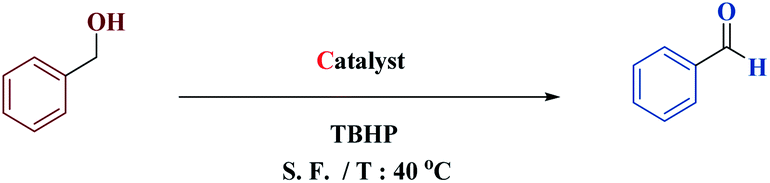
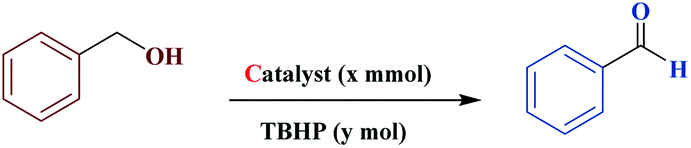DOI:
10.1039/D1RA00208B
(Paper)
RSC Adv., 2021,
11, 23192-23206
Retracted Article: Robust, highly active, and stable supported Co(II) nanoparticles on magnetic cellulose nanofiber-functionalized for the multi-component reactions of piperidines and alcohol oxidation†
Received
10th January 2021
, Accepted 27th May 2021
First published on 1st July 2021
Abstract
The new recyclable cobalt three-core magnetic catalyst obtained by anchoring a Schiff base ligand sector and cellulose nanofiber slings on MNP (Fe3O4) was prepared and named as MNP@CNF@ATSM–Co(II). Separately, MNPs and CNF have adsorbent properties of great interest. In this way, this catalyst was designed to synthesize piperidine derivatives under solvent-free conditions and alcohol oxidation reactions in EtOH as the solvent. It should be noted that this catalyst is environmentally safe and does not need an external base. This MNPs@CNF@ATSM–Co(II) separable catalyst has been evaluated using various characterization techniques such as FT-IR, XRD, FE-SEM, EDX, EDS, ICP, TGA, DLS, HRTEM, and VSM. The catalyst was compatible with a variety of benzyl alcohols, benzaldehydes, and amines derivatives, and gave complimentary coupling products with sufficient interest for all of them. The synergistic performance of Co (trinuclear) in the catalyst was demonstrated and its different homologs such as MNPs, MNPs@CNF, MNPs@CNF@ATS–Co(II), and MNPs@CNF@ATSM–Co(II) were separately synthesized and applied to a model reaction, and then their catalytic activity was investigated. Also, the performance of these components for the oxidation reaction of alcohols was evaluated. The advantages of the current protocol include the use of a sustainable and safe low temperature, eco-friendly solvent no additive, and long-term stability and magnetic recyclability of the catalyst for at least five successive runs, thus following green chemistry principles. This protocol is a benign and environment-friendly method for oxidation and heterocycle synthesis. This powerful super-magnetic catalyst can use its three arms to advance the reactions, displaying its power for multi-component reactions and oxidation.
Introduction
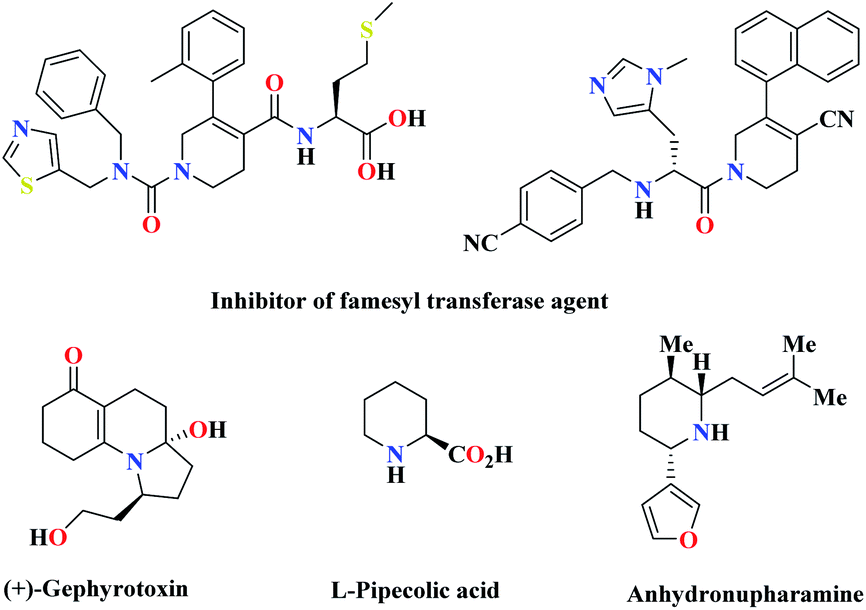
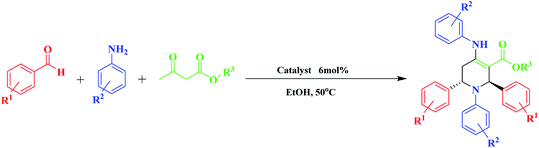
Green chemistry has played a key role in the development of human civilization. Green and sustainable chemistry is defined by the Environmental Protection Agency (EPA) as the design of chemical products that reduce or eliminate the use of hazardous substances.1 Organic synthesis through multifunctional reactions is a research area in organic chemistry. Multi-component reactions play a beneficial role in chemistry, especially in pharmaceuticals and drug discovery.2 Multi-component reaction (MCR) is a synthetic methodology in which three or more reactants come together in a single reaction vessel to form a new product. The characteristic aspect of MCRs is that the final products contain almost all portions of substrates, generating almost no by-products. This makes MCR an extremely ideal and eco-friendly reaction system. The target compounds can be obtained in one pot with fewer steps. These types of reactions have some advantages over conventional linear syntheses, including lower costs, shorter reaction times, high degree of atom economy, possibility for the combinatorial surveying of structural variations, and environmental friendliness.3 Among the multi-component reactions, polyfunctionalized heterocyclic compounds play important roles in the drug discovery process, analysis, and drug delivery.4 The development of new and efficient methods for the synthesis of N-heterocycles is a major interest of modern synthetic organic chemistry. Heterocyclic compounds have been noted for their biological and medicinal importance. Among the nitrogenous cyclic compounds, piperidines and their derivatives have shown impressive biological activity, such as antihypertensive,5 antibacterial,6 anticonvulsant, anti inflammatory agents,7 farnesyltransferase inhibitors,8 norepinephrine reuptake inhibitor,9 antipsychotic agents,10 and antidepressant.11 In addition, piperidine derivatives act as an important group of therapeutic agents in the treatment of Parkinson's disease,12 prolactinoma,13 schizophrenia,14 influenza infection,15 cancer metastasis,16 viral infections including AIDS,17 obesity, and diabetes,18 and also play key roles in many disease processes (Scheme 1).19 Due to the importance of these heterocyclic compounds in terms of the biological and medicinal properties, the introduction of a better and more efficient method for the preparation of piperidine derivatives is in demand. In recent years, organic catalysts, due to their good and unique performance, including the possibility of performing reactions for acid-sensitive substrates and selectivity, has attracted the attention of many scientists to perform reactions.20 Because of the great importance of piperidines, recently, the synthesis of these compounds has been reported using multi-component reactions in the presence of VCl3,21 BF3·SiO2,22 Ph3CCl,23 LaCl3·7H2O,24 tartaric acid,25 iodine,26 ZrOCl2·8H2O,27 ZrCl4,28 InCl3,29 tetrabutylammonium tribromide (TBATB),30 Bi(NO3)3·5H2O,31 L-proline,8 InCl3,7 Mg(HSO4)2,32 etc. So far, several catalysts that have been used, in addition to the advantages, also have disadvantages, such as long reaction time, low efficiency, use of high temperature, and expensive catalyst. In the last few years, several piperidine derivatives have been considered in clinical trials.9
 |
| | Scheme 1 Some biologically active and pharmaceutically active compounds containing the piperidine unit. | |
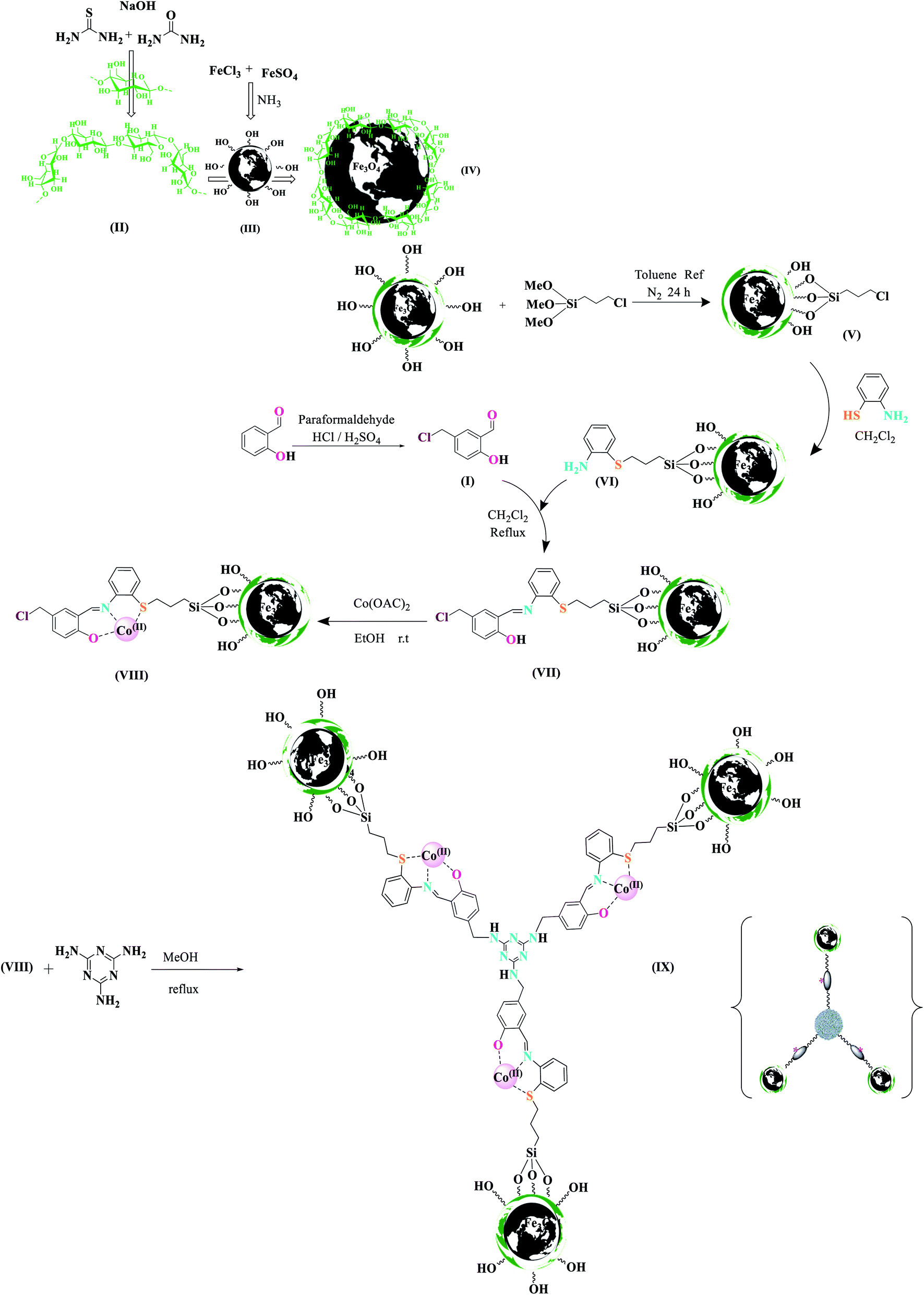
Although these protocols find certain merits of their own, they still suffer from several demerits such as multi-step conditions, use of toxic organic solvents or catalysts containing transition metals, hard work-up procedure, relatively expensive catalysts, troublesome waste disposal, and unsatisfactory yields. In order to improve the quality of the previous approach for the synthesis of pyridine derivatives, attention has been focused on methods that preserve the environment and protect it from pollution as much as possible. Heterogeneous catalysis is a significant approach toward environmental remediation because it decreases the cost and energy requirements of chemical reactions. In this regard, two-, three-, or multi-nuclear metal complexes have become a major research topic due to their very useful properties and multiple performances compared to single-nuclear catalysts. Metal catalysts, known as Lewis acid catalysts, have gained more attention due to their simplicity, low cost, high performance, easy preparation, and cost-effectiveness. One of the synthetic strategies for preparing a catalyst complex of two or more cores is the use of units such as Schiff base with active positions to bind high-performance metals. There is currently a great deal of interest in the synthesis and characterization of polynuclear cobalt complexes. Due to their potential applications, they are currently being employed in catalysis, transfer of electrons to solar cells, antiviral agents, and molecular magnets.33 Since the focus of our research team is on safe and nature-inspired compounds, it was decided to select magnetic cellulose and nanoparticles as the main working materials. Such particles commonly consist of two components, a magnetic material, often iron, nickel, and cobalt, and a chemical component that has a functionality. Magnetic nanoparticles have been the focus of much research recently because they possess attractive properties that could be potentially used in catalysis, including nanomaterial-based catalysts, biomedicine, tissue-specific targeting, magnetically-tunable colloidal photonic crystals, microfluidics, magnetic resonance imaging, magnetic particle imaging, environmental remediation, nanofluids, optical filters, defect sensors, magnetic cooling, and cation sensors.34 The metallic core of magnetic nanoparticles may be passivated by gentle oxidation, surfactants, polymers, and precious metals.35 MNPs maintain their scattered active sites using the outer shells such as polymers, zeolites, titanium dioxide, silica, and carbon.36 Recently, our research team has explored the synthesis and exchange effect in these Fe3O4 core nanoparticles with a cellulose outer shell. Nanoparticles with a magnetic core consisting either of iron with the shell of cellulose have been synthesized recently.37 CNF is a carbohydrate polymer made up of repeating β-D-glucopyranose units and consists of three hydroxyl groups per anhydrous glucose unit.38 This compound is environment-friendly; in addition, its advantages include low toxicity and its physical and biological properties. It has been used in several fields such as the medicine, electronics, and texture engineering.39 The cellulose structure, due to its high surface area, thermal stability, and most importantly, the presence of hydroxyl groups on its surface, make it a suitable support for the preparation of nanocatalysts based on biopolymers. Lately, biopolymers such as cellulose, chitosan, or wool have been used in heterogeneous catalyst systems.40 However, there are limitations in the use of these compounds in the industry, such as the dispersion stability, which make them difficult to be recycled. For this reason, they are coated with magnetic nanoparticles, which result in the formation of nanocomposites with an organic composite structure.41,42 Therefore, our main goal is to synthesize a new trinuclear catalyst and develop new methods for the synthesis of N-heterocycles. There have been no reports on the use of MNPs@CNF@ATSM–Co(II) as a trinuclear catalyst for organic reactions. In our ongoing research into the activity of the nanomagnetic catalyst, its successful use for alcohol oxidation reactions under green conditions has been reported. Either in the laboratory or in the chemical industry, the catalytic oxidation of benzyl alcohol to benzaldehyde is important. Eventually, this magnetic catalyst will show the performance and power of its three arms.
Results and discussion
Synthesis and characterization of MNPs@CNF@ATSM–Co(II)
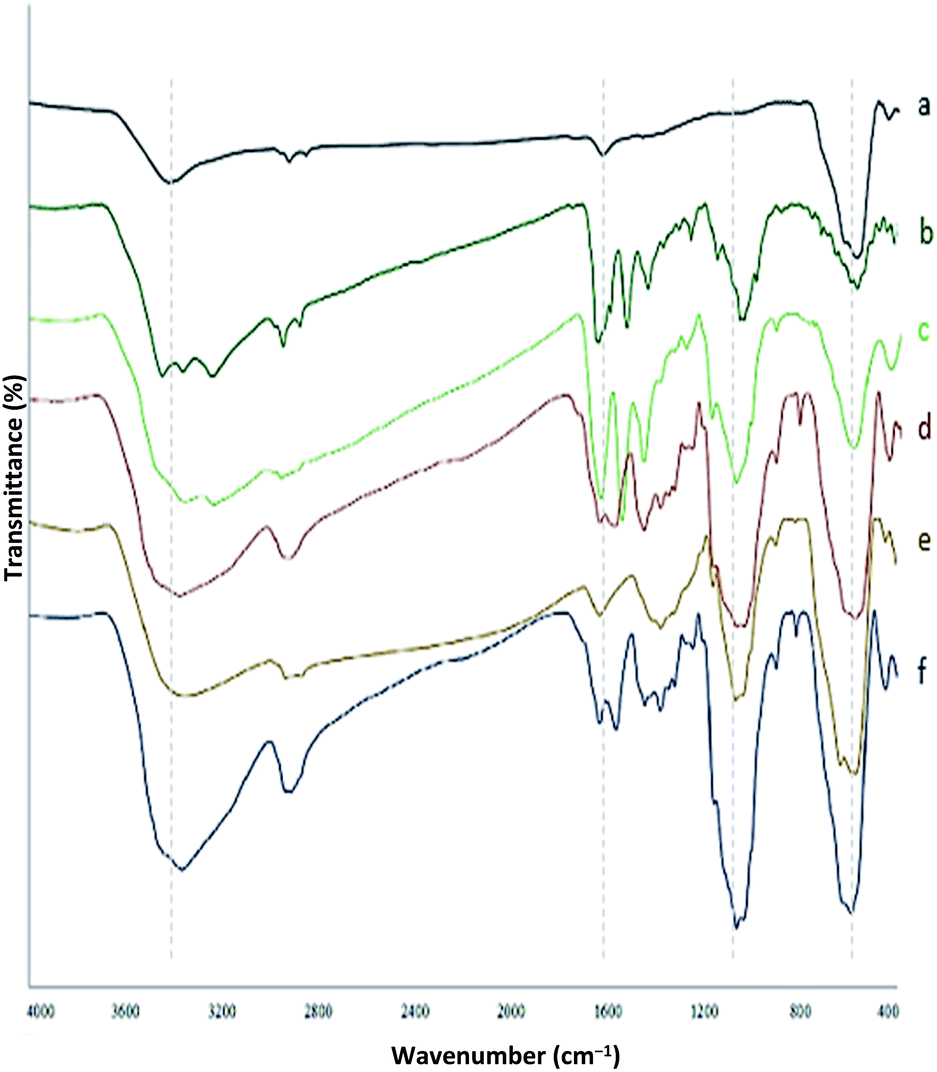
Chemistry is a unique and growing science that is very active, and new catalysts are growing and developing this science. One of the synthetic strategies for the preparation of polynuclear transition metal complexes is simple metal-ion complexes. Due to the excellence of this catalyst, powerful trinuclear catalysts with better performance over other previous catalysts, for example, easy recovery, non-toxicity, high catalytic efficiency, easy synthesis, and reusability, have been prepared. This catalyst can be a suitable selection in catalysis (Scheme 2). To identify the catalyst using different methods such as FT-IR, XRD, FE-SEM, EDX, EDS, ICP, TGA, DLS, HRTEM, and VSM were used. The FT-IR spectra of the compounds III, IV, V, VI, VII, and MNPs@CNF@ATSM–Co(II) are shown in (Fig. 1a–f). As shown in Fig. 1a, the FT-IR spectrum of compound III shows MNPs with two characteristic peaks at 3411 cm−1 and 588 cm−1 related to O–H and Fe–O stretching bands, respectively, which confirms the formation of MNPs. The characteristic bands that appeared at 1620 and 3980 cm−1 are ascribed to the bending and stretching vibrations of the surface hydroxyl groups and adsorbed water molecules, respectively. In addition to the MNPs peaks observed in the spectrum that confirms the MNPs' formation, the strong peaks at 1097, 2960, and 3328 cm−1 show the C–O, C–H, and O–H stretching vibrations of the cellulose nanofibers, respectively (Fig. 1b; IV).
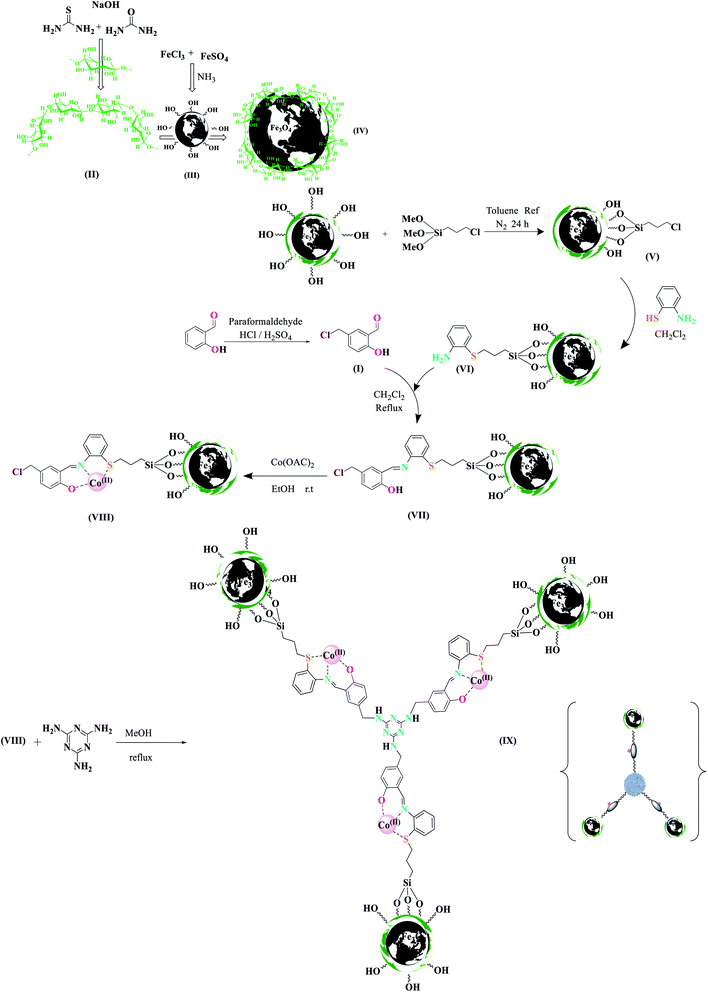 |
| | Scheme 2 Design and synthesis all stages of the MNPs@NFC@ATSM–Co(II) catalyst. | |
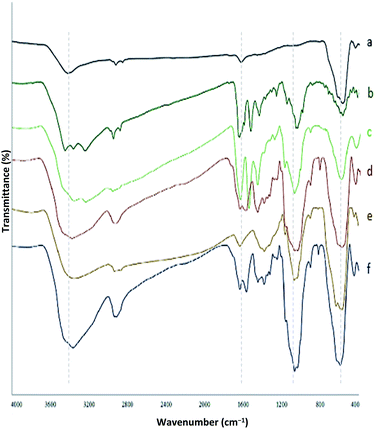 |
| | Fig. 1 FT-IR spectrum (a) MNPs, (b) MNPs@CNF, (c) MNPs@CNF–Cl, (d) MNPs@CNF@AT, (e) MNPs@CNF@ATS, (f) MNPs@CNF@ATSM–Co(II). | |
Fig. 1c shows the FT-IR spectrum with a strong peak of Si–O at 1026 cm−1 and also a new sharp peak at 1100 cm−1, which was assigned to the C–N stretching vibration, while 1450–1530 and 3120 cm−1 were assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H aromatic stretching vibration, respectively, referring to the 2-aminothiophenol linkage to the MNPs (Fig. 1d; VI). As shown in Fig. 1e, the FT-IR spectrum of compound VII (Fig. 1e) indicates the Schiff base reaction between 2-aminothiophenol and 5-chlorosalicylaldehyde; the characteristic peaks at 759 cm−1 (C–Cl stretching), 1473 cm−1 (CC aromatic stretching vibration), 1633 cm−1 (CN stretching vibration), 2970–3130 cm−1 (C–H aliphatic and aromatic stretching vibration), and 3380 cm−1 (O–H stretching) confirm the successful preparation of the Schiff base. The complexation of cobalt and melamine to MNPs@CNF@ATS causes the peak for the chlorine bond to be lost and causes the imine bond (1670 cm−1) peak to shift to a lower wavenumber by about 7 cm−1 (1663 cm−1), indicating the participation of the azomethine nitrogen in the bonding with the metal ion and confirming the coordination of ν(CN) stretching to the metal via a nitrogen atom. Also, absorption bands at 1113 and 1641 cm−1 could be assigned to C–N and CN melamine stretching, respectively (Fig. 1f).
C and C–H aromatic stretching vibration, respectively, referring to the 2-aminothiophenol linkage to the MNPs (Fig. 1d; VI). As shown in Fig. 1e, the FT-IR spectrum of compound VII (Fig. 1e) indicates the Schiff base reaction between 2-aminothiophenol and 5-chlorosalicylaldehyde; the characteristic peaks at 759 cm−1 (C–Cl stretching), 1473 cm−1 (CC aromatic stretching vibration), 1633 cm−1 (CN stretching vibration), 2970–3130 cm−1 (C–H aliphatic and aromatic stretching vibration), and 3380 cm−1 (O–H stretching) confirm the successful preparation of the Schiff base. The complexation of cobalt and melamine to MNPs@CNF@ATS causes the peak for the chlorine bond to be lost and causes the imine bond (1670 cm−1) peak to shift to a lower wavenumber by about 7 cm−1 (1663 cm−1), indicating the participation of the azomethine nitrogen in the bonding with the metal ion and confirming the coordination of ν(CN) stretching to the metal via a nitrogen atom. Also, absorption bands at 1113 and 1641 cm−1 could be assigned to C–N and CN melamine stretching, respectively (Fig. 1f).
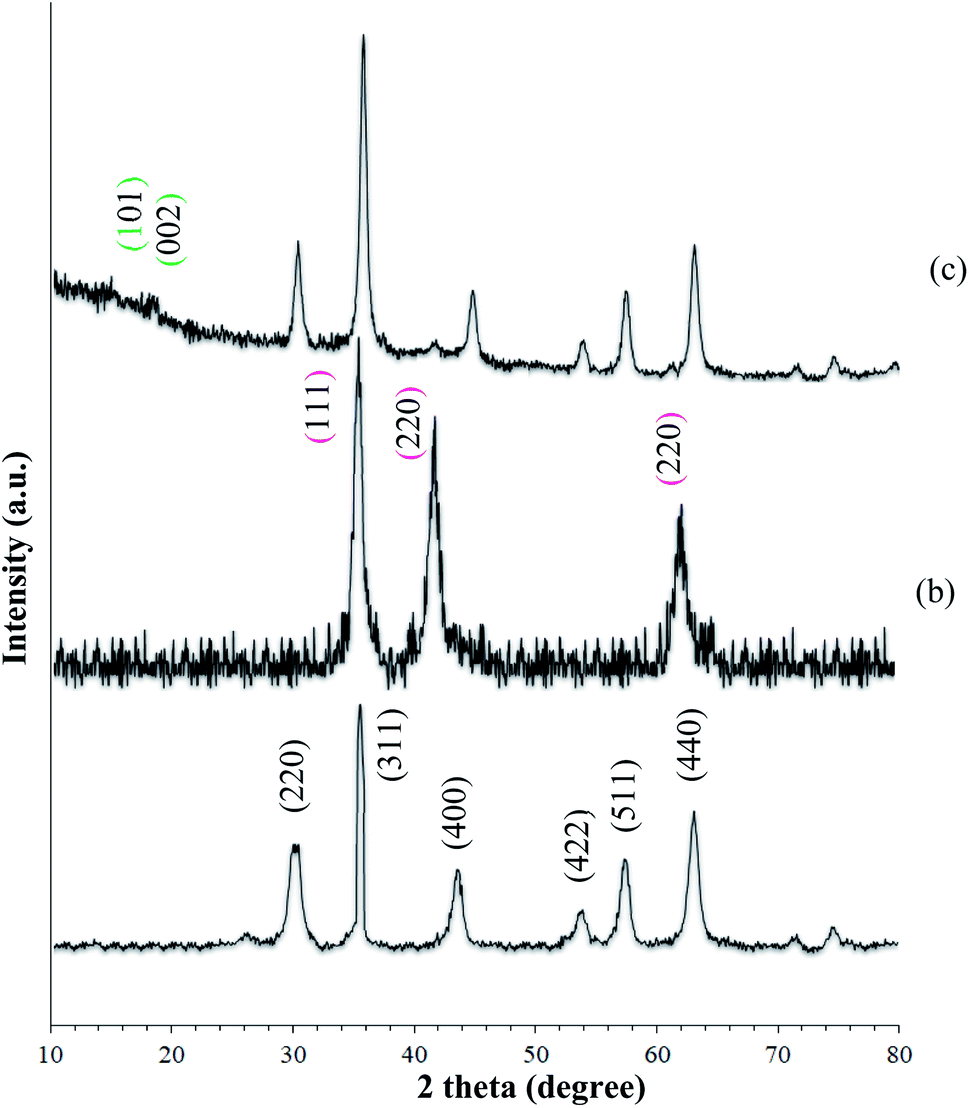
XRD was used to identify the crystal structure of the species. The patterns of the MNPs and MNPs@CNF@ATSM–Co(II) are shown in Fig. 2. The characteristic diffraction peaks at the 2θ values of 30.3°, 35.8°, 43.6°, 54.8°, 57.3°, and 63.2° correspond to the (220), (311), (400), (422), (511), and (440) planes, which can be indexed to the cubic inverse spinel-structure of Fe3O4 (JCPDS Card 19-0629).37 Strong peaks at 2θ = 36.71°, 42.47°, and 61.64° corresponding to the (111), (200), and (220) crystallographic phases in the XRD pattern are related to CoO.43 Furthermore, the broad conventional peaks related to cellulose at 2θ = 19.27° and 16.87° correspond to the (002) and (101) planes of cellulose, which are characteristic of cellulose.
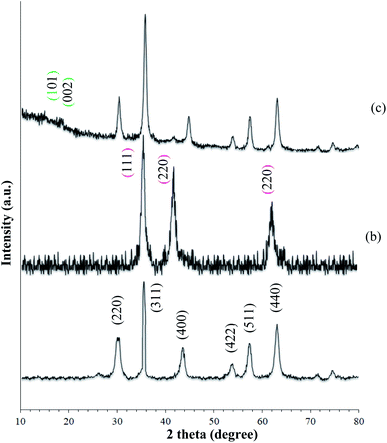 |
| | Fig. 2 XRD spectra of (a) MNP (Fe3O4), (b) CoO, and (c) MNPs@CNF@ATSM–Co(II). | |
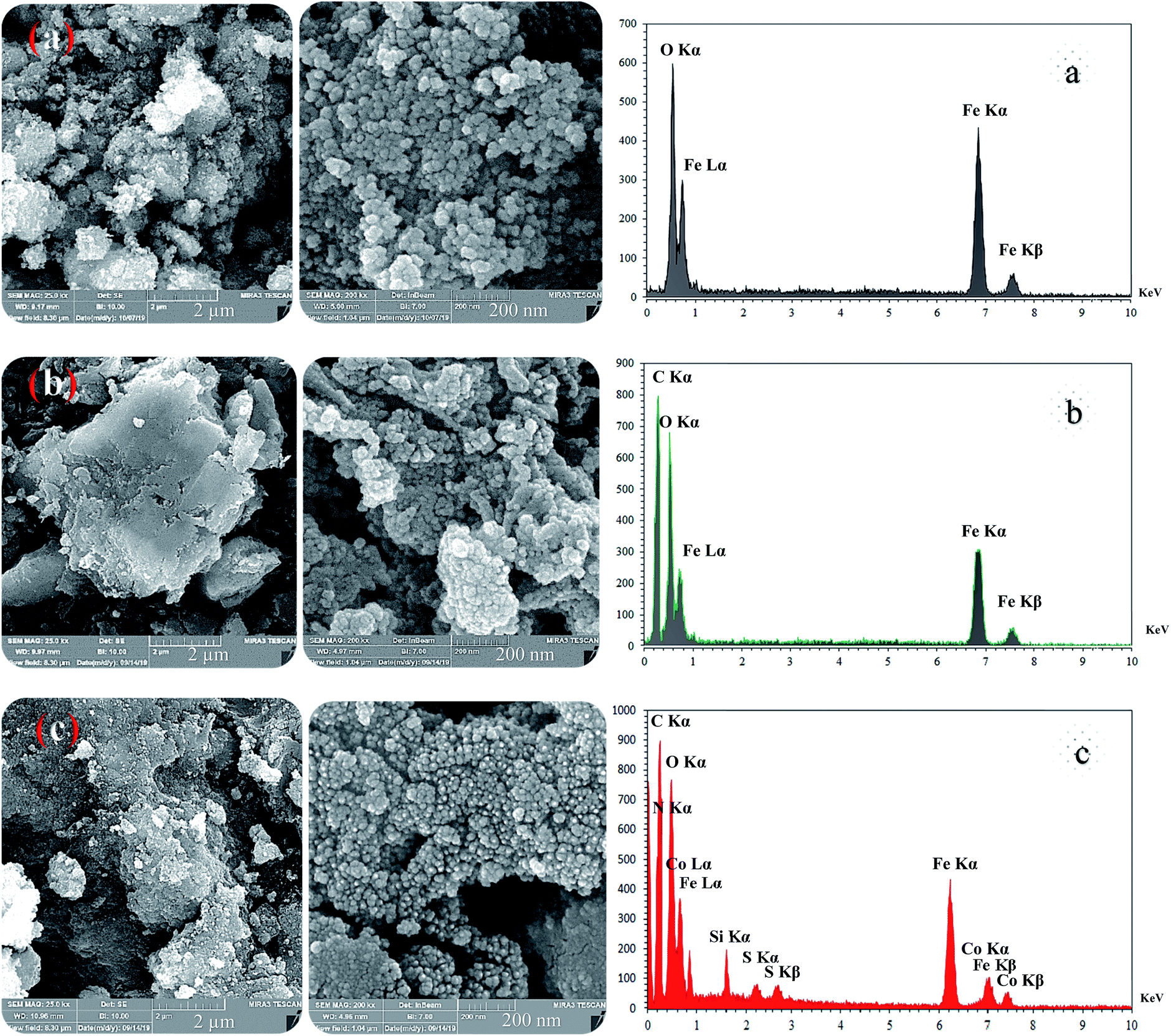
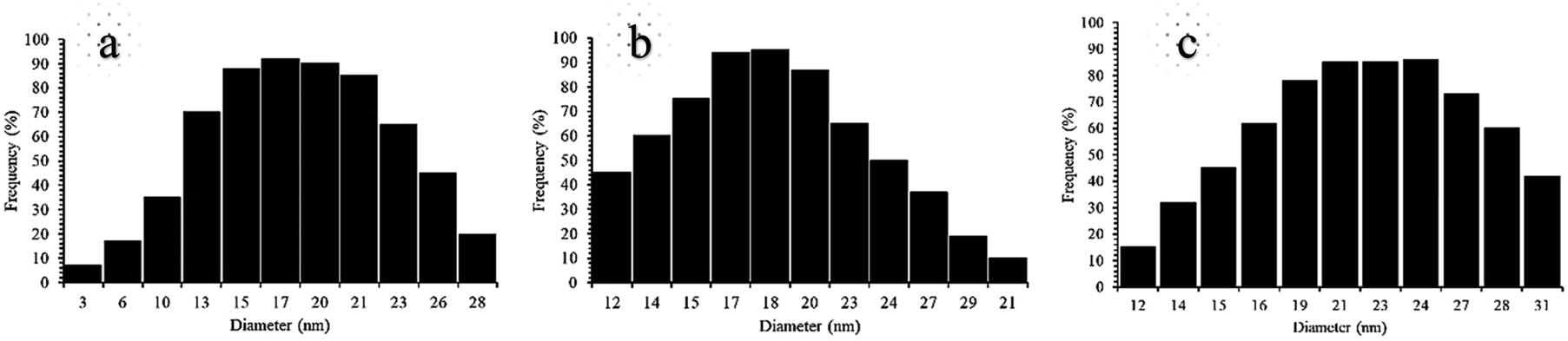
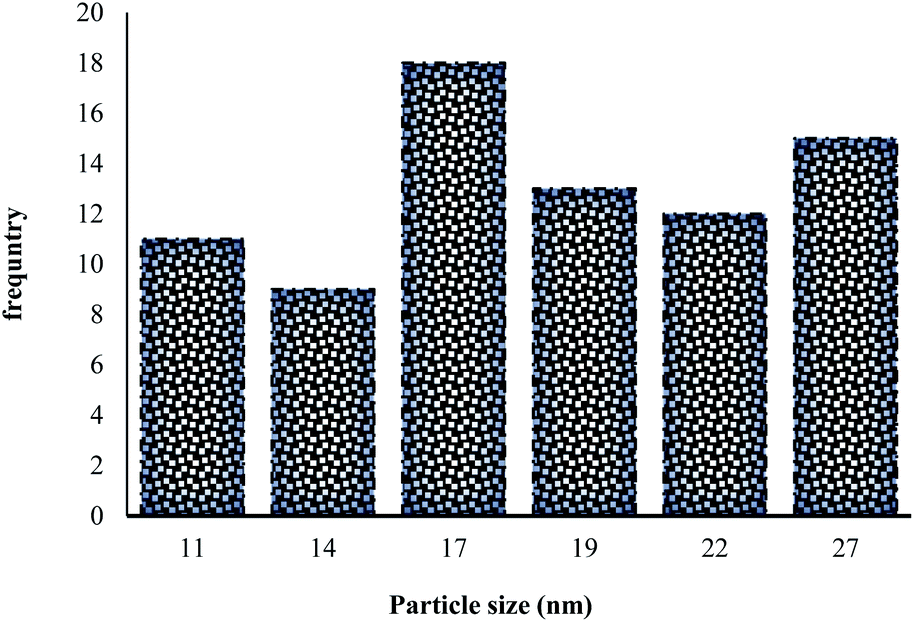
These absorption bonds support the successful preparation of the catalyst. SEM and DLS analyses were employed to obtain a visual image of the supported catalyst to determine the shape, morphology, particle distribution; EDX analysis was conducted to determine the elements in the prepared samples qualitatively. The SEM image, as shown in Fig. 3, in the 10 μ image reveals that MNP (Fe3O4), MNPs@CNF (core–shell), and MNPs@CNF@ATSM–Co(II) have an almost uniform and spherical morphology ranging in the size range from 4 to 31 nm, indicating good agreement with the calculated result from the Debye–Scherrer equation. Some aggregation occurs due to the magnetic property of the nanoparticles. In the EDX analysis of the MNPs (Fig. 3a, Fe3O4), MNPs@CNF (Fig. 3b, core–shell), and MNPs@CNF@ATSM–Co(II) (Fig. 3c, salen complex nanocatalyst), the results displayed the presence of Co, Fe, Si, O, N, S, and C elements, which could be acceptable evidence of the modification of the Fe3O4 surface by the nanocatalyst. Therefore, it can be inferred that the elements were loaded onto the magnetic surface (Fe3O4) of MNPs@CNF@ATSM–Co(II) (Fig. 3c). According to the results from ICP and EDX, the amount of copper in the MNPs@CNF@ATSM–Co(II) nanocatalyst was calculated to be 1.56 mmol g−1. In this study, to examine the size distribution of these nanoparticles, particle size histograms were prepared by DLS analysis (Fig. 4a–c). The average diameters of the particles were evaluated to be about 17 nm for the MNPs (Fig. 4a), 20 nm for the MNPs@CNF (Fig. 4b), and 24 nm for the MNPs@CNF@ATSM–Co(II) (Fig. 4c). The corresponding EDS mapping images of MNPs@CNF@ATSM–Co(II) showed that C, N, O, Si, S, Fe, and Co were dispersed uniformly in structure (S1, see ESI† for more details). The presence of these elements also confirmed using EDX (S1†). The structure of MNPs@CNF@ATSM–Co(II) was studied using transmission electron microscopy (HRTEM) (Fig. 5). These images are a suitable tool for determining the size and structure of the particles. The HRTEM images and histogram of MNPs@CNF@ATSM–Co(II) show the small particles of size 16–28 nm. The size and structure of the MNPs@CNF@ATSM–Co(II) were also evaluated using transmission electron microscopy (HRTEM) (Fig. 6).
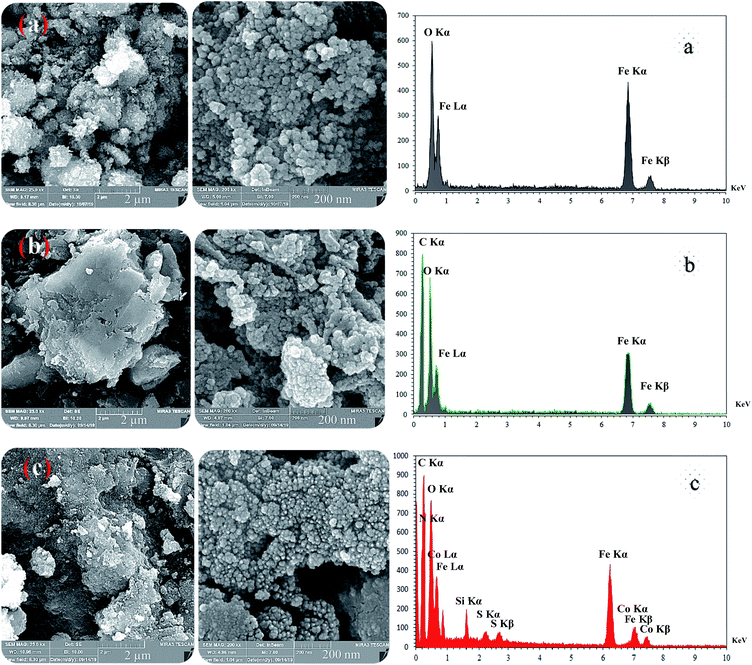 |
| | Fig. 3 FE-SEM (left) and EDX (right) images of the magnetic nanoparticles: (a) MNPs, (b) MNPs@CNF, and (c) MNPs@CNF@ATSM–Co(II). | |
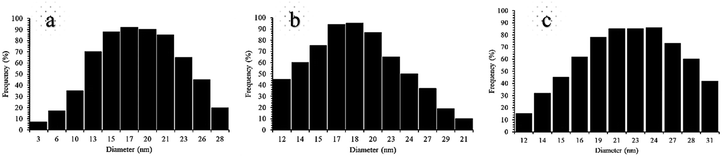 |
| | Fig. 4 DLS analysis of pure (a) MNPs, (b) MNPs@CNF, and (c) MNPs@CNF@ATSM–Co(II). | |
 |
| | Fig. 5 HRTEM image of the MNPs@CNF@ATSM–Co(II). | |
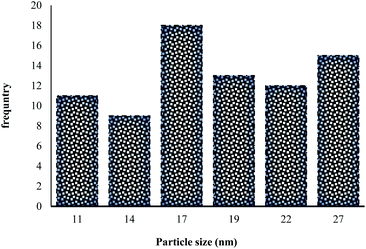 |
| | Fig. 6 Particle size distribution histogram of MNPs@CNF@ATSM–Co(II). | |
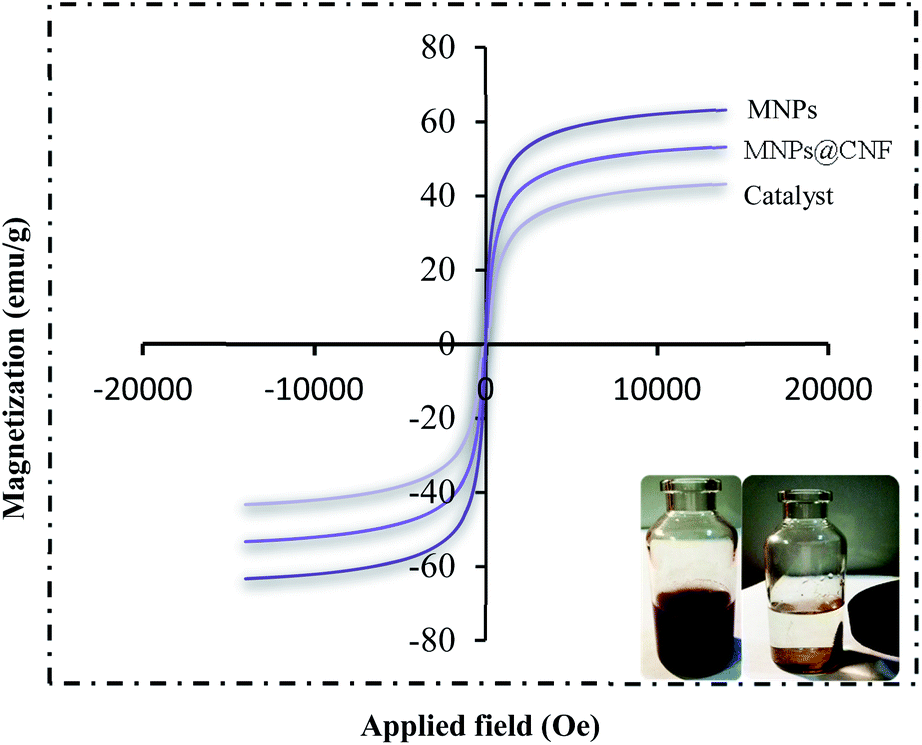
HRTEM analysis shows a dark MNPs core surrounded by a gray cellulose shell thick, and the average size of the obtained particles is 11–27 nm. The room temperature magnetization curves (Fig. 7) demonstrated that the MNPs@CNF@ATSM–Co(II) is superparamagnetic. The saturation magnetization of MNPs was 63.23 emu g−1, 52.9 emu g−1 for MNPs@CNF, and 41.3 emu g−1 for MNPs@CNF@ATSM–Co(II). In contrast with the uncoated MNPs, the saturation magnetization of the MNPs@CNF@ATSM–Co(II) reduced because the diamagnetic contribution of the thick cellulose and organic matter resulted in low mass deduction of the MNPs magnetic substance.
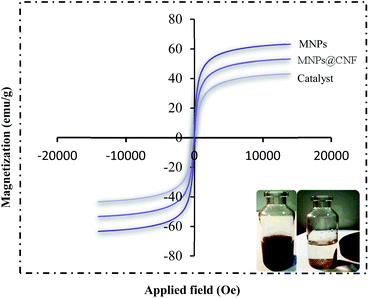 |
| | Fig. 7 VSM pattern (a) MNPs, (b) MNPs@CNF, and (c) MNPs@CNF@ATSM–Co(II). | |
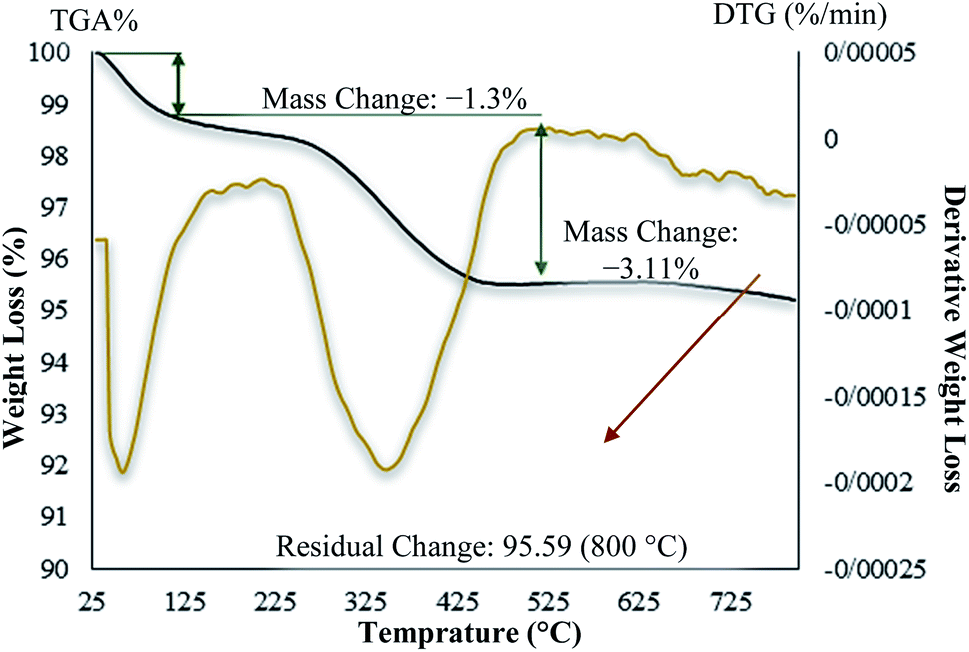
In contrast with the uncoated MNPs, the saturation magnetization of the MNPs@CNF@ATSM–Co(II) reduced because the diamagnetic contribution of the thick cellulose and organic matter resulted in low mass deduction of the MNPs magnetic substance. There is some reduction in the saturation magnetization but it is nevertheless possible to separate the material from the permanent magnetic solution. The thermal stability of the MNPs@CNF@ATSM–Co(II) was examined by TGA (Fig. 8). The analysis showed two decreasing peaks. As introduced in Fig. 8, a total weight loss of about 4.5% was determined from the TGA curve of the MNPs@CNF@ATSM–Co(II). Thermal gravimetric analysis of the MNPs@CNF@ATSM–Co(II) showed weight loss in the range of 25–150 °C, which is related to physisorbed water and solvents. Subsequently, the main weight loss step in the temperature range of 170–450 °C (3.11% reduction) is attributed to the decomposition of cellulose units through the formation of levoglucosan, and other volatile compounds or another mass loss, which shows higher thermal stability as MNPs@CNF@ATSM–Co(II) has been modified due to a reasonable reduction in the amount of oxygen-containing functional groups in cellulose.44 It is likely that due to the change in the phase of the thermal crystal from Fe3O4 to γ-Fe2O3, or due to the decomposition of particles at the organic moieties on the surface of the MNPs@CNF core–shell nanoparticles, weight loss of about 0.34% occurs between 600 and 800 °C.
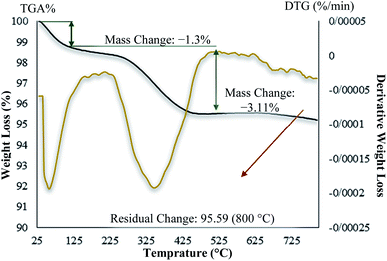 |
| | Fig. 8 TG/DTG thermogram for the MNPs@CNF@ATSM–Co(II) catalyst. | |
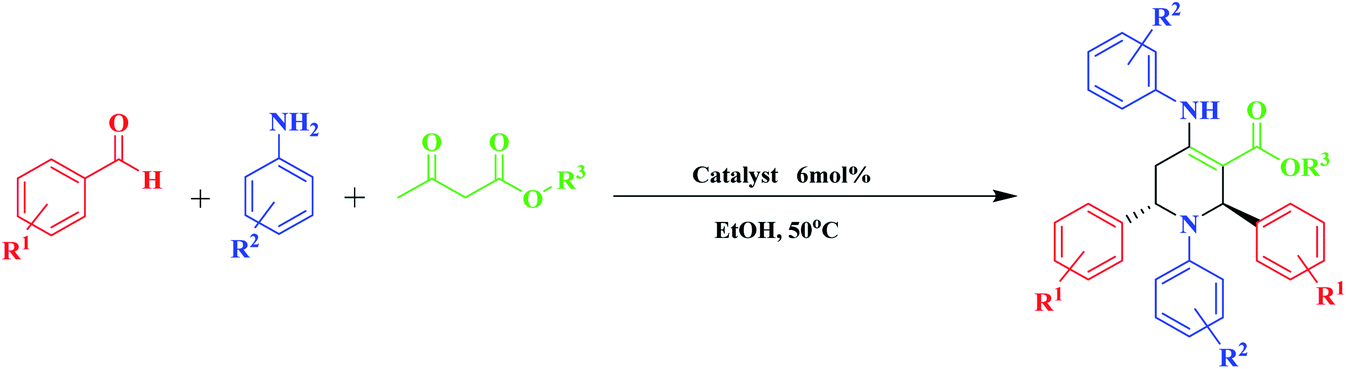
General experimental procedure for the synthesis of piperidine
Optimization of the reaction conditions is required to obtain the best catalyst activity. Firstly, the reaction of benzaldehyde (2 mmol), aniline (2 mmol), and methyl acetoacetate (1 mmol) was used as a model. It was conducted under different reaction parameters such as solvent, temperature, and amount of catalyst. Initially, when the reaction was carried out under solvent-free conditions, the product was obtained in a moderate yield of 43% due to the lack of effective interactions of the reactants with the catalyst. To illustrate the need for catalytic amounts of MNPs@CNF@ATSM–Co(II) in these reactions as a potentially powerful trinuclear catalyst, the direct synthesis of piperidine by the model reaction of benzaldehyde (2 mmol), aniline (2 mmol), and methyl acetoacetate (1 mmol) was carried out in EtOH at 40 °C, with the results listed in Table 1. MNPs@CNF@ATSM–Co(II) proved to be superior, producing the best yield of 4a; only trace amounts formed in the absence of the catalyst (Table 1, entry 9). Encouraged by this result, we then focused on optimizing the reaction conditions. A survey of solvents revealed ethanol to be the best choice, which was used directly without rigorous drying. Low yields were obtained when DMF, EtOAc, or water was employed as the solvent. Some dependence was also observed on the amount of MNPs@CNF@ATSM–Co(II) used. A satisfactory result was obtained in the presence of 1.5 mol% MNPs@CNF@ATSM–Co(II) (Table 1, entry 12). The reaction conditions were evaluated without the presence of the catalyst and up to 2 mol% catalyst. The process of product formation was investigated by TLC and it was observed that for the desired reaction without the catalyst, good efficiency was not achieved after 24 h (Table 1, entry 9). In the presence of the catalyst, this efficiency showed a significant change, and on increasing the catalyst up to two percent, it showed good efficiency, while the reaction time was also reduced (Table 1, entries 10–12). The presence of the catalyst in this reaction indicates its strong performance. Increasing the amount of the catalyst by more than 1.5 mol% did not show much growth. Following the investigation of the reaction conditions, we prioritized the temperature changes after identifying the amount of the catalyst. After the temperature studies, it was observed that 40 °C was the most suitable temperature for this reaction. Increasing the temperature above 40 °C did not show a significant improvement but when the temperature reached 60 °C, the product efficiency decreased (Table 1, entries 13–16). Various piperidine derivatives were successfully prepared from the reaction of benzaldehyde and aniline derivatives, and ethyl/methyl acetoacetate. This reaction was catalyzed by MNPs@CNF@ATSM–Co(II) and was successfully performed; in this way the efficiency of the catalyst was confirmed, the results of which are presented in Table 1, entry 2. To obtain a better insight into the catalytic efficiency of MNPs@CNF@ATSM–Co(II), the nano catalytic activity of MNPs, MNPs@CNF, MNPs@CNF@ATS–Co(II) (mononuclear), and MNPs@CNF@ATSM–Co(II) (trinuclear) was separately studied in the model reaction (Table 1, entries 17–19). As can be seen in Table 1, no product was obtained using MNPs and MNPs@CNF species. However, when the reaction was carried out in the presence of MNPs@CNF@ATS–Co(II), the result above satisfactory (Table 1, entry 19). These findings indicated that the enhanced nanocatalytic activity of MNPs@CNF@ATSM–Co(II) could be attributed to the synergistic catalytic effect of melamine and MNPs@CNF@ATS–Co(II) toward the piperidine reaction as a high-efficiency trinuclear catalyst. It can be imagined that the proper combination of melamine and MNPs@CNF@ATS–Co(II) in the nanocatalyst not only strengthens the catalyst but also increases the performance of the nanocatalyst. This property expedites and facilitates the reaction process in terms of time and other reaction conditions. These observations well proved the significant influence of the trinuclear catalyst in advancing the piperidine MCR reaction. Benzaldehyde derivatives have electron donors, and withdrawing groups in ortho, meta, and para positions provide the reaction high efficiency. Besides, the synthesis of piperidine derivatives from methyl acetoacetate and ethyl acetoacetate has been used successfully. Based on the provided data, the efficiency of our presented protocol and specifically the MNPs@CNF@ATSM–Co(II) was such that all the derivatives of piperidines were obtained in high yield without any dependence on the reactants. The NMR spectra are consistent with the past valid results, which is a confirmation of the efficiency of the catalyst (Table 2).
Table 1 Optimization of the solvent for the synthesis of piperidine in the model reactiona
Table 2 Synthesis of various poly-substituted piperidine derivativesa
|
| Entry |
R1 |
R2 |
R3 |
Product |
Time (h) |
Yieldb (%) |
m.p./°C |
Lit. m.p./°C [ref.] |
| Reaction conditions: benzaldehyde (2 mmol), aniline (2 mmol), β-ketoester (1 mmol), and catalyst (1.5 mol%) in 5 mL of EtOH at 50 °C. Isolated yield. |
| 1 |
H |
H |
Et |
4a |
2 |
92 |
173–174 |
174–175 [ref. 35] |
| 2 |
H |
H |
Me |
4b |
2 |
94 |
169–170 |
169–171 [ref. 35] |
| 3 |
H |
4-Br |
Et |
4c |
3 |
87 |
198–199 |
196–198 [ref. 36b] |
| 4 |
H |
4-Cl |
Et |
4d |
3 |
83 |
201–202 |
202 [ref. 36c] |
| 5 |
4-OMe |
4-Cl |
Me |
4e |
2.5 |
85 |
195–196 |
193–195 [ref. 36e] |
| 6 |
4-OMe |
4-Br |
Me |
4f |
2.5 |
78 |
177–178 |
177–179 [ref. 20] |
| 7 |
2-NO2 |
H |
Me |
4g |
2 |
70 |
217–218 |
217–219 [ref. 35] |
| 8 |
4-NO2 |
H |
Me |
4h |
3 |
82 |
240 |
239–241 [ref. 36d] |
| 9 |
4-NO2 |
H |
Et |
4i |
3 |
87 |
249–250 |
247–250 [ref. 36d] |
| 10 |
4-Br |
H |
Me |
4j |
4 |
84 |
245–246 |
245–247 [ref. 36c] |
| 11 |
4-Br |
4-Cl |
Me |
4k |
4 |
82 |
160–161 |
160–163 [ref. 36c] |
| 12 |
4-Cl |
H |
Me |
4l |
4 |
82 |
227 |
225–227 [ref. 36e] |
Magnetic catalytic activity of MNPs@CNF@ATSM–Co(II) in the alcohol oxidation reaction
Our research team, encouraged by the promising results obtained from the multi-component reactions of piperidine, next explored the magnetic catalytic activity of MNPs@CNF@ATSM–Co(II) toward the alcohol oxidation reaction under solvent-free conditions (Table 3). First, benzyl alcohol was selected as a model substrate, to demonstrate the optimal cases and main results, which are summarized in Table 3. It should be noted that there is little progress of the reaction under conditions without the MNPs@CNF@ATSM–Co(II) catalyst or in the presence of precursor salts. Oxidation of benzyl alcohol in different solvents was performed under solvent-free conditions (entries 1–4). The best product efficiency was obtained under solvent-free conditions. The reason behind this observation is the exposed catalytic sites under the solvent-free conditions.
Table 3 Screening the reaction conditionsa

|
| Entry |
Solvent |
Catalyst (mol%) |
Temp. (°C) |
TBHP (mmol) |
Yieldb (%) |
| Reaction conditions: benzyl alcohol (1 mmol), 30 min. Yields of product isolated. |
| 1 |
Water |
1 |
40 |
2 |
40 |
| 2 |
EtOH |
1 |
40 |
2 |
75 |
| 3 |
CH3CN |
1 |
40 |
2 |
60 |
| 4 |
Solvent free |
1 |
40 |
2 |
92 |
| 5 |
Solvent free |
1 |
RT |
2 |
50 |
| 6 |
Solvent free |
1 |
40 |
2 |
92 |
| 7 |
Solvent free |
1 |
60 |
2 |
65 |
| 8 |
Solvent free |
— |
40 |
2 |
25 |
| 9 |
Solvent free |
0.5 |
40 |
2 |
65 |
| 10 |
Solvent free |
1 |
40 |
2 |
92 |
| 11 |
Solvent free |
1.5 |
40 |
2 |
92 |
| 12 |
Solvent free |
2 |
40 |
2 |
94 |
| 13 |
Solvent free |
1 |
40 |
0.5 |
60 |
| 14 |
Solvent free |
1 |
40 |
1 |
92 |
| 15 |
Solvent free |
1 |
40 |
1.5 |
85 |
| 16 |
Solvent free |
1 |
40 |
2 |
70 |
Then, the model reaction was performed under solvent-free conditions at room temperature, 40, and 60 °C (entries 5–7). As the results show, the reaction at room temperature did not continue well but increasing the temperature to 40 °C resulted in a significant increase in the crop yields. Increasing the reaction temperature to more than 60 °C was found to be unfavorable for enhancing the reaction time and product yield. The reduction in the product yield could be due to the decomposition of TBHP to molecular water and tert-butanol in the presence of MNPs@CNF@ATSM–Co(II) at elevated temperatures. To determine the role of the catalyst, the model experiment confirmed that the favorable product was not formed in the catalyst's absence. It was found that increasing the catalyst content to 1 mol% resulted in a significant increase in the yield of the product, and the oxidation reaction of alcohol was evaluated with different oxidant values (entries 13–17). On increasing the amount of the oxidant, the efficiency of the product increased, while on increasing the oxidant beyond 1.5 mol%, the opposite effect was observed in the product yield, which could be due to excessive oxidation. Following the oxidation reaction of alcohol, in addition to TBHP, we also evaluated other oxidants, including H2O2, O2, and NaIO4, in the presence of MNPs@CNF@ATSM–Co(II) and a temperature of 40 °C without any organic and aqueous solvent, and it was observed that the best oxidant could be TBHP, which is the best source of oxygen (Table 4). After the optimized conditions were confirmed, this catalytic system was practical for the oxidation of a vast variety of primary and secondary alcohols to the corresponding carbonyl compounds.
Table 4 Screening the nature of oxidantsa
| Entry |
Oxidant |
Time (min) |
Yieldb (%) |
| Reaction conditions: benzyl alcohol (1 mmol), oxidant (1 mmol), catalyst (1 mol%), S. F., 40 °C. Yields of product isolated. |
| 1 |
H2O2 |
60 |
85 |
| 2 |
NaIO4 |
90 |
40 |
| 3 |
O2 |
90 |
65 |
| 4 |
TBHP |
30 |
92 |
As shown in Table 5, good to great yields were obtained for both primary and secondary alcohols. Clearly, the electronic nature of the substituents on the aromatic ring of primary alcohols illustrated no obvious effect due to this alteration because the products were obtained in high yields.
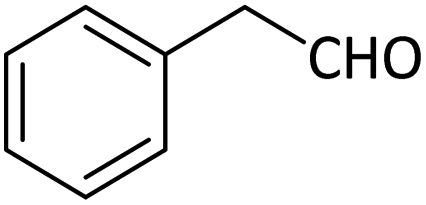
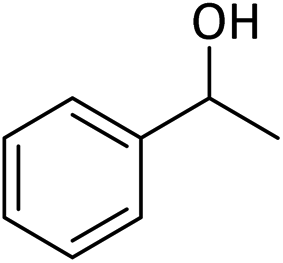
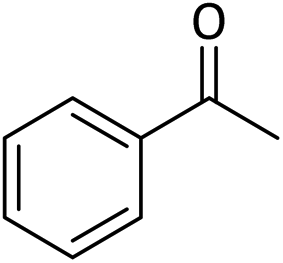
Table 5 Oxidation of alcohols to corresponding aldehydes or ketones in the presence of MNPs@CNF@ATSM–Co(II) catalysta
Stability and reusability of MNPs@CNF@ATSM–Co(II) as a trinuclear manganese catalyst in the piperidine and alcohol-oxidation reactions
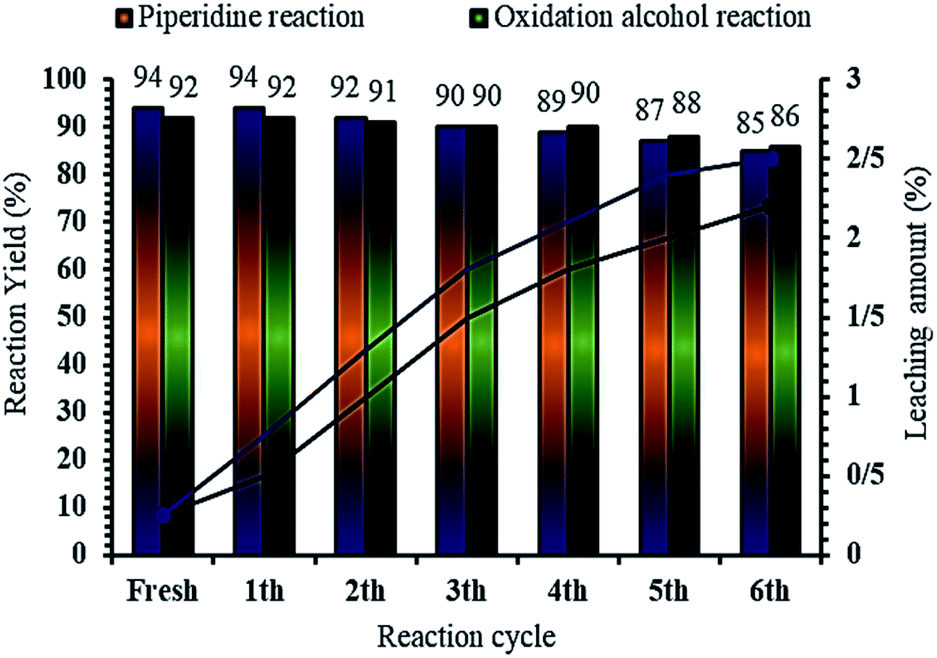
It is important to note that the magnetic property of MNPs@CNF@ATSM–Co(II) facilitates its effective recovery from the reaction mixture during the working steps. To further evaluate the potential of industrial applications of the MNPs@CNF@ATSM–Co(II) catalyst, the activity of the recycled catalyst was also investigated under freshly optimized conditions. Benzyl alcohol was used for alcohol oxidation reaction, while benzaldehyde, aniline, and acetyl acetate were used for the piperidine reaction. Both reactions were performed using the optimized conditions after completion of the reaction, the catalyst was separated by an external magnet, washed with ethanol, and dried. Residual activation was analyzed by ICP for measuring the amount of cobalt is lost from the catalyst to the solution. The recovered catalyst was reused for five consecutive periods without significant catalytic activity loss (Fig. 9). The piperidine and alcohol-oxidation reactions yields reached 85% and 86% in the 5th run, respectively. To show the durability and structure of the catalyst, the recovered catalyst after the 6th run was subjected to some analyses. Also, metal leaching from the catalyst was measured in each cycle. As shown in Fig. 9, very less leaching was observed for MNPs@CNF@ATSM–Co(II) as it was only 2.9% for the piperidine reaction and 2.2% for the alcohol oxidation reaction metal leaching after the 6th run. Moreover, ICP analysis of the catalyst for each run demonstrated an insignificant change in its weight percentage compared to the corresponding values for the fresh catalyst: Fe 44.28, Si 2.15, Co 6.48 w%. These results demonstrated insignificant changes. Due to the magnetic nature of the catalyst, after the first use of the product catalyst, it is simply extracted with ethanol, while the catalyst was separated by a magnet. The catalyst showed good recycling for five times. FT-IR, VSM, and FE-SEM of the catalyst after 6 reuses showed that the structure and morphology of the catalyst were preserved with extraordinarily little change during the recycling process for the piperidine reaction. Also, FT-IR analysis of the reused MNPs@CNF@ATSM–Co(II) for the alcohol oxidation reaction showed that there was no significant change in the internal structure (S2, see ESI†).
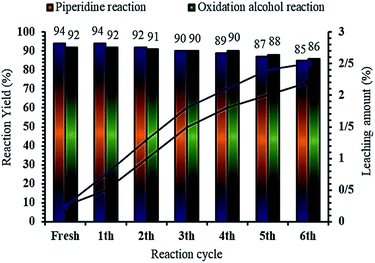 |
| | Fig. 9 Recyclability of MNPs@CNF@ATSM–Co(II) in the model alcohol oxidation and piperidine multi-component reaction, under the optimized reaction conditions; reaction times: 60 min (piperidine multi-component reaction) and 15 min (alcohol oxidation). | |
Experimental
General
2-Aminothiophenol, melamine, salicylaldehyde, and all solvents and reagents were obtained from Sigma-Aldrich. All chemicals were used without any further purification. The progress of the reactions and the purity of the products were determined by TLC on silica-gel Polygram SILG/UV254 plates. Fourier transform infrared (FT-IR) spectra were recorded with a Nicolet system 800 with beam splitter KBr SCAL = 800 in the range of 400–4000 cm−1. The FT-IR spectrometer used a room temperature detector. NMR spectra were recorded in DMSO-d6 using Bruker Advance DPX-400 and 250 instruments using tetramethyl silane as the internal standard. The powder X-ray diffraction pattern of MNPs@CNF@ATSM–Co(II) was obtained with an X'Pert Pro MPD diffractometer. Transmission electron microscopy (TEM) images were obtained using a Philips CM120 microscope. Thermogravimetric analysis (TGA) of the nano-powders was carried out using a TGA/DSC (Thermogravimetric analysis & Differential Scanning Calorimetry) between 25 and 800 °C with a heating rate of 15 °C min−1 under an air atmosphere. Elemental analysis was performed with a PerkinElmer OPTIMA 7300DV inductively coupled plasma (ICP) atomic emission spectrometer. Field emission scanning electron microscopy (FE-SEM) images were obtained on a Tescan MIRA3. EDX spectroscopy was performed using a field emission scanning electron microscope (JEOL 7600F). A Lake Shore Cryotronics 7407 vibrating sample magnetometer (VSM) was used at room temperature to study the magnetic properties of the catalyst.
Preparation of the new MNPs@CNF-anchored Co(II) aminothiophenol, salicylaldehyde, and melamine complex [MNPs@CNF@ATSM–Co(II)] of 5-(chloromethyl)-2-hydroxybenzaldehyde (I)
A mixture of salicylaldehyde (0.25 mol), paraformaldehyde (0.15 mol), and H2SO4 (2–3 drops) as catalyst in 150 mL HCl was stirred at 70 °C for 48 h. Then, the mixture was cooled to room temperature, and 20 mL each of distilled water and dichloromethane was added to the mixture. The organic layer was separated, dried over Na2SO4, and the solvent was removed under reduced pressure. The resultant purple sediment was washed with NaHCO3 (100 mL) and then dried under an air atmosphere. The pure product, 5-chloromethyl salicylaldehyde (I), was obtained by recrystallization from EtOH (m.p. 85–87 °C, 95% isolated yield).
Fabrication of the cellulose nanofiber (CNF)
We prepared nanofiber cellulose according to the procedure described.37 1.1 g of nanofiber cellulose was added to 50 mL of NaOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :urea:thiourea:H2O (4.8:4.8:3.88) (by weight) solution at −12 °C with stirring for 15 min to generate a homogenous cellulose solution.
:urea:thiourea:H2O (4.8:4.8:3.88) (by weight) solution at −12 °C with stirring for 15 min to generate a homogenous cellulose solution.
Nanoparticle synthesis (MNPs@CNF)
Nanoparticle synthesis was adapted from a previously reported method. FeSO4·7H2O and FeCl3·6H2O were added to deoxygenated water in the molar ratio of 2:3 (FeSO4:FeCl3) and stirred by a mechanical mixer at 250 rpm at a temperature of 70 °C under flowing N2 until the salts were fully dissolved, producing a transparent, brown solution. Next, NH3 was added. The flowing N2 gas was stopped, and the mixture was stirred at 70 °C for one h, after which the temperature was increased to 90 °C, and N2 flow was reinitiated, following which the mixture was stirred for an additional one h, and later stopped stirring again and cooled to room temperature. After a few min, a suspension of magnetic nanoparticles (MNPs), followed by the iron solution, was added to the pre-prepared cellulose. The solution was stirred vigorously at 30 °C for 2.5 h and washed three times using deoxygenated Millipore water and three times by Et2O before being dried under flowing argon.
Preparation of MNPs@CNF-CPTMS
Initially, 1.1 g of core–shell MNPs@CNF was dissolved in 15 mL toluene dry and sonicated for 30 min; then, 1 mL of 3-chloropropyl-trimethoxysilane was added slowly to the mixture. The mixture was then stirred for 24 h at room temperature under N2 atmosphere at reflux conditions. After completion of the reaction, the reaction mixture was washed with toluene and diethylether. The dark brown powder obtained was separated by a magnet, washed six times with distilled water and acetone, and dried at room temperature to afford MNPs@CNF-CPTMS as a dark brown powder.
Preparation of aminothiophenol-functionalized MNPs@CNF (MNPs@CNF@ATS)
MNPs@CNF–Cl (1.00 g) was refluxed with 2-aminothiophenol (4.0 mmol) in CH2Cl2 (20.0 mL) for 24 h to replace the terminal chlorine atoms. Then, 5-chloromethylsalicylaldehyde (4 mmol) was added, and the mixture was refluxed for 24 h. Finally, MNPs@CNF@ATS was filtered-off, washed three times with ethanol, and dried at 70 °C in a vacuum oven.
Loading with Co(OAc)2
MNPs@CNF@ATS (1.00 g) was mixed with Co (OAc)2 (0.21 g, 1.2 mmol) in ethanol (5 mL). The mixture was kept room temperature for 12 h. Then, it was filtered, and the solid obtained was washed with ethanol and dried at 70 °C overnight to obtain MNPs@CNF@ATS–Co(II). Cobalt loading of MNPs@CNF@ATS–Co(II) was determined by the inductively coupled plasma (ICP) technique.
Preparation of melamine-functionalized MNPs@CNF@ATS–Co(II) (MNPs@CNF@ATSM–Co(II))
At this stage, we first weighed melamine and transferred it to a round bottom flask containing methanol (10.0 mL) and K2CO3 (3.0 mmol). Then, using the molar ratio of 3:1, some of the MNPs@CNF@ATS–Co(II) complex (VIII) was weighed and added to the reaction mixture. The reaction mixture was refluxed under nitrogen gas for 12 h. After the reaction, the desired product MNPs@CNF@ATSM–Co(II) (9) was removed with a strong magnet and washed with water and ethanol, and finally placed in a vacuum oven at 70 °C for 8 h. Scheme 2 shows the complete route for the preparation of MNPs@CNF@ATSM–Co(II).
General procedure for the synthesis of highly substituted piperidine
First, the MNPs@CNF@ATSM–Co(II) catalyst (1.5 mol%) was added to a flask containing ethanol (5 mL) and was stirred for 10 min at room temperature.
Then, aromatic amine (2 mmol) and β-ketoester (1 mmol) were added to the reaction mixture and stirred for 30 min. Finally, aromatic aldehyde (2 mmol) was added to the reaction mixture and the reaction mixture was stirred at 50 °C. The progress of the reaction was monitored by TLC paper. After the end of the reaction, the reaction mixture was cooled to room temperature, the catalyst was removed with a magnet, and the residue was concentrated with ethanol and crystallized. The physical and spectral data of the selected products are represented in the ESI† file.
The general catalytic alcohol oxidation reaction method
In a general experiment, the catalyst MNPs@CNF@ATSM–Co(II) (1 mol%) and TBHP (1 mmol) were first added together in a flask under solvent-free conditions and stirred for a few minutes. Benzyl alcohol (1 mmol) was then added to the reaction vessel and stirred at 40 °C. The reaction process was evaluated by TLC, and considering that all final products are known, their physical data were compared with the reference samples.
Comparative study
To show the merit of the present work in comparison with the reported results in the literature, we compared the performance of MNPs@CNF@ATSM–Co(II) as a powerful trinuclear heterogeneous catalyst in this reaction. Our results and reaction conditions were compared with other conditions reported for heterogeneous catalysis (Table 6). The results obtained show that our proposed catalyst is quite comparable with other heterogeneous catalysts used with about the same yields. On the other hand, it has been observed that the synthesized catalyst has an excellent advantage over other reported catalysts, which can be due to suitable reaction conditions, short reaction time, high reaction efficiency, lower catalyst load, and the timely recovery of the magnetic catalyst.
Table 6 Comparison of MNPs@CNF@ATSM–Co(II) with reported catalysts for the synthesis of highly substituted piperidine derivativesa
| Entry |
Catalyst (mol%) |
Solvent |
Time (h) |
Temp. (°C) |
Yield (%) |
Ref. |
| Based on the three-component reaction of benzaldehyde (2 mmol), aniline (2 mmol), and methyl acetoacetate (1 mmol). |
| 1 |
BF3·SiO2 |
MeOH |
— |
65 |
— |
27 |
| 2 |
Ph3CCl |
MeOH |
5 |
50 |
79 |
18 |
| 3 |
Tartaric acid |
MeOH |
14 |
RT |
79 |
25 |
| 4 |
ZrOCl2·8H2O |
EtOH |
3.5 |
Reflux |
80 |
27 |
| 5 |
ZrCl4 |
EtOH |
9 |
RT |
90 |
28 |
| 6 |
BDMS |
EtOH |
3 |
RT |
75 |
45 |
| 7 |
Bi(NO3)3·5H2O |
EtOH |
12 |
RT |
81 |
46 |
| 8 |
p-TsOH·H2O |
EtOH |
10 |
RT |
78 |
20 |
| 9 |
TBATB |
EtOH |
24 |
RT |
74 |
47 |
| 10 |
InCl3 |
CH3CN |
24 |
RT |
60 |
31 |
| 11 |
Ascorbic acid |
EtOH |
11 |
RT |
86 |
48 |
| 12 |
I2 |
MeOH |
8 |
RT |
81 |
49 |
| 13 |
Glutamic acid |
EtOH |
7 |
RT |
89 |
50 |
| 14 |
MNPs@CNF@ATSM–Co(II) |
EtOH |
2 |
40 |
94 |
This work |
In addition to the reusability of the catalyst, most importantly, the use of a non-toxic green solvent that is environment friendly and does not require the use of any additives or toxic solvents supports the green chemistry approach well. These promising results should be attributed to the cooperation of the three metal positions: Lewis acid in MNPs@CNF@ATSM–Co(II).
Mechanism studies
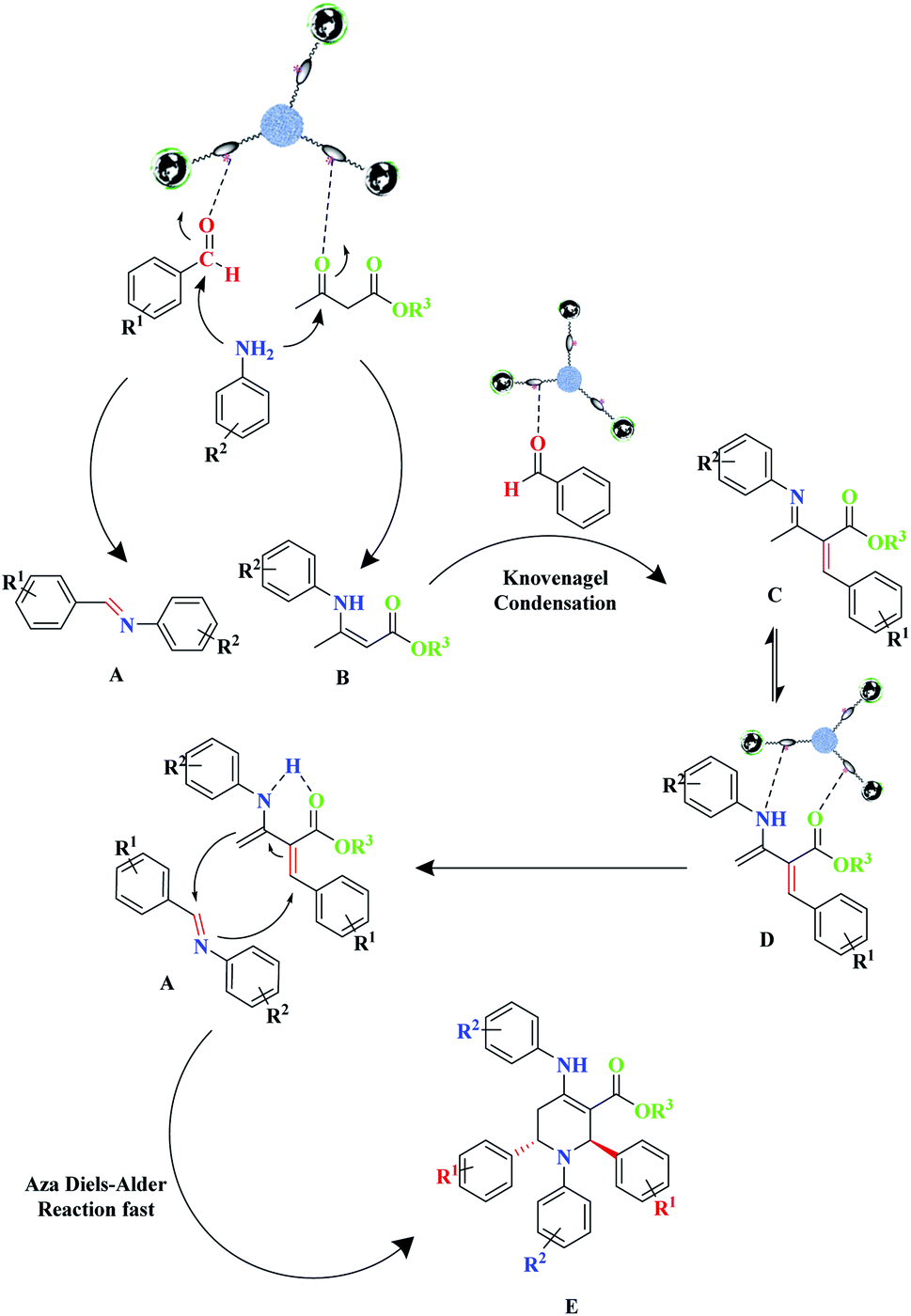
To demonstrate the benefit and uniqueness of the MNPs@CNF@ATSM–Co(II) catalyst as well as its application in the reactions of piperidine, a homolog of the MNPs@CNF@ATS–Co(II) as a mononuclear catalyst triwere synthesized and used in the model reaction. The two catalysts were compared in the piperidine reaction; the reaction did not progress much after 2 h using the mononuclear catalyst, whereas the catalyst trinuclear MNPs@CNF@ATSM–Co(II) showed 92% yield after 2 h. On the other hand, after 2 h, the MNPs@CNF@ATS–Co(II) catalyst did not perform. However, it can be concluded that the trinuclear form plays a key role in the catalyst. A proposed reaction mechanism for this three-component reaction for the formation of piperidine derivatives is depicted in Scheme 3.
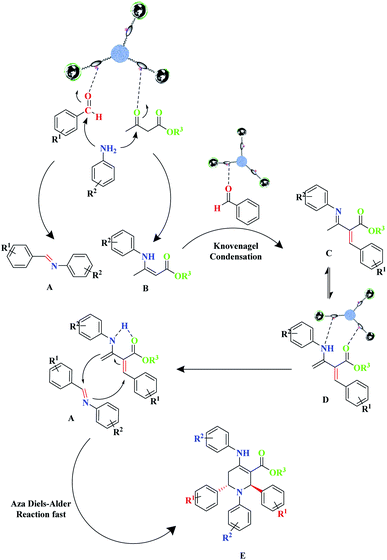 |
| | Scheme 3 The suggested mechanism for the synthesis of piperidine derivatives with MNPs@CNF@ATSM–Co(II). | |
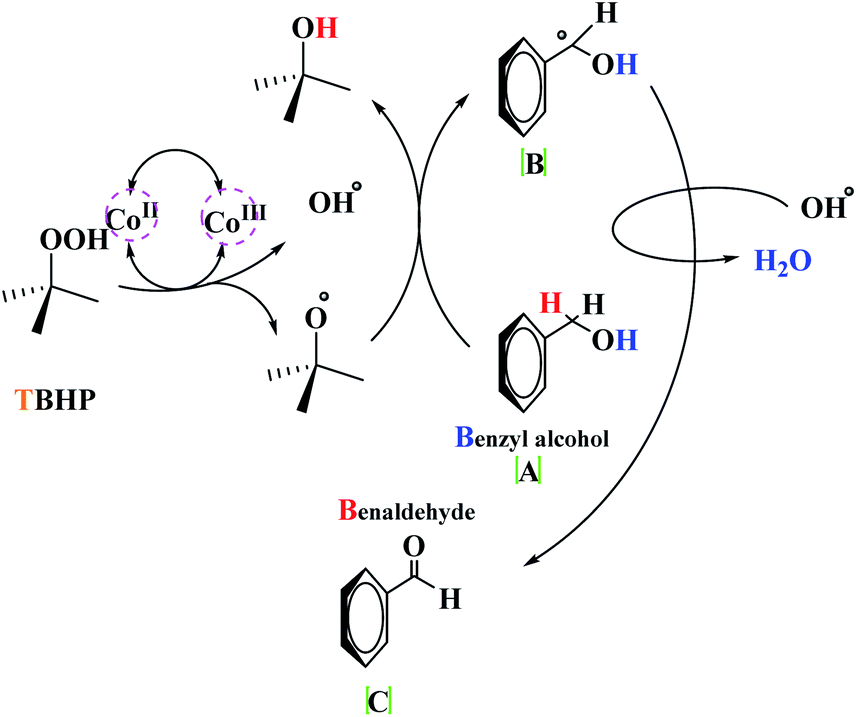
The detailed mechanism herein is based on a mechanism that has previously been presented in the literature.18 This proposed mechanism involves the Knoevenagel and Aza Diels–Alder reaction, wherein amine initially reacts with β-ketoester and aldehyde in the presence of the catalyst to form enamine (A) and imine (B), respectively. Benzaldehyde, which is retained in the reaction mixture, undergoes Knoevenagel condensation with enamine (A), leading to the formation of intermediate (C) and reactive form (D). In the next step, due to the diene core present in intermediate (D), it proceeds toward an intramolecular Aza Diels–Alder reaction with imine (A), which affords the expected piperidine (E) (Scheme 3). Besides, the decomposition of t-BuOOH on the surface of Co(II) generates the t-BuO˙ species. Next, t-BuOO˙ and t-BuO˙ radical species undergo hydrogen abstraction of the substrate (a) to produce intermediate (b). The reaction of intermediate (b) with HO results in (c), which ultimately gives the corresponding aldehyde (e) through the elimination of t-BuOH (Scheme 4).
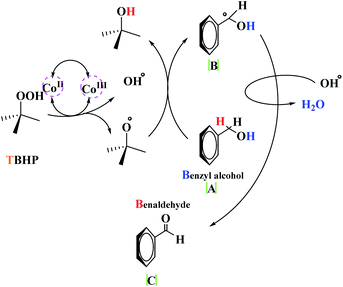 |
| | Scheme 4 The suggested mechanism for alcohol oxidation with MNPs@CNF@ATSM–Co(II). | |
Conclusion
We have introduced a new recyclable cobalt trinuclear magnetic catalyst) MNPs@CNF@ATSM–Co(II) (as a green trinuclear catalyst. Also, features such as high efficiency and biodegradability have made the synthesized catalyst an active compound. This catalyst has three strong arms for the oxidation of primary and secondary alcohol and highly substituted piperidines via multi-component reaction between aromatic aldehydes, anilines, and β-ketoesters in ethanol medium. To characterize the trinuclear catalyst, various techniques, including FT-IR, XRD, TGA, HRTEM, FE-SEM, EDX, EDS, VSM, and ICP analysis were employed. The use of this green, highly efficient, and readily available catalyst with simple and clean exaction profile, substantial yields, and short reaction times, besides the safe solvent ethanol, simple purification of products, and absence of hazardous organic solvents, provides a good example of a competitive and alternative synthetic methodology. It can be used to prepare important compounds such as piperidine derivatives and aldehydes. In the end, it can be claimed that this protocol is a benign, environment-friendly method for oxidation and heterocycle synthesis.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We gratefully acknowledge the financial support of the research Council of the University of Birjand.
References
-
(a) P. Ghamari Kargar, M. Noorian, E. Chamani, G. Bagherzade and Z. Kiani, RSC Adv., 2021, 11, 17413–17430 RSC;
(b) P. Ghamari Kargar, S. Aryanejad and G. Bagherzade, Appl. Organomet. Chem., 2020, e5965 CAS.
- P. Ghamari Kargar, M. Bakherad, A. Keivanloo and A. H. Amin, Iran. J. Catal., 2018, 8, 179–187 Search PubMed.
-
(a) N. Elders, D. van der Born, L. J. D. Hendrickx, B. J. J. Timmer, A. Krause, E. Janssen, F. J. J. de Kanter, E. Ruijter and R. V. A. Orru, Angew. Chem., 2009, 121, 5970–5973 CrossRef;
(b) E. R. Bonfield and C.-J. Li, Adv. Synth. Catal., 2008, 350, 370–374 CrossRef CAS.
- J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS PubMed.
- S. Petit, J. Nallet, M. Guillard, J. Dreux, R. Chermat, M. Poncelet, C. Bulach, P. Simon, C. Fontaine, M. Barthelmebs and J. Imbs, Eur. J. Med. Chem., 1991, 26, 19–32 CrossRef CAS.
- Y. Zhou, V. E. Gregor, B. K. Ayida, G. C. Winters, Z. Sun, D. Murphy, G. Haley, D. Bailey, J. M. Froelich, S. Fish, S. E. Webber, T. Hermann and D. Wall, Bioorg. Med. Chem. Lett., 2007, 17, 1206–1210 CrossRef CAS PubMed.
- B. Ho, A. Michael Crider and J. P. Stables, Eur. J. Med. Chem., 2001, 36, 265–286 CrossRef CAS PubMed.
- M. Misra, S. K. Pandey, V. P. Pandey, J. Pandey, R. Tripathi and R. P. Tripathi, Bioorg. Med. Chem., 2009, 17, 625–633 CrossRef CAS PubMed.
- B. I. Mobele, T. Kinahan, L. G. Ulysse, S. V. Gagnier, M. D. Ironside, G. S. Knox and F. Mohammadi, Org. Process Res. Dev., 2006, 10, 914–920 CrossRef CAS.
- L. A. Sorbera, J. Silvestre and J. Castañer, Drugs Future, 1998, 23, 955 CrossRef CAS.
- J. C. Pastre and C. R. Duarte Correia, Org. Lett., 2006, 8, 1657–1660 CrossRef CAS PubMed.
- J. Adams Jr, M.-L. Chang and L. Klaidman, Curr. Med. Chem., 2001, 8, 809–814 CrossRef CAS PubMed.
- C. Jaspers, G. Benker and D. Reinwein, Clin. Invest., 1994, 72(6), 451–456 CAS.
- G. Gründer, H. Wetzel, E. Hammes and O. Benkert, Psychopharmacology, 1993, 111, 123–126 CrossRef PubMed.
- P. Chand, P. L. Kotian, A. Dehghani, Y. El-Kattan, T.-H. Lin, T. L. Hutchison, Y. S. Babu, S. Bantia, A. J. Elliott and J. A. Montgomery, J. Med. Chem., 2001, 44, 4379–4392 CrossRef CAS PubMed.
- T. M. Block, N. Zitzmann, A. S. Mehta, S. Carrouée, T. D. Butters, F. M. Platt, J. McCauley, B. S. Blumberg and R. A. Dwek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11878–11882 CrossRef PubMed.
- J. E. Groopman, Clin. Infect. Dis., 1990, 12, 931–937 CrossRef CAS PubMed.
- S. S. Sajadikhah, N. Hazeri, M. T. Maghsoodlou, S. M. Habibi-Khorassani and A. C. Willis, Res. Chem. Intermed., 2014, 40, 723–736 CrossRef CAS.
- S. Källström and R. Leino, Bioorg. Med. Chem., 2008, 16, 601–635 CrossRef PubMed.
- Z. Madanifar and M. Kangani, Org. Chem. Res., 2017, 3(2), 119–125 Search PubMed.
- S. Pal, L. H. Choudhury and T. Parvin, Mol. Diversity, 2012, 16, 129–143 CrossRef CAS PubMed.
- R. Ramachandran, S. Jayanthi and Y. T. Jeong, Tetrahedron, 2012, 68, 363–369 CrossRef CAS.
- S. S. Sajadikhah, M. T. Maghsoodlou, N. Hazeri, S. M. Habibi-Khorassani and A. C. Willis, Chin. Chem. Lett., 2012, 23, 569–572 CrossRef CAS.
- B. Umamahesh, V. Sathesh, G. Ramachandran, M. Sathishkumar and K. Sathiyanarayanan, Catal. Lett., 2012, 142, 895–900 CrossRef CAS.
- J. Aboonajmi, M. T. Maghsoodlou, N. Hazeri, M. Lashkari and M. Kangani, Res. Chem. Intermed., 2015, 41, 8057–8065 CrossRef CAS.
- A. T. Khan, M. M. Khan and K. K. R. Bannuru, Tetrahedron, 2010, 66, 7762–7772 CrossRef CAS.
- S. Mishra and R. Ghosh, Tetrahedron Lett., 2011, 52, 2857–2861 CrossRef CAS.
- J. Aboonajmi, M. R. Mousavi, M. T. Maghsoodlou, N. Hazeri and A. Masoumnia, Res. Chem. Intermed., 2015, 41, 1925–1934 CrossRef CAS.
- P. A. Clarke, A. V. Zaytzev and A. C. Whitwood, Tetrahedron Lett., 2007, 48, 5209–5212 CrossRef CAS.
- P. Clarke, A. Zaytsev and A. Whitwood, Synthesis, 2008, 2008, 3530–3532 CrossRef.
- G. Brahmachari and S. Das, Tetrahedron Lett., 2012, 53, 1479–1484 CrossRef CAS.
- M. R. M. Shafiee, B. H. Najafabadi and M. Ghashang, Res. Chem. Intermed., 2013, 39, 3753–3762 CrossRef CAS.
-
(a) F. Karipcin, M. Cengiz and B. Dede, J. Chem. Sci., 2009, 121, 163–171 CrossRef;
(b) S. Pal, A. K. Barik, S. Gupta, S. Roy, T. N. Mandal, A. Hazra, M. S. El Fallah, R. J. Butcher, S.-M. Peng, G.-H. Lee and S. K. Kar, Polyhedron, 2008, 27, 357–365 CrossRef CAS.
-
(a) J. Philip, V. Mahendran and L. J. Felicia, J. Nanofluids, 2013, 2, 112–119 CrossRef CAS;
(b) H. Khashei Siuki, G. Bagherzade and P. Ghamari Kargar, ChemistrySelect, 2020, 5, 13537–13544 CrossRef CAS;
(c) A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed;
(d) B. Ramaswamy, S. D. Kulkarni, P. S. Villar, R. S. Smith, C. Eberly, R. C. Araneda, D. A. Depireux and B. Shapiro, Nanomedicine, 2015, 11, 1821–1829 CrossRef CAS PubMed;
(e) L. He, M. Wang, J. Ge and Y. Yin, Acc. Chem. Res., 2012, 45, 1431–1440 CrossRef CAS PubMed;
(f) I. Kavre, G. Kostevc, S. Kralj, A. Vilfan and D. Babič, RSC Adv., 2014, 4, 38316–38322 RSC;
(g) S. Mornet, S. Vasseur, F. Grasset, P. Veverka, G. Goglio, A. Demourgues, J. Portier, E. Pollert and E. Duguet, Prog. Solid State Chem., 2006, 34, 237–247 CrossRef CAS;
(h) B. Gleich and J. Weizenecker, Nature, 2005, 435, 1214–1217 CrossRef CAS PubMed;
(i) D. W. Elliott and W. Zhang, Environ. Sci. Technol., 2001, 35, 4922–4926 CrossRef CAS PubMed;
(j) J. Philip, P. D. Shima and B. Raj, Appl. Phys. Lett., 2008, 92, 043108 CrossRef;
(k) J. Philip, T. Jaykumar, P. Kalyanasundaram and B. Raj, Meas. Sci. Technol., 2003, 14, 1289–1294 CrossRef CAS;
(l) V. Mahendran and J. Philip, Appl. Phys. Lett., 2012, 100, 073104 CrossRef;
(m) V. Chaudhary and R. V. Ramanujan, Sci. Rep., 2016, 6, 35156 CrossRef CAS PubMed.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
-
(a) K. Missoum, M. N. Belgacem, J.-P. Barnes, M.-C. Brochier-Salon and J. Bras, Soft Matter, 2012, 8, 8338 RSC;
(b) X. Zhu, J. Niu, F. Zhang, J. Zhou, X. Li and J. Ma, New J. Chem., 2014, 38, 4622–4627 RSC;
(c) T. S. El-Din, A. A. Elzatahry, D. M. Aldhayan, A. M. El-Din and S. S. Al-Deyab, Int. J. Electrochem. Sci., 2011, 6, 6177–6183 Search PubMed;
(d) S. Xuan, W. Jiang, X. Gong, Y. Hu and Z. Chen, J. Phys. Chem. C, 2009, 113, 553–558 CrossRef CAS;
(e) Z. Chen, Z. Xue, L. Chen, Z. Geng, R. Yang, L. Chen and Z. Wang, New J. Chem., 2013, 37, 3731 RSC.
-
(a) P. Ghamari kargar, G. Bagherzade and H. Eshghi, RSC Adv., 2020, 10, 32927–32937 RSC;
(b) P. Ghamari Kargar, G. Bagherzade and H. Eshghi, RSC Adv., 2020, 10, 37086–37097 RSC;
(c) P. Ghamari kargar, G. Bagherzade and H. Eshghi, RSC Adv., 2021, 11, 4339–4355 RSC;
(d) P. Ghamari kargar and G. Bagherzade, RSC Adv., 2021, 11, 19203–19220 RSC.
- N. Lin and A. Dufresne, Eur. Polym. J., 2014, 59, 302–325 CrossRef CAS.
-
(a) M. Kaushik and A. Moores, Green Chem., 2016, 18, 622–637 RSC;
(b) M. Patchan, J. L. Graham, Z. Xia, J. P. Maranchi, R. McCally, O. Schein, J. H. Elisseeff and M. M. Trexler, Mater. Sci. Eng., C, 2013, 33, 3069–3076 CrossRef CAS PubMed;
(c) M. Jorfi and E. J. Foster, J. Appl. Polym. Sci., 2015, 14, 132 Search PubMed;
(d) J. O. Zoppe, V. Ruottinen, J. Ruotsalainen, S. Rönkkö, L.-S. Johansson, A. Hinkkanen, K. Järvinen and J. Seppälä, Biomacromolecules, 2014, 15, 1534–1542 CrossRef CAS PubMed.
-
(a) J.-E. Kim, J.-Y. Shin and M.-H. Cho, Arch. Toxicol., 2012, 86, 685–700 CrossRef CAS PubMed;
(b) J. Safari, S. H. Banitaba and S. D. Khalili, J. Mol. Catal. A: Chem., 2011, 335, 46–50 CrossRef CAS;
(c) S. Wu, H. Ma, X. Jia, Y. Zhong and Z. Lei, Tetrahedron, 2011, 67, 250–256 CrossRef CAS;
(d) Y.-X. An, N. Li, H. Wu, W.-Y. Lou and M.-H. Zong, ACS Sustainable Chem. Eng., 2015, 3, 2951–2958 CrossRef CAS.
- T. Cheng, Q. Zhao, D. Zhang and G. Liu, Green Chem., 2015, 17, 2100–2122 RSC.
-
(a) M. H. Mashhadizadeh and Z. Karami, J. Hazard. Mater., 2011, 190, 1023–1029 CrossRef CAS PubMed;
(b) A. E. Karatapanis, Y. Fiamegos and C. D. Stalikas, Talanta, 2011, 84, 834–839 CrossRef CAS PubMed.
- G. P. Glaspell, P. W. Jagodzinski and A. Manivannan, J. Phys. Chem. B, 2004, 108, 9604–9607 CrossRef CAS.
- N. Safajoo, B. B. F. Mirjalili and A. Bamoniri, RSC Adv., 2019, 9, 1278–1283 RSC.
- A. T. Khan, T. Parvin and L. H. Choudhury, J. Org. Chem., 2008, 73, 8398–8402 CrossRef CAS PubMed.
- T. Mukaiyama and S. Kobayashi, J. Organomet. Chem., 1990, 382, 39–52 CrossRef CAS.
- A. T. Khan, M. Lal and M. M. Khan, Tetrahedron Lett., 2010, 51, 4419–4424 CrossRef CAS.
- M. Lashkari, F. Mohamadpoura and N. Hazeri, Org. Chem. Res., 2020, 6, 82–91 Search PubMed.
- M. Shokoohian, N. Hazeri, M. Maghsoodlou and M. Lashkari, Chem. J. Mold., 2019, 14, 97–104 CAS.
- F. Mohamadpour, Polycyclic Aromat. Compd., 2020, 40, 681–692 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00208b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Ghodsieh Bagherzade
and
Ghodsieh Bagherzade