DOI:
10.1039/D1RA02345D
(Paper)
RSC Adv., 2021,
11, 16892-16905
Newly synthesized MAX phase Zr2SeC: DFT insights into physical properties towards possible applications†
Received
24th March 2021
, Accepted 30th April 2021
First published on 7th May 2021
Abstract
A DFT study of the synthesized MAX phase Zr2SeC has been carried out for the first time to explore its physical properties for possible applications in many sectors. The studied properties are compared with prior known MAX phase Zr2SC. The structural parameters (lattice constants, volume, and atomic positions) are observed to be consistent with earlier results. The band structure and density of states (DOS) are used to explore the metallic conductivity, anisotropic electrical conductivity, and the dominant role of Zr-d states to the electrical conductivity at the Fermi level. Analysis of the peaks in the DOS and charge density mapping (CDM) of Zr2SeC and Zr2SC revealed the possible variation of the mechanical properties and hardness among them. The mechanical stability has been checked using elastic constants. The values of the elastic constants, elastic moduli and hardness parameters of Zr2SeC are found to be lowered than those of Zr2SC. The anisotropic behavior of the mechanical properties has been studied and analyzed. Technologically important thermodynamic properties such as the thermal expansion coefficient (TEC), Debye temperature (ΘD), entropy (S), heat capacity at constant volume (Cv), Grüneisen parameter (γ) along with volume (V) and Gibbs free energy (G) are investigated as a function of both temperature (from 0 to 1600 K) and pressure (from 0 to 50 GPa). Besides, the ΘD, minimum thermal conductivity (Kmin), melting point (Tm), and γ have also been calculated at room temperature and found to be lowered for Zr2SeC compared to Zr2SC owing to their close relationship with the mechanical parameters. The value of the ΘD, Kmin, Tm, and TEC suggest Zr2SeC as a thermal barrier coating material. The optical properties such as dielectric constant (real and imaginary part), refractive index, extinction coefficient, absorption coefficient, photoconductivity, reflectivity, and loss function of Zr2SeC are computed and analyzed to reveal its possible applications.
1. Introduction
Since the 1990s, one of the most widely known classes of transition metal carbides or nitrides is the so-called MAX phase materials with general formula Mn+1AXn (M – early transition metal, A – A-group element, X – C or N; n = 1–4).1–4 MAX phase materials can attract great attention owing to their exceptional performance combining both ceramics (elastically rigid, lightweight, creep and fatigue resistant as ceramic materials) and metals (machinable, electrically and thermally conductive, not susceptible to thermal shock, plastic at high temperature and exceptionally damage-tolerant);1,2 consequently, they are promising candidates such as in high-temperature technology as components, sliding electrical contacts, and contacts for 2D electronic circuits, Li-ion batteries, wear and corrosion-resistant coatings, superconducting materials, spintronics, and nuclear industry.2,5–9 The hybrid properties of MAX phases are due to the existence of strong covalent M–X bonds and relatively weak metallic M–A bonds within their structure.2,10,11 These challenging properties are always motivating scientists; consequently, more than 150 MAX phases have been discovered.1 Moreover, researchers are also trying to manipulate the composition and structure to achieve better combination of the properties such as different alloys/solid solutions,12–20 M2A2X21–23 and M3A2X,24 rare-earth i-MAX phases,25,26 212 MAX phases,27–29 314 MAX phases,27,30 MAX phase borides6,31–37 and two-dimensional (2D) MAX phase derivatives termed MXenes.38,39
As an A element, chalcogen S containing MAX phases have drawn attention owing to their comparatively strong M–A(p–d) bonding which is usually weak for MAX phases. So far known, the S-containing ternary MAX phases are M2SC (M = Ti, Zr, Hf, and Nb) and M2SB (M = Zr, Hf, and Nb).1,13–15 The existence of strong M–A bonding for the S-containing MAX phases is reflected from the higher values of Young's modulus, bulk modulus, and shear modulus of M2SC (M = Ti, Zr, Hf) compared to that of M2AlC (M = Ti, Zr, Hf).34,40–42 All the S-containing phases have been studied completely by the different research groups. Amini et al.40 have synthesized and studied the mechanical properties of Ti2SC. Bouhemadou et al.41 have performed a first-principles investigation of M2SC (M = Ti, Zr, Hf) compounds. A comprehensive study of elastic properties of 211 MAX phases has been carried out by Cover et al.,42 where the Ti2SC and Zr2SC were included. M2SB (M = Zr, Hf, Nb) borides have been synthesized by Rackl et al.,32,33 which has been further subjected for the comprehensive study of the physical properties of M2SX (X = C and B).34 For each case, the S-containing MAX phases have enhanced mechanical properties compared to corresponding Al-containing MAX phases.
Recently, a new chalcogen (Se) containing MAX phase (Zr2SeC) has been synthesized by Chen et al.,43 where they have investigated the only electronic density of states, charge density, electrical resistivity, and thermal conductivity of Zr2SeC. These limited results are not enough to explore the Zr2SeC thoroughly for practical application in many sectors where many other MAX phases have already been used. For example, the MAX phases have the potential to be used as structural components at high-temperature1,44,45 where the knowledge of mechanical properties is essential. MAX phases are also using in high-temperature technology [e.g., as thermal barrier coating (TBC) material] where some prior knowledge of Debye temperature, minimum thermal conductivity as well as melting temperature is required.46,47 Moreover, the MAX phases are potential candidates for use as cover materials to diminish solar heating48 where the knowledge of optical properties is fundamental. Thus, to predict its possible relevance in other sectors, a detailed study of Zr2SeC is of scientific importance. Therefore, a detailed study of Zr2SeC needs to be performed to take the full advantages for possible use in many sectors.
Therefore, the structural, electronic, mechanical, thermal, and optical properties of Zr2SeC have been presented in this paper, and the properties of Zr2SeC have been compared with those of the Zr2SC MAX phase. It is found that the Zr2SeC is soft with low Vickers hardness like other phase materials. Moreover, it is suitable to be used as a TBC material. Furthermore, it can also be used as a cover material for spacecraft to reduce solar heating. The rest of the article is organized as follows: the detailed Computational methodology is given in Section 2, Results and discussion are presented in Section 3, and important Conclusions are drawn in Section 4.
2. Computational methodology
A density functional theory based on the plane-wave pseudopotential method was implemented in the Cambridge Serial Total Energy Package (CASTEP) code49,50 to calculate the physical properties of Zr2SeC. The exchange and correlation functions were treated by the generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE).51 The pseudo-atomic calculations were performed for C – -2s22p2, Se – 4s24p4, and Zr – 4d25s2 electronic orbitals. A k-point52 mesh of size 10 × 10 × 3 was selected to integrate the Brillouin zone, and the cutoff energy was set as 500 eV. The Broyden Fletcher Goldfarb Shanno (BFGS) technique53 was used for structure relaxation, and density mixing was used for electronic structure calculation. The structure was relaxed with the following parameters: the self-consistent convergence of the total energy is 5 × 10−6 eV per atom, the maximum force on the atom is 0.01 eV Å−1, the maximum ionic displacement is set to 5 × 10−4 Å, and maximum stress of 0.02 GPa. The optical properties of the Zr2SeC compound were calculated using the complex dielectric function ε(ω) = ε1(ω) + iε2(ω). The time-dependent perturbation theory (first-order) is used to obtain the imaginary part of the dielectric function. The equation used to calculate the imaginary part ε2(ω) is given by:| |
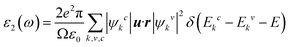 | (1) |
where Ω and ε0 correspond to the volume of the unit cell and dielectric constant of the free space, u and r stand for the incident electric field vector (polarized) and position vector. ω, e and ψkc and ψkv stand for the light frequency, electronic charge, and conduction and valence band wave functions at k, respectively. Here, the sum k was used to present the Brillouin zone’ sampling in the k space. The sums v and c were used to represent the contribution from the unoccupied conduction band (CB) and occupied valence band (VB). The Kramers–Kronig relations were used to estimate the real part ε1(ω) from the imaginary part ε2(ω). The real and imaginary parts of the dielectric function were further used to obtain the other optical constants: refractive index, extinction coefficient, absorption spectrum, reflectivity, and energy-loss spectrum based on the relations found elsewhere.54
3. Results and discussion
3.1 Structural properties
The unit of Zr2SeC is shown in Fig. 1, which is crystallized in the hexagonal system with space group P63/mmc (194).2 The unit cell contains two formula units, and there are eight atoms in the unit cell. The Zr6C octahedron interleaved between two atomic layers of the Se atom is shown in Fig. 1. For 211 MAX phases, the atomic positions of Zr, Se, and C atoms in the unit cell are (1/3, 2/3, 0.0965), (1/3, 2/3, 3/4), and (0, 0, 0). The unit of Zr2SeC is optimized geometrically to calculate the physical properties further. The lattice constants of the optimized cell are presented together with previous experimental and theoretical results43 that assure the parameters used for calculations. For example, our calculated values of a, c, and V are only 0.1%, 0.18%, and 1.08%, respectively, higher than those of experimental values (Table 1).
 |
| | Fig. 1 Crystal structure (unit cell) of the Zr2SeC compound. | |
Table 1 Calculated lattice parameters (a and c), c/a ratio, volume (V), and atomic positions of Zr2SeC MAX phase
| a (Å) |
c (Å) |
c/a |
V (Å3) |
Ref. |
Positions |
Zr |
Se |
C |
| 3.4655 |
12.5406 |
3.618 |
130.429 |
This study |
x |
1/3 |
1/3 |
0 |
| 3.462 |
12.518 |
3.615 |
129.029 |
Expt.43 |
y |
2/3 |
2/3 |
0 |
| 3.487 |
12.631 |
3.622 |
132.080 |
Theo.43 |
z |
0.0965, 0.0963 (ref. 43) |
3/4 |
0 |
3.2 Electronic properties and bonding nature
The electronic band structure of Zr2SeC of optimized structure within the GGA-PBE is estimated and analyzed as shown in Fig. 2(a) along the high symmetry lines of the Brillouin zone (Γ–A–H–K–Γ–M–L–H). None existence of a bandgap close to the Fermi level owing to the overlapping of the conduction and valence band reveals the metallic nature of the titled compound like other MAX phases.55 The band structure also reveals the anisotropic nature of electronic conductivity. It is seen that the energy dispersion along Γ–A, K–H, and L–M directions, which are along the c-direction, are small. The conductivity in the basal plane is exhibited by H–K, Γ–M, and L–H that are larger than those of c-direction. Thus, the conductivity is higher in the basal plane than that of c-direction for Zr2SeC, which is typical for MAX phases.20,29,56
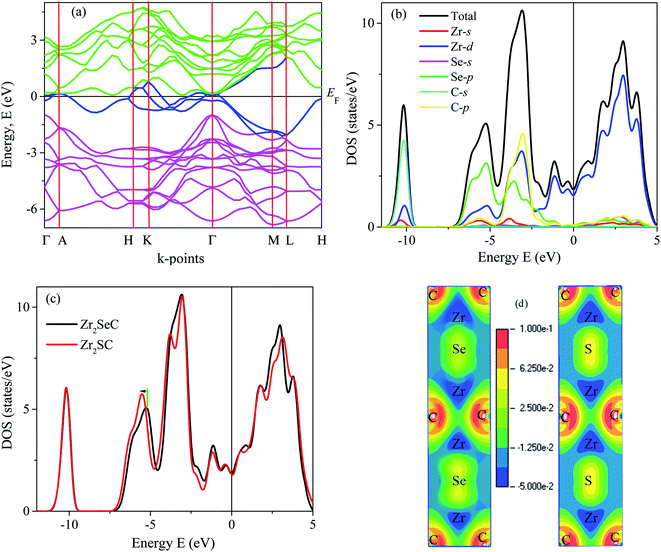 |
| | Fig. 2 The electronic band structure of (a) Zr2SeC, (b) total and partial DOS of Zr2SeC, (c) the total DOS of Zr2AC (A = S, Se) and (d) charge density mapping of Zr2AC (A = S, Se). | |
After band structure, the next step for insight into the electronic properties of Zr2SeC is to investigate its density of states (DOS). Fig. 2(b) shows the DOS of Zr2SeC with a finite value of DOS (1.84 states per unit cell per eV) at the Fermi level that is slightly higher than the value (1.48 states per unit cell per eV) obtained by Chen et al.,43 might due to the difference in the functionals used. The DOS of Zr2SeC is slightly higher than that of Zr2SC (1.79 states per unit cell per eV); a similar result is also reported by Chen et al.43 The value of DOS at the Fermi level (EF) can provide information regarding electrical conductivity as it is directly related to the DOS and electron mobility.2 The partial DOS is used to explore the contribution from different states to the total DOS. For instance, the DOS at the EF is contributed from Zr-d states and involved in the conduction of Zr2SeC. The C and Se do not contribute to the DOS at EF and are not involved in the conduction properties. The contribution of different atoms to the DOS at different energy states is also observed from the partial DOS (PDOS). The DOS profile is almost similar to that of the reported DOS.43 The hybridization among the different Zr, Se, and C electronic states is observed from PDOS. The lowest energy band (−9 to −12 eV) comes from the hybridization between Zr-d and C-s orbital's electrons with a dominating role of C-s states. The peak in the energy range −4.5 to −7.5 eV is accredited by the strong hybridization of Se-p and Zr-d states with a minor contribution from Zr-s and C-p states. The strongest peak in the range – 1.7 to 4.5 eV results from the hybridization between Zr-d, Se-p, and C-p states with dominant contribution from C-p states.
The highest valence band in the range 0 to 1.7 eV is dominated by Zr-d states with a small contribution from Se-p and C-p states. The PDOS of Zr2SC is not presented here due to similar nature. The TDOS of Zr2AC (A = S, Se) is shown in Fig. 2(c) to disclose the effect of the A element. As seen in Fig. 2(b), the S/Se contributed to the TDOS mostly in the energy range of −4.5 to −7.5 eV. The peak found in this region is shifted towards lower energy (indicated by the green line and black arrow) for Zr2SC compared to Zr2SeC, indicating a higher bonding M–A strength for Zr2SC compared to that in Zr2SeC. The peaks position due to Zr and C remain the same for both compounds. Thus, higher values of the mechanical properties characterizing parameters are expected owing to the presence of stronger M–A bonding in Zr2SC than in Zr2SeC. To strengthen this statement, we have calculated the charge density mapping (CDM) for both phases, as shown in Fig. 2(d). Fig. 2(d) shows the CDM for both Zr2AC (A = S, Se), where the red and blue colors indicate the highest and the lowest value, respectively. The directional covalent bonding occurs between Zr and C atoms where the charges are geometrically localized, and this boding is comparatively stronger, required more energy to break this bonding. As seen in the figure, the charge density at Zr and C position is unchanged for both Zr2SC and Zr2SeC. The charges are accumulated to a greater extent at S positions of Zr2SC than that of Se positions of Zr2SeC. The greater charges at the S position the greater bonding strength between Zr–S than Zr–Se which is in good agreement with DOS results. Now it's time to prove the statement that the Zr– S bonding stronger than Zr–Se which will be proven in the following section. The electronic band structure and DOS of Zr2SeC and Zr2SC are similar to that of M2SC (M = Hf, Nb).32,41
3.3 Mechanical properties
Some of the physical properties of solids, for example, the mechanical stability, bonding strength, deformation, failure mode, stiffness, anisotropic nature in bonding strength, etc., can be brought out by studying mechanical properties characterizing parameters that are subjected in this section. At first, the stiffness constants (Cij) have been calculated using the well-known strain–stress method.29,30,57–59 The stiffness constants, which are five in number as independent due to the hexagonal nature of Zr2SeC and presented in Table 2 together with those of other S-containing MAX phases Zr2SX (X = C, B). One of the prime importance of stiffness constants is the use of checking the mechanical stability of solids. Max Born60 proposed some conditions on the stiffness constants of solids for being mechanically stable, having a limitation that they are not enough to predict the stability for all crystal systems correctly. The limitation is overcome by Mouhat et al.61 and the stability conditions for a hexagonal systems becomes:| | |
C11 > 0, C11 > C12, C44 > 0, (C11 + C12)C33 − 2(C13)2 > 0.
| (2) |
Table 2 The elastic constants, Cij (GPa), bulk modulus, B (GPa), shear modulus, G (GPa), Young's modulus, Y (GPa), macro, Hmacro (GPa), micro hardness, Hmicro (GPa), Pugh ratio, G/B, Poisson ratio, ν and Cauchy pressure, CP (GPa) of Zr2SeC, together with those of M2SC (M = Zr, Hf, Nb)
| Phase |
C11 |
C12 |
C13 |
C33 |
C44 |
B |
G |
Y |
Hmacro |
Hmicro |
G/B |
ν |
CP |
Ref. |
| Zr2SeC |
260 |
97 |
96 |
293 |
128 |
154 |
100 |
247 |
14.85 |
17.79 |
0.65 |
0.23 |
−30 |
This study |
| Zr2SC |
295 |
89 |
102 |
315 |
138 |
166 |
115 |
280 |
17.89 |
21.57 |
0.69 |
0.22 |
−49 |
34 |
| Hf2SC |
311 |
97 |
121 |
327 |
149 |
181 |
120 |
295 |
17.35 |
21.72 |
0.66 |
0.23 |
−52 |
34 |
| Nb2SC |
316 |
108 |
151 |
325 |
124 |
197 |
105 |
267 |
11.58 |
15.84 |
0.53 |
0.27 |
−16 |
34 |
The Cij presented in Table 2 satisfy the above relations, and hence Zr2SeC is expected to be mechanically stable like M2SC (M = Zr, Hf, Nb). It should be noted here that Zr2SeC is already experimentally realized; thus, the question regarding stability is not expected at all. But, checking mechanical stability is necessary for practical application under load. The importance of Cij is not only limited to mechanical stability checking but also provides significant information. For example, the stiffness of solids along [100] and [001] directions are defined by C11 and C33, respectively, thus, comparatively low pressure is required to deform Zr2SeC along crystallographic a-axis than c-axis because of C11 < C33. The resistance to shear deformation is measured by the value of C44. It is evident from Table 2 that C44 < C33 and C11, indicating a low pressure required to shear deformation than axial deformation. Moreover, C44 is also a hardness predictor, in fact, directly related to the hardness compared to other elastic moduli.62 C44 of Zr2SeC is smaller than M2SC (M = Zr, Hf) but larger than Nb2SC, thus, the hardness of Zr2SeC is expected to be lower than M2SC (M = Zr, Hf) but higher than Nb2SC. Furthermore, the unequal values of C11 and C33 reveal the anisotropy in the bonding strength in the a and c-axis owing to the difference in the atomic arrangement in a and c-axes. The difference in the atomic arrangement is mainly responsible for the mechanical anisotropy that will be presented in the letter.
Bulk modulus (B) defines the property related to the incompressibility of the materials. The material's resistance to change its volume keeping shape unchanged under hydrostatic pressure is measured by its B. The resistance of solids to plastic deformation at constant volume is provided by its shear modulus (G). The stiffness of solids is measured by its Young's modulus (Y); the higher value of Y indicates stiffer solids and vice versa. These moduli are also important to realize the hardness, stiffness, brittleness/ductileness of solids. From these points of view, we have calculated the B and G using Hill's approximation63 based on the Voigt64 and Reuss65 models as follows:
where
BV = [2(
C11 +
C12) +
C33 + 4
C13]/9 and
BR =
C2/
M;
C2 = (
C11 +
C12)
C33 − 2
C132;
M =
C11 +
C12 + 2
C33 − 4
C13.
BV represents the upper limit of B (Voigt bulk modulus) and BR represents the lower limit of B (Reuss bulk modulus). Like B, average values of Voigt (GV) and Reuss (GR) were used to calculate G using the following equations:
where
GV = [
M + 12
C44 + 12
C66]/30 and
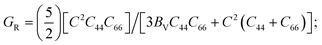 C66
C66 = (
C11 −
C12)/2.
The Young's modulus (Y) and Poisson's ratio (υ) were also calculated using their relationships with B and G:34
and
The lower values of B and G of Zr2SeC than those of Zr2SC revealed that low pressure is required to volume and plastic deformation for Zr2SeC than Zr2SC and lower value of Y for Zr2SeC than Zr2SC, indicating that Zr2SeC is less stiff than Zr2SC. The elastic moduli were further used to study the hardness of Zr2SeC using the equations:66,67
| | |
Hmacro = 2(k2G)0.585 − 3
| (7) |
(where Pugh's ratio,
k =
G/
B) and
| |
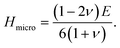 | (8) |
The study of the hardness parameter is beneficial for those materials which are used as structural components like MAX phases. The replacement of Se by S is enough to increase the M–A bonding strength that must improve the hardness of Zr2SC than Zr2SeC. The obtained values of Hmacro and Hmicro = 14.85 (17.89) and 17.79 (21.57) GPa for Zr2SeC (Zr2SC). Thus, the enhancement of hardness for Zr2SC is observed. Though the elastic moduli (B, G, Y) of Zr2SeC are smaller than that of Nb2SC but the hardness parameters of Zr2SeC are larger than those of Nb2SC owing to the larger value of C44 (ref. 62) (C44 of Zr2SeC and Nb2SC are 128 and 124 GPa, respectively). The elastic moduli and hardness parameters of Hf2SC are larger than those of Zr2SeC. The lowering of the bonds strength (Zr–C and Zr–Se bond) for Zr2SeC compared to Zr2SC (Zr–C and Zr–S bond) is responsible for the lowering of elastic moduli and hardness for the same. The bond length of Zr–C (2.339 Å) in Zr2SeC is higher than in Zr2SC (2.326 Å), and Zr–Se (2.776 Å) in Zr2SeC is higher than Zr–S in Zr2SC (2.687 Å) and hence the bonds (Zr–C, Zr–S) of Zr2SC are stronger than those of Zr2SeC (Zr–C, Zr–Se). Thus, lower hardness parameters for Zr2SeC compared to Zr2SC is expected as reflected from the hardness parameters presented in Table 2. To be more confirmed, we have calculated the Vickers hardness (Hv) using the Gou et al.68 formula that is based on the Mulliken bond population and geometrical averages of the bonds present within the crystal. The relevant formula can be found elsewhere.69 The calculated values of Hv for Zr2SeC and Zr2SC are 2.52 and 4.33 GPa, respectively. Again, the Vickers hardness of Zr2SeC is lower than that of Zr2SC as expected. The obtained elastic moduli and hardness are in good agreement with the DOS and CDM results.
3.4 Mechanical anisotropy
For hexagonal crystal, the elastic anisotropy is related to microcracks and anisotropic plastic deformation, which plays a vital role in understanding the mechanical stability of the material under service. The mechanical anisotropy in MAX phases is normally due to unequal elastic constants, i.e..C11 ≠ C33, which indicates that the mechanical properties in all crystallographic planes are not identical.70 Moreover, the knowledge of anisotropy offers the information necessary to enhance further the stability of solids for many applications.71 Therefore, it is necessary to study the anisotropic behavior of mechanical properties in Zr2SeC. For this purpose, the 2D and 3D visualization of Young's modulus, compressibility, shear modulus, and Poisson's ratio of Zr2SeC MAX phase is presented ESI [Fig. S1(a–d)†] in comparison with those of Zr2SC [Fig. S2(a–d)†] by using the open-source software packages ELATE and AnisoVis.72,73 The extent of anisotropy is estimated by the variable value of elastic properties in all directions. The 2D projections and 3D plots are perfectly circular and spherical for isotropic materials, and variation from circular/spherical shape demonstrates the degree of anisotropy of the corresponding elastic property. The obtained results for Zr2SeC and Zr2SC are tabulated in Table 3. The unit value of the anisotropic index indicates the identical mechanical properties in all directions where the value higher or lower than 1 (one) measures the degree of anisotropy. Table 3 confirms the anisotropic nature of the mechanical properties and Zr2SeC is more anisotropic compared to Zr2SC.
Table 3 The minimum and maximum values of Young's modulus, Y (GPa), linear compressibility, K (TPa−1), shear modulus, G (GPa), and Poisson's ratio, υ and their anisotropic indices, A of Zr2AC (A = Se, S)
| Phases |
Ymin |
Ymax |
AY |
Kmin |
Kmax |
AK |
Gmin |
Gmax |
AG |
υmin |
υmax |
Aυ |
Ref. |
| Zr2SeC |
208.93 |
277.50 |
1.32 |
1.913 |
2.288 |
1.19 |
81.071 |
127.59 |
1.57 |
0.078 |
0.343 |
4.394 |
This work |
| Zr2SC |
250.08 |
306.39 |
1.22 |
1.78 |
2.12 |
1.19 |
101.19 |
137.27 |
1.35 |
0.110 |
0.290 |
2.53 |
34 |
We have also investigated the different anisotropic factors for the {100}, {010} and {001} planes that are computed using the eqn (9)–(11), respectively and presented in Table 4:74
| |
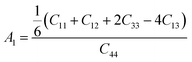 | (9) |
| |
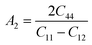 | (10) |
| |
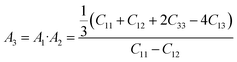 | (11) |
Table 4 The anisotropic factors, A1, A2, A3, Ba, Bc, kc/ka, and universal anisotropic index AU of Zr2AC (A = Se, S)
| Phase |
A1 |
A2 |
A3 |
kc/ka |
Ba |
Bc |
AU |
| Zr2SeC |
0.73 |
1.57 |
1.14 |
0.84 |
383 |
904 |
0.218 |
| Zr2SC |
0.73 |
1.34 |
0.98 |
0.85 |
412 |
955 |
0.113 |
Since the values of Ai's are not equal to 1 (one), thus Zr2SeC and Zr2SC are anisotropic because Ai = 1 implies the isotropic nature. The bulk modulus for a and c-direction are computed using the eqn (12) and (13), respectively:75
| |
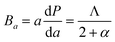 | (12) |
| |
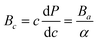 | (13) |
where
Λ = 2(
C11 +
C12) + 4
C13α +
C33α2 and
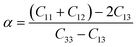
. The unequal values of
Ba and
Bc [
Table 4] indicates the anisotropic nature of Zr
2AC (A = Se, S). The linear compressibility (
k) along the
a and
c-axis are calculated by the equation:
76| |
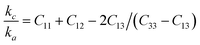 | (14) |
The obtained values of kc/ka are not equal to 1 (kc/ka = 1 for isotropic materials) and hence Zr2AC (A = Se, S) are anisotropic. Another important anisotropic factor, the universal anisotropic index AU is estimated by the following equation:77
| |
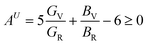 | (15) |
where
B and
G are obtained by Voigt and Reuss models. Since, the values of
AU are greater than zero, indicating the anisotropic nature of Zr
2AC (A = Se, S).
3.5 Thermal properties
The thermodynamic properties of the material at high temperatures and pressures are of scientific and technical significance, which help to predict the material's applications at elevated temperatures and pressure. Here we have investigated the thermodynamic properties of the newly synthesized Zr2SeC MAX phase in comparison with Zr2SC over the wide temperature (from 0 to 1600 K) and pressure (from 0 to 50 GPa) by using the quasi-harmonic Debye approximation.78,79 To disclose the important thermodynamic properties, the thermal expansion coefficient (TEC), heat capacity at constant volume (Cv), entropy (S), and Grüneisen parameter (γ) have been investigated in this temperature and pressure range. The quasi-harmonic Debye model remains valid in this given range of temperature and pressure and has been successfully used to calculate the thermodynamic properties of MAX phases.80,81
The temperature dependence of the thermal expansion coefficient (TEC) is displayed in Fig. 3. It is seen that the TEC increases rapidly with the increase in temperature up to 300 K. The increase in TEC of Zr2SeC and Zr2SC becomes less sensitive of temperature at the T ≥ 300 K. However, at the constant temperature, TEC decreases with the increase in pressure. The calculated value of TEC for Zr2SeC (3.88 × 10−5 K−1) < Zr2SC (4.03 × 10−5 K−1) at zero pressure and T = 300 K.
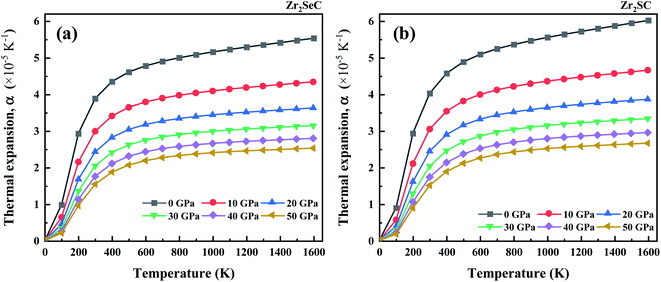 |
| | Fig. 3 The temperature effect on the thermal expansion coefficient of (a) Zr2SeC and (b) Zr2SC at different pressure. | |
The specific heat is significant to predict the density of states, band structure, and lattice vibrations. The effect of temperature on the specific heat at constant volume (Cv) for Zr2SeC and Zr2SC is given in Fig. 4. The heat capacity for both MAX phases satisfies the Debye T3 power-law at the low temperatures, which is mainly due to an exponential increase in the number of exciting phonon modes. At high temperatures, the specific heat curves become converged to get the classical Dulong-petit (DP) limit (Cv = 3nNAkB). The DP limit for Zr2SeC and Zr2SC is found to be at ∼197.12 J mol−1 K−1 and ∼196.80 J mol−1 K−1. The heat capacity decreases linearly with the increase in pressure. The calculated value of Cv for Zr2SeC (156.46 J mol−1 K−1) is greater than that of Zr2SC (151.32 J mol−1 K−1) at zero pressure, and T = 300 K. It is also observed in Fig. 4 that the heat capacity of Zr2SeC reached the DP limit at a lower temperature compared to Zr2SC. The values of ΘD (Table 5) and Hv of Zr2SeC are smaller than those of Zr2SC; thus, it is expected to reach the DP limit at a lower temperature for Zr2SeC than that of Zr2SC. A similar result is also found for other solids.82
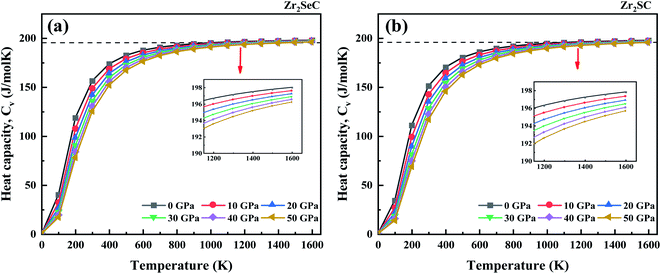 |
| | Fig. 4 The temperature on the heat capacity at constant volume, Cv of (a) Zr2SeC and (b) Zr2SC at different pressure. | |
Table 5 The calculated volume (V), Debye temperature (ΘD), melting temperature (Tm), TEC (α), Grüneisen parameter (γ), minimum thermal conductivity (Kmin), specific heat (Cv) and entropy (S) of Zr2SeC together with those of M2SC (M = Zr, Hf, Nb) MAX phases at temperature of 300 K and pressure of 0 Pa
| Phases |
V (Å3) |
ΘD (K) |
Tm (K) |
α (×10−5 K−1) |
γ |
Kmin (W m−1 K−1) |
Cv (J mol−1 K−1) |
S (J mol−1 K−1) |
| Ref. 34. |
| Zr2SeC |
129.7 |
679 |
1571 |
3.88 |
3.11 |
1.3 |
156.5 |
126.5 |
| Zr2SC |
126.3 |
727 |
1712a |
4.03 |
3.12 |
1.12a |
151.3 |
115.9 |
| Hf2SC |
130.1 |
598 |
1778a |
3.59 |
3.11 |
0.85a |
164.8 |
146.7 |
| Nb2SC |
113.7 |
821 |
1790a |
3.12 |
3.12 |
1.09a |
141.0 |
98.1 |
The Grüneisen parameter (γ) for Zr2SeC and Zr2SC as a function of temperature is depicted in Fig. 5. It is an important parameter that is used to calculate the thermal states and can also quantify the anharmonic effect. The Grüneisen parameter for Zr2SeC and Zr2SC increases with the increase in temperature and decreases with the increase in pressure. The effect of temperature on γ for Zr2SeC and Zr2SC at higher pressure is less significant. The trend of γ at 50 GPa is nearly constant with the increase in temperature. The calculated value of γ for Zr2SeC (3.11) is smaller than that of Zr2SC (3.12) at zero pressure and T = 300 K.
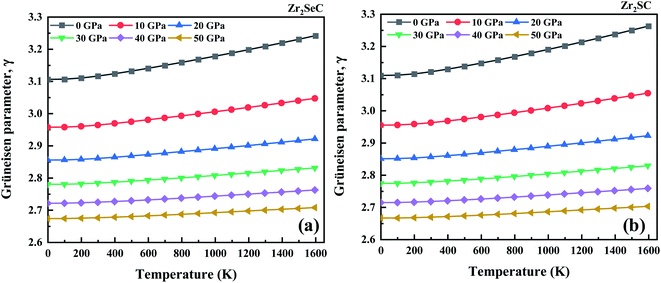 |
| | Fig. 5 The effect of temperature and pressure on the Grüneisen parameter of (a and c) Zr2SeC and (b and d) Zr2SC, respectively. | |
We have further calculated pressure and temperature dependence of volume (V), Gibbs free energy (G) Debye temperature (ΘD) and entropy (S) of Zr2SeC and the results are presented in ESI (see ESI Fig. S3–S6†). Moreover, we have also calculated the parameters discussed above for M2SC (M = Zr, Hf, Nb) and the results are presented in ESI† (to save the journal space) together with those of Zr2SeC (see Fig. S3–S9†).
Furthermore, we have also calculated three important parameters regarding the application at high temperatures, such as the Debye temperature (ΘD), minimum thermal conductivity (Kmin) and melting temperature (Tm) at room temperature. The ΘD is calculated using the quasi-harmonic Debye model. It is found that the ΘD for Zr2SeC (679 K) is smaller than that of Zr2SC (727 K) which is good agreement with statement that Debye temperature is higher for harder solids and vice versa. The thermal conductivity of the materials which are used at high temperatures (e.g., ceramics) reduces to the smallest value (Kmin) at high temperatures. The Kmin of Zr2SeC is computed by the equation:83
| |
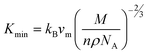 | (16) |
where
kB,
vm,
NA and
ρ are Boltzmann constant, average phonon velocity, Avogadro's number and density of crystal, respectively. The obtained value of
Kmin is 1.3 (W m
−1 K
−1) for Zr
2SeC which is comparable with those of M
2SC (M = Zr, Hf, Nb)
34 as well as other MAX phase materials.
7,29,30 The thermal conductivity of Zr
2SeC (18.30 W m
−1 K
−1) is also lower than that of Zr
2SC (21.10 W m
−1 K
−1) as measured by Chen
et al.43 They have also measured the electronic contribution to the electrical conductivity, which is important for predicting high-temperature application. Accordingly, the electronic contribution to the thermal conductivity is 25% for Zr
2SeC and 19.3% for Zr
2SC; thermal conductivity is dominated by phonon contribution. Thus,
Kmin of Zr
2SeC and Zr
2SC is also dominated by phonon contribution and the low values of
Kmin could be useful to predict its suitability as TBC material. Elastic constants can be used to calculate the melting temperature (
Tm) of Zr
2SeC and Zr
2SC by using the expression:
84| | |
Tm = 3C11 + 1.5C33 + 354
| (17) |
It is found that the melting temperature of Zr2SeC (1571 K) is lower than that of Zr2SC (1712 K), Hf2SC (1778 K) and Nb2SC (1790 K),34 and is comparable with the melting point of Zr2GaC (1481 K), Hf2GaC (1648 K), and Hf3SnC2 (1773 K).59,70 Recently, the research has been focused to prove the MAX phases as TBC materials.47,85 The calculated values of the parameters related to the TBC applications for Zr2SeC are: ΘD ∼ 679 K, TEC ∼ 2.81 × 10−6 K−1, Kmin – 1.3 W m−1 K−1, and Tm ∼ 1571 K. The values for a promising TBC material, Y4Al2O9 are: ΘD ∼ 564 K, TEC ∼ 7.51 × 10−6 K−1, Kmin – 1.13 W m−1 K−1, and Tm ∼ 2020 K.86,87 A comparison of these values indicates that the Zr2SeC MAX phase can be used as a TBC material for high-temperature applications. However, all the thermal properties characterizing parameters of Zr2SeC calculated at ambient condition are presented in Table 5 together with those of M2SC (M = Zr, Hf, Nb) for comparison.
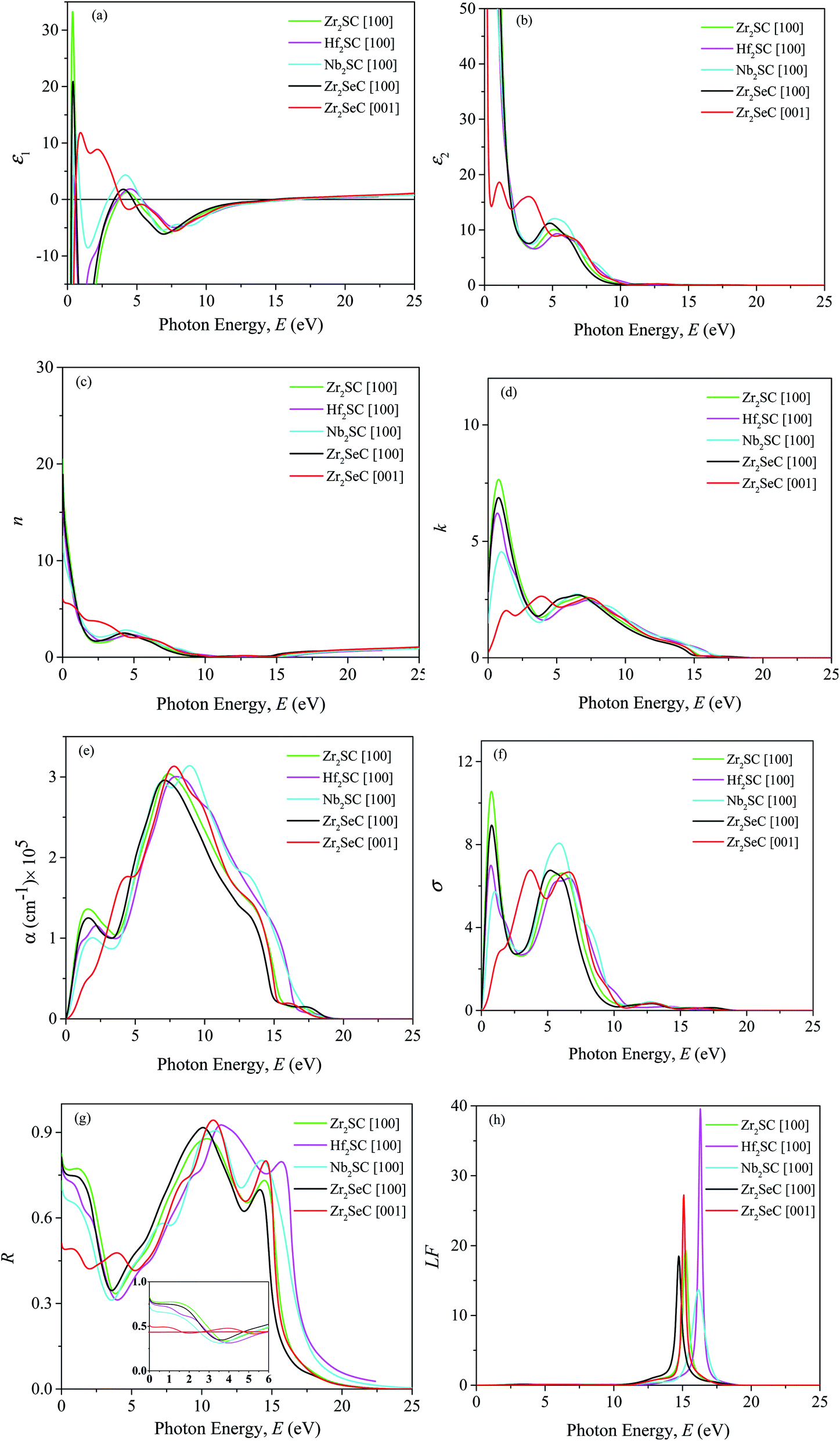
3.6 Optical properties
The optical properties of the Zr2SeC compound are calculated and presented in Fig. 6 for [100] and [001] polarization directions up to 25 eV incident phonon energy. These two polarization directions represent the related electric field which is in perpendicular and parallel directions to the c-axis of unit cell structure. As confirmed that the titled compound is metallic; thus, a correction (intra-band) is needed to the low energy region of the spectrum. This correction is done by setting up a plasma frequency of 3 eV and damping of 0.5 eV. Besides, Gaussian smearing of 0.5 eV was used with the intention that the k-points be more effective on the Fermi level. The optical constants of Zr2SeC are compared with those of M2SC (M = Zr, Hf, Nb) for [100] direction only.
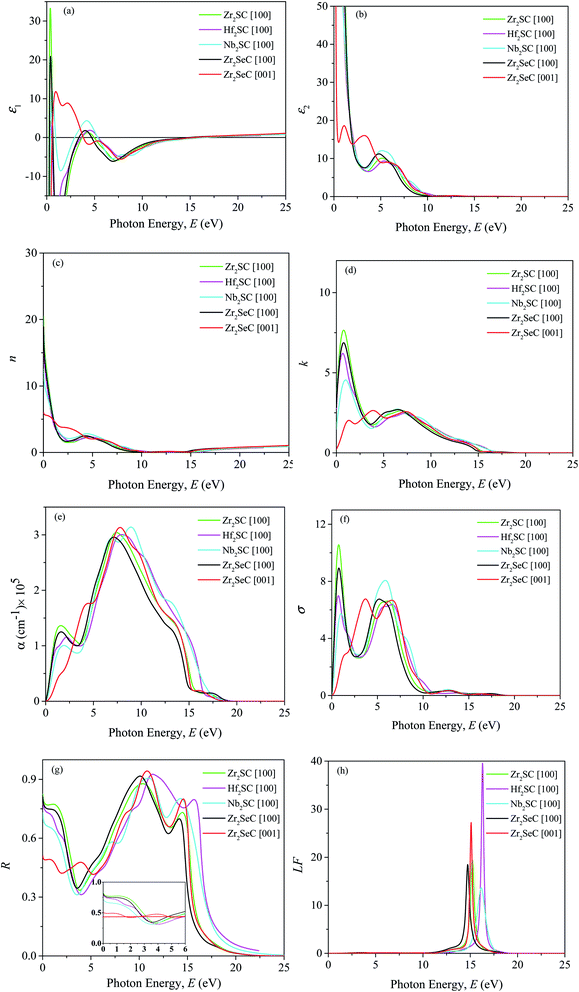 |
| | Fig. 6 (a) Real part ε1 and (b) imaginary part ε2 of dielectric function ε (ω), (c) refractive index n, (d) extinction coefficient k, (e) absorption coefficient α, (f) photoconductivity σ, (g) reflectivity R, and (h) loss function LF of Zr2SeC together with those of M2SC (M = Zr, Hf, Nb) MAX phases as a function of photon energy. | |
Fig. 6 shows the (a) real and (b) imaginary part of the complex dielectric function of Zr2SeC, which is very important and used to explain the material's response to the electric field.47 As seen, the high negative static value of the real part and high positive static value of the imaginary part of the dielectric function indicates the metallic nature of Zr2SeC; is consistent with the band structure result. The ε1(ω) and ε2(ω) of Zr2SeC are too much similar to those of M2SC (M = Zr, Hf, Nb). Strong anisotropy is observed in the low energy part, which tends to be isotropic in the high-energy region. The peaks in the ε2(ω) are the results of electrons excitation, such as intra-band transitions (within M-d states) and inter-band transitions due to absorption of photon incident on it. The first two peaks for [001] direction of Zr2SeC are due to electron transition within Zr-d states. The third peak for [001] and the only peak for [100] direction assumed to be due to the inter-band transition of electrons. Approaching of zero by ε1(ω) from below [at 14.91 and 15. 25 eV for [100] and [001] directions, respectively] and ε2(ω) from above [at 19 eV for both directions] is another indication for the metallic nature of Zr2SeC. The energy at which the ε1(ω) touches zero from below are 15.42, 16.5, 16.44 eV for M2SC (M = Zr, Hf, Nb), respectively. Fig. 6(c) shows the refractive index n(ω) of Zr2SeC. The value of static n(0) is 19 and 6 for [100] and [001] directions, respectively. As the value of n(ω) is an indication for the light velocity propagating the materials, thus, lower value of n(ω) is good for optical devices such as waveguides. The value of n(ω) is 6 for [001] direction implies the use of Zr2SeC in parallel to the field direction (parallel to the c-axis) is better than that of [100] direction (perpendicular to the c-axis). However, the velocity is reduced by the interaction with electrons during propagation through the Zr2SeC, as confirmed from values greater than the unit value. Fig. 6(d) shows the imaginary part of the refractive index, known as extinction coefficient, k(ω), also used to measure the absorption capability of solids. The spectra of k(ω) follow the trend of ε2(ω) for each of the MAX phases presented here. The static value of k(ω) is 2.8 and 0.23 for [100] and [001] directions, respectively. The k(ω) decreases to zero at ∼16 eV, indicating that the intrinsic oscillation frequency of Zr2SeC is 16.0 eV. The higher value of k(ω) than n(ω) indicates the energy range wherein light cannot propagate, and Zr2SeC exhibit the behavior belongs to the reflective metal.88,89
Fig. 6(e) shows the absorption coefficient (α) of Zr2SeC [100] and [001] for two polarization directions along with that of M2SC (M = Zr, Hf, Nb) for [100] direction only. The metallic behavior of Zr2SeC and M2SC (M = Zr, Hf, Nb) is reflected from the starting of the spectra, started at zero photon energy owing to no existence of the bandgap. For [100] directions, α increases with the rise in photon energy, exhibits a peak in the range of 1.5–1.9 eV, and then decreases slightly 3.3 to 3.6 eV, finally reaches maximum values at 7 to 9 eV. The energy range wherein α attains a value of greater than 105 cm−1 is the zone of strong absorption. The strong absorption zone of Zr2SeC is 3.5 to 14 eV and 2.8 to 14.4 eV for [100] and [001], respectively. The absorption coefficient of Zr2SeC is similar to that of M2SC (M = Zr, Hf, Nb) for [100] direction. The photoconductivity (σ) of Zr2SeC is shown in Fig. 6(f), in which the curve is noted to be started in association with the photon incident, which implies the zero bandgap of the titled compound. The σ is found to be similar to that of k(ω) in peaks position as σ is the results of photon absorption that is exhibited by k(ω). Strong anisotropy is also observed in the low energy region that tends to be lowered in the high region.
Fig. 6(g) demonstrates the reflectivity (R) Zr2SeC that is similar to that of M2SC (M = Zr, Hf, Nb) for [100] direction. The static values of R are 0.81 and 0.5 for [100] and [001] directions, respectively. The inset of Fig. 6(g) shows the R up to 6 eV in which a horizontal redline is drawn at 0.44 (44%) of R to demonstrate the suitability for use as a cover material to diminish solar heating. It was suggested by Li et al.48,90 that the MAX phase with reflectivity greater than 44% is suitable for use cover materials for the same mentioned before. As seen, R decreases to lower than 0.44 at 1.7 and 2.8 eV for [001] and [100] directions, respectively. However, R of Zr2SeC decreases to lower than 0.44 at slightly lower energy than that of M2SC (M = Zr, Hf), but the lowest value (34%) is higher than that of M2SC (M = Zr, Hf, Nb). R increases to a maximum of 0.91 at 10 eV, followed by another peak of 0.7 at 14.1 eV and finally declined sharply towards zero, and the titled compound becomes transparent for the incident light. The Zr2SeC shows reflective behavior in the energy range of 7.5 to 14 eV in which the refractive index is very low, and the reflectivity is greater than 70%. A similar reflectivity is also shown by M2SC (M = Zr, Hf, Nb) MAX phases. Fig. 6(h) shows the loss function of Zr2SeC that demonstrates how fast electrons losing their energy when traversing the Zr2SeC compound. A peak corresponding to the plasma frequency (ωp) is observed at the energy where ε1(ω) and ε2(ω) approaches zero from below and above respectively, and reflectivity shows trailing edges. The ωp of Zr2SeC are 14.7 and 15.1 eV for [001] and [100] directions, respectively. The ωp of M2SC (M = Zr, Hf, Nb) MAX phases are slightly higher than that of Zr2SeC for [100] direction. This plasma frequency defined a critical value at which the material changes its behavior from metallic to transparent dielectric.
4. Conclusions
A comparative study of the structural, electronic, mechanical, thermal, and optical properties of synthesized MAX phase Zr2SeC with prior known MAX phase Zr2SC has been performed by density functional theory for the first time. The obtained lattice constants, volume, and atomic positions are noticed to be agreed with experimental and theoretical results. The metallic behavior has been confirmed from the band structure and density of states (DOS). The less energy dispersion along the c-direction confirmed the anisotropic behavior of electrical conductivity, and partial DOS revealed the dominant role of Zr-d states to the electrical conductivity. Shifting of the peak in the DOS owing to the replacement of Se by S and difference in charge density mapping (CDM) revealed that the strength of bonds present in Zr2SeC is lower than the bonds in Zr2SC. The mechanical stability of the synthesized phase is further confirmed by the stiffness constants. The stiffness constants, elastic moduli, and hardness parameters of Zr2SeC are found lower than those of Zr2SC. The lowering of the parameters is explained based on the bond populations, and bond lengths present within them in an association with the DOS and CDM results. Both the directional projections of the elastic moduli and anisotropy indices confirm the anisotropic behavior of Zr2SeC and Zr2SC; Zr2SeC is more anisotropic than Zr2SC. The temperature and pressure-dependent properties exhibit the expected variation for the same. The value of ΘD, Kmin, Tm, and TEC of Zr2SeC are lower than those of Zr2SC, Cv of Zr2SeC reach classical DP limit at a lower temperature than that of Zr2SC; is consistent with the parameters used to characterize mechanical properties. A comparison of the values of ΘD, Kmin, Tm, and TEC for Zr2SeC with those of a promising TBC material (Y4Al2O9)87 reveals its possibility to be used as TBC material. The real and imaginary parts of the dielectric constant and the absorption and photoconductivity curves agree with the band structure results and confirm the metallic nature of Zr2SeC. The possible relevance of Zr2SeC for use as shielding material to diminish solar heating has been confirmed from the analysis of the studied optical properties. Like electrical conductivity and mechanical properties, the optical properties of Zr2SeC also exhibit anisotropic behavior.
Data availability
The data sets generated and/or analyzed in this study are available from the corresponding author on reasonable request.
Author contributions
M. A. A. designed and supervised the project, performed the calculation and analysis of a part of the results, and wrote a part of the draft manuscript. M. W. Q. performed the calculation and analysis of a part of the results and wrote a part of the draft manuscript. Both authors finalized the draft of the manuscript.
Conflicts of interest
There are no conflicts to declare.
References
- M. Sokol, V. Natu, S. Kota and M. W. Barsoum, On the Chemical Diversity of the MAX Phases, Trends Chem., 2019, 1, 210–223, DOI:10.1016/j.trechm.2019.02.016.
- M. W. Barsoum, MAX phases: Properties of machinable ternary carbides and nitrides, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013. DOI:10.1002/9783527654581.
- M. R. Khatun, M. A. Ali, F. Parvin and A. K. M. A. Islam, Elastic, thermodynamic and optical behavior of V2AC (A = Al, Ga) MAX phases, Results Phys., 2017, 7, 3634–3639, DOI:10.1016/j.rinp.2017.09.043.
- M. A. Ali, M. T. Nasir, M. R. Khatun, A. K. M. A. Islam and S. H. Naqib, An ab initio investigation of vibrational, thermodynamic, and optical properties of Sc2AlC MAX compound, Chin. Phys. B, 2016, 25, 103102, DOI:10.1088/1674-1056/25/10/103102.
- M. W. Barsoum and T. El-Raghy, Synthesis and Characterization of a Remarkable Ceramic: Ti3SiC2, J. Am. Ceram. Soc., 1996, 79, 1953–1956, DOI:10.1111/j.1151-2916.1996.tb08018.x.
- G. Surucu, Investigation of structural, electronic, anisotropic elastic, and lattice dynamical properties of MAX phases borides: An Ab-initio study on hypothetical M2AB (M = Ti, Zr, Hf; A = Al, Ga, In) compounds, Mater. Chem. Phys., 2018, 203, 106–117, DOI:10.1016/j.matchemphys.2017.09.050.
- M. A. Ali, M. M. Hossain, M. A. Hossain, M. T. Nasir, M. M. Uddin, M. Z. Hasan, A. K. M. A. Islam and S. H. Naqib, Recently synthesized (Zr1-xTix)2AlC (0 ≤ x ≤ 1) solid solutions: Theoretical study of the effects of M mixing on physical properties, J. Alloys Compd., 2018, 743, 146–154, DOI:10.1016/j.jallcom.2018.01.396.
- M. A. Ali, M. M. Hossain, N. Jahan, A. K. M. A. Islam and S. H. Naqib, Newly synthesized Zr2AlC, Zr2(Al0.58Bi0.42)C, Zr2(Al0.2Sn0.8)C, and Zr2(Al0.3Sb0.7)C MAX phases: A DFT based first-principles study, Comput. Mater. Sci., 2017, 131, 139–145, DOI:10.1016/j.commatsci.2017.01.048.
- I. R. Shein and A. L. Ivanovskii, Elastic properties of superconducting MAX phases from first-principles calculations, Phys. Status Solidi, 2011, 248, 228–232, DOI:10.1002/pssb.201046163.
- Y. Bai, N. Srikanth, C. K. Chua and K. Zhou, Density Functional Theory Study of Mn+1AXn Phases : A Review Density Functional Theory Study of Mn+1AXn Phases : A Review, Crit. Rev. Solid State Mater. Sci., 2017, 1–52, DOI:10.1080/10408436.2017.1370577.
- M. Barsoum and T. El-Raghy, The MAX Phases: Unique New Carbide and Nitride Materials, Am. Sci., 2001, 89, 334, DOI:10.1511/2001.4.334.
- B. Tunca, T. Lapauw, O. M. Karakulina, M. Batuk, T. Cabioc’h, J. Hadermann, R. Delville, K. Lambrinou and J. Vleugels, Synthesis of MAX Phases in the Zr-Ti-Al-C System, Inorg. Chem., 2017, 56, 3489–3498, DOI:10.1021/acs.inorgchem.6b03057.
- T. Lapauw, B. Tunca, D. Potashnikov, A. Pesach, O. Ozeri, J. Vleugels and K. Lambrinou, The double solid solution (Zr,Nb)2(Al, Sn)C MAX phase: a steric stability approach, Sci. Rep., 2018, 8, 12801, DOI:10.1038/s41598-018-31271-2.
- B. Tunca, T. Lapauw, R. Delville, D. R. Neuville, L. Hennet, D. Thiaudière, T. Ouisse, J. Hadermann, J. Vleugels and K. Lambrinou, Synthesis and Characterization of Double Solid Solution (Zr,Ti)2(Al,Sn)C MAX Phase Ceramics, Inorg. Chem., 2019, 58, 6669–6683, DOI:10.1021/acs.inorgchem.9b00065.
- M. Griseri, B. Tunca, S. Huang, M. Dahlqvist, J. Rosén, J. Lu, P. O. Å. Persson, L. Popescu, J. Vleugels and K. Lambrinou, Ta-based 413 and 211 MAX phase solid solutions with Hf and Nb, J. Eur. Ceram. Soc., 2020, 40, 1829–1838, DOI:10.1016/j.jeurceramsoc.2019.12.052.
- M. A. Ali, Newly Synthesized Ta based MAX phase (Ta1- xHfx)4AlC3 and (Ta1- xHfx)4Al0.5 Sn0.5C3 (0 ≤ x ≤ 0.25) Solid Solutions: Unravelling the Mechanical, Electronic and Thermodynamic Properties, Phys. Status Solidi B, 2021, 258(4), 2000307, DOI:10.1002/pssb.202000307.
- E. Z. Solvas, S. G. Christopoulos, N. Ni, D. C. Par, D. Horlait, M. E. Fitzpatrick, A. Chroneos and W. E. Lee, Experimental synthesis and density functional theory investigation of radiation tolerance of Zr3(Al1−xSix)C2 MAX phases, J. Am. Ceram. Soc., 2017, 100, 1377–1387, DOI:10.1111/jace.14742.
- R. Pan, J. Zhu and Y. Liu, Synthesis, microstructure and properties of (Ti1−x,Mox)2AlC phases, Mater. Sci. Technol., 2018, 34, 1064–1069, DOI:10.1080/02670836.2017.1419614.
- C. Zuo and C. Zhong, Screen the elastic and thermodynamic properties of MAX solid solution using DFT procedure: Case study on (Ti1-xVx)2AlC, Mater. Chem. Phys., 2020, 123059, DOI:10.1016/j.matchemphys.2020.123059.
- M. A. Ali and S. H. Naqib, Recently synthesized (Ti1−xMox)2AlC (0 ≤ x ≤ 0.20) solid solutions: deciphering the structural, electronic, mechanical and thermodynamic properties via ab initio simulations, RSC Adv., 2020, 10, 31535–31546, 10.1039/D0RA06435A.
- M. A. Ali, M. R. Khatun, N. Jahan and M. M. Hossain, Comparative study of Mo2Ga2C with superconducting MAX phase Mo2GaC: First-principles calculations, Chin. Phys. B, 2017, 26, 033102, DOI:10.1088/1674-1056/26/3/033102.
- C. Hu, C.-C. Lai, Q. Tao, J. Lu, J. Halim, L. Sun, J. Zhang, J. Yang, B. Anasori, J. Wang, Y. Sakka, L. Hultman, P. Eklund, J. Rosen and M. W. Barsoum, Mo2Ga2C: a new ternary nanolaminated carbide, Chem. Commun., 2015, 51, 6560–6563, 10.1039/C5CC00980D.
- H. He, S. Jin, G. Fan, L. Wang, Q. Hu and A. Zhou, Synthesis mechanisms and thermal stability of ternary carbide Mo2Ga2C, Ceram. Int., 2018, 44, 22289–22296, DOI:10.1016/j.ceramint.2018.08.353.
- H. Chen, D. Yang, Q. Zhang, S. Jin, L. Guo, J. Deng, X. Li and X. Chen, A Series of MAX Phases with MA-Triangular-Prism Bilayers and Elastic Properties, Angew. Chem., Int. Ed., 2019, 58, 4576–4580, DOI:10.1002/anie.201814128.
- Q. Tao, J. Lu, M. Dahlqvist, A. Mockute, S. Calder, A. Petruhins, R. Meshkian, O. Rivin, D. Potashnikov, E. N. Caspi, H. Shaked, A. Hoser, C. Opagiste, R.-M. Galera, R. Salikhov, U. Wiedwald, C. Ritter, A. R. Wildes, B. Johansson, L. Hultman, M. Farle, M. W. Barsoum and J. Rosen, Atomically Layered and Ordered Rare-Earth i -MAX Phases: A New Class of Magnetic Quaternary Compounds, Chem. Mater., 2019, 31, 2476–2485, DOI:10.1021/acs.chemmater.8b05298.
- L. Chen, M. Dahlqvist, T. Lapauw, B. Tunca, F. Wang, J. Lu, R. Meshkian, K. Lambrinou, B. Blanpain, J. Vleugels and J. Rosen, Theoretical Prediction and Synthesis of (Cr2/3Zr1/3)2AlC i -MAX Phase, Inorg. Chem., 2018, 57, 6237–6244, DOI:10.1021/acs.inorgchem.8b00021.
- N. Miao, J. Wang, Y. Gong, J. Wu, H. Niu, S. Wang, K. Li, A. R. Oganov, T. Tada and H. Hosono, Computational Prediction of Boron-Based MAX Phases and MXene Derivatives, Chem. Mater., 2020, 32, 6947–6957, DOI:10.1021/acs.chemmater.0c02139.
- J. Wang, T. N. Ye, Y. Gong, J. Wu, N. Miao, T. Tada and H. Hosono, Discovery of hexagonal ternary phase Ti2InB2 and its evolution to layered boride TiB, Nat. Commun., 2019, 10, 1–8, DOI:10.1038/s41467-019-10297-8.
- M. A. Ali, M. M. Hossain, M. M. Uddin, A. K. M. A. Islam, D. Jana and S. H. Naqib, DFT insights into new B-containing 212 MAX phases: Hf2AB2 (A = In, Sn), J. Alloys Compd., 2021, 860, 158408, DOI:10.1016/j.jallcom.2020.158408.
- M. A. Ali, M. M. Hossain, A. K. M. A. Islam and S. H. Naqib, Ternary boride Hf3PB4: Insights into the physical properties of the hardest possible boride MAX phase, J. Alloys Compd., 2021, 857, 158264, DOI:10.1016/j.jallcom.2020.158264.
- P. Chakraborty, A. Chakrabarty, A. Dutta and T. Saha-Dasgupta, Soft MAX phases with boron substitution: A computational prediction, Phys. Rev. Mater., 2018, 2, 103605, DOI:10.1103/PhysRevMaterials.2.103605.
- T. Rackl, L. Eisenburger, R. Niklaus and D. Johrendt, Syntheses and physical properties of the MAX phase boride Nb2SB and the solid solutions Nb2SBxC1-x(x=0-1), Phys. Rev. Mater., 2019, 3, 054001, DOI:10.1103/PhysRevMaterials.3.054001.
- T. Rackl and D. Johrendt, The MAX phase borides Zr2SB and Hf2SB, Solid State Sci, 2020, 106, 106316, DOI:10.1016/j.solidstatesciences.2020.106316.
- M. A. Ali, M. M. Hossain, M. M. Uddin, M. A. Hossain, A. K. M. A. Islam and S. H. Naqib, Physical properties of new MAX phase borides M2SB (M = Zr, Hf and Nb) in comparison with conventional MAX phase carbides M2SC (M = Zr, Hf and Nb): Comprehensive insights, J. Mater. Res. Technol., 2021, 11, 1000–1018, DOI:10.1016/j.jmrt.2021.01.068.
- M. Khazaei, M. Arai, T. Sasaki, M. Estili and Y. Sakka, Trends in electronic structures and structural properties of MAX phases: a first-principles study on M2AlC (M = Sc, Ti, Cr, Zr, Nb, Mo, Hf, or Ta), M2AlN, and hypothetical M2AlB phases, J. Phys.: Condens. Matter, 2014, 26, 505503, DOI:10.1088/0953-8984/26/50/505503.
- G. Surucu, B. Yildiz, A. Erkisi, X. Wang and O. Surucu, The investigation of electronic, anisotropic elastic and lattice dynamical properties of MAB phase nanolaminated ternary borides: M2AlB2 (M=Mn, Fe and Co) under spin effects, J. Alloys Compd., 2020, 838, 155436, DOI:10.1016/j.jallcom.2020.155436.
- A. Gencer and G. Surucu, Electronic and lattice dynamical properties of Ti 2 SiB MAX phase, Mater. Res. Express., 2018, 5, 076303, DOI:10.1088/2053-1591/aace7f.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Two-Dimensional Nanocrystals: Two-Dimensional Nanocrystals Produced by Exfoliation of Ti3AlC2., Adv. Mater., 2011, 23, 4248–4253, DOI:10.1002/adma.201190147.
- R. Meshkian, L. Å. Näslund, J. Halim, J. Lu, M. W. Barsoum and J. Rosen, Synthesis of two-dimensional molybdenum carbide, Mo2C, from the gallium based atomic laminate Mo2Ga2C, Scr. Mater., 2015, 108, 147–150, DOI:10.1016/j.scriptamat.2015.07.003.
- S. Amini, M. W. Barsoum and T. El-Raghy, Synthesis and mechanical properties of fully dense Ti2SC, J. Am. Ceram. Soc., 2007, 90(12), 3953–3958, DOI:10.1111/j.1551-2916.2007.01999.x.
- A. Bouhemadou and R. Khenata, Structural, electronic and elastic properties of M2SC (M=Ti, Zr, Hf) compounds, Phys. Lett. A., 2008, 372, 6448–6452, DOI:10.1016/j.physleta.2008.08.066.
- M. F. Cover, O. Warschkow, M. M. M. Bilek and D. R. McKenzie, A comprehensive survey of M2AX phase elastic properties, J. Phys.: Condens. Matter, 2009, 21, 305403, DOI:10.1088/0953-8984/21/30/305403.
- K. Chen, X. Bai, X. Mu, P. Yan, N. Qiu, Y. Li, J. Zhou, Y. Song, Y. Zhang, S. Du, Z. Chai and Q. Huang, MAX Phase Zr2SeC and Its Thermal Conduction Behavior, 2021, http://arxiv.org/abs/2102.02560 Search PubMed.
- W.-K. Pang and I. M. Low, Understanding and improving the thermal stability of layered ternary carbides and nitrides, Adv. Ceram. Matrix Compos., Elsevier, 2018, pp. 429–460, DOI:10.1016/B978-0-08-102166-8.00018-9.
- Y. Fu, B. Wang, Y. Teng, X. Zhu, X. Feng, M. Yan, P. Korzhavyi and W. Sun, The role of group III, IV elements in Nb4AC3 MAX phases (A = Al, Si, Ga, Ge) and the unusual anisotropic behavior of the electronic and optical properties, Phys. Chem. Chem. Phys., 2017, 19, 15471–15483, 10.1039/C7CP01375B.
- M. M. Ali, M. A. Hadi, I. Ahmed, A. F. M. Y. Haider and A. K. M. Islam, Physical properties of a novel boron-based ternary compound Ti2InB2, Mater. Today Commun., 2020, 25, 101600, DOI:10.1016/j.mtcomm.2020.101600.
- M. A. Hadi, M. Dahlqvist, S.-R. G. Christopoulos, S. H. Naqib, A. Chroneos and A. K. M. A. Islam, Chemically stable new MAX phase V2SnC: a damage and radiation tolerant TBC material, RSC Adv., 2020, 10, 43783–43798, 10.1039/D0RA07730E.
- S. Li, R. Ahuja, M. W. Barsoum, P. Jena and B. Johansson, Optical properties of Ti3SiC2 and Ti4AlN3, Appl. Phys. Lett., 2008, 92, 221907, DOI:10.1063/1.2938862.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, First-principles simulation: ideas, illustrations and the CASTEP code, J. Phys.: Condens. Matter, 2002, 14, 2717–2744, DOI:10.1088/0953-8984/14/11/301.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, First principles methods using CASTEP, Z. Kristallogr.–Cryst. Mater., 2005, 220, 567–570, DOI:10.1524/zkri.220.5.567.65075.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192, DOI:10.1103/PhysRevB.13.5188.
- T. H. Fischer and J. Almlof, General methods for geometry and wave function optimization, J. Phys. Chem., 1992, 96, 9768–9774, DOI:10.1021/j100203a036.
- R. John and B. Merlin, Optical properties of graphene, silicene, germanene, and stanene from IR to far UV – A first principles study, J. Phys. Chem. Solids., 2017, 110, 307–315, DOI:10.1016/j.jpcs.2017.06.026.
- Y. Zhou and Z. Sun, Electronic structure and bonding properties of layered machinable and ceramics, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 12570–12573, DOI:10.1103/PhysRevB.61.12570.
- J. Wang and Y. Zhou, Dependence of elastic stiffness on electronic band structure of nanolaminate M2AlC (M = Ti, V, Nb, and Cr) ceramics, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 214111, DOI:10.1103/PhysRevB.69.214111.
- Y. X. Wang, Z. X. Yan, W. Liu and G. L. Zhou, Structure stability, mechanical properties and thermal conductivity of the new hexagonal ternary phase Ti2InB2 under pressure, Philos. Mag., 2020, 100, 2054–2067, DOI:10.1080/14786435.2020.1754485.
- Y. Medkour, A. Bouhemadou and A. Roumili, Structural and electronic properties of M2InC (M = Ti, Zr, and Hf), Solid State Commun., 2008, 148, 459–463, DOI:10.1016/j.ssc.2008.09.006.
- M. W. Qureshi, X. Ma, G. Tang and R. Paudel, Ab initio predictions of structure and physical properties of the Zr2GaC and Hf2GaC MAX- phases under pressure, Sci. Rep., 2021, 11, 1–23, DOI:10.1038/s41598-021-82402-1.
- M. Born, On the stability of crystal lattices. I, Math. Proc. Cambridge Philos. Soc., 1940, 36, 160–172, DOI:10.1017/S0305004100017138.
- F. Mouhat and F.-X. Coudert, Necessary and sufficient elastic stability conditions in various crystal systems, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104, DOI:10.1103/PhysRevB.90.224104.
- S. Jhi, J. Ihm and S. G. Louie, Electronic mechanism of hardness enhancement in transition-metal carbonitrides, Nature, 1999, 399, 132–134 CrossRef CAS.
- R. Hill, The Elastic Behaviour of a Crystalline Aggregate, Proc. Phys. Soc., London, Sect. A, 1952, 65, 349–354, DOI:10.1088/0370-1298/65/5/307.
- W. Voigt, Lehrbuch der Kristallphysik, Vieweg+Teubner Verlag, Wiesbaden, 1966. DOI:10.1007/978-3-663-15884-4.
- A. Reuss, Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle, Z. Angew. Math. Mech., 1929, 9, 49–58, DOI:10.1002/zamm.19290090104.
- X.-Q. Chen, H. Niu, D. Li and Y. Li, Modeling hardness of polycrystalline materials and bulk metallic glasses, Intermetallics, 2011, 19, 1275–1281, DOI:10.1016/j.intermet.2011.03.026.
- A. Bouhemadou, First-principles study of structural, electronic and elastic properties of Nb4AlC3, Brazilian J. Phys., 2010, 40, 52–57, DOI:10.1590/S0103-97332010000100009.
- X. Guo, L. Li, Z. Liu, D. Yu, J. He, R. Liu, B. Xu, Y. Tian and H. T. Wang, Hardness of covalent compounds: Roles of metallic component and d valence electrons, J. Appl. Phys., 2008, 104, 023503, DOI:10.1063/1.2956594.
- M. A. Ali, M. A. Hadi, M. M. Hossain, S. H. Naqib and A. K. M. A. Islam, Theoretical investigation of structural, elastic, and electronic properties of ternary boride MoAlB, Phys. Status Solidi, 2017, 254, 1700010, DOI:10.1002/pssb.201700010.
- M. A. Hadi, S. R. G. Christopoulos, S. H. Naqib, A. Chroneos, M. E. Fitzpatrick and A. K. M. A. Islam, Physical properties and defect processes of M3SnC2 (M = Ti, Zr, Hf) MAX phases: Effect of M-elements, J. Alloys Compd., 2018, 748, 804–813, DOI:10.1016/j.jallcom.2018.03.182.
- J. Chang, G.-P. Zhao, X.-L. Zhou, K. Liu and L.-Y. Lu, Structure and mechanical properties of tantalum mononitride under high pressure: A first-principles study, J. Appl. Phys., 2012, 112, 083519, DOI:10.1063/1.4759279.
- D. Healy, AnisoVis, GitHub, 2021 Search PubMed.
- R. Gaillac, P. Pullumbi and F.-X. Coudert, ELATE: an open-source online application for analysis and visualization of elastic tensors, J. Phys.: Condens. Matter, 2016, 28, 275201, DOI:10.1088/0953-8984/28/27/275201.
- H. M. Ledbetter, Elastic properties of zinc: A compilation and a review, J. Phys. Chem. Ref. Data, 1977, 6, 1181–1203, DOI:10.1063/1.555564.
- A. K. M. A. Islam, A. S. Sikder and F. N. Islam, NbB2: a density functional study, Phys. Lett. A, 2006, 350, 288–292, DOI:10.1016/j.physleta.2005.09.085.
- J. Wang, Y. Zhou, T. Liao and Z. Lin, First-principles prediction of low shear-strain resistance of Al3BC3: A metal borocarbide containing short linear BC2 units, Appl. Phys. Lett., 2006, 89, 021917, DOI:10.1063/1.2220549.
- S. I. Ranganathan and M. Ostoja-Starzewski, Universal Elastic Anisotropy Index, Phys. Rev. Lett., 2008, 101, 055504, DOI:10.1103/PhysRevLett.101.055504.
- A. Otero-De-La-Roza, D. Abbasi-Pérez and V. Luaña, Gibbs2: A new version of the quasi harmonic model code. II. Models for solid-state thermodynamics, features and implementation, Comput. Phys. Commun., 2011, 182, 2232–2248, DOI:10.1016/j.cpc.2011.05.009.
- M. A. Blanco, E. Francisco and V. Luaña, GIBBS: Isothermal-isobaric thermodynamics of solids from energy curves using a quasi-harmonic Debye model, Comput. Phys. Commun., 2004, 158, 57–72, DOI:10.1016/j.comphy.2003.12.001.
- Y. Zhao, S. Deng, H. Liu, J. Zhang, Z. Guo and H. Hou, First-principle investigation of pressure and temperature influence on structural, mechanical and thermodynamic properties of Ti3AC2 (A = Al and Si), Comput. Mater. Sci., 2018, 154, 365–370, DOI:10.1016/j.commatsci.2018.07.007.
- M. W. Qureshi, X. Ma, G. Tang and R. Paudel, Structural stability, electronic, mechanical, phonon, and thermodynamic properties of the M2GaC (M = Zr, Hf) max phase: An ab initio calculation, Materials, 2020, 13, 1–18, DOI:10.3390/ma13225148.
- F. Sultana, M. M. Uddin, M. A. Ali, M. M. Hossain, S. H. Naqib and A. K. M. A. Islam, First principles study of M2InC (M = Zr, Hf and Ta) MAX phases: The effect of M atomic species, Results Phys., 2018, 11, 869–876, DOI:10.1016/j.rinp.2018.10.044.
- G. A. Slack, The Thermal Conductivity of Nonmetallic Crystals, Solid State Phys., 1979, 34, 1–71, DOI:10.1016/S0081-1947(08)60359-8.
- M. E. Fine, L. D. Brown and H. L. Marcus, Elastic constants versus melting temperature in metals, Scr. Metall., 1984, 18, 951–956, DOI:10.1016/0036-9748(84)90267-9.
- M. A. Hadi, N. Kelaidis, S. H. Naqib, A. Chroneos and A. K. M. A. Islam, Mechanical behaviors, lattice thermal conductivity and vibrational properties of a new MAX phase Lu2SnC, J. Phys. Chem. Solids., 2019, 129, 162–171, DOI:10.1016/j.jpcs.2019.01.009.
- Y. Zhou, X. Lu, H. Xiang and Z. Feng, Preparation, mechanical, and thermal properties of a promising thermal barrier material: Y4Al2O9, J. Adv. Ceram., 2015, 4, 94–102, DOI:10.1007/s40145-015-0141-5.
- Y. Zhou, H. Xiang, X. Lu, Z. Feng and Z. Li, Theoretical prediction on mechanical and thermal properties of a promising thermal barrier material: Y4Al2O9, J. Adv. Ceram., 2015, 4, 83–93, DOI:10.1007/s40145-015-0140-6.
- X. Li, H. Cui and R. Zhang, First-principles study of the electronic and optical properties of a new metallic MoAlB, Sci. Rep., 2016, 6, 39790, DOI:10.1038/srep39790.
- X.-H. Li, H.-L. Cui and R.-Z. Zhang, Structural, optical, and thermal properties of MAX-phase Cr2AlB2, Front. Phys., 2018, 13, 136501, DOI:10.1007/s11467-017-0743-1.
- M. A. Ali, M. S. Ali and M. M. Uddin, Structural, elastic, electronic and optical properties of metastable MAX phase Ti5SiC4 compound, Indian J. Pure Appl. Phys., 2016, 54, 386–390 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02345d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Muhammad Waqas Qureshi
*a and
Muhammad Waqas Qureshi


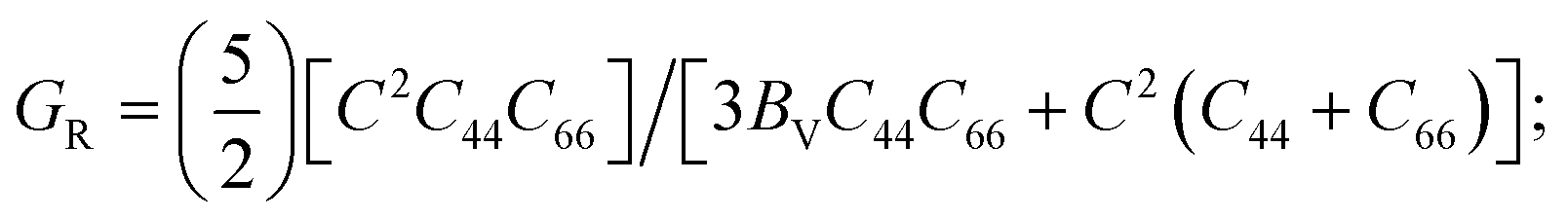 C66 = (C11 − C12)/2.
C66 = (C11 − C12)/2.






 . The unequal values of Ba and Bc [Table 4] indicates the anisotropic nature of Zr2AC (A = Se, S). The linear compressibility (k) along the a and c-axis are calculated by the equation:76
. The unequal values of Ba and Bc [Table 4] indicates the anisotropic nature of Zr2AC (A = Se, S). The linear compressibility (k) along the a and c-axis are calculated by the equation:76