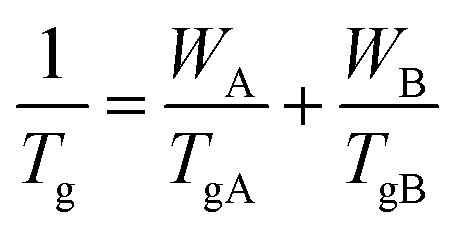DOI:
10.1039/D1RA02467A
(Paper)
RSC Adv., 2021,
11, 23184-23191
Copolymerization of 1,3-butadiene with phenyl/phenethyl substituted 1,3-butadienes: a direct strategy to access pendant phenyl functionalized polydienes†
Received
29th March 2021
, Accepted 18th June 2021
First published on 1st July 2021
Abstract
Copolymerization of 1,3-butadiene with various types of phenyl substituted 1,3-butadiene derivatives, including (E)-1-phenyl-1,3-butadiene (PBD), 1-phenethyl-1,3-butadiene (PEBD), 1-(4-methoxylphenyl)-1,3-butadiene (p-MEPBD), 1-(2-methoxylphenyl)-1,3-butadiene (o-MEPBD) and 1-(4-N,N-dimethylaminophenyl)-1,3-butadiene (p-DMPBD), by using a coordination polymerization system of CpTiCl3/MAO is reported herein. Comonomers PBD and PEBD can be copolymerized with 1,3-butadiene in a large range of comonomer feed ratios (0–44.6% for PBD, 0–30.2% for PEBD), affording the targeted copolymers with well-controlled comonomer incorporations, molecular weights, polydispersities and microstructure, whereas no corresponding copolymer products were obtained under identical conditions when p-MEPBD, o-MEPBD and p-DMPBD were employed. Moreover, different polymerization parameters, including temperature, Al/Ti ratio, etc., posed a significant influence on the polymerization behaviors, as well as the properties of the resultant copolymers. Microstructure analysis by NMR spectra revealed high 1,4-selectivities of the catalysts, and the glass transition temperature (Tg) of the resulted copolymer was found to be highly dependent on the incorporation content of the comonomers; with an increasing comonomer content, a gradually increasing Tg was demonstrated.
Introduction
Styrene-butadiene rubber (SBR) and cis-1,4-polybutadiene rubber (BR) are the two most commonly used synthetic general-purpose rubbers. Due to their different main-chain compositions, strikingly different mechanical performances are obtained and therefore they are employed for different applications. SBR displays superior abrasion resistance and wet-skid resistance, which is structurally caused by the presence of rigid polystyrene segments, whereas BR has much better elastic properties and lower rolling resistance due to the long flexible cis-1,4-sequences, but its abrasion resistance is inferior.1 Therefore, in practical applications, such as tire treads, shoe soles, etc., a proper combination of these two rubbers is generally applied. Inspired by the above-mentioned different chemical structures and the resultant different properties, we anticipate that if pendant phenyl groups were incorporated into the main chain of cis-1,4-polybutadienes, the produced polymers will be highly likely to balance the properties of SBR and BR, and thus demonstrate similar properties to SBR/BR blends. In addition, this chemical modification is also capable of avoiding the microphase separation that usually occurs in SBR/BR blends.
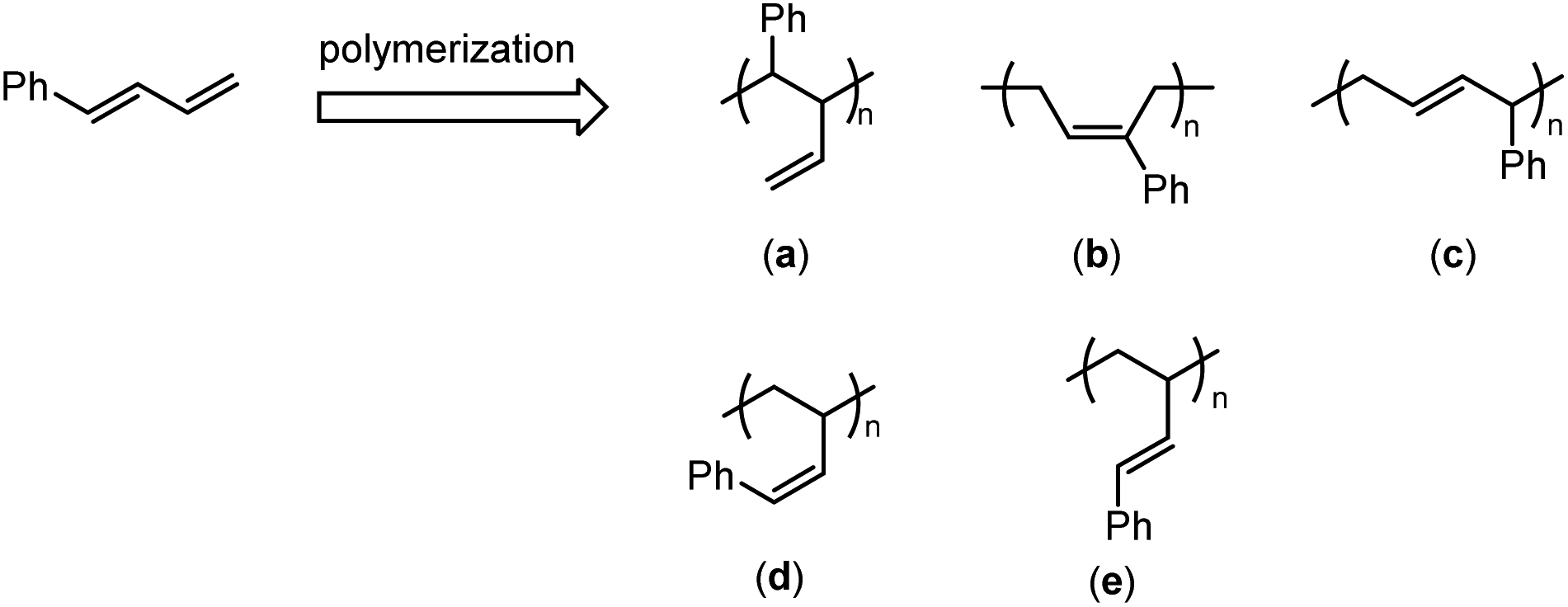
Despite colossal advances in the functionalization of polybutadienes in the past few decades,2–16 very few of them are concerned about pendant phenyl functionality. Theoretically, there are two possible approaches to access the pendant phenyl functionalized polybutadienes; one is the post-modification process by reacting readily available polybutadienes with phenyl derivatives, and the other is the direct process by copolymerization of 1,3-butadiene (BD) with phenyl substituted 1,3-butadiene (PSBD) derivatives. However, due to the poor mutual reactivities between the polydiene backbone and the benzene ring, the post-modification process usually relies on the conversion of main-chain C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds into reactive intermediates, such as alkyl halides, epoxides, etc., and thus sacrifices the flexibility of double bonds and causes irreversible damage to the rubber elasticity.17–20 In contrast, the second methodology leaves behind or newly generates a double bond that could maintain flexibility after copolymerization, and therefore is preferably adopted by us. PSBD is a kind of unique and interesting monomer, since it may follow two polymerization patterns and form theoretically five different types of isomers. Such as 1-phenyl-1,3-butadiene (PBD), it may follow the polymerization patterns of a 2-vinyl substituted styrene to form isomer “a” or of a 1-phenyl substituted 1,3-butadiene to form isomers “b–e” (Scheme 1). Up to date, many methodologies have been reported to polymerize PSBD, including anionic,21,22 cationic,23,24 and radical polymerizations.25–27 Whereas the coordination polymerization was less investigated, in spite of the high stereoselectivity it usually possessed. Vessel, Bruzzone and Longo reported the usage of titanium-based Ziegler–Natta to homo-/co-polymerize PSBDs.28–30 Cui et al. recently employed N-heterocyclic carbene lutecium and scandium complexes to promote highly 3,4-selective polymerization of 2-phenyl or 1-phenyl substituted 1,3-butadienes.31,32 Mecking et al. utilized nickel and neodymium precatalysts to achieve the cis-1,4-selective copolymerization of 1,3-butadiene and PSBD.33–35 It is obvious that these coordination polymerization system revealed great advantages in performing high regio- and stereo-selectivities. Herein, we report the copolymerization of 1,3-butadiene with various phenyl/phenethyl substituted 1,3-butadiene by half-titanocene precatalyst to access diversified types of pendant phenyl functionalized polydiene materials. Detailed analysis of the resulted copolymers was also carried out.
C double bonds into reactive intermediates, such as alkyl halides, epoxides, etc., and thus sacrifices the flexibility of double bonds and causes irreversible damage to the rubber elasticity.17–20 In contrast, the second methodology leaves behind or newly generates a double bond that could maintain flexibility after copolymerization, and therefore is preferably adopted by us. PSBD is a kind of unique and interesting monomer, since it may follow two polymerization patterns and form theoretically five different types of isomers. Such as 1-phenyl-1,3-butadiene (PBD), it may follow the polymerization patterns of a 2-vinyl substituted styrene to form isomer “a” or of a 1-phenyl substituted 1,3-butadiene to form isomers “b–e” (Scheme 1). Up to date, many methodologies have been reported to polymerize PSBD, including anionic,21,22 cationic,23,24 and radical polymerizations.25–27 Whereas the coordination polymerization was less investigated, in spite of the high stereoselectivity it usually possessed. Vessel, Bruzzone and Longo reported the usage of titanium-based Ziegler–Natta to homo-/co-polymerize PSBDs.28–30 Cui et al. recently employed N-heterocyclic carbene lutecium and scandium complexes to promote highly 3,4-selective polymerization of 2-phenyl or 1-phenyl substituted 1,3-butadienes.31,32 Mecking et al. utilized nickel and neodymium precatalysts to achieve the cis-1,4-selective copolymerization of 1,3-butadiene and PSBD.33–35 It is obvious that these coordination polymerization system revealed great advantages in performing high regio- and stereo-selectivities. Herein, we report the copolymerization of 1,3-butadiene with various phenyl/phenethyl substituted 1,3-butadiene by half-titanocene precatalyst to access diversified types of pendant phenyl functionalized polydiene materials. Detailed analysis of the resulted copolymers was also carried out.
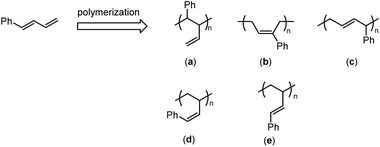 |
| | Scheme 1 The generated possible isomers for polymerization of PBD. | |
Experimental
General procedure and materials
All oxygen and moisture sensitive manipulations were carried out under nitrogen/argon atmosphere by using standard Schlenk techniques. Toluene, n-hexane, tetrahydrofuran were refluxed over sodium/diphenylketyl for twelve hours and distilled under nitrogen before use. Cyclopentadienyltitanium trichloride (CpTiCl3), triphenylmethyl-phosphonium bromide, trans-cinnamaldehyde and sodium bis(trimethylsilyl)amide (NaHMDS) were purchased from Alfa aesar and used without further purification. Methylaluminoxane (MAO, 10 wt% in toluene) was commercially available from Akzo Nobel Chemical. 1,3-Butadiene was supplied by Jinzhou Petrochemical Corporation and purified by passing through four columns packed with 4 Å molecular sieves and KOH prior to use. The monomers of 1-(4-methoxylphenyl)-1,3-butadiene, 1-(2-methoxylphenyl)-1,3-butadiene, and 1-(4-N,N-dimethylaminophenyl)-1,3-butadiene were prepared according to previous reports.36
The number average molecular weights (Mn) and molecular weight distributions (Mw/Mn) of polymers were measured at 30 °C by using gel permeation chromatography (GPC) equipped with a Waters 515 HPLC pump, four columns (HMW 7 THF, HMW 6E THF × 2, HMW 2 THF), and a Waters 2414 refractive index detector. Tetrahydrofuran (THF) was used as eluent at a flow rate of 1.0 mL min−1. Sample solutions were filtered through a 0.45 μm microfilter before injection. The values of Mn and Mw/Mn were calculated by using polystyrene calibration. FTIR spectra were performed on Thermo Nicolet is 10 FTIR spectrophotometer and films were cast from CS2 solution onto KBr plates. The isomer units proportions of polybutadiene were determined as reported in literatures.37,38 The NMR data were obtained on a Bruker 400 MHz spectrometer at ambient temperature, CDCl3 is used as solvent. Differential scanning calorimetry (DSC) analyses were performed on a Perkin Elmer Diamond DSC apparatus at a heating rate of 10 °C min−1 under a continuous flow of helium, using aluminum capsules. Glass transition temperatures were determined from the second cycle.
Synthesis of (E)-1-phenyl-1,3-butadiene comonomer (PBD)
The synthetic procedure was carried out according to previous literature.39 Under nitrogen atmosphere, to a solution of triphenylmethylphosphonium bromide (10.72 g, 30 mmol) in THF (150 mL) was slowly added NaHMDS (2.0 mol L−1 in THF, 15 mL, 30 mmol) at 0 °C. After stirring at 0 °C for 1 h, trans-cinnamaldehyde (3.41 g, 25.9 mmol) was slowly added dropwise at 0 °C. The reaction mixture was allowed to stir at 25 °C overnight. The resulting brown suspension was filtered through a short pad of silica gel with diethyl ether washings (100 mL × 3). The combined filtrate was concentrated under partial vacuum and the residue was purified by silica gel flash chromatograph (petroleum ether as eluent) to give the desired product as a colorless oil (2.34 g, 70.0%). Then a desired amount of toluene was added until the concentration of (E)-1-phenyl-1,3-butadiene reached 2 mol L−1, and the obtained solution was stored under nitrogen for further use. 1H NMR (400 MHz, CDCl3, δ, ppm): 7.41 (m, 2H), 7.32 (m, 2H), 7.25–7.21 (m, 1H), 6.79 (dd, 1H), 6.59–6.49 (m, 2H), 5.33 (d, J = 16.9 Hz, 1H), 5.17 (d, 1H). The NMR agreed with previous reports.40
Synthesis of 1-phenethyl-1,3-butadiene comonomer (PEBD)
Similar procedure was adopted to prepare PEBD but use 3-phenylpropionaldehyde and allyltriphenylphosphonium bromide as starting materials. Yield: 89%. 1H NMR (400 MHz), CDCl3, δ 7.40–7.30 (m, 1H), 7.30–7.20 (m, 3H), 6.78–6.56 (m, 0.45H), 6.48–6.30 (m, 0.55H), 6.23–6.00 (m, 1H), 5.86–5.70 (m, 0.55H), 5.67–5.46 (m, 0.45H), 5.24 (dd, 0.45H), 5.19–5.08 (m, 1H), 5.03 (dd, 0.55H), 2.88–2.72 (m, 2H), 2.63–2.52 (m, 1H), 2.52–2.41 (m, 1H). The NMR agreed with previous reports.41
Typical copolymerization procedure
Polymerizations were carried out in an oxygen- and moisture-free ampule capped with a rubber septum. A toluene solution (10 mL) of 1,3-butadiene and comonomer was placed in ampule at prescribed ratios, followed by the addition of MAO to the ampule. The total amount of monomer is 10.3 mmol. After equilibration of the solution at the desired temperature, the reaction was started by injection of a toluene solution of the CpTiCl3 catalyst (10.3 μmol). Polymerization was carried out for 5 h and quenched by adding 2.0 mL acidified ethanol containing 2,6-di-tert-butyl-4-methylphenol as a stabilizer. The polymer was washed with ethanol repeatedly, and then dried in vacuum at 40 °C to constant weight. The polymer yield was determined by gravimetric analysis.
Results and discussion
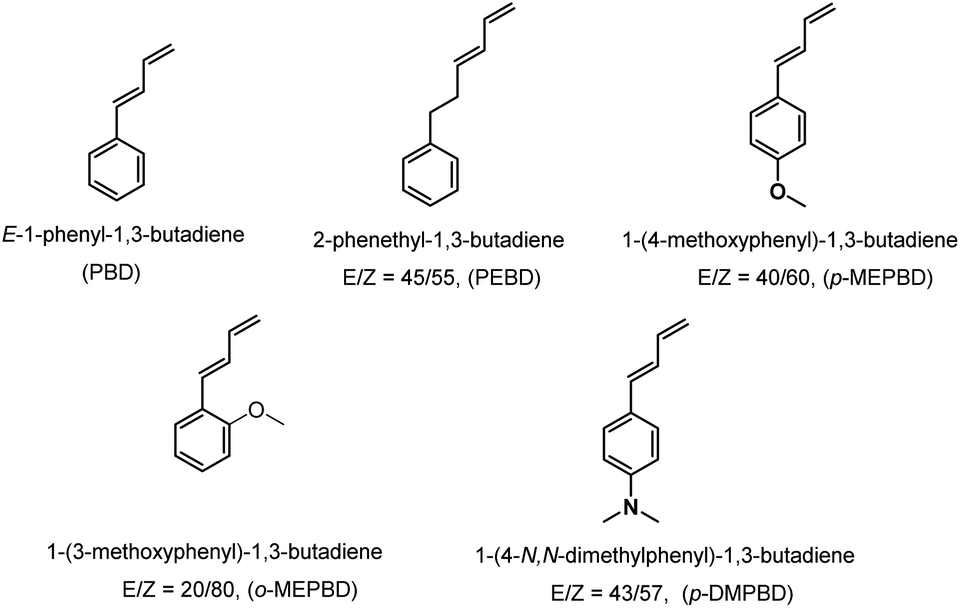
The PSBD comonomers (Chart 1), including 1-phenyl-1,3-butadiene (PBD), 1-phenethyl-1,3-butadiene (PEBD), 1-(4-methoxylphenyl)-1,3-butadiene (p-MEPBD), 1-(2-methoxylphenyl)-1,3-butadiene (o-MEPBD), 1-(4-N,N-dimethylaminophenyl)-1,3-butadiene (p-DMPBD), were prepared in high yields under mild conditions via Wittig reaction according to previously reported protocol.36 Further purification by flash column chromatography was undertaken to obtain the readily polymerizable comonomers. All the chemical structures and purities were confirmed by 1H NMR and 13C NMR. As evidenced from 1H NMR spectra, two isomers were obtained for comonomer PEBD, p-MEPBD, o-MEPBD, and p-DMPBD, with E/Z ratios of 45/55, 40/60, 20/80 and 43/57 respectively.
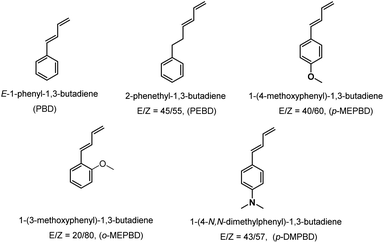 |
| | Chart 1 Specific structures for PSBD comonomers. | |
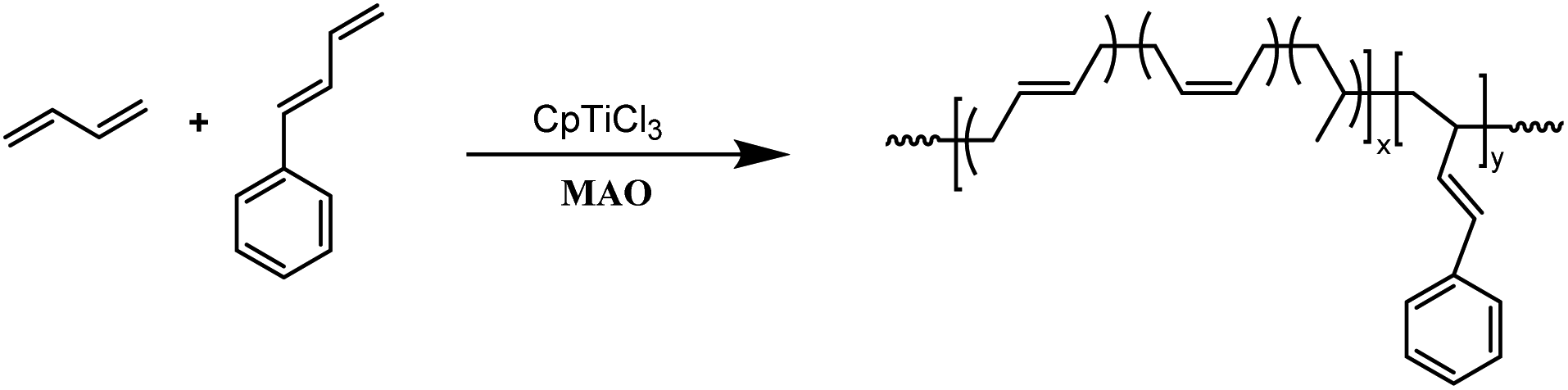
The copolymerization studies were commenced between BD monomer and PBD comonomer under various conditions by using CpTiCl3/MAO (where Cp is η5-cyclopentadienyl) catalytic system (Scheme 2). As the data summarized in Table 1, polymerization temperature brought significant influences on the catalytic performances. The optimized catalytic activity was observed at 20 °C, both lower and higher temperature would render the active species less active. Higher temperature also resulted in smaller molecular weights in this study, probably due to the accelerated chain transfer reactions. Despite of lower yields, the highest PBD incorporation was observed at 50 °C with a value of 7.84% when 10% feed ratio is applied. On this account, latter examinations of the effect of cocatalyst ratio on copolymerization behaviors were carried out at this temperature. Increasing the ratio of MAO/Ti from 100 to 200 resulted in a dramatical enhancement of the catalytic activity from 68.2% to 97.1%, simultaneous increasing PBD contents from 7.84% to 9.04% was also observed. Continuous increasing ratio to 300 brought little influences neither on the activities nor on microstructures. However, a much lower polymer yield (80.6%) was displayed when the ratio of MAO/Ti reached 500, which is usually attributed to over-reduction of the active species. Similar to other copolymerization studies, increasing the PBD/BD feed ratio led to remarkably increased PBD content in the copolymer, but accompanied by obviously decreased catalytic activities and molecular weights, which is probably caused by the lower reactivity of PBD. When fifty percent of PBD was applied to the polymerization system, its incorporation ratio in the resultant copolymers is able to reach as high as 44.6 mol%, which is the highest value in the present study. It deserves to mention that all of the GPC profiles in Table 1 are unimodal, indicating the formation of copolymers rather than a mixture of poly(1,3-butadiene)s and poly(1-phenyl-1,3-butadiene)s.
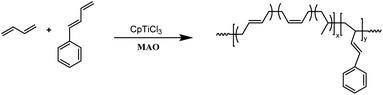 |
| | Scheme 2 Copolymerization of 1,3-butadiene and 1-phenyl-1,3-butadiene. | |
Table 1 Copolymerizations of BD and PBD with CpTiCl3/MAO systema
| Run |
Comon. in feedb |
MAO/Ti |
T (°C) |
Yield (%) |
Cont.c (mol%) |
Microstructure of PBDd |
Mne × 10−4 |
Đe |
Tgf (°C) |
| cis-1,4 |
trans-1,4 |
1,2- |
| Polymerization conditions: in toluene for 5 h, [BD] = 1.85 M, [BD + comonomer]/[Ti] = 1000. Molar ratio of comonomer to 1,3-butadiene. Comonomer content (mol%) in the copolymer that established by 1H NMR spectra. Microstructure determined by FTIR and NMR.42 Determined by GPC using polystyrene standards. Determined by DSC. Polymerization time 24 h. Styrene was used as comonomer instead of 1-phenyl-1,3-butadiene. |
| 1 |
10 |
100 |
0 |
33.7 |
6.28 |
74.5 |
9.3 |
16.1 |
2.29 |
3.05 |
−83.8 |
| 2 |
10 |
100 |
20 |
72.1 |
7.07 |
74.9 |
9.7 |
15.5 |
2.15 |
2.51 |
−78.2 |
| 3 |
10 |
100 |
50 |
68.2 |
7.84 |
73.4 |
11.2 |
15.4 |
1.48 |
2.15 |
−75.7 |
| 4 |
10 |
200 |
50 |
97.1 |
9.04 |
74.2 |
11.4 |
14.4 |
1.99 |
2.64 |
−74.0 |
| 5 |
10 |
300 |
50 |
98.9 |
9.38 |
74.1 |
11.3 |
14.7 |
2.03 |
2.56 |
−72.3 |
| 6 |
10 |
500 |
50 |
80.6 |
9.10 |
74.0 |
11.8 |
14.2 |
1.74 |
2.49 |
−73.8 |
| 7 |
20 |
200 |
50 |
75.3 |
17.3 |
79.5 |
14.1 |
6.4 |
1.56 |
3.41 |
−58.0 |
| 8 |
30 |
200 |
50 |
63.3 |
25.9 |
79.2 |
15.8 |
5.0 |
1.10 |
3.63 |
−42.9 |
| 9 |
50 |
200 |
50 |
44.9 |
44.6 |
79.8 |
15.7 |
4.5 |
0.44 |
1.96 |
−22.4 |
| 10g |
100 |
200 |
50 |
58.9 |
— |
— |
— |
— |
3.63 |
1.63 |
36.8 |
| 11 |
0 |
100 |
50 |
89.0 |
0 |
76.1 |
19.2 |
4.7 |
2.78 |
2.39 |
−93.7 |
| 12h |
10 |
200 |
50 |
89.4 |
1.2 |
77.2 |
17.4 |
5.5 |
2.42 |
2.96 |
— |
| 13h |
20 |
200 |
50 |
67.8 |
2.3 |
77.5 |
17.1 |
5.4 |
3.45 |
3.05 |
— |
| 14h |
30 |
200 |
50 |
40.6 |
3.6 |
76.7 |
17.4 |
5.9 |
2.33 |
2.83 |
— |
| 15h |
100 |
200 |
50 |
77.4 |
— |
— |
— |
— |
0.32 |
1.28 |
— |
Due to the reason that PBD polymerization has the chance to follow the pattern of a 2-vinyl substituted styrene, in order to contrast the differences of PBD and styrene, comparative copolymerization of BD and styrene (St) was carried out in subsequent studies. As the data shown in Table 1, runs 12–15, in comparison with BD/PBD copolymerizations, the catalytic activities and the St content in the resultant BD/St copolymers were much lower under identical feed ratios. When 30 mol% St was fed into the system, only 3.6 mol% was observed in the copolymers, which is remarkably lower than PBD counterpart. Two reasons might be accountable for this:
(1) The active species of the present CpTiCl3/MAO system is a strongly electrophilic, coordinatively unsaturated organotitanium cation [CpTiP]+ (where P is growing polymer chain). PBD is more electron-rich than St monomer, and may follow a stronger η4 fashion than η2 fashion in St, and thus have bigger chance to be incorporated;
(2) π–σ switching for the growing chain terminal is difficult for BD/St copolymerizations due to their different polymerization mechanisms, which adopts π-allylic unit and σ bond as growing species respectively. Whereas both BD and PBD use π-allylic unit as growing species.
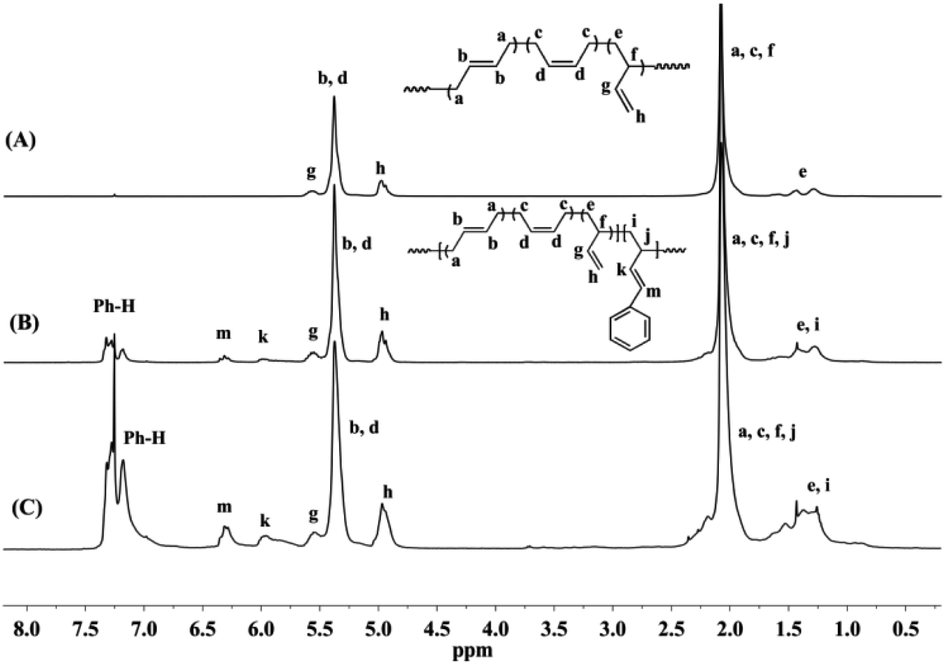
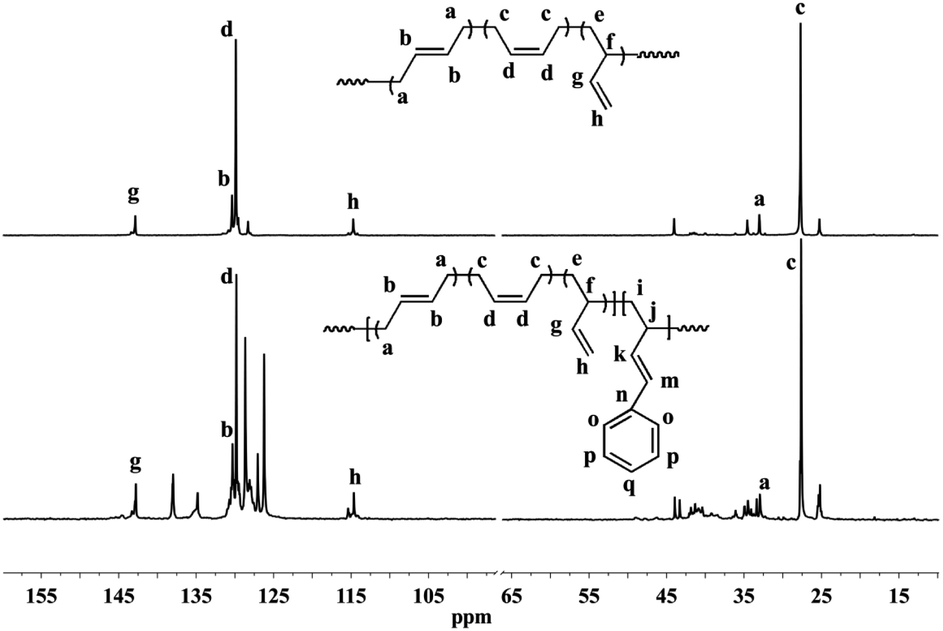
Detailed microstructures of BD/PBD copolymers were fully characterized by 1H and 13C NMR spectra. Representative 1H NMR and 13C NMR spectra along with the assignments are provided in Fig. 1–3. Peak assignments were confirmed by comparison with the data reported in the literature.43 As shown in Fig. 1, the peaks at 7.14–7.30 ppm are assigned to the protons of the aromatic rings. The olefin proton resonances at 5.60 ppm and 4.95 ppm are attributed to 3,4-units of poly(PBD). The methine (2.20 ppm) and methylene (1.36 ppm) proton resonances of 3,4-units of poly(PBD) were completely overlapped by polybutadiene protons. Additionally, the absence of proton resonances at 3.05 and 3.09 ppm that attributed to the methine proton resonances of 1,4-poly(PBD) indicates that there are only 3,4-moieties in the copolymer. Based on these analyses, PBD contents in the copolymers can therefore be calculated from the relative intensities of these protons using the following eqn (1):
| | |
PBD mol% = (I7.14–7.30/5)/[I7.14–7.30/5 + (I5.38–5.60 − 1/2I4.97)/2 + I4.97/2] × 100% = 2I7.14–7.30/[2I7.14–7.30 + 5(I5.38–5.60 + 1/2I4.97)] × 100%
| (1) |
where
I7.14–7.30 is the intensities of protons at 7.14–7.30 ppm and
I5.38–5.60 is the total intensity of protons at 5.38–5.60 ppm. Moreover, it was observed that, when the PBD incorporation increases, two new peaks at 3.05 and 3.09 ppm can be tracked in
Fig. 3, which are assignable to the methine protons of 1,4-poly(PBD), implying the enchainment differences of PBD at higher concentrations.
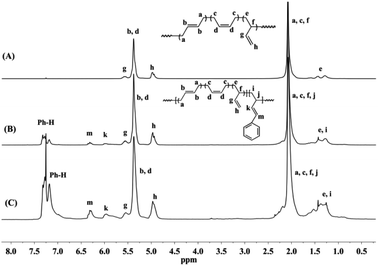 |
| | Fig. 1 Typical 1H NMR spectra of polybutadiene and BD/PBD copolymers (A): polybutadiene; (B): BD/PBD copolymer with 7.84 mol% PBD (Table 1, run 3); (C): BD/PBD copolymer with 25.9 mol% PBD (Table 1, run 8). | |
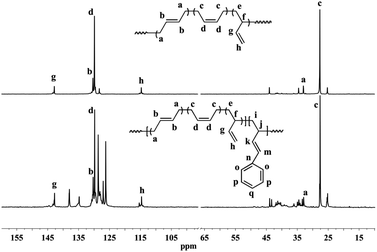 |
| | Fig. 2 Typical 13C NMR spectra of polybutadiene and BD/PBD copolymers (A): polybutadiene; (B): BD/PBD copolymer with 25.9 mol% PBD (Table 1, run 8). | |
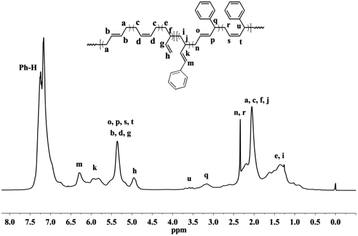 |
| | Fig. 3 1H NMR spectrum of BD/PBD copolymer with 44.6 mol% PBD (Table 1, run 9). | |
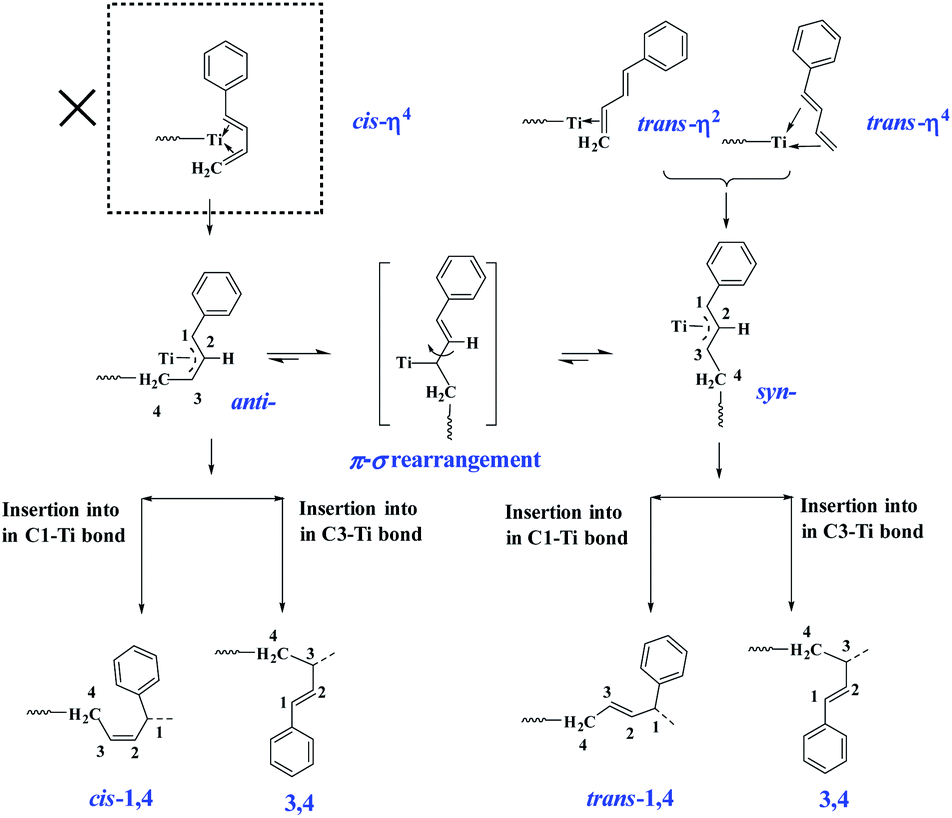
The above-observed regio- and stereoselectivity of poly(PBD) can be explained on the basis of the well-established mechanism for polymerization of conjugated dienes.44 As depicted in Scheme 3, the two different coordination modes (cis-η4- and trans-η4- or trans-η2-) of monomer to the metal center lead to a η3-coordinated allyl complex in syn- or anti-configuration, respectively. From these intermediates, the anti-isomer gives rise to cis-1,4- and 3,4-polymerization, while the syn-isomer produces trans-1,4- and 3,4-structures. Because all of the PBD monomer is E configuration, the cis-η4 coordination mode is absent in the present case, and all the monomer coordinates to the metal center via trans-η4- or trans-η2-fashions. Considering the insertion step, it is hypothesized that the presence of phenyl group causes a bulkier steric hindrance on C1 than C3, and thus the coordinated monomers preferably insert into the less congested C3 position, which resulted in predominant 3,4-unit especially under low concentration circumstances. When the feed ratio of PBD increased to 50 mol%, a small amount of trans-1,4- and cis-1,4-unit were detected, in which the cis-1,4-moiety is probably generated from the anti–syn isomerization through π–σ rearrangement.
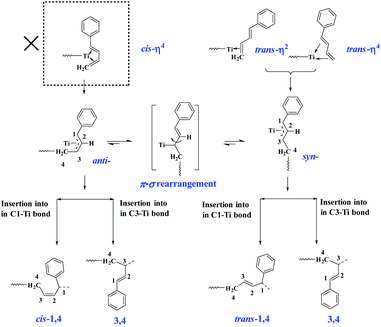 |
| | Scheme 3 The suggested mechanism for polymerization of PBD. | |
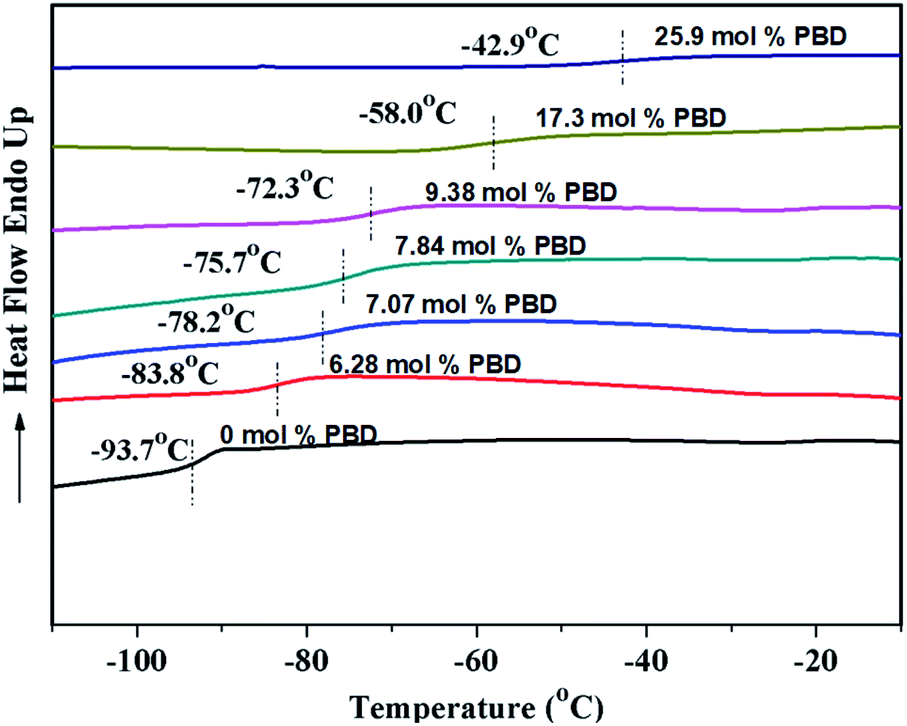
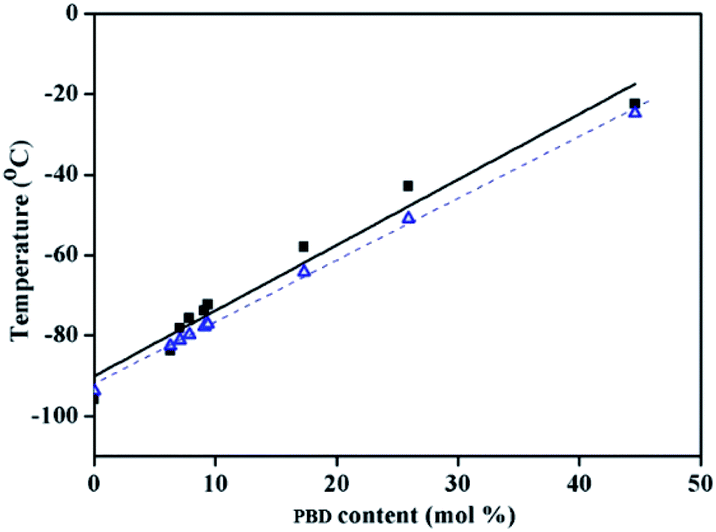
As shown in Table 2, regardless of polymerization conditions, the total cis-1,4-moiety always keeps around 75–80%, which is much higher than that obtained by ionic or radical polymerization.25,43 The thermal transitions of copolymers were determined by DSC. All the copolymers showed only one transition temperature (Tg) (Fig. 4), indicating the authenticity of copolymer rather than mixture of homopolymers. It is observed that the higher PBD incorporation is, the higher the glass (Tg) of the resulting copolymer is. Tg increased from −93.7 °C to −22.4 °C as the content of PBD in the copolymer increased from 0 to 44.6%. It is known that Tgs are strongly influenced by the chemical structure of the repeating unit, that is, Tg increases with the decreasing flexibility of the polymer chain. In the case of BD/PBD copolymer, the rotational energy increased with the incorporation of PBD due to the rigidity of the phenyl ring, resulting the eventual copolymer not completely amorphous anymore from appearance. The dependence of Tg of BD/PBD copolymers on the PBD content (mol%) is illustrated in Fig. 5. An almost linear increase of Tg with the PBD content was observed. Then the following experiential expression could be obtained:
| Tg (°C) = −90.07 + 1.629CPBD, (R2 = 0.9656) |
where
CPBD is the PBD content (mol%) in the copolymer. Thus, we can synthesize BD/PBD copolymer with needed
Tg via adjusting the PBD content through altering PBD feed ratios, polymerization temperature or cocatalyst loadings. It is worthy of note that the
Tgs of the copolymer is lower than zero when PBD content in copolymer is lower than 50 mol%, and meanwhile, it is absence of any
Tm at temperature higher than 200 °C. This means that the copolymers still possess the elasticity of rubber and could be processed similar as SBR.
Table 2 Copolymerizations of BD and PEBDs by CpTiCl3/MAO systema
| Run |
Comon. |
Comon. in feedb |
MAO/Ti |
Yield (%) |
Cont.c (mol%) |
Microstructure of PBDd |
Mne × 10−4 |
Đe |
Tgf (°C) |
| cis-1,4 |
trans-1,4 |
1,2- |
| Polymerization conditions: in toluene for 5 h, 50 °C, [BD] = 1.85 M, [BD + comonomer]/[Ti] = 1000. Molar ratio of comonomer to 1,3-butadiene. Comonomer content (mol%) in the copolymer that established by 1H NMR spectra. Microstructure determined by FTIR and NMR.42 Determined by GPC using polystyrene standards. Determined by DSC. Polymerization temperature of 0 and 20 °C for entries 16 and 17. |
| 16g |
PEBD |
10 |
100 |
31.2 |
4.70 |
75.8 |
11.3 |
12.9 |
5.23 |
2.03 |
−85.4 |
| 17g |
PEBD |
10 |
100 |
72.5 |
5.98 |
75.0 |
11.2 |
13.8 |
2.60 |
2.26 |
−83.2 |
| 18 |
PEBD |
10 |
100 |
78.9 |
6.72 |
74.9 |
12.3 |
12.8 |
3.96 |
2.58 |
−80.2 |
| 19 |
PEBD |
10 |
200 |
98.0 |
6.89 |
74.0 |
11.9 |
14.1 |
4.59 |
2.47 |
−81.7 |
| 20 |
PEBD |
10 |
300 |
100 |
7.34 |
74.4 |
12.0 |
13.6 |
3.82 |
2.63 |
−80.3 |
| 21 |
PEBD |
10 |
500 |
79.8 |
7.67 |
74.3 |
11.3 |
14.4 |
3.10 |
3.00 |
−79.7 |
| 22 |
PEBD |
20 |
200 |
23.5 |
12.5 |
78.6 |
14.9 |
6.5 |
2.04 |
2.27 |
−71.4 |
| 23 |
PEBD |
30 |
200 |
12.1 |
21.1 |
78.8 |
15.0 |
6.2 |
1.54 |
2.07 |
−57.7 |
| 24 |
PEBD |
50 |
200 |
15.5 |
30.2 |
79.3 |
14.1 |
6.6 |
0.76 |
1.75 |
−42.1 |
| 25 |
PEBD |
100 |
200 |
— |
— |
— |
— |
— |
— |
— |
— |
| 26 |
p-MEPBD |
10 |
200 |
— |
— |
— |
— |
— |
— |
— |
— |
| 27 |
m-MEPBD |
10 |
200 |
— |
— |
— |
— |
— |
— |
— |
— |
| 28 |
DMPBD |
10 |
200 |
— |
— |
— |
— |
— |
— |
— |
— |
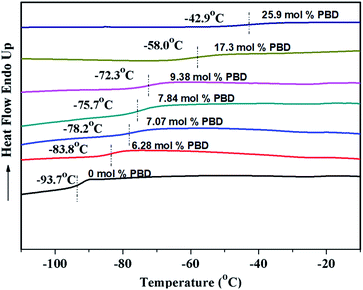 |
| | Fig. 4 DSC curves of BD/PBD copolymers. | |
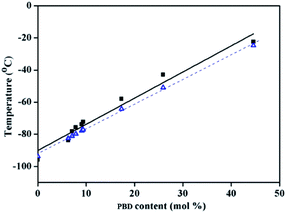 |
| | Fig. 5 Dependence of Tg of BD/PBD copolymers on the PBD incorporation content (mol%) (■: experimental Tg, Δ: theoretical Tg). | |
The dash line in Fig. 5 is fitted according to the Fox equation:45
| |
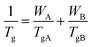 | (2) |
where the value
TgA is 179.3 K (−93.7 °C), the value of
TgB is 309.8 K (36.8 °C),
WA and
WB are the mass fraction of polybutadiene and poly(PBD) respectively. The experimental
Tgs are almost in agreement with the calculated
Tg values. It can be concluded that the copolymers are random. The monomer reactivity ratios of
rBD and
rPBD was calculated with the Fineman–Ross equation, and reactivity ratios of
rBD = 1.12 and
rPBD = 0.88 were obtained.
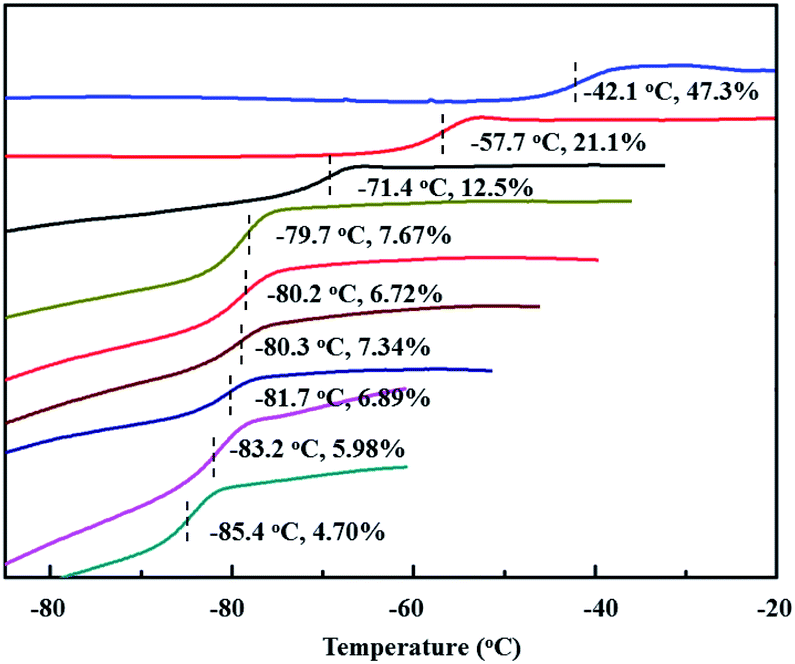
Encouraged by the above copolymerizations between BD and PBD, comonomers were next expanded to more phenyl substituted 1,3-butadienes, including 1-phenethyl-1,3-butadiene (PEBD), 1-(4-methoxylphenyl)-1,3-butadiene (p-MEPBD), 1-(2-methoxylphenyl)-1,3-butadiene (o-MEPBD), 1-(4-N,N-dimethylaminophenyl)-1,3-butadiene (p-DMPBD). As the results shown in Table 2, temperature posed significant influences on the copolymerization behaviors. When 10% PEBD was applied to the polymerization system, increasing polymerization temperature from 0 to 50 °C resulted in a monotonous increment of copolymer yield from 31.2% to 78.9% and PEBD incorporation content from 4.70% to 6.72% with 50 °C as the optimized temperature. Similar to BD/PBD copolymerization system, increasing the ratio of Al/Ti from 100 to 500 gave rise to firstly increased and then decreased monomer conversions, the relative lower copolymer yield at Al/Ti = 500 was probably due to the over-reduction of the active species. Meanwhile, an enhanced the PEBD incorporation content from 6.72% to 7.67% was observed when increasing the Al/Ti ratio from 100 to 500. Feeding more PEBD comonomer into the system can remarkably increase the PEBD content in the copolymer from 6.89% to 47.3%, albeit with obviously decreased catalytic activities and molecular weights, which implies the sluggish reactivity of comonomer PEBD than BD. When equimolar of BD and PEBD was applied to the system, as high as 47.3% PEBD was incorporated in the copolymer mainchain, which is the highest value in this study. Nevertheless, attempts to carry out PEBD homopolymerization proved to be a failure in the end. During DSC analysis of the obtained copolymers, it was observed that, with an increasing PEBD content, a gradually increased glass transition temperature (Tg) from −85.4 to −42.1 °C was observed (Fig. 6), which is similar to BD/PBD copolymers.
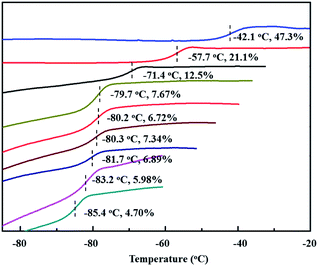 |
| | Fig. 6 DSC curves of BD/PEBD copolymers. | |
Besides PEBD, three other polar phenyl substituted 1,3-butadiene (PSBD) comonomers p-MEPBD, o-MEPBD, p-DMPBD were also attempted to be copolymerized with BD by CpTiCl3/MAO under identical conditions in order to prepare functionalized polybutadienes. However, due to the complete poisoning of active species by the heteroatoms that present in the comonomers, no copolymer products were obtained.
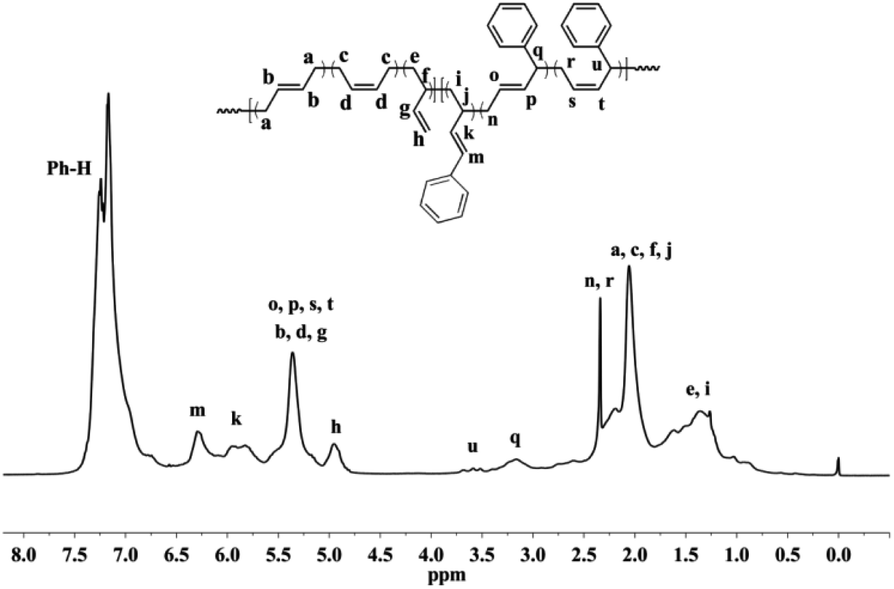
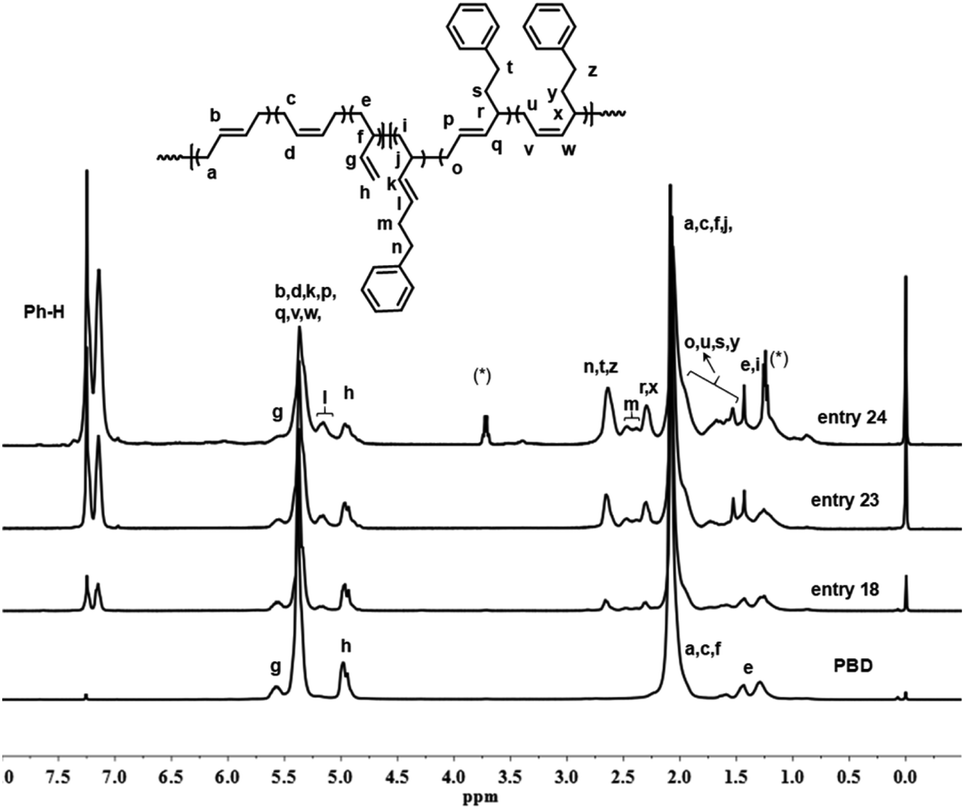
Detailed microstructures of the resultant BD/PEBD copolymers were characterized by 1H NMR spectra (Fig. 7). In the olefinic region, besides the two peaks (4.95 and 5.58 ppm) that are assigned to the 3,4-unites of polybutadiene, a small peak at 5.19 ppm is also observed, which belongs to proton ‘l’ in the 3,4-unit of PEBD. In the high field region, the peak at 2.60–2.71 ppm is attributed to the methylene protons adjacent to the phenyl ring; the broad peak at 2.37–2.53 ppm, whose intensity almost equals to peak ‘l’, is assigned to methylene proton ‘m’ that is adjacent to the double bond of 3,4-unit of PEBD. 1,4-Polymerized PEBD, with characteristic peaks of ‘r’ and ‘x’ in the regions of 2.25–2.33 ppm, can be also found in the resultant copolymers, of note is that these peaks appeared even when PEBD content is low, which shows a striking difference to PBD/BD copolymers. Based on these analyses, the PEBD contents in the resultant copolymers can be calculated from the following eqn (3):
| | |
PEBD mol% = (I7.10–7.30/5)/[I7.10–7.30/5 + (I5.21–5.60 − 1/2I4.95 − I5.19 − 2I2.25–2.33)/2 + I5.19 + I2.25–2.33] = 2(I7.10–7.30)/[2I7.10–7.30 + 5(I5.21–5.60 − 1/2I4.95 − I5.19 − 2I2.25–2.33) + 10I5.19 + 10I2.25–2.33]
| (3) |
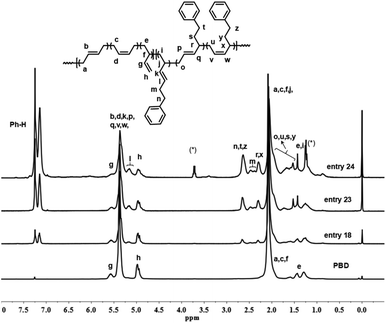 |
| | Fig. 7 1H NMR spectrum of BD/PEBD copolymer. | |
Moreover, the 1,4- and 3,4-inserted percentage of PEBD can be also calculated through comparing the peaks ‘m’ and ‘r,x’. For instance, for the copolymer obtained in entry 23, Table 2, the percentage of 1,4- and 3,4-units of PEBD are calculated as 12.3% and 8.8% respectively.
Conclusions
In summary, this research discloses the copolymerization of 1,3-butadiene with different types of phenyl substituted 1,3-butadiene copolymers by a coordination catalytic system of CpTiCl3/MAO. (E)-1-phenyl-1,3-butadiene (PBD) and 1-phenethyl-1,3-butadiene (PEBD) can be copolymerized with 1,3-butadiene with a broad range of comonomer feed ratio, giving rise to corresponding copolymers with well-regulated comonomer incorporations (0–44.6% for PBD, 0–30.2% for PEBD). For comonomers 1-(4-methoxylphenyl)-1,3-butadiene (p-MEPBD), 1-(2-methoxylphenyl)-1,3-butadiene (o-MEPBD), and 1-(4-N,N-dimethylaminophenyl)-1,3-butadiene (p-DMPBD), however, due to the complete poisoning of the active species by heteroatoms in the comonomer, complete inactive system were demonstrated. The copolymerization performances and the properties of the resultant copolymers were found to be highly dependent on the polymerization conditions, by employing different polymerization parameters, such as temperature, Al/Ti ratio, etc., copolymer properties, including microstructure, molecular weights, molecular weights distributions, can be facilely adjusted. Glass transition temperature (Tg) of the resulted copolymer was found to be highly dependent on the incorporation content of the comonomers, with an increasing comonomer content, a gradual increasing Tg was demonstrated, which fits well the reported Fox equation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Joint Funds of the National Natural Science Foundation of China (U1862206), National Natural Science Foundation of China (no. 21901020) and Jilin Provincial Science and Technology Development Program (20200201089JC), Jilin Province Department of Education (grant no. JJKH20200665KJ). Key Basic Research Topics of Foundation Strengthening Program (2019-JCJQ-ZD-271-01).
References
- J. A. Brydson, Rubber Chemistry, London, 1978 Search PubMed.
- J.-P. Dilcher, H. Jürgens and G. A. Luinstra, in Multi-Component and Sequential Reactions in Polymer Synthesis, ed. P. Theato, Springer International Publishing, Cham, 2015, vol. 269, pp. 163–201 Search PubMed.
- M. P. McGrath, E. D. Sall and S. J. Tremont, Chem. Rev., 1995, 95, 381–398 CrossRef CAS.
- Q. Zhou, S. Jie and B.-G. Li, Ind. Eng. Chem. Res., 2014, 53, 17884–17893 CrossRef CAS.
- J. Spanring, C. Buchgraber, M. F. Ebel, R. Svagera and W. Kern, Polymer, 2006, 47, 156–165 CrossRef CAS.
- H. Otsuka, T. Muta, M. Sakada, T. Maeda and A. Takahara, Chem. Commun., 2009, 1073–1075 RSC.
- S. W. Beavan, P. A. Hackett and D. Phillips, Eur. Polym. J., 1974, 10, 925–932 CrossRef CAS.
- M. Coquillat, J. Verdu, X. Colin, L. Audouin and R. Nevière, Polym. Degrad. Stab., 2007, 92, 1343–1349 CrossRef CAS.
- Q. Wang, X. Zhang, L. Wang and Z. Mi, J. Mol. Catal. A: Chem., 2009, 309, 89–94 CrossRef CAS.
- F. R. De Risi, L. D'Ilario and A. Martinelli, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3082–3090 CrossRef CAS.
- M. J. Tornaritis, E. Davoras, K. Vretzou and A. G. Coutsolelos, Polymer, 1994, 35, 2857–2858 CrossRef CAS.
- C.-C. Peng and V. Abetz, Macromolecules, 2005, 38, 5575–5580 CrossRef CAS.
- H. Zhang, Y. Li, C. Zhang, Z. Li, X. Li and Y. Wang, Macromolecules, 2009, 42, 5073–5079 CrossRef CAS.
- Y. Xu, J. Zhao, Q. Gan, W. Ying, Z. Hu, F. Tang, W. Luo, Y. Luo, Z. Jian and D. Gong, Polym. Chem., 2020, 2034–2043, 10.1039/c9py01808e.
- Y. Jiang, X. Kang, Z. Zhang, S. Li and D. Cui, ACS Catal., 2020, 5223–5229, DOI:10.1021/acscatal.9b04590.
- L. Cai, S. Long, C. Wu, S. Li, C. Yao, X. Hua, H. Na, D. Liu, T. Tang and D. Cui, Polym. Chem., 2020, 11, 1646–1652 RSC.
- C. M. Geiselhart, J. T. Offenloch, H. Mutlu and C. Barner-Kowollik, ACS Macro Lett., 2016, 5, 1146–1151 CrossRef CAS.
- M. A. Kayumova, O. S. Kukovinets, N. N. Sigaeva, R. R. Muslukhov, V. N. Zaboristov, V. P. Budtov and M. I. Abdullin, Vysokomol. Soedin., Ser. A Ser. B, 2008, 50, 1546–1552 CAS.
- J.-C. Soutif and J.-C. Brosse, Die Makromolekulare Chemie, 1984, 185, 839–846 CrossRef CAS.
- H. Leicht, S. Huber, I. Gottker-Schnetmann and S. Mecking, Polym. Chem., 2016, 7, 7195–7198 RSC.
- R. J. Ambrose and W. L. Hergenrother, Macromolecules, 1972, 5, 275–279 CrossRef CAS.
- T. Suzuki, Y. Tsuji, Y. Takegami and H. J. Harwood, Macromolecules, 1979, 12, 234–239 CrossRef CAS.
- T. Masuda, M. Otsuki and T. Higashimura, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 1385–1394 CrossRef CAS.
- R. Asami and K.-i. Hasegawa, Polym. J., 1976, 8, 67 CrossRef CAS.
- G. Friedmann and N. Brosse, Eur. Polym. J., 1991, 27, 747–749 CrossRef CAS.
- L. Wu and V. V. Sheares, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3227–3238 CrossRef CAS.
- Y. Yang and V. V. Sheares, Polymer, 2007, 48, 105–109 CrossRef CAS.
- J. K. Stille and E. D. Vessel, J. Polym. Sci., 1961, 49, 419–425 CrossRef CAS.
- W. Marconi, A. Mazzei, G. Lugli and M. Bruzzone, J. Polym. Sci., Polym. Symp., 1967, 16, 805–819 CrossRef.
- S. Pragliola, M. Cipriano, A. C. Boccia and P. Longo, Macromol. Rapid Commun., 2002, 23, 356–361 CrossRef CAS.
- C. Yao, F. Lin, M. Wang, D. Liu, B. Liu, N. Liu, Z. Wang, S. Long, C. Wu and D. Cui, Macromolecules, 2015, 48(7), 1999–2005 CrossRef CAS.
- C. Yao, H. Xie and D. Cui, RSC Adv., 2015, 5, 93507–93512 RSC.
- H. Leicht, I. Göttker-Schnetmann and S. Mecking, Macromolecules, 2017, 50, 8464–8468 CrossRef CAS.
- H. Leicht, I. Göttker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2017, 139, 6823–6826 CrossRef CAS PubMed.
- H. Leicht, I. Göttker-Schnetmann and S. Mecking, ACS Macro Lett., 2016, 5, 777–780 CrossRef CAS.
- S. Liang, H. Zhang, R. Cong, H. Liu, F. Wang, Y. Hu and X. Zhang, RSC Adv., 2019, 9, 33465–33471 RSC.
- W. Gronski, G. Quack, N. Murayama and K. F. Elgert, Ber. Bunsen-Ges., 1975, 79, 1166 Search PubMed.
- G. Ricci, A. Forni, A. Boglia, A. Sommazzi and F. Masi, J. Organomet. Chem., 2005, 690, 1845–1854 CrossRef CAS.
- Q. Wu, J. Hu, X. Ren and J. Zhou, Chem.–Eur. J., 2011, 17, 11553–11558 CrossRef CAS PubMed.
- H. Lebel and V. Paquet, Organometallics, 2004, 23, 1187–1190 CrossRef CAS.
- C. Qiao, A. Chen, B. Gao, Y. Liu and H. Huang, Chin. J. Chem., 2018, 36, 929–933 CrossRef CAS.
- Y. Hu, W. Dong and T. Masuda, Macromol. Chem. Phys., 2013, 214, 2172–2180 CrossRef CAS.
- T. Suzuki, Y. Tsuji and Y. Takegami, Macromolecules, 1978, 11, 639–644 CrossRef CAS.
- S. K. H. Thiele and D. R. Wilson, J. Macromol. Sci., Part C: Polym. Rev., 2003, 43, 581–628 CrossRef.
- G. Odian, Principles of Polymerization, New York, 1991 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02467a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc,
Chunyu Zhangb,
Heng Liu
*bc,
Chunyu Zhangb,
Heng Liu