
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamically driven self-formation of Ag nanoparticles in Zn-embedded carbon nanofibers for efficient electrochemical CO2 reduction†
Gi-Baek Leea,
In-Kyoung Ahna,
Won-Hyo Jooa,
Jae-Chan Leea,
Ji-Yong Kima,
Deokgi Honga,
Hyoung Gyun Kima,
Jusang Leea,
Miyoung Kima,
Dae-Hyun Nam *b and
Young-Chang Joo*acd
*b and
Young-Chang Joo*acd
aDepartment of Materials Science & Engineering, Seoul National University, Seoul 151-744, Republic of Korea
bDepartment of Energy Science & Engineering, Daegu Gyeongbuk Institute of Science & Technology (DGIST), Daegu 42988, Republic of Korea. E-mail: dhnam@dgist.ac.kr
cResearch Institute of Advanced Materials (RIAM), Seoul National University, Seoul 08826, Republic of Korea
dAdvanced Institute of Convergence Technology, 145 Gwanggyo-ro, Yeongtong-gu, Suwon 16229, Republic of Korea. E-mail: ycjoo@snu.ac.kr
First published on 15th July 2021
Abstract
The electrochemical CO2 reduction reaction (CO2RR), which converts CO2 into value-added feedstocks and renewable fuels, has been increasingly studied as a next-generation energy and environmental solution. Here, we report that single-atom metal sites distributed around active materials can enhance the CO2RR performance by controlling the Lewis acidity-based local CO2 concentration. By utilizing the oxidation Gibbs free energy difference between silver (Ag), zinc (Zn), and carbon (C), we can produce Ag nanoparticle-embedded carbon nanofibers (CNFs) where Zn is atomically dispersed by a one-pot, self-forming thermal calcination process. The CO2RR performance of AgZn–CNF was investigated by a flow cell with a gas diffusion electrode (GDE). Compared to Ag–CNFs without Zn species (53% at −0.85 V vs. RHE), the faradaic efficiency (FE) of carbon monoxide (CO) was approximately 20% higher in AgZn–CNF (75% at −0.82 V vs. RHE) with 1 M KOH electrolyte.
Introduction
Powering the CO2 reduction reaction (CO2RR) with renewable electricity is a promising way to recycle CO2 into high-value added chemicals, such as carbon monoxide (CO),1–3 methane (CH4),4–6 methanol (CH3OH),6–8 ethylene (C2H4),9–13 ethanol (C2H5OH),14–17 and n-propanol (C3H7OH).18,19 For industrial applications, the CO2RR product selectivity and energy efficiency need to be enhanced to achieve economic profitability.20–23 Although previous studies have focused on the surface treatments of catalysts, alloying, and physical structural design of catalyst electrodes, the activity of catalysts still needs to be improved before industrial implementation.Local environmental control around the active materials in catalysts can play an important role in enhancing the performance of catalysts.24–26 For instance, the secondary-sphere effect can reduce the energy barrier for CO2-to-CO conversion by the interaction between the negatively charged oxygen (O) of CO2 and the positively charged secondary atom. This behavior can be induced by the stabilization of nearby CO2, which is located between the active sites of the catalyst and the secondary sphere, such as pendant protons, positive charges, and other metal atoms (ions).27 In previous approaches to apply Lewis acidity control in heterogeneous catalysts, a single-atom catalyst was normally fabricated on a defective support; however, these materials suffered from aggregation or detachment.28–30
To realize the secondary-sphere effect in heterogeneous CO2RR electrocatalysts, it is critical to fabricate secondary spheres, such as Lewis acidic metal atoms and primary catalysts, with good electrical conductivity and chemically stable supports. Here, we report a fabrication methodology for producing primary metal nanoparticles surrounded by Lewis acidic metal in a C matrix through a one-pot self-formation process; this method has been designed by considering the thermodynamics of the oxidation tendency between metals and C during calcination. We selected Ag, which is a widely used CO-producing catalyst, as the primary metal and Zn as a single-atom Lewis acidic metal, which is also a CO-producing catalyst that has a strong affinity for O.31 Based on thermodynamic calculations, we investigated the temperature dependence of the standard Gibbs free energy for compound formation and performed calcination based on the calculation-derived processing parameters to induce the targeted reaction. In this work, we applied a strategy to control the Lewis acidity within a structure containing a stable C support and metal nanoparticle catalysts. Previously, Lewis acid control was utilized in molecular catalysis, which holds Lewis acidic metal ions near the catalyst through a ligand or pendant group. However, we simplified the structure and manufacturing process by distributing the Lewis acid sites in the catalyst containing support through a thermodynamically designed one pot self-formation method. Carbon nanofiber (CNF)s, which are widely used as a conductive and porous support, were used herein. As a consequence, we have gained insights into enhancing the local CO2 concentration effect of Lewis acidic metals in a C matrix.
Experimental
Electrospinning and calcination of AgZn–CNFs
Electrospinning was used to form catalyst-embedded CNFs. A precursor solution of AgNO3/ZnN2O6/N–CNF was prepared by mixing PAN (Mw = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Sigma-Aldrich), AgNO3 (Mw = 169.87 g mol−1, Sigma-Aldrich), and Zn(NO3)2·6H2O (Mw = 297.49 g mol−1, Sigma-Aldrich). Next, 85 mg of AgNO3 and 149 mg of Zn(NO3)2·6H2O, as metal precursors, were dissolved in dimethylformamide (DMF, C3H7NO, Sigma-Aldrich). Samples were split into 4 groups: (1) pure PAN without metal (bare CNF), (2) PAN with Ag (Ag–CNF), (3) PAN with Zn (Zn–CNF), and (4) PAN with AgZn (AgZn–CNF). The solution was stirred for 12 h at room temperature. After stirring, the solution was loaded in a syringe for electrospinning. The solution-loaded syringe was pressed by a syringe pump (KDS 100, KD scientific) at a constant rate of 0.4 ml h−1 and an applied voltage of 15 kV. The distance between the needle tip of the syringe and the collector was 15 cm. The as-spun metal nitrate/PAN nanofibers were first stabilized in an air atmosphere at 280 °C for 2 h at a heating rate of 5 °C min−1. After evacuation by a rotary pump and turbo molecular pump, the samples were carbonized at 800 °C for 3 h in a N2 flow of 200 mTorr at a heating rate of 5 °C min−1.
000 g mol−1, Sigma-Aldrich), AgNO3 (Mw = 169.87 g mol−1, Sigma-Aldrich), and Zn(NO3)2·6H2O (Mw = 297.49 g mol−1, Sigma-Aldrich). Next, 85 mg of AgNO3 and 149 mg of Zn(NO3)2·6H2O, as metal precursors, were dissolved in dimethylformamide (DMF, C3H7NO, Sigma-Aldrich). Samples were split into 4 groups: (1) pure PAN without metal (bare CNF), (2) PAN with Ag (Ag–CNF), (3) PAN with Zn (Zn–CNF), and (4) PAN with AgZn (AgZn–CNF). The solution was stirred for 12 h at room temperature. After stirring, the solution was loaded in a syringe for electrospinning. The solution-loaded syringe was pressed by a syringe pump (KDS 100, KD scientific) at a constant rate of 0.4 ml h−1 and an applied voltage of 15 kV. The distance between the needle tip of the syringe and the collector was 15 cm. The as-spun metal nitrate/PAN nanofibers were first stabilized in an air atmosphere at 280 °C for 2 h at a heating rate of 5 °C min−1. After evacuation by a rotary pump and turbo molecular pump, the samples were carbonized at 800 °C for 3 h in a N2 flow of 200 mTorr at a heating rate of 5 °C min−1.
Materials characterization
The morphology of the samples was analyzed by field-emission scanning electron microscopy (FE-SEM, SIGMA, Carl Zeiss), high-resolution transmission electron microscopy (HRTEM, JEM-2100F, JEOL) and Cs-corrected scanning transmission electron microscopy (JEM-ARM200F, JEOL). The metal phase was analyzed using X-ray diffraction (XRD, New D8 Advance, Bruker) with a Cu Kα 1 radiation source (λ = 1.54 Å) and X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe, Ulvac-PHI). Additionally, X-ray absorption spectroscopy (XAS) was conducted at Pohang Light Source (PLS-II) in Korea.Electrochemical CO2 reduction
A gas diffusion electrode (GDE) was prepared by spray coating the catalyst ink onto a gas diffusion layer (GDL-39BC, SIGRACET) using an air spray gun. For the catalyst ink, 40 mg of as-prepared catalyst (bare CNF, Ag–CNF, Zn–CNF, AgZn–CNF) was added to a solution of 9 ml of IPA (isopropyl alcohol, DAEJOONG Chem.) and 200 μl of Nafion (5 wt%, Sigma-Aldrich). The mixed solution was then sonicated for 30 minutes. After sonication, the dispersed ink was air sprayed as a gas diffusion layer (2.5 cm × 2.5 cm) with a loading amount of 0.3 mg cm−2 and then dried in vacuum for 6 h. The fabricated GDEs were electrochemically tested in a flow cell configuration using a potentiostat with a three-electrode electrochemical cell configuration consisting of a Hg/HgO electrode as the reference electrode and Pt foil as the counter electrode. The flow rate of the electrolyte was set to 0.85 ml min−1 by a peristaltic pump, and the CO2 flow rate at the inlet was fixed to 20 ml min−1 by a mass flow controller (MFC). The flow rate of the outlet was confirmed by a mass flow meter (MFM). During the electrochemical reaction, ejected gas products were analyzed by gas chromatography (GC, INFICON, Inc.), which was directly connected to the outlet of the flow cell. We measured the solution resistance by electrochemical impedance spectroscopy (EIS) before the reaction (Ru,s) to precisely calculate the applied potential. Then, we calculated the IR corrected potential using the following equation: Ecor = Eapp + 0.8IavgRu,s, where Ecor is the cathode potential after correction and Iavg is the average current of the overall reaction. We applied each potential to electrochemical cell for 40 minutes. During CO2RR, we analyzed the quantities of gaseous products three times (15, 25, 35 min) using gas chromatography (GC) for a single cathode. Then, the measurements proceeded with the three electrodes with same batch, and the average and standard deviation were calculated and displayed on the faradaic efficiency data of each potential.Results and discussion
Thermodynamic design of the catalyst and fabrication process
We proceeded thermochemical calculation-based process design before the thermal calcination of electrospun nanofibers. In the calculation, the element that exhibits a lower Gibbs free energy for oxidation can act as a reducing agent for the other elements with higher Gibbs free energies (Fig. 1a). Partially positively charged single-atom Zn can come from Zn–nitrogen (N) bond formation in the calcination process. We can anticipate the increased concentration of local CO2 on the surface of Ag nanoparticles due to the surrounding Lewis acidic Zn atoms in the C matrix (Fig. 1b). N-bonded Zn can attract the partially negatively charged O of CO2 to increase the local CO2 concentration on the surface of Ag nanoparticles. The increased concentration of local CO2 from Lewis acidic Zn enhances the CO producing ability of the Ag nanoparticles.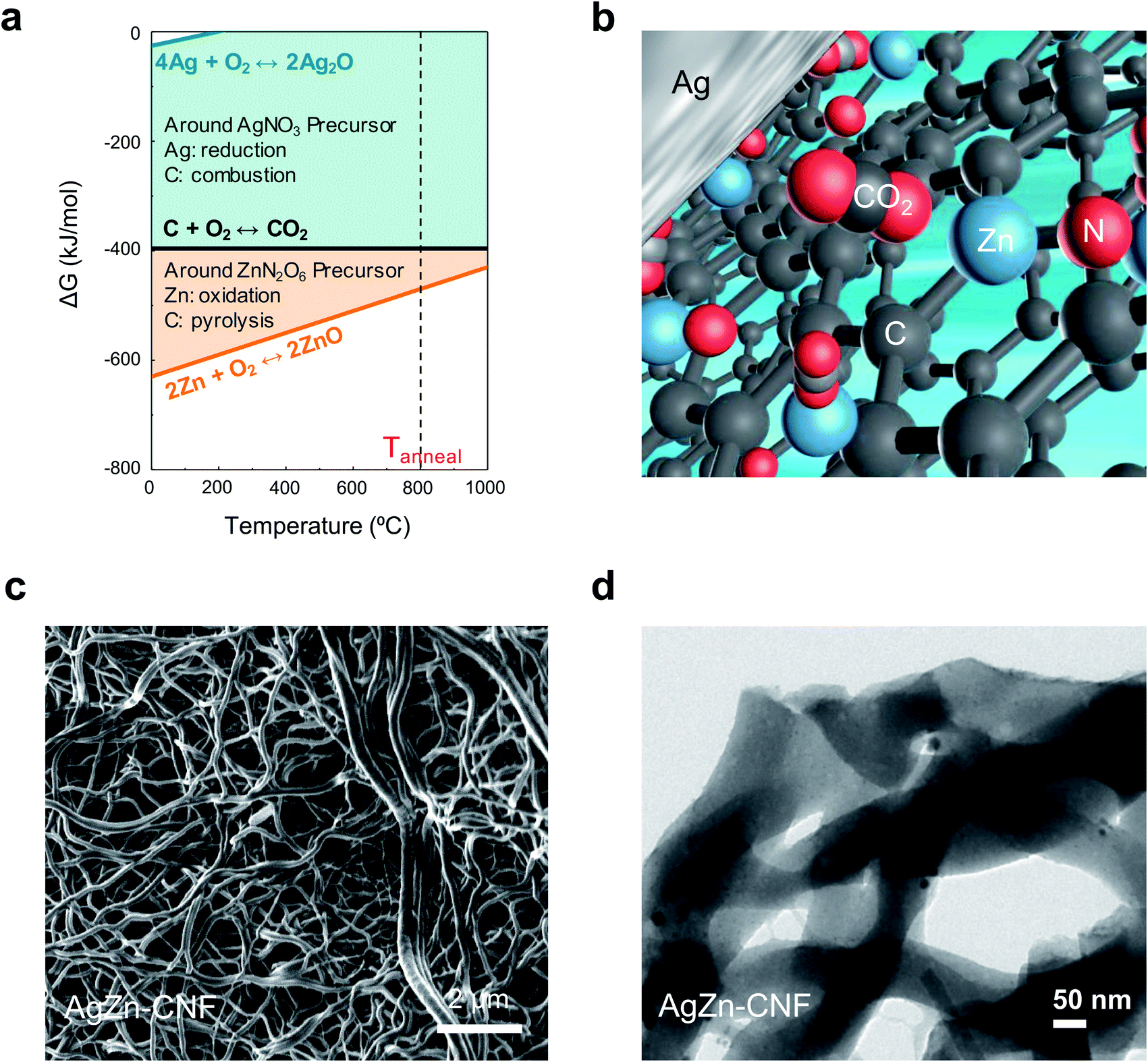 | ||
| Fig. 1 Graphical image of the fabricated catalyst. (a) Ellingham diagram for thermodynamically designed calcination and (b) 3D atomic illustration of the local CO2 concentration between the Ag nanoparticles and single-atom Zn in the C matrix. (c) FESEM images of AgZn–CNF and (d) TEM images of AgZn–CNF. | ||
We fabricated Ag nanoparticles embedded in atomically dispersed Zn–CNFs by a stabilization and calcination process. The stabilization process in ambient air was performed for 2 h at 280 °C. Calcination in a N2 flow proceeded for 3 h at 800 °C. In this work, metal nitrates were used as metal precursors for Ag and Zn to induce N doping in the CNFs, and a direct supply of O to the metals was provided during the calcination process. The morphology and metal distribution of the samples were investigated via FE-SEM (Fig. 1c) and TEM (Fig. 1d). Considering the TEM image of AgZn–CNF with the SEM image, we confirmed that Ag nanoparticles and Zn species were formed within the CNFs.
In the Ag–Zn–C ternary system, Ag has a lower oxidation reactivity than C, so Ag ions in the metal precursor will be reduced into metal within the C matrix. As a result, Ag nanoparticles embedded in the C matrix are fabricated by Ag-ion reduction and agglomeration. On the other hand, Zn has a higher oxidation reactivity than C. In this case, at the beginning of calcination, the agglomeration of Zn ions from the metal precursor will be prevented. As calcination proceeds, Zn will be atomically fixed within the C matrix as Zn–N–C, which is a stable structure of a single-atom transition metal catalyst.32–37 It seems that there would be a concentration limit of Zn in the C matrix to maintain the fixation effect above the inherent agglomeration tendency of metals. If the concentration of Zn is low enough, atomic Zn will be dispersed in the C matrix structure as Lewis acidic sites with N bonds. These processes independently occur even when Ag and Zn are mixed together in the C matrix because of the difference in oxidation reactivity of each metal precursor with C during calcination. Therefore, Ag nanoparticle-embedded CNFs where Zn is atomically dispersed catalysts could be fabricated.
Characterization of the structure of Ag nanoparticle-embedded CNFs where Zn is atomically dispersed catalyst
The composition and structure of annealed nanofiber was investigated as a function of annealing temperature in Fig. S1 and S2.† In the cross-sectional EDS mapping results of Fig. S2 and Table S1,† we found the decrease of Ag concentration as the temperature increases. As shown in Fig. S3,† the Ag concentration was too low to detect at 900 °C. It seems that Ag nanoparticles in the fiber network had gathered to form agglomerated Ag particle outside of CNF network induced by high temperature. Based on the energy-dispersive X-ray spectroscopy (EDS) analysis of AgZn–CNF, we found that the Ag nanoparticles are well embedded in the CNFs; however, Zn is distributed not in the form of nanoparticles over all the CNFs but rather like atomic dispersion (Fig. 2a). This result tends to coincide with the thermodynamic prediction considering the oxidation tendency between C and metal. To analyze the distribution of single-atom Zn more directly and precisely, we conducted a condenser lens corrected scanning transmission electron microscopy (cs-STEM) analysis. These results confirm that single-atom Zn is atomically dispersed throughout the nanofibers (Fig. S4 in the ESI†).11 In this case, single-atom Zn can surround the Ag nanoparticles from every direction, which can enhance the local CO2 concentration near the surface of all Ag nanoparticles.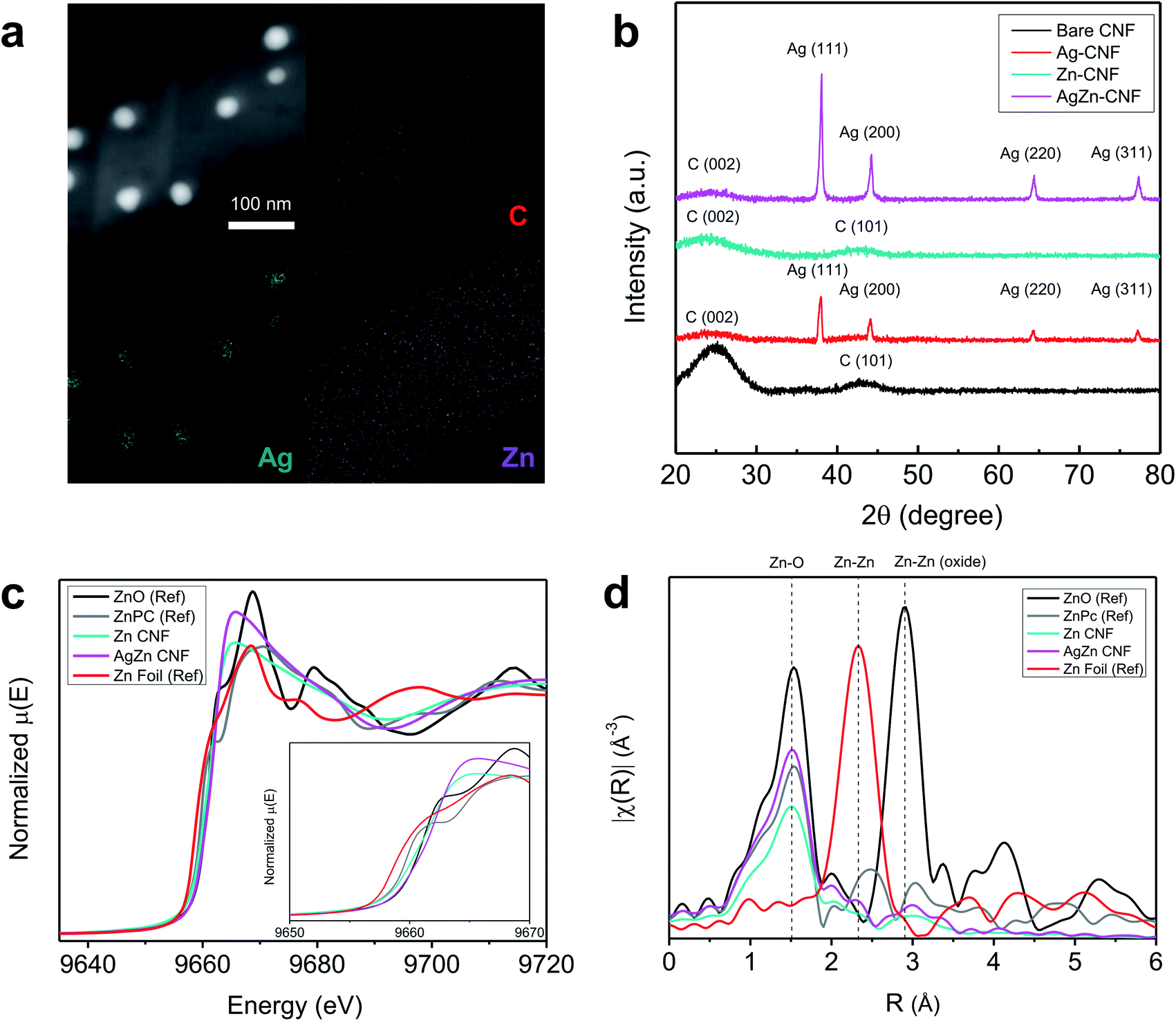 | ||
| Fig. 2 (a) EDS analysis of AgZn–CNF and (b) XRD patterns of the samples. (c) Zn K-edge X-ray absorption near edge structure spectra of Zn foil, ZnO, ZnPc, Zn–CNF and AgZn–CNF. (d) Fourier transform (FT) of the Zn K-edge extended (EXAFS) spectra of the Zn foil, ZnO, ZnPc, Zn–CNF and AgZn–CNF. | ||
These microstructures can be confirmed by the XRD results. As a result of the XRD analysis, a broad (002) diffraction peak of amorphous C is observed at 2θ = 25°, and other peaks correspond to the Ag reference card (no. 01-987-0719) (Fig. 2b).38,39 This result proves the formation of crystalline Ag nanoparticles, which corresponds with the TEM image results. On the other hand, Zn-related species are not detected by XRD, indicating that Zn does not form nanoparticles.40
To ensure the atomic structure of AgZn–CNF, we conducted an XAS analysis of the fabricated nanofibers. Additionally, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) provide information on the oxidation states and coordinative structures. Fig. 2c shows the XANES results of the Zn K-edge in the Zn–CNF and AgZn–CNF samples, with ZnO, zinc phthalocyanine (ZnPc) and Zn foil as references. In the ZnPc reference sample, a single atom of Zn is surrounded by four N atoms.41 The low Zn K-edge energies of Zn–CNF and AgZn–CNF (9665 eV) shifted lower than those of all references. Based on the first derivative of normalized energy, the oxidation state of Zn in AgZn–CNF is between that of Zn foil and ZnO. (Fig. S5 in the ESI†). It means that the Zn in Zn–CNF and AgZn–CNF has an oxidation state of more than 0 and less than 2+. It seems that the coordinative structure around Zn affects the oxidation state of Zn. Fig. 2d shows the EXAFS results of Zn–CNF and AgZn–CNF. The R (Å) range of the main peaks of Zn–CNF and AgZn–CNF is at approximately similar with the Zn–O bond of ZnO and the Zn–N bond of ZnPc. Neither of the samples exhibit a major peak for Zn–Zn bonding. From the XAS analysis, we expected that the Zn in Zn–CNF and AgZn–CNF would be bonded with N or O and it seems that Zn atoms are dispersed with enough distance to prevent bond formation between Zn atoms. In the study of identifying the structure of Zn–N2, theoretical XANES spectra of the Zn K-edge were proposed for Zn–N2 which is single atom Zn bonded with two Ns.37 Based on this paper, we think that the Zn in Zn–CNF and AgZn–CNF might be bonded with N because the XANES and EXAFS of the Zn K-edge of theoretical Zn–N2 results was similar to our Zn–CNF and AgZn–CNF samples.
To investigate the chemical states of the components in AgZn–CNF, we conducted XPS analysis. Regardless of the metal species, the concentrations of O and N were higher than those of bare CNF (Table S2 in the ESI†). It seem that the increased contents of N and O compared to bare CNF may be originated from the decomposition of metal precursors. The higher O concentration of Zn–CNF compared to Ag–CNF may come from oxidized Zn during calcination. When we analyzed the N 1s spectra with deconvolution, we found that the ratio of graphitic N was lower for the metal-included CNF sample than for the bare CNF sample (Fig. 3b, S6 and S7 in the ESI†). On the other hand, the ratio of pyridinic N and pyridinic N oxide increased. Samples containing Zn, such as Zn–CNF and AgZn–CNF, exhibited the existence of pyridinic N oxide, unlike bare CNF or Ag–CNF. This pyridinic N–O peak can be interpreted as an enhanced potential for forming single-atom metals surrounded by N; this concept has also been reported in other studies of transition metal–N–C catalysts.42–45 From these results, we think that single-atom Zn can be coordinated with N species.
 | ||
| Fig. 3 XPS analysis of bare CNF, Ag–CNF, Zn–CNF and AgZn–CNF: (a) atomic concentration of each element and (b) deconvoluted N 1s results. | ||
CO2 electroreduction performance
To study the effect of the AgZn ensemble on enhancing the catalytic activity, the product selectivity of the CO2RR depending on the presence or type of metal was investigated. The electrochemical CO2RR proceeded under the condition of a flow cell with 1 M KOH electrolyte. To discover the catalytically active sites, we measured the CO2RR performance of the bare CNF and compared it with the CNFs which include metallic species. In the case of the bare CNF, the faradaic efficiency (FE) of H2 is more than 90% and CNF does not show CO2RR activity. (Fig. S8 in the ESI†). Zn–CNF exhibits low CO FE of lower than 20% and high H2 FE of higher than 60% on the other hand, Ag–CNF exhibits 65% FE for CO and 22% FE for H2. As the potential decreases, the FE of CO decreases to 53%, and the FE of H2 increases to 46% (Fig. 4c). CO2RR product distribution according to the potential was similar with other CO2RR metal catalysts, which produce CO.46–48 When the Ag nanoparticles were embedded in Zn–CNF, CO FE increased to 75% and potentials for CO production decreased compared to Ag–CNF (Fig. 4d). The enhanced CO2RR performance of AgZn–CNF can be explained by the existence of Zn single atoms near Ag active materials. If Ag and Zn act individually in AgZn–CNF, the overall CO selectivity would decrease due to the low CO selectivity of Zn–CNF. | ||
| Fig. 4 (a) Current density versus applied potential of Zn–CNF, Ag–CNF and AgZn–CNF and the faradaic efficiency of electrocatalytic CO2 reduction of (b) Zn–CNF, (c) Ag–CNF, and (d) AgZn–CNF. | ||
We think that the Ag nanoparticle-embedded CNFs where Zn is atomically dispersed structures may increase the local CO2 concentration by CO2 attraction of Lewis acidic Zn on the surface of Ag nanoparticles. Increased local CO2 concentration around active materials can lower the thermodynamic energy barrier for CO production in CO2RR.49 In the AgZn–CNFs, increase of local CO2 concentration could be induced by the interaction between Lewis acidic sites and CO2, supplied from the backside of GDL.50 The interaction between O lone pair in distorted CO2 and Lewis acidic sites has been reported in previous research.51 Within the operation time of 3 hours for maintaining the hydrophobicity of GDL, we can see the stable CO production from our catalysts (Fig. S9 in the ESI†).52,53
Conclusions
We designed a synthetic pathway for the fabrication of metal single atoms-dispersed C matrix with embedded metal nanoparticles by harnessing the different oxidation tendencies of atomic elements. It is expected that the metal single atom dispersed C matrix can be achieved for the metals, whose oxidation states is below C on the Ellingham diagram. We demonstrated this principle to the Ag–Zn–C system for the fabrication of AgZn–CNF. AgZn–CNF exhibited improved CO selectivity compared to Ag–CNF. The enhanced CO2RR performance could be induced by the increased local CO2 concentration around Ag nanoparticles. This thermodynamic design and synthesis methodology of metal single atoms-dispersed C matrix with embedded metal nanoparticles can be extended to the fabrication of various materials by considering the thermodynamic Gibbs free energy for oxidation of the elements and this can provide the tailored material synthesis for various applications.Author contributions
Gi-Baek Lee: conceptualization, methodology, validation, investigation, writing – original draft, In-Kyoung Ahn: resources, investigation, writing – review and editing Won-Hyo Joo, Jae-Chan Lee, Ji-Yong Kim: resources, investigation, Hyoung Gyun Kim, Jusang Lee, Miyoung Kim: visualization, resources Dae-Hyun Nam: conceptualization, methodology, supervision, writing – review and editing and Young-Chang Joo: conceptualization, methodology, supervision, writing – review and editing, funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Creative Materials Discovery Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2017 M3D1A1040688 and 2019R1A2C2090859). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2020R1F1A1077411). The XRD and TEM analysis was supported by the Research Institute of Advanced Materials (RIAM) at Seoul National University. Experiments conducted at PLS-II were supported in part by MSIT and POSTECH.References
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS.
- Q. Yu, X. Meng, L. Shi, H. Liu and J. Ye, Chem. Commun., 2016, 52, 14105–14108 RSC.
- S. Back, M. S. Yeom and Y. Jung, ACS Catal., 2015, 5, 5089–5096 CrossRef CAS.
- C. J. Chang, S. C. Lin, H. C. Chen, J. Wang, K. J. Zheng, Y. Zhu and H. M. Chen, J. Am. Chem. Soc., 2020, 142, 12119–12132 CrossRef CAS PubMed.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- A. Prašnikar, A. Pavlišič, F. Ruiz-Zepeda, J. Kovač and B. Likozar, Ind. Eng. Chem. Res., 2019, 58, 13021–13029 CrossRef.
- Q. H. Low, N. W. X. Loo, F. Calle-Vallejo and B. S. Yeo, Angew. Chem., 2019, 58, 2256–2260 CrossRef CAS PubMed.
- T. C. Chou, C. C. Chang, H. L. Yu, W. Y. Yu, C. L. Dong, J. J. Velasco-Velez, C. H. Chuang, L. C. Chen, J. F. Lee, J. M. Chen and H. L. Wu, J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef CAS PubMed.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed.
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, ACS Catal., 2017, 8, 531–535 CrossRef.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- S. Y. Lee, H. Jung, N. K. Kim, H. S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681–8689 CrossRef CAS PubMed.
- N. Theaker, J. M. Strain, B. Kumar, J. P. Brian, S. Kumari and J. M. Spurgeon, Electrochim. Acta, 2018, 274, 1–8 CrossRef CAS.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- Z. Chen, T. Wang, B. Liu, D. Cheng, C. Hu, G. Zhang, W. Zhu, H. Wang, Z. J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 6878–6883 CrossRef CAS PubMed.
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS.
- R. A. Geioushy, M. M. Khaled, K. Alhooshani, A. S. Hakeem and A. Rinaldi, Electrochim. Acta, 2017, 245, 456–462 CrossRef CAS.
- M. Rahaman, K. Kiran, I. Z. Montiel, V. Grozovski, A. Dutta and P. Broekmann, Green Chem., 2020, 22, 6497–6509 RSC.
- L. C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS.
- J.-P. Jones, G. K. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- A. S. Malkani, J. Anibal and B. Xu, ACS Catal., 2020, 10, 14871–14876 CrossRef CAS.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- A. W. Nichols and C. W. Machan, Front. Chem., 2019, 7, 397 CrossRef CAS PubMed.
- C. Mondelli, G. Gozaydin, N. Yan and J. Perez-Ramirez, Chem. Soc. Rev., 2020, 49, 3764–3782 RSC.
- D. Yang, P. Xu, N. D. Browning and B. C. Gates, J. Phys. Chem. Lett., 2016, 7, 2537–2543 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- D. Hong, T. Kawanishi, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2019, 141, 20309–20317 CrossRef CAS PubMed.
- C. Tang, B. Wang, H. F. Wang and Q. Zhang, Adv. Mater., 2017, 29, 1703185 CrossRef.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., 2017, 56, 610–614 CrossRef CAS PubMed.
- S. H. Ahn, X. Yu and A. Manthiram, Adv. Mater., 2017, 29, 1606534 CrossRef.
- H. W. Liang, S. Bruller, R. Dong, J. Zhang, X. Feng and K. Mullen, Nat. Commun., 2015, 6, 7992 CrossRef CAS.
- Q. Wang, Z. Y. Zhou, Y. J. Lai, Y. You, J. G. Liu, X. L. Wu, E. Terefe, C. Chen, L. Song, M. Rauf, N. Tian and S. G. Sun, J. Am. Chem. Soc., 2014, 136, 10882–10885 CrossRef CAS.
- F. Li, Y. Bu, G. F. Han, H. J. Noh, S. J. Kim, I. Ahmad, Y. Lu, P. Zhang, H. Y. Jeong, Z. Fu, Q. Zhong and J. B. Baek, Nat. Commun., 2019, 10, 2623 CrossRef PubMed.
- K. Shameli, M. B. Ahmad, A. Zamanian, P. Sangpour, P. Shabanzadeh, Y. Abdollahi and M. Zargar, Int. J. Nanomed., 2012, 7, 5603–5610 CrossRef CAS PubMed.
- K. Jurkiewicz, M. Pawlyta and A. Burian, C, 2018, 4, 68 CAS.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- A. Ogunsipe, J.-Y. Chen and T. Nyokong, New J. Chem., 2004, 28, 822–827 RSC.
- L. Zhang, S. Zhu, S. Dong, N. J. Woo, Z. Xu, J. Huang, J.-K. Kim and M. Shao, J. Electrochem. Soc., 2018, 165, J3271–J3275 CrossRef CAS.
- P. Xu, W. Chen, Q. Wang, T. Zhu, M. Wu, J. Qiao, Z. Chen and J. Zhang, RSC Adv., 2015, 5, 6195–6206 RSC.
- J. Li, S. Chen, N. Yang, M. Deng, S. Ibraheem, J. Deng, J. Li, L. Li and Z. Wei, Angew. Chem., 2019, 58, 7035–7039 CrossRef CAS PubMed.
- J. Zang, F. Wang, Q. Cheng, G. Wang, L. Ma, C. Chen, L. Yang, Z. Zou, D. Xie and H. Yang, J. Mater. Chem. A, 2020, 8, 3686–3691 RSC.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed. Engl., 2018, 57, 12303–12307 CrossRef CAS.
- B. Zhang, J. Zhang, J. Shi, D. Tan, L. Liu, F. Zhang, C. Lu, Z. Su, X. Tan, X. Cheng, B. Han, L. Zheng and J. Zhang, Nat. Commun., 2019, 10, 2980 CrossRef.
- K. Jiang, G. Chen and H. Wang, J. Visualized Exp., 2018, 134, 57380 Search PubMed.
- C. B. Hiragond, H. Kim, J. Lee, S. Sorcar, C. Erkey and S.-I. In, Catalysts, 2020, 10, 98 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- W. Thongnuam, T. Maihom, S. Choomwattana, Y. Injongkol, B. Boekfa, P. Treesukol and J. Limtrakul, Phys. Chem. Chem. Phys., 2018, 20, 25179–25185 RSC.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2020, 6, 33–40 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02463a |
| This journal is © The Royal Society of Chemistry 2021 |