-IR spectra of CoFe2O4-HMS, CoFe2O4 MNPs and HMS. | ||
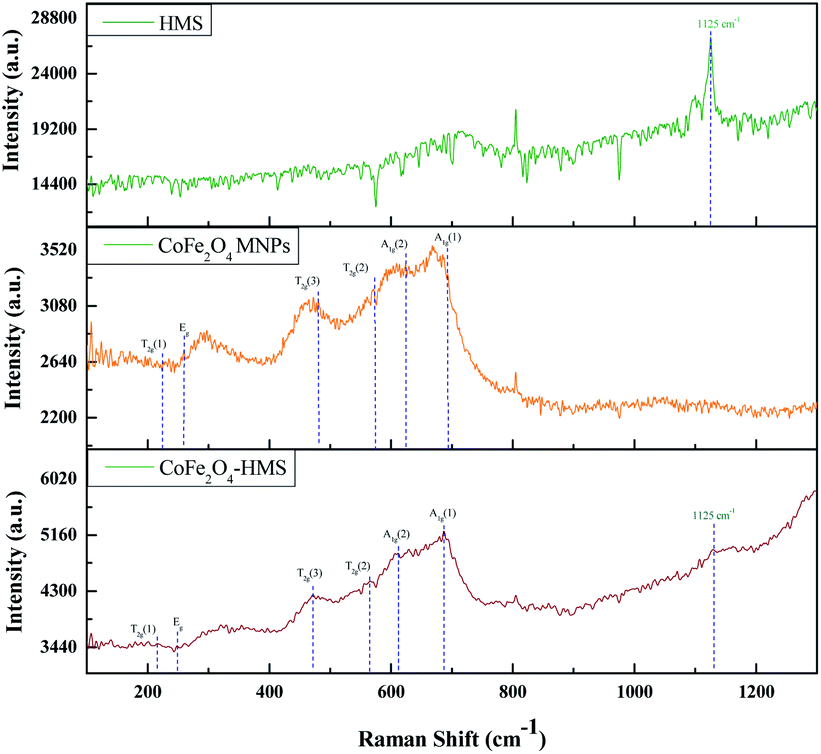 | ||
| Fig. 7 Raman spectra of CoFe2O4-HMS, CoFe2O4 MNPs and HMS. | ||
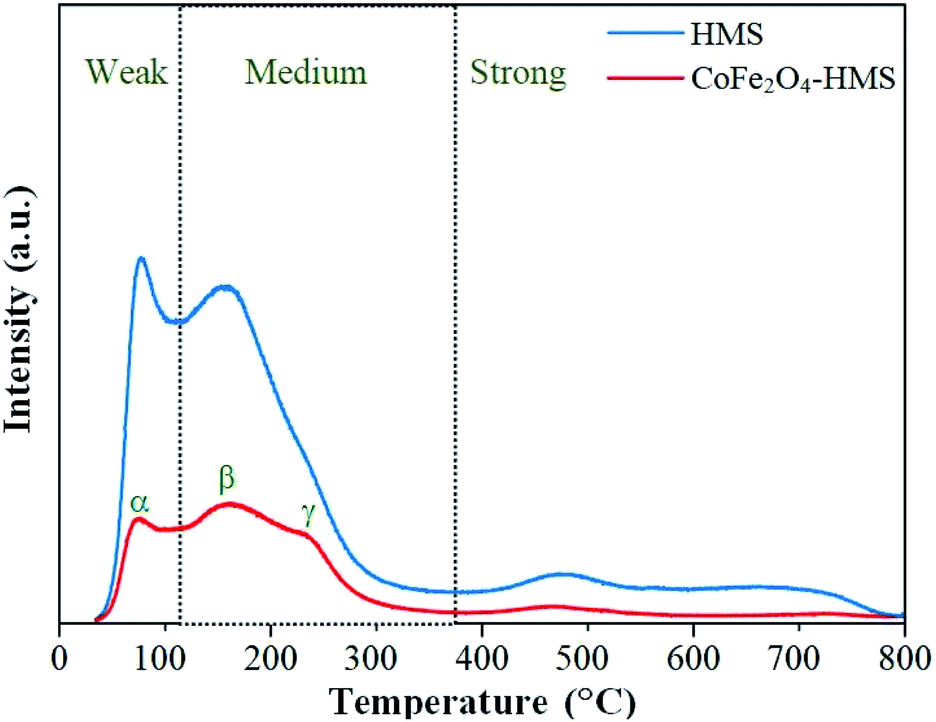 | ||
| Fig. 8 CO2-TPD profiles of HMS and CoFe2O4-HMS. | ||
After impregnation of the cobalt ferrite magnetic nanoparticles on the hydromagnesite sheets, the positions of the desorption peaks slightly changed. This implies that the strength of the basic sites on the surface of CoFe2O4-HMS and HMS remained the same, where the increase in the area of the desorption peaks after impregnation is attributed to the increment in surface area and coercivity with a decrease in the crystalline size of CoFe2O4-HMS.
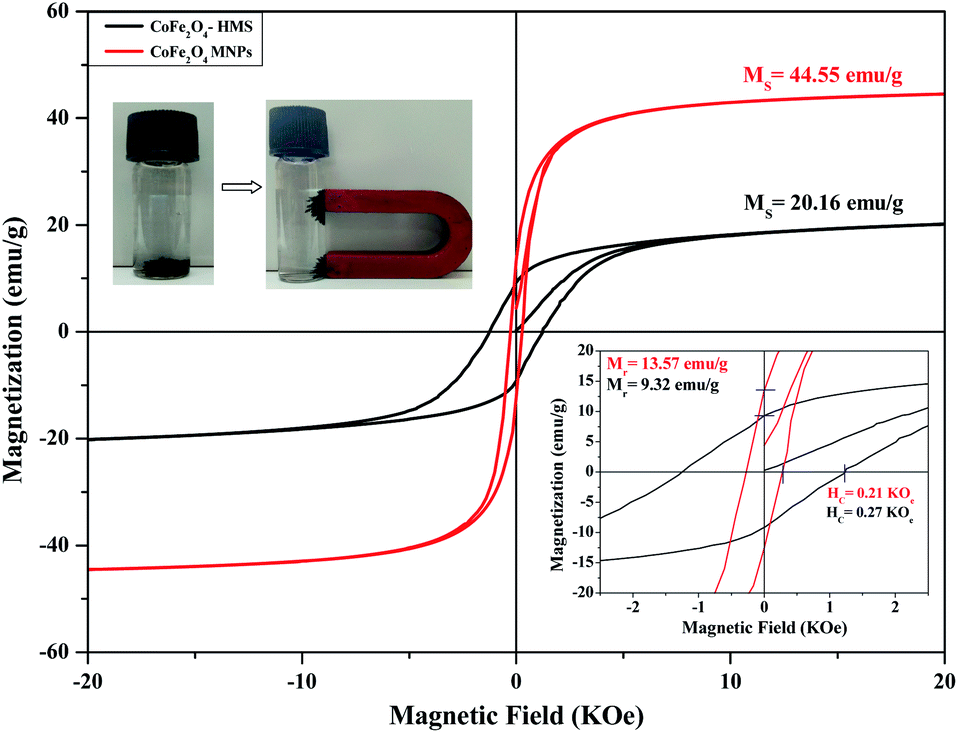 | ||
| Fig. 9 Hysteresis curves of CoFe2O4-HMS and CoFe2O4 MNPs at room temperature (300 K). | ||
The decrease in the saturation magnetisation can be attributed to the non-collinear spin arrangement at or near the surface of the particles. We observed that the saturation magnetisation for the smaller crystals has a significantly lower value for the ferrite impregnated in HMS than for the bare cobalt ferrite. The values of Ms for the samples are attributed to the greater fraction of surface spin glasslike state with a smaller net moment. The hysteresis curve reveals an increment in the coercivity value for CoFe2O4-HMS compared to that of the bare CoFe2O4 magnetic nanoparticles. This increment may be due to the decrease in the particle size with less magnetization. The decrease in magnetization and increase in coercivity for smaller particles is attributed to a decrease in domain wall motion and different anisotropies of the surface and bulk material.39,41 The trends for the various magnetic parameters are summarized in Table 5.
| S. no. | Composition | Saturation magnetization Ms (emu g−1) | Retentivity Mr (emu g−1) | Coercivity Hc (kOe) |
|---|---|---|---|---|
| 1 | CoFe2O4-HMS | 20.16 | 9.32 | 0.27 |
| 2 | CoFe2O4 MNPs | 44.55 | 13.57 | 0.21 |
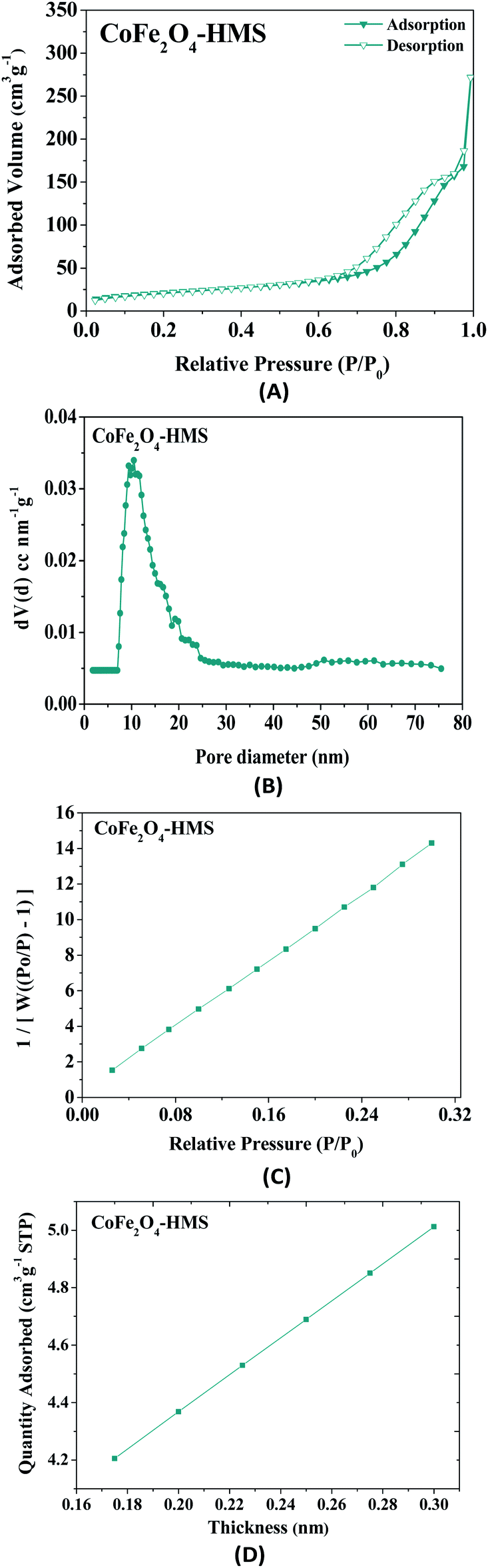 | ||
| Fig. 10 (A) Nitrogen adsorption isotherm pattern of CoFe2O4-HMS, (B) pore size distribution, (C) BET surface area and (D) t-plot analysis. | ||
The BET surface area of CoFe2O4-HMS was found to be 75 m2 g−1. The samples exhibited mesoporosity with a pore volume of 0.4207 cm3 g−1. The experimental point of the t-plot, as shown in Fig. 10D is consistent with the Harkins and Jura isotherm equation.46 It is evident from the plot that the experimental data points fall in a straight line for t = 0.17–0.30 nm (linear portion of the curve). Thus, the hydromagnesite sheets impregnated with cobalt–ferrite magnetic nanoparticles were found to be porous in nature, given that the t-plot did not pass through the origin. The fitted linear line exhibits a positive intercept, which further substantiates the presence of mesopores in nanocomposites.46–48 The values of the surface area, pore diameter, and pore volume obtained from the above-mentioned studies indicate that CoFe2O4-HMS possessed a wide mesopore size distribution with the maximum centred at 10.50 nm.
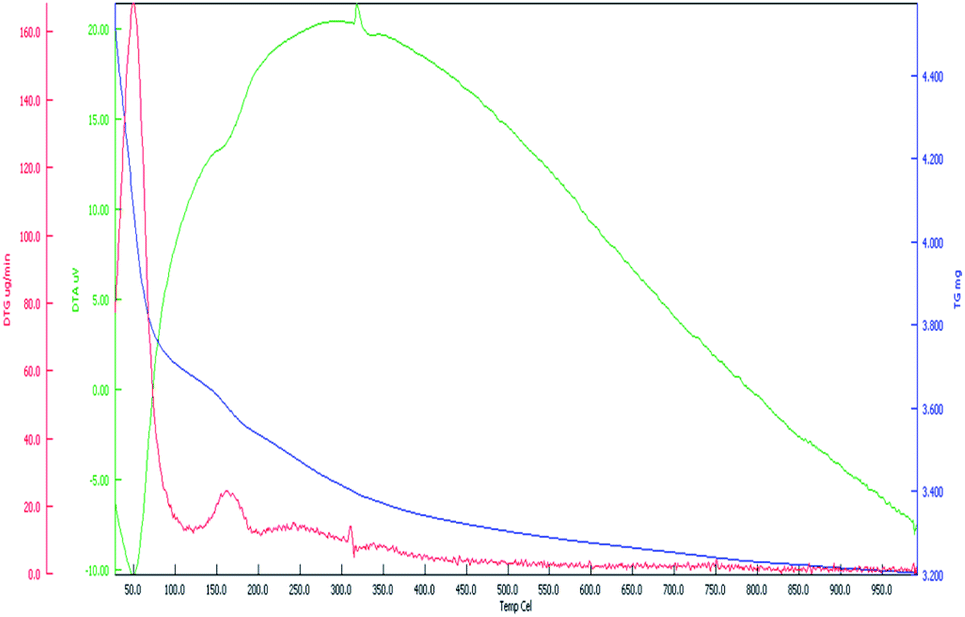 | ||
| Fig. 11 TGA, DTA and DTG patterns for CoFe2O4-HMS. | ||
The first stage occurred in the temperature range of 100–160 °C, which is attributed to the dehydration of crystalline adsorbed water on the catalyst surface with a weight loss of 17.80%, corresponding to the very feeble endothermic peak at 130–170 °C in the DTA (green line) curve. Besides, there was a further weight loss of about 36.70%, which is associated with the evolution of CO2, in the temperature range of 170 °C to 250 °C. The second endothermic peak at around 250–340 °C corresponds to the decomposition of amorphous hydrocarbonates, M2CO3(OH)2, which were generated at high temperature before the production of metal oxide. Consequently, the total weight loss over the CoFe2O4-HMs magnetic nanocatalyst in both steps was determined to be 54.50%. The DTA curve indicates that the decomposition took place faster between 100 °C to 340 °C and slowly until the maximum temperature of 1000 °C. Therefore, given that the sample weight remained constant after 1000 °C, this temperature is considered a suitable calcination temperature for CoFe2O4-HMS in this trans-esterification reaction.25,49
3. Experimental
All chemicals and solvents were purchased from Sigma Aldrich and Merck India. The reactions were performed in an aerobic atmosphere without any specific precautions. Melting points were determined in open capillary tubes on a Veego melting-point apparatus and are uncorrected. The ultrasound-promoted reactions were performed using a Cole Parmer ultrasonic processor Model CPX 130, with a maximum power of 130 W, operating at an amplitude of 60% and frequency of 20 kHz. All products were characterized using spectroscopic data (FTIR, 1H and 13C NMR). The 1H and 13C NMR spectra of the synthesized compounds were recorded at 400 and 100 MHz respectively, using a Bruker Advance II 400 NMR spectrometer in chloroform (CDCl3) solvent, and the chemical shifts (δ) are expressed in parts per million. Spin multiplicities are described as s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Fourier transform infrared (FTIR) spectra were recorded as ATR spectra in the range of 4000–650 cm−1 using a Frontier PerkinElmer FTIR SP 10 STD. Thin-layer chromatography (TLC) was performed using precoated aluminum sheets with Silica Gel 60 F254. The Brunauer–Emmett–Teller (BET) surface area analysis was carried out using a QuantachromeAutosorb iQ2 TPX gas sorption system equipped with highly accurate pressure transducers and a thermostatic bath. The pore size distribution was calculated using density functional theory (DFT). Raman spectra were recorded on a Micro Raman microscope (Jobin Yvon Horibra LABRAM-HR visible 800) equipped with a 532 nm Nd:YAG solid state laser (10 mW). The PXRD measurements were carried out on a Bruker D8 Advance X-ray diffractometer using Cu Kα radiation with a wavelength of 0.154 nm. Field-emission scanning electron microscopy (FE-SEM) images and the corresponding electron dispersive X-ray analysis (EDAX) data were recorded using a Jeol microscope (JSM7100F). The surface basic property was determined through the temperature-programmed desorption of CO2 (CO2-TPD) using a Chemisorb 2750 (Micromeritics) instrument. Thermogravimetric analysis-differential thermal analysis (TGA-DTA) was performed on a Hitachi (STA 7300) thermal analysis instrument under an air flow in the temperature range of 25 °C to 1000 °C at a heating rate of 10 °C min−1. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis was performed on a PerkinElmer, Optima 2000. Magnetic measurements were carried out on a vibrating sample magnetometer (VSM, JDM-13) at room temperature. A 2100F high-resolution transmission electronic microscope (HR-TEM) operating at 200 kV was used to examine the grain size and morphology of the nanosheets.25,263.1. Preparation of hydromagnesite sheets (HMS)
The hydromagnesite sheets were synthesized according to the procedure available in the literature.22 In a typical procedure, 120 mL of aqueous 1 M Na2CO3 was slowly added from a burette to a 500 mL round-bottomed flask containing 200 mL of aqueous 1 M MgCl2·6H2O solution under vigorous stirring at room temperature to afford a white precipitate.3.2. Preparation of hydromagnesite sheets impregnated with cobalt-ferrite magnetic nanoparticles (CoFe2O4-HMS)
The synthesis of hydromagnesite sheets impregnated with cobalt–ferrite magnetic nanoparticles was executed via the chemical co-precipitation method according to a literature protocol.19,25 The acquired HMS (0.16 g) were dispersed in 40 mL of distilled water and sonicated at room temperature for 30 min. A homogenous dispersion of CoCl2·6H2O (1.19 g), FeCl3·6H2O (2.70 g) and 10 mL of distilled water was added to the solution of HMS. Afterward, 25 mL of 3 mol L−1 NaOH solution was slowly added to the above-mentioned solution under vigorous stirring. The mixture was stirred continuously at 100 °C for one hour. The obtained black solid of CoFe2O4-HMS was magnetically separated, washed with water and ethanol until the pH reached 7.0, and dried under vacuum at 60 °C for 24 h. The structural, morphological and magnetic properties of HMS, CoFe2O4 MNPs and CoFe2O4-HMS were characterized via PXRD (Fig. 2), FE-SEM (Fig. 3), EDAX analysis (Fig. 4), HR-TEM (Fig. 5), FTIR analysis (Fig. 6), Raman analysis (Fig. 7), CO2-TPD (Fig. 8), magnetic studies (Fig. 9), nitrogen BET adsorption–desorption pattern (Fig. 10), and TGA-DTA-DTG pattern (Fig. 11).3.3. General procedure for the ultrasonic-assisted synthesis of imidazo[1,2-a]pyridines derivatives (4a–j)
A mixture of 2-aminopyridine derivatives (1 mmol) (1a–b), pertinent aryl aldehyde (1 mmol) (2a–e), phenyl acetylene (1 mmol) (3) and a catalytic amount (7 mol% or 0.07 g) of CoFe2O4-HMS in PEG 400 (5 mL) was irradiated employing an ultrasonicator at room temperature for the optimal time (25–40 min). Considering that the reaction time was very short, there was no substantial increase in temperature due to ultrasonic shock. The ultrasonic apparatus used showed the temperature automatically, and thus the temperature was controlled and fixed at room temperature using a water circulator in the case of any temperature increase.20 The progress of the reaction was monitored through TLC (on aluminum sheets precoated with silica) using n-hexane/ethyl acetate (4:1) as the eluting system. After the reaction was complete, the mixture was diluted with ethyl acetate (2 mL). The organic layer was washed with water and dried over MgSO4 and concentrated under vacuum. The catalyst was separated via a magnetic bar to obtain pure products (4a–j) (Table 2).25
The analytical and spectroscopic data for the synthesized imidazo[1,2-a]pyridines derivatives (4a–j) are given below:
C/CN), 1611, 1480, 1434 (C⋯C ring str.); 1H NMR (400 MHz, CDCl3): δH ppm 4.53 (s, 2H, CH2), 6.92–6.96 (m, 1H), 7.17–7.22 (m, 2H), 7.23–7.26 (m, 1H), 7.29–7.40 (m, 4H), 7.43–7.48 (m, 2H), 7.70–7.73 (m, 2H), 7.80–7.83 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 29.9 (C-16), 112.5 (C-9), 117.5 (C-7), 117.8 (C-6), 123.5 (C-8), 124.6 (C-5), 127.0 (C-13, 20), 127.8 (C-11, 15), 128.4 (C-19, 21), 128.8 (C-18, 22), 129.2 (C-12, 14), 134.3 (C-10), 136.8 (C-17), 144.0 (C-4), 145.0 (C-2); HRMS (ESI) m/z: 284.1306 [M + H]+; Anal. Cald. for C20H16N2: C, 84.48; H, 5.67; N, 9.85%. Found: C, 84.49; H, 5.69; N, 9.89%.C/CN), 1613, 1481, 1436 (C⋯C ring str.), 698 (C–Cl); 1H NMR (400 MHz, CDCl3): δH ppm 4.58 (s, 2H, CH2), 6.91–6.94 (m, 1H), 7.16–7.21 (m, 2H), 7.22–7.26 (m, 1H), 7.27–7.29 (m, 3H), 7.30–7.38 (m, 2H), 7.69–7.73 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 29.9 (C-16), 112.5 (C-9), 117.4 (C-7), 117.6 (C-6), 123.5 (C-8), 127.2 (C-20), 127.2 (C-5), 128.6 (C-19, 21), 129.1 (C-11, 15), 129.2 (C-18, 22), 129.5 (C-12, 14), 129.8 (C-10), 134.4 (C-13), 136.6 (C-17), 144.0 (C-4), 144.8 (C-2); HRMS (ESI) m/z: 318.0915 [M + H]+; Anal. Cald. for C20H15ClN2: C, 75.35; H, 4.74; N, 8.79%. Found: C, 75.38; H, 4.77; N, 8.82%.C/CN), 1613, 1482, 1453 (C⋯C ring str.), 1087 (C–O); 1H NMR (400 MHz, CDCl3): δH ppm 4.52 (s, 2H), 6.28 (s, 1H), 6.92–6.95 (m, 1H), 7.07–7.12 (m, 2H), 7.17–7.21 (m, 2H), 7.23–7.26 (m, 1H), 7.27–7.30 (m, 3H), 7.31–7.38 (m, 2H), 7.69–7.73 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 29.9 (C-16), 112.5 (C-9), 116.5 (C-12, 14), 117.4 (C-7), 117.8 (C-6), 123.6 (C-8), 125.5 (C-10), 127.1 (C-20), 127.2 (C-5), 129.1 (C-19, 21), 129.3 (C-11, 15), 130.0 (C-18, 22), 136.8 (C-17), 144.1 (C-4), 144.8 (C-2), 158.6 (C-13); HRMS (ESI) m/z: 300.1256 [M + H]+; Anal. Cald. for C20H16N2O: C, 79.98; H, 5.37; N, 9.33%. Found C, 80.02; H, 5.41; N, 9.38%.C/CN), 1543 (N–O), 1611, 1481, 1453 (C⋯C ring str.); 1H NMR (400 MHz, CDCl3): δH ppm 4.51 (s, 2H, CH2), 6.91–6.93 (m, 1H), 7.16–7.19 (m, 2H), 7.21–7.24 (m, 1H), 7.27–7.31 (m, 3H), 7.67–7.71 (m, 2H), 7.78–7.81 (m, 1H), 7.93–7.96 (m, 1H), 8.08–8.11 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 29.9 (C-16), 112.6 (C-9), 117.4 (C-7), 117.8 (C-6), 123.6 (C-8), 124.5 (C-12), 124.9 (C-5), 125.3 (C-10), 127.1 (C-20), 128.5 (C-19, 21), 128.9 (C-18, 22), 129.7 (C-13), 132.7 (C-15), 135.4 (C-14), 136.8 (C-17), 144.2 (C-4), 144.9 (C-2), 148.9 (C-11); HRMS (ESI) m/z: 329.1156 [M + H]+; Anal. Cald. for C20H15N3O2: C, 72.94; H, 4.59; N, 12.76%. Found: C, 72.97; H, 4.62; N, 12.79%.C/CN), 1613, 1483, 1439 (C⋯C ring str.), 1287, 1082 (C–O); 1H NMR (400 MHz, CDCl3): δH ppm 3.94 (s, 3H, OCH3), 4.53 (s, 2H, CH2), 6.30–6.74 (s, 2H), 6.96–6.99 (m, 1H), 7.07–7.11 (m, 1H), 7.17–7.20 (m, 2H), 7.21–7.23 (m, 1H), 7.25–7.34 (m, 3H), 7.38–7.41 (m, 2H), 7.71–7.74 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 29.9 (C-16), 56.1 (C-11′), 106.2 (C-12), 108.8 (C-14), 112.6 (C-9), 115.6 (C-10), 117.4 (C-7), 117.9 (C-6), 124.3 (C-8), 126.0 (C-20), 127.8 (C-5), 129.1 (C-19, 21), 130.1 (C-18, 22), 132.0 (C-15), 137.0 (C-17), 144.1 (C-4), 144.8 (C-2), 154.8 (C-13), 158.7 (C-11); HRMS (ESI) m/z: 330.1354 [M + H]+; Anal. Cald. for C21H18N2O2: C, 76.34; H, 5.49; N, 8.48%. Found C, 76.37; H, 5.51; N, 8.52%.C/CN), 1612, 1487, 1449 (C⋯C ring str.); 1H NMR (400 MHz, CDCl3): δH ppm 2.47 (s, 3H, CH3), 4.57 (s, 2H, CH2), 7.16–7.19 (m, 1H), 7.29–7.32 (m, 1H), 7.34–7.40 (m, 3H), 7.43–7.48 (m, 2H), 7.52 (s, 1H), 7.70–7.73 (m, 3H), 7.81–7.84 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 21.4 (C-8′), 29.9 (C-16), 116.9 (C-9), 117.5 (C-7), 122.1 (C-6), 124.9 (C-5), 125.7 (C-20), 127.7 (C-11, 15), 128.7 (C-19, 21), 128.8 (C-13), 128.9 (C-18, 22), 129.3 (C-12, 14), 134.1 (C-10), 136.9 (C-17), 139.8 (C-8), 144.1 (C-4), 144.8 (C-2); HRMS (ESI) m/z: 298.1462 [M + H]+; Anal. Cald. for C21H18N2: C, 84.53; H, 6.08; N, 9.39%. Found: C, 84.56; H, 6.11; N, 9.44%.C/CN), 1612, 1482, 1438 (C⋯C ring str.), 686 (C–Cl); 1H NMR (400 MHz, CDCl3): δH ppm 2.48 (s, 3H, CH3), 4.55 (s, 2H, CH2), 7.17–7.19 (m, 1H), 7.28–7.31 (m, 1H), 7.33–7.41 (m, 3H), 7.43–7.49 (m, 2H), 7.53 (s, 1H), 7.71–7.76 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 21.4 (C-8′), 29.9 (C-16), 116.9 (C-9), 117.5 (C-7), 122.2 (C-6), 124.9 (C-5), 125.8 (C-20), 128.7 (C-19, 21), 129.1 (C-11, 15), 129.2 (C-18, 22), 129.4 (C-12, 14), 131.2 (C-10), 134.4 (C-13), 137.0 (C-17), 139.8 (C-8), 144.2 (C-4), 144.9 (C-2); HRMS (ESI) m/z: 332.1068 [M + H]+; Anal. Cald. for C21H17ClN2: C, 75.78; H, 5.15; N, 8.42%. Found: C, 75.81; H, 5.18; N, 8.46%.C/CN), 1613, 1483, 1451 (C⋯C ring str.), 1089 (C–O); 1H NMR (400 MHz, CDCl3): δH ppm 2.47 (s, 3H, CH3), 4.53 (s, 2H, CH2), 6.29 (s, 1H), 7.06–7.11 (m, 2H), 7.16–7.19 (m, 1H), 7.21–7.25 (m, 1H), 7.29–7.34 (m, 3H), 7.43–7.48 (m, 2H), 7.51 (s, 1H), 7.71–7.74 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 21.4 (C-8′), 29.9 (C-16), 116.6 (C-12, 14), 116.9 (C-9), 117.5 (C-7), 122.3 (C-6), 125.8 (C-10), 127.2 (C-20), 127.2 (C-5), 129.2 (C-19, 21), 129.2 (C-11, 15), 130.1 (C-18, 22), 136.9 (C-17), 139.8 (C-8), 144.2 (C-4), 144.9 (C-2), 158.6 (C-13); HRMS (ESI) m/z: 314.1406 [M + H]+; Anal. Cald. for C21H18N2O: C, 80.23; H, 5.77; N, 8.91%. Found: C, 80.26; H, 5.79; N, 8.94%.C/CN), 1537 (N–O), 1611, 1487, 1448 (C⋯C ring str.); 1H NMR (400 MHz, CDCl3): δH ppm 2.46 (s, 3H, CH3), 4.56 (s, 2H, CH2), 7.17–7.19 (m, 1H), 7.21–7.24 (m, 1H), 7.28–7.33 (m, 3H), 7.59 (s, 1H), 7.68–7.71 (m, 2H), 7.76–7.80 (m, 1H), 7.92–7.95 (m, 1H), 8.08–8.10 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 21.4 (C-8′), 29.9 (C-16), 117.5 (C-7), 117.9 (C-9), 122.2 (C-6), 124.6 (C-12), 125.0 (C-5), 125.3 (C-10), 127.1 (C-20), 128.7 (C-19, 21), 128.9 (C-18, 22), 129.8 (C-13), 132.8 (C-15), 135.5 (C-14), 136.8 (C-17), 139.7 (C-8), 144.2 (C-4), 145.0 (C-2), 149.0 (C-11); HRMS (ESI) m/z: 343.1314 [M + H]+; Anal. Cald. for C21H17N3O2: C, 73.45; H, 4.99; N, 12.24%. Found: C, 73.48; H, 5.02; N, 12.29%.C/CN), 1613, 1488, 1437 (C⋯C ring str.), 1126, 1085 (C–O); 1H NMR (400 MHz, CDCl3): δH ppm 2.47 (s, 3H, CH3), 3.95 (s, 3H, OCH3), 4.52 (s, 2H, CH2), 6.30–6.73 (s, 2H), 7.08–7.13 (m, 1H), 7.17–7.19 (m, 1H), 7.24–7.29 (m, 1H), 7.34–7.40 (m, 3H), 7.43–7.48 (m, 2H), 7.56 (s, 1H), 7.73–7.78 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 21.5 (C-8′), 29.9 (C-16), 56.2 (C-11′), 106.0 (C-12), 108.9 (C-14), 112.6 (C-9), 115.6 (C-10), 117.5 (C-7), 122.2 (C-6), 126.1 (C-20), 128.0 (C-5), 128.8 (C-19, 21), 130.2 (C-18, 22), 132.9 (C-15), 137.0 (C-17), 139.9 (C-8), 144.2 (C-4), 144.8 (C-2), 154.9 (C-13), 158.8 (C-11); HRMS (ESI) m/z: 344.1519 [M + H]+; Anal. Cald. for C22H20N2O24: C, 76.72; H, 5.85; N, 8.13%. Found: C, 76.75; H, 5.89; N, 8.17%.3.4. General procedure for the ultrasonic-assisted synthesis of 2-benzyl-3-(4-chlorophenyl)-7-methylimidazo[1,2-a]pyridine (4g) (with maximum yield) on a gram-scale level
A mixture of 2-amino-4-methyl pyridine (10 mmol) (1b), 4-chlorobenzaldehyde (10 mmol) (2b), phenyl acetylene (10 mmol) (3) and a catalytic amount (7 mol% or 0.07 g) of CoFe2O4-HMS in PEG 400 (50 mL) was irradiated using an ultrasonicator at room temperature for the optimum time (25 min). The progress of the reaction was monitored through TLC (on aluminum sheets precoated with silica) using n-hexane/ethyl acetate (4:1) as the eluting system. After the completion of the reaction, the mixture was diluted with ethyl acetate (20 mL). The organic layer was washed with water and dried over MgSO4 and concentrated under vacuum. The catalyst was separated using a magnetic bar to give the anticipated product (4g) in 90% yield.25
3.5. Recovery of CoFe2O4-HMS magnetic nanocatalyst
To evaluate the reusability and recovery of the magnetic nanocatalyst, the reaction of 2-amino-4-methyl pyridine (1 mmol) (1b), 4-chlorobenzaldehyde (1 mmol) (2b) and phenyl acetylene (1 mmol) (3) with a catalytic amount (7 mol% or 0.07 g) of CoFe2O4-HMS in PEG 400 (5 mL) was selected as the model reaction to acquire the desired product (4g). The reaction mixture was ultrasonically irradiated at room temperature for 25 min. The progress of the reaction was monitored through TLC (on aluminum sheets precoated with silica) using n-hexane/ethyl acetate (4:1) as the eluting system. According to the knowledge from the literature,25,36 after the completion of the reaction, the catalyst was separated from the reaction mixture by using an external magnet. The isolated catalyst was washed with dichloromethane (DCM) and dried for 3 h at 60 °C. The magnetically recovered nanocatalyst was further subjected to the model reaction and this process was repeated for up to five runs under similar conditions to afford the product (4g) in 94%, 93%, 90%, 89%, and 87% yield.
4. Conclusions
An efficient ultrasonic-assisted and environmentally sustainable methodology for the synthesis of pharmacologically significant imidazo[1,2-a]pyridine scaffolds through a domino A3-coupling strategy catalyzed by hydromagnesite sheets impregnated with cobalt-ferrite magnetic nanoparticles was developed using green media. The role of CoFe2O4-HMS as a heterogeneous catalyst was envisaged to intensify the viability and yield of the products. This novel approach affords several advantages for achieving sustainable chemistry and opens up a new scope to advance the catalytic potential of hydromagnesite sheets impregnated with cobalt-ferrite magnetic nanoparticles (CoFe2O4-HMS) for the synthesis of biologically active synthetic equivalents. The high environmental compatibility and sustainability factors including a high eco-scale score, small E-factor, and considerable atom economy and carbon efficiency place this protocol under the umbrella of green chemistry principles. The reaction protocol is also feasible for multi-gram scale, which presents an economically affordable methodology on a large scale.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank MPCST-JRF (File no. A/R&D/RP-2/Phy&Engg./2017-18/269 Dated 31/03/18) for the fellowship to D. Geedkar. The authors wish to thank SAP (no. F.540/13/DRS-I/2016 (SAP-I) dated 7 Nov., 2016) UGC, New Delhi research grant to the department for developing SIC facilities. We would like to thank UGC-DAE CSR, Indore for their great support in providing HR-TEM, VSM and Raman analyses facilities.References
- C. E. Gueiffier and A. Gueiffier, Mini-Rev. Med. Chem., 2007, 7, 888–899 CrossRef PubMed.
- A. Kamal, J. S. Reddy, M. J. Ramaiah, D. Dastagiri, E. V. Bharathi, M. V. PremSagar, S. N. C. V. L. Pushpavalli, P. Ray and M. P. Bhadra, Med. Chem. Commun., 2010, 1, 355–360 RSC.
- S. C. Goodacre, L. J. Street, D. J. Hallett, J. M. Crawforth, S. Kelly, A. P. Owens, W. P. Blackaby, R. T. Lewis, J. Stanley, A. J. Smith, P. Ferris, B. Sohal, S. M. Cook, A. Pike, N. Brown, K. A. Wafford, G. Marshall, J. L. Castro and J. R. Atack, J. Med. Chem., 2006, 49, 35–38 CrossRef CAS PubMed.
- S. S. Kataev, B. Y. Syropyatov, N. E. Gavrilova and V. V. Zalesov, Pharm. Chem. J., 2004, 38, 181–182 CrossRef CAS.
- Y. Rival, G. Grassy, A. Taudou and R. Ecalle, Eur. J. Med. Chem., 1991, 26, 13–18 CrossRef CAS.
- L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305–312 CrossRef CAS PubMed.
- A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555–1575 RSC.
- N. Nagarajan, G. Velmurugan, A. Prakash, N. Shakti, M. Katiyar, P. Venuvanalingam and R. Renganathan, Chem.–Asian J., 2014, 9, 294–304 CrossRef CAS PubMed.
- J. Y. Lee, J. Y. Shim, H. K. Kim, D. Ko, M. H. Baik and E. J. Yoo, J. Org. Chem., 2017, 82, 4352–4361 CrossRef CAS PubMed.
- Y. Prostota, O. D. Kachkovsky, L. V. Reis and P. F. Santos, Dyes Pigm., 2013, 96, 554–562 CrossRef CAS.
- Z. T. Bhutia, D. Das, A. Chatterjee and M. Banerjee, ACS Omega, 2019, 4, 4481–4490 CrossRef CAS PubMed.
- M. Tajbakhsh, M. Farhang, R. Hosseinzadeh and Y. Sarrafi, RSC Adv., 2014, 4, 23116–23124 RSC.
- U. Balijapalli and S. K. Iyer, Dyes Pigm., 2015, 121, 88–98 CrossRef CAS.
- A. Maleki, Helv. Chim. Acta, 2014, 97, 587–593 CrossRef CAS.
- E. Allahabadi, S. Ebrahimi, M. Soheilizad, M. Khoshneviszadeh and M. Mahdavi, Tetrahedron Lett., 2017, 58, 121–124 CrossRef CAS.
- C. Zong, R. Zeng and J. Zou, Chem. Res. Chin. Univ., 2014, 30, 632–638 CrossRef CAS.
- T. R. Mandlimatha and K. I. Sathiyanarayanan, RSC Adv., 2016, 6, 3117–3125 RSC.
- G. Purohit, A. Kharkwal and D. S. Rawat, ACS Sustainable Chem. Eng., 2020, 8, 5544–5557 CrossRef CAS.
- M. Zhang, J. Lu, J. N. Zhang and Z. H. Zhang, Catal. Commun., 2016, 78, 26–32 CrossRef CAS.
- S. Sadjadi and M. Eskandari, Ultrason. Sonochem., 2013, 20, 640–643 CrossRef CAS PubMed.
- A. Maleki and M. Aghaie, Ultrason. Sonochem., 2017, 39, 534–539 CrossRef CAS PubMed.
- U. C. Rajesh, U. Gulati and D. S. Rawat, ACS Sustainable Chem. Eng., 2016, 4, 3409–3419 CrossRef.
- M. Akao, F. Marumo and S. Iwai, Acta Crystallogr., 1974, 30, 2670–2672 CrossRef.
- S. Verma, R. B. N. Baig, C. Han, M. N. Nadagouda and R. S. Varma, Chem. Commun., 2015, 51, 15554–15557 RSC.
- D. Geedkar, A. Kumar and P. Sharma, J. Heterocycl. Chem., 2020, 57, 4331–4347 CrossRef CAS.
- D. Geedkar, A. Kumar, G. K. Reen and P. Sharma, J. Heterocycl. Chem., 2020, 57, 1963–1973 CrossRef CAS.
- R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233–1246 CAS.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- K. V. Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 1–7 Search PubMed.
- R. A. Sheldon, Chem. Commun., 2008, 39, 3352–3365 RSC.
- O. Ebenezer, P. Awolade, N. Koorbanally and P. Singh, Chem. Biol. Drug Des., 2020, 95, 162–173 CrossRef CAS PubMed.
- J. B. Bharate, S. K. Guru, S. K. Jain, S. Meena, P. P. Singh, S. Bhushan, B. Singh, S. B. Bharate and R. A. Vishwakarma, RSC Adv., 2013, 3, 20869–20876 RSC.
- N. Chernyak and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2743–2746 CrossRef CAS PubMed.
- B. V. S. Reddy, P. S. Reddy, Y. J. Reddy and J. S. Yadav, Tetrahedron Lett., 2011, 52, 5789–5793 CrossRef CAS.
- Y. Volkova and V. Gevorgyan, Chem. Heterocycl. Compd., 2017, 53, 409–412 CrossRef CAS.
- N. Basavegowda, K. Mishra and Y. R. Lee, Mater. Technol., 2019, 34, 558–569 CrossRef CAS.
- B. D. Cullity and S. R. Stock, Elements of X-ray Diffraction Boston, Prentice-Hall, Addison-Wesley, MA, USA, 3rd edn, 2001 Search PubMed.
- T. Selvamani, T. Yagyu, S. Kawasaki and I. Mukhopadhyay, Catal. Commun., 2010, 11, 537–541 CrossRef CAS.
- S. Iftikhara, M. F. Warsia, S. Haiderb, S. Musaddiqc, I. Shakird and M. Shahid, Ceram. Int., 2019, 45, 21150–21161 CrossRef.
- S. Qu, J. Wang, J. L. Kong, P. Y. Yang and G. Chen, Talanta, 2007, 71, 1096–1102 CrossRef CAS PubMed.
- P. Sivakumar, R. Ramesh, A. Ramanand, S. Ponnusamy and C. Muthamizhchelvan, Appl. Surf. Sci., 2012, 258, 6648–6652 CrossRef CAS.
- Z. Wang, R. T. Downs, V. Pischedda, R. Shetty, S. K. Saxena, C. S. Zha, Y. S. Zhao, D. Schiferl and A. Waskowska, Phys. Rev. B, 2003, 68, 941011–941016 Search PubMed.
- P. Chandramohan, M. P. Srinivasan, S. Velmurugan and S. V. Narasimhan, J. Solid State Chem., 2011, 184, 89–96 CrossRef CAS.
- V. Bartunek, D. Sedmidubsky, S. Huber, M. Svecova, P. Ulbrich and O. Jankovsky, Materials, 2018, 11, 1241–1252 CrossRef PubMed.
- J. Hu, K. Zhu, L. Chen, C. Kubel and R. Richards, J. Phys. Chem. C, 2007, 32, 12038–12044 CrossRef.
- W. D. Harkins and G. Jura, J. Am. Chem. Soc., 1944, 66, 1366–1372 CrossRef CAS.
- C. Santhosh, P. Kollu, S. Felix, S. K. Jeong and A. N. Grace, RSC Adv., 2015, 5, 28965–28972 RSC.
- S. Chella, P. Kollu, E. V. P. R. Komarala, S. Doshi, M. Saranya, S. Felix, R. Ramachandrana, P. Saravanand, V. L. Konerue, V. Venugopal, S. K. Jeongf and A. N. Grace, Appl. Surf. Sci., 2015, 327, 27–36 CrossRef CAS.
- N. F. Sulaiman, W. A. W. A. Bakar, S. Toemen, N. M. Kamal and R. Nadarajan, Renewable Energy, 2019, 135, 408–416 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02516c |
| This journal is © The Royal Society of Chemistry 2021 |