
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe coordination chemistry of benzhydrazide with lanthanide(III) ions: hydrothermal in situ ligand formation, structures, magnetic and photoluminescence sensing properties†
Chatphorn Theppitakab,
Filip Kielarc,
Winya Dungkaewd,
Mongkol Sukwattanasinitte,
Laongdao Kangkaewf,
Somboon Sahasithiwatf,
Hikaru Zennog,
Shinya Hayami g and
Kittipong Chainok*a
g and
Kittipong Chainok*a
aThammasat University Research Unit in Multifunctional Crystalline Materials and Applications (TU-McMa), Faculty of Science and Technology, Thammasat University, Pathum Thani 12121, Thailand. E-mail: kc@tu.ac.th
bDepartment of Chemistry, Faculty of Science and Technology, Thammasat University, Pathum Thani 12121, Thailand
cDepartment of Chemistry, Faculty of Science, Naresuan University, Phitsanulok 65000, Thailand
dDepartment of Chemistry, Faculty of Science, Mahasarakham University, Maha Sarakham, 44150, Thailand
eDepartment of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand
fNational Metal and Materials Technology Center (MTEC), The National Science and Technology Development Agency, Pathum Thani 12121, Thailand
gDepartment of Chemistry, Graduate School of Science and Technology and Institute of Pulsed Power Science, Ku-mamoto University, 2-39-1 Kurokami, Chuoku, Kumamoto, 860-8555 Japan
First published on 15th July 2021
Abstract
The influence of synthetic conditions on the solid-state structural formation of lanthanide(III) complexes based on a hydrazide ligand have been investigated and reported. Depending on the solvents and reaction temperatures, the reactions of hydrated Ln(NO3)3 with a benzohydrazide (bzz) ligand afforded three classes of lanthanide(III) coordination complexes viz. [Ln(bzz)(NO3)](NO3)2 (1Ln; Ln = Sm (1), Eu (2), Gd (3), Tb (4), Dy (5)), [Ln(bzz)(ben)3(H2O)]·H2O (2Ln; Ln = Pr (6), Nd (7), Sm (8), Eu (9), Gd (10), Tb (11), Dy (12), Er (13)), and [Ln3(ben)3] (3Ln; Ln = Eu (14), Gd (15), Tb (16), Dy (17), Er (18), Tm (19), Yb (20), Lu (21)). Complexes 1–5 in series 1Ln were isolated by slow evaporation of their isopropanol solutions at ambient temperature, and the complexes display similar discrete structures bearing distinct intermolecular N–H⋯O hydrogen bonds to generate a three-dimensional (3D) supramolecular architecture. Complexes 6–13 in series 2Ln were obtained under hydrothermal conditions at 110 °C where the in situ generated benzoate (ben) ligands participated in the formation of one-dimensional (1D) coordination polymers (CPs) with the bzz ligands. At a temperature of 145 °C the hydrothermal conditions result in the formation of the thermodynamically more stable products of 14–21 in series 3Ln, in which the bzz ligand underwent complete in situ hydrolysis to create the ben ligand. These coordination assemblies feature 1D zigzag chains that are formed by unusual low coordination numbers of the six- and seven-fold coordinated Ln3+ centers bridged by the ben ligands in μ2- and μ3-coordination modes. Notably, the chain structures of 2Ln can be transformed into the zigzag tape-like structures of 3Ln upon heating the crystalline samples to 400 °C in air. In the solid state at room temperature, the Eu- (2, 9, 14) and Tb- (4, 11, 16) containing complexes emit red and green light, respectively. The luminescence investigations show that the Eu- (9, 14) and Tb-(11, 16) based CPs could be used as fluorescent probes for acetone and Co2+ ions via an energy competition mechanism. Meanwhile, the Gd- (10, 15) and Dy- (12, 17) based CPs show typical antiferromagnetic interactions.
Introduction
The coordination chemistry of lanthanides has been extensively studied during the last two decades due to their fascinating structures coupled with their potential applications in the fields of luminescence, magnetism, catalysis, and gas adsorption.1 These materials can be prepared from a large number of organic ligands and lanthanide metal ions through straightforward solvothermal synthetic methods and their structures can be characterized by laboratory X-ray crystallography. In fact, the design and controllable synthesis of lanthanide complexes is more difficult since the harder lanthanide ions have high coordination numbers and flexible coordination geometries compared to transition metal ions.2 To some extent, the nature of organic ligands (i.e. conformation, binding modes or configurations, and ability to form intermolecular interactions) and the synthetic parameters (i.e. metal–ligand ratio, solvent, temperature, time, and pH) may also affect the final structure and coordination number of lanthanide assemblies.3 A large number of lanthanide coordination complexes with fascinating structures and interesting properties have been reported. However, establishing a structure–property relationship in these materials is of great importance in the field of materials science and still remains a challenging task in crystal engineering.Since the lanthanide ions exhibit a high affinity and prefer to bind to hard donor atoms, most of the reported lanthanide complexes are constructed using organic ligands containing oxygen-donor atoms. In this context, polycarboxylic acids have been extensively used in the creation of various lanthanide complexes, especially for the development of optical, magnetic, and energy materials.4 Meanwhile, ligands containing both oxygen- and nitrogen-donors, for example, pyridine dicarboxylic acid and nicotinic acid, have also been employed to construct luminescent lanthanide based porous coordination polymers.5 It appears that some of the complexes with this type of ligands can exhibit highly selective recognition of chemical analytes and can show selective gas adsorption, due to the existence of the non-coordinated nitrogen atoms in the framework structures.6
On the other hand, hydrothermal in situ ligand formation has been demonstrated to be an effective crystal growth strategy in the isolated pure phases of novel coordination assemblies, particularly, those unattainable through conventional methods.7 This strategy involves a series of consecutive bond breaking and bond reforming steps at the molecular level, allowing for the formation of the more stable thermodynamic products. With the development of this strategy, a variety of novel coordination assemblies with in situ-generated ligands have been successfully synthesized, especially for the tetrazole and triazine based complexes.8
We are interested in the synthesis of new crystalline coordination materials9 with the aim of understanding their structure–property relationships that helps to better rationalize the structures and to achieve desired functionalities. In continuation of our ongoing research on this topic, we selected benzohydrazide (bzz) as the ligand for the complexation with lanthanide(III) ions. This choice was made by considering that this ligand contains hydrazide nitrogen and carbonyl oxygen atoms, which can help to saturate lanthanide coordination sites. Furthermore, we were attracted by the chelating ability of the bzz ligand and its potential for forming strong hydrogen bonds. Hitherto, there are only a few reported lanthanide coordination complexes incorporating this kind of ligand, and these structures are discrete mononuclear species.10 In this work, we report the synthesis, structural characterization, fluorescence sensing and magnetic properties of three classes of lanthanide coordination complexes viz. [Ln(bzz)(NO3)](NO3)2 (1Ln; Ln = Sm (1), Eu (2), Gd (3), Tb (4), Dy (5)), [Ln(bzz)(ben)3(H2O)]·H2O (2Ln; Ln = Pr (6), Nd (7), Sm (8), Eu (9), Gd (10), Tb (11), Dy (12), Er (13)), and [Ln3(ben)3] (3Ln; Ln = Eu (14), Gd (15), Tb (16), Dy (17), Er (18), Tm (19), Yb (20), Lu (21)). To the best of our knowledge, the polymeric coordination assemblies of 2Ln and 3Ln represent the first examples in which the hydrazide-based ligands were used to develop novel lanthanide coordination polymers (LnCPs) through hydrothermal in situ carboxylate ligand formation. Interestingly, upon heating the crystalline samples to 400 °C in air, the structures of 2Ln can be transformed into the more stable thermodynamic phase 3Ln. In the solid-state, the Eu-(9, 14) and Tb-(11, 16) based CPs exhibit intense emission in the visible region, and all of these LnCPs behave as selective fluorescent probes for acetone as well as Co2+ ions with good recyclability. Moreover, the temperature-dependent investigation of the magnetic properties of Gd-(10, 15) and Dy-(12, 17) based LnCPs reveal that they have typical antiferromagnetic interactions between adjacent Ln(III) ions in these compounds.
Experimental
Materials and methods
All starting materials were of reagent grade quality, and were obtained from commercial sources without further purification. Elemental (C H N) analysis was determined with a LECO CHNS 932 elemental analyser. IR spectra were recorded on a Perkin-Elmer model Spectrum 100 spectrometer using ATR mode, in the range of 650–4000 cm−1. Variable-temperature powder X-ray diffraction (VT-PXRD) measurements were carried out on a Bruker D8 ADVANCE X-ray powder diffractometer using Cu-Kα (λ = 1.5418 Å) in the temperature range 30–800 °C. The luminescence spectra and emission decay curves were measured at room temperature using a Horiba Scientific model FluoroMax-4 spectrofluorometer. Thermogravimetric analyses (TGA) were performed by a Netzsch STA 449C thermal analyzer at a heating rate of 10 °C min−1 in air. X-ray fluorescence spectroscopy (XRF) was performed on a Bruker S6 JAGUAR WDXRF spectrometer under helium. Magnetic susceptibility data were collected using a Quantum Design SQUID magnetometer calibrated against a standard palladium sample. The data were collected between 2 and 300 K and the scan rate of the temperature was fixed at 2.0 K min−1. Measurements were taken continuously under an applied field of 5000 Oe.Synthesis of [Ln(bzz)(NO3)](NO3)2 (1Ln; Ln = Sm (1), Eu (2), Gd (3), Tb (4), Dy (5))
Complexes 1–5 were prepared by slow evaporation from isopropanol solutions at room temperature. Typically, an isopropanol solution (5 mL) of bzz (27.3 mg, 0.2 mmol) was added dropwise to an isopropanol solution (5 mL) of Ln(NO3)3·6H2O (0.1 mmol) with constant stirring for 1 h and then filtered to remove any undissolved solid. The filtrate was allowed to stand for slow evaporation. Crystals were harvested from the mother liquid after two weeks. XRF spectra were then collected with an Orbis PC micro-XRF spectrometer equipped with a 50 W Rhodium anode X-ray tube.Synthesis of [Ln(bzz)(ben)3(H2O)]·H2O (2Ln; Ln = Pr (6), Nd (7), Sm (8), Eu (9), Gd (10), Tb (11), Dy (12), Er (13))
Complexes 6–13 were prepared with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 lanthanide-to-ligand stoichiometry under hydrothermal conditions at 110 °C for 48 h. Typically, a mixture of Ln(NO3)3·6H2O (0.1 mmol) and bzz (27.3 mg, 0.2 mmol) in distilled H2O (4 mL) were placed in a Teflon lined reactor, stirred at room temperature for 10 min, sealed in a 15 mL stainless steel autoclave, placed in an oven, and then heated to 110 °C under autogenous pressure for 48 h. The reaction mixture was cooled to room temperature. After filtration, the product was washed with distilled H2O and dried in air at room temperature.
:2 lanthanide-to-ligand stoichiometry under hydrothermal conditions at 110 °C for 48 h. Typically, a mixture of Ln(NO3)3·6H2O (0.1 mmol) and bzz (27.3 mg, 0.2 mmol) in distilled H2O (4 mL) were placed in a Teflon lined reactor, stirred at room temperature for 10 min, sealed in a 15 mL stainless steel autoclave, placed in an oven, and then heated to 110 °C under autogenous pressure for 48 h. The reaction mixture was cooled to room temperature. After filtration, the product was washed with distilled H2O and dried in air at room temperature.
Synthesis of [Ln3(ben)3] (3Ln; Ln = Eu (14), Gd (15), Tb (16), Dy (17), Er (18), Tm (19), Yb (20), Lu (21))
Complexes 14–21 were prepared hydrothermally at 140 °C for 24 h by a procedure analogous to 2Ln above.Crystallographic data collection and structure refinement
Suitable crystals of complexes 1–21 were mounted on MiTeGen micromounts using paratone oil. X-ray diffraction data were collected using a Bruker D8 Quest Cmos Photon II operating at T = 296(2) K. Data were collected using ω and ϕ scans and using Mo-Kα radiation (λ = 0.71073 Å). The total number of runs and images was based on the strategy calculation from the program APEX3 and unit cell indexing was refined using SAINT.11 Data reduction was performed using SAINT and SADABS were used for absorption correction. The integrity of the symmetry was checked by using PLATON.12 The structure was solved using the algorithm implemented in ShelXT.13 The structure was refined by least squares using ShelXL.14 In the final refinement cycles, all non-hydrogen atoms were refined anisotropically. All carbon-bound hydrogen atoms were placed in calculated positions and refined using a riding-model approximation with C–H = 0.93 Å and Uiso(H) = 1.2Ueq(C). While, all nitrogen- and oxygen-bound hydrogen atoms were located in a difference-Fourier map and refined with N–H = 0.88 ± 0.01 Å and O–H = 0.84 ± 0.01 Å, respectively. For 1Ln, two nitrate anions and one benzene ring of the bzz ligand are disordered, and were refined with PART instruction with split over two sites with a total occupancy of 1 for each pair of disordered atoms. The lattice water molecule in 2Ln presents a positional disorder that was refined as two contributions with a 0.80:0.20 occupancy ratio. Four benzene rings from different ben ligands in 3Ln are highly disordered and were split over two fragments with a total occupancy of 1. The crystallographic data for 1–21 are listed in Tables 1 and 2.
| Identification code (CCDC no.) | 1 (2064160) | 2 (2064161) | 3 (2064162) | 4 (2064163) | 5 (2064164) |
| Empirical formula | C28H32N12O16Sm | C28H32EuN12O16 | C28H32GdN12O16 | C28H32N12O16Tb | C28H32DyN12O16 |
| Formula weight | 943.00 | 944.61 | 949.90 | 951.57 | 955.15 |
| Temperature (K) | 296 | 296 | 296 | 296 | 296 |
| Crystal system, space group | Monoclinic, C2/c | Monoclinic, C2/c | Monoclinic, C2/c | Monoclinic, C2/c | Monoclinic, C2/c |
| a (Å) | 24.726(8) | 24.7339(8) | 24.6998(8) | 24.6726(9) | 24.6473(7) |
| b (Å) | 15.346(5) | 15.3481(6) | 15.3676(5) | 15.3731(5) | 15.3972(5) |
| c (Å) | 10.498(3) | 10.4654(4) | 10.4550(4) | 10.4434(4) | 10.4290(3) |
| β (°) | 99.982(10) | 100.1570(10) | 100.1530(10) | 100.1700(10) | 100.2710(10) |
| V (Å3) | 3923(2) | 3910.6(2) | 3906.3(2) | 3898.9(2) | 3894.4(2) |
| Z | 4 | 4 | 4 | 4 | 4 |
| ρcalcd (g cm−3) | 1.597 | 1.604 | 1.615 | 1.621 | 1.629 |
| GOF on F2 | 1.069 | 1.051 | 1.115 | 1.072 | 1.124 |
| R1, wR2 [I > 2σ(I)] | 0.0389, 0.0895 | 0.0460, 0.0985 | 0.0337, 0.0812 | 0.0440, 0.1085 | 0.0336, 0.0799 |
| R1, wR2 [all data] | 0.0594, 0.1018 | 0.0910, 0.1274 | 0.0479, 0.0939 | 0.0569, 0.1233 | 0.0435, 0.0925 |
| Identification code (CCDC no.) | 6 (2064165) | 7 (2064166) | 8 (2064167) | 9 (2064168) |
| Empirical formula | C28H27N2O9Pr | C28H27N2NdO9 | C28H27N2O9Sm | C28H27EuN2O9 |
| Formula weight | 676.42 | 679.75 | 685.86 | 687.47 |
| Temperature (K) | 296 | 296 | 296 | 296 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/c | Monoclinic, P21/c | Monoclinic, P21/c |
| a (Å) | 14.5239(7) | 14.5092(4) | 14.496(3) | 14.4680(5) |
| b (Å) | 19.8515(10) | 19.8107(6) | 19.771(4) | 19.7315(6) |
| c (Å) | 9.9728(4) | 9.9678(3) | 9.9597(19) | 9.9544(3) |
| β (°) | 91.725(2) | 91.7130(10) | 91.561(7) | 91.5680(10) |
| V (Å3) | 2874.1(2) | 2863.84(15) | 2853.4(10) | 2840.67(16) |
| Z | 4 | 4 | 4 | 4 |
| ρcalcd (g cm−3) | 1.563 | 1.577 | 1.597 | 1.607 |
| GOF on F2 | 1.034 | 1.034 | 1.041 | 1.044 |
| R1, wR2 [I > 2σ(I)] | 0.0211, 0.0468 | 0.0296, 0.0488 | 0.0270, 0.0512 | 0.0245, 0.0485 |
| R1, wR2 [all data] | 0.0303, 0.0500 | 0.0509, 0.0536 | 0.0409, 0.0554 | 0.0346, 0.0516 |
| Identification code (CCDC no.) | 10 (2064169) | 11 (2064170) | 12 (2064171) | 13 (2064172) |
| Empirical formula | C28H27GdN2O9 | C28H27N2O9Tb | C28H27DyN2O9 | C28H27ErN2O9 |
| Formula weight | 692.76 | 694.43 | 698.01 | 702.77 |
| Temperature (K) | 296 | 296 | 296 | 296 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/c | Monoclinic, P21/c | Monoclinic, P21/c |
| a (Å) | 14.4470(6) | 14.4340(5) | 14.4572(5) | 14.3923(8) |
| b (Å) | 19.6992(9) | 19.7062(7) | 19.7227(7) | 19.6216(11) |
| c (Å) | 9.9464(3) | 9.9308(3) | 9.9440(3) | 9.8933(5) |
| β (°) | 91.5220(10) | 91.4340(10) | 91.4680(10) | 91.287(2) |
| V (Å3) | 2829.69(19) | 2823.82(16) | 2834.45(16) | 2793.2(3) |
| Z | 4 | 4 | 4 | 4 |
| ρcalcd (g cm−3) | 1.626 | 1.633 | 1.636 | 1.671 |
| GOF on F2 | 1.062 | 1.047 | 1.077 | 1.136 |
| R1, wR2 [I > 2σ(I)] | 0.0247, 0.0442 | 0.0262, 0.0472 | 0.0240, 0.0552 | 0.0349, 0.0768 |
| R1, wR2 [all data] | 0.0392, 0.0478 | 0.0422, 0.0515 | 0.0328, 0.0591 | 0.0492, 0.0811 |
| Identification code (CCDC no.) | 14 (2064173) | 15 (2064174) | 16 (2064175) | 17 (2064176) |
| Empirical formula | C63H45Eu3O18 | C63H45Gd3O18 | C63H45O18Tb3 | C63H45Dy3O18 |
| Formula weight | 1545.87 | 1561.74 | 1566.75 | 1577.49 |
| Temperature (K) | 296 | 296 | 296 | 296 |
| Crystal system, space group | Monoclinic, P21/n | Monoclinic, P21/n | Monoclinic, P21/n | Monoclinic, P21/n |
| a (Å) | 11.4654(4) | 11.4601(3) | 11.4419(4) | 11.4293(6) |
| b (Å) | 22.6148(8) | 22.5734(4) | 22.5595(7) | 22.5089(10) |
| c (Å) | 22.7507(8) | 22.7365(5) | 22.7629(8) | 22.7805(11) |
| β (°) | 95.350(2) | 95.3270(10) | 95.3380(10) | 95.250(2) |
| V (Å3) | 5873.3(4) | 5856.4(2) | 5850.2(3) | 5835.9(5) |
| Z | 4 | 4 | 4 | 4 |
| ρcalcd (g cm−3) | 1.748 | 1.771 | 1.779 | 1.795 |
| GOF on F2 | 1.040 | 1.062 | 1.031 | 1.054 |
| R1, wR2 [I > 2σ(I)] | 0.0297, 0.0529 | 0.0245, 0.0410 | 0.0294, 0.0485 | 0.0189, 0.0390 |
| R1, wR2 [all data] | 0.0492, 0.0584 | 0.0365, 0.0446 | 0.0534, 0.0542 | 0.0249, 0.0410 |
| Identification code (CCDC no.) | 18 (2064177) | 19 (2064178) | 20 (2064179) | 21 (2064180) |
| Empirical formula | C63H45Er3O18 | C63H45O18Tm3 | C63H48O18Yb3 | C63H45Lu3O18 |
| Formula weight | 1591.77 | 1596.78 | 1609.11 | 1614.90 |
| Temperature (K) | 296 | 296 | 296 | 296 |
| Crystal system, space group | Monoclinic, P21/n | Monoclinic, P21/n | Monoclinic, P21/n | Monoclinic, P21/n |
| a (Å) | 11.3816(4) | 11.3218(8) | 11.2721(3) | 11.2439(14) |
| b (Å) | 22.4859(8) | 22.5530(15) | 22.5893(7) | 22.591(2) |
| c (Å) | 22.8587(7) | 22.9222(18) | 23.0294(6) | 23.111(3) |
| β (°) | 95.3050(10) | 95.884(3) | 96.1110(10) | 96.311(5) |
| V (Å3) | 5825.1(3) | 5822.1(7) | 5830.6(3) | 5834.9(12) |
| Z | 4 | 4 | 4 | 4 |
| ρcalcd (g cm−3) | 1.815 | 1.822 | 1.833 | 1.838 |
| GOF on F2 | 1.038 | 1.040 | 1.030 | 1.124 |
| R1, wR2 [I > 2σ(I)] | 0.0279, 0.0468 | 0.0288, 0.0467 | 0.0287, 0.0495 | 0.0256, 0.0504 |
| R1, wR2 [all data] | 0.0491, 0.0517 | 0.0500, 0.0536 | 0.0458, 0.0551 | 0.0370, 0.0572 |
Results and discussion
Synthesis and characterization
In this work, three classes containing a total of twenty-one lanthanide coordination complexes were obtained from the reaction of bzz ligand with hydrated Ln(NO3)3 under different synthetic conditions, Scheme 1. The results clearly demonstrated the important roles of solvents, reaction temperatures, and the synthetic methods in determining the structure of the coordination assemblies. For the discrete complexes 1–5 in series 1Ln, good quality crystals for the single crystal structural determination could only be grown by slow evaporation in isopropanol at ambient temperature. Complexes in series 1Ln are moisture-sensitive and can form solid powders when exposed to air for ∼10 h. Meanwhile, the formation of the polymeric complexes in series 2Ln (6–13) and 3Ln (14–21) can be readily controlled via in situ formation of a new ligand by carefully selecting the reaction temperature under hydrothermal conditions. When the reactions were carried out at 110 °C, the initial material bzz was partly converted into ben through in situ hydrolysis. Apparently, the cooperativity of bzz and ben ligands results in the assembly of the stable products in the form of polymeric species 2Ln. Finally, the complete hydrolysis of bzz into ben was observed in the structures of 3Ln upon increasing the reaction temperature to 145 °C. Indeed, heating the crystalline samples 2Ln at 400 °C in air, the structures were completely transformed into 3Ln. This was confirmed by the PXRD patterns of 2Ln and 3Ln collected at 400 °C, which show good match in the main peak positions (see discussion below). This observation implies that the complexes in series 3Ln are more thermodynamically stable than those in series 2Ln. Interestingly, all the polymeric complexes in series 2Ln and 3Ln are very stable in the solid state upon extended exposure to air for over a period of ten months (Fig. S1†), and are insoluble in water as well as in various organic solvents (Fig. S2†). | ||
| Scheme 1 Synthetic routes for complexes in series 1Ln, 2Ln, and 3Ln. | ||
The phase purity of all complexes was verified by PXRD analysis and the results showed that the as-synthesized and the simulated patterns of PXRD of complexes are in good agreement with each other (Fig. S3†), indicating the phase purity of the bulk samples. It should be noted that the PXRD patterns of complexes in each series are similar due to their isostructural natures. Analogously, the IR spectra of complexes in each series are also almost identical, further reflecting their isomorphous natures (Fig. S4†). For 1Ln and 2Ln, the absorption band observed at around 3100 cm−1 demonstrates the existence of hydrazine group. Whereas, the expected strong characteristic absorptions for the symmetric and asymmetric stretching vibrations of carboxylate groups in 2Ln and 3Ln are observed in the ranges of 1360–1438 and 1530–1640 cm−1, respectively. In addition, the absorption bands between 739 and 1448 cm−1 reveal the presence of coordinated and isolated nitrate groups in the structures of 1Ln, while the broad bands in the region 3474–3266 cm−1 indicate the presence of coordinated and lattice water molecules in the structures of 2Ln.
Structural description
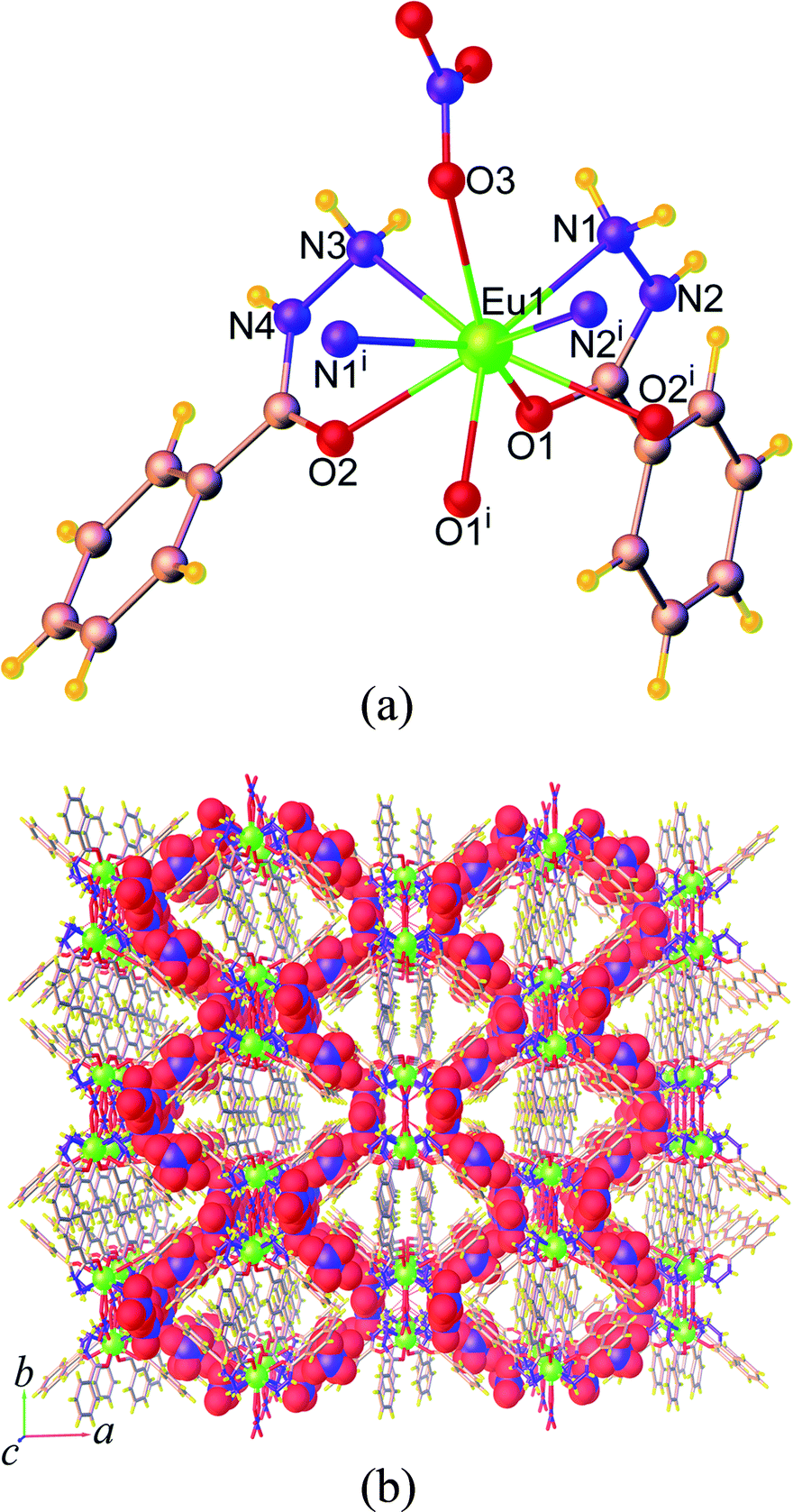 | ||
| Fig. 1 (a) Coordination environment of the Eu3+ ion and (b) perspective view of packing diagram along the c axis in 2. Symmetry code: (i) 1 – x, y, 3/2 – z. | ||
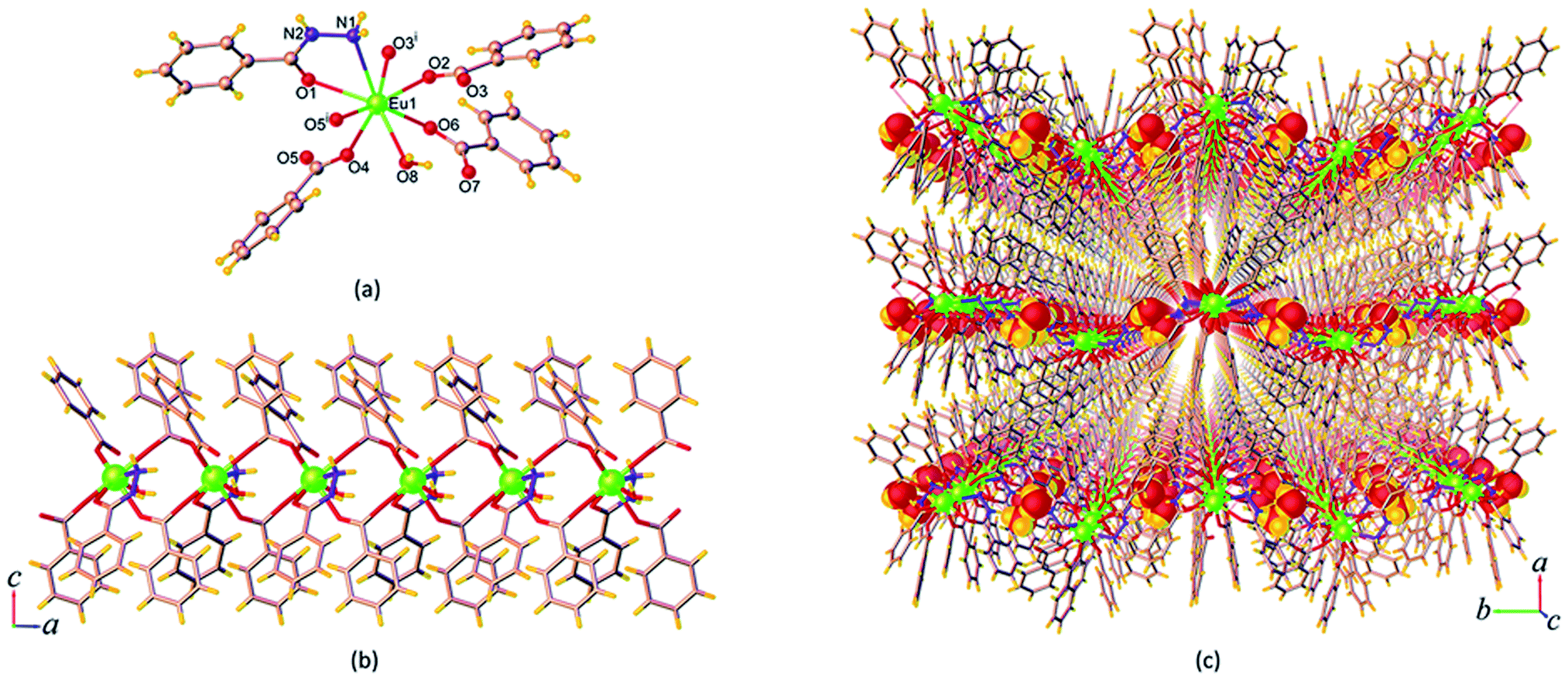 | ||
| Fig. 2 (a) Coordination environment of Eu3+ ion, (b) the 1D chain spread parallel to the a axis, and (c) perspective view of the packing diagram along the c axis of 9. Symmetry code: (i) x, 3/2 – y, z – 1/2. | ||
In 9, the bzz ligand adopts a μ2-κ1:κ1 bidentate chelating coordination mode, whereas the ben ligand exhibits two kinds of coordination fashions viz. the μ2-κ1:κ1 bidentate chelating and κ1 monodentate that leave an uncoordinated carboxylate group. On the basis of these coordination modes, the central Eu3+ ion is ligated by a bidentate chelate bzz ligand and a monodentate ben ligand. Subsequently, the bidentate ben ligands further bridge two symmetry-related Eu3+ ions through the carboxylate group to result in the formation of a 1D chain-like structure running parallel to the crystallographic a axis as shown in Fig. 2b. The Eu⋯Eu separation within the chains is 4.9816(2) Å. There are classical O–H⋯O (d[O⋯O] = 2.638(2)–3.117(4) Å) and N–H⋯O (d[N⋯O] = 2.817(14)–3.122(2) Å) hydrogen bonds involving the amine group and water molecules as the donors and the acyl and carboxylate groups as acceptors (Fig. S6a†), which help in spatially stabilizing the 1D chain. Along the a axis, the lattice water molecules are located between the chains and behave as both hydrogen bond donors and acceptors capable of participating noncovalently with the amine groups and the coordinated water molecules as well as the acyl groups of neighbouring chains. As a result, a 2D corrugated layer is created. The shortest interlayer separation between the Eu3+ centres is 10.824(2) Å. In addition, edge-to-face (ef) N–H⋯π interaction involving the amine hydrogen atom and the aromatic centroid (Cg) of ben molecule (H⋯Cg = 3.410(4) Å) also appears to further reinforce this layer structure. The 2D layers are stacked together through ef C–H⋯π interactions (C⋯Cg = 3.193(4) Å) (Fig. S6b†), which exist between the hydrogen atom of the bzz molecule and the aromatic ben molecule, giving rise to an overall 3D supramolecular structure, Fig. 2c. It should be noted that no aromatic π–π interactions between the aromatic rings of the ligands in 9 are evident.
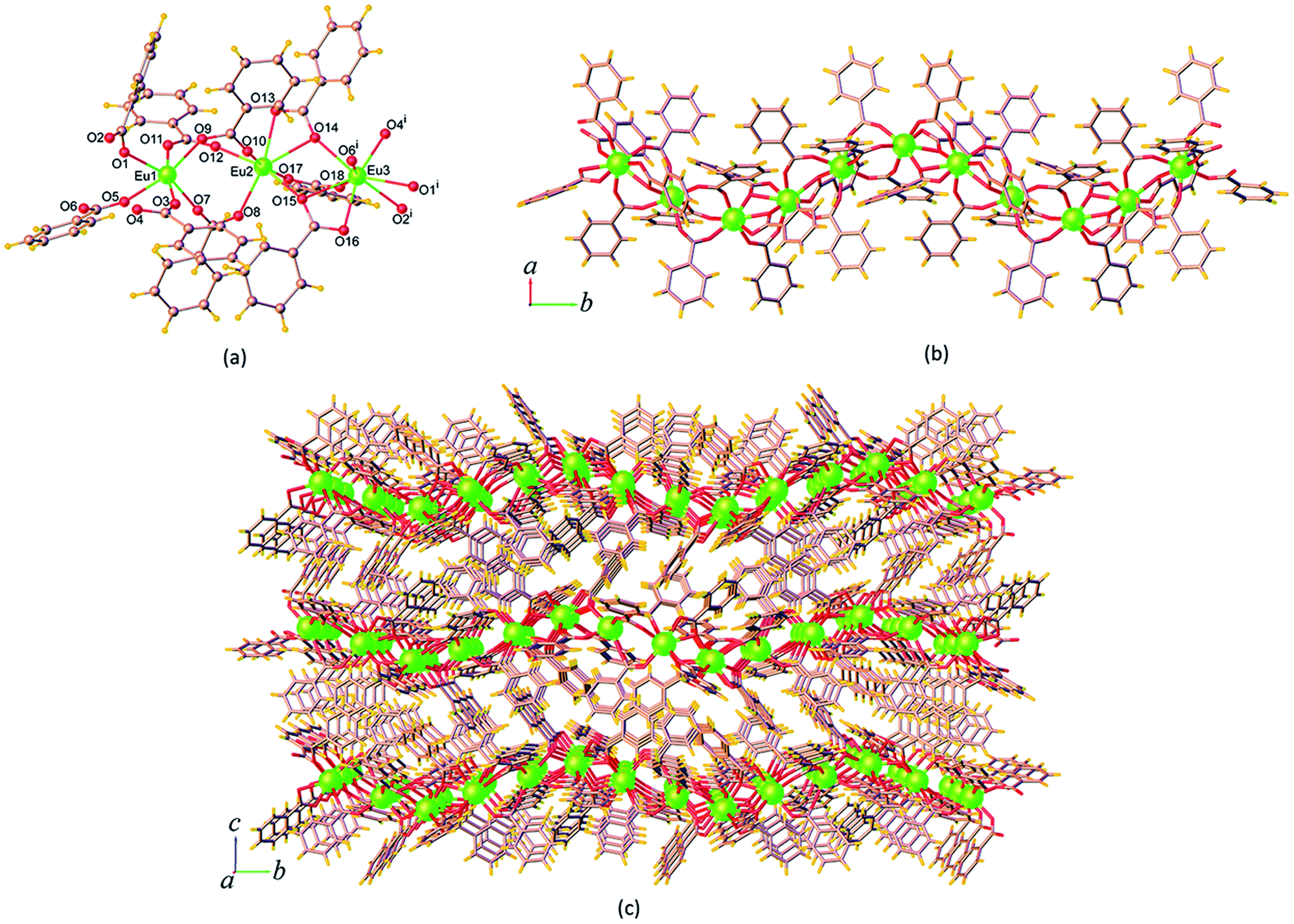 | ||
| Fig. 3 (a) Coordination environment of Eu3+ ion, (b) the 1D chain, and (c) perspective view of the packing diagram along the a axis of 14. Symmetry code: (i) x, 3/2 – y, z – 1/2. | ||
The ben ligands in 14 adopt two kinds of coordination modes, the μ2-κ1:κ1 chelating bidentate and μ3-κ1:κ2 bridging-chelating tridentate. As seen in Fig. 3b, these ben ligands are connected with adjacent Eu3+ ions to form a 1D ribbon chain structure running parallel to the b axis. The Eu⋯Eu separations along the ribbon chains in 14 are 4.5549(3) Å (Eu1⋯Eu2), 4.0139(3) Å (Eu2⋯Eu3), and 4.3537(3) Å (Eu1⋯Eu3). These values are all shorter than the Eu⋯Eu separations found in the chains of 9 described above. The chains are reinforced by ef C–H⋯π (H⋯Cg = 3.023(5) Å) interactions (Fig. S7a†), and are stacked along the a direction by face-to-face (ff) π–π interactions (Cg⋯Cg = 3.768(4) Å) (Fig. S7b†), resulting in the formation of a 2D sheet. Further stabilization of the structure is achieved by another ef C–H⋯π (H⋯Cg = 3.127(4) Å) and π–π interactions (Cg⋯Cg = 4.175(4) Å), which link the sheets along the c axis to form a supramolecular 3D network as shown in Fig. 3c. The intersheet Eu⋯Eu separation in 14 is 11.103(4) Å.
Structural stability and transformation
While the complexes fall into three classes, the crystals of complexes in series 1Ln were not stable when exposed to air as mentioned above. Therefore, only the Eu-based CPs 9 and 14 are selected as the representative examples for the exploration of their thermal stability by means of TGA and in situ VT-PXRD. As shown in Fig. 4a, the TGA curve of 9 merely undergoes three steps of weight loss. The first weight loss of 5.3% between 90 and 160 °C can be attributed to one lattice and one coordinated water molecules (calcd 5.2%). The second weight loss of 21.6% observed in the range 200–355 °C corresponds to the loss of one bzz ligand (calcd 19.8%). The structure of 9 then remained stable until 415 °C, and beyond this temperature, a decomposition of the structure takes place.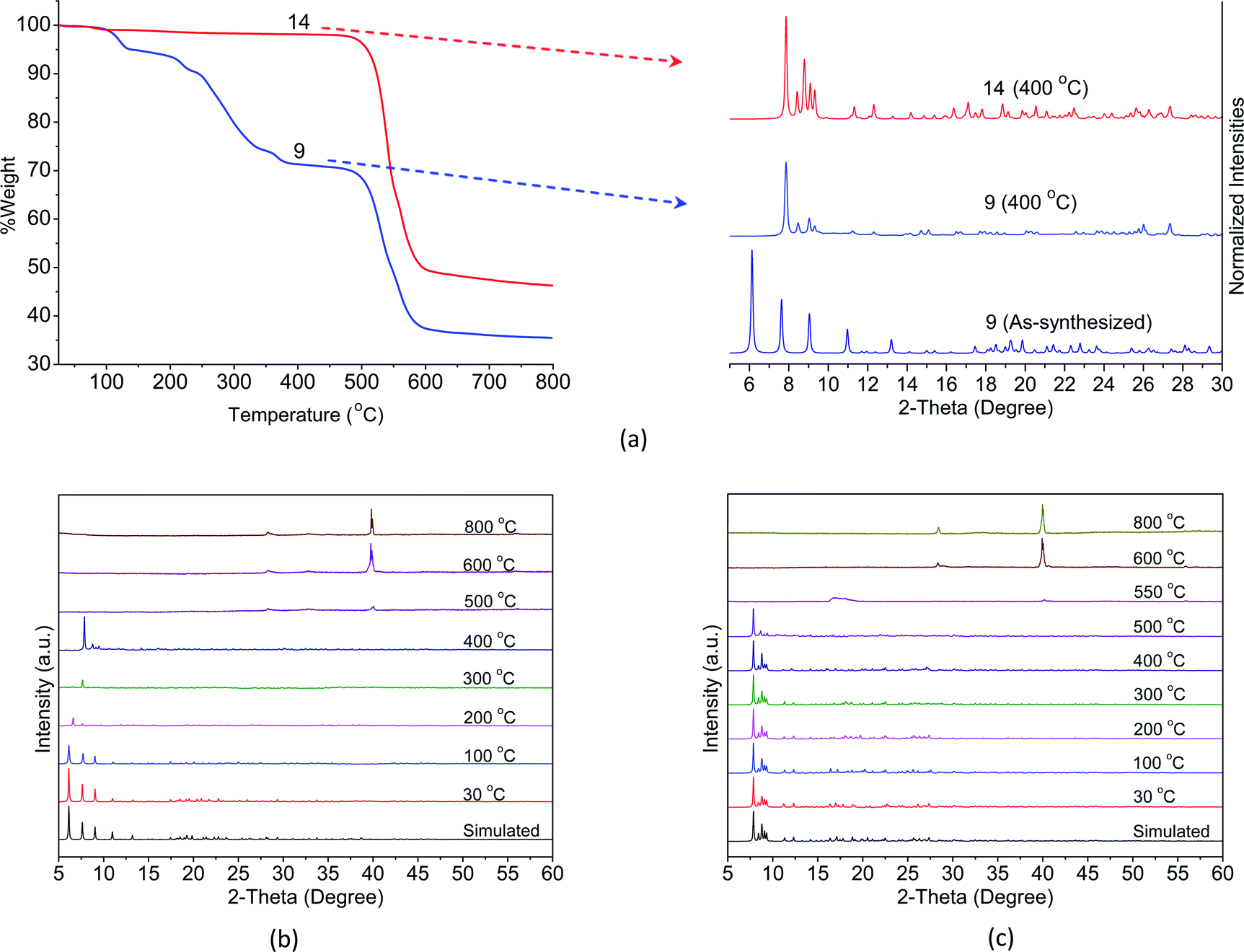 | ||
| Fig. 4 (a) TGA curves (left) and PXRD patterns (right) at 400 °C for 9 and 14. VT-PXRD (30–800 °C) patterns for (b) 9 and (c) 14. | ||
According to the results of VT-XRD, Fig. 4b, three different phases of 9 can be recognized from the patterns. Initially, the patterns from 30 to 100 °C can be identified as the as-synthesized crystal structure. Between 200 and 300 °C shift in the patterns found in the 6.48–7.85 of the 2θ range are observed, suggesting that some structural deformation has occurred upon the removal of the water molecules and the bzz ligands. Finally, when the temperature reached 400 °C, peaks at 2θ = 7.91, 8.64, 8.95, and 9.46° were observed, implying that a new phase is produced. The crystal structure of 9 has clearly decomposed and became amorphous at 500 °C. Upon further heating to 800 °C, new peaks have appeared at 2θ = 28.30, 32.80, and 55.85°, which possibly results from solid-state reactivity. These peaks are consistent with the pattern of Eu2O3 (PDF: 00-034-0392).
For 14, the TGA curve shows no weight loss up to 500 °C. Thereafter, the decomposition of ben ligands and collapse of the structure begins to take place, leaving the final residue of Eu2O3 (PDF: 00-034-0392). It was evident from the PXRD patterns that no phase transition occurs and the structure remains intact up to 500 °C, Fig. 4c. This result is in good agreement with the results of TGA described above. Such an outstanding thermal stability might be attributed to the diverse coordination environment of the lanthanide metal centres, which effectively increases the rigidity of the aromatic backbones of the ligands. This is further enhanced by the intermolecular interactions among the ligands.
From these results, it can be noticed that a complete transformation of crystal 9 to crystal 14 occurs in the solid-state when the as-synthesized samples of 9 are heated at 400 °C in air. This is clearly demonstrated by their similar PXRD patterns and the shape of the TGA curves after removing the water molecules and the bzz ligands (Fig. 4a). In addition, the absence of the hydrazine group is also confirmed by the disappearance of the vibrations band at 3000–3200 cm−1 in the IR spectra16 (Fig. S8†).
Solid state photoluminescence properties
The solid-state photoluminescence properties and lifetime decay behaviours of the Eu- and Tb-based complexes in the UV-visible region were examined at room temperature. Notably, luminescence spectra of the free ligands were also determined in the solid-state and exhibit two broad emissions at 417 and 437 nm (λexc = 290 nm) for the bzz and 333 and 450 nm (λexc = 305 nm) for the ben, due to π → π* and/or n → π* transitions of the molecules (Fig. S9†). As shown in Fig. 5, under the excitation at 290 nm the Eu-based complexes (2, 9, 14) exhibit four characteristic emission bands for f–f transitions for the Eu3+ ion, which are attributed to the 5D0 → 7FJ (J = 1–4) transitions.17 Among these transitions, the most intense emissions are attributed to the electric dipole 5D0 → 7F2 transition at 614, 617, and 615 for 2, 9, and 14, respectively. The 5D0 → 7F2 transition is hypersensitive to the coordination environment of the Eu3+ ion, and is responsible for the bright red emission of the complexes. Clearly, the intensity of the electric dipole transitions in these complexes are much higher than the magnetic dipole transitions (5D0 → 7F1), indicating that the Eu3+ ion occupies a low-symmetry coordination site without inversion centre.18 This agrees with the results of X-ray structural analyses described above. For the Tb-containing complexes 4, 11, and 16, four emission bands are observed upon excitation at 300 nm, which can be attributed to the 5D4 → 7FJ (J = 6−3) transitions of the Tb3+ ions.19 The strongest emission bands from the 5D4 → 7F5 transition are observed at 544, 545, and 542 nm for 4, 11, and 16, respectively, resulting in a green luminescence emission. It should be noted that the lack of ligand-centred emission bands around 380–450 nm in all luminescence spectra of these complexes demonstrates that Eu3+ and Tb3+ centres can be efficiently sensitized by the ligands. The decay lifetimes (τ) for 2, 4, 9, 11, 14, and 16 are 0.18, 0.57, 0.40, 1.01, 2.27, and 1.43 ms, respectively (Table S4 and Fig. 10†).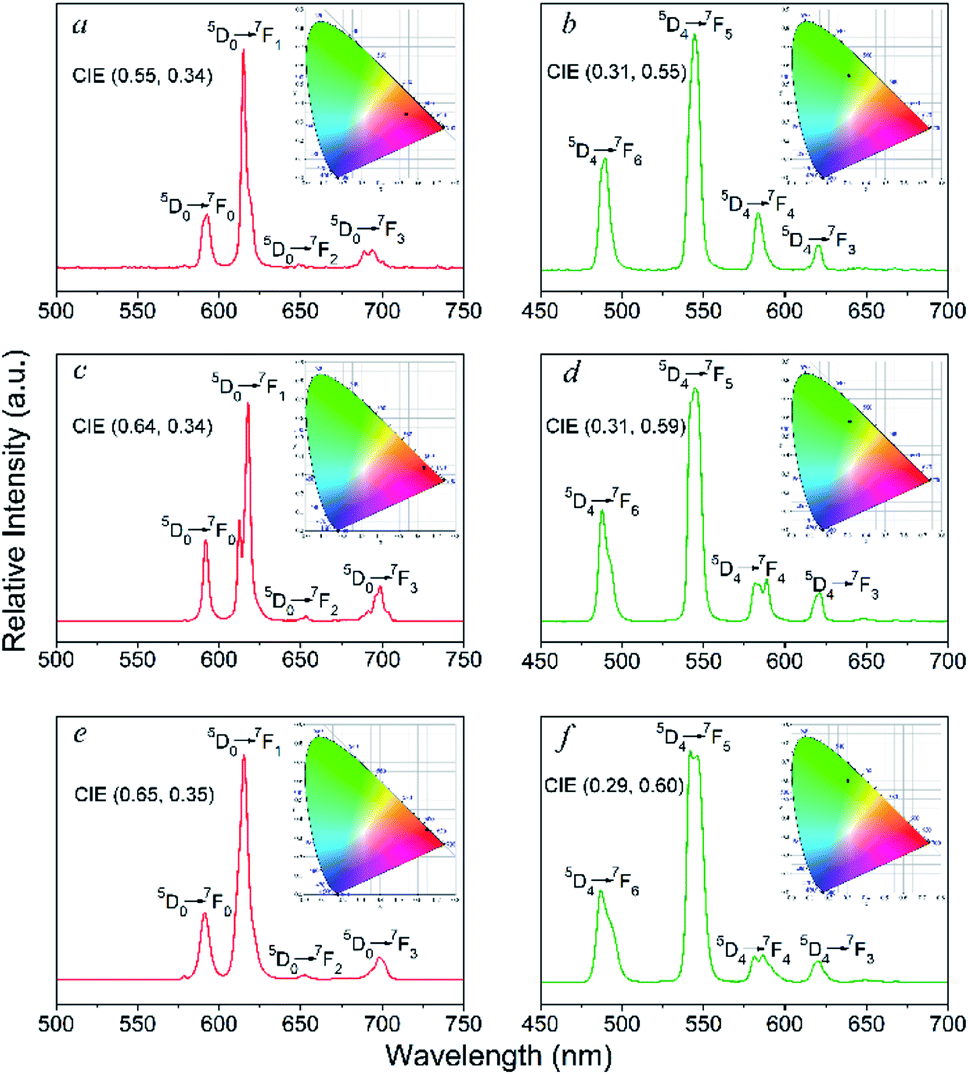 | ||
| Fig. 5 Emission spectra and colorimetric coordinates (insert) of 2 (a), 4 (b), 9 (c), 11 (d), 14 (e), and 16 (f). | ||
Fluorescent sensing properties
Encouraged by the intense red and green luminescence emissions coupled with chemical stability of the Eu- and Tb-based LnCPs, their application as chemical sensors for molecules and ions was investigated. In order to identify the potential of 9, 11, 14, and 16 toward sensing of organic molecules, their crystalline samples (3 mg) were simply immersed in different solvents (3 mL) including acetone, acetonitrile (MeCN), benzene, dioxane, chloroform (CHCl3), dichloromethane (DCM), DMF, DMSO, toluene, THF, MeOH, EtOH, isopropanol (iPrOH), ethyl acetate (EtOAc), and distilled H2O, and subsequently ultrasonicated for 10 min to form suspensions for luminescence measurements. As can be seen in Fig. 6, these complexes exhibit distinct solvent-dependent luminescence intensities, and their respective emissions are completely quenched by acetone. The possible reason of quenching by acetone molecules might be attributed to the competitive energy absorption.20 This was evidenced by the observation of an overlap of the absorption band of acetone with the excitation spectra of 9, 11, 14, and 16 in the UV-Visible spectra (Fig. S11†). In addition, the PXRD patterns of the samples after sensing tests are consistent with that of the original samples (Fig. S12†), suggesting that the basic 1D chain structures of these CPs remain unchanged. This has also proved that the luminescence quenching by acetone was not caused by the collapse of the structure.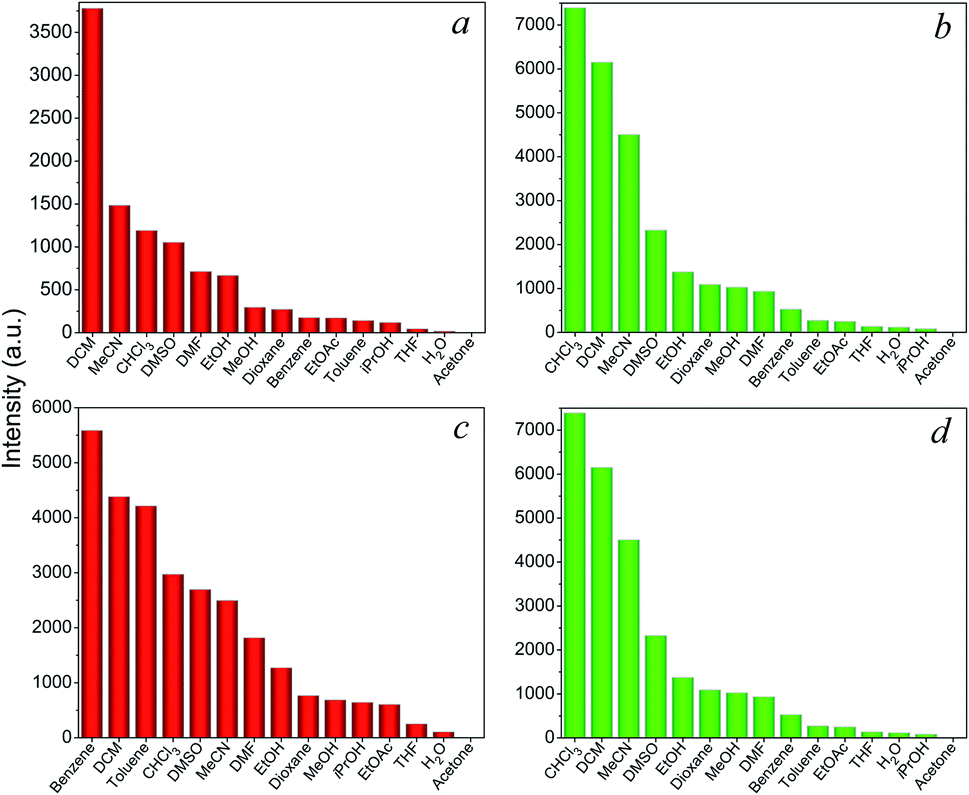 | ||
| Fig. 6 Emission intensities for (a) 9, (b) 11, (c) 14, and (d) 16 in different organic solvents. | ||
The above complexes show intense photoluminescence emission in DCM and MeCN. Hence, further exploration of their sensing properties toward a trace amount of metal ions was examined in MeCN solutions, since metal nitrate salts are highly soluble in this solvent. The crystalline samples (3 mg) of 9, 11, 14, and 16 were ground and immersed in individual MeCN solutions of Mx+ ions (3 mL) with a concentration of 5 × 10−4 M (M = K+, Na+ Ag+, Ca2+, Cd2+, Co2+, Cu2+, Fe2+, Hg2+, Mn2+, Ni2+, Zn2+, Al3+, Co3+, Cr3+, and Fe3+) for 30 min at ambient temperature and the suspensions were then ultrasonicated for 10 min. As shown in Fig. 7, metal ion incorporated 9, 11, 14, and 16 in MeCN suspensions (Mx+@9, Mx+@11, Mx+@14, Mx+@16) show distinct metal-dependent luminescence intensities and their emissions are strongly quenched by the Co2+ ion even in the presence of other metal ions (Fig. S13†). In addition, it can be clearly seen that the luminescence intensity of the suspension shows little changes with the addition of Co3+ ions. This observation suggested that the Eu-(9, 14) and Tb-(11, 16) based CPs may act as luminescent probes for selective sensing of Co2+ ions in the Co2+/Co3+ solution mixtures, which is important for applications in environmental, pharmaceutical, and biological systems.21
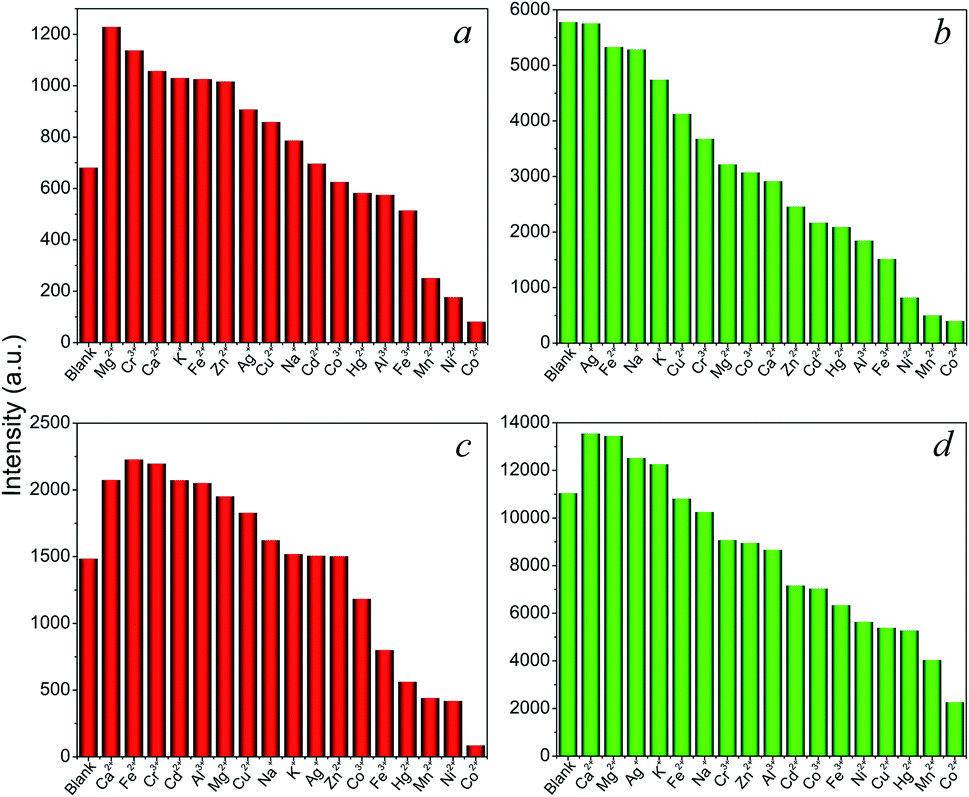 | ||
| Fig. 7 Emission intensities for (a) 9, (b) 11, (c) 14, and (d) 16 in MeCN solutions of different metal ions. | ||
Quantitatively, the quenching effect can be determined by the Stern–Volmer (S–V) equation: I0/I = 1 + KSV[M].22 The values of I0 and I are the luminescence intensities of the suspension of the Eu-(9, 14) and Tb-(11, 16) based CPs without and with the addition of Co2+, respectively. [M] is the molar concentration of Co2+, and KSV is the quenching effect coefficient (M−1). From the linear fitting of the S–V plots, the calculated KSV values for Co2+ was found to be 1.29 × 104, 1.02 × 104, 8.9 × 103, and 8.2 × 103 M−1 for 9, 11, 14, and 16, respectively. Following the IUPAC recommendation for the limit of detection (LOD) determination at a signal-to-noise ratio (S/N) of 3 (LOD = 3σblank/KSV),23 the LOD values for Co2+ in 9, 11, 14, and 16 were estimated to be 20.1, 12.7, 6.1, and 6.5 μM, respectively (Fig. S14†). It should be noted that the S–V plots are all nearly linear, suggesting that the mechanism of the luminescence quenching by Co2+ ions can be attributed to the dynamic quenching process.24
The possible sensing mechanism of luminescence quenching by the Co2+ ions was studied. The PXRD patterns of the sample after sensing of Co2+ ions were measured and compared with the corresponding as-synthesized patterns (Fig. S15†). This indicates that the luminescence quenching does not stem from the collapse of the structure. Furthermore, UV-Vis absorption spectra for metal ions in MeCN solutions were recorded. It was found that the absorption spectrum of Co2+ solution (250–400 nm) originating from the d–d transition has a large overlap with the excitation spectra of 9, 11, 14, and 16. While among other MeCN solutions of metal ions those of Ni2+ and Mn2+ have a slight spectra overlap (Fig. S16†). When excited at 290 nm (9, 14) or 300 nm (11, 16), the competition for energy of excitation and absorption between the lanthanide CPs and analyte will occur. This may be responsible for the quenching phenomenon.20 In addition, the sensing properties of 9 and 11 for anions were also explored by recording their emissive spectra of MeCN solutions containing potassium salts at the concentration of 5.0 × 10−4 M (Fig. S17†). The results indicate that the anionic analytes have negligible effects on the luminescence intensity.
Furthermore, experiments testing the sensing performance after recycling were carried out to study the recyclability and stability of 9, 11, 14, and 16 for the luminescent sensing detection of Co2+ ions. For this purpose, the recovered solid samples after detection of Co2+ were collected from the solution through centrifugation and washed ten times with MeCN. Subsequently, the luminescence intensities of the recovered solids were recorded and compared. It is clear that the luminescence intensities for three recycles are almost unchanged compared to the initial emission intensities of the as-synthesized compounds (Fig. S18†). Notably, PXRD patterns of the recovered samples after three cycles are also nearly identical to that of the original one (Fig. S19†), which indicates that the materials maintain their crystal structure and crystallinity after these experiments. This fact implies that compounds 9, 11, 14, and 16 represent recyclable luminescent probes for Co2+ ions as they can be reused at least three times without significant changes in performance. In addition, XRF analysis was also performed on the recovered solid samples after the detection of Co2+. The results reveal the presence of only the Eu or Tb signals in the XRF profiles (Fig. S20†), indicating that the Co2+ ion was not incorporated into the crystal structures. This observation further consolidates the conclusion that the binding interactions between the Co2+ analyte and the luminescent LnCP species in MeCN solution are somewhat weak.
Magnetic properties
The temperature dependence of the magnetic susceptibilities of the Gd- and Dy-based complexes were recorded in the range of 2–300 K at 5000 Oe, and the results in the form of the χmT product as a function of the temperature (χm is the magnetic susceptibility per mole of Ln3+ ion) are presented in Fig. 8. For 10 and 12, the χmT values at 300 K are 7.92 and 14.10 cm3 K mol−1, respectively, which is close to the theoretical values (7.88 (10) and 14.17 (12) cm3 K mol−1) for one magnetically isolated Ln3+ ion (Gd3+, 8S7/2, g = 2; Dy3+, 6H15/2, g = 4/3).25 Meanwhile, the χmT values of 23.57 and 41.65 cm3 K mol−1 at 300 K are also close to the values expected (23.64 (15) and 42.51 (17) cm3 K mol−1) for three isolated Ln3+ ions. The χmT values of complexes 10, 12, 15, and 17 gradually decrease with decreasing temperature and down to 6.79, 8.20, 21.97, and 31.90 cm3 K mol−1 at 2 K, respectively. Such magnetic behaviours in the Gd- and Dy-based CPs are attributed to the thermal depopulation of the Stark sublevels and weak antiferromagnetic interactions between the Ln3+ centres.26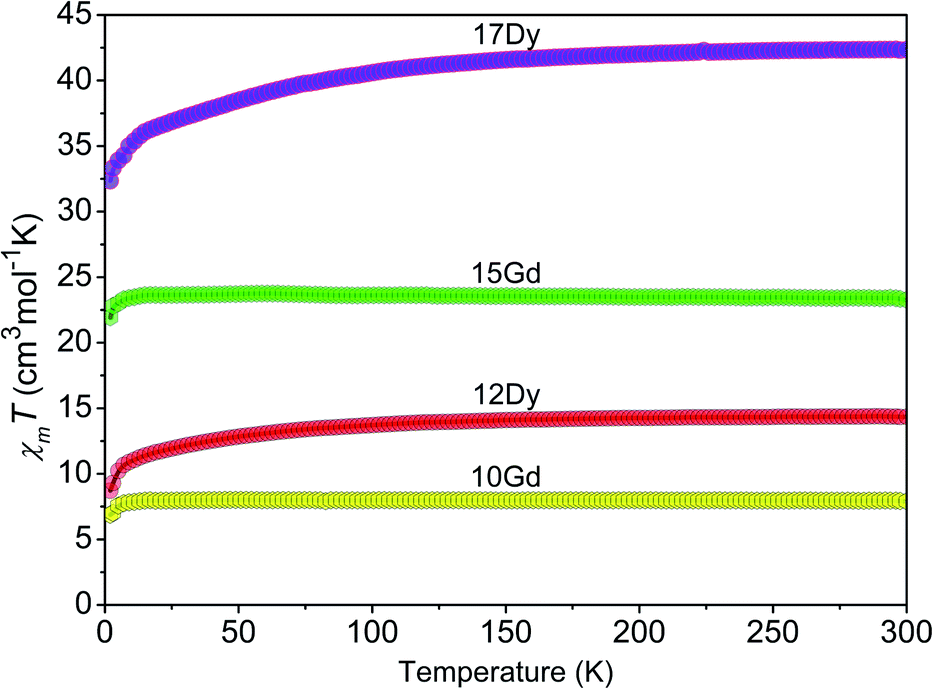 | ||
| Fig. 8 Plots of χMT as a function of temperature for the Gd-(10, 15) and Dy-(12, 17) containing complexes under a 5000 Oe field. | ||
Conclusions
In summary, the single crystal samples of twenty-one lanthanide coordination complexes have been successfully prepared by reacting the bzz ligand with different Ln(III) nitrate salts under different reaction conditions. In this work, the hydrothermal in situ formation of ben ligand through the hydrolysis of the bzz molecule has an important effect on the formation of the thermodynamically stable lanthanide-based CPs 2Ln and 3Ln. Indeed, these CPs are stable in air for long periods of time (up to ten months). Meanwhile, the discrete mononuclear complexes 1Ln containing only chelated bzz ligands tend to become unstable upon exposed to air for 6 h. Interestingly the structures of 2Ln can be transformed into the thermodynamically more stable phase 3Ln upon heating the crystalline samples at 400 °C in air. Notably, as a representative example of the 3Ln series, the structure of 14 shows good thermal stability up to 500 °C. The solid-state photoluminescence properties of the Eu- and Tb-based CPs demonstrate that the bzz and ben ligands provide good sensitization of the Eu3+ and Tb3+ luminescence via ligand-to-lanthanide energy transfer process. The Eu-(9, 14) and Tb-(11, 16) based CPs not only show intense luminescence emissions with long lifetimes at the microsecond scale but also can act as luminescent sensors for acetone and Co2+ ion through an energy competition mechanism with good recyclability. Additionally, temperature-dependent magnetic susceptibility measurements reveal that the Gd-(10, 15) and Dy-(12, 17) based CPs exhibit antiferromagnetic coupling between the corresponding Ln3+ ions. This work demonstrates for the first time that the in situ hydrolysis of hydrazide based organic ligand can be used to prepare lanthanide-based CPs with 1D chain structures and shows their interesting thermal, luminescence, and magnetic properties. Further studies will focus on the construction of new functional lanthanide-based CPs with the bzz derivatives by taking advantage of the in situ hydrolysis of hydrazide group under hydrothermal conditions to generate carboxylate-based ligands, and such investigations are now ongoing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Thammasat University Research Fund (Fast Track), Contract No. TUFT 044/2563, and was also partially funded by the Thailand Research Fund (RTA6180007) and Thammasat University Research Unit in Multifunctional Crystalline Materials and Applications (TU-McMa). C. T. would like to acknowledge the Graduate Development Scholarship 2020, National Research Council of Thailand (Contact No. 15/2563) and the NSTDA STEM Workforce (scholarship No. SCA-CO-2561-6014-TH).References
-
(a) L. Feng, J. Pang, P. She, J.-L. Li, J.-S. Qin, D.-Y. Du and H.-C. Zhou, Adv. Mater., 2020, 32, 2004414 CrossRef
F. Saraci, V. Quezada-Novoa, P. R. Donnarumma and A. J. Howarth, Chem. Soc. Rev., 2020, 49, 7949–7977 RSC
-
(a) B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS
-
(a) M. Kumar, L.-Q. Li, J. K. Zaręba, L. Tashi, S. C. Sahoo, M. Nyk, S.-J. Liu and H. N. Sheikh, Cryst. Growth Des., 2020, 20, 6430–6452 CrossRef CAS
-
(a) X. Zhang, V. Vieru, X. Feng, J.-L. Liu, Z. Zhang, B. Na, W. Shi, B.-W. Wang, A. K. Powell, L. F. Chibotaru, S. Gao, P. Cheng and J. R. Long, Angew. Chem., Int. Ed., 2015, 54, 9861–9865 CrossRef CAS PubMed
-
(a) S. Sairenji, S. Akine and T. Nabeshima, Dalton Trans., 2016, 45, 14902–14906 RSC
-
(a) B. Chen, L. Wang, Y. Xiao, F. R. Fronczek, M. Xue, Y. Cui and G. Qian, Angew. Chem., Int. Ed., 2009, 48, 500–503 CrossRef CAS
- X.-M. Chen and M.-L. Tong, Acc. Chem. Res., 2007, 40, 162–170 CrossRef CAS
-
(a) F. Debatin, A. Thomas, A. Kelling, N. Hedin, Z. Bacsik, I. Senkovska, S. Kaskel, M. Junginger, H. Müller, U. Schilde, C. Jäger, A. Friedrich and H.-J. Holdt, Angew. Chem., Int. Ed., 2010, 49, 1258–1262 CrossRef CAS PubMed
-
(a) K. Chainok, N. Ponjan, C. Theppitak, P. Khemthong, F. Kielar, W. Dungkaew, Y. Zhou and S. R. Batten, CrystEngComm, 2018, 20, 7446–7457 RSC
-
(a) L. B. Zinner, D. E. Crotty, T. J. Anderson and M. D. Glick, Inorg. Chem., 1979, 18, 2045–2048 CrossRef CAS
- Bruker, APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
-
(a) M. Seitz, A. G. Oliver and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 11153–11160 CrossRef CAS PubMed
-
(a) I. Gulaczyk, M. Kręglewski and A. Valentin, J. Mol. Spectrosc., 2003, 220, 132–136 CrossRef CAS
-
(a) D. Parker and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 1996, 3613–3628 RSC
-
(a) S. I. Klink, L. Grave, D. N. Reinhoudt, F. C. van Veggel, M. H. Werts, F. A. J. Geurts and J. W. Hofstraat, J. Phys. Chem. A, 2000, 104, 5457–5468 CrossRef CAS
-
(a) S. Aime, M. Botta, R. S. Dickins, C. L. Maupin, D. Parker, J. P. Riehl and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 1998, 881–892 RSC
-
(a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. V. Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed
-
(a) E. L. Baldwin, J. A. W. Byl and N. Osheroff, Biochemistry, 2004, 43, 728–735 CrossRef CAS
- A. Rose, Z. Zhu, C. F. Madigan, T. M. Swager and V. Bulovic, Nature, 2005, 434, 876–879 CrossRef CAS
-
(a) H. Xu, F. Liu, Y. Cui, B. Chen and G. Qian, Chem. Commun., 2011, 47, 3153–3155 RSC
- X.-H. Zhou, L. Li, H.-H. Li, A. Li, T. Yanga and W. Huang, Dalton Trans., 2013, 42, 12403–12409 RSC
-
(a) S. T. Hatscher and W. Urland, Angew. Chem., Int. Ed., 2003, 42, 2862–2864 CrossRef CAS PubMed
-
(a) L. Zhao, J. Wu, H. Ke and J. Tang, CrystEngComm, 2013, 15, 5301–5306 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Additional PL spectra, FT-IR spectra, PXRD patterns, XRF spectra, UV-Vis spectra, selected bond lengths and angles. CCDC 2064160–2064180. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03106f |
| This journal is © The Royal Society of Chemistry 2021 |